
Abstract
Mobilisation of Damage-Associated Molecular Patterns (DAMPs) determines the immunogenic properties of apoptosis, but the mechanisms that control DAMP exposure are still unclear. Here we describe an unconventional autophagic pathway that inhibits the release of ATP, a critical DAMP in immunogenic apoptosis, from dying cells. Mitochondrial BAK activated by BH3-only molecules interacts with prohibitins and stomatin-1 through its latch domain, indicating the existence of an interactome specifically assembled by unfolded BAK. This complex engages the WD40 domain of the autophagic effector ATG16L1 to induce unconventional autophagy, and the resulting LC3-positive vesicles contain ATP. Functional interference with the pathway increases ATP release during cell death, reduces ATP levels remaining in the apoptotic bodies, and improves phagocyte activation. These results reveal that an unconventional component of the autophagic burst that often accompanies apoptosis sequesters intracellular ATP to prevent its release, thus favouring the immunosilent nature of apoptotic cell death.
Similar content being viewed by others

Introduction
The BCL2 protein family plays critical roles in the intrinsic apoptotic pathway, which has important developmental and homoeostatic functions1,2,3. This route is initiated by the activation of BH3-only proteins, a BCL2-family subgroup including just one of the prototypical BCL2-Homology (BH) domains. Induced BH3-only effectors activate the multidomain effectors BAK and BAX to cause their partial unfolding and multimerization, thus opening high-conductance pores on the outer mitochondrial membrane (OMM) for cytochrome c release and caspase activation4,5,6.
Intrinsic apoptosis in homoeostatic conditions is normally immunosilent, in such a way that specialised phagocytes remove the cellular remains without triggering an immune response7. However, in reaction to certain drugs and/or conditions, apoptosis can be strongly immunogenic, thus sparking an adaptive immune reaction against neoantigens that may be present in the dying cells8. Immunogenic cell death favours long-lasting tumour remission by promoting anti-tumour immunity, and therefore plays a critical role in cancer therapy9,10. Induction of immunogenic cell death requires the release of adjuvant factors by the dying cells that stimulate the pro-inflammatory properties of the phagocytes that eliminate the cellular remains11. Such factors are known as Damage-Associated Molecular Patterns (DAMPs), and include exposure of calreticulin at the surface of the apoptotic cell, release of nuclear HMGB1 and ATP secretion12,13. Secreted ATP acts through purinergic receptors as a chemotactic agent for phagocyte recruitment and as a phagocyte activation factor for pro-inflammatory cytokine secretion14, and plays a central role in immunogenic cell death. The mechanisms that regulate DAMP emission, and how they are connected to the apoptotic machinery, are still unclear, but autophagy is known to participate in both HMGB1 and ATP secretion15,16,17,18,19, with a particularly relevant function described for the latter in anti-cancer immunity induced by chemotherapy16.
Autophagy, a catabolic process that degrades cytoplasmic components through the action of AuTophaGy-related proteins (ATGs)20, is frequently induced during cell death, suggesting that its degradative activity may contribute to the cell’s suicide. However, autophagy does not usually play an execution role, but rather it accompanies apoptosis with both contributory or inhibitory roles previously described21. While the canonical autophagic pathway is well characterised, different ATGs participate in a variety of atypical autophagic activities that are mechanistically unconventional and/or unrelated to the degradative function of autophagy22,23,24,25. These processes are collectively known as unconventional autophagy and serve a variety of purposes, like elimination of invading agents26, detection of lysosomal dysfunction27, protein secretion28,29,30 and trafficking31,32,33, inflammatory control34,35 or receptor signalling36. Notably, some of these activities involve lipidation of the autophagic marker LC3 in single-membrane vesicles unrelated to canonical double-membrane autophagosomes26,31,32,33,37. This atypical LC3 behaviour requires the C-terminal domain of the critical effector ATG16L1, a region of the molecule that includes seven WD40-type repeats (WD40 domain, WDD) and is unnecessary for canonical autophagy31,37,38,39. Given this functional plurality, detection of a certain autophagic activity may reflect a variety of underlying mechanisms and physiological roles. The exact nature and functional implications of the autophagic burst that often accompanies apoptosis are issues that remain to be fully elucidated.
Here we show that mitochondrial BAK activated by BH3-only molecules induces an unconventional autophagic response that suppresses the release of ATP during apoptotic cell death by sequestering it into LC3-positive vesicles, thus limiting the immunogenic potential of the dying cells. We provide evidence of a mitochondrial complex assembled by activated BAK to induce this activity.
Results
BAK mediates autophagy activated during apoptotic cell death
To study how autophagy is modulated during apoptosis we retrovirally expressed BH3-only molecules in mouse embryonic fibroblasts (MEFs) as a way to dissect the molecular consequences of exciting a single pathway and avoid the noise caused by chemicals that often stimulate a variety of stress signalling routes. We first transduced different BH3-only members into wild-type (WT) MEFs in the presence of the caspase inhibitor zVAD.fmk to delay cell death. Out of the eight BH3-only molecules tested, only BIM, tBID and PUMA induced robust accumulation of LC3-II, a common autophagic reporter (Supplementary Fig. 1a). Expression of the positive effectors did not cause LC3 activation in Bak/Bax-double knock-out MEFs (DKO; Fig. 1a, b), indicating that the autophagic response requires the presence of multidomain pro-apoptotic proteins. Surprisingly, BIM, tBID and PUMA induced more robust LC3 lipidation (Fig. 1a) and accumulation of LC3-positive vesicles (mainly in perinuclear clusters; Fig. 1b for GFP-LC3 and Supplementary Fig. 1b for endogenous LC3) in Bax-/- MEFs compared to their Bak-/- counterparts, arguing that BAK plays a more predominant role in this system. Similar results were obtained using the pro-apoptotic chemicals staurosporine (STS; Supplementary Fig. 1c, d) or mitoxantrone (MTX; Supplementary Fig. 1e, f). Notably, neither DKO nor BAK-deficient MEFs showed alterations in basal (Supplementary Fig. 2a, b) or canonical (Supplementary Fig. 2c, d) autophagy, indicating that their inability to induce autophagy in response to apoptotic inducers is not due to a defective autophagic machinery.

a The indicated strains of MEFs were transduced with retroviruses expressing the shown BH3-only molecules and treated with 25 μM zVAD.fmk 7 h later. Cells were lysed 22 h post-transduction for Western blot against the indicated molecules. Expression levels of HA tagged tBID, BIM and PUMA were measured in DKO cells to minimise the possible impact of cell death (right panel). b MEFs expressing GFP-LC3 were retrovirally transduced as in (a) and fixed 22 h later for microscopy. Shown are representative confocal pictures (left panel; green channel: GFP-LC3, blue channel: DAPI; scale bars represent 10 μm), and quantification of the number of GFP-LC3 puncta per cell (right panel, top) and the percentage of cells showing induced GFP-LC3 (right panel, bottom). Graph bars indicate control vector (pink), tBID (red), BIM (blue) or PUMA (green) retroviral transduction. Graphs represent mean values −/+ s.d. of triplicate scoring points (n = 3 microscopy fields, each including at least 25 cells per field; **P < 0.01, ***P < 0.001, ****P < 0.0001 two-tailed Student’s t-test). Numeric P-values are shown. c Bax-/- MEFs were retrovirally transduced as in (a) and treated with E64d and pepstatin (10 μg/ml each) for the last 16 h of culture. Cells were lysed 22 h post-transduction for Western blot using the indicated antibodies. d BAK expression in reconstituted DKO MEFs. The indicated MEF strains along with DKO MEFs retrovirally transduced with WT or L78E human BAK versions and subsequently selected in puromycin were lysed for Western blotting. e The reconstituted MEFs shown in (d) were retrovirally transduced as in (a) and lysed 22 h later for Western blot against the shown molecules. Source data are provided as a Source Data file.
The extent of cytochrome c mobilisation in response to BIM and tBID was comparable in Bak-/- and Bax-/- cells (Supplementary Fig. 3a). In addition, although there is a tight correlation between the ability of the BH3-only inducers to cause cytochrome c release and LC3 lipidation (Supplementary Fig. 3b), simultaneous expression of BAD and NOXA promoted strong cytochrome c release but limited LC3 lipidation (Supplementary Fig. 3c). These results show that cytochrome c release and LC3 lipidation can be uncoupled in this system, suggesting that the autophagic response mediated by BAK is unrelated to its ability to induce mitochondrial outer membrane permeabilization (MOMP).
The autophagic route activated by BAK is unconventional
To study the nature of the autophagic response in this context, we focused on Bax-/- MEFs. Previous publications show that absence of caspase activity during apoptosis enables the STING signalling pathway40, and separate studies indicate that this route is able to activate autophagy41,42. However, expression of BIM and tBID caused substantial LC3 lipidation in Bax-/- cells even without zVAD.fmk treatment (Supplementary Fig. 3d), indicating that the autophagic burst in this case is not inhibited by caspases. In addition, while we observed STING phosphorylation caused by BIM and tBID (Supplementary Fig. 3e), STING depletion did not blunt LC3 lipidation induced by these effectors (Supplementary Fig. 3e). These results argue that the STING route is unlikely to mediate the autophagic response in this experimental system.
LC3 lipidation caused by BH3-only inducers in Bax-/- cells reflects an enhanced autophagic flux, as the induced levels of LC3-II were further stimulated by lysosomal inhibition (Fig. 1c) and were accompanied by reduced p62 expression (Fig. 1c). Reconstitution of DKO cells with AU-BAK (Fig. 1d) restored the LC3 lipidation response (Fig. 1e), indicating that BAK is the molecular entity that mediates autophagy in this context. Pathway activation requires the BH3 domains of both BAK and BH3-only proteins, since mutant forms where a critical L in the BH3 motif was changed to E43 were inactive (Fig. 1d, e). Knock-down of the core autophagic effectors ATG7 or ATG16L1 via siRNAs inhibited LC3 lipidation caused by BIM, tBID and PUMA (Supplementary Fig. 4a). However, depletion of upstream initiators of canonical autophagy like VPS34 or ATG14 in Bax-/- cells (Supplementary Fig. 4b), or BECLIN1 in DKO cells restored with AU-BAK (Supplementary Fig. 4c), had no effect. These results suggest that the autophagic pathway induced by these effectors is atypical, since it does not require part of the canonical machinery, and therefore is probably different from a previously described autophagic response triggered by MOMP that requires the conventional autophagic route involving AMPK and ULK144 to promote mitophagy44,45.
Published evidence shows that the ATG16L1 WDD mediates unconventional autophagic activities where LC3 becomes lipidated in intracellular membranes unrelated to canonical autophagosomes31,37,38,39. Bax-/- MEFs depleted of ATG16L1 using CRISPR/Cas9 and reconstituted with ATG16L1 lacking the WDD (Nt; residues 1-299; Fig. 2a, b) showed unaffected basal (Fig. 2c, left panel) and canonical (Fig. 2c, right panel) autophagy, but displayed reduced LC3 activation in response to BH3-only inducers (Fig. 2d, e) or MTX (Fig. 2f, g). Similar results were obtained in Atg16l1-/- MEFs reconstituted with full-length ATG16L1 or just the N-terminal domain (Supplementary Fig. 5a–c), and also in Atg16l1-/- HCT116 colon carcinoma cells expressing full-length ATG16L1 or separate N- and C-terminal domains (split ATG16L1; Supplementary Fig. 5d–f). We previously described that Atg16l1-/- MEFs expressing the Nt domain of ATG16L136 and HCT116 cells harbouring split ATG16L139 cannot activate unconventional autophagy mediated by the WDD (in the latter case because the sensing (WDD) and effector (Nt region) domains are uncoupled), although they are fully able to sustain basal and canonical autophagy36,39. The WDD dependency of the autophagic response induced by BIM and tBID is conserved in the absence of zVAD.fmk (Supplementary Fig. 5g). Together, these results show that a large portion of the autophagic response triggered during apoptosis is unconventional, as it does not require some of the upstream effectors of canonical autophagy (VPS34, ATG14, BECLIN) and is mediated by the WD40 domain of ATG16L1.

a Bax-/- MEFs were transduced with a CRISPR/Cas9 lentivirus expressing the indicated sgRNAs against Atg16l1 (#1 and #2), selected in puromycin and lysed for Western blot. b Cells targeted by sgRNA #2 were transduced with a retrovirus expressing the ATG16L1 Nt region (1-299), and lysed along CRISPR/Cas9 control cells for Western blot. c The engineered Bax-/- MEFs shown in (a, b) were treated with bafilomycin (80 nM; left panel) or rapamycin (2 μM) plus E64d/pepstatin (10 μg/ml each; right panel) for 8 h and lysed for Western blot. d The engineered Bax-/- MEFs shown in (a, b) were retrovirally transduced with the shown BH3-only activators, supplemented with 25 μM zVAD.fmk 7 h later, and lysed 20 h post-transduction for Western blot against the indicated molecules. e The indicated Bax-/- MEFs engineered for ATG16L1 expression were transduced as in (d) and fixed for anti-LC3 immunofluorescence 17 h later. Shown are representative confocal pictures (left; red: LC3, blue: DAPI; scale bars represent 10 μm), and quantification of the number of LC3-positive puncta per cell (right). Data are presented as box-plots where the central line represents the median value, the box shows percentiles 25-75 and the whiskers include the most extreme values (n = 25 cells; ****P < 0.0001 two-tailed Student’s t-test). Numeric P-values are shown. f The indicated Bax-/- MEFs engineered for ATG16L1 expression were treated with 2 μM MTX in the presence of 25 μM zVAD.fmk and lysed 14 h later for Western blot against the shown molecules. g The indicated engineered Bax-/- MEFs expressing Cherry-LC3 were treated as in (f) and processed for microscopy 14 h later. Shown are representative confocal pictures (left; red: Cherry-LC3, blue: DAPI; scale bars represent 10 μm), and quantification of the number of Cherry-LC3-positive puncta per cell (right). Graph bars indicate control (yellow) and MTX (orange) treatment. Data are presented as in (e) (n = 25 cells; ****P < 0.0001 two-tailed Student’s t-test). Numeric P-values are shown. Source data are provided as a Source Data file.
Confocal microscopy studies carried out in Bax-/- MEFs expressing GFP-LC3 and mitochondrial RFP (mRFP) showed that most of the LC3-positive vesicles generated by BH3-only expression or MTX do not contain mRFP (Supplementary Fig. 6a), arguing that the unleashed autophagic reaction does not reflect the induction of mitophagy. Mitophagic events were sporadically and distinctively observed in the same samples as a GFP-LC3-positive signal that grows tightly around, and encloses, the mitochondrial marker mRFP (Supplementary Fig. 6b; less than 1% of the GFP-LC3-positive puncta). In addition, while LC3 lipidation was evident 16 h post-transduction (Supplementary Fig. 6c, top panel), the levels of the mitochondrial marker VDAC1 remained unchanged in Bax-/- cells 24 h after BH3-only transduction (Supplementary Fig. 6c, bottom panel), indicating no significant mitochondrial degradation. Although most GFP-LC3-positive vesicles were isolated in the cytoplasm, some of the GFP-LC3-positive puncta localised in close apposition to the mitochondrial surface to the point of overlapping with mRFP (white signal; insets in Supplementary Fig. 6a).
Closer inspection by anti-GFP immuno-electron (EM) microscopy revealed that the GFP-LC3-positive structures generated by BIM correspond to small vesicles and tubules devoid of discernible cytoplasmic content (Fig. 3a; control images shown in Supplementary Fig. 6d), although some of them appear to contain smaller vesicles that could reflect real contents or topological complexity (Fig. 3a). These vesicles are bound by a single membrane (Fig. 3a, insets), and therefore are different from canonical double-membrane autophagosomes. Interestingly, we found mitochondria directly labelled with a GFP-LC3 signal that was not associated to other LC3-positive membranous structures (Fig. 3a, bottom panels, left), arguing that these are not mitophagic events. Consistent with the confocal data, we detected rare examples of mitophagy (Fig. 3a, bottom panels, middle). Tubular compartments likely representing endoplasmic reticulum (ER) cisternae were sporadically labelled with GFP-LC3 (Fig. 3a, bottom panels, right), suggesting that reticular BAK46 may also be able to transmit autophagic signals. Correlative Light Electron Microscopy (CLEM) studies confirmed that the GFP-LC3-positive structures induced by BIM are small, irregular vesicles and tubules localised to perinuclear clusters and limited by just one membrane (Fig. 3b), with a general appearance that is completely different from canonical autophagosomes (examples of the latter found in the same samples are shown in Supplementary Fig. 6e).

a Anti-GFP immuno-EM of GFP-LC3-positive vesicles. Bax-/- MEFs expressing GFP-LC3 were transduced with BIM, treated 7 h later with 25 μM zVAD.fmk, and processed 17 h post-transduction for anti-GFP immuno-EM microscopy. Panels display four examples of clusters of GFP-LC3-positive, small, single-membrane vesicles and tubules. Two insets are presented for each example. White arrows point to single-membrane structures. The bottom panels show mitochondria labelled with GFP-LC3 (left), mitochondria engulfed by a GFP-LC3-positive autophagosome (mitophagy; middle) and GFP-LC3-positive tubes consistent with ER cisternae (right). Images of control Bax-/- MEFs transduced with empty vector and Bak-/- MEFs transduced with vector or BIM are shown in Supplementary Fig. 6d. b CLEM of the GFP-LC3-positive vesicles. Bax-/- MEFs expressing GFP-LC3 were treated as in (a) and processed for CLEM. Shown are three examples of perinuclear GFP-LC3-positive vesicle clusters present in the same cell at different z positions. The cell perimeter revealed in GFP-overexposed images was used for initial alignment between the optical and EM images (left panels). The top plane (height 1120 nm) includes de nucleus (DAPI), which was used for further alignment (top panels). The middle (360 nm) and bottom (260 nm) planes did not include the nucleus and shared one of the clusters. Peculiar surface cell features were used for finer alignment (black arrows). Random canonical autophagosomes colocalize with GFP-LC3 (white arrows; shown in Supplementary Fig. 6e). An incipient autophagosomal structure appears in the periphery of the top cluster (red asterisk). Yellow arrows point to single-membrane structures. The three examples show that the GFP-LC3-positive clusters are formed by small, irregular vesicles and tubules limited by one membrane.
Immunofluorescence assays showed that the GFP-LC3-positive vesicles induced by BIM do not colocalize with RAB11, RAB5, CD63, EEA1, HSP70 or HSP90 (Supplementary Fig. 7a), marginally overlap with STING (Supplementary Fig. 7a) and tightly colocalize with p62 and ubiquitin (Supplementary Fig. 7a), revealing similarities with previously-described vacuoles induced by dsDNA that become p62 and ubiquitin positive to regulate STING trafficking47,48,49,50. The GFP-LC3-positive vesicles partially overlap with LAMP1, RAB7 and lysotracker (Supplementary Fig. 7a) but not with the unconventional secretory marker RAB8 (Supplementary Fig. 7a), and are undetectable as GFP-LC3-II-positive entities in the supernatant of Bax-/- MEFs transduced with BIM (Supplementary Fig. 7b; only GFP-LC3-I is detected). These results point to a degradative outcome of the vesicles instead of their extracellular release. Consistent with the immunofluorescence data, purified GFP-LC3-positive vesicles show reactivity for p62 and ubiquitin, but not HSP70, HSP90 or STING (Supplementary Fig. 7c).
The atypical route inhibits ATP release during apoptosis
Bak-/- and Bax-/- cells died to a similar extent in response to BH3-only activators (Supplementary Fig. 8a) or MTX (Supplementary Fig. 8b), and BIM, tBID and MTX induced comparable levels of cell death in cells expressing full-length ATG16L1 or just the Nt domain (Supplementary Fig. 8c-e). Therefore, the physiological role of the atypical autophagic response in this setting seems unrelated to the ability to modulate apoptosis that autophagy has in other cell death paradigms21. In search for a possible function, we found that most LC3-positive vesicles generated in WT and Bax-/- MEFs by BH3-only expression were stained by quinacrine (Supplementary Fig. 9a–d), a lysosomotropic dye that emits green fluorescence in the presence of high ATP concentrations13. As expected, the quinacrine-positive signal was completely excluded from mRFP-labelled mitochondria (Supplementary Fig. 9e). A similar double LC3/quinacrine-staining phenotype was observed in response to MTX (Supplementary Fig. 10a–d). The number of quinacrine-positive and LC3/quinacrine-double positive vesicles was reduced in BH3-only and MTX-treated Bak-/- MEFs (see Supplementary Figs. 9a–d and 10a–d) and also in engineered Bax-/- cells lacking the WDD of ATG16L1 (Fig. 4a–d for BH3-only, and Supplementary Fig. 10e–h for MTX). The use of Bodipy-ATP as an alternative method to reveal vesicular ATP recapitulated the quinacrine data in engineered MEFs (Supplementary Fig. 11a–c). Together, these results raise the notion that the LC3-positive vesicles generated by the unconventional BAK-dependent autophagic pathway could be sequestering ATP.

a–d Induction of Cherry-LC3/quinacrine double-positive vesicles in Bax-/- cells expressing full-length ATG16L1 but not in those expressing ATG16L1-ΔWDD. a Bax-/- MEFs engineered for ATG16L1 expression and expressing Cherry-LC3 were transduced with the indicated BH3-only molecules and treated with 25 μM zVAD.fmk 7 h later. Cells were stained with quinacrine 17 h post-transduction and analysed in vivo by confocal microscopy. Shown are representative confocal pictures (Ch-LC3: Cherry-LC3 (magenta); Quin.: quinacrine (green)). Scale bars represent 10 μm. b–d Quantification of the phenotypes shown in (a). Graphs display the number of Cherry-LC3-positive puncta per cell (b), the number of quinacrine-positive puncta per cell (c), and the percentage of Cherry-LC3-positive signal that colocalizes with the quinacrine-positive signal (d). Data are presented as box-plots where the central line represents the median value, the box shows percentiles 25-75 and the whiskers include the most extreme values (n = 25 cells; ****P < 0.0001 two-tailed Student’s t-test). Numeric P-values are shown. e Increased levels of ATP released by apoptotic Bak-/- MEFs. The shown MEF strains were transduced with the indicated BH3-only molecules for 22 h (left), or treated with MTX (2 μM) for 24 h (right), and the levels of extracellular ATP measured at 14 and 22 h (BH3-only molecules) or 16 and 24 h (MTX). Graphs show mean values −/+ s.d. of cumulative data resulting from the sum of the luciferase activity units obtained at both time points from triplicate experimental points (n = 3 biological replicas; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 two-tailed Student’s t-test). Numeric P-values are shown. f Increased levels of ATP released by apoptotic Bak-/- MEFs expressing ATG16L1-ΔWDD. Bax-/- MEFs engineered for ATG16L1 expression were transduced with the shown BH3-only molecules (left) or treated with MTX (right), and the levels of extracellular ATP measured and displayed as in (e). Source data are provided as a Source Data file.
Previous reports showed that autophagy is required to promote redistribution of intracellular ATP from lysosomes to LC3-positive autophagosomes during immunogenic apoptosis as a prerequisite for ATP secretion19. Unexpectedly, however, we found that Bak-/- cells (where the autophagic pathway is impaired) secrete more ATP during apoptosis compared to WT and Bax-/- cells (where the autophagic pathway is active), both in response to BH3-only expression (Fig. 4e, left) and MTX (Fig. 4e, right), and retain less ATP remaining in the apoptotic bodies than Bax-/- cells (Supplementary Fig. 12a, b). These data argue that the atypical autophagic response induced by BAK might in fact prevent ATP secretion. Consistently, engineered Bax-/- cells lacking the WDD showed increased ATP liberation in response to BH3-only molecules (Fig. 4f, left) and MTX (Fig. 4f, right), and reduced ATP levels remaining in the apoptotic bodies (Supplementary Fig. 12c, d), compared to their control counterparts. Similar observations were made in Atg16l1-/- MEFs reconstituted with full-length ATG16L1 or the Nt domain (Supplementary Fig. 12e, f for BH3-only molecules, and 12 g, h for MTX), and also in ATG16l1-/- HCT116 cells harbouring full-length ATG16L1 or separated Nt and Ct domains (Supplementary Fig. 12i for BH3-only molecules, and 12j for MTX). Therefore, data obtained in different experimental systems reveal the existence of an unconventional autophagic route that emanates from activated BAK and prevents ATP secretion during apoptosis, perhaps by sequestering it into LC3-positive vesicles.
Treatment with clodronate (CLD) or DIDS, two chemically-unrelated inhibitors of the vesicular nucleotide transporter VNUT (which pumps ATP into vesicular compartments)51,52, reduced the intensity of quinacrine staining exhibited by the Cherry-LC3-positive vesicles (Supplementary Fig. 13a-d), suggesting that an active ATP transportation activity performed by VNUT is required to accumulate ATP in this compartment. However, we could not detect VNUT expression in these vesicles (Supplementary Fig. 13e, f), pointing to the idea that a different CLD- and DIDS-sensitive transporter is probably involved in ATP delivery in this context. Importantly, treatment with both inhibitors caused increased ATP secretion during BIM-induced apoptosis (Supplementary Fig. 13g), thus functionally linking accumulation of ATP in the LC3-positive vesicles with inhibition of ATP release. None of the inhibitors influenced the course of cell death (Supplementary Fig. 13h).
The latch domain of BAK induces LC3 lipidation
In search for the molecular mechanism that links activated BAK to the induction of unconventional autophagy, we found that a mutated version of BAK lacking the BH3 proapoptotic domain (BAK-ΔBH3) causes LC3 activation in a straight overexpression assay (Supplementary Fig. 14a, b) without significantly inducing cell death compared to wild-type BAK (Supplementary Fig. 14c). In contrast, BAX-ΔBH3 was inactive (see Supplementary Fig. 14a, b). These results suggest that BAK may have an intrinsic autophagic activity that might reflect its ability to trigger unconventional autophagy in response to BH3-only effectors. We reasoned that characterisation of this function could help identify downstream molecular machinery engaged by activated BAK to launch the noncanonical pathway.
In the native structure of BAK, the BH3 domain is buried into a hydrophobic core53, so BAK-ΔBH3 could be partially unfolded. Since this mutant strongly induces autophagy, there is a chance that its tertiary structure is not required for the autophagic function, and therefore it might be possible to identify a region of the primary sequence that retains the activity. To this end, we initially broke down the BAK open reading frame into portions that make structural sense (Fig. 5a). Upon activation, BAK sequentially unfolds, first, to expose its N-terminal region (helix 1) and, second, to separate the core (helices 2-5) and latch (helices 6-8) domains before dimerising/multimerizing to generate OMM pores4,54,55. Since BAK needs the hydrophobic C-terminal region (helix 9) for mitochondrial targeting, we generated constructs expressing these different domains fused to the TM helix for mitochondrial localisation (see Fig. 5a). Overexpression studies indicated that the autophagic activity is exclusively retained by the latch domain (G146-G186; Fig. 5b, c, and scheme in Fig. 5d). Expression of this region did not cause cell death (Supplementary Fig. 14d). Atg16l1-/- HCT116 cells reconstituted with separate N- and C-terminal domains of ATG16L1 did not respond to latch expression with LC3 lipidation (Supplementary Fig. 14e), thus confirming the unconventional nature of the autophagic route involved. The latch domain induced robust LC3 lipidation in HCT116 cells lacking both BAK and BAX (Supplementary Fig. 14f, left and right panels), indicating that its autophagic activity is not channelled through activation of the endogenous pool of these effectors.

a BAK scheme showing relevant amino acid positions for the constructs used in (b). b HEK-293T cells were transfected with AU-BAK-ΔBH3 constructs (Nt: M1-M71; Nt-core-ΔBH3: M1-G146 lacking G72-R88 (BH3); latch: G146-G186; all including the transmembrane (TM) domain: P187-S211) and HA-LC3, and lysed 36 h later for Western blot. Relative densitometric quantifications of LC3-II are shown (bottom). c HeLa cells were transfected with the indicated BAK constructs and GFP-LC3, and processed for microscopy 36 h later. Shown are representative confocal pictures (left; green: GFP-LC3, blue: DAPI; scale bars represent 10 μm), and quantification of the number of GFP-LC3-positive puncta per cell (right). Graph bars indicate control vector (white), BAK-ΔBH3 (light grey) and BAK-latch domain (dark grey). Data are displayed as box-plots where the central line represents the median value, the box shows percentiles 25-75 and the whiskers include the most extreme values (n = 25 cells; n.s. P > 0.05, two-tailed Student’s t-test). The numeric P-value is shown. d Alignment of the BAK and BCLXL latch domains showing functionally conserved (red font) and identical (red boxes) amino acids. Positions of the BH2 domains and α6 and α7 helices of BAK are indicated. e HEK-293T cells were transfected with the shown GFP-BAK/BCLXL latch chimeric constructs (right) and HA-LC3, and lysed 36 h later for Western blotting (left). f Scheme showing wild-type and 5M BAK latch domain versions. Boxed positions in the 5M version indicate BCLXL residues (ND-LEP) that substitute those present in BAK (LH-CIA). g HEK-293T cells were transfected with WT and 5M GFP-BAK-latch constructs and HA-LC3, and lysed 36 h later for Western blot. h HeLa cells were transfected with WT and 5M GST-BAK latch versions and GFP-LC3, and processed for microscopy 36 h later. Shown are representative confocal pictures (left; green: GFP-LC3, blue: DAPI; scale bars represent 10 μm), and quantification of the number of GFP-LC3-positive puncta per cell (right). Graph bars indicate control vector (white), BAK-latch-WT (light grey) and BAK-latch-5M (dark grey). Data displayed as in (c) (n = 25 cells; ****P < 0.0001 two-tailed Student’s t-test). Source data are provided as a Source Data file.
Systematic N- and C-terminal deletion studies showed that the minimal latch version that retains the activity lies between amino acids G146-Q173 (α6-α7) (Supplementary Fig. 14g, top and bottom panels, and scheme in Supplementary Fig. 14h). An equivalent region of the multidomain anti-apoptotic homologue BCLXL (E158-E184; Supplementary Fig. 14h) was inactive even when fused to the TM domain of BAK (Supplementary Fig. 14i, left panel), as it was the full-length BCLXL latch domain (E158-N197; Supplementary Fig. 14i, right panel; scheme in Fig. 5d). Studies using chimeric constructs between the minimal BAK and BCLXL latch fragments pointed to a region of the former that is critical for the activity (residues L163-Q173; Fig. 5e, left and right panels). This region overlaps with the BH2 domain of BAK that is highly homologous between BCL2 family members, so we hypothesised that the most relevant residues might be those excluded from the BH2 sequence (L163-A168; see schemes in Fig. 5d, f). Consistently, changing these residues to those present in BCLXL (BAK-latch-5M; Fig. 5f) impaired the autophagic activity of both the full-length BAK latch domain (Fig. 5g, h) and BAK-ΔBH3 (Supplementary Fig. 14j, k).
A complex including PHBs and STOM binds the WD40 domain
To identify the proximal signalling machinery engaged by the latch domain to induce unconventional autophagy, fusion constructs between GST and wild-type or mutant (5M) latch versions were transfected into HEK-293T cells and purified to identify co-precipitating proteins by mass spectrometry. Out of a total of 661 proteins identified (Supplementary Data 1), 239 were preferentially present in the wild-type sample at least by one more identified peptide compared to the 5M version (Supplementary Data 2). We further selected a short list of 35 candidates that fulfilled the arbitrary threshold of being overrepresented by more than 4 peptides in the wild-type sample (highlighted candidates; Supplementary Data 2 file). Members of the importin/exportin family of transporters across the nuclear membrane were present in this short list (Supplementary Fig. 15a), but co-immunoprecipitation studies between GST-latch and the highest-ranked family candidate (EXPORTIN-4) revealed no signs of interaction (Supplementary Fig. 15b). Intriguingly, the short list of candidates also included 4 members of the prohibitin and stomatin family: prohibitins 1 and 2 (PHB1, PHB2), stomatin (STOM) and stomatin-like protein 2 (SLP2)56, as well as the mitochondrial porin VDAC1 (Supplementary Fig. 15a and Fig. 6a). PHB1 and PHB2 are multifunctional, heterodimeric proteins located in the mitochondrial intermembrane space and critical for the mitochondrial metabolism and function57. STOM is a membrane-associated protein located in lipid rafts that regulates channel activity56, and SLP2 interacts with PHBs and controls the stability of respiratory complexes and mitochondrial fusion56. VDAC1 was also selected because the family member VDAC2 was previously described as a BAK interactor58.

a Selected list of prohibitin-family proteins and VDAC1 identified as potential latch interactors. The number of peptides identified in the WT and 5M proteomics samples, and their difference between both samples (DIFF.), are indicated. b Co-immunoprecipitation assay showing interaction between activated GST-BAK and PHB2/STOM in response to BIM. DKO MEFs depleted of CASP3 and CASP9 and expressing GST-BAK (and also human PHBs and STOM for increased detectability) were retrovirally transduced with HA-BIM and, 15 h later, crosslinked with 1% paraformaldehyde and lysed. Lysates were incubated with control or GSH-agarose beads and the resulting precipitates subjected to Western blot (left panel, IPs). Expression levels of all contenders in total protein lysates are shown on the right panel (TLs). c PHBs mediate the autophagic response induced by BIM (left panel) and tBID (right panel). Bax-/- MEFs were transduced with the shown CRISPR/Cas9 constructs and, 40 h later, with the indicated BH3-only molecules. Cells were treated with 25 μM zVAD.fmk 7 h later and lysed 20 h post-transduction for Western blot. Relative densitometric quantifications of LC3-II are shown at the bottom. d PHBs inhibit ATP secretion during BH3-only-induced apoptosis. Bax-/- MEFs were transduced with the shown CRISPR/Cas9 constructs and, subsequently, with the indicated BH3-only molecules (as in c) for 22 h, and the levels of extracellular ATP were measured at 14 and 22 h. Graph shows mean values −/+ s.d. of cumulative data resulting from the sum of the luciferase activity units obtained at both time points from triplicate experimental points (n = 3 biological replicas; *P < 0.05, ***P < 0.001, two-tailed Student’s t-test). Numeric P-values are shown. Control points were lysed 48 h post-CRISPR/Cas9 transduction for Western blot to determine PHB2 depletion (right panel). Source data are provided as a Source Data file.
Co-precipitation studies confirmed the specific interaction between the BAK latch domain and PHB1/2, STOM and VDAC1, both in a co-transfection modality (Supplementary Fig. 15c, top, middle and bottom panels, respectively) and also after expression of the GST-latch constructs and detection of co-precipitating endogenous proteins (Supplementary Fig. 15d, top, middle and bottom). However, SLP2 did not bind the latch domain in any of the experimental configurations (co-transfected, Supplementary Fig. 16a; endogenous, Supplementary Fig. 16b). CRISPR/Cas9-mediated depletion of PHB2 or STOM (Supplementary Fig. 16c, d), but not that of VDAC1 or SLP2 (Supplementary Fig. 16e, f), inhibited LC3 lipidation caused by the latch domain, demonstrating that they mediate the autophagic response in this context. In light of these results, VDAC1 and SLP2 were excluded from further studies.
Both PHB1/2 and STOM specifically co-precipitated with the latch domain after co-expression (Supplementary Fig. 17a, left and right panels), arguing that the three contenders could assemble into a complex nucleated by the latter. To establish the hierarchy of such possible complex we performed co-precipitation studies between GST-latch and endogenous PHBs and STOM in cells depleted for the different candidates using CRISPR/Cas9. Depletion of STOM did not alter the ability of the latch domain to bind endogenous PHBs (Supplementary Fig. 17b), but reduction of PHB levels impaired co-precipitation between GST-latch and endogenous STOM (Supplementary Fig. 17c). These results indicate that the interaction between STOM and the latch domain is probably bridged by PHBs. Consistently, PHB1/2 were found in STOM-GST precipitates (Supplementary Fig. 17d), arguing that these proteins can bind each other. The interaction between PHBs and the latch region is probably direct, since a pre-purified wild-type version of the latch domain fused to GST was able to specifically pull-down both PHBs from a crude cell lysate (Supplementary Fig. 17e).
Co-immunoprecipitation assays carried out in DKO MEFs reconstituted with GST-BAK (Fig. 6b, left and right panels) or untagged BAK (Supplementary Fig. 18a, left and right panels) (and also expressing human PHBs and STOM for increased detectability), showed that, under crosslinking conditions to stabilise the interactions, BAK activated by BIM coprecipitates with PHB2 and STOM. These results suggest that, upon activation by BH3-only effectors, BAK unfolds to expose the latch domain for binding to PHBs/STOM.
CRISPR/Cas9-mediated depletion of PHBs in Bax-/- MEFs inhibited LC3 lipidation induced by BH3-only molecules (Fig. 6c), indicating a critical role in the pathway. Importantly, such inhibition occurred without impairment of cytochrome c release or STING phosphorylation (Supplementary Fig. 18b), thus dissociating again the autophagic response from MOMP. Reduced PHB expression also caused increased ATP secretion in both BAX-deficient (Fig. 6d) and wild-type MEFs (Supplementary Fig. 18c) treated with the same activators. Together, these results confirm that the unconventional autophagic activity induced during apoptosis to inhibit ATP release is mediated by PHBs. Therefore, as observed in the overexpression experiments done with the isolated latch domain (see Supplementary Figs. 15c, d and 16c), BAK activated by BH3-only inducers also physically and functionally interacts with PHBs to induce unconventional autophagy, arguing that the functional properties of the isolated latch domain faithfully recapitulate the activity of native BAK in this autophagic pathway.
To try to dissociate the autophagic activity of BAK from its ability to induce MOMP, we generated a BAK version including the 5M mutation. This mutant was expressed at lower levels compared to wild-type BAK in DKO MEFs (Supplementary Fig. 19a, left), but even after repeated transduction steps to increase its expression this mutant was unable to trigger cell death in response to BIM (Supplementary Fig. 19a, right). These results indicate that the 5M mutation is probably too aggressive to preserve the MOMP activity of BAK. However, we identified a point mutant in the same region of BAK (L163A, see Fig. 5d) that impairs the ability of the isolated latch domain to both bind PHBs (Supplementary Fig. 19b) and cause LC3 lipidation in overexpression assays (Supplementary Fig. 19c). Notably, BAK including this mutation, and repeatedly transduced into DKO MEFs (Supplementary Fig. 19d, left), responds to BIM with robust cytochrome c mobilisation (Supplementary Fig. 19d, right), but promotes less LC3 lipidation compared to wild-type BAK (Supplementary Fig. 19d, right). These results underscore the idea that the molecular determinants required by BAK to induce autophagy and MOMP are different, as both activities can be uncoupled by the L163A mutation. In addition, these data link the autophagic activity of the isolated latch domain with its capacity to induce the pathway in the context of full-length BAK.
We previously published a proteomics study directed to identify proteins that may interact with the WD40 domain of ATG16L159. Notably, PHB1 and STOM were included in the candidate list (Supplementary Fig. 20a and Supplementary Data 3), suggesting that both proteins could be WDD interactors. Consistently, PHB1/2 (Supplementary Fig. 20b) and STOM (Supplementary Fig. 20c) bind GST-ATG16L1-320-607 (WDD) in co-immunoprecipitation studies. Since the autophagic activity of the latch domain requires a functional WDD (see Supplementary Fig. 14e), we hypothesised that the PHBs/STOM complex could help link the latch domain to ATG16L1 through the WDD. Consistently, while the latch region is by itself unable to interact with ATG16L1 (Supplementary Fig. 20d, left and right panels), the presence of PHBs and STOM facilitated detection of a co-expressed GFP-latch fusion protein in GST-ATG16L1-(320-607) precipitates (Supplementary Fig. 20e, left and right panels), suggesting that the PHB/STOM complex engaged by the BAK latch domain may help recruit ATG16L1 to mitochondrial membrane sites. In agreement with this notion, GFP-ATG16L1 is recruited to the mitochondrial surface in Bax-/- MEFs transduced with BIM or tBID (Supplementary Fig. 21a, b). Together, these data argue that BAK, once activated by BH3-only molecules, assembles a complex including PHBs and STOM that could interact with ATG16L1 through the WDD to facilitate the induction of unconventional autophagy.
The atypical autophagic route inhibits IL-1β release by BMDMs
Previous evidence shows that ATP released during cell death promotes activation and differentiation of the phagocytes that remove the cellular remains60. These results suggest that the unconventional autophagic route stimulated by BAK might inhibit phagocyte activation by suppressing ATP mobilisation. Consistently, we found that murine bone marrow-derived macrophages (BMDMs) pre-stimulated with LPS secrete increased amounts of IL-1β when they are co-incubated with engineered BAX-deficient apoptotic cells lacking the WDD (Fig. 7a, b), whereas the supernatant directly harvested from dying MEFs provided no IL-1β signal (Supplementary Fig. 22a). Similar observations were made using reconstituted Atg16l1-/- MEFs expressing full-length ATG16L1 or just the Nt domain (Supplementary Fig. 22b), and also in ATG16l1-/- HCT116 cells harbouring full-length or split ATG16L1 (Supplementary Fig. 22c), although the effect in the latter case was more discrete, perhaps because cells expressing FL ATG16L1 already responded quite strongly. Treatment with two different inhibitors of the purinergic P2X7 receptors (KN-62 and JNJ-47965567) blunted IL-1β liberation caused by Bax-/- cells expressing ATG16L1-ΔWDD (Fig. 7c), thus confirming that ATP is the molecular mediator that induces IL-1β secretion in this context. However, the supernatant of dying MEFs was insufficient to induce IL-1β secretion by co-cultured BMDMs (Supplementary Fig. 22d), as the presence of the apoptotic bodies was required for maximal activity (Supplementary Fig. 22d). These results are consistent with previous reports showing that phagocytosis of the apoptotic bodies is required for the released ATP to cause full macrophage activation17.

a, b The indicated Bax-/- MEFs engineered for ATG16L1 expression were transduced with BH3-only proteins (a) or treated with 2 μM MTX (b) and, 7 h later, supplemented with the ecto-ATPase inhibitor ARL67156 (3 μM). Apoptotic cells were collected 20 h (a) or 22 h (b) later and added to BMDMs pre-activated with LPS (100 ng/ml, 4 h). After 6 h, the supernatant was tested for IL-1β (ELISA). Graphs show mean values of IL-1β concentration −/+ s.d. of triplicate experimental points (n = 3 biological replicas; n.s. P > 0.05, *P < 0.05, ***P < 0.001 two-tailed Student’s t-test). Numeric P-values are shown. c P2X7 inhibitors reduce IL-1β secretion by BMDMs. Engineered Bax-/- MEFs expressing ATG16L1-Nt were treated as in (a) and the apoptotic bodies added to BMDMs pre-incubated for 45 min with control medium (condition 1) or the P2X7 channel inhibitors KN-62 (condition 2) and JNJ (condition 3), as indicated. After 6 h, supernatants were tested for IL-1β. Graphs show data as in (a) (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 two-tailed Student’s t-test). Numeric P-values are shown. d Secretion of mature IL-1β from BMDMs cultured with dying MEFs expressing ATG16L1-Nt (top) or HCT116 cells harbouring split ATG16L1 (bottom). BMDMs were incubated as in (a) with apoptotic bodies of the indicated cells transduced with tBID. Supernatants were collected 6 h later for anti-IL1β immunoprecipitation plus anti-IL-1β Western blot (top, MEFs) or directly subjected to anti-IL-1β Western blot (bottom; HCT116 cells). ATP-treated BMDMs (5 mM, 30 min) provide a positive control. Crossreactive bands of the immunoprecipitating antibody (Ig) confirm equal loading (top). e Increased caspase-1 (Casp-1) cleavage in BMDMs cultured with dying MEFs expressing ATG16L1-Nt. BMDMs were treated as in (d) and lysed for Western blot. f Gasdermin D inhibitor DMF reduces IL-1β secretion by BMDMs. Bax-/- MEFs expressing ATG16L1-Nt were treated as in (c) and the apoptotic bodies tested for IL-1β secretion by BMDMs in the absence or presence of DMF. Data are displayed as in (c). Green bars represent ATP treatment. Source data are provided as a Source Data file.
The pool of IL-1β secreted in these conditions includes the active 17 kDa fragment that results from caspase-1-dependent processing (Fig. 7d; top and bottom panels), and we detected increased caspase-1 activation (cleaved p20 form) in BMDMs incubated with apoptotic bodies expressing ATG16L1-ΔWDD (Fig. 7e). Both results are consistent with a scenario where ATP secreted by the dying MEFs lacking the WDD causes enhanced inflammasome activation in the co-cultured BMDMs. Previous publications showed that proteolyzed IL-1β is released by gasdermin D pores enabled by the inflammasome to induce pyroptotic cell death61. Treatment with the gasdermin D inhibitor dimethyl fumarate (DMF)62 inhibited IL-1β release in response to dying Bax-/- cells lacking the WDD (Fig. 7f), but we did not detect macrophage cell death at the end of the co-incubation period (Supplementary Fig. 22e). These data suggest that IL-1β is secreted through a gasdermin D-dependent route in the absence of pyroptotic death, a possibility that has previously been described63. Considered together, these results indicate that the unconventional autophagic activity that emanates from BAK truly represses the immunogenic potential of cell death, and underscore the notion that the secreted ATP fraction, and not the pool that remains sequestered inside the cellular remains, is the one that has immunogenic properties.
Discussion
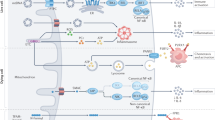
Our results reveal a signalling route that links the intrinsic apoptotic machinery to the induction of unconventional autophagy (Fig. 8). This pathway is initiated by the multidomain pro-apoptotic effector BAK once it is activated by BH3-only molecules. The autophagic activity appears unrelated to the induction of MOMP and cell death, since both activities can be uncoupled in a variety of experimental paradigms. Thus, Bak-/- and Bax-/- MEFs release cytochrome c and die to a similar extent in response to the BH3-only inducers while showing dissimilar autophagic responses, simultaneous expression of BAD and NOXA causes robust cytochrome c release but limited LC3 lipidation, and BAK-L163A induces MOMP but not the autophagic pathway.

BH3-only activators like BIM or BID engage mitochondrial BAK and trigger its unfolding at the mitochondrial surface. This initial activation step exposes the latch domain (orange), which likely crosses the OMM to bind PHBs and STOM. The resulting protein complex recruits ATG16L1 through interaction between PHBs/STOM and the ATG16L1 WD40 domain to induce formation of single-membrane, unconventional, LC3-positive vesicles. The origin of these vesicles and the detailed mechanisms of their formation remain unclear. The LC3-labelled vesicles sequester ATP and prevent ATP release during the apoptotic process, thus inhibiting the ability of the dying cells to stimulate the innate immune pathways of the recruited phagocytes.
Notably, BAK induces this route by binding the multifunctional mitochondrial effectors PHB1/2 and STOM, revealing the existence of a previously unappreciated interactome specifically assembled by unfolded BAK to trigger alternative activities not directly linked to MOMP. Since PHBs are located at the inner mitochondrial membrane facing the intermembrane space57, our data may have implications regarding the topology of activated, unfolded BAK at the surface of the OMM. One view suggests that the latch domain remains flat on the OMM, whereas an alternative theory argues that it penetrates into the OMM to favour pore formation4,6,64,65. Given that the latch domain likely needs to cross the OMM to bind both PHBs, our results seem to favour the second model. It is also conceivable that PHBs may change their location during apoptosis to become more accessible to the latch domain and STOM. While STOM has been described as a major lipid raft component66, it has also been found in mitochondria-associated structures during viral infection67, so a mitochondrial localisation of a STOM fraction for binding to the BAK latch/PHB complex is plausible.
Our data sustain the idea that the molecular assembly formed by BAK, PHBs and STOM interacts with the WDD of ATG16L1 to facilitate formation of single-membrane LC3-positive vesicles that are phenotypically very different from canonical autophagosomes. However, the exact origin of these vesicles (for instance, whether they are formed de novo or by fragmentation of an existing organelle) is unclear. Mitochondria have been shown to act as membrane donors for the generation of autophagic vesicles68, but we did not detect vesicular structures emanating from the mitochondrial surface in our EM studies. Mitochondria are also known to undergo fragmentation during apoptosis69, but the GFP-LC3-positive vesicles detected in our assays were devoid of the mitochondrial marker mRFP, thus arguing against a direct mitochondrial origin. Interestingly, PHB2 is a receptor for selective autophagy of the inner mitochondrial membrane once the OMM has been damaged70. This phenomenon may explain the direct labelling of apoptotic mitochondria with GFP-LC3 that we detected in both confocal and immuno-EM assays. However, these incipient events did not progress to obvious mitophagy to a significant extent in our experimental system, so their relevance in the pathway is unclear. While our results suggest that direct recruitment of ATG16L1 to the BAK/PHB/STOM complex may help generate the LC3-positive vesicles, it is possible that a diffusible element released by porated mitochondria could also play a role. The detected vesicles share some features with those induced in response to dsDNA, as they strongly colocalize with the adaptor protein p62 and ubiquitin chains. However, they only partially colocalize with STING and are not generated in a STING-dependent manner. These observations point to the notion that the vesicles we describe here are different from those previously identified, although they might share similar ubiquitin and p62-mediated regulatory mechanisms that control their function.
Previous publications show that MOMP, and the consequent decreased levels of ATP, activate autophagy through a canonical AMPK and ULK1-dependent pathway that degrades mitochondria44,45. However, BAK- and WDD-induced autophagy is probably unrelated to this activity, since it does not require upstream inducers of the canonical route (ATG14, VPS34, BECLIN) and it does not result in substantial mitophagy. In addition, previous results show that MOMP causes the release of mitochondrial DNA during apoptosis40,71,72, which in turn activates the STING pathway provided that caspase activity is blunted40. Separate studies indicate that STING stimulation induces autophagy, although the nature of this response is unclear, with canonical41 and unconventional42 components suggested. Our results showing that BAK-induced autophagy occurs without the need of caspase inhibition, and also that STING depletion does not inhibit LC3 lipidation, appear to exclude a relevant role of STING in the pathway. Together, these considerations argue that the route we describe here is different from previously identified mechanisms.
We found that the autophagic pathway activated by BAK inhibits ATP release during apoptosis (Fig. 8), thus repressing immunogenic cell death. Mechanistically, our results favour a model where ATP is sequestered inside single-membrane LC3-positive vesicles to prevent its secretion during the course of apoptosis. This scenario is supported by data showing that inhibition of ATP sequestration by CLD or DIDS increases ATP secretion by dying cells. ATP likely accumulates in these vesicles through the action of an ATP transporter mechanistically related to, though different from, VNUT, since this protein is not present in the ATP-rich, LC3-positive vesicles. It is also possible that ATP-binding proteins present in these vesicles may help retain ATP.
Different autophagic activities are known to occur during apoptotic death to, for example, sustain ATP levels for execution of cell death73, induce HMGB1 secretion15 or promote mitophagy44,74. Therefore, the unconventional autophagic process that we describe here may be induced along with other autophagic responses during physiological cell death, supporting a scenario where phenotypically-alike but functionally-different LC3-positive vesicles originated in various subcellular locations, containing different cargoes and performing different functions probably coexist. How these different mechanisms may overlap and interact with each other is an interesting question that remains to be investigated.
More generally, our results point to the idea that cell death immunogenicity is the default outcome of the apoptotic programme that is actively repressed by the same machinery that promotes the cell’s suicide in order to favour an immunosilent process. This view implies that inhibition of unconventional autophagy may boost the immunogenic potential of cell death, an attractive property that could be used with therapeutic purposes as an anti-cancer strategy. In fact, since the autophagic route that regulates this activity has a number of atypical features, it could conceivably be inhibited without altering canonical autophagy, thus avoiding any unwanted side effects that may be associated to impairment of this important housekeeping process.
Methods
Cell lines and reagents
HEK-293T and HeLa (S3) cells were purchased from the ATCC (Refs. CRL-3216 and CCL-2.2, respectively). Details regarding the origin of WT, Bax-/-, Bak-/- and DKO (Bax-/-, Bak-/-) SV40-transformed MEFs, Atg16l1-/- MEFs and ATG16L1-/- HCT116 cells are provided in Supplementary Methods. Cells were cultured at 37 °C and a humidified 5% CO2 atmosphere, in DMEM (Invitrogen, 52100-047) containing 10% heat-inactivated FBS (Invitrogen, 10437028) and 100 U/ml of penicillin/streptomycin (Invitrogen, 15140130). Other chemicals are described in Supplementary Methods.
DNA constructs, transfections and retroviral transductions
Constructs were expressed from the mammalian expression vector Peak12 or its retroviral derivative Peak12-MMP. To express BAK in DKO MEFs, wild-type human BAK and mutants were cloned into Peak12-MMP versions containing IRES-hygromycin or IRES-puromycin cassettes for selection in 200 μg/ml hygromycin B (Invitrogen, 10687010) or 1 μg/ml puromycin (Sigma, P8833). Detailed information about cDNA sources and DNA constructs is shown in Supplementary Methods. Transfections were done using jetPEI (Polyplus, PPLU10-40). Retroviral transductions were done using virus-containing supernatants generated by co-transfecting HEK-293T cells with the relevant P12-MMP constructs and helper plasmids expressing gag-pol (pMD.gag-pol) and env (VSV-G; pMD-G). Supernatants were filtered against 0.45 μM, diluted with fresh medium (1:1), supplemented with polybrene (8 μg/ml) and the resulting mix spun onto the target cells (1 h, 2000 rpm, 32 °C). ZVAD.fmk was added 5-7 h post-transduction.
CRISPR/Cas9 and siRNAs
For protein depletion using the CRISPR/Cas9 system we used the lentiCRISPRv2 vector (puromycin selection; Dr. Feng Zhang, Addgene 52961), and its blasticidin or hygromycin derivatives (Dr. Brett Stringer, Addgene 98293 and 98291, respectively). Guides were selected using the BreakingCas server (Centro Nacional de Biotecnologia, Madrid, Spain; https://bioinfogp.cnb.csic.es/tools/breakingcas/; Supplementary Methods, Oligonucleotide sequences section). Lentiviral constructs containing the guides were co-transfected with helper plasmids psPAX2 (Addgene, 12260) and pCMV-VSV-G (Addgene, 8454) into HEK-293T cells. Viral supernatants were used for infection as described above for retroviral transductions. Cells were selected for 5-7 days (puromycin 1 μg/ml; hygromycin 200 μg/ml; blasticidin (Invivogen, ant-bl-1) 2 μg/ml (MEFs), 4 μg/ml (HEK-293T)). Cells were used for the experiments as polyclonal populations to avoid the risk of phenotypic bias resulting from the selection of random clones. Specific methods for ATG16L1 and PHB depletions are provided in Supplementary Methods.
siRNAs against murine VPS34, ATG14, ATG7, BECLIN and ATG16L1 were mixtures of four siRNAs targeting the same molecule of the ON-TARGETplus SMART pools collection (Dharmacon, L-063416-00, L-172696-00, L-049953-00, L-055895-00, L-051669-01, respectively). siRNAs were transfected at a final concentration of 0.5 μM using DharmaFECT1 reagent (Dharmacon, T-2001-02), according to the manufacturer’s instructions. Control siRNAs were a mix of duplexes with no perfect match with any human or mouse gene (Dharmacon, D-001810-10).
Cell lysis, Western blot and immunoprecipitation
To obtain total lysates for Western blot, cells were collected, washed in PBS, resuspended in 2x SDS sample buffer (SB) lacking β-mercaptoethanol and bromophenol blue (100 mM Tris-HCl pH 6.8, 4% SDS and 20% de glycerol) supplemented with PMSF (1 mM; Merck, 10837091001) and a Protease Inhibitor Cocktail (PIC, 1/100, P8340, Sigma), boiled for 15 min and centrifuged (16100 × g, 5 min, 4 °C) to remove debris. Protein concentrations were measured using the DC Protein Assay Kit (Lowry method, BioRad, 5000116). Equal amounts of protein were volume normalised with SB, diluted 1:1 with 2x SDS reducing sample buffer (2x RSB; SB containing β-mercaptoethanol (10%) and bromophenol blue (0,3 %)), boiled and subjected to SDS-PAGE and Western blot.
For co-immunoprecipitation assays carried out in transfected HEK-293T cells, cells were harvested, washed and lysed for 30 min (in ice, occasional vortexing) in a buffer containing 1% Igepal CA-630 detergent (Sigma, I3021), 50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, PIC (1/100), PMSF (1 mM), orthovanadate (200 nM; New England Biolabs, P0758S) and β-glycerophosphate (25 mM; Sigma, G9422). After a 10 min centrifugation step (16200 × g, 4 °C), lysates were diluted to a final detergent concentration of 0.2% to preserve protein-protein interactions, precleared with agarose beads (Thermo Fisher, 26150; 30 min, 4 °C, rotation) and isolated using agarose-GSH (GE-Healthcare, 17-0756-01) for 2 h (4 °C, rotation). Samples were washed 2–3 times in the same immunoprecipitation buffer, resuspended in 2x RSB, and boiled for 10 min. A portion of the initial lysates was saved as control total lysates whose concentrations were measured using the DC Protein Assay Kit.
Co-immunoprecipitations in DKO MEFs reconstituted with GST-BAK or BAK were carried out in a derivative DKO cell line where both caspase-3 and caspase-9 were depleted using lentiviral CRISPR/Cas9 (as above). These assays were challenging due to high nonspecific signal provided by PHBs and STOM. To solve this difficulty, cells were first crosslinked in PBS containing 1% paraformaldehyde (5 min, RT) followed by quenching to block free aldehyde groups in Tris HCl pH 7.4, 150 mM NaCl (10 min, RT). Cells were lysed in the same 1% Igepal-based buffer described above and centrifuged to remove nuclei. Lysates were precleared (30 min, 4 °C, rotation) with agarose beads (for GST-BAK) or magnetic beads (Dynabeads, Thermo Fisher, 11201D) coupled to a control mouse IgG1 mouse (Thermo Fisher, 14-4714-82; for untagged BAK). Immunoprecipitations were done for 4 h (4 °C, rotation) with agarose-GSH (cells expressing GST-BAK) or magnetic beads coupled to an antibody that recognises the Nt region of BAK only when activated54 (mouse mAb G317.2, BD Biosciences, 556382, cells expressing untagged BAK). GST-BAK precipitates were washed 6 times (5 min, 4 °C, rotation) in the same 1% Igepal-based buffer used for co-precipitations in HEK-293T cells. Anti-BAK precipitates were washed 2 times in RIPA (5 min, 4 °C, rotation) and 4 times in 1% Igepal (5 min, 4 °C, rotation). Beads were resuspended in 2x RSB and boiled for 15 min.
Methods to evaluate cytochrome c release by cellular fractionation, to purify GFP-LC3-positive vesicles for Western blot and to conduct pull-down assays are described in Supplementary Methods.
Equal amounts of protein or equal immunoprecipitation volumes were resolved by SDS-PAGE, transferred to a polyvinilidene difluoride membrane (PVDF, Millipore, IPVH00010), and probed with the primary and secondary antibodies shown in Supplementary Methods. Blots were developed by chemiluminescence (ECL, Amersham, RPN2134). In case of a molecular weight conflict with a previous probing, membranes were stripped in a 7 M guanidinium hydrochloride solution (10 min, RT), or samples run in parallel on a different gel using the same protein concentration measurements. Signal quantifications were carried out using ImageJ. Uncropped images of all blots are included in the Source Data file.
Immunofluorescence and confocal microscopy
Cells were seeded on poly-L-lysine (Sigma, P4832)-coated coverslips and transfected or transduced the next day. Cells expressing chimeric proteins fused to fluorescent proteins were washed with PBS and fixed in 4% paraformaldehyde (PFA) for 10 min. For anti-LC3 staining, coverslips were washed with PBS, fixed in 4% PFA (10 min) and then methanol (100%, −20 °C, 5 min). Lysotracker staining was done by incubating the cells in growth medium containing 100 nM Lysotracker Red DND-99 (Molecular Probes, L7528, 30 min, 37 °C) followed by overnight fixation in 4% paraformaldehyde. Samples were quenched/permeabilized in PBS containing 100 mM glycine and 0.5% Igepal CA-630 (RT, 30 min), blocked in PBS/3% BSA (RT, 45 min) and stained with the primary and secondary antibodies indicated in Supplementary Methods, for 1 h, RT. Coverslips were incubated with DAPI (1 μg/ml, 5 min) and mounted in Prolong Gold anti-fade medium (Thermo Fisher, P10144). Pictures were taken in a STELLARIS 8 STED confocal microscope (Leica; LAS X 4.2.0.23645 software) using the 488 nm (GFP-LC3, GFP-ATG16L1, Alexa 488 and mGFP; detection window 505-545 nm), 553 nm (Cy3, Alexa-568, mRFP, lysotracker and Cherry-LC3; detection 563-630 nm) and 405 nm (DAPI; detection 415-475 nm) laser lines (10–20% power; gains: 2–10; 16 bit), and are displayed in an RGB mode. The blue signal represents DAPI-stained nuclei in all pictures. Scale bars represent 10 μm.
For quinacrine staining, cells expressing Cherry-LC3 were seeded on coverslips, treated and, at the end of the experiment, washed in Krebs-Ringer buffer (25 mM Hepes pH 7.4; 125 mM NaCl; 5 mM KCl; 1 mM MgSO4; 0.7 mM KH2PO4; 2 mM CaCl2; 6 mM glucose). Staining was carried out in Krebs-Ringer buffer containing 5 μM quinacrine for 15 min, 37 °C. Staining with Bodipy-ATP (Thermo Fisher, A12410) was done by incubating the cells with 30 μM Bodipy-ATP in culture medium (3 h, 37 °C). Coverslips were washed, placed upside down on glass-bottom Wilco plates (GWSt-3522) and analysed in vivo on an Okolab chamber at 37 °C and 5% CO2 coupled to the confocal microscope mentioned above. Pictures were taken using the 488 nm (quinacrine, Bodipy-ATP; detection 500-550 nm) and 553 nm (Cherry-LC3; detection 563-630 nm) laser lines. Assessment of Cherry-LC3 and quinacrine intensity was done in cells exhibiting a similar robust LC3 activation phenotype using the quantification module of the LAS X 4.2.0.23645 software package. Scale bars represent 10 μm.
Vesicles were blindly counted on confocal micrographs. Colocalizations were evaluated using the Fiji programme for image analysis (JACoP plugin). To quantify GFP-ATG16L1 recruitment to the mitochondrial surface, pictures were adjusted for identical red and green intensities and the percentage of GFP-ATG16L1-positive dots colocalizing with mitochondrial RFP was evaluated by counting over the adjusted images. Red images were pseudocoloured to magenta for colocalization with the green channel. All immunofluorescence experiments were repeated at least three times with qualitatively similar results.
Measurement of cell death
Both floating and attached cells were collected from each well (pipetting and trypsin treatment, respectively), centrifuged for 10 min (3300 × g, 4 °C) and resuspended in medium containing 1 μg/ml propidium iodide (PIE). DAPI (1 μg/ml) was used to measure cell death induced by MTX, since this drug emits red fluorescence. Samples were incubated for 10 min and then diluted 1:5 in medium before flow cytometry using a FACSCanto II device (Becton Dickinson) operating with the FACSDiva 8.0.1 software. Cells were gated in the FS/SS window to include both live and dead cells (and exclude small debris) and assessed for PIE or DAPI fluorescence (FL3). To measure BMDM death caused by dying MEFs, cells were resuspended in culture medium containing 0.1% azide, pre-incubated with an anti-Fc receptor antibody (TruStain FcX PLUS, Biolegend, 156604, 1/300, 15 min in ice), washed and incubated with an anti-F4/80 antibody coupled to Alexa Fluor 647 (Becton Dickinson, 565853; 1/100, 30 min, ice). Cells were then washed and stained with PIE as above. The percentage of PIE-positive cells (FL3) in the F4/80-positive compartment (FL5) was established using the FlowJo v10 software (Becton Dickinson). Gating strategies are shown in Supplementary Fig. 23.
Cell death induced by BAK constructs was measured in HEK-293T cells upon co-transfection with GFP to identify transfected cells. After 36 h, cultures were blindly scored for the percentage of green cells showing signs of cell death (membrane blebbing, cell shrinkage and detachment) in the different samples using an inverted fluorescence microscope (Axiovert200, Zeiss). A total of 10 fields including at least 20 cells per field were scored in all conditions.
Electron microscopy and CLEM
For anti-GFP-LC3 immuno-EM, cells were fixed in 4% PFA and 0.05% glutaraldehyde in 0.1 M PHEM buffer (25 mM Hepes, 60 mM Pipes, 2 mM MgCl2, 10 mM EGTA, pH 6.9), for 2 h, RT and then 16 h, 4 °C. After washing, samples were embedded in 10% gelatine. Sample blocks (< 1 mm3) were cryoprotected with 2.3 M sucrose (4 °C, 16 h) and flash frozen in liquid nitrogen. Samples were sectioned on an EM FCS cryoultramicrotome (Ultracut UCT, Leica) at −120 °C and collected in a mixture of 2.3 M sucrose and 2% methylcellulose solution (vol/vol 1:1). Thawed 90-nm-thick cryosections were incubated (30 min, RT) with a rabbit anti-GFP antibody (Chromotek, PABG1, 1:200) followed by protein A conjugated to 10 nm gold particles. Labelled sections were stained with a mix of 1.8% methylcellulose and 0.4% uranyl acetate and visualised at 80 kV with a JEOL JEM-1400 Flash electron microscope equipped with a One View CMOS 4 K camera (Gatan, US).
For CLEM, Bax-/- MEFs stably expressing GFP-LC3 cells were seeded on 35 mm gridded glass bottom plates (MatTek, P35G-1.5-14-C-GRD), retrovirally transduced with HA-BIM, fixed at the end of the experiment in a solution of 4% PFA prepared in phosphate buffer (PB, 0.1 M) and stained with DAPI. Confocal image acquisition, deconvolution and ultrastructure EM protocols are described in Supplementary Methods. Electron micrographs at the target depth were aligned with the corresponding deconvolved optical sections overexposed in the green channel to reveal cellular edges and peculiar morphological features that could serve as guides for accurate alignment. A picture of exactly the same size adjusted to the optimal exposure was then overlaid for alignment with the overexposed optical picture and the EM image. When the relevant EM section included a portion of the nucleus, the DAPI signal was also used for alignment. Accurate alignment was confirmed by colocalization of GFP-LC3 dots with random canonical autophagosomes detected in the EM images.
Measurement of secreted and intracellular ATP
Cells were seeded in 24-well plates, transduced or treated with MTX the next day, and supplemented 7–9 h later with 3 μM ARL67156 (Sigma, A265). To exclude variations in ATP secretion due to dissimilar death kinetics in the different cells, supernatants were sampled at two different times along the death process: 14–16 h and 20–26 h post-treatment (40–60% and 85–95% cell death, respectively), and measured using the Enliten ATP assay kit (Promega, FF2000). ATP secretion is shown as the integrated, cumulative data arising from the sum of the luciferase activity units obtained at early and late times, and reflects more accurately the total amount of ATP liberated along the death process.
To measure the ATP pool that remains in the apoptotic bodies, cellular remains were harvested by mild pipetting, spun at 3300 × g and the resulting pellet lysed in 20 μl of a buffer containing 1% Igepal CA-630, 150 mM NaCl, 50 mM Tris-HCl pH 7.5 and 5 mM EDTA. Control samples were trypsinized to harvest living cells and lysed following the same procedure. Lysates was spun (16100 × g, 4 °C) and the resulting supernatants collected and treated with a final concentration of 0.5% trichloroacetic acid (1 h, ice) for protein precipitation. Samples were spun (16100 × g, 4 °C), and the resulting supernatants neutralised with 5x volumes of 20 mM Tris-HCl pH 7. Samples were measured for ATP content using the Enliten ATP assay kit.
Proteomics
HEK-293T cells (20 × 10 cm plates) were transfected (jetPEI) with AU-GST-BAK-latch-WT or AU-GST-BAK-latch-5M (n = 1), lysed 36 h later and processed for precipitation with agarose-GSH beads as described above (co-immunoprecipitations in HEK-293T cells). Samples were resolved by SDS-PAGE and the gel was silver stained. Broad regions showing more intense bands in the AU-GST-BAK-latch-WT sample compared to the 5M control were excised and processed for proteomics identification. Detailed mass spectrometry methods are described in Supplementary Methods. Data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the identifiers PXD045985 and https://doi.org/10.6019/PXD045985.
To identify proteins commonly present in the BAK latch proteomics list and our previous ATG16L1-WDD proteomics approach59, we resorted to a Venn diagram (Venny 2.1 sever; https://bioinfogp.cnb.csic.es/tools/venny/). We confronted a list of ATG16L1-WDD hits including 1646 candidates (curated as Uniprot reviewed references) with the BAK latch results shown in Supplementary Data 2. A total of 117 proteins were present in both lists, including PHB1 and STOM (Supplementary Data 3).
IL-1β release by bone marrow-derived macrophages (BMDMs)
BMDMs were obtained from wild-type C57BL6/J mice (Envigo) as described in Supplementary Methods. Cells were detached with Accutase (Sigma, A6964), plated in 24-well plates (3-3.5 × 105 cells per well) and treated next day with LPS (ultrapure, Invivogen, Tlrl-3pelps; 100 ng/ml) for 4 h, and then with a 1:5 proportion of apoptotic bodies for 6 h, or ATP (5 mM, 30 min) as a positive control. Supernatants were harvested for IL-1β measurement by ELISA (Thermo Fisher, 88-7621-22). Apoptotic cells were generated by retroviral transduction or MTX treatment during 20-24 h, when cell death is almost complete. ARL67156 (3 μM) was added 7 h post-treatment to prevent ATP degradation. Apoptotic bodies were harvested by soft pipetting, spun at 3300 × g (4 °C, 10 min), resuspended in 350 μl of their own supernatant (which contains secreted ATP), and added to macrophage cultures pre-stimulated with LPS. Supernatants of live cells transduced with irrelevant vector or treated with vehicle were taken as control samples. To test the effect of the P2X7 (KN-62 and JNJ-47965567) and gasdermin D (DMF) inhibitors, macrophages were pre-incubated for 45 min and the apoptotic bodies added to the BMDM cultures in medium containing the inhibitors. IL-1β concentrations were calculated using a standard curve created following manufacturer’s instructions.
To identify the species of IL-1β secreted by HCT116 cells expressing split ATG16L1 (Nt + Ct), supernatants were harvested, clarified by centrifugation (3300 × g; 4 °C, 10 min) and directly subjected to anti-IL-1β Western blotting (goat anti-IL-1β antibody, R&D, AF-401-NA, 1 μg/ml). For supernatants generated from Bax-/- MEFs engineered to express ATG16L1-ΔWDD, a previous immunoprecipitation step was required to increase detectability. Clarified supernatants (150 μl) were incubated (overnight, 4 °C) with magnetic beads coupled to an anti-rabbit antibody (Dynabeads, Thermo Scientific, 11203D) and preloaded with a rabbit anti-IL-1β antibody (Abcam, ab234437, 1/30, 3 h, 4 °C). After three washes with PBS/0.1% Igepal, beads were treated with 2x RSB for Western blotting (goat anti-IL1β).
Ethics statement
Wild-type C57BL6/J mice (purchased from Envigo, female, 4–8 weeks old,) used for BMDM isolation were hosted in a standard animal facility including ventilated racks under controlled temperature (23 °C), humidity (50%) and light/dark cycles (12 h/12 h), following European Union regulations. Maintenance conditions and procedures were approved by the Ethics and Animal Wellbeing committees of both the research centre (Centro de Biología Molecular Severo Ochoa, CBMSO) and the hosting institution (Consejo Superior de Investigaciones Científicas, CSIC), and strictly follow European Union regulations. Animal sex was not considered in the studies.
Statistics and reproducibility
Graphs displaying less than 10 data points show means −/+ standard deviations obtained from the indicated number of experimental points (n; biological replicas). Results displaying more than 10 data points are presented as box-plots where the central line represents the median value, the box shows percentiles 25–75 and the whiskers include the most extreme values. Graphs were created using GraphPad Prism 8. Statistical significance (P-values) was calculated using two-tailed Student’s t-tests. Raw numeric values used for all graphs are included in the Source Data file. All experiments were repeated at least three times with qualitatively similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data and materials are freely available upon request to the corresponding author (fxp@cbm.csic.es). The mass spectrometry proteomics data generated in this study have been deposited in the ProteomeXchange Consortium database via the PRIDE partner repository under the accession code PXD045985. The previously published proteomics data to identify proteins that bind the ATG16L1 WD40 domain, which are used in this study, are also available from the ProteomeXchange Consortium database via the PRIDE partner repository under the accession code PXD013180. Source data generated in this study, including the processed proteomics data shown as Supplementary Data 1, the original numeric data underlying all graphs and uncropped versions of all Western blot images, have been deposited in the Figshare database [https://doi.org/10.6084/m9.figshare.28297229]. The main EM images shown in Fig. 3a and b have been submitted in a lossless format to the BioImage archive with accession numbers S-BIAD1711 and S-BIAD1712, respectively. Source data are provided with this paper. The raw numeric data underlying all graphs and uncropped versions of all Western blots are provided in the Source Data file. Source data are provided with this paper.
References
Czabotar, P. E., Lessene, G., Strasser, A. & Adams, J. M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 (2014).
Luna-Vargas, M. P. A. & Chipuk, J. E. The deadly landscape of pro-apoptotic BCL-2 proteins in the outer mitochondrial membrane. FEBS J. 283, 2676–2689 (2016).
Green, D. R. The mitochondrial pathway of apoptosis part ii: The bcl-2 protein family. Cold Spring Harb. Perspect. Biol. 14, a041046 (2022).
Westphal, D., Kluck, R. M. & Dewson, G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 21, 196–205 (2014).
Adams, J. M. & Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36 (2018).
Czabotar, P. E. & Garcia-Saez, A. J. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat. Rev. Mol. Cell Biol. 24, 732–748 (2023).
Green, D. R., Ferguson, T., Zitvogel, L. & Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363 (2009).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Kroemer, G., Galluzzi, L., Kepp, O. & Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013).
Galluzzi, L., Kepp, O., Hett, E., Kroemer, G. & Marincola, F. M. Immunogenic cell death in cancer: concept and therapeutic implications. J. Transl. Med. 21, 1–8 (2023).
Garg, A. D., Dudek-Peric, A. M., Romano, E. & Agostinis, P. Immunogenic cell death. Int. J. Dev. Biol. 59, 131–140 (2015).
Krysko, O., Aaes, T. L., Bachert, C., Vandenabeele, P. & Krysko, D. V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 4, 1–7 (2013).
Kepp, O. et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3, 1–19 (2014).
Kepp, O. et al. ATP and cancer immunosurveillance. EMBO J. 40, 1–18 (2021).
Thorburn, J. et al. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16, 175–183 (2009).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1578 (2011).
Ayna, G. et al. ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One 7, e40069 (2012).
Hou, W. et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 4, 1–10 (2013).
Martins, I. et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 21, 79–91 (2014).
Bento, C. F. et al. Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713 (2016).
Kroemer, G. & Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 (2008).
Codogno, P., Mehrpour, M. & Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 13, 7–12 (2012).
Bestebroer, J., V’kovski, P., Mauthe, M. & Reggiori, F. Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 14, 1029–1041 (2013).
Pimentel-Muiños, F. X. & Boada-Romero, E. Selective autophagy against membranous compartments: canonical and unconventional purposes and mechanisms. Autophagy 10, 397–407 (2014).
Cadwell, K. & Debnath, J. Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol. 217, 813–822 (2018).
Sanjuan, M. A. et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257 (2007).
Durgan, J. & Florey, O. Many roads lead to CASM: diverse stimuli of noncanonical autophagy share a unifying molecular mechanism. Sci. Adv. 8, eabo1274 (2022).
DeSelm, C. J. et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 21, 966–974 (2011).
Bel, S. et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052 (2017).
Dupont, N. et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 (2011).
Boada-Romero, E. et al. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J. 32, 566–582 (2013).
Wu, X. et al. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 7, 10533 (2016).
Heckmann, B. L. et al. Noncanonical function of an autophagy protein prevents spontaneous Alzheimer’s disease. Sci. Adv. 6, eabb9036 (2020).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 (2008).
Sorbara, M. T. et al. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 39, 858–873 (2013).
Serramito-Gómez, I. et al. Regulation of cytokine signaling through direct interaction between cytokine receptors and the ATG16L1 WD40 domain. Nat. Commun. 11, 1–15 (2020).
Fletcher, K. et al. The WD40 domain of ATG 16L1 is required for its non‐canonical role in lipidation of LC3 at single membranes. EMBO J. 37, 1–17 (2018).
Fujita, N. et al. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: Analysis of the organization of the Atg16L1 complex in fibroblasts. J. Biol. Chem. 284, 32602–32609 (2009).
Boada-Romero, E. et al. The T300A Crohn’s disease risk polymorphism impairs function of the WD40 domain of ATG16L1. Nat. Commun. 7, 11821 (2016).
Rongvaux, A. et al. Apoptotic caspases prevent the induction of type i interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014).
Gui, X. et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266 (2019).
Fischer, T. D., Wang, C., Padman, B. S., Lazarou, M. & Youle, R. J. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J. Cell Biol. 219, 1–13 (2020).
Lomonosova, E. & Chinnadurai, G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 27, S2–S19 (2009).
Lindqvist, L. M. et al. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ. 25, 782–794 (2017).
Colell, A. et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129, 983–997 (2007).
Scorrano, L. et al. BAX and BAK regulation of endoplasmic reticulum Ca 2 : a control point for apoptosis. Science 300, 135–139 (2003).
Gonugunta, V. K. et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. 21, 3234–3242 (2017).
Prabakaran, T. et al. Attenuation of cGAS-STING signaling is mediated by a p 62 / SQSTM 1 -dependent autophagy pathway activated by TBK 1. EMBO J. 37, e97858 (2018).
Gentili, M. et al. ESCRT-dependent STING degradation inhibits steady-state and cGAMP-induced signalling. Nat. Commun. 14, 611 (2023).
Kuchitsu, Y. et al. STING signalling is terminated through ESCRT-dependent microautophagy of vesicles originating from recycling endosomes. Nat. Cell Biol. 25, 453–466 (2023).
Sawada, K. et al. Identification of a vesicular nucleotide transporter. Proc. Natl Acad. Sci. USA 105, 15–18 (2008).
Kato, Y. et al. Identification of a vesicular ATP release inhibitor for the treatment of neuropathic and inflammatory pain. Proc. Natl Acad. Sci. USA. 114, E6297–E6305 (2017).
Moldoveanu, T. et al. The X-Ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol. Cell 24, 677–688 (2006).
Alsop, A. E. et al. Dissociation of Bak α 1 helix from the core and latch domains is required for apoptosis. Nat. Commun. 6, 1–13 (2015).
Brouwer, J. M. et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol. Cell 55, 938–946 (2014).
Lapatsina, L., Brand, J., Poole, K., Daumke, O. & Lewin, G. R. Stomatin-domain proteins. Eur. J. Cell Biol. 91, 240–245 (2012).
Signorile, A., Sgaramella, G., Bellomo, F. & Rasmo, D. De. prohibitins: a critical role in mitochondrial functions and implication in diseases. Cells 8, 71 (2019).
Cheng, E. H. Y., Sheiko, T. V., Fisher, J. K., Craigen, W. J. & Korsmeyer, S. J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517 (2003).
Slowicka, K. et al. Physical and functional interaction between A20 and ATG16L1-WD40 domain in the control of intestinal homeostasis. Nat. Commun. 10, 1834 (2019).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1179 (2009).
Vandenabeele, P., Bultynck, G. & Savvides, S. N. Pore-forming proteins as drivers of membrane permeabilization in cell death pathways. Nat. Rev. Mol. Cell Biol. 24, 312–333 (2023).
Humphries, F. et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 77, 1633–1637 (2020).
Evavold, C. L. et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages report the pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44.e6 (2018).
Westphal, D. et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl Acad. Sci. Usa. 111, E4076–E4085 (2014).
Dai, H. et al. A BAK subdomain that binds mitochondrial lipids selectively and releases cytochrome C. Cell Death Differ. 30, 794–808 (2023).
Mairhofer, M., Steiner, M., Mosgoeller, W., Prohaska, R. & Salzer, U. Stomatin is a major lipid-raft component of platelet alpha granules. Blood 100, 897–904 (2002).
Kim, J.-H., Rhee, J.-K., Ahn, D.-G., Kim, K. P. & Oh, J.-W. Interaction of stomatin with hepatitis C virus RNA polymerase stabilizes the viral RNA replicase complexes on detergent-resistant membranes. J. Microbiol. Biotechnol. 24, 1744–1754 (2014).
Hailey, D. W. et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667 (2010).
Sun, M. G. et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat. Cell Biol. 9, 1057–1065 (2007).
Wei, Y., Chiang, W., Sumpter, R., Mishra, P. & Levine, B. Prohibitin 2 Is an inner mitochondrial membrane mitophagy receptor. Cell 168, 224–238 (2017).
Riley, J. S. et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37, 1–16 (2018).
McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, 1–12 (2018).
Qu, X. et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931–946 (2007).
Yee, K. S., Wilkinson, S., James, J., Ryan, K. M. & Vousden, K. H. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 16, 1135–1145 (2009).
Acknowledgements
We thank Drs. Mario Mairhofer (University of Applied Sciences, Austria), Joaquín Mádrenas (UCLA Medical School, Los Angeles, USA), Dirk Goerlich (Max Plack Institute, Gottingen, Germany), Craig Thompson (Sloan Kettering Institute, New York, USA) and Bert Vogelstein (Johns Hopkins School of Medicine, Baltimore, Maryland, USA) for the STOM, SLP2, EXP4, BCLXL and PUMA cDNAs, respectively. We also thank Drs. Bert Vogelstein for the HCT116 Bax-/- cells, Stanley Korsmeyer (Dana Farber Cancer Institute, Boston, MA, USA) for the BAK and/or BAX-deficient MEFs, David Boone (Indiana University School of Medicine, USA) for the ATG16L1-/- HCT116 cells, Ramnik Xavier (Broad Institute MIT and Harvard, USA) for the Atg16l1-/- MEFs, Dr. Javier Traba for advice on macrophage protocols, Dr. Emilio Lecona and Rodrigo Martín for help with the co-precipitation studies, the confocal and electron microscopy units of the CBM for their excellent technical service, and CBM facilities for support. This work was funded by grants SAF2017-88390-R (F.X.P), PID2020-114699RB-I00 (F.X.P) and PID2023-148747OB-I00 (F.X.P) from the Ministerio de Ciencia y Tecnología (Spanish Government; MCIN/AEI/10.13039/501100011033, FEDER, UE), IDEAS18093PIME from the Asociación Española Contra el Cáncer (AECC) and SA042P17 from the Junta de Castilla y León local government. Additional funding comes from the Excellence Severo Ochoa Grant CEX2021-001154-S from MCIN/AEI/10.13039/501100011033. The CBM receives institutional support by Fundación Ramon Areces. E.T. and J.B.L. are recipients of predoctoral fellowships PRE2018-085243 and PRE2021-097461, respectively, from the Formación de Personal Investigador (FPI) programme of the Spanish Ministry of Science and Technology (MCIU/AEI/10.13039/501100011033; Spanish Government and FSE + ). F.X.P. holds a tenured position at CSIC.
Author information
Authors and Affiliations
Contributions
E.T.S., R.V., J.B.L., M.L., D.O.S., C.R.B., and C.M.J. performed the experiments. F.X.P. conceived and designed the study, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Terraza-Silvestre, E., Villamuera, R., Bandera-Linero, J. et al. An unconventional autophagic pathway that inhibits ATP secretion during apoptotic cell death. Nat Commun 16, 3409 (2025). https://doi.org/10.1038/s41467-025-58619-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58619-3
