
Abstract
Obesity is a major and increasingly prevalent chronic metabolic disease with numerous comorbidities. While recent incretin-based therapies have provided pharmaceutical inroads into treatment of obesity, there remains an ongoing need for additional medicines with distinct modes of action as independent or complementary therapeutics. Among the most promising candidates, supported by phase 1 and 2 clinical trials, is cagrilintide, a long-acting amylin and calcitonin receptor agonist. As such, understanding how cagrilintide functionally engages target receptors is critical for future development of this target class. Here, we determine structures of cagrilintide bound to Gs-coupled, active, amylin receptors (AMY1R, AMY2R, AMY3R) and calcitonin receptor (CTR) and compare cagrilintide interactions and the dynamics of receptor complexes with previously reported structures of receptors bound to rat amylin, salmon calcitonin or recently developed amylin-based peptides. These data reveal that cagrilintide has an amylin-like binding mode but, compared to other peptides, induces distinct conformational dynamics at calcitonin-family receptors that could contribute to its clinical efficacy.
Similar content being viewed by others

Introduction
Obesity is a major chronic disease that continues to increase in prevalence worldwide and constitutes a key risk factor for development and progression of numerous related diseases including hypertension, dyslipidaemia and type 2 diabetes1. As such, it creates a substantial health burden and a parallel need for medicines to support weight reduction. The last 5 years have seen the emergence of efficacious peptide-based therapies for obesity treatment that target glucagon-like peptide-1 receptor (GLP-1R), either selectively or via co-activation with related receptors, such as the glucose-dependent insulinotropic polypeptide and/or glucagon receptors1. Nonetheless, there remains an ongoing need for additional medicines, particularly those with a distinct mode of action that could be used independently or in combination with the current incretin-based therapeutics. Among the most promising non-incretin receptor targets are amylin (Amy) receptors (AMYRs), supported by data from phase I and phase II clinical trials showing efficacy of the long-acting amylin and calcitonin receptor agonist, cagrilinitide (Cagri), both as monotherapy2,3 and in combination with the selective GLP-1R agonist, semaglutide4,5.
AMYRs are heterodimers of the class B1 G protein-coupled receptor, calcitonin (CT) receptor (CTR), and receptor activity-modifying proteins (RAMPs)6,7. All three RAMPs can interact with CTR, and this gives rise to three discrete Amy receptor phenotypes, AMY1R, AMY2R and AMY3R, with RAMP1, RAMP2, and RAMP3, respectively7,8. The CTR is also expressed at the cell surface independently of RAMPs and has a distinct phenotype, exhibiting potent responses to human, and other species of CT peptides, but weak responses to amylin. In contrast, AMYRs have high affinity and potency in response to amylin peptides, but weak response to human CT (hCT)7,8. Both selective AMYR agonists, and dual CTR and AMYR agonists such as sCT5,6,7, have demonstrated efficacy in controlling appetite and body weight, however this is not seen with a CTR-selective agonist9. This led to the assumption that the primary receptors mediating weight loss for this target class were AMYRs, yet, dual agonists are reported to have higher efficacy than AMYR selective agonists2,10,11,12. Emerging data suggests that co-activation of both CTR and AMYRs may confer greater efficacy in metabolic control than selective activation of AMYRs13.
Dual CTR and AMYR agonist peptides have been developed on either a sCT backbone (32 amino acids) or an amylin backbone (37 amino acids) (Fig. 1a). While agonist peptides based on either scaffold are potent in rat obesity models2,10,11,12,13, only a transient weight loss effect is observed with sCT in mouse diet-induced obesity (DIO) models14, indicating that there may be distinctions in the mode of action of individual dual agonists. Recent studies of peptide-bound, active, Gs-coupled, structures of CTR or AMYRs provide evidence supporting distinct conformations of receptors when bound to CT peptides versus amylin peptides15 that may contribute to pharmacological differences between dual CTR and AMYR agonists. These studies emphasize the need to understand how the clinically efficacious dual agonist, Cagri (Fig. 1a), engages with AMYRs and CTR.

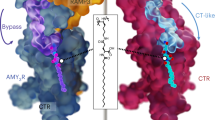
a Amino acid sequence alignment of CT and Amy agonists. Red type indicates residues that differ from pramlintide, the backbone peptide used for generation of cagrilintide. K[Cagri] indicates the position of the lipid modification of cagrilintide with the chemical structure of the conjugated lipid shown below the sequence alignment. There is a disulphide bridge between Cys2 and Cys7 of amylin-based peptides and between Cys1 and Cys7 of sCT. All peptides are C-terminally amidated. b–h Cryo-EM maps and model comparison of active, Gs-coupled complexes of AMYRs and CTR bound to cagrilintide (Cagri). b–e cryo-EM maps shown in surface representation of AMY1R-Gs (b), AMY2R-Gs (c), AMY3R-Gs(d) and CTR-Gs (e) in complex with Cagri. f–h Model comparison of Cagri-AMY1R and Cagri-CTR (f), Cagri-AMY1R, Cagri-AMY2R, and Cagri-AMY3R (g), Cagri-CTR and sCT-CTR (h). Structures were aligned on the TMD (residues 139-409) of the CTR protomers with protein backbone displayed in ribbon format. In (f–h) the Gs protein is not shown. EM map surface and protein chains are coloured according to the labelling on the panel, except for (g) in which Cagri was coloured as same as the corresponding RAMP protomer. The displayed maps were contoured to the following levels: (a) AMY1R, 0.0144, (b) AMY2R, 0.0188, (c) AMY3R, 0.406/0.173 (ECD focused map), (d) CTR, 0.0129.
In this study, we used cryogenic electron microscopy (cryo-EM) to determine structures of Cagri bound to Gs-coupled, active, AMY1R, AMY2R, AMY3R and CTR and compared the mode of interaction of Cagri and the associated dynamics of the Cagri-bound receptor complexes with previously reported cryo-EM structures and dynamics of receptors bound to rat amylin (rAmy, all receptor subtypes), sCT (CTR, AMY1R, AMY2R)15 or recently developed amylin-based peptides (CTR and/or AMY3R)16. These data revealed that Cagri generally had a similar binding mode to amylin peptides but distinct receptor complex dynamics at the calcitonin-family receptors compared to other peptides.
Results and discussion
Structure determination
Human CTR, plus or minus individual RAMPs, and a stabilised form of Gαs (dominant negative Gs (DNGs)) and Gβ1γ2, were co-expressed in Trichoplusia ni insect cells. The assembly of complexes with Gs protein was stimulated with 10 μM peptide and stabilised by the addition of nanobody 35 (Nb35) and removal of nucleotides with apyrase. Affinity purified protein samples were further resolved by size exclusion chromatography and assessed by SDS-PAGE and negative stain TEM before vitrification and cryo-EM imaging (Supplementary Fig. 1). The resolutions of the final consensus maps were 2.2 Å, 2.2 Å, 2.7 Å and 3.0 Å for the CTR, AMY1R, AMY2R and AMY3R, respectively, determined at gold standard Fourier shell correlation (FSC) 0.143 (Fig. 1b–e; Supplementary Fig. 1, Supplementary Table 1). Apart from the α-helical domains of the DNGαs that were poorly resolved and masked out, the bound peptide, individual receptor components and heterotrimeric DNGs protein were visible in all maps, allowing robust modelling for all major components of the complexes. To improve the local resolution of CTR-RAMP extracellular domains (ECDs), focused refinements (masking around receptors or ECD domains) were performed. For these regions, the side chains were stubbed in the final PDB models if no robust density was present in the EM maps. In particular, the Cagri-AMY3R complex maps displayed weaker density of the RAMP3 ECD, and the complete ECD is only visible in its entirety in the ECD-focused map at high contour level, indicative of a higher dynamics of the AMY3R ECD relative to the receptor core (Fig. 1d). Therefore, only the backbone is modelled in the final pdb file. Cagri is N-terminally acylated with a γGlu linker and a C20 fatty diacid moiety. There was limited density for the conjugated lipid, which could only be resolved in high-resolution receptor-focused maps of Cagri-AMY1R-Gs and Cagri-CTR-Gs structures, albeit modelling was restricted to the γGlu linker and an adjacent short segment of the fatty diacid as the remainder of the modification was not resolved. These postprocessed focused maps with high-resolution features also enabled structural waters to be modelled ab initio. To interrogate the potential effect of the lipidation on the Cagri-bound structures, we also determined structures of the CTR and AMY1R bound to a Cagri analogue lacking the N-terminal lipidation (CagriBB). The biochemistry, processing and maps are shown in Supplementary Fig. 2. The final consensus maps for both complexes had resolutions of 3.0 Å, enabling confident modelling of all the major features of the complexes. Interactions formed by Cagri, CagriBB (and compared with rAmy) with AMYRs and CTR are reported in Supplementary Tables 2 and 3.
Structural features of active, Cagri-bound, AMY1R, AMY2R and AMY3R complexes
Overall, the consensus structures of AMYRs bound to Cagri or CagriBB exhibited highly similar backbone conformations of the peptide and CTR subunit (Fig. 1g and Supplementary Fig. 3), and the backbone of each complex also closely resembled the previously determined active structures of individual AMYRs bound to rat Amy (rAmy)15 (Fig. 2a, c, e Supplementary Figs. 4a,c and 5a,c), as well as the AMY3R bound to San385 [S19K-pramlintide]16. As expected, the engagement mode of Cagri with AMYRs is mainly conserved with that of rAmy and characterized by the formation of a ‘bypass’ motif (residues S19Cagri-P25Cagri) (Fig. 2b and Supplementary Fig. 4b, 5b), however notable conformational differences were observed around residues (acylated K1Cagri, 14ECagri, 17RCagri and 37PCagri) that diverge from rAmy and pramlintide, an Amy peptide approved clinically for diabetes6,7.

a Model comparison of the Cagri-AMY1R and rAmy-AMY1R at the global backbone level (Gs protein not shown). b interactions within the bypass motif of Cagri (residues 19-25). c interactions in Cagri-AMY1R arising from the Cagri ionic lock (E14Cagri-R17Cagri) in comparison with rAmy-AMY1R interactions, with (d) the corresponding map to model for Cagri-AMY1R. (e) Cagri interactions in AMY1R as a result of Cagri N-terminal lipidation, in comparison with equivalent rAmy-AMY1R interactions, with (f) corresponding map to model for Cagri-AMY1R. The displayed maps were contoured to the following levels: (d) 0.01555, (f) 0.0138. Roman numerals indicate transmembrane helix numbers, H8-helix 8, ICL- intracellular loop, ECL- extracellular loop.
As noted above, the mid-region residues (S19Cagri-P25Cagri) form the prototypical ‘bypass’ motif that was observed for rAmy/San385, constrained by conserved intramolecular hydrogen bonds between the backbone of L16Cagri/F23Cagri and S20Cagri, and the backbone of P25Cagri and N21Cagri (Fig. 2b and Supplementary Fig. 4b). The phenyl ring of F23Cagri forms extensive van der Waals contact with L16Cagri, Y1491.40, Y1461.37(superscript numbers for receptor residues refer to the class B1 GPCR numbering scheme17), anchoring the peptide segment with the CTR transmembrane (TM) bundle level with the micelle interface. Thus, these conserved interactions promote the formation and stability of the bypass motif, which is a key structural determinant for the high potency of Amy analogues at AMYRs15,16.
To counteract amyloid fibril formation that occurs with human Amy, a pair of helix-stabilizing mutations (N14E and V17R) were incorporated into Cagri2. This was predicted to stabilise the N-terminal α-helix (5-18) of the peptide through formation of an intramolecular salt bridge, as observed in a crystal structure of CagriBB fused at the C-terminal end of maltose-binding protein (PDB:7BG0)2. This intramolecular salt bridge is present on the same face of the peptide α-helix in the active state AMYR structures and rotamers of both residues constrain each other with their sidechains located close to ECL2 of the CTR protomer (Fig. 2c–d; Supplementary Fig. 4c, d, Fig. 5c, d). Specifically, E14Cagri adopts a downward rotamer to form hydrogen bonds and electrostatic interactions with R17Cagri. In contrast, in AMYRs bound to rAmy15 or the pramlintide analogue, San38516, an Asn at the equivalent peptide position (N14rAmy) adopts an upward rotamer that is exposed to the solvent environment and forms a H-bond with the backbone of L291ECL2 (Fig. 2c). Steric hindrance from the increased side-chain volume of E14Cagri and R17Cagri promotes a shift of the α-helix of Cagri away from ECL2 and a slight expansion of the peptide binding pocket at the extracellular side of the CTR TM domain, relative to those observed with rAmy/San385-bound AMYR complexes.
The lipid modification at the Cagri N-terminus is poorly resolved in the cryo-EM maps for AMYR complexes, but some limited density was visible for the γGlu linker and a very short segment of the acyl chain in the AMY1R complex (Fig. 2e and f). The γGlu linker exits the receptor TM bundle between H296ECL2 and W3616.58 and the following short acyl segment merges into the detergent micelle, adjacent to a conserved lipid density (Fig. 2f) that is also observed in the rAmy-AMY1R complex15. Comparison with structures of the AMY1R bound to non-lipidated peptides (CagriBB and rAmy) revealed that the lipid conjugation does not affect the conformation of the peptide, nor the receptor when Cagri is stably engaged. The exception to this was K1Cagri, the site of lipid conjugation, which adopts a different rotamer to K1rAmy. However, CagriBB has equivalent potency to Cagri, in CRE-reporter assays in mammalian cells expressing AMY3Rs (Supplementary Table 4), suggesting there is negligible impact of this rotamer difference. This is consistent with tolerance of K1A substitution in human Amy (hAmy) for cAMP potency at AMYRs18.
The C-terminal ~10 amino acids of CT-family peptides, including CT, rAmy, and Cagri, bind to the same groove within the ECD of their cognate receptors (Fig. 3). The amidated C-terminal residue differs across CT-family peptides and this residue forms the only direct contact with RAMPs in previously reported AMYR complexes15,16. Moreover, the chemical nature of the C-terminal residue side chain determines ligand contact with RAMP and can influence activity at AMYRs and CTR18,19. An amidated C-terminal tyrosine is not tolerated for high affinity peptide interactions with the CTR in the absence of RAMPs. This is evidenced by the negligible affinity of amylin or pramlintide for the isolated CTR ECD19,20, and by loss of affinity and/or potency of P32Y substituted CT peptides at the CTR18,20. In contrast, substitution to an amidated proline instead of an amidated tyrosine in the C-terminus of rAmy is well tolerated, resulting in enhanced affinity and activity at both the CTR and AMYRs, but, in some cases, reduced selectivity between these receptors18.

a–e model comparison of the interactions of C-terminal residues of Cagri, rAmy and sCT with receptor ECDs: (a) P37Cagri-AMY1R vs Y37rAmy-AMY1R, (b) P37Cagri-AMY1R vs P32sCT-AMY1R, (c) P37Cagri-AMY2R vs Y37rAmy-AMY2R, and (d) P37Cagri-AMY2R vs P32sCT-AMY2R, (e) P37Cagri-AMY3R vs Y37rAmy-AMY3R. Structures were aligned on the ECD (residues 41-138) of the CTR protomer with protein backbone displayed in ribbon format. H-bonds are shown as dashed lines. The rigid-body translocations of RAMP ECD in the presence of Cagri relative to the rAmy are highlighted by the red arrows. f Map to model representation of the ECD interface of RAMP3 and CTR in complex with Cagri. The displayed map was contoured to 0.234.
A structural explanation for these findings was provided from our previous cryo-EM structures of CT(Pro32) and rAmy(Tyr37)-bound AMYRs structures where the bulky phenolic side chain of Y37rAmy forms perpendicular π stacking interactions with W79ECD 15, and additional interactions with the respective RAMP protomer that are likely important for selectivity of rAmy for AMYRs over CTR. The nature of these interactions is likely also dependent upon the ability of the CTR ECD to adopt the conformation induced by the bypass motif of Amy-template peptides; sCT when bound to AMYRs induce a CT-like receptor conformation15, while P32Y substitutions of CT analogues are also detrimental to affinity and potency at AMYRs18,19. Our structures of Cagri-AMYR-Gs complexes allow direct examination of the influence of a C-terminal proline in an amylin-template peptide through comparison of the interactions formed by P37Cagri to that of Y37rAmy with the CTR and AMYRs.
In all three AMYRs, P37Cagri and Y37rAmy occupy the same pocket above W79ECD of the CTR ECD where they form conserved interactions with surrounding residues (Fig. 3), including H-bonds with the backbone of S129ECD, and hydrophobic interactions with D77ECD, G78ECD, W79ECD, Y131ECD and W128ECD, consistent with interactions also formed by Pro32 of sCT. In the AMY1R and AMY3R Y37rAmy interacts with residues in the respective RAMP and the CTR ECD, however the small side chain of P37Cagri is completely buried by the surrounding CTR ECD residues, forming extensive hydrophobic contacts with W79ECD and no direct interactions with either RAMP1 (AMY1R) or RAMP3 (AMY3R), again consistent with the restrictive interactions formed by Pro32sCT with the CTR ECD. The lack of interactions of P37Cagri with RAMP 1 and 3 is consistent with the ability of Cagri to form high-affinity interactions with the CTR protomer alone (Fig. 3a). In both Cagri- and rAmy-bound AMY1R structures, F83RAMP1 packs over R126ECD stabilising the same interface of the RAMP1-CTR ECD when bound by both peptides15. There is also good alignment in the structures of the Cagri C-terminal residues and those of sCT in complex with AMY1R, and of the RAMP1 and CTR ECDs and the interface between them, when aligned by the CTR protomer ECD (Fig. 3b), albeit that the overall CTR/RAMP1 ECD orientation relative to the core differs between these structures. At the AMY3R, the RAMP3/CTR interface for the Cagri-bound AMY3R appears to be weaker than the rAmy-AMY3R where Y37rAmy forms a H-bond interaction with E74RAMP3 that stabilises the position of the ECD and the interface between the RAMP3 and CTR ECDs. In contrast, in the absence of direct interactions the RAMP3 ECD moves away from P37Cagri and from the ECD of the CTR protomer. While the specific details of the ECD interface of Cagri-AMY3R could not be determined due to the low-resolution maps, these findings are indicative of substantial loss of RAMP3/CTR ECD interface interactions and considerable dynamic motions within the RAMP3 and CTR ECD protomers (Fig. 3f).
In the Cagri-bound AMY2R complex the small G110RAMP2, positionally equivalent of F83RAMP1/Y83RAMP3 allows the RAMP2 ECD to orient closer to P37Cagri (<4 Å) than the ECD of RAMP1 or 3, facilitating direct hydrophobic contacts between N35Cagri and the backbone of G110RAMP2 and F111RAMP2 (Fig. 3c). This proximal position of the RAMP2 ECD is conserved in AMY2R complexes bound to CT peptides that also have a C-terminal Pro (P32CT) (Fig. 3d), however, this is in the context of the CT-AMY2R complex adopting a “CT” conformation, compared to the “bypass” conformation stabilised by Cagri. In contrast, the bulkier Y37rAmy in rAmy-bound AMY2R prevents equivalent interactions of N35rAmy with RAMP2, and instead the bulkier Y37rAmy forms interactions with E101RAMP2 and F111RAMP2 stabilising the peptide at the CTR/RAMP ECD interface in the “bypass” conformation.
3D variability analysis (3DVA) of the AMYR complexes revealed that the Cagri-AMY1R-Gs and Cagri-AMY3R-Gs complexes had increased conformational dynamics at the extracellular side relative to equivalent rAmy-AMYR complexes (Fig. 4). As discussed above, unlike Y37rAmy, P37Cagri does not interact directly with RAMP1 or RAMP3, which likely decreases the stability of the ECD interface, leading to increased dynamics relative to the rAmy-bound AMY1R and rAmy-bound AMY3R (Fig. 4a, b, e and f). The reverse was true for the AMY2R complexes (Fig. 4c and d). Here, the previously determined rAmy-AMY2R-Gs was highly dynamic at the extracellular side, transitioning between the consensus (bypass) AMYR conformation and a CT-like conformation (Fig. 4d15). This was associated with substantive mobility of the RAMP2 linker and TM helix, which had weaker interaction with the CTR TM bundle compared to RAMP1 and RAMP3 in AMY1R and AMY3R complexes. In contrast, the Cagri-AMY2R-Gs complex was stable in the consensus AMYR conformation with stable TM interaction between RAMP2 and CTR, and the peptide maintained a bypass motif. This difference is likely to be related to the more tightly packed ECD interface of RAMP2 and CTR, driven by the interactions with P37Cagri, and the preferential CTR engagement by Cagri via a bypass conformation, as discussed below in the context of CTR complexes.

a–g Overlay of backbone models of receptor and peptide fitted to the two extreme maps: frame 000 and 019 from the main principal component of the 3DVA. The degree of ECD motion was calculated through measurement of the distance between the Cα of E123CTR between the two frames revealing differential movement of the CTR ECD in all complexes. a Cagri-AMY1R, (b) rAmy-AMY1R, (c) Cagri-AMY2R, (d) rAmy-AMY2R, (e) Cagri-AMY3R, (f) rAmy-AMY3R, (g) San385-AMY3R.
The major conformational dynamics of AMY1R-Gs and AMY2R-Gs bound to individual peptide agonists were primarily related to motions of the combined RAMP:CTR ECD relative to the TM core, and this was also true for previously determined AMY3R-Gs complexes with rAmy (Fig. 4f15) or selective (San385, Fig. 4g16) and dual agonist (San45) pramlintide analogues16. Intriguingly, the Cagri-AMY3R complex exhibited more distinct dynamics when compared to other complexes, with the RAMP3 ECD displaying substantive motion relative to the CTR ECD; this loss of stable ECD-ECD interaction was associated with increased motions of the RAMP-proximal loops of the CTR ECD (7-angstrom motion, measured at Cɑ of E123ECD; Fig. 4e). These motions are consistent with the loss of key interactions mediated by Tyr37 of amylin peptides and the poor resolution of the RAMP3 ECD in the 3D EM map reconstructions. This hyper ECD motion was propagated to the receptor TMD, with motions within TMs 3 and 7, creating a wider TM pocket that also influences the positioning of the N-terminus of the Cagri peptide (Fig. 4e) and was not seen in other Cagri-AMYR complexes that had stable TM interactions. This dynamic feature also causes disengagement of F23Cagri, and a loss of map density for the peptide residues proximal to Y1491.40 (Supplementary Fig. 5). In the AMY3R consensus maps, density allows potential modelling of an alternate rotamers of F23Cagri in antiparallel with Y1461.37 (Supplementary Fig. 5c and d). A similar rotamer change was observed in the San385-AMY3R complex that displayed a 9 Å ECD motion in 3DVA (Fig. 4g). However, for San385, the higher ECD dynamics were driven by increased flexibility of the bypass motif induced by the S19KSan385 substitution; the RAMP3 and CTR ECDs move in parallel with stable interactions at the interface that are well resolved in focused cryo-EM maps16. Despite the substantial conformational dynamics when bound to AMY3R, Cagri maintains the bypass conformation across the motions, likely arising from the consistent TMD and linker interactions between RAMP3 and CTR that enable robust engagement of residues within the Cagri bypass motif.
To further interrogate the underlying dynamics of the Cagri-AMY3R complex, relative to rAmy-AMY3R complex, we performed molecular dynamics (MD) simulations of Cagri:AMY3R:Gαs and rAmy:AMY3R:Gαs (each system in triplicate, for a total of 6 μs). Results are summarised in Supplementary Figs. 6 and 7, Supplementary Tables 4-8 and Supplementary Movie 1. Overall, the MD data was consistent with the markedly different dynamics observed in the cryo-EM 3DVA with the ECDs undergoing substantially greater dynamic motions in the Cagri-AMY3R complex than observed in the parallel rAmy-AMY3R complex (Supplementary Fig. 6a–e). Moreover, the simulations confirmed that the CagriP37 had weaker interactions with the ECDs leading to greater uncoupling of the peptide C-terminus from the ECDs (Supplementary Movie 1, Supplementary Tables 4, 5), and parallel increases in the dynamics of the peptide as it engaged with the CTR TM bundle, including F23. Within the peptide mid-region, the introduced ionic lock CagriR17-E14 introduced increased stability to this region of the peptide (Supplementary Fig. 7a–c, Supplementary Tables 7 and 8) and led to greater relative engagement of this peptide with TMs 5, 6 and 7, relative to rAmy, which interacted more with ECL2 (Supplementary Fig. 6h and i).
Collectively, our data reveal that Y37P substitution in Cagri (from Amy) promotes differential engagement of the Cagri peptide with each RAMP that contributes to distinct conformational dynamics from those observed for each individual rAmy-bound AMYR complex.
Structural features of Cagri-bound active CTR complexes
Previous structural analysis of CTR-Gs bound to rAmy revealed that the receptor complex was dynamic, adopting distinct states where the receptor was resolved into two major conformations, a bypass (55%, PDB: 7TYL) and CT-like (45%, PDB: 7TYI) conformation15. In the latter conformation, the rAmy bypass motif becomes disordered, enabling the receptor ECD to adopt a position similar to CT-bound CTR or AMYR complexes15. The structural data supports an ability of the bypass motif to transition between structured and unstructured conformations in the absence of the allosteric constraint from a RAMP protein. In contrast to rAmy, Cagri primarily engages the CTR in a bypass conformation, equivalent to that observed when bound to AMYRs, with 85% of particles in the final particle stack stabilised in this conformation (Fig. 1e–f, Supplementary Fig. 8a). This includes the peptide and CTR (including the ECD position) that adopt the same conformation to those observed in Cagri-bound AMYR-Gs complexes, assessed by overlaying the backbones of the structures (Fig. 1f), and distinct from the ECD orientation of CTR-Gs bound to sCT (Fig. 1h). Thus, Cagri appears to increase CTR engagement via a distinct mechanism to San45 that adopts a stable CT-like ECD conformation16. This enables Cagri to form conserved interactions when bound to CTR and AMYRs, including the inter-peptide contacts that facilitate the formation and stabilisation of the bypass motif and the intramolecular salt bridge (E14Cagri and R17Cagri). Like at AMYRs, these features require the expansion of extracellular TM bundle of CTR through outward movements of TM1, ECL1/TM3, TM4/ECL2 and a small shift in the positioning of the mid-region of the peptide at the start of the N-terminal helix, relative to rAmy when bound to the CTR in the bypass conformation (Fig. 5a–d).

a Model comparison of the Cagri-CTR and rAmy-CTR (bypass) at the global backbone level. b conserved interactions within the bypass motif of Cagri (residues 19-25). c interactions in the Cagri-CTR, arising from the Cagri ionic lock (E14Cagri-R17Cagri), in comparison to rAmy-CTR interactions in the bypass conformation; (d) the corresponding map to model for Cagri-CTR. e Cagri interactions in CTR as a result of Cagri N-terminal lipidation, in comparison with equivalent Cagri-AMY1R interactions in the bypass conformation; (f) corresponding map to model for Cagri-CTR. The displayed maps were contoured to the following levels: (d) 0.022 (receptor-focused map), (f) 0.011 (consensus map). Roman numerals indicate transmembrane helix numbers, H8-helix 8, ICL—intracellular loop, ECL—extracellular loop.
Further ECD-focused 3D classification revealed an alternate CT-like conformation induced by Cagri when bound to the CTR, however, this conformation was only observed in 8% of the particles (Fig. 6a–g, and Supplementary Fig. 8a), indicating that while the Cagri-CTR complex can sample this conformation, it occurs at lower frequency than rAmy-CTR. In the CT-like conformation, the bypass motif becomes unstructured, and residues (S21-P28) are not well resolved, suggesting this flexible segment is exposed to the solvent environment. The TM1 stalk (135-138) of CTR in the Cagri CT-like conformation also undergoes rearrangement relative to the bypass conformation, in which the phenyl of F1371.28 packs against F15Cagri, H18Cagri and S19Cagri (Fig. 6c and d). These structure features are conserved in the CTR bound to amylin-based peptides (rAmy and San45) in CT-like conformations (Fig. 6d). However, this conformational change reduces the space available to accommodate the α-helix of the peptide increasing the steric hindrance between the bulky ionic lock (R17Cagri-E14Cagri) and ECL2. As such, the side chain of E14Cagri rotates slightly downwards and reduces the number of H-bonds with R17Cagri. Relative to rAmy bound to CTR, Cagri is likely to require extra energy to weaken the salt bridge to attain a CT-like state, thus providing a structural rationale for the smaller population of Cagri-bound particles sampling a CT-like conformation. To further confirm this potential shift in the conformational equilibrium, we collected a second cryo-EM dataset for Cagri-CTR (N = 2) from a separate protein purification. While the number of particles in the second reconstruction was much lower than the first, the data were consistent with a lower number of particles in a CT-like conformation (~ 17% of particles) relative to the bypass conformation (55% of particles) compared to the percentage of particles previously observed for the Amy-CTR-Gs complex (note, in the repeat experiment 28% of the particles could not be confidently classified, due to poorly resolved in ECD features) (Supplementary Fig. 8b).

a, b Model comparison of (a) the Cagri-CTR CT-like conformation and the Cagri-CTR bypass conformation, and (b) Cagri-CTR CT-like conformation vs rAmy-CTR CT-like conformation. c, d the conserved rotamer rearrangement of F137 in the CT-like conformation showing the influence of the Cagri ionic lock rearrangements (E14Cagri-R17Cagri) when comparing (c) the Cagri-CTR CT-like vs bypass conformations and (d) the Cagri-CTR CT-like conformation vs the rAmy-CTR CT-like conformation. e the conserved position of the N-terminal lipidation in Cagri-CTR in the CT-like and bypass states, with (f, g) showing the corresponding map to model representation. The displayed maps were contoured to the following levels: f 0.912 (receptor-focused map), (g) 0.553 (receptor-focused map). Roman numerals indicate transmembrane helix numbers, H8-helix 8, ICL- intracellular loop, ECL- extracellular loop.
In the initial larger, higher resolution, dataset of Cagri-CTR-Gs (N = 1), density was observed for the proximal region of the conjugated lipid at the peptide N-terminus that was consistent regardless of peptide conformation (bypass or CT-like) and supports modelling the γGlu linker and a short segment of acyl chain in a different orientation to that observed in the AMY1R complex (Figs. 5e–f, 6e and g). The K1Cagri rotamer resides in a similar position to the non-acylated K1rAmy 15, and this contrasts with the orientation observed in the Cagri-AMY1R-Gs complex. H296ECL2 adopts a distinct rotamer in Cagri-bound CTR relative to Cagri-bound-AMY1R to accommodate the γGlu linker that extends proximal to the top segment of TM5. The following acyl chain segment merges back into the micelle where density can be observed running along the TM5 helix. ECL3 and the top of TM7 are tilted slightly away from the peptide, adopting a marginally more open TM bundle than observed in AMYRs. This is likely due to a reduction of peptide interactions (mainly A5Cagri) with ECL3 that is a consequence of the engagement of the lipidated N-terminus with the top segment of TM5/ECL2, facilitating flexibility of ECL3. In contrast, in the Cagri-AMY1R-Gs structure, L119RAMP1 is likely to clash with a rearranged imidazole side chain of H296ECL2 that is observed when Cagri is bound to CTR and likely accounts for the different orientation of the linker in the Cagri bound AMY1R (Fig. 5e). While density for the N-terminal lipidation was not visible in the lower resolution cryo-EM map of the repeat Cagri-CTR-Gs complex (N = 2), the outward rotamer of H296ECL2 was observed, congruent with similar interactions between the N-terminal acylation and ECL2. However, the consensus map from the repeat experiment revealed that the ECL3 conformation differed very slightly from the initial experiment (Supplementary Figs. 8 and 9), potentially due to the weaker interactions between Cagri and ECL3. 3DVA performed on the particle stacks from the initial larger dataset revealed that ECL3 is dynamic, consistent with the identification of two slightly differing ECL3 conformations from individual experiments (discussed in more detail below). Interestingly, the position of ECL3 in the two consensus maps correlated with the presence (more open ECL3) or absence (more closed ECL3) of an intact ionic lock between R17Cagri and E14Cagri, whether in the bypass or CT-like conformation, suggesting that both the ionic lock and the positional anchoring of the peptide by the lipid interactions play a role in favouring the Cagri bypass conformation in the Cagri-CTR-Gs complex. Moreover, the greater stability of the Cagri-induced bypass conformation relative to the rAmy-induced bypass conformation, when engaged with the CTR, is likely a major contributor to the increased conformational stability of the Cagri-AMY2R-Gs complex relative to the rAmy-AMY2R-Gs complex.
Due to the distinct position of the conjugated N-terminal lipid between Cagri-CTR and Cagri-AMY1R, we also determined a structure of CTR-Gs in complex with an analogue of Cagri that lacked the lipid modification (CagriBB) to interrogate the contribution of the acylation to the conformation of Cagri-CTR complex. Like Cagri-CTR-Gs complexes, CagriBB-CTR-Gs complexes could also be resolved into two major clusters based on the Cagri conformation: bypass (80%), CT-like (20%). A smaller experimental repeat dataset yielded a similar result with a higher percentage of particles in the bypass (50%) vs the CT-like (25%) conformation. As also observed in the repeat experiment of Cagri-CTR-Gs, due the smaller dataset and lower resolution imaging, 25% of the particles could not be classified due to poor resolution features in the ECD (Supplementary Fig. 10). Overall, across the two repeat experiments for Cagri and CagriBB bound to the CTR, the conformational equilibrium between the bypass vs CT-like state was similar, suggesting that the salt bridge/ionic lock is sufficient to preferentially stabilize the bypass conformation at CTR (Supplementary Fig. 11). Moreover, the R17Cagri-E14Cagri ionic lock was present in the bypass conformation but absent in the CT-like conformation in both CagriBB-bound structures, consistent with a higher energy requirement to form the CT-like conformation (Supplementary Fig. 11c and f). Unlike the Cagri-CTR-Gs complexes, in the CagriBB-bound CTR H296ECL2 adopted the inward rotamer position that was observed when bound to other non-lipidated peptides, and ECL3 formed the more closed conformation in both experimental repeats. Nonetheless, 3DVA of CTR complexes bound to either Cagri or CagriBB revealed transitions between the two major alternative conformations (Supplementary Fig. 12). In addition, after excluding particles of CT-like clusters, the bypass conformation of Cagri-CTR complexes displayed a wider range (7.6 Å) of ECD motion than the Cagri-AMYR complexes (Supplementary Fig. 12b). The absence of allosteric modulation from the RAMP ECD allows the CTR ECD to adopt a conformation that sits further from the receptor TMD than the position observed in the consensus cryo-EM map.
Pharmacological responses to Cagri and related peptides
Cagri has been broadly profiled in pharmacological assays of cells transiently transfected with CTR, CTR/RAMP1 or CTR/RAMP321. To assess the importance of lipidation modification to Cagri, pharmacological comparison of Cagri and its non-lipidated backbone (CagriBB) was undertaken using a cAMP response element (CRE)-reporter assay. These studies revealed that these peptides had similar potency and maximal response in cells co-expressing CTR and RAMP3 (Supplementary Fig. 13, Supplementary Table 9). Interestingly, CagriBB exhibited a reduced potency relative to Cagri in cells expressing CTR alone (Supplementary Fig. 13a, Supplementary Table 9), suggesting that the distinct rotamer orientation of H296ECL2 in Cagri-bound CTR (to accommodate the γGlu linker) and/or the observed more open ECL3 conformation induced by the lipidated peptide may facilitate or stabilize an active CTR conformation (Supplementary Fig. 11d and 11g)
Given the differentiation in conformational dynamics of Cagri-bound versus rAmy-bound to CTR or AMYRs and the likely role that the P37Cagri played in these differences, we also assessed in the assay an analogue of Cagri that contained a P37Y substitution. As expected, and consistent with a reduced interaction of the amidated C-terminal tyrosine with the CTR ECD, CagriP37Y presented a lower potency than Cagri in cells expressing CTR alone (Supplementary Fig. 13a, Supplementary Table 9). However, in cells co-expressing CTR and RAMP3, similar potency was observed for the analogues (Supplementary Fig. 13b, Supplementary Table 9), indicating that the interactions between the C-terminal tyrosine and RAMP3 can compensate, in this peptide context, the reduced CTR ECD affinity derived from the P37Y substitution.
We recently reported the development of a Gs protein proximity assay that provides a direct, AMYR-selective, measurement of Gs protein recruitment and activation22. This enabled us to assess whether the structural differences and conformational dynamics observed in the Cagri and rAmy bound AMYR structures alter the nature of Gs protein interaction (Supplementary Fig. 14a and b). In this system, Cagri had a distinct pattern of behaviour at CTR and each individual AMYR relative to previously reported control peptides (rAmy, hCT and sCT). This was particularly evident when comparing the maximal response across the different receptors (Supplementary Fig. 14a). The magnitude of response in this system is an integrated composite of the rate of Gs-Venus recruitment and activation (that will release the G protein from the activated receptor). While it is not clear how this translates to the kinetics of cAMP production in cells expressing individual receptors, the divergence in peptide responses across each receptor is consistent with the dynamic conformational differences that were observed structurally for individual AMYRs bound to distinct peptides. The distinct mode of CTR and AMYR engagement by Cagri revealed in the current study, together with the correlated distinctions in functional responses at individual receptors, is likely to be important in the differentiated, high efficacy, of Cagri as a first-in-class obesity drug in both preclinical animal models and in treatment of obese patients in clinical trials2,3,4.
Collectively, our data provides mechanistic insight into the behaviour of Cagri as a dual CTR and AMYR agonist peptide. The introduction of the R17Cagri-E14Cagri intra-peptide ionic lock energetically favours the formation of a predominant bypass conformation at the CTR, which, together with the P37 substitution and the effects induced by the N-terminal acylation, likely underlies its improved potency over rAmy at this receptor. At the AMYRs, the peptide adopts the conserved bypass motif and a common orientation of the ECD observed with amylin peptides. In parallel, the C-terminal P37Cagri contributes to altered dynamics of the ECD protomers in an AMYR-subtype-specific manner. These structural distinctions, in turn, may contribute to the distinct pharmacological profile of Cagri when assessed in proximal, selective, assays of primary transducer interaction.
Methods
Peptide agonists and general reagents
Rat amylin (rAmy), salmon calcitonin (sCT) and human calcitonin (hCT) were purchased from China Peptides. Cagrilintide, Cagri[P37Y] and the analogue CagriBB lacking the N-terminal lipidation were obtained from Novo Nordisk. Polyethylenimine (PEI) Max (mol. wt. 40,000) was purchased from Polysciences. 3-Isobutyl-1-methylxanthine (IBMX), poly(D-lysine), and forskolin were from Sigma-Aldrich. FUGENE HD transfection reagent for generation of P0 virus and Wizard® Plus SV Minipreps DNA Purification System were purchased from Promega. QIAGEN Plasmid Midi Kit was from QIAGEN. Precision Plus Protein™ Dual Color Standards were purchased from BioRad. Lauryl Maltose Neopentyl Glycol (LMNG) and Cholesteryl Hemisuccinate (CHS) Tris Salt were obtained from Anatrace. Uranyl formate was from Bioscientific. Superdex 200 Increase 10/300 column was from Cytiva. pFastBacTM Dual vector was from Invitrogen. ESF 921 Insect Cell Culture Medium, pVL dual vector and DH10Bac™ competent cells were purchased from Expression System. Nanobody-35 (Nb35) and HRV 3 C protease were in-house purified from E. coli strain BL21 as previously described23.
Constructs
The human CTRaleu (hCTR), RAMPs and G protein constructs used in the structure study have been previously described24,25. The human CTR was modified to contain an N-terminal Flag tag epitope and a C-terminal 8× histidine tag epitope, each flanked by a HRV 3 C protease cleavage sites. Human RAMPs were modified with an N-terminal Flag tag epitope. To improve expression in insect cell culture, the native signal peptides were replaced by that of hemagglutinin (HA) in both the CTR and RAMPs constructs built in pFastBac vector. Dominant negative Gαs (DNGαs) in pFastBac vector, and poly-his tagged Gβ1 and Gγ2 in a single pVL dual vector construct were used as previously described. Poly-his tagged Nb35 in pET-20b vector was obtained from Dr. Brian Kobilka26.
Purification of CTR/AMYR-DNGs complex in complex with peptide ligands
High-titre baculovirus was generated by transfecting recombinant bacmid (CTR, RAMP and DNGαs) or plasmid (Gβ1γ2) into Sf9 cells using the Bac-to-bac baculovirus system (Invitrogen). Each component for the ternary complex formation was co-expressed in Trichoplusia ni cells (Expression systems) with a combination of individual baculoviruses as previously described15.
AMYR-DNGs complex formation and purification was conducted as described15,16,27. Trichoplusia ni cell pellets were suspended in 30 mM HEPES (pH 7.4), 50 mM NaCl, 5 mM CaCl2, 2 mM MgCl2 supplemented with protease inhibitors (REF118361 45001, Roche) and benzonase (Merk Millipore). Complex was formed by adding excess peptide and stabilized in the presence of Nb35 (10 μg/mL, a nanobody that binds across Gαs and Gβ26 and apyrase (25 mU/mL, New England Biolabs). 3 C protease (10 μg/mL) was also supplemented to remove Flag and His tags from CTR. The cell suspension was incubated for 1 h at room temperature. The complexes were solubilized from membrane using detergent (1% (w/v) LMNG and 0.06% (w/v) CHS) for 1 h at 4 °C. The insoluble portion was subsequently removed by centrifugation and the supernatant was incubated with Ni-NTA resin (GE Healthcare) for 2 h batch binding at 4 °C. The Ni-NTA resin was packed into a glass column and washed with 25 column volumes of 20 mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 0.01% (w/v) LMNG and 0.0006% (w/v) CHS, 1 μM peptide, followed by elution using the same buffer supplemented with 250 mM imidazole and 3 mM CaCl2. The solubilized complex in the elution was immobilized by 2 h batch binding to M1 anti-Flag affinity resin at room temperature. The M1 resin was pack into a column and washed with 20 column volumes of 20 mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 3 mM CaCl2, 0.01% (w/v) LMNG and 0.0006% (w/v) CHS, 1 μM peptide. The bound protein was eluted using 20 mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 0.01% (w/v) LMNG and 0.0006% (w/v) CHS, 1 μM peptide, and 5 mM EGTA and 0.1 mg/mL Flag peptide. The elution was concentrated using an Amicon Ultra Centrifugal Filter (MWCO 100 kDa) and subjected to size-exclusion chromatography (SEC) on a Superdex 200 Increase 10/300 column that was pre-equilibrated with 20 mM HEPES pH 7.4, 100 mM NaCl, 2 mM MgCl2, 1 μM peptide, 0.01% (w/v) LMNG and 0.0006% (w/v) CHS. Eluted fractions consisting of complex were pooled and concentrated to ~4 mg/mL and flash frozen by liquid nitrogen as aliquots and stored at −80 °C until use. Similarly, the purification of CTR-DNGs complex was performed according to the previous described protocol24. Briefly, the complex was assembled in the excess peptides and solubilized in LMNG/CHS and purified using M1 anti-Flag affinity resin and SEC. The purity and integrity of complex were assessed by SDS-PAGE with at least 5% SDS to ensure solubilization of components. Gradient TGX gels (Bio-Rad) with 4–20% polyacrylamide were used to separate proteins within samples by electrophoresis at 200 V for 30 min. Gels were subsequently stained by Instant Blue (Expedeon).
Vitrified specimens and data collection
Holey TEM grids (Ultrafoil R1.2/1.3 Au 300 gold foil grids, Quantifoil GmbH) were pre-washed with chloroform prior to glow discharged. Purified complex (3 μL, ~4 mg/mL) was applied to the grids that were subsequently vitrified in liquid ethane using Vitrobot Mark IV (Thermo Fisher Scientific) under 100% humidity at 4 °C. Optimal grids for AMYR:Cagri complexes and CTR: Cagri (N = 1) were imaged by conventional cryo-TEM on a Thermo Fisher Scientific Titan Krios microscope operated at an accelerating voltage of 300 kV with a 50 μm C2 aperture and a 100 μm objective aperture at an indicated magnification of 105kx in nanoprobe EFTEM mode, yielding a calibrated pixel sampling of 0.85 Å.pix-1. A Gatan K3 direct electron camera operating in correlated double sampling (CDS) mode equipped post a Gatan Quantum energy filter (Gatan, Pleasanton, CA, USA) was to record movies with a total dose of ~60 e− per Å2. Movies were comprised of 60 sub-frames and recorded with a dose rate of 10.8 e− Å−2 s−1, except for the movie for AMY2R:Cagri recorded with a dose rate of 12.43 e− Å−2 s−1. To achieve a higher throughput data collection, aberration-free image shift (AFIS) was used as implemented in the Thermo EPU software package. See Supplementary Table 1 for data collection parameters. Datasets for the four remaining complexes were collected by a conventional cryo-TEM on a Thermo Fisher Scientific Glacios microscope operated at an accelerating voltage of 200 kV with a 50 μm C2 aperture and a 100-μm objective aperture, at an indicated magnification of 120 k× in nanoprobe TEM mode. A bottom-mounted Falcon 4 direct electron detector operated in Electron Event Representation (EER) mode was used to acquire images of the samples. Data were collected using aberration-free image shift implemented in EPU. Movies were recorded with a total dose of ~50 e− per Å2.
Cryo-EM data processing
RELION-3.1.2 were used for all data processing to yield the polished particle stack that was subjected into cryoSPARC v4.4.1 for 3D reconstitution28,29. Movies recorded in LZW-compressed TIFFs from the Krios microscope were subjected to beam-induced motion correction using MotionCor230 and contrast transfer function (CTF) estimation using CTFFIND-4.1 software package31 in the RELION software package. Particle auto-picking was performed using a pretrained model in the crYOLO software package, followed by particle extraction in a box-size equivalent to 320 Å and Fourier down-sampled to 64 pixels. After a 2D classification with a binned dataset in CRYOSPARC, a homogeneous subset of particles was re-extracted as the native pixel sampling and subjected to Bayesian particle polishing in RELION, followed by another further 2D classification in cryoSPARC. The resulting particles were subjected to non-uniform 3D refinement, followed by a final local Euler angle local refinement to generate the final consensus maps from which the α-helical domain (AHD) of the Gαs protein and the detergent micelle were masked out. The global resolutions were calculated according to the gold standard Fourier shell correlation (GSFSC) criterion of 0.143. Local resolution estimations were conducted with half-reconstructions as input maps. To improve confidence in the de novo modelling of ECD region, additional focused refinements were performed through specific masking of the receptor or ECD. Representative micrographs and data processing workflows for each structure are shown in Supplementary Fig. 15.
Atomic model refinement
The initial template for the AMYR:Cagri/CagriBB complex was generated by changing the divergent resides of rAmy in the corresponding AMYR:rAmy complex (PDB: 7TYF,7TYX, 7TZF). The initial model was rigid-body fitted into the consensus EM density map via UCSF Chimera 1.1432, followed by manual adjustment using ISOLDE33 embedded in UCSF ChimeraX 1.634. The fitted model was refined by iterative of manual adjustment in Coot 0.935 and real-space-refinement as implemented in PHENIX 1.236. The ECD regions were modelled manually against the ECD- and receptor-focused maps due to the poorly resolved density in the consensus map. The final model was subjected to comprehensive validation in MolProbity37. The restraints for the N-terminal lipid modification of Cagri were generated by the electronic Ligand Builder and Optimization Workbench (eLBOW) in PHENIX. Moreover, the initial model of CTR: Cagri/CagNN in the bypass conformation was generated from AMY1R:Cagri by removal of RAMP1. The model of CTR: rAmy (CT-like, PDB: 7TYI) was used as the initial template for the CTR:Cagri/CagriBB complex in the CT-like conformation by replacement of the peptide.
The placement of waters was confined to AMY1R:Cagri and CTR:Cagri (N = 1) within the deep binding pocket in order to avoid over interpretation of the cryo-EM maps. Water molecules were modelled manually into clear spherical density within reasonable hydrogen bonding distance to amino-acids or other waters. The positions of water were primarily refined in Coot based on the receptor focused maps and further inspected with the density in the consensus maps. The N-terminal lipid modification of Cagri was modelled into the strip density extended from the K1 towards the detergent micelle in the receptor-focused maps. Similarly, other possible endogenous lipids and CHS were also modelled into the strips of density that decorated the TM helix.
Model residue interaction analysis
Interactions of peptides with receptors or RAMPs were analysed using the “Dimplot” module within the Ligplot+ v2.2438. Hydrogen bonds were additionally analysed using the COOT, and the UCSF Chimera package, with relaxed distance and angle criteria (0.4 Å and 25-degree tolerance, respectively).
Cryo EM dynamics analysis and post-3D classification of CTR:Cagri/CagriBB complex
3D variability analysis (3DVA) implemented in cryoSPARC v4.4.129,39 was performed to interrogate and visualize the dynamics in the complexes as previously described25. The final particle stack and generous mask from the consensus non-uniform refinement were used in 3DVA to ensure any possible motions could be captured. 3 principal components were considered in the 3DVA that accounted for the most common motions. Output files were visualized in UCSF Chimera using the volume series function and captured as movies. To further distinguish heterogeneous conformations observed in the datasets of CTR:Cagri/CagriBB, the authentic particles of complexes were separated into multiple clusters by 3D Classification in cryoSPARC with approximately 50–100k particles for each cluster. By comparing reconstructed 3D volumes of each cluster, the particle stacks representing distinct states were identified and subsequently subjected into the non-uniform consensus refinement and the receptor-focused refinement.
Cyclic AMP response element (CRE)-reporter assay
A BHK cell line was stably transfected with the human calcitonin (a) receptor (hCTRa) and a cAMP responsive element (CRE) luciferase reporter gene. The cell line was further stably transfected with receptor modifying protein 3 (RAMP3) thus generating the human amylin 3(a) receptor (hAMY3(a)R). Cell culture was performed under aseptic conditions in Class II biosafety cabinets and incubation took place at 37 oC and 5% CO2 in a humidified incubator. The hCTR cells were cultured in growth medium consisting of Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, #31966-021) supplemented with 10% v/v foetal bovine serum (FBS, Gibco, #16140-071), 1% v/v Penicillin-Streptomycin (Gibco, #15140-122), 0.6 mg/mL Geneticin G418 (Gibco, #10131-027) and 0.2 mg/mL Hygromycin (Invitrogen, #10687010). The hAMY3R cells were cultured in growth medium consisting of DMEM (Gibco, #31966-021) supplemented with 10% v/v FBS (Gibco, #16140-071), 1% v/v Penicillin-Streptomycin (Gibco, #15140-122), 0.5 mg/mL Geneticin G418 (Gibco, #10131-027), 0.4 mg/mL Hygromycin (Invitrogen, #10687010) and 250 nM Methotrexate (Sigma, A6770). Cells were harvested, washed once with phosphate buffered saline (PBS, Gibco, #14190-094) and loosened from the cell flasks with Versene (Gibco, #15040-033) or TrypLETM (Gibco, #12605-010). When fully dissociated, cells were centrifuged (230 × g, 3 min) and the cell pellet resuspended and diluted to approximately 2.5E06 cells/mL in RecoveryTM Cell Culture Freezing Medium (Gibco, #12648-010). Cells were aliquoted and stored at −180 °C until use.
The day before the assay, frozen aliquots of the above-mentioned BHK cell lines were thawed, washed once in PBS (Gibco, #14190-094) and seeded in 40 µL medium in a white 384-well culture plate (PerkinElmer, #6007688) at a cell density of 4000 cells/well and incubated in plastic bags over night at 37 °C and 5% CO2. The seeding medium consisted of DMEM (Gibco, #31966-021) supplemented with 10% v/v FBS (Gibco, #16140-071), 1% v/v Penicillin-Streptomycin (Gibco, #15140-122). On the day of the assay, serial dilutions (7-fold dilutions, 7 concentrations per compound and one well containing only assay buffer) of test compounds were performed in duplicate in assay buffer to appropriate concentrations in 96-well plates (Greiner, U-shape #GR-650201) using a Biomek i7 liquid handler. The assay buffer consisted of DMEM without phenol red (Gibco #11880-028) supplemented with 10 mM HEPES (Gibco #15630-056), 1X GlutaMAX™ (Gibco #35020-038), and 1% (w/v) ovalbumin (Sigma A5503), 0.1% (v/v) Pluronic F-68 (Gibco, #24040-032). The cells were washed once in with 50 µL PBS (Gibco, #14190-094) after which 30 µL of each concentration from the dilution series was added to the 384-well assay plate with cells. The assay plate was incubated for 3 h at 37 °C in 5% CO2 and allowed to equilibrate at room temperature for 5 min after which 30 µL SteadyLite Plus (PerkinElmer, 6066759) was added to each well. The assay plate was sealed and incubated at room temperature with gentle shaking for 30 min (300 rpm) while protected from light. Luminescence was detected on a luminescence plate reader e.g. a Synergy 2 (BioTek). All data were imported into GraphPad Prism version 9.0.1 (GraphPad Software Inc., San Diego, CA). EC50-values were determined from the concentration-response curves using a three-parameter logistic equation.
Gαs recruitment assay
G protein recruitment to CTR or individual AMYR subtypes was measured in COS-7 cells (ATCC, CRL-1651) transfected with c-myc-hCTRaleu-Rluc8:Gαs-Venus:Gβ1:Gγ2, c-myc-hCTRaleu:RAMP1 Nluc:Gαs-Venus:Gβ1:Gγ2, c-myc-hCTRaleu:RAMP2-Nluc:Gαs-Venus:Gβ1:Gγ2 or c-myc-hCTRaleu:RAMP3-Nluc:Gαs-Venus:Gβ1:Gγ2 at a ratio of 2:0.1:1:1:1, in a total of 51 ng DNA per well using PEI at a ratio of 1:6 of DNA to PEI. Cells were mixed with transfection reagents and plated at 13,000 cells per well into 96-well Greiner CELLSTAR white-walled plates (Sigma-Aldrich), and assays were performed 48 h later. At the time of assay, the growth medium was replaced with HBSS, supplemented with 10 mM HEPES and 0.1% (w/v) ovalbumin and incubated at room temperature for 30 min. Furimazine (NanoBRET™ Nano-Glo® substrate; Promega) (for AMYRs) or coelenterazine h (Nanolight Technologies, Pinetop, AZ) (for CTR) was then added to cells at a final concentration of 1:1000 or 5 µM, respectively, and incubated for a further 10–15 min. Cell plates were transferred to a LUMIstar plate reader (BMG Labtech) at 30 °C for BRET measurements. Luminescence measurements were performed using 475/30 nm and 535/30 nm filters with baseline measurements taken for ~3 min before addition of vehicle or peptide and reading resumed for a further 10 min. BRET signal was calculated as the ratio of the 535/30 nm emission over the 475/30 nm emission. This ratio was vehicle-corrected by subtracting the response of vehicle-treated wells from the ratio of ligand-treated wells and then baseline-corrected by subtracting to the mean BRET ratio of baseline values (prestimulation) for each well. From experimental data of kinetic measurements of BRET, following vehicle and baseline corrections, quantification of induced responses from individual peptide concentrations was determined by calculating the net area under curve from the 10 min response (AUC0-10min)) in GraphPad Prism version 9.0.1 (GraphPad Software Inc., San Diego, CA). These were used to plot concentration-response curves, which were analysed using a three-parameter logistic equation.
Cagrilintide N-terminal γ-Glu-fatty acid parameterization
The cagrilintide N-terminal γ-Glu-Fatty acid (γ-Glu-FA) coordinates were extracted from the Cagri:AMY3R:Gαs complex, retaining K1Cagri Cα and N atoms. Hydrogen atoms were added using Chimera 1.1432. This ensured that the chemical environment of all the atoms of (γ-Glu-FA) was retained, hence their atom type. The terminal carboxy group was protonated because is exposed to a lipid environment, generating a formal charge -1. The resulting capped γ-Glu-FA residue was submitted to CGenFF40,41,42 to retrieve topology and parameter files. Charges belonging to atoms inherited from K1Cagri were removed, and their sum (0.049e) was equally redistributed to the seven sp3 carbon atoms bound to the terminal carboxy group to restore formal charge -1. This allowed us to build the γ-Glu-FA topology (available at https://zenodo.org/records/14825993) using the CHARMM36 force field43 format (named using a three-letter name CG9). Atom types belonging to the backbone were consistent with standard amino acids (e.g. C, O, CT1).
For γ-Glu-FA, the following parameters with mixed CgenFF/ CHARMM36 atom types were assigned by homology and added to the CHARMM36 parameters file for proteins (parameters available at https://zenodo.org/records/14825993).
MD simulations of the Cagrilintide and rat-Amylin AMY3R structures
The cryo-EM-derived models of Cagri:AMY3R:Gαs and rAmy:AMY3R:Gαs were superimposed with the PDB 6×18 OPM44 to orient the receptor to the membrane and parameterized with the CHARMM3643 force field. The resulting systems were prepared for simulations using in-house scripts based on HTMD2.3.245 and VMD1.9.4 frameworks. This multistep procedure performs the preliminary hydrogen atoms addition employing the pdb2pqr46 and PROPKA347 software combination through the systemPrepare HTMD implementation48, considering a simulated pH of 7.4. For Cagri:AMY3R:Gαs, histidine side chains were predicted in the tautomeric state δ except H223, H302 (CTR), H18 (Cagri), H97 (RAMP3), H142, and H183 (Gβ), which were modelled as ε tautomer. In the case of rAmy:AMY3R:Gαs, the histidine residues predicted as ε tautomer were: H226 (CTR), H41, H357 (Gα), 54, 183, 311 (Gβ). D57 (CTR) and D254 (Gβ), were predicted protonated, however, visual inspection did not support this prediction because of their solvent-exposed location and, therefore, were modelled in the anionic side chain form. Following this, the receptors were then embedded in a rectangular 126 Å x 126 Å 1-palmitoyl-2-oleyl-sn-glycerol-3-phosphocholine (POPC) bilayer (previously built by using the VMD Membrane Builder plugin 1.1 at http://www.ks.uiuc.edu/Research/vmd/plugins/membrane/) considering the coordinates retrieved from the OPM database to gain the correct orientation within the membrane, while removing the lipid molecules overlapping the receptor TMD bundle. TIP3P water molecules49 were added to the simulation box (final dimensions 126 Å x 126 Å x 186 Å) using the VMD Solvate plugin 1.5 (VMD Solvate plugin, Version 1.5; http://www.ks.uiuc.edu/Research/vmd/plugins/solvate/). Finally, the overall charge neutrality was reached by adding Na+/Cl− counter ions (final ionic concentration of 0.150 M) using the VMD Autoionize plugin 1.3 (Autoionize Plugin, Version 1.3; http://www.ks.uiuc.edu/Research/vmd/plugins/autoionize/). For both systems, the equilibration and production simulations were computed using the ACEMD350 MD engine.
Systems were equilibrated in isothermal-isobaric conditions (NPT) using the Berendsen barostat51 with a target pressure of 1 atm, the Langevin thermostat52 with a target temperature of 310 K, along with a low damping factor of 1 ps−1 and an integration time step of 2 fs. Clashes between protein and lipid atoms were reduced through 2500 conjugate-gradient minimization steps, followed by a 6 ns long MD simulation with a linearly-released positional constraint of 1 kcal mol−1 Å−2 on protein and lipid phosphorus atoms. Subsequently, 100 ns of MD simulation were performed, constraining only the protein atoms. Lastly, positional constraints were applied only to the protein backbone alpha carbons for a further 20 ns. Productive trajectories (1 ms for each replica) were computed with an integration time step of 4 fs in the canonical ensemble (NVT). The temperature was set at 310 K, using a thermostat damping of 0.1 ps−1 and the M-SHAKE algorithm53 to constrain the bond lengths involving hydrogen atoms. The cut-off distance for electrostatic interactions was set at 9 Å, with a switching function applied beyond 7.5 Å. Long-range Coulomb interactions were handled using the particle mesh Ewald summation method (PME)54 by setting the mesh spacing to 1.0 Å. Frames were saved every 200 ps. The composition of the simulated systems is detailed in Supplementary Table S10.
MD analysis
Interatomic distances, root mean square deviations (RMSD), root mean square fluctuations, and centroids were computed using VMD1.9.455. Interatomic contacts and hydrogen bonds were detected using the GetContacts scripts tool (https://getcontacts.github.io), setting a hydrogen bond donor-acceptor distance of 3.3 Å and an angle value of 120° as geometrical cut-offs. Contacts and hydrogen bond persistency are quantified as the percentage of frames (over all the frames obtained by merging the different replicas) in which protein residues formed contacts or hydrogen bonds with the ligand.
The Nature Communications MD checklist is provided as Supplementary Table 11.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates and electron microscopy maps have been deposited in the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) under accession codes: 9BP3/EMD-44760 [https://www.ebi.ac.uk/emdb/EMD-44760] (Cagri-AMY1R-DNGs complex), 9BLW/EMD-44678 [https://www.ebi.ac.uk/emdb/EMD-44678] (CagriBB-AMY1R-DNGs complex), 9BQ3/EMD-44796 [https://www.ebi.ac.uk/emdb/EMD-44796] (Cagri-AMY2R-DNGs complex), 9BTW]/EMD-44898 [https://www.ebi.ac.uk/emdb/EMD-44898] (Cagri-AMY3R-DNGs complex), 9BUB/EMD-44904 [https://www.ebi.ac.uk/emdb/EMD-44904] (Cagri-CTR-DNGs complex 1, bypass conformation), 9BUD/EMD-44906 [https://www.ebi.ac.uk/emdb/EMD-44906] (Cagri-CTR-DNGs complex 1, CT-like conformation), 9BUC/EMD-44905 [https://www.ebi.ac.uk/emdb/EMD-44905] (Cagri-CTR-DNGs complex 2, bypass conformation), 9BUE/EMD-44907 [https://www.ebi.ac.uk/emdb/EMD-44907] (Cagri-CTR-DNGs complex 2, CT-like conformation), 9BLC/EMD-44653 [https://www.ebi.ac.uk/emdb/EMD-44653] (CagriBB-CTR-DNGs complex 1, CT-like conformation), 9BLB/EMD-44652 [https://www.ebi.ac.uk/emdb/EMD-44652] (CagriBB-CTR-DNGs complex 1, bypass conformation). Topology and paramaterisation files for the γ-Glu-FA are available in Zenodo [https://zenodo.org/records/14825993]. MD trajectories are available in Zenodo [https://zenodo.org/records/14260587]. Source data files for gels are available within the Supplementary Information file included with this manuscript. Source data files for pharmacological experiments are available as a Supplementary Information Excel file included with this manuscript. Source data are provided with this paper.
References
Nogueiras, R., Nauck, M. A. & Tschop, M. H. Gut hormone co-agonists for the treatment of obesity: from bench to bedside. Nat. Metab. 5, 933–944 (2023).
Kruse, T. et al. Development of cagrilintide, a long-acting amylin analogue. J. Med. Chem. 64, 11183–11194 (2021).
Lau, D. C. W. et al. Once-weekly cagrilintide for weight management in people with overweight and obesity: a multicentre, randomised, double-blind, placebo-controlled and active-controlled, dose-finding phase 2 trial. Lancet 398, 2160–2172 (2021).
Frias, J. P. et al. Efficacy and safety of co-administered once-weekly cagrilintide 2.4 mg with once-weekly semaglutide 2.4 mg in type 2 diabetes: a multicentre, randomised, double-blind, active-controlled, phase 2 trial. Lancet 402, 720–730 (2023).
Enebo, L. B. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet 397, 1736–1748 (2021).
Hay, D. L., Chen, S., Lutz, T. A., Parkes, D. G. & Roth, J. D. Amylin: pharmacology, physiology, and clinical potential. Pharm. Rev. 67, 564–600 (2015).
Hay, D. L., Garelja, M. L., Poyner, D. R. & Walker, C. S. Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br. J. Pharm. 175, 3–17 (2018).
Christopoulos, G. et al. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol. Pharm. 56, 235–242 (1999).
Larsen, A. T., Sonne, N., Andreassen, K. V., Karsdal, M. A. & Henriksen, K. The calcitonin receptor plays a major role in glucose regulation as a function of dual amylin and calcitonin receptor agonist therapy. J. Pharm. Exp. Ther. 374, 74–83 (2020).
Andreassen, K. V. et al. KBP-066A, a long-acting dual amylin and calcitonin receptor agonist, induces weight loss and improves glycemic control in obese and diabetic rats. Mol. Metab. 53, 101282 (2021).
Mathiesen, D. S., Lund, A., Vilsboll, T., Knop, F. K. & Bagger, J. I. Amylin and Calcitonin: Potential Therapeutic Strategies to Reduce Body Weight and Liver Fat. Front. Endocrinol. 11, 617400 (2020).
Sonne, N., Karsdal, M. A. & Henriksen, K. Mono and dual agonists of the amylin, calcitonin, and CGRP receptors and their potential in metabolic diseases. Mol. Metab. 46, 101109 (2021).
Larsen, A. T. et al. Does receptor balance matter?—comparing the efficacies of the dual amylin and calcitonin receptor agonists cagrilintide and KBP-336 on metabolic parameters in preclinical models. Biomed. Pharmacother. 156, 113842 (2022).
Arrigoni, S. et al. A selective role for receptor activity-modifying proteins in subchronic action of the amylin selective receptor agonist NN1213 compared with salmon calcitonin on body weight and food intake in male mice. Eur. J. Neurosci. 54, 4863–4876 (2021).
Cao, J. et al. A structural basis for amylin receptor phenotype. Science 375, eabm9609 (2022).
Cao, J. et al. Structural insight into selectivity of amylin and calcitonin receptor agonists. Nat. Chem. Biol. 20, 162–169 (2024).
Wootten, D., Simms, J., Miller, L. J., Christopoulos, A. & Sexton, P. M. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. USA 110, 5211–5216 (2013).
Bower, R. L. et al. Molecular signature for receptor engagement in the metabolic peptide hormone amylin. ACS Pharm. Transl. Sci. 1, 32–49 (2018).
Lee, S. M., Hay, D. L. & Pioszak, A. A. Calcitonin and amylin receptor peptide interaction mechanisms. Insights into peptide-binding modes and allosteric modulation of the calcitonin receptor by receptor activity-modifying proteins. J. Biol. Chem. 291, 16416 (2016).
Johansson, E. et al. Type II turn of receptor-bound salmon calcitonin revealed by X-ray crystallography. J. Biol. Chem. 291, 13689–13698 (2016).
Fletcher, M. M. et al. AM833 is a novel agonist of calcitonin family G protein-coupled receptors: Pharmacological comparison with six selective and nonselective agonists. J. Pharm. Exp. Ther. 377, 417–440 (2021).
Keov, P. et al. Development of a novel assay for direct assessment of selective amylin receptor activation reveals novel differences in behavior of selective and nonselective peptide agonists. Mol. Pharmacol. 105, 359–373 (2024).
Zhang, X. et al. Differential GLP-1R binding and activation by peptide and non-peptide agonists. Mol. Cell 80, 485–500.e487 (2020).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017).
Liang, Y. L. et al. Structure and dynamics of adrenomedullin receptors AM1 and AM2 reveal key mechanisms in the control of receptor phenotype by receptor activity-modifying proteins. ACS Pharm. Transl. Sci. 3, 263–284 (2020).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Liang, Y. L. et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D. Struct. Biol. 74, 519–530 (2018).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. 66, 213–221 (2010).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Punjani, A. & Fleet, D. J. 3D variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Vanommeslaeghe, K. & MacKerell, A. D. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 52, 3144–3154 (2012).
Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 52, 3155–3168 (2012).
Yu, W., He, X., Vanommeslaeghe, K. & MacKerell, A. D. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 33, 2451–2468 (2012).
Huang, J. & MacKerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Doerr, S., Harvey, M. J., Noé, F. & De Fabritiis, G. HTMD: high-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput. 12, 1845–1852 (2016).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pK predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Doerr, S., Giorgino, T., Martínez-Rosell, G., Damas, J. M. & De Fabritiis, G. High-throughput automated preparation and simulation of membrane proteins with HTMD. J. Chem. Theory Comput. 13, 4003–4011 (2017).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 (1983).
Harvey, M. J., Giupponi, G. & Fabritiis, G. D. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 5, 1632–1639 (2009).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684 (1984).
Loncharich, R. J., Brooks, B. R. & Pastor, R. W. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 32, 523–535 (1992).
Kräutler, V., van Gunsteren, W. F. & Hünenberger, P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 (2001).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577 (1995).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996).
Acknowledgements
P.M.S. received a NHMRC Senior Principal Research Fellowship (grant ID: 1154434), a NHMRC Leadership grant (ID: 2025694), and an ARC Discovery Project grant (DP210101504). D.W. received a NHMRC Senior Research Fellowship (grant ID: 1155302) and a NHMRC Leadership grant (ID: 2026300). P.M.S. is Director and D.W. is the Monash University Node leader of the ARC Industrial Transformation Training Centre for Cryo-electron Microscopy of Membrane Proteins (CCeMMP) (grant ID: IC200100052), which received support from Novo Nordisk for the current project. The work was supported by the Monash University Ramaciotti Centre for Cryo-Electron Microscopy, the Bio21 Ian Holmes Imaging Centre (The University of Melbourne) and the Monash eResearch capabilities, including M3 High-performance computing. Figures were created with UCSF Chimera and ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Contributions
D.W., P.M.S., S.R.-R., K.R. and T.G. conceptualized the study. G.C. generated constructs for the study. J.C., R.M.J. generated proteins and formed complexes used in structure determination. J.C. and M.J.B. collected and/or processed cryo-EM data. P.K., C.A.H., T.G. and B.B.-G. performed pharmacological assays. Z.M. and G.D. performed and analysed molecular dynamics simulations. J.C., C.B.-J., B.B.-G., Z.M., G.D., D.W., P.M.S. and M.J.B. analysed and interpreted the data. J.C., Z.M., G.D. and M.J.B. contributed to data visualisation. P.M.S. and D.W. acquired funding for the research. P.M.S., D.W., M.J.B., S.R.-R. and T.G. provided project supervision and administration. J.C., D.W. and P.M.S. wrote the original draft and all authors contributed to reviewing and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.M.S. is a co-founder and P.M.S. and D.W. are shareholders of Septerna Inc. P.M.S. and D.W. are co-founders and shareholders of Dacra Tx. T.G., K.R., B.B.-G., and S.R.-R. are employees and shareholders of Novo Nordisk. The work was funded, in part, by Novo Nordisk. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Toni Giorgino and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, J., Belousoff, M.J., Johnson, R.M. et al. Structural and dynamic features of cagrilintide binding to calcitonin and amylin receptors. Nat Commun 16, 3389 (2025). https://doi.org/10.1038/s41467-025-58680-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58680-y
This article is cited by
-
The story of amylin: from physiology to therapy
Nature Metabolism (2026)
-
Structural and mechanistic insights into dual activation of cagrilintide in amylin and calcitonin receptors
Acta Pharmacologica Sinica (2026)
