
Abstract
Single-atom catalysts (SACs) have attracted significant interest due to their exceptional and tunable performance, enabled by diverse coordination environments achieved through innovative synthetic strategies. However, various local structures of active sites pose significant challenges for precise characterization, a prerequisite for developing structure-activity relationships. Here, we combine 17O solid-state NMR spectroscopy and DFT calculations to elucidate the detailed structural information of Pt/CeO2 SACs and their catalytic behaviors. The NMR data reveal that single Pt atoms, dispersed from clusters with water vapor, exhibit a square planar geometry embedded in CeO2 (111) surface, distinct from the original clusters and other conventionally generated Pt single atoms. The square planar Pt/CeO2 SAC demonstrates improved CO oxidation performance compared to Pt/CeO2 SAC with octahedral coordination, due to moderately strong CO adsorption and low energy barriers. This approach can be extended to other oxide-supported SACs, enabling spatially resolved characterization and offering comprehensive insights into their structure-activity relationships.
Similar content being viewed by others

Introduction
Single-atom catalysts (SACs) are increasingly recognized for their pivotal role in catalysis due to their maximized atomic economy (especially for noble metals), superior catalytic activity, and selectivity1,2,3,4,5,6. By employing tailored synthesis strategies7, oxide-supported SACs can achieve tunable active sites resulting from the distinct spatial and coordination environments of metal atoms8,9,10,11,12, as well as specially engineered anchoring sites and various defects in the metal oxide support13,14,15,16,17. Therefore, these catalysts provide significant potential for addressing a myriad of challenges in catalysis. At the same time, the significant structural diversity of these catalysts poses substantial characterization challenges, creating difficulties in establishing structure-activity relationships, which are the basis for the rational design of better catalysts. For example, in single-atom Pt/CeO2 catalysts, which often serve as model systems18,19,20 and are widely studied for their applications in environmental contaminant removal and energy conversion21,22,23,24, Pt sites can undergo dynamic transformations, maintaining their single-atom status while evolving into diverse structures with distinct catalytic properties under controlled atmospheres and/or annealing temperatures21,25,26,27. Current characterization techniques, such as aberration-corrected scanning transmission electron microscopy and X-ray absorption fine structure (XAFS)28,29, primarily focus on the metal sites, probing the isolated states of individual metal atoms, their oxidation states and coordination numbers, while falling short of capturing the detailed local coordination environment around the metal atoms30. This environment, which includes not only the metal sites but also the surrounding atoms (e.g., coordinating species and coordination geometry), fundamentally dictates catalytic performance yet remains ambiguous6,31. Therefore, in order to fully elucidate the structure-activity relationship of oxide-supported SACs, developing high-resolution analytical techniques is urgently required.
Solid-state nuclear magnetic resonance (NMR) plays an indispensable role in investigating the structure and properties of materials across a wide variety of applications32,33,34,35. In particular, rich information has been obtained by applying 17O NMR spectroscopy for oxygen-containing catalysts, taking advantage of the large 17O chemical shift range (>1000 ppm)36,37,38,39. More recently, with the assistance of surface-selective 17O labeling and DFT calculations, 17O NMR has been successfully employed to distinguish oxygen sites at different surface layers and facets of oxide nanomaterials, and explore gas adsorption sites and gas-surface interactions40,41,42,43. However, there have been very few 17O NMR studies on the structure of the supported catalysts44,45, especially the important noble-metal-based catalysts, a key category of catalysts where single-atom dispersion is desired to reduce costs, not to mention structure-activity relationships. Here, we developed an approach based on 17O solid-state NMR spectroscopy and DFT calculations, to reveal the detailed local structures of oxide-supported SACs, using Pt1/CeO2 as a model system. The precise structural information and reaction mechanisms obtained shed light on the factors responsible for the different CO oxidation performances of two Pt/CeO2 SACs.
Results
Traditional characterization
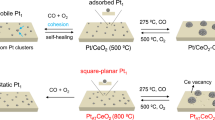
An impregnation method (see Methods for details) was used to prepare the parent oxidized Pt cluster catalyst (denoted as PtnOx/CeO2, vide infra) using CeO2 nanoparticles as the support which expose the thermodynamically stable (111) facets as determined by high-resolution transmission electron microscopy (HRTEM) (Supplementary Fig. 1a) and 17O solid-state NMR (Supplementary Fig. 2). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image shows obvious Pt clusters (blue circles) on the surface with a diameter of approximately 5 nm (Fig. 1a). PtnOx/CeO2 was treated with 8.4 vol.% H2O(g)/Ar at 750 °C for 9 h, during which no further sintering occurred (Supplementary Figs. 1c and 3). Only isolated Pt atoms (orange circles) are observed in the HAADF-STEM image of the resulting material (Fig. 1a). The Pt single atoms turn out to be embedded in the CeO2 lattice with a low-coordination number and the material is therefore denoted as PtLC/CeO2 (LC for low-coordinated, vide infra). For a clearer illustration of the structure-activity relationship of SACs, we prepared PtHC/CeO2 (HC for high-coordinated, vide infra), a CeO2-nanoparticles-supported single atom Pt catalyst with a higher coordination number, by using a conventional atom trapping method, as reported previously15,46.

a HAADF-STEM images for PtnOx/CeO2, PtLC/CeO2 and PtHC/CeO2. Blue circles highlight the PtnOx cluster, while orange and green circles highlight single-atom Pt species in the SACs. b k2-weighted Fourier-transformed EXAFS and c normalized Pt L3-edge XANES spectra of PtnOx/CeO2, PtLC/CeO2, PtHC/CeO2, Pt foil and PtO2.
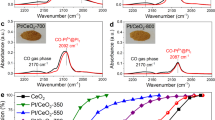
For PtnOx/CeO2, the extended X-ray absorption fine structure (EXAFS) spectrum exhibits contributions from Pt-O at 1.6 Å as well as Pt-Pt environments ranging from 2.4 to 3.0 Å (Fig. 1b, Supplementary Fig. 4 and Supplementary Table 1). Normalized X-ray absorption near-edge structure (XANES) results indicate that the oxidation state of Pt in this material is higher than that in Pt foil (Fig. 1c)47. The two peaks at 72.9 and 72.0 eV in the Pt 4f X-ray photoelectron spectroscopy (XPS) data can be attributed to Pt2+ and Pt+, respectively (Supplementary Fig. 5 and Supplementary Table 2)48. X-band electron paramagnetic resonance (EPR) spectra further confirm the presence of Pt+ (Supplementary Fig. 6)49. In addition, diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) data shows only a CO adsorption band at 2093 cm−1, which is typically associated with CO adsorption on ionic Pt species, in addition to a weaker band at 2170 cm−1 owing to CO in the gas phase (Supplementary Fig. 7). Similar CO-DRIFTS results have been previously observed on oxidized Pt clusters50. Therefore, the presence of oxidized Pt clusters on the surface of the sample can be confirmed.
The absence of the peak related to Pt-Pt in the EXAFS spectrum of PtLC/CeO2 in Fig. 1b substantiates the primary presence of single Pt atoms, confirming the formation of a SAC along with the results of HAADF-STEM (Fig. 1a). Therefore, this water vapor treatment disperses the cluster catalyst into a single-atom Pt catalyst, which is similar to the dispersion of Cu nanoparticles into Cu single atoms with the assistance of water in a very recent study by Fan et al. 51. The structural change from PtnOx to Pt single atoms was observed using EXAFS (Supplementary Fig. 8). Notably, the EXAFS spectra of both PtLC/CeO2 and PtHC/CeO2 show exclusively Pt-O scattering (Fig. 1b), with PtLC/CeO2 exhibiting a lower coordination number of 4.0 ± 0.3 and PtHC/CeO2 a higher coordination number of 6.0 ± 0.5 (Supplementary Fig. 4 and Supplementary Table 1). XANES data shows that Pt in PtLC/CeO2 has a significantly higher oxidation state than PtnOx/CeO2, but is slightly lower than PtHC/CeO2 (Fig. 1c). Similar XPS and EPR spectra are observed for both SACs, which suggest the presence of only Pt2+ and higher oxidation states than PtnOx/CeO2 (Supplementary Figs. 5, 6, and Supplementary Table 2) as well as the low concentrations of Ov on their surface (Supplementary Figs. 6 and 9, and related discussion in Supplementary Information). Bands at 2093 cm−1 are observed in DRIFTS data, again corresponding to CO on the top of ionic Pt species (Supplementary Fig. 7)12,21. These results indicate that two SACs with different coordination numbers have been obtained.
Precise local environment determined by 17O solid-state NMR
Since the precise local environment of these Pt/CeO2 catalysts is crucial for their catalytic performances, we combine 17O solid-state NMR spectroscopy and DFT calculations to extract detailed structural information. Strong resonances ranging from 790 to 1080 ppm can be observed for all three samples, which are attributed to the oxygen ions at the first, second, third surface layers and in the bulk part of CeO2 (111) (Fig. 2)40. This confirms that the predominantly exposed crystal facets of CeO2 remained unchanged after thermal treatment in air or steam, consistent with observations in Supplementary Fig. 1. The major difference in NMR spectra is the signals at lower frequencies, which is expected considering the lower resonant frequencies of PtO2 compared to CeO2 (Supplementary Fig. 10). For PtnOx/CeO2 (Fig. 2a), three different sets of peaks are observed, centered at approximately 500, 300, and 50 ppm, respectively. The wide distribution of chemical shifts implies multiple binding modes between oxygen and the various Pt atoms in PtnOx clusters. In contrast, the distribution of the 17O NMR shifts in PtLC/CeO2 and PtHC/CeO2 is much narrower, suggesting more uniform oxygen environment in Pt-related sites in the two SACs (Fig. 2b, c). One relatively broad peak centered at 490 ppm, and two peaks centered at 500 and 655 ppm are observed for PtLC/CeO2 and PtHC/CeO2, respectively. At the same time, very different structures are expected for PtLC/CeO2 and PtHC/CeO2 due to their distinct NMR shifts.

17O solid-state NMR spectra of (a) PtnOx/CeO2, (b) PtLC/CeO2, and (c) PtHC/CeO2 and the summary of calculated NMR shifts (center of gravities) of O atoms connected directly to Pt, as well as the corresponding structures. The spectra were recorded at a spinning speed of 53 kHz with a recycle delay of 0.2 s at 14.1 T. A rotor synchronized Hahn-echo pulse sequence (π/6-τ-π/3-τ-acquisition) with 1H decoupling was applied. Asterisks denote spinning sidebands.
DFT calculations of NMR parameters (i.e., isotropic chemical shift (δiso), quadrupolar coupling constant (CQ), asymmetry parameter (ηQ), and center of gravity (δCG)) were performed to assist the spectral assignments and extract more structural information of these catalysts. We use a model of adsorbed Pt4O5 cluster on CeO2 (111) surface (denoted as A-Pt4O5) to represent PtnOx/CeO2 catalyst (see Supplementary Fig. 11 and additional discussion in the Supplementary Information). Oxygen ions connected to one, two and three Pt ions are associated with different NMR shifts and their calculated center of gravities (δCGs) follow the order: OPt1 > OPt2 > OPt3 (see Fig. 2a, Supplementary Figs. 12–14, and Supplementary Tables 3–5). Thus, the signals centered at 500, 300, and 50 ppm obtained at 14.1 T can be assigned to OPt1, OPt2, and OPt3, respectively. For SACs, several different structure models were developed, including the following possible Pt single atom structures (Fig. 3): different Pt1 species adsorbed on CeO2 (111) surface (denoted as A-Pt1, A-Pt1O2, and A-Pt1(OH)2), Pt ion trapped into the CeO2 (111) surface with steps (denoted as S-Pt1)14,46,52, and Pt ion embedded into the CeO2 (111) surface with different coordination numbers (denoted as E-Pt1(4c)13, E-Pt1(4c’)27,52, E-Pt1(6c)12,27,52). Comparing the calculated NMR shifts (δCGs) for the oxygen ions directly connected to Pt (see Fig. 3, Supplementary Figs. 15–21 and Supplementary Tables 6–12, and related discussion in the Supplementary Information) to the experimental NMR spectrum of PtLC/CeO2, only the models E-Pt1(4c) and S-Pt1 give rise to the peaks close to 490 ppm without additional peaks at other frequencies (also see Supplementary Figs. 22–27 and Supplementary Tables 13–18, and related discussion in the Supplementary Information). However, according to calculations, resonances with a relatively wide shift distribution in the range of 655–745 ppm are expected for oxygen ions (not connected to Pt) at the first and second layers in the model S-Pt1 (Supplementary Fig. 28). The absence of such signals in the experimental NMR data confirms that PtLC/CeO2 should be associated with the model E-Pt1(4c). In this structure, a single Pt atom is trapped into the CeO2 (111) surface, connecting to two O ions each from the first (δCGs ~ 478 ppm) and second surface layers (δCGs ~ 506 ppm), forming a square planar structure (Fig. 2b, Supplementary Fig. 19, and Supplementary Table 10). Furthermore, the experimental data obtained at three different external magnetic fields cannot be fitted with a single site (Supplementary Fig. 29, Supplementary Table 19 and related discussion in the Supplementary Information). Instead, two sites must be included in the fitting, further supporting the spectral assignments (Supplementary Fig. 30 and Supplementary Table 20 and related discussion in the Supplementary Information). The embedded Pt atoms are expected to contribute to the excellent thermal stability of PtLC/CeO2. Similarly, only the calculated shifts from the model E-Pt1(6c) are in agreement with the experimental NMR spectrum of PtHC/CeO2, indicating a 6-coordinated Pt environment in this sample. The peaks at 655 and 500 ppm can be attributed to oxygen ions directly connected Pt (Pt-O-Ce) at the first and second layers, respectively (Fig. 2c, Supplementary Fig. 21 and Supplementary Table 12). The capability of 17O NMR spectroscopy to distinguish a variety of different structures around Pt single atoms (Fig. 3) can be attributed to the fact that 17O chemical shifts are dependent on atoms at the third coordination shell42. Therefore, the precise local environment, including coordination geometries, can be elucidated by combining 17O NMR with DFT calculations, based on which an energetically feasible pathway for the dispersion process by water molecules was proposed (Supplementary Figs. 31 and 32, and related discussion in the Supplementary Information). Water molecules play a crucial role in dispersing platinum oxide clusters into single atoms on the support and in the final formation of the square planar structure of Pt species.

The light orange-marked area indicates the region where the new NMR signal corresponds to PtLC/CeO2. The models include adsorbed Pt1 species on the CeO2 surface (A-Pt1, A-Pt1O2 and A-Pt1(OH)2), Pt ions trapped in the steps of CeO2 surface (S-Pt1) and embedded Pt ions in the CeO2 surface (E-Pt1(4c), E-Pt1(4c’), and E-Pt1(6c)).
Structure-activity relationship
The Pt/CeO2 catalysts were evaluated for CO oxidation and the light-off curves show that PtLC/CeO2 demonstrates a marked improvement in catalysis compared to PtHC/CeO2 (Fig. 4a). Notably, the onset temperature decreases from ~205 °C (PtHC/CeO2, green line) to ~120 °C (PtLC/CeO2, orange line) and the apparent activation energy becomes lower (Fig. 4b). A long-term duration test for PtLC/CeO2 was also conducted at 200 and 230 °C, with CO conversion maintained at 23% and 100%, respectively, for at least 18 h (Fig. 4c, d). Furthermore, to investigate the thermal stability of PtLC/CeO2, the catalyst was aged at 800 °C for an additional 24 h (denoted as PtLC/CeO2-a, see Supplementary Figs. 3, 33, 34 and Supplementary Table 21, and related discussion in the Supplementary Information) and the catalytic performance for CO oxidation does not change much (Fig. 4a). These results indicate that the newly formed PtLC/CeO2 possesses both high activity and thermal stability.

a The light-off curves for CO oxidation performances of PtnOx/CeO2, PtLC/CeO2, PtLC/CeO2-a, and PtHC/CeO2. b Reaction rates of catalysts as a function of reaction temperature. PtLC/CeO2 (orange line and circles), PtLC/CeO2-a (grey line and circles) and PtHC/CeO2 (green line and triangles). CO oxidation durability tests for PtLC/CeO2 at (c) 200 °C and (d) 230 °C.
With detailed structures of the two SACs, the structure-activity relationship of the catalysts is explored. The CO oxidation process on Pt/CeO2 is widely recognized to follow a Mars-van Krevelen (MvK) reaction mechanism53. Calculations show a CO adsorption energy of −2.09 eV at 4-coordinated Pt site in PtLC/CeO2, and the following reaction between adsorbed CO and the vicinal bridging O only requires a minimal energy barrier of 0.10 eV (Fig. 5a). In contrast, the 6-coordinated Pt (PtHC/CeO2) is associated with a much higher CO adsorption energy (−2.75 eV) (Fig. 5b). The difference in calculated CO adsorption energies for the two SACs is consistent with the in-situ CO-DRIFTS results (Supplementary Fig. 35), where CO molecules are found to be completely desorbed from PtLC/CeO2 at 150 °C, whereas the PtHC/CeO2 catalyst still exhibits strong adsorption at this temperature, suggesting the excessively strong bonding of CO on the PtHC/CeO2 surface. Furthermore, a relatively high activation barrier of 1.10 eV needs to be overcome for adsorbed CO to react with the nearby surface lattice oxygen ion. This indicates that the surface of PtHC/CeO2 is susceptible to poisoning by CO at low temperatures. Subsequently, an O2 molecule fills the oxygen vacancy and the second CO molecule interacts with the adsorbed O2 species, resulting in the formation of CO2, which similar barriers are found in both catalysts. These data show that the low-coordinated embedded single atoms in PtLC/CeO2 are more effective as a CO oxidation catalyst than those with a higher coordination number in PtHC/CeO2.

Calculated reaction pathways over (a) the model E-Pt1(4c) and (b) the model E-Pt1(6c). The insets show the calculated energy profiles and the structures of intermediates and transition states (TSs, see Supplementary Fig. 36 for details) of the key elementary steps.
Discussion
We combine high-resolution 17O solid-state NMR spectroscopy, along with DFT calculation, to reveal the precise local structure of ceria-supported Pt SACs, including coordination species and geometries, which is evidenced by characteristic 17O NMR shifts. Owing to its very fine resolution, this method can resolve different Pt species including Pt clusters, and 4-and 6-coordinated Pt ions with unique geometries, thereby enabling the development of significantly improved structural models for DFT calculations of reaction mechanisms. On this basis, the enhanced performances of PtLC/CeO2 can be ascribed to the square planar geometry of the Pt single atoms embedded into the ordered CeO2 (111) surface, which is associated with moderately strong CO adsorption and low energy barriers. Our results demonstrate the establishment of structure-activity relationship of oxide-supported Pt SACs, highlighting the importance of extracting key local environment around single atoms for a better understanding of the properties of the catalysts and emphasizing that SACs require more detailed structural characterization. This method can also be extended to study other oxide-supported SACs, which have potential in solving critical challenges in sustainable development3,4,54.
Methods
Synthesis of CeO2 nanoparticles
Ce(NO3)3·6H2O (99%; Sigma-Aldrich) was heated in air at 300 °C for 3 h to obtain CeO2 nanoparticles (NPs).
Synthesis of PtnOx/CeO2
PtnOx/CeO2 was prepared by incipient wetness impregnation with H2PtCl6·6H2O (≥37.50% Pt basis; Sigma-Aldrich) as the precursor. In a typical procedure, 1.0 g of CeO2 NPs was slowly immersed in 0.5 mL of H2PtCl6 solution with a concentration of 0.02 g/mL, drop by drop. After drying overnight at 80 °C, the sample was calcined at 800 °C for 12 h in a tube furnace in dry air with a flow rate of 50 mL/min. The Pt content in the sample was determined by inductively coupled plasma atomic emission spectrometer (ICP-OES) to be 0.94 wt.%.
Synthesis of PtLC/CeO2
The treatment was carried out in a fixed-bed reactor. 0.34 g of PtnOx/CeO2 sample (40–60 mesh) was packed into a U-type quartz tube and heated by programmable tube furnace from room temperature to 750 °C in Ar atmosphere with a flow rate of 50 mL/min. The whole stainless-steel gas path was kept at 90 °C. The sample was treated by 8.4 vol.% H2O(g)/Ar at 750 °C for 9 h and then cooled down to 300 °C. Steam flow was discontinued and the sample was dried for an additional 2 h before cooling down to room temperature naturally. The obtained sample was denoted as PtLC/CeO2. The Pt content in the sample was determined by ICP to be 0.92 wt.%.
Synthesis of PtHC/CeO2
PtHC/CeO2 was prepared using the conventional atom trapping method according to the literature15,21. In a typical procedure, 1.0 g of CeO2 nanoparticle was immersed in 0.5 mL of H2PtCl6 solution with a concentration of 0.02 g/mL and the mixture was ground in a mortar. After drying overnight at 80 °C, the sample was calcined at 800 °C for 12 h in the muffle furnace. The Pt content in the sample was determined by ICP to be 0.97 wt.%.
Synthesis of CeO2 Nanorods
The CeO2 nanorods (NRs) were synthesized using the hydrothermal method. First, 0.868 g of Ce(NO3)3·6H2O (Sigma-Aldrich, 99%) was dissolved in 10 mL of deionized water and stirred for 10 min. The solution was then mixed with 70 mL of aqueous NaOH (9 M), stirred for an additional 30 min, and transferred into a Teflon bottle. Subsequently, it was placed in a stainless-steel vessel autoclave and heated at 100 °C for 24 h in an oven. After cooling down to room temperature, a purple milky slurry was obtained. The precipitates were collected by centrifugation, washed three times with deionized water and ethanol, and then dried at 60 °C overnight. Finally, the powder was calcined in a tube furnace at 700 °C for 5 h in flowing air to obtain CeO2 NRs.
Synthesis of Pt/CeO2_S
Pt/CeO2_S was prepared according to literature21.
Basic characterization
The mass loading of Pt ions was determined by ICP-OES on an Optima 4300 DV spectrometer. X-ray photoelectron spectra (XPS) were recorded on a Thermo ESCALAB 250Xi instrument with Al Kα radiator (hν = 1486.6 eV). The binding energies were calibrated using C1s at 284.8 eV. X-band EPR spectra were obtained at 107 K using a Bruker EMXplus spectrometer with the sample mass of 150 mg. Brunaure−Emmet−Teller (BET) specific surface area was measured by nitrogen adsorption at 77 K on a Micromeritics Tristar 2020 apparatus.
Electron microscopy
HRTEM images were acquired on a Talos F200X G2 TEM with an acceleration voltage of 200 kV. The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were acquired on a FEI Titan G2 60–300 STEM equipped with a probe corrector and HF5000.
X-ray absorption fine structure (XAFS) spectroscopy
XANES and EXAFS data were conducted at the Pt L3-edge on BL11B beamline at Shanghai Synchrotron Radiation Facility (SSRF), China, operated at 3.5 GeV with injection currents of 140–210 mA. A Si (111) double-crystal monochromator was used for the energy selection. The energy was calibrated by Pt foil (E0 = 11564 eV). Pt foil and PtO2 were used as reference samples and measured in the transmission mode. Other samples were measured in the fluorescence mode using a Lytle detector. XAS data were processed and analyzed using the Demeter software package55.
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)
In-situ DRIFTS data of CO on catalysts were conducted on a VERTEX 70 spectrometer equipped with an in-situ Praying Mantis diffuse reflection reaction cell (Harrick Scientific) and a liquid nitrogen cooled mercury–cadmium–telluride (MCT) narrow-band detector with a resolution of 4 cm−1. For each sample, about 30 mg Pt/CeO2 was packed smoothly in the reaction cell and then pretreated with air for 1 h at 200 °C. After cooling to room temperature, the atmosphere was switched to argon for 10 min and the background spectra were recorded. Subsequently, the atmosphere was switched to 5 vol.% CO/Ar and CO-DRIFTS data were collected after allowing CO adsorption for 20 min. Then, the sample was purged with argon at room temperature for CO desorption until no further change could be observed in the spectra. Finally, while continuing the argon purge, the temperature was gradually increased to 160 °C at a rate of 2 °C/min. During the progress, spectra were collected to observe the temperature-dependent CO desorption behaviors from the catalysts.
17O Isotopic Labeling
Samples (typically 150 mg) were put into a glass tube and heated at 120 °C under vacuum for 1 h to remove physically adsorbed water on the surface, then cooled to room temperature before 17O2 gas (90% 17O, Cambridge Isotope Laboratories) was introduced. The tube was sealed and heated at 270 °C for 24 h. This labeling temperature was carefully chosen such that surface enrichment of the interface sites (Pt-17O-Ce) was optimized, balancing efficient labeling with minimal oxygen diffusion to ensure high 17O concentration at the catalyst surface.
Solid-State NMR Measurement
17O magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were collected on a 9.4 T Bruker Avance III spectrometer at a Larmor frequency of 54.2 MHz with 1.3 mm HX and 3.2 mm HX MAS probes, a 14.1 T Bruker Avance III spectrometer at a Larmor frequency of 81.3 MHz with a 1.3 mm HX MAS probe, and a 20.0 T Bruker Avance NEO spectrometer at a Larmor frequency of 115.2 MHz with a 1.6 mm HX MAS probe. 1H MAS NMR spectra were performed on a 9.4 T Bruker Avance III spectrometer at a Larmor frequency of 400 MHz with a 3.2 mm HX MAS probe. 17O and 1H chemical shifts are referenced to H2O at 0.0 ppm and adamantane at 1.92 ppm, respectively. All samples were packed into laser marked rotors in a N2 glove box. Dmfit package was used to simulate the 17O NMR spectra.
CO Oxidation Performance
The CO oxidation reaction was performed in a fixed-bed reactor, with a gas mixture of 1 vol% CO and 8 vol% O2 balanced with Ar at a total flow rate of 100 mL/min. Thirty milligrams of the catalyst with a particle size of 40–60 mesh, was diluted with inert SiC powder (0.3 g) to prevent the temperature gradients, was packed into a U-type quartz tube and pretreated with 10 vol% O2/Ar at 300 °C for 1 h. After cooling down to room temperature, reactant gas was introduced with the temperature ramped at 2 °C/min until 450 °C. The concentration of outlet gas composition was monitored by on-line MS and the conversion of CO was determined by the concentration of outlet CO2. The turnover frequency is calculated by using formula reported in ref. 46.
Flow rate of CO: 100 mL/min × 1 vol.% = 1 mL /min = 6.82 × 10−7 mol/s
where XCO and VCO are the CO conversion and the flow rate of CO (6.82 × 10−7 mol/s), respectively. mcatalyst is the mass of catalysts (0.03 g). wPt is the weight percentage of Pt in catalysts determined by ICP-OES. MPt is the molar mass of Pt (195 g/mol).
DFT Calculation
Spin-polarized calculations were performed by using the Vienna Ab initio Simulation Package (VASP)56, applying the generalized gradient approximation with the Perdew-Burke-Ernzerhof (PBE) functional57. To account for strong correlation effects, we implemented a Hubbard U correction58, applying an effective U value of 5.0 eV to the localized Ce 4f orbitals. For all calculations, plane-wave kinetic energy cutoff was set at 500 eV59. Geometry optimizations were finished when the Hellman-Feynman force on each relaxed ion fell below 0.02 eV·Å−1. The convergence thresholds were set at 10−5 eV for geometric optimizations and 10−8 eV for calculating chemical shifts and electric field gradients40. The van der Waals (vdW) interactions were incorporated by using Grimme et al.’s DFT-D3 method60. The optimized lattice parameter for cerium oxide is 5.448 Å, which is in agreement with the experimental value of 5.411 Å61.
We used the CeO2 (111) surface to model the CeO2 substrate, consisting of six O−Ce−O trilayers with a (4 × 4) surface cell used in A-Pt4O5, A-Pt9O4, A-Pt6O7, S-Pt1, E-Pt1(4c), E-Pt1(4c’), E-Pt1(6c), E-Pt1H2 and A-Pt3O3, and a (3 × 3) surface cell used in A-Pt1, A-Pt1O2, A-Pt1(OH)2, and A-Pt3. A 2 × 2 × 1 Monkhorst-Pack grid was used to sample the k-point mesh. To eliminate slab-slab interactions, a 15 Å vacuum gap was introduced along the [111] direction.
The 17O NMR parameter calculations were carried out in accordance with the methodology outlined in our previous work40. To calculate the quadrupolar parameters (CQ and ηQ) for 17O, the experimental quadrupole moment (Q) of −0.02558 barns62 was used. The isotropic chemical shift (δiso) is defined as δiso = δref + δcal, where δref is the reference chemical shift and δcal is the chemical shift value calculated using VASP. The δref for each model was determined by aligning the average δcal of middle layers (O layers 4–9) to the experimental value of 877 ppm. All determined δref values are in the range of 50–60 ppm (given in title of Supplementary Tables 3–13, 16 and 17).
For calculations of CO oxidation and Pt4O5 cluster decomposition, a (4 × 4) CeO2 (111) surface with three O−Ce−O trilayers was used to model the substrate and the bottom CeO2 trilayers were fixed for the geometric optimizations. The transition states for the CO oxidation reaction over E-Pt1(4c) and E-Pt1(6c) models were determined by employing the climbing-image nudged elastic band technique63. Further frequency calculations were performed to confirm the obtained TS structures.
The adsorption energies (Eads) of H2O and CO were calculated as follows:
where Eadsorbate/slab is the total energy of the adsorption complex, Eslab is the energy of the Pt/CeO2 substrate, and Eadsorbate is the energy of the gas-phase molecule.
The Gibbs free energy change for the dissociative water adsorption process of Pt4O5/CeO2 were calculated as follows:
where Eabs, ∆EZPE, and ∆S are the adsorption energy of intermediate, change in zero-point energy and entropy change of the reaction, respectively. ∆U is the internal energy difference between 0 K and T. The thermal correction for adsorbate was calculated by using the VASPKIT package64.
Data availability
The authors declare that the data generated in this study are provided in the Supplementary Information/Source Data files. Source data are provided with this paper.
References
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Yang, X.-F. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Lang, R. et al. Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 120, 11986–12043 (2020).
Li, Z. et al. Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites. Chem. Rev. 120, 623–682 (2020).
Kaiser, S. K. et al. Single-atom catalysts across the periodic table. Chem. Rev. 120, 11703–11809 (2020).
Ji, S. et al. Chemical synthesis of single atomic site catalysts. Chem. Rev. 120, 11900–11955 (2020).
Yang, M. et al. Catalytically active Au-O(OH)x- species stabilized by alkali ions on zeolites and mesoporous oxides. Science 346, 1498–1501 (2014).
Liu, J. Catalysis by supported single metal atoms. ACS Catal. 7, 34–59 (2017).
Ma, Y. et al. Tailoring of the proximity of platinum single atoms on CeO2 using phosphorus boosts the hydrogenation activity. ACS Catal. 9, 8404–8412 (2019).
Qin, R. et al. Surface coordination chemistry of atomically dispersed metal catalysts. Chem. Rev. 120, 11810–11899 (2020).
Zhang, Z. et al. Memory-dictated dynamics of single-atom Pt on CeO2 for CO oxidation. Nat. Commun. 14, 2664 (2023).
Bruix, A. et al. Maximum noble-metal efficiency in catalytic materials: atomically dispersed surface platinum. Angew. Chem. Int. Ed. 53, 10525–10530 (2014).
Dvorák, F. et al. Creating single-atom Pt-ceria catalysts by surface step decoration. Nat. Commun. 7, 10801 (2016).
Jones, J. et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 353, 150–154 (2016).
Liu, P. et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 352, 797–801 (2016).
Wan, W. et al. Highly stable and reactive platinum single atoms on oxygen plasma-functionalized CeO2 surfaces: nanostructuring and peroxo effects. Angew. Chem. Int. Ed. 61, e202112640 (2022).
Slavinskaya, E. M. et al. States of Pt/CeO2 catalysts for CO oxidation below room temperature. J. Catal. 421, 285–299 (2023).
Resasco, J. et al. Uniformity is key in defining structure–function relationships for atomically dispersed metal catalysts: the case of Pt/CeO2. J. Am. Chem. Soc. 142, 169–184 (2020).
Kottwitz, M. et al. Local structure and electronic state of atomically dispersed Pt supported on nanosized CeO2. ACS Catal. 9, 8738–8748 (2019).
Nie, L. et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 358, 1419–1423 (2017).
Chen, L.-N. et al. Efficient hydrogen production from methanol using a single-site Pt1/CeO2 catalyst. J. Am. Chem. Soc. 141, 17995–17999 (2019).
Jiang, Z. et al. Stabilizing platinum atoms on CeO2 oxygen vacancies by metal-support interaction induced interface distortion: mechanism and application. Appl. Catal. B 278, 119304 (2020).
Zhang, S. et al. Insights into the mechanism of n-Hexane reforming over a single-site platinum catalyst. J. Am. Chem. Soc. 142, 16533–16537 (2020).
DeRita, L. et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 18, 746–751 (2019).
Tan, W. et al. Fine-tuned local coordination environment of Pt single atoms on ceria controls catalytic reactivity. Nat. Commun. 13, 7070 (2022).
Ding, C. et al. Reversible transformation and distribution determination of diverse Pt single-atom species. J. Am. Chem. Soc. 145, 2523–2531 (2023).
Tieu, P. et al. Directly probing the local coordination, charge state, and stability of single atom catalysts by advanced electron microscopy: a review. Small 17, 2006482 (2021).
Chen, Z. et al. Structural analysis of single-atom catalysts by X-ray absorption spectroscopy. Acc. Chem. Res. 57, 521–532 (2024).
Wang, A. et al. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65–81 (2018).
Hülsey, M. J. et al. Approaching molecular definition on oxide-supported single-atom catalysts. Acc. Chem. Res. 56, 561–572 (2023).
Zhang, W. et al. In situ solid-state NMR for heterogeneous catalysis: a joint experimental and theoretical approach. Chem. Soc. Rev. 41, 192–210 (2012).
Ashbrook, S. E. et al. Recent advances in solid-state nuclear magnetic resonance spectroscopy. Annu. Rev. Anal. Chem. 11, 485–508 (2018).
Xu, J. et al. Metal active sites and their catalytic functions in zeolites: insights from solid-state NMR spectroscopy. Acc. Chem. Res. 52, 2179–2189 (2019).
Reif, B. et al. Solid-state NMR spectroscopy. Nat. Rev. Method Prime 1, 2 (2021).
Peng, L. et al. Detection of Brønsted acid sites in zeolite HY with high-field 17O-MAS-NMR techniques. Nat. Mater. 4, 216–219 (2005).
Huo, H. et al. Solid-state MAS NMR studies of Brønsted acid sites in zeolite H-Mordenite. J. Am. Chem. Soc. 134, 9708–9720 (2012).
Ashbrook, S. E. & Smith, M. E. Solid state 17O NMR—an introduction to the background principles and applications to inorganic materials. Chem. Soc. Rev. 35, 718–735 (2006).
Ashbrook, S. E. et al. 17O NMR spectroscopy of crystalline microporous materials. Chem. Sci. 12, 5016–5036 (2021).
Wang, M. et al. Identification of different oxygen species in oxide nanostructures with 17O solid-state NMR spectroscopy. Sci. Adv. 1, e1400133 (2015).
Wang, Q. et al. Mapping the oxygen structure of γ-Al2O3 by high-field solid-state NMR spectroscopy. Nat. Commun. 11, 3620 (2020).
Chen, J. et al. Emerging applications of 17O solid-state NMR spectroscopy for catalytic oxides. ACS Catal. 13, 3485–3500 (2023).
Li, Y. et al. Distinguishing faceted oxide nanocrystals with 17O solid-state NMR spectroscopy. Nat. Commun. 8, 581 (2017).
Merle, N. et al. 17O NMR gives unprecedented insights into the structure of supported catalysts and their interaction with the silica carrier. J. Am. Chem. Soc. 134, 9263–9275 (2012).
Hope, M. A. et al. A 17O paramagnetic NMR study of Sm2O3, Eu2O3, and Sm/Eu-substituted CeO2. Solid State Nucl. Magn. Reson. 102, 21–30 (2019).
Kunwar, D. et al. Stabilizing high metal loadings of thermally stable platinum single atoms on an industrial catalyst support. ACS Catal. 9, 3978–3990 (2019).
Yoshida, H. et al. XANES study of the support effect on the state of platinum catalysts. J. Synchrotron Radiat. 6, 471–473 (1999).
Lykhach, Y. et al. Reactivity of atomically dispersed Pt2+ species towards H2: model Pt-CeO2 fuel cell catalyst. Phys. Chem. Chem. Phys. 18, 7672–7679 (2016).
Hejazi, S. et al. On the controlled loading of single platinum atoms as a Co-catalyst on TiO2 anatase for optimized photocatalytic H2 generation. Adv. Mater. 32, 1908505 (2020).
Pereira-Hernández, X. I. et al. Tuning Pt-CeO2 interactions by high-temperature vapor-phase synthesis for improved reducibility of lattice oxygen. Nat. Commun. 10, 1358 (2019).
Fan, Y. et al. Water-assisted oxidative redispersion of Cu particles through formation of Cu hydroxide at room temperature. Nat. Commun. 15, 3046 (2024).
Su, Y.-Q. et al. Theoretical approach to predict the stability of supported single-atom catalysts. ACS Catal. 9, 3289–3297 (2019).
Beniya, A. & Higashi, S. Towards dense single-atom catalysts for future automotive applications. Nat. Catal. 2, 590–602 (2019).
He, C. et al. Recent advances in the catalytic oxidation of volatile organic compounds: a review based on pollutant sorts and sources. Chem. Rev. 119, 4471–4568 (2019).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Kresse, G. & Hafner, J. Ab Initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Perdew, J. P. et al. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Nolan, M. et al. The electronic structure of oxygen vacancy defects at the low index surfaces of ceria. Surf. Sci. 595, 223–232 (2005).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Kümmerle, E. A. & Heger, G. The structures of C–Ce2O3+δ, Ce7O12, and Ce11O20. J. Solid State Chem. 147, 485–500 (1999).
Sundholm, D. & Olsen, J. Finite element multiconfiguration Hartree-Fock calculations on carbon, oxygen, and neon: the nuclear quadrupole moments of 11C, 17O, and 21. Ne. J. Phys. Chem. 96, 627–630 (1992).
Henkelman, G. et al. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Wang, V. et al. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Acknowledgements
This work was supported by National Key R&D Program of China (Grant No. 2021YFA1502803) to L.P., the National Natural Science Foundation of China (NSFC) (Grant Nos. 22472075, 22272075, W2421041, 21972066, and 91745202) to L.P., NSFC—Royal Society Joint Program (Grant No. 21661130149) to L.P., the NSFC (Grant Nos. 21773047 and U1832180) to Z.Z., Zhejiang Provincial Natural Science Foundation (ZJNSF) (Grant No. LGC22B050025) to H.G., and the NSFC (Grant No. 22402119) to J.C.L.P. thanks the Royal Society and Newton Fund for a Royal Society—Newton Advanced Fellowship. This work was also supported by the Research Funds for the Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. We thank the BL11B station in the Shanghai Synchrotron Radiation Facility (SSRF). We thank Prof. Arno P.M. Kentgens and Dr. Ernst R.H. van Eck in the Radboud University for invaluable discussions and help in this work. We are grateful to the High Performance Computing Center of Nanjing University for doing the numerical calculations in this paper on its blade cluster system.
Author information
Authors and Affiliations
Contributions
Y.W., J.C., J.Z., X.K., H.G., and L.P. performed 17O isotope enrichment, collected as well as analyzed the NMR spectra; Y.W., Q.W., X.X., J.W., J.-H.D., and M.W. conducted other characterizations of the catalysts and analyzed the results; Y.W., C.D., and L.N. carried out the preparation of SACs and the reaction performance; F.W. and W.L. conducted the DFT calculations; Z.Z., W.H., W.T., and W.D. provided valuable suggestions; L.P. conceived the idea, designed the study and coordinated the project. Y.W., J.C., and L.P. wrote the manuscript, and all authors discussed the experiments and final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Olga Lapina, Talat Shahnaz Rahman, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wen, Y., Wang, F., Zhu, J. et al. Revealing the structure-activity relationship of Pt1/CeO2 with 17O solid-state NMR spectroscopy and DFT calculations. Nat Commun 16, 3537 (2025). https://doi.org/10.1038/s41467-025-58709-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58709-2
