
Abstract
Developing ruthenium-based oxide catalysts capable of suppressing lattice oxygen participation in the catalytic reaction process is crucial for maintaining stable oxygen evolution reaction (OER) under acidic conditions. Herein, we delicately construct a RuO2 nanoparticle-anchored LiCoO2 nanosheet electrocatalyst (RuO2/LiCoO2), achieving dynamic optimization of RuO2 during the reaction process and improving catalytic stability. Benefiting from the unique electrochemical delithiation characteristics of the LiCoO2 support, the covalency of the Ru-O bond is effectively regulated during the OER process. The weakened Ru-O covalent bond inhibits the participation of lattice oxygen in the catalytic reaction and ensures the continuous operation of the Ru active sites. Moreover, the extended Ru-O bond in the optimized RuO2/LiCoO2 catalyst reduces the formation energy barrier of the *OOH intermediates, accelerating the progress of the OER. As a result, the RuO2/LiCoO2 catalyst requires only an overpotential of 150 ± 2 mV at 10 mA cm−2 in 0.5 M H2SO4 and operates stably for 2000 h at 1 A cm−2 in a proton exchange membrane water electrolysis. This work opens new avenues for designing efficient ruthenium-based catalysts.
Similar content being viewed by others

Introduction
Proton exchange membrane water electrolysis (PEMWE) with high current density and low resistance loss is regarded as a promising hydrogen production technology in the future1. Currently, noble metal ruthenium (Ru) and iridium (Ir) oxide catalysts are extensively used in the anodes of PEMWE2,3. Compared to IrO2, RuO2 offers high activity and cost-effectiveness, presenting significant potential for application in acidic oxygen evolution reactions (OER)4,5. However, RuO2 catalysts tend to follow the lattice oxygen mechanism (LOM) and generate numerous oxygen defects during the OER reaction, leading to crystal structure collapse6,7. Additionally, the metal Ru sites can be over-oxidized into soluble RuO4 species, which separate from the crystal lattice under high oxidation potential, resulting in poor catalytic stability8,9. From a crystal structure perspective, the instability of RuO2 is closely related to the charge distribution of the Ru-O bond10,11,12,13. Therefore, regulating the electronic state of the Ru-O bond to suppress the involvement of lattice oxygen in the OER process is an effective strategy for enhancing the activity and stability of RuO2 catalysts.
Generally, electron-rich Ru sites in RuO2 activate lattice oxygen and generate defects, while electron-deficient states tend to oxidize to excessively high valence states and dissolve14,15. Traditional electron-donating support strategies modulate the electron distribution of the Ru-O bond, preventing the dissolution of the metal center and following a relatively stable adsorption oxygen mechanism (AEM)16,17. At present, electron-donating supports with limited regulatory capacity cannot meet the demand for manipulating dynamically changing Ru-O bonds under complex high-potential OER conditions18,19. Therefore, designing supports capable of dynamically regulating the electronic structure of RuO2 in response to changes in the catalytic reaction process is highly attractive. To address this challenge, transition metal oxides with cation intercalation dynamically optimize the local microenvironment of metal-oxygen bonds during the catalytic reaction through the extraction and insertion of cations20,21. Notably, lithium cobalt oxide (LiCoO2) with a unique layered structure exhibits an ordered cation arrangement and superior thermodynamic stability22,23,24. The tight edge-shared CoO6 octahedral structure in LiCoO2 reduces the migration energy barrier of Li+ and ensures two-dimensional diffusion of Li+ in the plane, facilitating dynamic reconstruction during the catalytic process25,26. However, the multi-layered bulk structure of LiCoO2 exhibits slow electron transport capability and limited surface area, which cannot meet the requirements for efficient catalyst support25. In this regard, two-dimensional LiCoO2 nanosheets markedly shorten the intercrystalline electron transmission path and enhance conductivity27. Additionally, the two-dimensional nanosheet structure forms a fast electron transmission channel perpendicular to the exposure plane, resulting in strong metal-support interaction28,29. Therefore, using two-dimensional LiCoO2 nanosheets as a support for RuO2 is expected to achieve dynamic regulation of the electronic structure during catalytic reactions and exhibit high catalytic activity.
Herein, we propose an effective strategy to improve the charge distribution of RuO2 through the unique dynamic evolution process of the LiCoO2 support (RuO2/LiCoO2). The LiCoO2 nanosheet support with electron-donating ability induces electron transfer from Co to Ru sites, providing electron compensation to stabilize the valence state of RuO2. More importantly, the two-dimensional diffusion and extraction of Li ions within the interlayer of the LiCoO2 nanosheet under OER potential cause the dynamic reconstruction and evolution of the RuO2/LiCoO2 catalyst interface. The unique dynamic self-optimization process moderately weakens the covalency of the Ru-O bond, suppressing the participation of lattice oxygen and achieving a good balance between catalytic activity and stability. The optimized Ru sites facilitate the formation of the *OOH intermediate, significantly lowering the catalytic energy barrier of the rate-determining step. Consequently, the RuO2/LiCoO2 electrocatalyst provides a current density of 10 mA cm−2 at an overpotential of 150 ± 2 mV and maintains stability for over 2300 h in 0.5 M H2SO4. In addition, RuO2/LiCoO2 as anode can operate continuously for 2000 h at 1 A cm−2 in a PEM electrolyzer. This work advances the application of ruthenium-based catalysts in PEMWE.
Results
Reaction mechanism and structural analysis
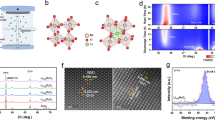
Due to the differences in the covalency of Ru-O bonds, Ru oxides follow either the lattice oxygen mechanism (LOM) or the adsorption oxygen mechanism (AEM) during the acidic OER process (Fig. 1a, b)7,30. In the LOM pathway, activated lattice oxygen with stronger covalency participates in the OER process, resulting in the generation of oxygen defects31. An excessive number of oxygen vacancies can induce detachment from the crystal lattice at the Ru metal sites, which greatly reduces the catalytic stability. However, Ru oxides with weak covalent Ru-O bonds follow the AEM mechanism, achieving a stable acidic OER process13. Therefore, dynamically regulating the covalency of Ru-O bonds during the catalytic reaction is crucial for enhancing catalyst stability. The O3-type LiCoO2 material with Li element intercalation exhibits superior conductivity and structural stability. More importantly, the extraction of interlayer lithium ions at the OER potential triggers electron transfer, enabling real-time regulation of the covalency of metal-oxygen bonds (Fig. 1c). Additionally, we employ density functional theory (DFT) calculations to study the stability of different Co-based oxides in acidic environments. LiCoO2 presents a thermodynamically unfavorable dissolution barrier, indicating that it can still maintain stability in an acidic environment (Fig. 1d). Inspired by these results, dynamically regulating the Ru-O bond covalency in RuO2 using the layered LiCoO2 support is expected to achieve high catalytic stability.

a LOM schematic of RuO2 in acidic OER. b AEM schematic of RuO2 in acidic OER. c Structural models of LiCoO2 and Li1−xCoO2. d Dissolution energy barriers of cobalt-based oxides.
Materials characterization
A facile adsorption calcination strategy is employed to prepare LiCoO2 nanosheets with loaded RuO2 nanoparticles (RuO2/LiCoO2). Briefly, LiCoO2 nanosheets are obtained through ultrasonic-assisted exfoliation of solid-phase synthesized bulk LiCoO2 (Supplementary Figs. 1, 2). Subsequently, ruthenium ions are adsorbed on the LiCoO2 surface through electrostatic binding and then calcined to form the RuO2/LiCoO2 electrocatalyst. According to the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, RuO2/LiCoO2 exhibits an ultrathin nanosheet structure (Fig. 2a and Supplementary Figs. 3–5). Interestingly, the RuO2 nanoparticles are tightly confined in the LiCoO2 lattice in the high-resolution TEM (HRTEM) image, which is different from simple physical adsorption (Fig. 2b). A clear lattice contact exists between RuO2 nanoparticles and the LiCoO2 support, in which lattice stripes with interplanar spacings of 0.31 and 0.20 nm correspond to the RuO2 (110) and LiCoO2 (104) planes, respectively (Fig. 2c, h)25. The LiCoO2 support provides lattice confinement for the RuO2 nanoparticles to form a unique metal-support interaction. Atomic force microscopy (AFM) shows that the thickness of the RuO2/LiCoO2 nanosheet is 5.1 nm in Fig. 2d. The ultrathin nanosheet structure of RuO2/LiCoO2 maximizes the exposure of more Ru active sites. Atomic-resolution aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) is used to analyze the atomic structure of the RuO2/LiCoO2 composite. The Co atoms are arranged in an αβγ stacking mode in the HAADF-STEM image along the [010] zone axis, indicating the formation of O3-type LiCoO2 and consistent with the XRD results (Fig. 2e and Supplementary Figs. 6, 7)23. The spacing of about 0.47 nm corresponds to the typical interlayer spacing of LiCoO2. In addition, the bright Ru atomic array is embedded in the lattice of LiCoO2 and exhibits superb lattice matching at the interface (Fig. 2f). The distribution of Ru, Co, and O elements in the energy dispersive spectroscopy (EDS) spectrum further confirms the uniformity of the heterostructure (Fig. 2g). The above analysis shows that the structurally ordered LiCoO2 nanosheet support is closely connected to the RuO2 nanoparticles, which provides the possibility for the optimization of the electronic structure.

a TEM, (b, c) HRTEM, and (d) AFM images of RuO2/LiCoO2 (inset: height profile of RuO2/LiCoO2). e, f HAADF-STEM images of RuO2/LiCoO2. g The EDS elemental mapping of RuO2/LiCoO2. h The atomic model of RuO2 and LiCoO2. i Normalized Ru K-edge XANES spectra and (j) Ru K-edge FT-EXAFS spectra of RuO2/LiCoO2, RuO2, and Ru foil, respectively. k Normalized Co K-edge XANES spectra and (l) Co K-edge FT-EXAFS spectra of RuO2/LiCoO2, LiCoO2, Co3O4, and Co foil, respectively.
To further understand the coordination structure and electronic states of RuO2/LiCoO2, X-ray absorption spectroscopy (XAS) was performed. As shown in Fig. 2i, the oxidation state of RuO2/LiCoO2 is lower than that of RuO2 in the Ru K-edge X-ray absorption near-edge structure (XANES) spectra, implying the formation of an electron transfer channel between RuO2 and LiCoO232. This is also consistent with the results of the first derivative of the Ru K-edge XANES spectra and high-resolution X-ray photoelectron spectroscopy (XPS) (Supplementary Figs. 8, 9). The LiCoO2 nanosheet support with charge compensation effectively regulates the oxidation state of Ru sites, preventing excessive oxidation during the catalytic process. According to the Fourier transformed Ru K-edge extended X-ray absorption fine structure (FT-EXAFS) spectra, the Ru-O bond length is slightly shortened after contact between RuO2 and LiCoO2 (from 1.93 to 1.91 Å), which is attributed to the formation of interfacial interactions (Fig. 2j and Supplementary Figs. 10, 11 and Supplementary Tables 1, 2)33. The Co K-edge XANES of RuO2/LiCoO2 is transferred to higher energies compared to LiCoO2 in Fig. 2k, which is consistent with the XPS valence band spectrum and electron paramagnetic resonance (EPR) results. This verifies the electron-donating ability of the LiCoO2 carrier (Supplementary Figs. 12, 13). Moreover, the main peaks of RuO2/LiCoO2 at 1.4 and 2.4 Å correspond to Co-O and Co-Co coordination shells in the Co K-edge FT-EXAFS spectra, respectively (Fig. 2l)34,35. It is worth noting that the Co-O bonds in RuO2/LiCoO2 are stretched more than LiCoO2, which is consistent with the wavelet transform spectral results (Supplementary Figs. 14–16). The electron transfer from Co to Ru are verified by collecting electron energy loss spectra (EELS) of Co-L2,3 and Ru-M2,3 at the RuO2/LiCoO2 interface (Supplementary Fig. 17). Therefore, the interfacial interaction between RuO2 and LiCoO2 electronically compensates the Ru sites and improves the stability of the electrocatalyst.
OER electrocatalytic performance
Inspired by the structural advantages of the RuO2/LiCoO2 catalyst, the OER performance was evaluated via a three-electrode system in 0.5 M H2SO4. In Fig. 3a, the RuO2/LiCoO2 electrocatalyst exhibits an overpotential of 150 ± 2 mV at 10 mA cm−2, which is much smaller than commercial RuO2 (260 ± 4 mV) (denoted Com-RuO2) and RuO2 (210 ± 5 mV) (Supplementary Figs. 18–24). In comparison, the highly acidic OER activity of RuO2/LiCoO2 also surpasses most previously reported noble metal-based electrocatalysts (Supplementary Figs. 25 and Supplementary Table 3). The Tafel slope of RuO2/LiCoO2 (51.97 mV dec−1) shows improved reaction kinetics compared to RuO2 (62.06 mV dec−1) and Com-RuO2 (92.80 mV dec−1) (Fig. 3b)36. At the OER potential, RuO2/LiCoO2 exhibits fast intermediate conversion efficiency. Meanwhile, the phase angle of RuO2/LiCoO2 rapidly decreases at different potentials in the Bode plot, further confirming the rapid charge diffusion on the catalyst surface (Supplementary Figs. 26–28 and Supplementary Table 4). The mass activity of the RuO2/LiCoO2 catalyst is 41.81 times that of RuO2 at an overpotential of 240 mV in Fig. 3c. In addition, the turnover frequency (TOF) and OER current normalized by the electrochemically active surface area (ECSA) of RuO2/LiCoO2 electrocatalysts are significantly higher than RuO2 (Supplementary Figs. 29–32 and Supplementary Table 5). The catalyst durability is tested using chronopotentiometry to evaluate the potential for practical applications. The RuO2/LiCoO2 electrocatalyst can operate stably for 2300 h at a current density of 10 mA cm−2, while RuO2 almost loses its activity after 400 h (Fig. 3d and Supplementary Fig. 33). The above results show that the LiCoO2 support in the RuO2/LiCoO2 electrocatalyst avoids the dissolution of RuO2 in acidic OER to achieve sustained acidic OER catalytic activity.

a OER polarization curves and (b) Tafel plots of RuO2/LiCoO2, RuO2, Com-RuO2, and LiCoO2 in 0.5 M H2SO4 electrolyte, respectively. The voltage is corrected by an automatic 90% of iR compensation (R is 1.10 ± 0.02 Ω). c Mass activity of Ru atoms in RuO2/LiCoO2 and RuO2 as a function of overpotential. d Chronopotentiometric curves of RuO2/LiCoO2 and RuO2 at 10 mA cm−2, respectively (Inset: Potential changes of RuO2/LiCoO2 before and after stabilization). e The polarization curves of PEMWE with RuO2/LiCoO2 | |Pt/C and RuO2 | |Pt/C catalyst in pure water at 80 °C without iR-correction. f Chronopotentiometric curve of PEMEW using RuO2/LiCoO2 | |Pt/C catalyst at 1 A cm−2.
To evaluate the industrial application potential of RuO2/LiCoO2, a PEM electrolyzer was assembled with RuO2/LiCoO2 and Pt/C as anode and cathode catalysts, respectively. Specifically, RuO2/LiCoO2 | |Pt/C only requires a cell voltage of 1.68 V to reach a current density of 1 A cm−2, which is significantly lower than RuO2 | |Pt/C (1.84 V) (Fig. 3e). Overpotential analysis shows that improved mass transport alleviates concentration polarization effects in the local reaction environment, indirectly enhancing catalytic kinetics (Supplementary Fig. 34). Furthermore, the mass activity of RuO2/LiCoO2 | |Pt/C (2.56 A mgRu−1) is approximately 21.33 times greater than that of RuO2 | |Pt/C (0.12 A mgRu−1) at a cell voltage of 1.7 V (Supplementary Fig. 35). The mass activity and cost of RuO2/LiCoO2 | |Pt/C are also much lower than those of commercial IrO2 | |Pt/C (Supplementary Fig. 36). Strikingly, the PEM electrolyzer using RuO2/LiCoO2 | |Pt/C can operate stably for 2000 h at a current density of 1 A cm−2 with negligible decay in Fig. 3f, indicating the potential of the RuO2/LiCoO2 catalyst for practical applications. The long-term durability of RuO2/LiCoO2 | |Pt/C in PEMWE also exceeds that of most recently reported various high-performance electrocatalysts (Supplementary Table 6). These findings demonstrate that the RuO2/LiCoO2 electrocatalyst exhibits outstanding performance as a choice for hydrogen production in the PEM electrolyzer, providing strong support for sustainable energy production.
Structural transformation after stability
The physical structure of the RuO2/LiCoO2 electrocatalyst after OER stability testing was investigated to explore the influencing factors for the improved catalytic activity. SEM and TEM images show that RuO2 nanoparticles remain tightly supported on the surface of LiCoO2 nanosheets in the RuO2/LiCoO2 electrocatalyst after stability testing (Supplementary Figs. 37, 38). Besides, a reconstructed amorphous layer can be clearly observed on the surface of LiCoO2 nanosheets in RuO2/LiCoO2. This layer is attributed to the migration and extraction of interlayer Li-ions during voltage application, resulting in the formation of a surface Li1−xCoO2 amorphous layer. Compared with the original RuO2/LiCoO2, the (003) crystal plane peak of RuO2/LiCoO2 after OER stability shifts to a lower angle, which may be attributed to the lattice distortion caused by the extraction of Li ions (Fig. 4a and Supplementary Fig. 39). Additionally, the Raman spectrum of RuO2/LiCoO2 detects two characteristic peaks of Eg and A1g modes at 484 and 594 cm−1, corresponding to O-Co-O bending and Co-O stretching, respectively (Fig. 4b and Supplementary Fig. 40). Due to the enhanced polarity of the Co-O bond induced by Li extraction, the Eg and A1g characteristic peaks of RuO2/LiCoO2 exhibit a blue shift after OER stability (Supplementary Fig. 41)23. In particular, the new peak at 665 cm−1 after stabilization is attributed to the Co-O bonds of the spinel structure Li1−xCoO2 (LiCo2O4) of the surface reconstruction layer, which enhances the polarity of the Co-O bonds. The interlayer Li ions detach from RuO2/LiCoO2 through a two-dimensional diffusion path under the OER potential, performing dynamic structural reconstruction and altering the interface coordination environment of the catalyst.

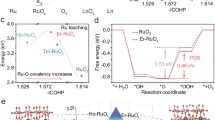
a XRD patterns and (b) Raman spectra of RuO2/LiCoO2 and RuO2/LiCoO2 after stability, respectively. c The content of Co2+, Co3+, Ru3+, and Ru4+ in RuO2/LiCoO2 before and after OER stability. d Bader charge analysis of RuO2, LiCoO2, RuO2/LiCoO2, and RuO2/Li1−xCoO2, respectively. e Comparison of charge amounts of RuO2, LiCoO2, RuO2/LiCoO2, and RuO2/Li1−xCoO2, respectively. f COHP of Ru-O in RuO2/LiCoO2 and RuO2/Li1−xCoO2, respectively. g Schematic diagram of the dynamic evolution of RuO2/LiCoO2.
The electronic states of RuO2/LiCoO2 catalysts before and after OER stability are further analyzed to understand the influence of the reconstruction process. The Co3+ ratio of RuO2/LiCoO2 after OER stabilization increases compared with the pristine RuO2/LiCoO2, implying the reduction of electron density for Co sites (Fig. 4c and Supplementary Fig. 42)37,38. The dynamic extraction of Li ions induces electron transfer, leading to partial oxidation of Co sites and increased covalency in the Co-O bond. Additionally, the proportion of Ru4+ in RuO2/LiCoO2 slightly increases from 58.02% to 61.17% after the stability measurement39. As a comparison, the proportion of Ru4+ in RuO2 after the stability measurement is 85.24%, which is much higher than that of RuO2/LiCoO2 (Supplementary Figs. 43, 44). Despite the slight oxidation of the LiCoO2 support, it can maintain the oxidation state of RuO2 by continuously donating electrons, thereby preventing the dissolution of Ru sites (Supplementary Figs. 45, 46). A structural model of RuO2/LiCoO2 after partial delithiation (RuO2/Li1−xCoO2) is constructed to reveal the changes in electronic structure by DFT calculations (Supplementary Fig. 47). The Bader charge indicates that Ru in RuO2/LiCoO2 obtains about 0.169 e of electrons from the LiCoO2 support (Fig. 4d)40,41. Due to the extraction of Li ions, the charge density at the Ru sites in RuO2/Li1−xCoO2 (6.584 e) is lower than that of RuO2/LiCoO2 (6.626 e), while remaining higher than that of RuO2 (6.457 e) (Fig. 4e). Crystal orbital Hamiltonian population (COHP) analysis shows that the Ru-O bond covalency of RuO2/Li1−xCoO2 (−4.07 eV) is lower than that of RuO2/LiCoO2 (−4.35 eV) (Fig. 4f)42,43. The unique delithiation process ensures dynamic regulation of the covalency of the Ru-O bond during the catalytic process to suppress the participation of lattice oxygen (Fig. 4g).
In situ characterization of catalyst structural changes
In-situ XAS was implemented to explore the dynamic evolution mechanism of electrocatalysts in the OER process (Supplementary Fig. 48). In the fitted Ru K-edge FT-EXAFS spectra, the Ru-O bond of RuO2/LiCoO2 is appropriately extended as the potential increases from open circuit voltage (OCV) to 1.7 V (from 1.90 to 1.99 Å), indicating that the covalency of Ru-O is weakened (Fig. 5a, b and Supplementary Fig. 49 and Supplementary Table 7)44. In contrast, the Co-O bond tends to be more covalent during the delithiation process, resulting in a negative shift of the Co-O coordination shell in RuO2/LiCoO2 and the formation of a surface reconstruction layer (from 1.92 to 1.82 Å) (Supplementary Figs. 50, 51 and Supplementary Table 8)45. The wavelet transform of Ru K-edge and Co K-edge EXAFS spectra also intuitively confirm this result (Supplementary Figs. 52–54). Thus, the dynamic extraction of lithium ions regulates the covalency of the Ru-O bond through interfacial interactions, which can restrict the participation of lattice oxygen and maintain the structural stability of RuO2 (Fig. 5c). Notably, the Ru-O bond in RuO2 undergoes a significant shortening from 1.93 Å to 1.81 Å as the voltage transitions from OCV to 1.7 V (Fig. 5d, e and Supplementary Figs. 55–57 and Supplementary Table 9). The excessive enhancement of the covalency of the Ru-O bond provides conditions for triggering lattice oxygen to participate in the OER reaction44. Moreover, the slight change in the Ru-O coordination number in RuO2/LiCoO2 indicates the stability of the coordination structure of RuO2. As a comparison, a reduced coordination number can be observed for the Ru-O bonds in RuO2, which is attributed to the presence of defective oxygen (Fig. 5f). Due to the strong covalency of the Ru-O bond, RuO2 follows the LOM path and generates a large number of oxygen defects during the OER process, leading to the collapse of the catalyst surface structure.

a In situ Ru K-edge EXAFS spectra of RuO2/LiCoO2 with applied potentials from OCV to 1.7 V. b Changes in bond length and intensity of the Ru-O shell of RuO2/LiCoO2 at different potentials. c Schematic diagram of the structural evolution of RuO2/LiCoO2 at different potentials. d In situ Ru K-edge EXAFS spectra of RuO2 with applied potentials from OCV to 1.7 V. e Changes in bond length and intensity of the Ru-O shell of RuO2 at different potentials. f Schematic diagram of the structural evolution of RuO2 at different potentials. g In situ Ru K-edge XANES spectra of RuO2/LiCoO2. Inset: Oxidation state changes of Ru in RuO2/LiCoO2. h In situ Ru K-edge XANES spectra of RuO2. Inset: Oxidation state changes of Ru in RuO2.
To elucidate the electronic structure changes of the electrocatalyst at high potentials, the oxidation states of RuO2/LiCoO2 and RuO2 are detected by in situ XANES spectra. The average valence state of Ru species in RuO2/LiCoO2 rises gently from +3.82 to +4.21 as the potential transitions from OCV to 1.7 V vs. RHE in Fig. 5g (Supplementary Fig. 58). This further confirms that the strong electronic interaction between the LiCoO2 and RuO2 interface inhibits the excessive oxidation of RuO2. In addition, the Co species in the RuO2/LiCoO2 catalyst can still maintain an oxidation state of +3.16 at a high voltage of 1.7 V (Supplementary Figs. 59, 60). As a result, the extraction of lithium ions can moderately regulate the electron distribution of LiCoO2 support without destroying the main structure. Remarkably, the valence state of Ru rapidly increases from +4.02 to +5.57 in RuO2 switching the voltage from OCV to 1.7 V vs. RHE, which is attributed to the excessive oxidation originating from the damage of the RuO2 structure (Fig. 5h, Supplementary Figs. 61, 62). Based on the above results, the dynamic reconstruction process of RuO2/LiCoO2 can effectively modify the charge environment of the Ru-O bond and stabilize the lattice oxygen, resulting in improved OER performance.
Origin of high catalytic performance
To investigate the transfer phenomena of reaction intermediates on the electrocatalyst surface, a series of electrochemical measurements are performed. The cyclic voltammetry curves of RuO2/LiCoO2 and RuO2 can observe two pairs of redox peaks around 0.5 V and 1.0 V respectively, which correspond to the formation of *OH reaction intermediates with H2O deprotonation (Fig. 6a). Obviously, the redox peak of RuO2/LiCoO2 moves downwards than that of RuO2, indicating favorable H2O deprotonation and promoting the reaction process46. Subsequently, methanol is used as a probe molecule to detect the coverage of *OH intermediates on the catalyst surface (Supplementary Fig. 63). The methanol oxidation reaction (MOR) is more active on OH*-dominated catalyst surfaces due to the mechanism of nucleophilic attack on electrophilic sites12. RuO2/LiCoO2 exhibits high surface OH* coverage compared to RuO2 in Fig. 6b, implying that RuO2/LiCoO2 contains more effective OER active sites. Furthermore, the change in catalytic activity of the RuO2/LiCoO2 catalyst at different pH values is negligible, which is a characteristic of the typical AEM mechanism (Fig. 6c and Supplementary Fig. 64)47. In contrast, RuO2 exhibits a pH-dependent behavior of OER activity, demonstrating the occurrence of an unstable LOM mechanism. The rotating ring-disk electrode test and the adsorption/desorption energy barrier analysis of Li-containing intermediates indicate that it is difficult for the extracted Li ions to participate in the OER process of RuO2/LiCoO2 (Supplementary Figs. 65–68). Furthermore, RuO2/LiCoO2 maintains the same potential in electrolytes with different Li-ion concentrations after 50 h of stability testing, further confirming that the small amount of extracted Li has a negligible effect on the performance of the catalyst (Supplementary Fig. 69).

a Cyclic voltammetry curves of RuO2/LiCoO2 and RuO2 catalysts in 0.5 M H2SO4. b The function plot between MOR current density and methanol concentration on RuO2/LiCoO2 and RuO2 catalysts. c The pH dependence of the OER potential at various current densities for RuO2/LiCoO2 and RuO2. Operando ATR-SEIRAS spectra of d RuO2/LiCoO2 and e RuO2 at various applied potentials. f Schematic diagrams of the OER mechanism on RuO2/LiCoO2 and RuO2, respectively. g OER Gibbs free energy diagrams of RuO2/Li1−xCoO2, RuO2/LiCoO2, and RuO2, respectively.
The mechanism of the acidic OER reaction of RuO2/LiCoO2 and RuO2 is further monitored by Operando attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) (Supplementary Fig. 70)48. A strong absorption band can be observed in RuO2/LiCoO2 at about 1033 cm−1, which is identified as O-O stretching in the *OOH intermediate in Fig. 6d49. Since *OOH is a typical intermediate of the AEM mechanism, the OER process of RuO2/LiCoO2 is mainly dominated by AEM. In contrast, the characteristic peaks at 1033 and 1122 cm−1 in RuO2 correspond to *OOH and *OO intermediates, respectively, indicating a combined path of LOM and AEM (Fig. 6e)50. Thus, the dynamic reconstruction effect realizes the complete transformation of RuO2/LiCoO2 electrocatalysis from LOM to AEM mechanism, thereby preventing the dissolution of the catalyst and enhancing OER stability (Fig. 6f). The Gibbs free energy barriers of OER reaction intermediates on the electrocatalyst are calculated to explore the activity differences (Supplementary Figs. 71–73). According to Fig. 6g, the OER rate-determining step (RDS) of RuO2/Li1−xCoO2, RuO2/LiCoO2, and RuO2 is the formation energy barrier of the intermediate *OOH. The RuO2/Li1−xCoO2 electrocatalyst only requires an energy barrier of 1.77 eV to overcome RDS, which is much lower than that of RuO2 (2.19 eV). The formation barrier of the *OO intermediate of RuO2/Li1−xCoO2 is much higher than that of *OOH, which is more conducive to the AEM mechanism (Supplementary Figs. 74–81). In addition, the overlap between Ru d and O p orbitals is reduced, further confirming the weakened covalency of the Ru-O bond. This hinders lattice oxygen from participating in the OER reaction process, thereby enhancing structural stability (Supplementary Figs. 82, 83). As a result, the modification of Li1−xCoO2 support optimizes the binding energy of RuO2 with key intermediates, resulting in a favorable thermodynamic process and enhanced intrinsic activity.
Discussion
In summary, we have successfully manipulated the bonding environment of RuO2 during the catalytic reaction by utilizing the dynamic reconstruction of the LiCoO2 support to achieve a balance between activity and stability in acidic OER. Dynamic electrochemical delithiation regulates the electron distribution and coordination structure at the interface, promoting the self-optimization of the RuO2/LiCoO2 catalyst during the OER process. The weakened covalency of the Ru-O bond triggers a complete transition of the RuO2/LiCoO2 electrocatalyst from the LOM to the AEM reaction pathway, improving the catalytic stability. As a result, the RuO2/LiCoO2 electrocatalyst reaches a current density of 10 mA cm−2 at a low overpotential of 150 ± 2 mV. In particular, the PEM electrolyzer using RuO2/LiCoO2 operates stably for 2000 h at a high current density of 1 A cm−2. This work provides a method to solve the balance between the activity and stability of ruthenium-based oxide electrocatalysts in acidic OER.
Methods
Experimental section
Materials preparation
Preparation of LiCoO2 Nanosheets
The stoichiometric amount of Co3O4 (solid, ACS reagent, ≥ 98%) and 4% excess Li2CO3 (solid, ACS reagent, ≥ 98%) was mixed by ball milling for 6 h at 4.47 × g. Subsequently, the precursors were calcined for 4 h at 950 °C in air with a heating rate of 5 °C min−1 to obtain bulk LiCoO2 powder. The bulk LiCoO2 powders were dispersed in distilled water and sonicated for 4 h in an ice-water bath, maintaining the temperature at 15 °C. The supernatant was collected by centrifugation at 698.75 × g. Finally, LiCoO2 nanosheet powders were obtained by freeze-drying the supernatant at −40 °C.
Preparation of RuO2/LiCoO2
The 50 mg of LiCoO2 and 10 mg of RuCl3 (99.9%) were dissolved in 20 mL of deionized water and magnetically stirred at room temperature for 12 h. Then, the precursor powder was collected by centrifugation at 4025 × g. Finally, the obtained powder was calcined in air at 300 °C for 3 h with a heating rate of 5 °C min−1 to obtain RuO2/LiCoO2. For comparison, the amount of RuCl3 was changed to 5 mg and 15 mg to obtain a RuO2/LiCoO2 electrocatalyst with different RuO2 loadings.
Characterization
The morphology of the catalysts was characterized by a scanning electron microscope (SEM JEOL JSM-6700F) and transmission electron microscope (TEM FEI Tecnai G2 F20). Aberration-corrected high-angle annular dark-field scanning transmission electron microscope (AC HAADF-STEM) images were taken at JEM-ARM200F equipped with a JED-2300T SDD. X-ray diffraction (XRD) data obtained from Bruker D8 Advance equipment was used to analyze the crystal structure. The elemental compositions were analyzed by ICP (ICP-MS, Inductively coupled plasma-mass spectrometry). X-ray photoelectron spectroscopy (XPS) analysis was performed on an Escalab 250Xi system using Al Kα X-rays. The Co K-edge and Ru K-edge X-ray absorption spectroscopy (XAS) was measured at the beamline of TLS 17C1 and TPS 44A1 at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan.
Electrochemical measurements
The electrochemical performance was tested on an electrochemical workstation using a three-electrode system (Autolab PGSTAT302, Metrohm). Graphite rod and saturated Hg/HgSO4 electrode were used as counter electrode (CE) and reference electrode (RE), respectively. The catalyst, carbon black, and polyvinylidene fluoride (PVDF) were mixed in a weight ratio of 7:2:1, and N-methyl-2-pyrrolidone (NMP) was used as the solvent. The viscous slurry was evenly coated on carbon paper and dried under a vacuum. The catalyst loading is 1 mgcat cm−2 and the loading area is 1 cm2. The measured potential was converted into a reversible hydrogen electrode according to the equation ERHE = EHg/Hg2SO4 + 0.656 V + 0.0591 pH. Subsequently, CV was measured at a scan rate of 1 mV s−1. The linear sweep voltammetry (LSV) was performed in N2-saturated 0.5 M H2SO4 solution (pH is 0.3 ± 0.1) at a scan rate of 5 mV s−1. The electrolyte was prepared and used immediately and stored in a glass bottle at room temperature. The potential was corrected according to the E=Eapplied - iR formula. The electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 0.01-100 kHz with an amplitude of 5 mV. The double-layer capacitance (Cdl) was obtained by collecting CV curves with scan rates of 10 to 60 mV s−1. OCP tests were performed with H2SO4 saturated with high-purity hydrogen to confirm the reference electrode potential. Calibration experiments were performed at room temperature (25 °C) to reduce temperature effects.
Rotating ring disk electrode (RRDE) test of RuO2/LiCoO2 catalyst. The 5 mg RuO2/LiCoO2 catalyst was dispersed in a mixed solution of 980 μL isopropyl alcohol and 20 μL Nafion (5 wt%), followed by ultrasonication for 1 h to obtain a uniformly dispersed catalyst ink. 20 μL of catalyst ink was dropped onto a disk electrode (area 0.2475 cm2) and dried under vacuum at room temperature to obtain a working electrode. A carbon rod and Hg/HgSO4 were used as the counter electrode and reference electrode, respectively. The linear sweep voltammetry was carried out in 0.5 M H2SO4 solution with different Li-ion concentrations at a scan rate of 5 mV s−1 and a rotation speed of 1600 rpm.
The electrochemical surface area (ECSA) of an electrocatalyst can be evaluated by electrochemical double capacitance (Cdl) according to the following equation:
where Cdl was determined by taking half the slope of the current differences (Δj = janodic - jcathodic) that were plotted as a function of the scan rate in a CV experiment. Cs is the general surface specific capacitance (0.04 mF cm−2).
The turnover frequency (TOF) of the electrocatalyst was calculated by the following equation:
The formate turnover per geometric area was obtained from the geometric current density for the LSV polarization curves according to the equation:
Electrochemical in situ ATR-SEIRAS experiments
ATR-SEIRAS measurements were conducted using a Nicolet iS50 FT-IR spectrometer, with each spectrum obtained by accumulating 32 interferograms, achieving a spectral resolution of 8 cm−1. The working electrode preparation involved two key steps. First, an ultra-thin Au film was chemically deposited onto a silicon crystal to enhance infrared signal sensitivity and electron conductivity. Then, a catalyst slurry with a loading of 0.1 mg cm−2 was applied onto the Au surface. This slurry was prepared by dispersing 7 mg of catalyst and 3 mg of carbon black in 1 mL ethanol, followed by the addition of 50 μL of Nafion after 30 min of sonication. The assembled working electrodes were placed in a three-electrode electrochemical cell, where Hg/HgSO4 served as the reference electrode, a graphite rod as the counter electrode, and Ar-saturated 0.5 M H2SO4 as the electrolyte for the OER reaction51. All measurements were performed using linear sweep voltammetry (LSV) to investigate OER reaction intermediates at various applied potentials.
In situ XAFS measurements
In-situ X-ray absorption spectroscopy (XAS), including both XANES and EXAFS at the Ru and Co K-edges, was conducted in total fluorescence yield mode under ambient conditions at BL-12B2 of SPring-8, NSRRC. The measurements were carried out using a custom-designed Teflon container equipped with a Kepton tape-sealed window, allowing X-rays to pass through both the tape and electrolyte. This setup ensured that XAS signals were effectively captured in total fluorescence yield mode at the National Synchrotron Radiation Research Center (NSRRC), SPring-8. The experiments were performed under a three-electrode configuration, consistent with the electrochemical characterization conditions52. For data processing, spectral normalization was achieved by removing the pre-edge baseline and adjusting the post-edge region. The k2-weighted EXAFS oscillations underwent Fourier transformation to facilitate EXAFS analysis, with all EXAFS spectra presented without phase correction. The Fourier-transformed (FT) data fitting was conducted using Artemis (version 0.9.25), employing a k3 weighting factor with a k-range of 3–12 Å−1 and an R-range of 1.0–4.0 Å. The coordination number, bond length, Debye-Waller factor, and energy shift (CN, R, σ2, and ΔE0) were determined through fitting without any fixed parameters, while the amplitude reduction factor (S02) was set to 0.85.
PEMWE measurements
RuO2/LiCoO2 was used as anode catalysts in PEM electrolyzers. Commercial Pt/C was used as a cathode catalyst. The membrane electrode assembly was fabricated via the catalyst-coated membrane technique, covering a geometric area of 2 cm × 2 cm (4 cm2). The catalyst powder was dispersed in isopropanol, deionized water, and Nafion solution to prepare the ink. The uniformly dispersed ink was obtained by slice emulsification and ultrasonic cell disruption. The well-dispersed RuO2/LiCoO2 and Pt/C catalyst inks were sprayed on both sides of the PEM, respectively. The loadings of RuO2/LiCoO2 anode and Pt/C cathode are 4 mgcat cm−2 and 1 mgcat cm−2, respectively. Nafion 115 was used as a proton exchange membrane (PEM) and was treated with H2O2 and 0.5 M H2SO4 at 80 °C for 1 h in sequence. The size of the proton exchange membrane was 2.6 cm2 and the membrane thickness was 127 μm. The sprayed membrane, anode gas diffusion layer (Ti felt), and cathode gas diffusion layer (carbon paper) were hot-pressed at 130 °C and 10 MPa pressure to obtain a membrane electrode assembly. Subsequently, a PEM water electrolyzer was assembled and tested at 80 °C using pure water as the electrolyte. The polarization curve of PEMWE was obtained at a scan rate of 5 mV s−1, and a chronopotentiometric test was performed at 1 A cm−2 to evaluate the stability.
DFT calculations
All density functional theory (DFT) calculations were conducted using the Vienna Ab initio Simulation Package (VASP)53. The projector augmented wave (PAW) pseudopotential was applied in combination with the PBE generalized gradient approximation (GGA) exchange-correlation functional54. To appropriately describe the localized d-electrons of Co, the DFT + U method was employed, incorporating a Hubbard-U correction of Ueff(Co) = 3.32 eV, determined via linear response theory. The calculations were performed with a plane wave basis set energy cutoff of 500 eV, and a Monkhorst-Pack k-point grid of 3 × 3 × 1 was used for Brillouin zone sampling. Spin polarization was considered, and full structural relaxation was conducted until the energy convergence criterion reached 10−5 eV per atom, with the final force acting on each atom kept below 0.05 eV Å−1. Additionally, Pourbaix diagrams were generated using the Atomic Simulation Environment (ASE), where input formation energies were derived from DFT calculations of bulk and surface models55.
The adsorption energy of reaction intermediates can be computed using the following Equation:
Where ads = OH*, O*, OOH*, an \({\text{E}}_{\text{ads}}-{\text{E}}_{*}\) is the binding energy, \(\Delta {\text{E}}_{\text{ZPE}}\) is the zero-point energy change, \(\Delta \text{S}\) is the entropy change. In this work, the values of \(\Delta {\text{E}}_{\text{ZPE}}\) and \(\Delta \text{S}\) were obtained by vibration frequency calculation.
The Gibbs free energy of the reaction steps can be calculated by the following four Equations:
In this work, \(\Delta {\text{G}}_{1-4}\) were calculated at U = 0.
Data availability
The source data underlying Figures are provided as a Source Data file. Source data are provided with this paper.
References
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science 384, 666–670 (2024).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Wang, L. et al. Optimizing edge active sites via intrinsic in-plane iridium deficiency in layered iridium oxides for oxygen evolution electrocatalysis. Adv. Mater. 36, 2312608 (2024).
Qin, Y. et al. RuO2 electronic structure and lattice strain dual engineering for enhanced acidic oxygen evolution reaction performance. Nat. Commun. 13, 3784 (2022).
Zhu, J. et al. Regulative electronic states around ruthenium/ruthenium disulphide heterointerfaces for efficient water splitting in acidic media. Angew. Chem. Int. Ed. 60, 12328–12334 (2021).
Song, H. et al. RuO2–CeO2 lattice matching strategy enables robust water oxidation electrocatalysis in acidic media via two distinct oxygen evolution mechanisms. ACS Catal. 14, 3298–3307 (2024).
Jin, H. et al. Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution. Nat. Commun. 14, 354 (2023).
Deng, L. et al. Valence oscillation of Ru active sites for efficient and robust acidic water oxidation. Adv. Mater. 35, 2305939 (2023).
Lee, K. et al. Modulating the valence electronic structure using earth-abundant aluminum for high-performance acidic oxygen evolution reaction. Chem. 9, 3600–3612 (2023).
Chen, D. et al. Heteroanion induced structural asymmetricity centered on Ru sites switches the rate-determining step of acid water oxidation. Energy Environ. Sci. 17, 1885–1893 (2024).
Xi, W. et al. Accelerating Ru0/Ru4+ adjacent dual sites construction by copper switch for efficient alkaline hydrogen evolution. Adv. Energy Mater. 13, 2302668 (2023).
Li, L. et al. Lanthanide-regulating Ru-O covalency optimizes acidic oxygen evolution electrocatalysis. Nat. Commun. 15, 4974 (2024).
Xu, Y. et al. Strain-modulated Ru-O covalency in Ru-Sn oxide enabling efficient and stable water oxidation in acidic solution. Angew. Chem. Int Ed. Engl. 63, e202316029 (2024).
Liang, X. et al. Electrocatalytic water oxidation activity-stability maps for perovskite oxides containing 3d, 4d and 5d transition metals. Angew. Chem. Int. Ed. 62, e202311606 (2023).
Qin, Y. et al. Orthorhombic (Ru, Mn)2O3: a superior electrocatalyst for acidic oxygen evolution reaction. Nano Energy 115, 108727 (2023).
Xiao, K., Wang, Y., Wu, P., Hou, L. & Liu, Z.-Q. Activating lattice oxygen in spinel ZnCo2O4 through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023).
Lee, G. R. et al. Efficient and sustainable water electrolysis achieved by excess electron reservoir enabling charge replenishment to catalysts. Nat. Commun. 14, 5402 (2023).
Long, X. et al. Ru-RuO2 nano-heterostructures stabilized by the sacrificing oxidation strategy of Mn3O4 substrate for boosting acidic oxygen evolution reaction. Appl. Catal. B Environ. 343, 123559 (2024).
Du, K. et al. Interface engineering breaks both stability and activity limits of RuO2 for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Jia, H., Yao, N., Yu, C., Cong, H. & Luo, W. Unveiling the electrolyte cations dependent kinetics on CoOOH-catalyzed oxygen evolution reaction. Angew. Chem. Int. Ed. 62, e202313886 (2023).
Sun, Y. et al. Navigating surface reconstruction of spinel oxides for electrochemical water oxidation. Nat. Commun. 14, 2467 (2023).
Zheng, X. et al. Ru–Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212–222 (2021).
Hu, E. et al. Oxygen-redox reactions in LiCoO2 cathode without O–O bonding during charge-discharge. Joule 5, 720–736 (2021).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Funct. Mater. 32, 2200663 (2022).
Zheng, X. et al. Electronic structure engineering of LiCoO2 toward enhanced oxygen electrocatalysis. Adv. Energy Mater. 9, 1803482 (2019).
Zheng, X. et al. Multifunctional active-center-transferable platinum/lithium cobalt oxide heterostructured electrocatalysts towards superior water splitting. Angew. Chem. Int. Ed. 59, 14533–14540 (2020).
Yan, G. et al. Ultrathin two-dimensional medium-entropy oxide as a highly efficient and stable electrocatalyst for oxygen evolution reaction. Nano Res 17, 2555–2562 (2024).
Zhu, W. et al. Stable and oxidative charged Ru enhance the acidic oxygen evolution reaction activity in two-dimensional ruthenium-iridium oxide. Nat. Commun. 14, 5365 (2023).
Ping, X. et al. Locking the lattice oxygen in RuO(2) to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Yao, N. et al. Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem. 9, 1882–1896 (2023).
Deng, L. et al. Activity-stability balance: the role of electron supply effect of support in acidic oxygen evolution. Small 19, e2302238 (2023).
Zhang, X.-L. et al. Efficient acidic hydrogen evolution in proton exchange membrane electrolyzers over a sulfur-doped marcasite-type electrocatalyst. Sci. Adv. 9, eadh2885 (2023).
Rao, P. et al. Single atomic cobalt electrocatalyst for efficient oxygen reduction reaction. eScience 2, 399–404 (2022).
Dan, M. et al. Dual-axial engineering on atomically dispersed catalysts for ultrastable oxygen reduction in acidic and alkaline solutions. Proc. Natl Acad. Sci. USA 121, e2318174121 (2024).
Jian, L., Wang, G., Liu, X. & Ma, H. Unveiling an S-scheme F–Co3O4@Bi2WO6 heterojunction for robust water purification. eScience 4, 100206 (2024).
Wang, Z. et al. Optimizing the oxygen-catalytic performance of Zn–Mn–Co spinel by regulating the bond competition at octahedral sites. Adv. Funct. Mater. 33, 2214275 (2023).
Sun, K. et al. Manipulating the spin state of Co sites in metal–organic frameworks for boosting CO2 photoreduction. J. Am. Chem. Soc. 146, 3241–3249 (2024).
Yang, F. et al. Sub-3 nm Pt@Ru toward outstanding hydrogen oxidation reaction performance in alkaline media. J. Am. Chem. Soc. 145, 27500–27511 (2023).
Tian, X. et al. Synergy of dendrites-impeded atomic clusters dissociation and side reactions suppressed inert interface protection for ultrastable Zn anode. Adv. Mater. 36, 2400237 (2024).
Sun, Z. et al. Lattice strain and mott–schottky effect of the charge-asymmetry Pd1Fe single-atom alloy catalyst for semi-hydrogenation of alkynes with high efficiency. ACS Nano 18, 13286–13297 (2024).
Yu, Z.-Y. et al. General synthesis of tube-like nanostructured perovskite oxides with tunable transition metal–oxygen covalency for efficient water electrooxidation in neutral media. J. Am. Chem. Soc. 144, 13163–13173 (2022).
Kuang, J., Deng, B., Jiang, Z., Wang, Y. & Jiang, Z.-J. Sr-stabilized IrMnO2 solid solution nano-electrocatalysts with superior activity and excellent durability for oxygen evolution reaction in acid Media. Adv. Mater. 36, 2306934 (2024).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Wei, Y. et al. Triggered lattice-oxygen oxidation with active-site generation and self-termination of surface reconstruction during water oxidation. Proc. Natl Acad. Sci. USA 120, e2312224120 (2023).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659–23669 (2023).
Rong, C. et al. Defect-balanced active and stable Co3O4−x for proton exchange membrane water electrolysis at ampere-level current density. Energy Environ. Sci. 17, 4196–4204 (2024).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical CO2 reduction and C–C coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935–1945 (2024).
Lin, Y. et al. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 12, 5345–5355 (2022).
Zhang, T. et al. Spatial configuration of Fe–Co dual-sites boosting catalytic intermediates coupling toward oxygen evolution reaction. Proc. Natl Acad. Sci. USA 121, e2317247121 (2024).
Hao, Y. et al. Designing neighboring-site activation of single atom via tunnel ions for boosting acidic oxygen evolution. Nat. Commun. 15, 8015 (2024).
Hung, S.-F. et al. Unraveling geometrical site confinement in highly efficient iron-doped electrocatalysts toward oxygen evolution reaction. Adv. Energy Mater. 8, 1701686 (2018).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Hjorth Larsen, A. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52371226, L.L.L and 52371226, S.J.P), and the Scientific and Technological Innovation Special Fund for Carbon Peak and Carbon Neutrality of Jiangsu Province (BK20220039, S.J.P). This work was also supported by the 2030 Cross-Generation Young Scholars Program of the National Science and Technology Council in Taiwan under the grant NSTC 112-2628-E-007-014-MY4 to Dr. Han Yi-Chen. The authors thank the National Synchrotron Radiation Research Center, Hsinchu, Taiwan, for providing synchrotron XAS beamline TLS 17C1 and TPS 44A1.
Author information
Authors and Affiliations
Contributions
L.W., S.Z., L.L., F.H. and S.P. designed the study. L.W. and S.P. conducted the experiments. L.W., S.F.H., J.J.M., C.Z., Y.Z., T.Y.C. and H.Y.C. participated in the characterization of the samples. L.W., Y.W., S.B., S.L., Y.W. and S.P. analyzed data. L.W. wrote the paper. S. P. conceived the idea and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sung Jong Yoo, Ronghui Qi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Hung, SF., Zhao, S. et al. Modulating the covalency of Ru-O bonds by dynamic reconstruction for efficient acidic oxygen evolution. Nat Commun 16, 3502 (2025). https://doi.org/10.1038/s41467-025-58654-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58654-0
This article is cited by
-
Electronic tuning of RuO₂ polarizes metal–oxygen redox for proton exchange membrane water electrolysis
Nature Communications (2025)
