
Abstract
Electrochemical conversion of petrochemical-derived hydrocarbons to high-value oxygenates can utilize renewable energy and reduce carbon emissions. However, this involves the challenging activation of inert C(sp3)-H bonds at room temperature. Here, we introduce an electrocatalyst:mediator assembly in which CoOx clusters-decorated IrO2 electrocatalyst activates NO3- mediator to a highly reactive radical capable of abstracting a hydrogen atom from benzylic C-H. The interface between CoOx and IrO2 promotes NO3- activation by facilitating the desorption of NO3● radical for subsequent reaction. Our strategy is demonstrated through the selective oxidation of toluene to benzaldehyde with high Faradaic efficiency of 86( ±1)% at 25 mA/cm2, a factor of >3 times higher than the bare electrocatalyst. The electrocatalyst:mediator assembly is operated stably for 100 h, with minimal decline in performance. When translated into a flow system, a Faradaic efficiency of 60( ±4)% at 200 mA/cm2 was achieved.
Similar content being viewed by others


Introduction
The partial oxidation of petrochemical-derived hydrocarbons to oxygenates is important for the plastics and textiles industries. This involves the activation of inert C(sp3)-H bonds in the hydrocarbons, which are characterized by high bond dissociation energies of 70–130 kcal/mol1. Thus, the partial oxidations of hydrocarbons typically occur with high temperatures, pressures and harsh oxidants, such as chromium or selenium-based reagents, thereby leading to significant carbon emissions and environmental consequences2. For example, the partial oxidation of toluene is important to the production of benzaldehyde, a pharmaceuticals and plastics precursor with an annual demand of 170 million kg3,4. The reaction is conducted at high temperatures (i.e., 250–650 oC) and pressures due to the relative stability of C(sp3)-H bond adjacent to aromatic rings, and requires extensive cooling due to its highly exothermic nature, thus resulting in high energy requirements and CO2 emissions5.
The electrification of hydrocarbon partial oxidations offers the potential for benign conditions and utilization of renewable energy, which can significantly reduce the carbon footprint6. These reactions can be coupled to a cathodic hydrogen evolution reaction (HER) from water, thereby increasing the chemical value that can be derived from a given amount of electricity input. An example is the electrochemical oxidation of ethylene and propylene to the respective epoxides/glycols by the direct oxidation or chlorohydrin routes7,8,9,10. Nevertheless, the scope of electrochemical partial oxidations remains limited, especially for reactions that involve C(sp3)-H activation11. While electrochemical C-H functionalization, such as amination12 and arylation13 have been developed, these are mainly for the synthesis of specialty chemicals in a fully organic environment and tend to operate in a restricted current density window (i.e., 0.5–5 mA/cm2)14. These reactions rely on homogeneous catalysts such as organometallics12,13,15 and organic mediators5, while non-targeted commercial electrodes, such as platinum and reticulated vitreous carbon, serve as current collectors (Fig. 1a). In the context of electro-refining petrochemicals-derived hydrocarbons to commodity chemicals, however, parameters such as Faradaic efficiency, current density and the ability to interface with cathodic HER become critical to enable technical scale application and maximize the chemical output of renewable electricity. In this case, materials-driven electrocatalysts are important to reduce reaction overpotential and provide adequate active sites to funnel electron selectivity towards the desired redox reaction2,16, but lack the ability to selectively target inert C(sp3)-H bonds (Fig. 1b). We believe that combining the insights from organic electrosynthesis and materials-driven electrocatalysis can help to overcome the low reactivity of C(sp3)-H bonds while enabling the partial oxidation of hydrocarbons at high Faradaic efficiencies and current densities.

Illustration of anodic toluene oxidation by a, homogeneous catalysts with non-targeted commercial electrodes as current collectors, b, materials-driven electrocatalysis and c, the electrocatalyst:mediator assembly. d Schematic of anodic toluene oxidation coupled to hydrogen evolution from water at cathode. e Faradaic efficiencies of anodic toluene oxidation with different mediators. f LSV of the reaction system with and without NO3-. All error bars represent standard deviation based on three independent samples. No iR correction was conducted. Source data for Fig. 1e, f are provided as a Source Data file.
In this work, we present the concept of an electrocatalyst:mediator assembly, in which materials-driven electrocatalysis funnels electron selectivity towards the desired redox reaction, while the mediator selectively targets inert C(sp3)-H bonds (Fig. 1c). We demonstrate this through the selective oxidation of toluene to benzaldehyde; CoOx clusters-decorated IrO2 (CoOx/IrO2) electrocatalyst electrochemically oxidizes NO3- mediator to the NO3● radical, which can abstract a hydrogen atom from benzylic C-H in toluene. The electrocatalyst:mediator assembly yielded a high Faradaic efficiency of 86( ±1)% towards benzaldehyde at 25 mA/cm2, which is more than 3 times higher than direct oxidation on the bare electrocatalyst. The electrocatalyst:mediator assembly is operated stably for 100 h with minimal decline in performance throughout, and can also be broadly applied to the electrochemical oxidation of other toluene derivatives. The interface between CoOx and IrO2 promotes NO3- activation by facilitating the desorption of NO3● radical for subsequent reaction, yielding 2x higher Faradaic efficiency compared to commercial electrodes such as glassy carbon, boron doped diamond and Pt, and 20% higher compared to IrO2. When translated into a flow system, a Faradaic efficiency of 60( ±4)% at 200 mA/cm2 was achieved.
Results
We demonstrated the concept of our electrocatalyst:mediator assembly through the partial oxidation of toluene to benzaldehyde (Fig. 1d, configuration of the H-type cell in Supplementary Fig. 1). This is conducted on the CoOx/IrO2 electrocatalyst on Ti support with 0.24 M toluene in an anolyte comprising 0.1 M LiBF4 in acetonitrile (Supplementary Fig. 2a). At the cathode, hydrogen evolution from water is conducted on a Pt plate in aqueous LiBF4 solution (Supplementary Fig. 2b). Both reactions are separated by a proton exchange membrane. The direct oxidation of toluene on the bare CoOx/IrO2 electrocatalyst yielded a low Faradaic efficiency of 26( ±4)% at 25 mA/cm2 (Fig. 1b, potential versus reaction progress and linear sweep voltammogram can be found in Supplementary Fig. 3). With the introduction of the NO3- mediator, the Faradaic efficiency increased by a factor of >3x to 86%. We compared the electrochemical performance with that of other mediator candidates with known oxidative properties, such as NHPI, ClO4-, Br- and Cl- (Fig. 1e, the corresponding amount of produced benzaldehyde and LSVs can be found in Supplementary Fig. 4 and Fig. 1f, respectively)17,18,19,20. The Faradaic efficiencies with N-Hydroxyphthalimide (NHPI) and ClO4- are higher than that of the bare electrocatalyst at 46( ±13) and 30( ±2)%, but far lower than that obtained with NO3-. No difference in Faradaic efficiency was observed whether NH4NO3, LiNO3 or NaNO3 was used, which indicates that the cation does not play a role in this reaction (Supplementary Fig. 5a). The optimal NO3- concentration was 0.06 M; further increments in NO3- concentration led to slightly lower Faradaic efficiencies (Supplementary Fig. 5b). The volume ratio of toluene to acetonitrile was also optimized (Supplementary Fig. 5c).
To understand the role of NO3-, we introduced 2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO) radical scavenger into the reaction mix (Fig. 2a). This drastically reduced the Faradaic efficiency to 4.9( ±2.6)%, which suggests that the reaction involves the formation of radicals, which are quenched through the addition of TEMPO. We removed aliquots of anolyte over the course of the reaction for electron paramagnetic resonance (EPR) studies, which showed that the TEMPO signal decreased in intensity and was completely extinct at 20 min (Fig. 2b). This confirmed the formation of radicals during reaction, suggesting that NO3- was promoted to the highly reactive NO3● radical on the CoOx/IrO2 electrocatalyst. Cyclic voltammetry (CV) revealed an oxidation peak at about 1.7 V versus Ag/Ag+, which can be attributed to the conversion of NO3- to NO3● (Supplementary Fig. 6). This conversion is reversible, as evidenced by the reduction peak in reverse scan at about 1.8 V versus Ag/Ag+. The UV-vis spectra of the anolyte before the reaction showed a peak at 300 nm, which corresponds to the n→π* transition in acetonitrile21. This peak disappeared after the reaction, and a new peak was formed at 268 nm, which can be attributed to HNO3 (Fig. 2c). This suggests that the NO3● radical abstracted a hydrogen atom from toluene to form a benzyl radical, which reacted with O2 from air to form benzaldehyde. This is confirmed by the drop in Faradaic efficiency to 11( ±2)% when Ar was sparged into the anolyte (Supplementary Fig. 7). It also means that the Faradaic efficiency can be improved by increasing the presence of O2 in the anolyte, i.e., 86( ±1)% with the sparging of air versus 65( ±10)% at ambient conditions.

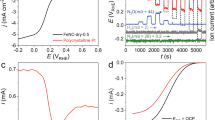
a Quenching of reaction with the addition of TEMPO radical scavenger. All error bars represent standard deviation based on three independent samples. b EPR spectra of the anolyte with and without NO3-, using TEMPO to track the formation of radical intermediates. c UV-vis spectra of the anolyte before and after reaction. d Half-cell potential and Faradaic efficiency of benzaldehyde over 100 h. e Composition of the CoOx/IrO2 anode before and after 100 h of reaction, based on EDX analysis. Source data for Fig. 2a-e are provided as a Source Data file.
Meanwhile, the Faradaic efficiency for bare CoOx/IrO2 electrocatalyst decreased to 3.0% with the addition of TEMPO, which suggests that the reaction pathway for direct oxidation also involves the formation of a benzyl radical (Fig. 2a). The time taken for the TEMPO signals to disappear, as observed by EPR spectroscopy, was longer at 40 min, which indicates that this reaction pathway was not as robust as with a NO3- mediator (Fig. 2b). This explains why the Faradaic efficiency achieved with the electrocatalyst:mediator assembly is much higher than direct oxidation on the bare electrocatalyst.
The CoOx/IrO2:NO3- electrocatalyst:mediator assembly was highly stable under the electrochemical settings required for the oxidation of toluene to benzaldehyde. We demonstrated this through 100 h of continuous operation at a current density of 25 mA/cm2, during which portions of the electrolyte were periodically removed for analysis and replaced with fresh electrolyte. The system maintained a stable applied potential of 2.17( ±0.06) V versus Ag/Ag+ and Faradaic efficiency averaging 81( ±7)% (Fig. 2d). Post-reaction analysis of the anode through energy-dispersive X-ray spectroscopy (EDX) revealed no obvious changes in composition of the CoOx/IrO2 electrocatalyst (Fig. 2e, Supplementary Fig. 8), which suggests that electrocatalyst:mediator assembly is suitable for long-term operation.
The CoOx/IrO2:NO3- electrocatalyst:mediator assembly can also be broadly applied to the electrochemical oxidation of other toluene derivatives (Supplementary Fig. 9). The Faradaic efficiency is higher when the electron-donating methyl group is present in the benzene ring (i.e., 97( ±7)% for p-xylene), which stabilizes the benzyl radical intermediate. The converse is true for the electron-withdrawing chloro and carboxylic groups (i.e., 64( ±7)% for p-chlorotoluene and 52( ±6)% for p-toluic acid). We observed lower Faradaic efficiencies when bulky benzyl or ethyl groups are adjacent to the benzylic C-H (i.e., 44( ±4)% for diphenylmethane and 34( ±7)% for propylbenzene). This indicates that the oxidation reaction occurs at the electrocatalyst surface where the bulky groups present a large steric hindrance, which suggests that CoOx/IrO2 plays a significant role in the electrocatalyst:mediator assembly.
We thus proceeded to characterize the CoOx/IrO2 electrocatalyst. CoOx/IrO2 was deposited on a Ti mesh through dip coating in electrocatalyst ink comprising Co and Ir precursors, followed by thermal decomposition22. The CoOx loading amount was optimized by varying the ratio of Co to Ir precursors in the electrocatalyst ink (see Supplementary Figs. 10-16 for characterization of the CoOx/IrO2 samples with different CoOx loadings). The CoOx/IrO2 anode synthesized with Co/Ir ratio of 20 wt% showed the highest double layer capacitance (Cdl) in the reaction medium, which indicates highest electrochemical active surface area (ECSA, Supplementary Fig. 10h, CV curves of anodes synthesized from inks of Co/Ir ratio in Supplementary Fig. 10b-g) and therefore highest catalytic activity (Supplementary Fig. 10a). LSV scans performed in reaction medium (Supplementary Fig. 10i) revealed an obvious oxidation peak at around 1.6-1.7 V versus Ag/Ag+ for the anode synthesized from ink of Co/Ir ratio 20 wt%. This peak corresponds to the oxidation peak of NO3- (see Supplementary Fig. 6), which indicates greater affinity towards NO3-. This means that NO3- can be oxidized more easily to the NO3● radical to facilitate the electrochemical toluene oxidation reaction, as evidenced by the higher corresponding current density. Scanning electron microscopy (SEM) showed that CoOx/IrO2 particles were well-coated on the Ti mesh (Supplementary Fig. 13a-b). X-ray photoelectron spectroscopy (XPS) confirmed the presence of Co in +2 and +3 oxidation states, as well as Ir in +4 oxidation state (Fig. 3a-c). X-ray diffraction (XRD) showed that the CoOx/IrO2 electrocatalyst consisted of (110), (101), (200), (210), (220) and (301) planes of rutile IrO2 at 28.1o, 34.7o, 40.1o, 45.0o, 58.5o and 69.3o respectively, as well as the (400) plane of Co3O4 at 44.8o (Fig. 3d)23,24. EDX showed that Co, Ir and O were well-distributed in the CoOx/IrO2 electrocatalyst (Fig. 3e). High-resolution transmission electron microscopy (HRTEM) revealed the CoOx cluster-on-IrO2 structure of the CoOx/IrO2 electrocatalyst (Fig. 3f). The small and dark particles, with an average size approximating 2 nm, are CoOx clusters distributed over the IrO2 substrate. The corresponding FFT pattern showed that the clusters comprised face-centered cubic lattice with spinel structure (inset, Fig. 3f). IrO2 existed in continuous phase across a relatively large area, which was crystalline and comprised tetragonal lattice with rutile structure, which is clearly distinguished from that of the CoOx phase. (Supplementary Fig. 13c). As no distortion was observed in IrO2 lattice, it can be concluded that Co was neither doped into IrO2 lattice at interstitial sites nor by replacing Ir atoms.

XPS spectra of a, Co b, Ir and c, O in CoOx/IrO2. d XRD pattern of CoOx/IrO2. e EDX images showing the distribution of Co, Ir, and O on CoOx/IrO2. f HRTEM image of the CoOx cluster of the CoOx/IrO2 electrocatalyst. Inset: The corresponding FFT image of the CoOx cluster. g Co K-edge XANES spectrum and h, Fourier transformation of the k2-weighted EXAFS of CoOx/IrO2. i Ir L3 edge XANES spectrum and j, Fourier transformation of k2-weighted EXAFS of IrO2 and CoOx/IrO2 catalyst. Source data for Fig. 2a–d and g–j are provided as a Source Data file.
To understand the influence of CoOx clusters on the electronic structure and local atomic geometric environment of IrO2, we performed X-ray absorption fine structure (XAFS) experiment on the CoOx/IrO2 electrocatalyst and on IrO2 synthesized through the same method. The Co K edge X-ray absorption near edge structure (XANES) spectrum of CoOx/IrO2 showed an identical edge energy compared to that of a commercial Co3O4 standard, which implies that Co possess similar oxidation states as Co3O4 (Fig. 3g). The Fourier transformation of extended X-ray absorption fine structure (EXAFS) revealed three peaks with positions similar to those of Co3O4 but with smaller amplitudes. These peaks correspond to the Co-O path, the Co-Co path between octahedral centers, and the Co-Co path between octahedral and tetrahedral centers, indicating the presence of small Co3O4 clusters highly distributed across the IrO2 surface (Fig. 3h). This observation is consistent with the findings from the HRTEM analysis. The low intensity of Co-Co scattering paths compared with those of the Co3O4 standard suggests higher atomic disorder due to the Co3O4 cluster configuration. Meanwhile, Ir L3 edge XANES spectrum of CoOx/IrO2 revealed a higher white line than those of both standard rutile IrO2 and our synthesized IrO2, implying that the Ir in CoOx/IrO2 exists in >4+ oxidation state and is therefore capable of enhanced electro-oxidation performance (Fig. 3i-j)25,26.
The importance of CoOx/IrO2 in the electrocatalyst:mediator assembly is demonstrated by comparing its electrochemical performance with electrocatalyst:mediator assemblies comprising other electrocatalysts (Fig. 4a, LSVs in Supplementary Fig. 17a). No reaction occurred on the Ti mesh, which indicates that it functions as a support and does not contribute to the reaction. Poor Faradaic efficiencies (5.4–34%) were obtained with commercial electrocatalysts such as glassy carbon, boron-doped diamond (BDD) and Pt. The highest Faradaic efficiency was obtained with IrO2 at 66( ±4)%. The modification of IrO2 with CoOx clusters led to an increase in Faradaic efficiencies even under different current densities (10–40 mA/cm2). As little reaction was observed with Co3O4, this suggests that the CoOx clusters may not be the reaction centers of the CoOx/IrO2 electrocatalyst. Meanwhile, modification with Au (Au/IrO2) yielded lower Faradaic efficiencies (Fig. 4b, see Supplementary Figs. 16 and 18 for characterization of IrO2 and Au/IrO2 and LSV in Supplementary Fig 17b). EPR spectroscopy performed on a reaction with TEMPO scavenger showed that, when IrO2 was used as the electrocatalyst, the TEMPO signal took a longer time to become extinct at 30 min, compared to 20 min with the CoOx/IrO2 electrocatalyst (Fig. 4c). When Au/IrO2 was used as the electrocatalyst, the TEMPO signal became extinct at 35 min. This indicates that the rate of radical formation was highest with CoOx/IrO2, followed by IrO2 and Au/IrO2, which explains why Faradaic efficiency is the highest with CoOx/IrO2 electrocatalyst.

Faradaic efficiencies of anodic toluene oxidation with electrocatalyst:mediator assemblies comprising a commercial electrocatalysts and b IrO2-based electrocatalysts. All error bars represent standard deviation based on three independent samples. The reaction was run in a H-type cell, in which 0.5 mL toluene was dissolved in 19.5 mL of anolyte comprising 0.1 M LiBF4 and 0.06 M LiNO3 in acetonitrile solution. The catholyte is 20 mL of 0.1 M LiBF4 MilliQ water (18.2 Ω) solution. The anolyte and catholyte were separated with a Nafion® 117 membrane. c EPR spectra of the anolyte with electrocatalyst:mediator assemblies comprising IrO2-based electrocatalysts. d Adsorption energies of the NO3● radical on the IrO2(100)-Ov, CoOx/IrO2(100)-Ov and Co3O4(100) slabs. Side views of the NO3● radical adsorbed on the e, Co and f, Ir atom of CoOx/IrO2(100)-Ov. Source data for Fig. 4a-c are provided as a Source Data file.
We further investigated the CoOx cluster-promoted NO3- activation through density function theory (DFT) analysis, by modelling the adsorption of NO3● radical on the IrO2 versus CoOx/IrO2 electrocatalysts. The IrO2 electrocatalyst was modelled through an IrO2(100) slab containing an oxygen vacancy, termed here as IrO2(100)-Ov, and the NO3● radical adsorption energy is -2.15 eV (Fig. 4d, Supplementary Fig. 19). This suggests that it is difficult for NO3● radicals formed on the IrO2 surface to desorb and abstract a hydrogen atom from toluene. A Co3O4(100) slab was built to simulate NO3● radical adsorption energy on the surface (Supplementary Fig. 20), and the NO3● radical adsorption energy is -1.02 eV. To simulate the interface between the CoOx cluster and IrO2 surface, we built a cluster-on-surface model reported in previous studies22,27, referred to as CoOx/IrO2(100)-Ov (Fig. 4e-f, Supplementary Fig. 21). The synergistic effect between the CoOx cluster and IrO2 surface is such that the NO3● radical adsorption energy is -2.18 eV on the Irinterfacial site and -0.81 eV on the Cointerfacial site. This suggests that it is easier for the NO3● radicals formed on the interfacial Co site to desorb for subsequent reaction, thereby regenerating the active site for another NO3- activation and furbishing the electrochemical system with higher concentration of NO3● radicals.
We also employed DFT to investigate the role of the NO3● radical in enabling C–H bond cleavage (the cartesian coordinates of intermediates and transition states are in Supplementary data 1). We simulated initial states where toluene binds, in different configurations, to an IrO2(100)-Ov slab (Supplementary Fig. 22). In the A0 initial state where the methyl group in toluene binds with an Ir atom, the methyl group points toward the substrate and allows the hydrogen atom to adsorb onto the surface to stabilize itself upon C-H bond cleavage. In this configuration, the C-H bond is elongated to 1.21 Å compared to the typical 1.10 Å in the methyl group of toluene, thereby weakening the bond and facilitating its breakage (Fig. 5a, Supplementary Fig. 23). The released hydrogen atom is transferred to an oxygen atom on the IrO2(100)-Ov surface via the transition state A1-TS, resulting in the formation of an adsorbed benzylic radical and OH group in A2. The overall reaction step from A0 to A2 is characterized by an energy barrier of 0.74 eV and releases -1.07 eV of energy (Fig. 5c).

Proposed reaction pathways of C-H bond cleavage a directly on the IrO2(100)-Ov surface and b mediated by a NO3● radical. Calculated energy profiles of the proposed reaction pathways c, without and d, with the NO3● radical. e Side view and f top view of the B0 state.
On the other hand, the NO3● radical facilitates the cleavage of the C–H bond by abstracting a hydrogen atom from toluene via the transition state B1-TS, thereby generating an adsorbed benzylic radical in B2 (Fig. 5b, e-f and Supplementary Fig. 24). The overall reaction step from B0 to B2 has an energy barrier of 0.26 eV and releases -1.31 eV of energy (Fig. 5d), making it more favorable than the first pathway. This means that the pathway with the NO3● radical (B0-B2) has an energy barrier that is 0.48 eV lower and releases -0.24 eV more reaction energy, compared to the pathway without NO3● radical (A0-A2). Thus C-H bond cleavage is kinetically and thermodynamically more favorable in the presence of the NO3● radical.
To compare with the electrochemical toluene oxidations reported in literature15,20,28,29, we conducted a batch reaction on 0.12 M toluene in an anolyte comprising 0.1 M LiBF4 in acetonitrile, under a current density of 25 mA/cm2 over a reaction time of 800 mins. This is because Faradaic efficiency can decrease towards the end of the batch reaction as the toluene reactant gets depleted; our average Faradaic efficiency over the course of the reaction is about 56.5%. We obtained a yield of 75% and overall conversion rate of 0.15 mmol per cm2 catalyst per hour of reaction, which corresponds to an overall benzaldehyde generation rate of 0.13 mmol per cm2 catalyst per hour of reaction (Fig. 6a). The average Faradaic efficiency of the partial ethylbenzene-to-acetophenone conversion is about 67.2%, and we obtained a yield of 89% and overall conversion rate of 0.17 mmol per cm2 catalyst per hour of reaction, which corresponds to an overall acetophenone generation rate of 0.16 mmol per cm2 catalyst per hour of reaction (Fig. 6b)15,20,28,29.

Comparison of the partial current density, Faradaic efficiency, yield, selectivity, overall conversion rate and product generation rate against state-of-art a electrochemical toluene-to-benzaldehyde conversions20,28, and b electrochemical ethylbenzene-to-acetophenone conversions15,29. c Schematic of flow cell setup that conducts anodic toluene oxidation and cathodic HER. d Faradaic efficiencies of partial toluene-to-benzaldehyde conversion at different current densities with and without the addition of HFIP. The reaction in flow was conducted in a 3-compartment flow cell system. 340 µL toluene (or ethylbenzene or 4-chlorotoluene) was dissolved in 25 mL of anolyte comprising 0.1 M LiBF4 and 0.06 M LiNO3 in pure acetonitrile solvent or 4:1 acetonitrile:HFIP mixture. The catholyte was 0.1 M LiBF4 aqueous solution. The flow rate was 60 mL/s. All error bars represent standard deviation based on three independent samples. e Breakdown of costs at current densities of 20 and 200 mA/cm2, as calculated by TEA. Source data for Fig. 2a-b, d and e are provided as a Source Data file.
Finally, we translated our reaction into a flow cell system based on the obtained insights on the reaction mechanism (Fig. 6c, cell configuration in Supplementary Fig. 25). We postulated that an additive such as hexafluoro-2-propanol (HFIP) can help to stabilize the electron-deficient radicals and funnel selectivity towards the benzaldehyde product14. Fig. 6d shows that without HFIP, the Faradaic efficiency decreases dramatically as the current density increases. The addition of HFIP enabled high Faradaic efficiencies of 60–82% for the electrochemical oxidation of toluene derivatives even at higher current densities of up to 200 mA/cm2. This is significant as prior electrochemical oxidations of benzylic C-H tended to be reported at low current densities <20 mA/cm2 (Supplementary Table 1)20,28,30. Technoeconomic analysis shows that higher current densities (i.e., 200 mA/cm2) lead to profitable plant-gate levelized costs due to reduced capital costs, as the same production rates can be enabled by smaller electrolyzer areas (Fig. 6f, Supplementary Fig. 26). Our electrochemical system also fulfils the minimum Faradaic efficiencies required to be profitable for different electricity costs up to 10 ¢/kWh (Supplementary Fig. 27).
Discussion
We developed an electrocatalyst:mediator assembly, in which the electrocatalyst promotes the mediator to highly reactive state capable of activating the inert hydrocarbons. This is demonstrated in the selective oxidation of toluene to benzaldehyde; CoOx/IrO2 electrocatalyst electrochemically activates NO3- mediator to a NO3● radical that can abstract a hydrogen atom from benzylic C-H in toluene. The electrocatalyst:mediator assembly a high Faradaic efficiency of 86( ±1)% towards benzaldehyde at 25 mA/cm2, a factor of >3 times higher than direct oxidation on the bare electrocatalyst, as the reaction pathway with NO3● radical has an energy barrier that is 0.48 eV lower than the reaction pathway without. The synergistic effect between the CoOx cluster and IrO2 surface reduces the NO3● radical adsorption energy on the Cointerfacial site, which allows NO3● formed on the Cointerfacial site to desorb more readily after activation on the CoOx/IrO2 electrocatalyst. When translated into a flow system, a Faradaic efficiency of 60( ±4)% was achieved at the higher current density of 200 mA/cm2, which can significantly reduce capital costs by achieving the same production rates with smaller electrolyzer areas.
Methods
Materials
All chemicals were used as purchased without further purification. Acetonitrile (suitable for HPLC, gradient grade, ≥99.9%, MeCN), ethylbenzene (ReagentPlus®, 99%), 4-chlorotoluene (98%), Iridium (III) chloride hydrate (reagent grade, 98%, IrCl3·xH2O), Cobalt(II) chloride hexahydrate (ACS reagent, 98%, CoCl2·6H2O), Gold (III) chloride trihydrate (≥99.9% trace metals basis), Lithium nitrate (ReagentPlus®, LiNO3), oxalic acid (ReagentPlus®, ≥99%) and Lithium tetrafluoroborate (98%, LiBF4), N-Hydroxyphthalimide (97%, for peptide synthesis, NHPI), 2,2,6,6-tetramethyl−1-piperidinyloxy (98%, TEMPO), Lithium bromide (ReagentPlus®, ≥99%, LiBr), Lithium chloride (ACS reagent, ≥99%, LiCl), Lithium perchlorate (99.99% trace metals basis, LiClO4), p-Xylene (ReagentPlus®, 99%), 2,4-Dichlorotoluene (99%), 4-Chlorotoluene (98%), p-Toluic acid (98%), Ethylbenzene (ReagentPlus®, 99%), Diphenylmethane (99%), Propylbenzene (98%) 1,2,3,4-Tetrahydronaphthalene (ReagentPlus®, 99%) Tetrabutylammonium perchlorate (for electrochemical analysis, ≥99.0%, TBAP) were purchased from Sigma Aldrich. Dichloromethane (anhydrous (max. 0.005% H2O) ≥99.8% stabilised, AnalaR NORMAPUR® analytical reagent, CH2Cl2), isopropanol (≥99.5%, CMOS for microelectronic, J.T.Baker®) were purchased from VWR Chemicals. Ti screen (0.002 inches thickness, Ti mesh) and Nafion® 117 membrane were purchased from FuelCellStore. Hydrochloric acid (Puriss. p.a., ACS Reagent, Reag. ISO, Reag. Ph. Eur., fuming, ≥37%, APHA: ≤10) and Nitric acid (Puriss. p.a., Reag. ISO, Reag. Ph. Eur., for determinations with dithizone, ≥65%) were purchased from Honeywell. Toluene (ACS Reagent) was purchased from TEDIA. Silver nitrate (99.9%-Ag, ACS, AgNO3) was purchased from Strem Chemicals. The standard calibration gas mixtures for calibrating gas product at gas chromatography system and nitrogen gas (99.99%) were purchased from Air Liquide Singapore Pte. Ltd.
Synthesis of modified IrO2 electrode
The CoOx/IrO2 electrocatalysts were synthesized through dip-coating followed by thermal decomposition22. Specifically, Ti mesh (cut into 10×10 mm2) etched in boiling 0.5 M oxalic acid solution for one hour. The etched Ti mesh was then soaked in a solution of 24 mg IrCl3·xH2O and 6 mg CoCl2·6H2O (or AuCl3·3H2O, total metal chloride salt amounting to 3 mg/mL ink) with 10% concentrated HCl in 10 mL isopropanol solution. The resulting Ti-mesh was then dried at 100 °C for 10 min and calcined at 500 °C for 10 min. The soaking and calcination cycle was repeated for 15 times, such that the catalyst loading is about 1.5 mg/cm2 and the resultant materials were denoted as CoOx/IrO2 (or Au/IrO2 when modified with gold).
Electrocatalyst characterization
Scan electron microscopy (SEM) images were obtained on a Hitachi SU8220 FESEM, at an accelerating voltage of 10 kV. Energy-dispersive X-ray (EDX) spectroscopy was performed with an on-board AEMTEK® Octane Elite EDS System, with an accelerating voltage of 20 kV. Transmission electron microscopy (TEM) images were obtained on TEM JEOL 2100 F. X-ray photoelectron spectroscopy (XPS) was conducted on a PHI Quantum 2000 XPS system. X-ray diffraction (XRD) analysis was operated on a Bruker® D8 ADVANCE system with an increment of 0.1°. X-Ray absorption spectroscopy (XAS) was conducted on XAFCA beamline at the Singapore Synchrotron Light Source (SSLS).
For electron paramagnetic resonance (EPR) measurements, electrochemical toluene oxidation was conducted on a reaction mix containing 0.01 M of 2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO). Samples were periodically collected with a capillary tube and, within 30 s of sample collection, analyzed using a JEOL JES-X320 system with center field of 327 mT and sweep depth of 10 mT.
Evaluation of electrochemical performance
Electrochemical toluene oxidation experiments were carried out in a H-type cell (Tianjin Aida) equipped with anode (CoOx/IrO2 electrocatalyst on Ti mesh support) Ag/Ag+ reference electrode (Tianjin Aida), Pt plate cathode, and Nafion® 117 membrane (size 2.5 cm by 2.5 cm, thickness 0.18 mm). The Nafion® 117 membrane was activated in MilliQ water (18.2 Ω) at 70 °C for overnight before use. Unless otherwise stated, the anolyte is 19.5 mL of 0.1 M LiBF4 and 0.06 M LiNO3 acetonitrile solution and 0.5 mL toluene, and the catholyte is 20 mL of 0.1 M LiBF4 MilliQ water (18.2 Ω) solution. All experiments were conducted using a Metrohm Autolab potentiostat PGSTAT204 without iR correction. Each electrochemical toluene oxidation experiment was conducted at 25 mA/cm2 for 4800 s (120 C) unless otherwise stated. The electrolyte for each test was prepared individually directly before the testing.
The calibration of Ag/Ag+ reference electrode was carried out in a 3-electrode electrochemical cell31. Two glassy carbon electrodes with 0.5 mm diameter were employed as working and counter electrode; the to-be-calibrated Ag/Ag+ reference electrode (0.1 M TBAP and 0.01 M AgNO3 acetonitrile solution) was employed as a reference electrode. The potentials of Fc/Fc+ (2.5 mM) redox pair were measured in 0.1 M TBAP acetonitrile solution and 0.1 M TBAP / 0.5 M [BMIM]PF6 acetonitrile solution with a scan rate of 10 mV/s, respectively.
Cyclic Voltammetry (CV) and Linear Scan Voltammetry (LSV) were obtained in a one-pot electrochemical cell equipped with a 3-electrode system, including different materials as working electrode, Pt plate as counter electrode and Ag/Ag+ as reference electrode. Acetonitrile was used as solvent with 0.1 M Lithium tetrafluoroborate and 0.06 M Lithium nitrate as electrolyte. 0.5 mL toluene was added into the scan if required.
The double layer capacitance (\({C}_{{dl}}\)) values of CoOx/IrO2 anode from different Co/Ir ratios were determined by collecting cyclic voltammetry at different scan rates (\(v\)) of 10, 20, 40, 60, 80 and 100 mV/s. The potential range was -0.40 – -0.30 V vs. Ag/Ag+. The current density (\(\Delta J\)) at -0.35 V vs. Ag/Ag+ was then plotted against the corresponding \(v\). The double-layer capacitance (\({C}_{{dl}}\)) was calculated from \(\Delta J-v\) plot according to the formula:
The slopes for the \(\Delta J-v\) plots are \({C}_{{dl}}\) values of CoOx/IrO2 anode from different Co/Ir ratios.
Flow cell tests were carried out in a standard 2-compartment flow cell system (purchased from Tianjin Aida) equipped with anode (CoOx/IrO2 electrocatalyst), Ag/Ag+ reference electrode, Ni mesh cathode, and Nafion® 117 membrane. The anolyte was 0.1 M LiBF4 0.06 M LiNO3 and 340 µL toluene in 25 mL of pure acetonitrile solvent or 4:1 acetonitrile:HFIP mixture. The catholyte was 0.1 M aqueous LiBF4 solution.
The generated benzaldehyde, benzyl alcohol and benzoic acid, as well as remaining toluene in anolyte was quantified with quantitative NMR (qNMR) spectroscopy using CH2Cl2 as internal standard at a JOEL JNM-ECA500II FT NMR System. Calibration curves were generated from qNMR spectra of standards containing different concentrations of the target chemicals, by integrating the specified peak proportional to the peak area of CH2Cl2. The resultant calibration curves showed R2 > 0.991.
The calculation of Faradaic efficiency was then calculated through the following equation:
where \(n\) is the amount of product; \(Z\) is the number of electrons transferred to obtain one molecule of product; \(F\) is the Faraday constant (96485 C/mol) and \(Q\) stands for the total passed charge in the reaction, which is 120 C unless otherwise stated.
Faradaic efficiency of hydrogen evolution reaction (HER) at the cathode side was evaluated through gas chromatography, using an on-line 990 micro GC system equipped with a thermal conductivity detector and a flame ionization detector. The measurement was using 10 sccm N2 as carrier gas, and a sample was taken every 10 min. The calculation of FE was processed through the following quotation:
where \(Z\) is the number of electrons transferred to require one molecule of product, in this hydrogen evolution reaction is 2; and \(F\) stands for the Faraday constant (96485 C/mol); \(v\) is the gas flow rate, in our measurement is 10 sccm, which is measured with a mass flow controller (Alicat Scientific); \(c\) is the concentration of H2 measured by GC; \(i\) is the current applied and \({V}_{m}\) is the unit molar volume of gas.
Data availability
The data supporting the findings of this work are available within the articles and its Supplementary Information files. Source data are provided with this paper. Additional data can be obtained from the corresponding author upon request. Source data are provided with this paper.
References
Shi, Y. et al. Photocatalytic toluene oxidation with nickel-mediated cascaded active units over Ni/Bi2WO6 monolayers. Nat. Commun. 15, 4641 (2024).
Lin, X. et al. Electrochemical activation of C–H by electron-deficient W2C nanocrystals for simultaneous alkoxylation and hydrogen evolution. Nat. Commun. 12, 3882 (2021).
Kesavan, L. et al. Solvent-Free Oxidation of Primary Carbon-Hydrogen Bonds in Toluene Using Au-Pd Alloy Nanoparticles. Science (1979) 331, 195–199 (2011).
Cao, X. et al. A photochromic composite with enhanced carrier separation for the photocatalytic activation of benzylic C–H bonds in toluene. Nat. Catal. 1, 704–710 (2018).
Horn, E. J. et al. Scalable and sustainable electrochemical allylic C-H oxidation. Nature 533, 77–81 (2016).
Leow, W. R. et al. Electrified hydrocarbon-to-oxygenates coupled to hydrogen evolution for efficient greenhouse gas mitigation. Nat. Commun. 14, 1954 (2023).
Chung, M. et al. Direct propylene epoxidation via water activation over Pd-Pt electrocatalysts. Science (1979) 383, 49–55 (2024).
Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science (1979) 368, 1228–1233 (2020).
Fan, L. et al. Selective production of ethylene glycol at high rate via cascade catalysis. Nat. Catal. 6, 585–595 (2023).
Chung, M., Jin, K., Zeng, J. S. & Manthiram, K. Mechanism of Chlorine-Mediated Electrochemical Ethylene Oxidation in Saline Water. ACS Catal. 10, 14015–14023 (2020).
Li, T. et al. Photoelectrochemical oxidation of organic substrates in organic media. Nat. Commun. 8, 390 (2017).
Sauermann, N., Mei, R. & Ackermann, L. Electrochemical C−H Amination by Cobalt Catalysis in a Renewable Solvent. Angew. Chem. 130, 5184–5188 (2018).
Zhang, L. & Hu, X. Nickel catalysis enables convergent paired electrolysis for direct arylation of benzylic C-H bonds. Chem. Sci. 11, 10786–10791 (2020).
Wiebe, A., Riehl, B., Lips, S., Franke, R. & Waldvogel, S. R. Unexpected high robustness of electrochemical cross-coupling for a broad range of current density. Sci. Adv. 3, eaao3920 (2017).
Das, A., Nutting, J. E. & Stahl, S. S. Electrochemical C-H oxygenation and alcohol dehydrogenation involving Fe-oxo species using water as the oxygen source. Chem. Sci. 10, 7542–7548 (2019).
Winiwarter, A. et al. Towards an atomistic understanding of electrocatalytic partial hydrocarbon oxidation: Propene on palladium. Energy Environ. Sci. 12, 1055–1067 (2019).
Hoque, M. A. et al. Electrochemical PINOylation of Methylarenes: Improving the Scope and Utility of Benzylic Oxidation through Mediated Electrolysis. J. Am. Chem. Soc. 144, 15295–15302 (2022).
Li, Z. et al. Photoelectrocatalytic C–H halogenation over an oxygen vacancy-rich TiO2 photoanode. Nat. Commun. 12, 6698 (2021).
Wang, Q. et al. Electrocatalytic Methane Oxidation Greatly Promoted by Chlorine Intermediates. Angew. Chem. Int. Ed. 60, 17398–17403 (2021).
Nikl, J. et al. Electrochemical oxo-functionalization of cyclic alkanes and alkenes using nitrate and oxygen. Nat. Commun. 14, 4565 (2023).
Dimeglio, J. L., Terry, B. D., Breuhaus-Alvarez, A. G., Whalen, M. J. & Bartlett, B. M. Base-Assisted Nitrate Mediation as the Mechanism of Electrochemical Benzyl Alcohol Oxidation. J. Phys. Chem. C 125, 8148–8154 (2021).
Li, Y. et al. Redox-mediated electrosynthesis of ethylene oxide from CO2 and water. Nat. Catal. 5, 185–192 (2022).
Pfeifer, V. et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 18, 2292–2296 (2016).
Prabaharan, D. D. M., Sadaiyandi, K., Mahendran, M. & Sagadevan, S. Precipitation method and characterization of cobalt oxide nanoparticles. Appl. Phys. A 123, 264 (2017).
Cai, C. et al. Ultrahigh Oxygen Evolution Reaction Activity Achieved Using Ir Single Atoms on Amorphous CoOx Nanosheets. ACS Catal. 11, 123–130 (2021).
Zhu, Y. et al. Iridium single atoms incorporated in Co3O4 efficiently catalyze the oxygen evolution in acidic conditions. Nat. Commun. 13, 7754 (2022).
Xin, X. et al. Large electronegativity differences between adjacent atomic sites activate and stabilize ZnIn2S4 for efficient photocatalytic overall water splitting. Nat. Commun. 15, 337 (2024).
Seo, B. et al. Electrochemical oxidation of toluene with controlled selectivity: The effect of carbon anode. Appl. Surf. Sci. 534, 147517 (2020).
Li, Z. et al. Electrocatalytic ethylbenzene valorization using a polyoxometalate@covalent triazine framework with water as the oxygen source. ChemComm 57, 7430–7433 (2021).
Yin, Z. et al. A Green and Efficient Electrocatalytic Route for the Highly-Selective Oxidation of C−H Bonds in Aromatics over 1D Co3O4-Based Nanoarrays. Angew. Chem., Int. Ed. 64, e202415044 (2025).
Shi, J. et al. Highly selective and efficient reduction of CO2 to CO on cadmium electrodes derived from cadmium hydroxide. ChemComm 54, 5450–5453 (2018).
Acknowledgements
This work is supported by the A*STAR Career Development Award (C210112053), Young Individual Research Grant (A2084c0180) and National Research Foundation Fellowship (NRFF16-2024-0034). Y. L and J. Z. acknowledge support and funding from A*STAR under its LCERFI program (Award No U2102d2002). Y.L. and J.Z. also thank the high-performance computational facilities from the National Supercomputing Centre (NSCC) Singapore and A*STAR Computational Resource Centre (A*CRC). We are also thankful to the Chemical, Molecular and Materials Analysis Centre (CMMAC) in the Department of Chemistry, National University of Singapore (NUS) for conducting the EPR studies.
Author information
Authors and Affiliations
Contributions
W.R.L. conceived the idea, supervised the project and wrote the manuscript. Z.M. designed and carried out the electrochemical experiments, material synthesis and characterization. Y.L. and J.Z. conducted the theoretical simulations. C.W. and S.X. performed the XAS measurements. M.Z. performed XPS measurements. X.C. collected the TEM images. All authors discussed the results and assisted during manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wei Li, Yan Zhao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mi, Z., Li, Y., Wu, C. et al. CoOx clusters-decorated IrO2 electrocatalyst activates NO3- mediator for benzylic C-H activation. Nat Commun 16, 3424 (2025). https://doi.org/10.1038/s41467-025-58733-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58733-2
