
Abstract
Reducing Pt loading in the anodic hydrogen oxidation reaction (HOR) and concurrently increasing mass activity and CO tolerance are essential for advancing proton exchange membrane fuel cells (PEMFCs). Here, an electrocatalyst of 1.7 wt% Pt clusters loaded on a hollow bowl-like W3O/WC heterostructure is designed to increase the anti-CO-poisoning HOR. Due to its unique electron delocalization effect, the W3O/WC heterostructure serves as a warehouse to share electrons with Pt; this simultaneously lowers the HOR barrier and accumulates the hydroxyl radicals (•OH) to accelerate CO oxidation. The Pt‒O bond originating from the oxygen spillover effect of W3O promotes hydrogen and CO oxidation, whereas the lattice‒O consumed in W3O replenished through water dissociation. The resultant electrocatalyst exhibits mass activity of 469 A g‒1 at 50 mV and anti-toxicity even at 2000 ppm CO. The PEMEC delivers a peak power density of 1.63 W cm‒2 and maintains considerable anti-CO poisoning performance.
Similar content being viewed by others


Introduction
Proton exchange membrane fuel cells (PEMFCs) are essential for establishing the hydrogen energy industry and are critical to the transition from traditional to sustainable energy systems1,2,3. With the rapid development of cost-effective cathode catalysts and various proton exchange membranes, the irreplaceability of the precious metal Pt used in anode catalysts has become increasingly prominent4,5,6,7. Additionally, the high mass activity of the Pt catalyst during the anodic hydrogen oxidation reaction (HOR) is susceptible to CO poisoning since the feed H2 is primarily sourced from reformed gas and inevitably contains CO impurities8,9. Specifically, the strong CO adsorption on Pt results in significant degradation of device performance even at concentrations as low as 10 ppm CO10. Under these circumstances, exploiting lower Pt loading, highly active and anti-CO poisoning anodic catalysts is significant for the continued advancement of PEMFCs.
The incorporation of foreign components to form heterostructural catalysts can optimize the adsorption and oxidation kinetics for both hydrogen binding energy (HBE) and CO oxidation11,12,13,14. Early transition metal oxides, such as tungsten oxides, can provide Lewis acidic sites that facilitate the adsorption of hydroxyl (•OH) radicals from water in acidic environments, thereby increasing the CO oxidation efficiency15. These oxides likely synergize with Pt that improve both the HOR activity and anti-CO poisoning capability16. Recent studies have proposed that lattice active species act as overflow active substances, and these substances could participate in various catalytic reactions17,18. For example, the overflow of lattice-O can synergize with Pt to enhance CO oxidation19,20. Moreover, tungsten carbides are classified as transition metal interstitial compounds (TMICs), exhibit Pt-like properties and electronic delocalization effects, which can act as cocatalysts to improve the microenvironment of the active sites to regulate intermediate adsorption21,22,23. Therefore, the collaboration between tungsten oxide and tungsten carbide species could reduce the reliance on Pt while enhancing both the HOR activity and the anti-CO poisoning capability. Despite substantial efforts to develop highly efficient HOR catalysts, the underlying mechanisms remain insufficiently explored and primarily rely on theoretical calculations and/or off-line characterizations; moreover, whether the lattice‒O of the composite material is involved in the reaction has not been examined. This leads to uncertainty due to the absence of direct and comprehensive experimental data on electrocatalytic processes. Consequently, our focus needs to shift toward a deeper understanding of the reaction mechanisms involved.
Herein, we design a hollow bowl carbon skeleton-supported Pt cluster-W3O/WC heterojunction with a low Pt loading of 1.7 wt% (denoted H-Pt-W3O/WC) as a highly active HOR and anti-CO electrocatalyst. The hollow carbon shell effectively enhances both mass and electron transfer during the electrocatalytic process. Moreover, the electron modulation effect of the W species results in a downward shift of the d-band center for Pt, which may lower the reaction barriers to reduce the HBE and COad energy. H-Pt-W3O/WC achieves well acidic HOR activity, as evidenced by the mass activity of 469 A g‒1 at 50 mV; this value is 15.6 times greater than that of commercial Pt/C. More importantly, it exhibits an improved anti-poisoning capability and maintains 94.5% of its initial mass activity in the presence of 2000 ppm CO/H2. H-Pt-W3O/WC can deliver a peak power density of 1.63 W cm‒2 with a current density of 3.4 A cm–2 and an outstanding performance of 0.77 W cm‒2 in 100 ppm CO-H2/O2 for assembling PEMFCs. Furthermore, W3O triggers its own structural changes through the oxygen spillover effect to continuously generate a Pt‒O environment and shares electrons to stabilize the well activity of the Pt clusters during the reaction, as evidenced by operando X-ray absorption spectra; this reaction can promote the HOR activity by attacking *H and accelerate CO oxidation. By combining operando diffuse reflectance infrared Fourier transform spectroscopy and isotope-labeled mass spectrometry, the lattice‒O can be replenished by 18O from H218O. Moreover, the adsorption of •OH originating from the water dissociation on the W Lewis acidic sites also plays a role in accelerating the CO oxidation process; this process promotes the exposure of the Pt active sites to facilitate HOR. The water produced in this process can replenish the consumed water to achieve a continuous oxygen overflow effect in the water cycle. Our study provides a direction for the subsequent rational design of advanced HOR and anti-poisoning electrocatalysts.
Results
Structural characterization
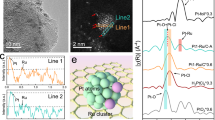
Figure 1a illustrates the synthetic scheme of H-Pt-W3O/WC. First, polystyrene was used as a template for synthesizing a hollow carbon bowl (HCB) by combining a coordination and pyrolytic strategy. The HCB structures had a diameter of approximately 200 nm, as evidenced by scanning electron microscopy (SEM) (Supplementary Fig. 1a). Subsequently, the tungsten species were introduced using an impregnation method, which was followed by heat treatment under an inert atmosphere to prepare H-W3O/WC (Supplementary Fig. 1b). X-ray diffraction (XRD) patterns demonstrated that W3O (PDF#41-1230) and WC (PDF#51-0939) formed (Supplementary Fig. 2). H-Pt-W3O/WC was subsequently obtained using a sodium borohydride reduction process. The Pt content in H-Pt-W3O/WC was approximately 1.7 wt%, as determined by inductively coupled plasma emission spectrometry (ICP‒AES). Furthermore, the absence of the diffraction peaks for Pt in the XRD pattern indicated that Pt could exist as clusters. As displayed in Fig. 1b-d and Supplementary Fig. 3, the distribution of nanoparticle sizes on the surface of HCB is primarily concentrated at approximately 3 nm and 17 nm, and the edges of H-Pt-W3O/WC have a thin laminar-like structure. The high-resolution transmission electron microscopy (HRTEM) image clearly displays two lattice spacings, the average lattice spacing was obtained by selecting seven stripe spacings of 0.23 nm and 0.25 nm (Fig. 1e, f and Supplementary Fig. 4); these spacings are assigned to the WC (100) and W3O (012) facets, respectively.

a Synthetic scheme of H-Pt-W3O/WC. b SEM, c TEM, d, e HRTEM, f Lattice fringe distribution of the selecting seven stripe spacings of WC (pink boxes) and W3O (yellow boxes), g HAADF-STEM. The large blue circles’ areas represent Pt clusters, while the small circles’ areas represent Pt single atoms. The pink and yellow lines represent the WC and W3O lattice fringes, respectively. h, EDS element mapping images of H-Pt-W3O/WC.
The HAADF-STEM image and corresponding atomic phase analyses clearly show that all bright spots originate from the Pt atoms and that W is present throughout the entire area (Fig. 1g and Supplementary Fig. 5). Thus, the Pt clusters are situated at the W3O/WC interface, with a small quantity of monodisperse Pt atoms. Furthermore, the Rietveld analysis results indicate that the sizes of WC and W3O in H-Pt-W3O/WC are approximately 12.8 nm and 4.5 nm (Supplementary Fig. 6), respectively. The results are in acceptable agreement with the particle size distribution observed in the TEM image mentioned above. Moreover, W, Pt and O are homogeneously dispersed on the carbon support, as confirmed by the corresponding energy dispersive X-ray spectroscopy (EDS) mappings (Fig. 1h). The semiquantitative EDS elemental analysis performed using the line sweep method is further utilized to evaluate the distribution of Pt (Supplementary Fig. 7). The considerable overlap between the W and Pt signals indicates that Pt is deposited primarily at the W3O/WC interface rather than on the carbon support, benefitting the synergistic effect between the Pt and W species. In addition, the alcoholysis process primarily yields tungsten oxide24, which plays a crucial role in the formation of lattice−O in H-Pt-W3O/WC. The XPS analysis depicted in Supplementary Fig. 8 reveals that the proportion of lattice−O steadily increases with prolonged alcoholysis and surpasses 50% after 36 h of reaction. Lattice−O is notably stable and remains intact even during heat treatment in high-temperature inert atmospheres. Consequently, at a lower lattice−O content (after 24 h of alcoholysis), the pyrolytic treatment causes the formation of tungsten carbide and tungsten oxide heterostructures. In contrast, a higher lattice−O content (after 36 h of alcoholysis) leads to the formation of pure tungsten oxide after pyrolytic treatment.
The elemental compositions were also investigated by using X-ray photoelectron spectroscopy (XPS). As shown in Supplementary Fig. 9a, a weak signal for Zn 2p at 1030 eV was observed in the H-Pt-C sample containing only 0.2 wt% zinc, as determined from the ICP‒AES results. These results indicate that a small amount of zinc may still be present in HCB even after high-temperature pyrolysis at 900 °C. However, after the alcoholysis reaction, the peak related to the zinc element is almost completely eliminated (Supplementary Fig. 8a, c), and no zinc species are observed in the H-Pt-W3O/WC and H-W3O/WC samples (Supplementary Fig. 9b, c). These results indicate that the acid produced during the alcoholysis step may effectively remove the residual zinc in HCB. The presence of the C‒N bonds in the deconvoluted C 1 s XPS spectra indicates that N is doped into the carbon support (Supplementary Fig. 10). In addition to the Pt, W, C, and O species, the presence of the N species in H-Pt-W3O/WC originates from the pyrolysis of 2-methylimidazole25. This leads to the formation of N-doped carbon structures, thereby enhancing electron transfer and electrocatalytic activity (Supplementary Fig. 10). The electronic structure of the individual metal atoms is redistributed due to the proximity effect exerted by adjacent metal atoms and is closely related to the adsorption behavior of the oxidation intermediates.
The deconvoluted W 4 f XPS spectra of H-Pt-W3O/WC reveal three prominent bimodal peaks corresponding to W6+, W4+ and W0; these peaks exhibit a positive shift of 0.31 eV relative to H-W3O/WC (Fig. 2a and Supplementary Table 1)26,27,28. Conversely, the Pt 4 f XPS spectra can be deconvoluted into four peaks corresponding to Pt0 and Pt2+; these peaks shift negatively approximately 0.25 eV compared with those observed for H-Pt-C (Fig. 2b)29,30. Because chloroplatinic acid has a high oxidation state of Pt+4 and was utilized as the source for the synthesis of H-Pt-W3O/WC, the oxidation state of Pt in H-Pt-W3O/WC was lower than that of the H-Pt-C due to the electron transfer from W in the W3O/WC heterostructure to Pt. Moreover, in H-Pt-W3O/WC, the relative intensities/areas of W6+ in the W 4 f XPS spectra are greater than those in H-W3O/WC. Conversely, the Pt 4 f XPS spectra exhibit reduced relative intensities/areas of Pt0 compared with those observed in H-Pt-C; these results confirm electron transfer from W to Pt. This occurs because the W sites on the surface of the W3O/WC heterojunction are abundant in spin electrons; these spin electrons exhibit delocalization effects that can be shared with adjacent Pt atoms, thereby enhancing HOR activity31. As shown in the deconvoluted O 1 s XPS spectra in Fig. 2c, lattice‒O and defect‒O are present and can potentially be used to adjust the HOR activity32.

XPS results of a W 4 f for H-Pt-W3O/WC and H-W3O/WC; b Pt 4 f for H-Pt-W3O/WC and H-Pt-C; and c O 1 s for H-Pt-W3O/WC, H-W3O/WC and H-Pt-C. d Pt L3-edge HERFD-XANES, e corresponding FT-EXAFS and f WT-EXAFS of Pt foil, H-Pt-W3O/WC and H-Pt-C. The different color (yellow/ blue and pink) shadow means first and second shell for Pt and W, respectively. g W L3-edge HERFD-XANES, h corresponding FT-EXAFS (the shadow means different shell) and i WT-EXAFS spectra of W foil, H-Pt-W3O/WC, and H-W3O/WC. Source data for Fig. 2 are provided as a Source Data file.
The electronic structures and local configurations were further probed by X-ray absorption fine spectroscopy (XAFS). Since the energy of the W L2-edge is only 20 eV different from that of the Pt L3-edge, we employed high-energy resolution fluorescence detection XAFS (HERFD-XAFS) technology to eliminate the signal interference from W on Pt, thereby ensuring the accuracy of the test data. The Pt L3-edge HERFD X-ray absorption near-edge structure (XANES) spectra are shown in Fig. 2d. Since the intensity of the white line peak in XANES is proportional to the valence state of the metal element, the valence state order of Pt is as follows: Pt foil <H-Pt-W3O/WC < H-Pt-C < PtO2. The lower valence state of Pt in H-Pt-W3O/WC with respect to H-Pt-C is attributed to electron donation from the W3O/WC heterostructure to Pt. Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra were further used to investigate the localized geometry of Pt. As shown in Fig. 2e, H-Pt-C and H-Pt-W3O/WC exhibit a first shell peak and a second shell peak characteristic of Pt foil; these peaks are likely attributed to the Pt–O/C/N and Pt–Pt/W scattering paths, respectively33. The detailed structural parameters were further examined through quantitative curve fitting analyses of k2-weighted EXAFS, as displayed in Supplementary Fig. 11 and Supplementary Table 2. Notably, the Pt‒O/C/N bond distance of 2.04 Å for H-Pt-W3O/WC is longer than the 1.97 Å of H-Pt-C; this difference is attributed to the increased electron density around Pt in H-Pt-W3O/WC. The Pt‒Pt bond distance in H-Pt-W3O/WC is 2.75 Å; this value is slightly longer than the 2.69 Å of H-Pt-C. This increase can be attributed to the influence of the Pt‒W coordination and is likely caused by the interfacial bonding between Pt and W3O/WC. The Pt‒O site is susceptible to proton attack and can be used to facilitate the formation of the *H intermediate state. This site is critical for sustaining the optimal adsorption and desorption dynamics of *H. Significantly, the average coordination number (CN) for the Pt‒Pt scattering path in H-Pt-W3O/WC is 4.47 (Supplementary Table 2); this value is much lower than that of 12 in Pt foil and indicates that Pt in H-Pt-W3O/WC predominantly exists as clusters of approximately six atoms. Moreover, the wavelet transform (WT) was employed because of its high-resolution capabilities in both R and k spaces. With respect to the corresponding metal reference, the WT for the k2-weighted EXAFS (WT-EXAFS) signal of the Pt L3-edge was obtained for H-Pt-W3O/WC and H-Pt-C. The intense WT signals of the Pt‒O/C/N and Pt-Pt/W bonds for H-Pt-W3O/WC and the Pt‒O/C/N and Pt‒Pt bonds for H-Pt-C (Fig. 2f) were consistent with the above EXAFS results.
As illustrated in Fig. 2g, the intensity of the white line peak in the W L3-edge XANES spectra of H-Pt-W3O/WC is significantly greater than that of H-W3O/WC, and its values fall between those for WC and WO3. These results indicate that the valence state of W in H-Pt-W3O/WC is greater than that in H-W3O/WC and that electron transfer occurs from W3O/WC to Pt. Additionally, the FT-EXAFS spectra at the W L3 edge depicted in Fig. 2h exhibit two non-phased corrected EXAFS distinct peaks at approximately 1.5 Å and 2.3 Å; these peaks correspond to the W − O/C/N and W − W scattering paths, respectively. The detailed fitting structural parameters of the W L3-edge FT-EXAFS spectra are shown in Supplementary Fig. 12 and Supplementary Table 3. Notably, compared with those of H-W3O/WC, the bond distances in H-Pt-W3O/WC shift as the W − O/C/N and the W − W bond distances decrease. The W‒O/C/N and W‒W bond distances are approximately 1.89 and 2.89 Å for H-Pt-W3O/WC are shorter than the 1.91 and 2.92 Å for H-Pt-C, respectively. The alteration in the bond distance is attributed to the reduced electron density around W, thus enhancing its interactions with neighboring atoms. Furthermore, the lower CN of W‒W with respect to that of the bulk-phase W foil confirms its smaller particle size. The W L3-edge WT-EXAFS spectra also show intense signals at the W‒C/O/N and W‒W bonds for both H-Pt-W3O/WC and H-W3O/WC (Fig. 2i). The high degree of consistency between the experimental and fitted data for the Pt L3-edge and W L3-edge in k space confirms the validity of the fitting results, as shown in Supplementary Figs. 13-14. For comparison, H-WC with a pure WC phase was prepared using a higher pyrolytic treatment of the same precursor as H-Pt-W3O/WC (Supplementary Fig. 15). As shown in Supplementary Fig. 16, the intensity of the white line peak at the Pt L3-edge for H-Pt-W3O/WC is lower than that for H-Pt-WC, whereas the intensity of the white line peak at the W L3-edge for H-Pt-W3O/WC is greater than that for H-Pt-WC. These results indicate an increase in the electron density at the Pt site and a decrease in the electron density at the W site in H-Pt-W3O/WC with respect to H-Pt-WC. Based on this observation, electron delocalization is more effective at the W3O/WC heterostructure interface than at the WC interface. Thus, the electron rearrangement in H-Pt-W3O/WC results from the interfacial effect between Pt and W, and contributes to a reservoir of spin-delocalized electrons. The electron transfer from W to Pt can enhance the d-orbital interactions and adjust the d-band center occupancy of Pt with regulating the binding energy of *H and COad under acidic conditions, thereby enhancing HOR kinetics29.
Evaluation of the HOR and CO tolerance performance
The electrocatalytic HOR performance of H-Pt-W3O/WC was investigated in H2-saturated 0.1 M HClO4 by using a rotating disk electrode (RDE) at 1600 rpm with a standard three-electrode system. As shown in the polarization curves in Fig. 3a, H-Pt-W3O/WC has a steeply increased anodic current density and achieves a higher limiting current density of 3.3 mA cm−2; this value is much higher than that of Pt/C (2.4 mA cm−2). Moreover, H-W3O/WC (0.9 mA cm−2), H-Pt-WC (2.5 mA cm−2) and H-Pt-C (1.8 mA cm−2) exhibit much lower limited current density values; these results further indicate the better activity of H-Pt-W3O/WC and are attributed to the synergy between the W3O/WC heterostructure and the Pt cluster. Moreover, the HOR polarization curves at different rotational speeds were further estimated (Supplementary Fig. 17). The limiting current density increased with increasing speed; these results were attributed to the acceleration of mass and electronic transfer and conformed to the first-order kinetic equation. By fitting with the Koutecky‒Levich (K‒L) equation established at an overpotential of 100 mV (Supplementary Fig. 18), slopes of 11.28, 9.96, 11.25, and 11.13 cm2 mA‒1 rpm‒1/2 were obtained for H-Pt-W3O/WC, H-Pt-WC, H-Pt-C, and Pt/C, respectively. Thus, the anodic current in H-Pt-W3O/WC predominantly originated from a two-electron HOR process34,35. For comparison, the sample synthesized without a polystyrene template was designated CC-Pt-W3O/WC. As shown in Supplementary Fig. 19, H-Pt-W3O/WC exhibited a higher limiting current density of 3.3 mA cm⁻2 than CC-Pt-W3O/WC (2.4 mA cm⁻2) toward the HOR. Furthermore, H-Pt-W3O/WC demonstrated a significantly greater specific surface area of 852.7 cm2 g⁻1 compared with 614.8 cm2 g⁻1 for CC-Pt-W3O/WC (Supplementary Fig. 20); these results indicated the improved mass transport capability of the hollow carbon shell. Additionally, the intrinsic conductivity of carbon nanostructures was beneficial for electron transfer; thus, the hollow carbon sphere structures in H-Pt-W3O/WC could enhance both the mass and electron transfer during the electrocatalytic process.
As displayed in Fig. 3b, the K‒L slope of H-Pt-W3O/WC was superior to those of the other catalysts from the kinetic to diffusion-limited regions; the result indicated well HOR kinetics due to the improved electronic structure. Furthermore, H-Pt-W3O/WC consistently exhibited the highest current density in both the kinetic and diffusion regions (Supplementary Fig. 21), showing the fastest current response rate. The exchange current density (j0) from the micro-polarization region (‒5 ~ 5 mV) was estimated according to Butler‒Volmer linear fitting (Supplementary Fig. 22). H-Pt-W3O/WC had a j0 value of 3.68 mA cm‒2; this value was approximately 1.1, 1.5, and 1.4 times greater than those of H-Pt-WC, H-Pt-C and Pt/C and confirmed the greater utilization of Pt in H-Pt-W3O/WC. To better compare the Pt utilization and comprehensively evaluate the HOR activity, the mass activity (MA) was assessed based on the limiting current density normalized by the usage of the precious metal Pt on the RDE. As a result, the MA of H-Pt-W3O/WC (465 mA mg Pt‒1) at an overpotential of 100 mV was approximately 1.3, 2.5, and 15.6 times greater than those of H-Pt-WC, H-Pt-C and Pt/C; these results highlight the advantage of promoting Pt utilization owing to the interaction effects between the W3O/WC heterostructure and the Pt cluster. The stability of the HOR performance was evaluated using an accelerated durability test (ADT). After 15,000 cycles of cyclic voltammetry (CV) within a potential window of 0 to 0.5 V and at a scanning rate of 0.1 V s‒1, H-Pt-W3O/WC exhibited negligible decay in current density (Fig. 3c). In contrast, Pt/C exhibited a significant 28.3% decrease in current density after only 10,000 cycles (Supplementary Fig. 23). These results demonstrated the robust stability of H-Pt-W3O/WC and indicated that W3O could maintain its stability in an acidic environment36. Moreover, H-Pt-W3O/WC displayed only 16% attenuation in activity after 22 h of testing, further confirming its well stability (Supplementary Fig. 24).

a Polarization curves in H2-saturated and b Tafel plots of the kinetics plots for all compared catalysts. c Stability test of H-Pt-W3O/WC before and after ADT. Polarization curves in H2 with or without 1000 ppm and 2000 ppm CO-saturated of d, H-Pt-W3O/WC, e H-Pt-C and f Pt/C. g Chronoamperometry response at 0.05 V vs. RHE in H2/1000 ppm CO-saturated. h MA, jL, jo and K-L slope values of all compared catalysts. All polarization curves were obtained with scan rate of 5 mV s-1 at 1600 rpm in 0.1 M HClO4 (pH~1.1) under 25 °C saturated gas conditions with the load amount 0.1 mg. PEMFC polarization and power density curves for H-Pt-W3O/WC and 60 wt% Pt/C tested in i, H2-O2 and j, H2-Air. k PEMFC performance for four times 100 ppm CO/H2-O2 poisoning tests. The yellow, blue and pink shadow means H2-O2, H2-AIR and 100ppm CO/H2-O2 atmosphere test conditions, respectively. The PEMFC curves were all measured under conditions of 100% humidity, 80 °C, and 1.0 bar. The Pt loadings on the anode and cathode are 0.05 and 0.25 mg cm–2, respectively. All the tests were conducted without iR correction. Source data for Fig. 3 are provided as a Source Data file.
Industrially produced H2 inevitably contains trace amounts of CO, and this trace CO amount tends to adsorb on the Pt surface and adversely affects HOR activity; therefore, we further investigated the resistance to CO poisoning. As shown in Fig. 3d, after introducing 1000 ppm CO in H2, the limiting current density of H-Pt-W3O/WC decreased by only 5.4%, and a 5.5% decrease was observed at 2000 ppm CO; these results indicated well anti-CO ability. In sharp contrast, for H-Pt-C and Pt/C (Fig. 3e, f), the limiting current density decreased by 20.2% and 18.1% at 1000 ppm CO, respectively, and by 25.6% and 36.1% at 2000 ppm CO. To further investigate the role of the lattice‒O, we extended the alcoholysis duration in the synthesis process, successfully synthesized the support containing only the WO3 crystalline phase, and subsequently used it to prepare the H-Pt-WO3 catalyst (Supplementary Fig. 25). The anti-CO capability of H-Pt-WO3 was clearly comparable to that of H-Pt-W3O/WC and significantly better than that of H-Pt-WC (Supplementary Figs. 26,27). These results indicated the well anti-CO poisoning effect of H-Pt-W3O/WC potentially originated from the lattice‒O involved in the CO oxidation process. Importantly, all the catalysts exhibited CO oxidation peaks at 2000 ppm, confirming their ability to oxidize CO. Specifically, the lowest oxidation potential observed in H-Pt-W3O/WC indicated its superior CO oxidation capacity. To further confirm the resistance of H-Pt-W3O/WC to CO poisoning, the stability was assessed using chronoamperometry tests at 0.05 V and 1000 ppm CO. As depicted in Fig. 3g, the HOR current density of H-Pt-W3O/WC decreased by only 2% after 3500 s of continuous operation, whereas a significant 69% loss was observed with H-Pt-WC. Conversely, H-Pt-C and Pt/C were completely deactivated after 3250 s. Additionally, H-Pt-W3O/WC demonstrated significant endurance to CO poisoning and retained 80% of its initial activity after 7000 s (Supplementary Fig. 28). Even under H2/2000 ppm CO conditions, the catalyst maintained approximately 53% of its initial activity after 8 h (Supplementary Fig. 29), demonstrating significant resistance to poisoning at high concentrations of CO. The MA, JL, Jo and K-L slope values of all compared catalysts toward the HOR are summarized in Fig. 3h. The corresponding testing and calculation errors can be found in the supporting information (Supplementary Fig. 30).
PEMFC performance
Due to its the remarkable HOR and anti-CO tolerance, H-Pt-W3O/WC was further applied as an anode for the membrane electrode assembly (MEA) in practical PEMFCs (Supplementary Fig. 31). Figure 3i shows the polarization and power density in H2–O2 under 1.0 bar backpressure; here, a peak power density (PPD) of 1.63 W cm–2 and a current density of 4.0 A cm–2 was attained across the entire range of polarization curves. The performance of the H-Pt-W3O/WC fuel cell is comparable to that of commercial 60 wt% Pt/C (1.43 W cm–2 at 3.4 A cm–2), H-Pt/WC (1.32 W cm–2 at 3.4 A cm–2) and H-Pt-C (1.08 W cm–2 at 2.8 A cm–2) fuel cell. Their open-circuit voltages are: 1.0 V, 0.99 V, 0.99 V and 0.96 V, respectively. (Fig. 3i). Notably, H-Pt-W3O/WC fuel cell could deliver a PPD of 0.85 W cm−2 in a H2-Air atmosphere; this PPD was also better than those of the other fuel cells (Fig. 3j). Thus, the catalyst had well intrinsic activity and good practical application value. In addition, it could approach a PPD of 1.74 W cm–2 in H2-O2 and could even reach approximately 0.93 W cm–2 in H2-Air at 2.0 bar backpressure (Supplementary Fig. 32a, b). To further assess the resistance to CO poisoning, the PEMFC was operated in a 100 ppm CO/H2-O2 environment at 1.0 bar backpressure. Notably, its PPD was able to recover to 49% for the primary poisoning test; this showed almost no performance degradation in three subsequent cycle tests (Fig. 3k). However, H-Pt-C fuel cell could even recover 15% performance in the primary poisoning test (Supplementary Fig. 32c). Thus, the introduction of W3O/WC could effectively improve the ability of PEMFC to resist CO poisoning in practical applications. In addition, PEMFC was initially operated at a constant potential of 0.6 V in a 10 ppm CO environment for 1 h, followed by testing in a H2–O2 atmosphere at 1.0 bar. Notably, its PPD was able to recover to 90% after five cycles; this value was higher than the 88% recovery observed with Pt/C (Supplementary Fig. 32d). These results indicated that the introduction of W3O/WC heterostructures enhanced both the PEMFC performance and anti-CO poisoning capability.
Study of the adsorption mechanism
Because the hydrogen binding energy (HBE) is a direct indicator of the origin of the HOR activity, *H adsorption was further investigated using underpotential deposition. Hydrogen underpotential deposition (Hupd) tests were performed in H2-saturated 0.1 M HClO4. As illustrated in Fig. 4a, the Hupd peak of H-Pt-W3O/WC was negatively shifted compared with that of Pt/C, H-Pt-WC and H-Pt-C indicated a reduction in HBE. However, the positive shift of the Hupd peak in relation to H-W3O/WC confirmed the enhancement of HBE. These results indicated that H-Pt-W3O/WC exhibited a moderate HBE, whereas H-W3O/WC showed weaker *H adsorption. The active site needed a moderate adsorption energy for the intermediate to efficiently accelerate the reaction; both strong and weak HBE could impede the HOR. Furthermore, based on the hydrogen temperature‒programmed desorption (H2-TPD) curves presented in Fig. 4b, the H2 desorption temperature for the H-Pt-W3O/WC sample was approximately 211 °C; this temperature was accompanied by a significant intensity and peak area. This temperature was approximately 19 °C higher than that observed for H-Pt-C and 42 °C lower than that for H-W3O/WC. A higher H2 desorption temperature indicated a more challenging desorption process and corresponded to a stronger HBE. In addition, a larger peak area correlated to a greater number of catalytic active sites. Therefore, H-Pt-W3O/WC facilitated easier desorption of *H for enhancing the HOR, and the increase in peak area at elevated temperatures could be attributed to the reduction of the oxidized state in the Pt cluster, and more active sites conducive to the HOR were generated (Fig. 4f)37.

a Hupd electrochemical desorption and b H2-TPD curves of the compared catalysts. c COad and d CO-TPD curves of the compared catalysts. e Bond length and orbital overlap state of the Pt 5d-orbital coordinated with CO adsorption. Atoms of W: 007DFF (blue), Pt: FBC723 (yellow), O: FF44AB (pink) and C: 808080 (gray). The different shadow (blue and pink) means different atmosphere test conditions (H2 an CO). f Adsorption mechanism of the delocalization effect for the HOR and anti-CO poisoning. All CV curves were obtained with scan rate of 50 mV s-1 at 1600 rpm in 0.1 M 0.1 M HClO4 under 25 °C saturated gas conditions. Source data for Fig. 4 are provided as a Source Data file.
Understanding the mechanism of CO oxidation is crucial for designing anti-poisoning catalysts. The CO adsorption (COad) analyses illustrated in Fig. 4b revealed that H-Pt-W3O/WC had the lowest COad desorption potential, confirming its superior resistance to CO poisoning. The positive shift in potential and increased intensity of the COad peak indicated that H-Pt-WC had strong CO adsorption capabilities. Therefore, in the presence of a high CO concentration of 2000 ppm, CO adsorption by H-Pt-WC was difficult, leading to CO poisoning and resulting in a pronounced oxidation peak at high potential (Supplementary Fig. 26). Notably, compared to H-Pt-C, both H-Pt-W3O/WC and H-W3O/WC demonstrated a lower CO desorption temperature of approximately 150 °C (Fig. 4d), indicating that COad was weak and more easily desorbed. However, a reduced CO desorption area in H-W3O/WC relative to H-Pt-W3O/WC and H-Pt-C indicated that the Pt clusters and not the W sites acted as the primary COad sites. Moreover, compared with that of H-Pt-C, the decreased CO desorption area for H-Pt-W3O/WC indicated a reduced number of adsorption Pt sites, which was attributed to electron transfer from W to Pt (Fig. 4e). This significantly enhanced the anti-CO poisoning capability of H-Pt-W3O/WC. Based on the above structural analyses, a Pt cluster was positioned at the interface of the W3O/WC heterostructure to construct the Pt-W3O/WC model, whereas a Pt cluster was placed on the carbon support to build the Pt-C model. Additionally, all theoretical calculation models employed in this study utilized the WC (100) and W3O (012) planes. Density functional theory (DFT) calculations indicated that COad on Pt sites led to an increased Pt‒C bond length in the Pt-W3O/WC model (1.887 Å) with respect to the Pt‒C model (1.860 Å), although the C‒O bond length remained unchanged. This phenomenon could be attributed to the nature of the bonding between Pt and CO, which primarily involved the donation of electrons from the HOMO 5σ orbitals of carbon to the 5 d orbitals of Pt. The electron donation from the 5σ orbitals was weaker in Pt-W3O/WC than in H-Pt-C, thereby leading to a weaker COad. Furthermore, the empty LUMO 2π* antibonding orbital of CO overlapped with the 5 d orbital of Pt and formed a feedback π-bond that was predominantly electron-supplied by Pt and did not alter the bond order of the C‒O bond. However, electron donation to the 2π* antibonding orbital could still weaken the C‒O bond, potentially facilitating its dissociation and oxidation processes38.
To more accurately determine the effect of the Pt valence state on the electrocatalytic activity, H-Pt-W3O/WC was subjected to a thermal reduction treatment at 400 °C at a heating rate of 5 °C min−1 and maintained for 3 h in a 10% H2/Ar ambient environment. After the thermal reduction treatment, Pt0 was present, Pt2+ was absent, W0 was reduced and W6+ was increased (Supplementary Fig. 33a); these results further supported electron transfer from W to Pt. Moreover, the HOR activity and anti-CO poisoning capability were clearly lower than those of H-Pt-W3O/WC (Supplementary Fig. 34a). The positive shift in the Hupd and COad peaks indicated the stronger adsorption of *H and CO on Pt0 (Supplementary Fig. 34b, c); this hindered desorption and subsequent oxidation processes and reduced the HOR activity and resistance to CO poisoning. Therefore, Pt in a low oxidation state but not zero valence had a moderate adsorption capacity for reactant molecules, which was more beneficial to the progression of the electrocatalytic reaction.
Operando mechanism study
Operando HERFD-XAFS was further used to investigate the HOR mechanism of H-Pt-W3O/WC in H2-saturated 0.1 M HClO4 (Supplementary Fig. 35). The Pt L3-edge HERFD-XANES spectra are shown in Fig. 5a and Supplementary Fig. 36a. The wet state characterization was performed by placing the catalyst in a 0.1 M HClO4 electrolyte, whereas testing under open circuit potential (OCP) was conducted in a natural state in H2-saturated 0.1 M HClO4 without applying an external voltage between the working electrode and counter electrode (Fig. 5d). Compared with the wet state, the noticeable increase in the white line peak under OCP conditions was mainly caused by the increased Had39. Moreover, the spectral features at 11574 and 11583 eV persisted in all the simulated spectra in the H2 atmosphere, indicating that H-Pt-W3O/WC had a moderate HBE to actuate HOR40. At the initial stage of the reaction, when the voltage increased from the OCP state to 0.05 V, the intensity of the white line peak significantly weakened; these results indicated a reduction in the oxidation state of Pt with strong *H adsorption. A similar trend was observed at the W L3-edge, as depicted in Fig. 5b and Supplementary Fig. 35b. Upon reaching a potential of 0.025 V, the intensity of the white line peak for the W L3 edge decreased, indicating that the W sites could sustain the electron supply state to Pt during the reaction. As the potential further increased from 0.025 V to 0.05 V, the white line peak of the W L3-edge intensified, showing W became slightly oxidized. These results further demonstrated that the W sites could play a pivotal role in redistributing the electrons at the Pt sites to maintain high activity for *H adsorption and subsequent oxidation reaction. Subsequently, as the potential increased from 0.05 V to 0.4 V, the intensity of the Pt L3-edge white line peak gradually increased to a state close to the OCV, indicating an increase in the oxidation of Pt. Moreover, the W sites continued to receive electrons from Pt to maintain the high activity of the Pt sites. Thus, after the limiting current in the HOR was reached, the valence state of Pt gradually returned to its initial valence state. Although the change in the oxidation state of W was similar with that of Pt, the final oxidation state of W deviated more significantly from its initial oxidation state than Pt. These results further indicated that W continuously modulated the electronic state of Pt during the reaction; this ensured that the Pt sites maintained an optimally active electronic state for efficient HOR.

a Operando Pt L3-edge HERFD-XANES, b W L3-edge XANES, c Pt L3-edge HERFD FT-EXAFS and d W L3-edge FT-EXAFS of H-Pt-W3O/WC in an H2-saturated HClO4 solution. e MS results of the CO and CO2 products for H-Pt-W3O/WC after CO electrooxidation in CO-saturated 0.1 M HClO4. f Operando W L3-edge XANES and g FT-EXAFS of H-Pt-W3O/WC in CO-saturated 0.1 M HClO4. The blue shadows correspond to the first shell for Pt and W L3-edge. h Schematic illustration of the oxygen spillover mechanism for HOR and CO oxidation on H-Pt-W3O/WC. Source data for Fig. 5 are provided as a Source Data file.
The FT-EXAFS spectra of the Pt L3 edge are displayed in Fig. 5c. The non-phase corrected EXAFS distance of the Pt‒O/C/N bonds was approximately 1.5 Å for the first shell, and that of the Pt‒Pt/W bonds was approximately 2.0–3.0 Å for the second shell. Further fitting results indicated that the bond length of Pt‒O/C/N was approximately 1.9 Å, whereas that of Pt‒Pt/W was approximately 2.73/2.83 Å (Supplementary Fig. 37 and Supplementary Table 4). Because Pt was loaded at the W site on the heterojunction instead of as a carbon carrier, the coordination changes in the first shell were attributed primarily to changes in Pt‒O. As the potential shifted from the OCV to 0.05 V, the stretched bond length of Pt‒O was attributed to the electron filling of the Pt orbital (Supplementary Table 4); these results indicated that the O sites were susceptible to *H attack, thereby increasing the HOR activity. This replenishment diminished the mass transfer capability of the catalyst as the reaction proceeded. In the potential range of 0.05 ~ 0.4 V, the bond length gradually returned to a closer state to the OCP, indicating that Pt could maintain high catalytic activity. The oxidation potential of Pt was greater than 1 V vs. RHE; thus, the voltage application process only slightly changed the electron configuration without affecting the oxidation coordination structure. Therefore, the CN of the Pt‒O bond gradually increased during this process because of the lattice‒O spillover effect from W sites. The Pt‒O bonds in the elevated oxidation state were susceptible to attack by *H, leading to a predominance of the protonation step (Volmer step: Had → H+ +e- + *) in the HOR mechanism41.
The FT-EXAFS spectra of the W L3‒edge is shown in Fig. 5d. According to the fitting curve in Supplementary Fig. 38, the main W‒O/C/N bonds were approximately 1.5 Å in the first shell for non-phased corrected EXAFS (Supplementary Table 5). Owing to the stable configuration of the carbon carrier, changes in the coordination within the first shell were attributed primarily to variations in the W–O interaction. Owing to the reduction in the valence state of W from the OCP to 0.025 V, the W‒O bond length increased, and the CN of W‒O decreased. These further supported the occurrence of an oxygen spillover effect from W to Pt; this increased the CN of Pt‒O and enhanced the HOR activity. Additionally, an increase in the W‒O coordination and a reduction in the bond length indicated that lattice‒O could be continuously replenished when the potential increased to 0.4 V. During the HOR process, the constant state of saturated H2 led to the continuous consumption of lattice‒O on the surface of Pt and was attributed to strong HBE at the O site. Moreover, the continuous oxygen spillover from the W sites to Pt ensured the sustained adsorption and oxidation activity of *H on Pt.
After the chronoamperometry test in H2/1000 ppm CO-saturated 0.1 M HClO4, H-Pt-W3O/WC was further characterized. As depicted in Supplementary Fig. 39, the bowl-like structure was effectively preserved, and the crystalline phase showed almost no change. After the stability test, the XPS analysis of H-Pt-W3O/WC is presented in Supplementary Fig. 40. W underwent a negative shift of 0.20 eV, and Pt showed a slight positive shift of 0.06 eV. These results revealed a decrease in the valence state of W and an increase in the valence state of Pt and were in agreement with the operando HERFD-XAFS measurements. Additionally, the replenishment of lattice‒O and subsequent oxidation processes resulted in the disappearance of W0. Conversely, after the stability test, the Pt 4 f spectra of H-Pt-C exhibited a negative shift of 0.13 eV and a reduction in the peak area of Pt2+ (Supplementary Fig. 40), indicating the absence of an oxygen spillover effect in H-Pt-C.
To further investigate the influence of lattice‒O on anti-poisoning, we tested the CO oxidation intermediates in the reaction process. A sealed three-electrode system was constructed by using Pt-W3O/WC as the anode in CO-saturated 0.1 M HClO4 via a constant potential test. Gas chromatography analysis revealed that exhaust gases in the potential range of 0.05 ~ 0.7 V contained both CO and CO2; these results indicated that the CO adsorbed on the catalyst surface could be oxidized to CO2. Diffuse reflectance infrared Fourier transform spectroscopy with CO as a probe molecule (CO-DRIFTS) revealed adsorption peaks for Pt0‒CO at 2172 and 2117 cm‒1, as shown in Supplementary Fig. 42a. Notably, the appearance of the Pt2+‒CO peak at 2060 cm‒1 in both H-Pt-W3O/WC and H-Pt/C indicated that Pt2+ was also the active site for COad19. Furthermore, the slightly lower intensity of the Pt2+‒CO peak in H-Pt-W3O/WC could be attributed to the higher electron density of Pt, which led to a reduced Pt2+ content and weaker COad. Operando DRIFTS was further tested in CO-saturated 0.1 M HClO4 (Supplementary Fig. 43). As shown in Supplementary Fig. 42b, the strong Pt0‒CO peak and the weaker Pt2+‒CO peak were evident in the working potential range of 0.01 to 0.05 V, demonstrating that the Pt sites rather than the W sites predominantly facilitated COad. Adsorption peaks of H2O (O‒H) were observed at approximately 1545, 1700 cm‒1 and 3250 cm‒1, whereas the *OH free radical peaks occurred at 3550 cm‒142,43. The adsorption peaks for CO32‒ were detected in the range of 1350 to 1400 cm‒1, and O‒C‒O appeared at 1400 ~ 1420 cm‒1 with a pronounced peak; these results indicated the efficient oxidation of Pt‒CO to CO2 through the lattice‒O or *OH oxidation path44. The oxidation rate of CO significantly increased with increasing working potential. Therefore, the adsorption peak of Pt‒CO gradually weakened due to the slow adsorption kinetics of CO and the fast oxidation rate. As displayed in Supplementary Fig. 42c, the DRIFTS spectra of the NH3 adsorption peak at 1257 and 1423 cm−1 showed many more Lewis acid sites on H-Pt-W3O/WC than on W3O/WC. However, H-Pt-C did not have Lewis acid sites, further confirming that the Lewis acid sites in H-Pt- W3O/WC mainly originated from the W3O/WC heterostructure. The Lewis acid sites facilitated the accumulation of hydroxyl radicals (•OH), thereby accelerating the oxidation of CO. Consequently, H-Pt-W3O/WC could increase the accumulation of •OH, thereby improving resistance to CO poisoning. Moreover, EPR radical trapping revealed a 1:2:2:1 peak originating from 5,5-dimethyl-1-pyrroline N-oxide (DMPO)‒OH at various working potentials; these results confirmed that the •OH radicals derived from water were critical for CO oxidation (Supplementary Fig. 42d).
The isotope labeling method of mass spectrometry (MS) was employed to examine the CO oxidation mechanism of H-Pt-W3O/WC by analyzing the gas products. Initial non-isotopic MS analyses confirmed the presence of a gas mixture with a molecular weight of 40; this mixture was identified as CO and CO2. Additionally, CO2 was separately detected, with a molecular weight of 44. Water was subsequently labeled using H218O with 98% atomic abundance to trace the source of oxygen in the reaction. As shown in Fig. 5e, a mixture of CO and CO2 (molecular weight 40) and C16O2 were detected at the initial reaction stage. As the reaction progressed (t = 5 min), the presence of C16O18O was observed; these results indicated that the lattice‒O from H-Pt-W3O/WC participated in the oxidation of CO, and CO was subsequently replenished by oxygen derived from water. The detection of C18O2 further ensured that the •OH radical in water was involved in the CO oxidation process. With increasing reaction time, the decreasing intensity of C16O2 indicated that the exhausted lattice‒O was gradually replenished by the oxygen source from water (Supplementary Fig. 44). The above analyses revealed that the O used for CO oxidation originated from both •OH in water and lattice‒O in H-Pt-W3O/WC. Additionally, the *OH oxidation pathway generated H2O and established a water cycle that facilitated sustained CO oxidation, thereby enhancing resistance to poisoning in PEMFC. As shown in the Raman spectra in Supplementary Fig. 45, after CO oxidation, the lattice‒O in W3O was entirely replaced by 18O, further substantiating that the lattice‒O was replenished by water45,46,47.
To elucidate the role of the W sites in the CO oxidation, operando W L3-edge HERFD-XAFS analyses were performed in a CO-saturated 0.1 M HClO4. As evidenced by the variation in the white line peak intensity shown in Fig. 5f and Supplementary Fig. 35c, W actively participated in CO oxidation. Additionally, the FT-EXAFS spectra and corresponding fitting results are presented in Fig. 5g, Supplementary Fig. 46 and Supplementary Table 5. In the initial stage of the reaction at 0.005 V, the CN of W‒O significantly decreased, indicating that the lattice‒O in W3O participated in CO oxidation through the oxygen spillover effect. Additionally, the oxyphilic sites could effectively capture *OH during the HOR process22. As the voltage increased to 0.025 V, the CN of W‒O clearly increased due to the replenishment of oxygen from water and the concurrent adsorption of *OH. Upon increasing the potential to 0.05 V, the CN of W‒O again decreased; this decrease was attributed to the accelerated kinetics of CO oxidation at higher potentials, necessitating greater oxygen availability. As the potential continued to increase, the rate of water dissociation accelerated and resulted in increased *OH adsorption at the W sites. Concurrently, the increase in the CN of W‒O eventually approached the wet state because of the oxygen spillover effect and the slow adsorption rate of CO, which could not consume lattice-O promptly. Therefore, H-Pt-W3O/WC enhanced its anti-poisoning ability in acidic media by facilitating CO oxidation through two pathways, namely, the hydroxyl ion *OH and lattice‒O oxidation mechanisms (Fig. 5h). Moreover, water dissociation not only replenished lattice‒O and supplied *OH but also effectively provided additional *H intermediates, thus increasing the intrinsic HOR activity at the Pt sites.
DFT investigation
To elucidate the intrinsic role of the electron redistribution between W and Pt in enhancing HOR and anti-CO poisoning, we constructed optimal models for DFT calculations. In addition to simple interface studies (namely, Pt-W3O/WC), the Pt clusters were also positioned on the W3O (012) and WC (100) planes of the W3O/WC heterojunction, labeled Pt-W3O/WC (WC site) and Pt-W3O/WC (W3O site) for comparative purposes (atomic coordinates as shown in Supplementary Data 1). Additionally, a Pt-WC model was built to align with previous experiments (atomic coordinates as shown in Supplementary Data 1). The Mulliken population analysis revealed that the electric charges on the Pt clusters in the Pt-W3O/WC and Pt-WC models were approximately −0.77 e and −0.47 e, respectively; these values were significantly lower than those in the Pt-C model of 1.54 e (Fig. 6a and Supplementary Fig. 47). The different atomic states of Pt were caused by varying degrees of electron delocalization effects between the electron donors of W3O/WC and WC. Furthermore, the atomic state of the W sites in Pt-W3O/WC was 29.08 e; this value was higher than that in W3O/WC (20.79 e) and Pt-WC (14.24 e), as shown in Supplementary Fig. 47. The work function (WF) values measured by a scanning Kelvin probe (SKP) for different catalysts were in the following order: H-Pt-W3O/WC (5.28 eV) > H-W3O/WC (5.19 eV) > H-Pt-C (5.12 eV) (Supplementary Fig. 48). A higher WF value indicated an increased electron delocalization capability and promoted the escape of electrons, thereby facilitating their participation in HOR. Consequently, the formation of a spin-excluded electron pool between the W3O/WC surface and the interface with Pt could lead to electronic redistribution and synergistically accelerate HOR of the Pt sites. Therefore, the redistribution of the orbitally hybridized electrons lowered the Ed of Pt while increasing that of W (Fig. 6b).

a Atomic Mulliken population and b PDOS of Pt and W 5 d for Pt-W3O/WC, H-Pt-C, H-W3O/WC and H-Pt-WC. c Schematic Pt PDOS diagrams illustrating Pt cluster doping at the d-band position of H-Pt-W3O/WC. d Free energy of H-Pt-W3O/WC, Pt/WC and Pt-C for *H, *OH and COad adsorption. e Schematic illustration of the regulation process for the coupling strength between the surface W 5d-band and *OH. f Schematic illustration of the HOR mechanism on H-Pt-W3O/WC. g Free energy profiles of the HOR on H-Pt-W3O/WC, H-Pt-C and H-Pt-WC. h Free energy profiles of CO oxidation on H-Pt-W3O/WC, H-Pt-C and H-Pt-WC. Source data for Fig. 6 are provided as a Source Data file. The yellow and blue shades correspond to the different rate determination steps (RDS) of the reaction respectively.
In addition, according to Nørskov’s d-band center theory, when the adsorption intermediate approached the metal surface, the Ed and the adsorption intermediate reforming band were coupled48,49. Due to the high degree of electron filling of the antibonding band of Pt-W3O/WC, the part above the Fermi level (EF) decreased to reduce the *H and COad adsorption energy of the Pt site (Fig. 6c).Therefore, HBE theory was crucial for acidic HOR, and the Gibbs free energy of *H adsorption (ΔG*H) was further calculated. The ΔG*H values of the W3O/WC, W3O/WC (WC site) and W3O/WC (W3O site) models were approximately −0.18, 0.45 and 0.35 eV, respectively, indicating relatively weak adsorption for *H (Supplementary Fig. 49). After introducing the Pt cluster, the ΔG*H values for the Pt-W3O/WC (Interface), Pt-W3O/WC (WC site), Pt-W3O/WC (W3O site) and Pt-WC modes were −0.32, −0.38, −0.47 and −0.39 eV, respectively. They had a lower HBE than Pt-C at −0.68 eV (Fig. 6d, and Supplementary Fig. 50). In addition, the ΔG*H values for the different Pt sites of the Pt-W3O/WC model were also much lower than those of the Pt-C model (Supplementary Fig. 51). The single W site weakened the *H adsorption, whereas the Pt sites with high electron cloud density could accelerate HOR of the Volmer pathway with moderate *H adsorption/desorption energy. These results indicated that the *H adsorption of the Pt sites was weakened by interactions with the W sites, whereas the Pt sites with high electron cloud density could accelerate HOR of the Volmer pathway with moderate *H adsorption/desorption energy. To clarify the rate-determining step (RDS) in the HOR, comprehensive calculations of the reaction barriers were conducted. As shown in Fig. 6g, the RDSs for the Pt-W3O/WC (WC site), Pt-W3O/WC (W3O site) and Pt-C models were associated with Tafel-step energy barriers of 0.41, 0.36 and 0.75 eV, respectively. In contrast, the Pt-W3O/WC and Pt-WC models exhibited Volmer-step energy barriers of 0.47 and 0.39 eV, respectively. Notably, the Pt-W3O/WC model lowered the energy barriers for both Tafel and Volmer steps. Due to the adsorption bonding theory, the internal charge distribution of active sites may change the stability of H2 bonding, accelerate the activation of H2 (Tafel step) and regulate the adsorption of *H. Pt-W3O/WC may be able to enhance the HOR efficiency through various delocalization effects. Additionally, considering the oxygen spillover effect, oxygen sites were engineered on the Pt surface in the Pt-W3O/WC model (denoted as PtO-W3O/WC). As illustrated in Supplementary Fig. 52, the Ed of Pt shifted upward, and the HBE at the oxygen site was -0.81 eV. This could compensate for the lack of mass transfer caused by long-term reactions, thereby enhancing hydrogen bonding. Consequently, the results are similar to those of previous in situ analyses. the continuous replenishment of oxygen via the spillover effect consistently maintained high activity during HOR (Fig. 6f).
Previous DRIFTS analyses confirmed that Pt was the COad site in acidic environments; thus, we calculated the CO adsorption energies with different models. The adsorption energies of the Pt-W3O/WC (Interface), Pt-W3O/WC (WC site), Pt-W3O/WC (W3O site) and Pt-WC models were −0.83, −1.02, −1.38 and −1.21 eV, respectively; these values were lower than that for the Pt-C model (−1.90 eV) (Fig. 6h). These results indicated that the presence of W could weaken CO adsorption at the Pt site, thereby increasing resistance to poisoning. Additionally, the adsorption of OH was crucial for anti-CO poisoning. Since only the W sites near Pt-CO played the main role in CO oxidation, these specific sites were targeted in the model construction. In addition, the electron-deficient W could serve as a location to attract *OH (σ-giving electrons)50, and the lower electron filling of the antibonding band increased the portion above EF to increase the *OH adsorption of W sites (Fig. 6e)51. The dissociation energy of water at the W site in the Pt-W3O/WC model (0.67 eV) was lower than that in the W3O/WC model (0.88 eV) (Supplementary Fig. 53). These results indicated that the production of *OH through water dissociation was more favorable in the Pt-W3O/WC model. Furthermore, the subsequent increase in the *OH adsorption energy at different W sites in the Pt-W3O/WC model promoted the oxidation of CO adsorbed at the Pt sites (Supplementary Figs. 54-56). For the CO oxidation process, the RDS for Pt-W3O/WC (Interface), Pt-W3O/WC (W3O site), and Pt-C was CO adsorption, whereas the RDS for Pt-W3O/WC (WC site) and Pt-WC was the Langmuir-Hinshelwood (L-H) step. Furthermore, the lower oxidative barriers of Pt-W3O/WC indicated that CO may be able to be rapidly oxidized, thereby effectively exposing the Pt sites to enhance the HOR activity. On the basis of the above analyses, the theoretical models of the HOR and CO oxidation processes for all compared models are shown in Supplementary Figs. 57-58.
Discussion
In this study, we synthesized H-Pt-W3O/WC with low-content Pt clusters (Pt 1.7 wt%) to increase the HOR activity. Operando XAFS combined with DFT demonstrated that W3O/WC could act as a unique electron carrier to redistribute the electrons between Pt and W by reorganizing the d-band center. This strategy effectively reduced the energy barriers of HOR and anti-CO of the active site Pt. The upshifted Ed of the W sites also effectively trapped •OH from the water dissolution to oxidize the COad on the Pt sites; this resulted in the full exposure of the Pt sites to improve the HOR activity. Moreover, W3O could spillover the lattice‒O to the Pt sites; these sites were attacked by *H with well mass transfer ability to promote HOR activity and were involved in the oxidation of COad. The W sites could promote the lattice‒O consumption from water, as demonstrated by the isotope method. In the process of the CO oxidation reaction, in addition to obtaining O and free radicals in water to participate in oxidation, water was also generated such that the water needed for the oxidation process could be supplemented to achieve the water cycle. Importantly, H-Pt-W3O/WC delivered a high limiting density of 3.3 mA cm‒2 and showed well CO resistance, with only 5.4 and 5.5% current attenuation at 1000 and 2000 ppm CO, respectively. In addition, H-Pt-W3O/WC had a high peak power density of 1.63 W cm‒2 as an anode for PEMFC in H2/O2; here, a PPD of 774 mW cm‒2 in 100 ppm CO-H2/O2 could also be maintained. Thus, our work provided guidance for designing efficient HOR electrocatalysts with enhanced anti-CO poisoning ability in PEMFC.
Methods
Synthesis of HCB
Typically, 3.5 mL of styrene, 185 µL of methyl methacrylate, 153 µL of acrylic acid, and 0.0816 g of ammonium bicarbonate were dissolved in 130 mL of deionized water (DW) under ambient nitrogen. Subsequently, a 10 mL solution containing 0.078 g of potassium persulfate was added. The reaction temperature was elevated to 80 °C and maintained for 10 h. After centrifugation and washing with deionized water and ethanol, a polystyrene (PS) template was prepared. Then, 200 mg of PS was dispersed in 25 mL of methanol, followed by the addition of 1.98 g of 2-methylimidazole, and the mixture was sonicated for 1 h. Afterward, 25 mL of 0.16 M Zn(NO₃)₂·6H₂O methanol solution was added, and the mixture was stirred for 24 h at room temperature to prepare PS@ZIF-8. The mixture was heated at a rate of 5 °C min−1 from room temperature to 950 °C under ambient nitrogen. Afterward, it was held at 950 °C for an extra 2 h, and a hollow carbon bowl (denoted as HCB) was obtained.
Synthesis of H-W3O/WC
For the synthesis, 120 mg of HCB and 1.5 g of WCl₆·6H₂O were evenly dispersed in 125 mL of methanol and then subjected to alcoholysis for 24 h at 30 °C under stirring. After being washed with ethanol, filtered, and dried, the sample was heated to 900 °C at a rate of 5 °C min⁻1 and held at this target temperature for 2 h under a nitrogen atmosphere to prepare H-W3O/WC. The mass contents of W in the samples before and after pyrolysis were approximately 8.9 and 7.7%, respectively, as evidenced by inductively coupled plasma emission spectrometry (ICP‒AES) analysis. To synthesize pure WC for comparative analysis, the resulting H-W3O/WC was further treated using a heating rate of 5 °C min⁻1 to 1000 °C, and this temperature was maintained for 2 h, resulting in the formation of H-WC. Moreover, when the alcoholysis duration was extended to 36 h, H-WO3 could be prepared.
Synthesis of H-Pt-W3O/WC
First, 40 mg of H-W3O/WC was uniformly dispersed in 50 mL of DW, and then 0.4 mL of 0.13 mM H2PtCl6 solution was added. The pH value of the solution was adjusted to approximately 8 by using 1 M NaOH. Next, 10 mL of 0.13 mM NaBH4 solution was rapidly added to the above solution and allowed to react for 2 h. The obtained catalyst was denoted H-Pt-W3O/WC. Additionally, H-WC, H-WO3 and HCB were utilized as supports to prepare H-Pt-WC, H-Pt-WO3 and H-Pt-C by the same synthetic procedure.
Preparation and storage of the electrolyte
Using a HClO4 with a mass fraction of 72% as the stock solution, 8.07 ml of the stock solution was uniformly dispersed in deionized water (DW). The solution was then diluted to a final volume of 1000 ml in a volumetric flask, with the entire process conducted at room temperature. Subsequently, the prepared electrolyte solution was transferred to a brown sealed glass container and stored at a controlled temperature of 25°C in a refrigerator. This process was carried out 12 hours prior to testing to avoid any changes in the electrolyte concentration.
Characterizations
XRD patterns were showed using a Rigaku D/max-IIIB diffractometer equipped with Cu Kα radiation (λ = 1.5406 Å) at an accelerating voltage of 40 kV. Scanning electron microscopy (SEM) images were acquired with a Hitachi S-4800 instrument operating at 5 kV and 15 kV. Transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and energy-dispersive X-ray spectroscopy (EDS) elemental mapping were performed on a JEM-2100 electron microscope (JEOL, Japan) at an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) analyses were performed on a VG ESCALABMK II instrument using Mg Kα radiation (1253.6 eV). The CO oxidation process tail gas was detected using a Scion 4560c gas chromatograph. Fourier transform infrared spectroscopy (FT-IR) was performed using a PE Spectrum One B IR spectrometer across the range of 400–4000 cm‒1 with KBr pellet samples. Operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed on a Nicolet Is50 FT-IR instrument (Thermos Scientific). The electron paramagnetic resonance (EPR) measurements were carried out on a Bruker EMX plus model spectrometer operating at the X-band frequency. Mass spectrometry (MS) was performed by Trace 1300 and ISQ LT of THERMO SCIENTIFIC. The metal content within the samples was determined via ICP‒AES with an Agilent 725 instrument. X-ray absorption fine structure (XAFS) measurements were collected at the BL20U beamline of the Shanghai Synchrotron Radiation Facility (SSRF), China, utilizing a Si (111) double-crystal monochromator in fluorescent mode.
ICP‒AES measurement method
To prepare the test solution for ICP‒AES analysis, 50 mg of the sample was dissolved in 10 mL of aqua regia, which consisted of HNO₃ and HCl at a 3:1 volume ratio. The solution was continuously stirred for 4 h at 65 °C. After filtering, the filtrate was diluted to a total volume of 100 mL in a volumetric flask for further metal content testing.
Electrochemical measurements
The electrochemical performance was assessed using a Pine Instrument (US) with a three-electrode system, in which a Pt foil (1.0 cm2) and a reversible hydrogen electrode (RHE) served as the counter and reference electrodes, respectively. The working electrode was a rotating disk electrode (RDE) featuring a glassy carbon disk (diameter: 5.61 mm). For the preparation of the working electrode, 5 mg of the catalyst was dispersed into 2 mL of 0.0125 wt% Nafion ethanol solution and sonicated for one hour. Subsequently, 40 µL of the catalyst ink was carefully applied onto the disk surface of the RDE. Therefore, the catalyst loading on the electrode was 0.1 mg. The testing device we used is a combined testing apparatus connected to a rotating disk electrode device (PINE AFMSRCE) and an electrochemical workstation (PINE WaveNow Wireless), thereby collecting the data effectively. The HOR performance was measured in a H2-saturated 0.1 M HClO4 electrolyte, and the anti-CO poisoning performance was determined using H2/1000 ppm CO-saturated and H2/2000 ppm CO-saturated 0.1 M HClO4 electrolytes. Polarization curves were collected over a potential range of −0.1 to 1.2 V versus RHE at a scan rate of 5 mV s‒1 and a rotation rate of 1600 rpm without iR compensation. Chronoamperometry (i-t) curves were obtained at a potential of 0.05 V versus RHE at 400 rpm. Additionally, CO stripping curves were obtained via cyclic voltammetry (CV) tests with a potential range of −0.3 to 1.2 V vs. RHE at a scan rate of 50 mV s‒1.
Reference electrode calibration details
A platinum sheet was used as the working electrode, and hydrogen gas was introduced into a 0.1 M HClO4 electrolyte for a period of time, typically exceeding half an hour, to ensure that the solution near the electrode reached hydrogen saturation. Subsequently, the hydrogen gas flow was positioned close to the solution surface to saturate the hydrogen concentration in the atmosphere above the electrochemical cell. Following this, a cyclic voltammetry (CV) test was conducted within a selected range near the open circuit voltage, with a scan rate of 1 mV s⁻¹, allowing the reaction system to reach equilibrium. The two electrode potentials corresponding to the zero current density were selected, and their average value was taken as the calibrated reference electrode potential.
EXAFS fitting parameters
The Fourier transformation of the k2-weighted EXAFS oscillations was performed over a range of 2.5–8.5 Å−1 (W in H2/CO saturated) and 2.5 − 10 Å−1 (Pt in H2 saturated). The best fits to the EXAFS signal were made in R-space in the intervals of 1.0–3.0 Å (W) and 1.0–3.3 Å (Pt). The overall amplitude reduction factor, S02, was fixed at the best-fit values of 0.86 (W) and 0.82 (Pt); these values were determined from fitting the data of the bulk phase W/Pt foil. The error bounds (accuracies) characterizing the structural parameters obtained by EXAFS spectroscopy were estimated as follows: CN, ±20%; R, ±5%; σ2, ±20%; and ΔE0, ±20%. The variable are defined as follows: CN: coordination number; R: distance between the absorber and backscatter atoms; σ2: Debye‒Waller factor; ΔE0: inner potential correction. R-factor: indicator of goodness of fit.
Calculation processes of mass activity and current density
The mass activity (MA) is a significant indicator of the performance of the PGM catalysts for HOR. The corresponding calculation formulas are as follows:
where jK is the kinetic current (mA), jL is the limiting current (mA), and MPt is the absolute Pt loading (mg). The jK was calculated using the Koutecky‒Levich equation as follows:
where jd is the diffusion limiting current, B is the Levich constant, ω represents the rotation speed, c0 is the solubility of H2 (7.33 × 10‒4 mol L‒1), n represents the electron transfer number, F represents the Faraday constant (96485 C mol‒1), D represents the diffusion coefficient of H2 (3.7 × 10‒5 cm2 s‒1), and v represents the viscosity coefficient.
The specific exchange current density (j0) is calculated by the following equation:
where F is the Faraday constant (96485 C mol‒1), R is the universal gas constant (8.314 J mol‒1 K‒1), T is the Kelvin and η is the overpotential.
PEMFC preparation and test
The synthesized sample served as the anode catalyst, whereas commercial 60 wt% Pt/C from Johnson-Matthey was used at the cathode. For the preparation of the catalyst ink, 20 mg of the catalyst was mixed with 80.0 mg of 5 wt% Nafion ionomer solution, 25 mL of isopropanol, and 4.5 mL of DW by ultrasonication for 12 h to form a uniform ink. The catalyst ink was subsequently spray-coated onto a 2 cm × 2 cm Gore proton exchange membrane with a thickness of 12 µm by using an ultrasonic sprayer (model SP202). The loading of the anode catalyst was set at 0.05 mgPt cm−2, whereas the cathode catalyst loading was 0.25 mgPt cm−2. The membrane electrode assemblies (MEAs) were assembled using commercial gas diffusion layers (GDLs, Freudenberg H24Cx483) without hot pressing. The H2-O2 fuel cells were examined under 0.5/1.0/2.0 bar H2/O2 gas at 100 % relative humidity and 80 °C in galvanic mode, and the flow rate of H2/O2 gas was 0.5 SLPM/2 SLPM. The PEMFC polarization curve was measured using a hydrogen-oxygen fuel cell test system (IKKEM-P-22001).
Operando XAFS test measurement
For the Pt-L3 edge, X-ray absorption spectroscopy with high-energy resolution fluorescence was performed by adjusting the spot (HERFD-XAFS) to filter out the interference of the W L2 edge. For the W-L3 edge, fluorescence mode was used for information collection. A three-electrode system fluorescence in situ cell was used for detection. Then, the in situ cell was connected to an electrochemical working station. The catalyst was coated on hydrophobic carbon paper as the working electrode. The reference electrode and the counter electrode were AgCl/Ag and a carbon rod, respectively.
Here, so it is necessary to convert the test voltage measured with this electrode to the corresponding voltage relative to the standard hydrogen electrode (RHE). The conversion process is as follows:
Where E(RHE) is the potential vs reversible hydrogen electrode, E0(Ag/AgCl) and E(Ag/AgCl) are the measured potential with Ag/AgCl and Ag/AgCl as reference electrodes. pH is the hydrogen ion concentration of electrolyte. The electrolyte was measured by pH instruments to ensure a constant test environment (pH = 1 for acidic medium). The sample was immersed in 0.1 M HClO4 in the in situ cell for testing once. The applied voltage of the electrochemical workstation is then set from the open-circuit potential to a higher potential. To set different potentials and run stably at the potential for 30 min, the spectrum was collected. In addition, the saturated reaction gas was continuously injected into the entire process.
Operando DRIFTS test measurement
The DRIFTS in situ cell was connected with an electrochemical working station. The catalyst was coated on the RDE as the working electrode. For the preparation of the working electrode, 5 mg of the catalyst was dispersed into 2 mL of 0.0125 wt% Nafion ethanol solution and sonicated for one hour. Subsequently, 40 µL of the catalyst ink was carefully applied onto the disk surface of the RDE. The reference electrode and the counter electrode were AgCl/Ag and a carbon rod, respectively.
The sample was immersed in 0.1 M HClO4 in the in situ cell for an initial test to remove the back sole. The applied voltage of the electrochemical workstation was then set from the open circuit potential to a higher potential of 0.1 M HClO4. To set different potentials and run stably at the potential for 30 min, the spectrum was collected.
Quasi-in situ EPR measurement
First, a deionized water (DW) solution with a DMPO concentration of 100 mM was prepared. Prior to the test, nitrogen (N2) needed to be continuously introduced into the solution for deoxygenation. This process was essential to prevent oxygen in the solution from interfering with the reaction. The catalyst was subsequently tested using a three-electrode system under constant potential conditions as part of an I-t experiment in solution. The reference electrode and the counter electrode were AgCl/Ag and a carbon rod, respectively. And the potential conversion was carried out according to Eq. 6. The reaction was conducted in a sealed three-electrode cell. Stability tests were performed at various voltages for one hour, after which the solution was rapidly extracted for electron paramagnetic resonance (EPR) analysis. The EPR measurements were carried out on a Bruker EMX plus model spectrometer operating at the X-band frequency.
Methods of exhaust gas detection and isotope labeling
First, the catalyst was tested with a three-electrode system at a constant potential for the I-t experiment in 0.1 M HClO4, and the reaction pool was a sealed three-electrode pool with an air inlet and outlet. The reference electrode and the counter electrode were AgCl/Ag and a carbon rod, respectively. And the potential conversion was carried out according to Eq. 6. The process continued to inject saturated CO, and the tail gas was connected to the gas partial pressure device through the tail pipe and then into the gas chromatograph for gas detection. Before performing this operation, the tightness of the reaction tank needed to be checked to prevent interference from the presence of CO2 in the air. Furthermore, the saturated reaction gas was continuously injected into the entire process. The isotope labeling method used H218O (98 at%) to prepare 0.1 M HClO4 as the electrolyte. The catalyst was then tested with a three-electrode system at a constant 100 mV potential in an I-t experiment in 0.1 M HClO4 under saturated CO atmosphere conditions. Gas chromatography and mass spectrometry were employed for the detection of the exhaust gases.
DFT calculations
Density functional theory (DFT) calculations were conducted using the CASTEP module within Materials Studio. We employed a plane-wave basis set with a cutoff energy of 380 eV and a self-consistent field (SCF) tolerance of 2 × 10−6 eV. The geometry optimization was carried out using a maximum force of 0.05 eV Å‒1, a maximum displacement of 0.002 Å, and a Gaussian electron smearing width of 0.005 eV. To investigate the potential active site of H-Pt-W3O/WC for HOR, informed by HAADF-STEM imaging data, we performed additional DFT calculations. During the structural optimization of the surface models, a single gamma-point k-grid was utilized for the geometry optimization, and a denser 2 × 1 × 1 k-point grid was employed for the electronic structure calculations. The topmost atomic layer was allowed to fully relax, whereas the bottommost atomic layer was kept fixed to replicate the bulk properties. To more accurately assess the adsorption states of hydrogen and intermediates on the active sites, Pt was situated at various sites on W3O and WC. Moreover, a vacuum layer extending 15 Å was introduced into the supercell to mitigate the interactions between the periodic images. For the adsorption studies, the top two atomic layers were permitted to fully relax, whereas the bottommost layer remained fixed.
The HORs for the two-electron pathway in acidic media are as follows:
Tafel: 2 M (*) + H2 — 2M-Hads
Volmer: 2M-Hads ‒ 2 M (*) + 2H++2e‒
The Gibbs-free energy of H (ΔGH*) is considered an effective descriptor of HOR activity. ΔGH* can be calculated by the following equation:
where ∆EH*, ∆EZPE, and ∆S are the adsorption energies of the H atom on the given unit cell, the difference corresponding to the zero-point energy between the adsorbed H2 and gas phased H2 and the entropy change of H* adsorption, respectively. ∆S can be obtained by the following: ∆S = S(H*) ‒ 1/2S0(H2), where S0 (H2) is the entropy of gas phased H2 under standard conditions. In addition, according to the entropy of H2 in the absorbed state is negligible, ∆S can be calculated as ‒1/2S0 H2. Therefore, the ΔGH* can be calculated as follows:
The adsorption energies of OH and CO were calculated by using the following equations:
Data availability
The details data generated in this study are provided in the Supplementary Information. Source data are provided with this paper.
Change history
03 October 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-64767-3
References
Li, W. et al. Defect engineering for fuel‐cell electrocatalysts. Adv. Mater. 32, 1907879 (2020).
Setzler, B. P., Zhuang, Z., Wittkopf, J. A. & Yan, Y. Activity targets for nanostructured platinum-group-metal-free catalysts in hydroxide exchange membrane fuel cells. Nat. Nanotechnol. 11, 1020–1025 (2016).
Durst, J. et al. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 7, 2255–2260 (2014).
Zhang, X. et al. Toward the fast and durable alkaline hydrogen oxidation reaction on ruthenium. Energ Environ Sci 15, 4511–4526 (2022).
Yang, Z. et al. Pt single atoms on CrN Nanoparticles deliver outstanding activity and CO tolerance in the hydrogen oxidation reaction. Adv. Mater. 35, 2208799 (2023).
Xiao, F. et al. Recent advances in electrocatalysts for proton exchange membrane fuel cells and alkaline membrane fuel cells. Adv. Mater. 33, 2006292 (2021).
Ott, S. et al. Ionomer distribution control in porous carbon-supported catalyst layers for high-power and low Pt-loaded proton exchange membrane fuel cells. Nat. Mater. 19, 77–85 (2019).
Su, L. et al. Identifying the role of hydroxyl binding energy in a non‐monotonous behavior of Pd‐Pd4S for hydrogen oxidation reaction. Adv. Funct. Mater. 32, 2113047 (2022).
Zhu, J. et al. Recent advances in electrocatalytic hydrogen evolution using nanoparticles. Chem. Rev. 120, 851–918 (2019).
Cai, J. et al. Host–guest ensemble effect on dual-Pt atom-on-Rh nanosheets enables high-efficiency and anti-CO alkaline hydrogen oxidation. ACS Catal 13, 6974–6982 (2023).
Zhang, J. et al. Atomic-thick metastable phase RhMo nanosheets for hydrogen oxidation catalysis. Nat. Commun. 14, 1761 (2023).
Qin, S. et al. Ternary nickel–tungsten–copper alloy rivals platinum for catalyzing alkaline hydrogen oxidation. Nat. Commun. 12, 2686 (2021).
Zhang, B. et al. Lattice‐confined Ir clusters on Pd nanosheets with charge redistribution for the hydrogen oxidation reaction under alkaline conditions. Adv. Mater. 33, 2105400 (2021).
Su, L. et al. Modification of the intermediate binding energies on Ni/Ni3N heterostructure for enhanced alkaline hydrogen oxidation reaction. Adv. Funct. Mater. 31, 2106156 (2021).
Chen, Z. et al. Metallic W/WO2 solid-acid catalyst boosts hydrogen evolution reaction in alkaline electrolyte. Nat. Commun. 14, 5363 (2023).
Li, M. et al. Sub‐monolayer YOx/MoOx on ultrathin Pt nanowires boosts alcohol oxidation electrocatalysis. Adv. Mater. 33, 2103762 (2021).
Lin, H. et al. The study of oxygen spillover and back spillover on Pt/TiO2 by a potential dynamic sweep method. J. Mol. Catal. A 144, 189–197 (1999).
Vayssilov, G. et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles. Nat. Mater. 10, 310–315 (2011).
Chen, J. et al. Reverse oxygen spillover triggered by CO adsorption on Sn-doped Pt/TiO2 for low-temperature CO oxidation. Nat. Commun. 14, 3477 (2023).
Jin, J. et al. Unveiling the Complementary Manganese and Oxygen Redox Chemistry for Stabilizing the Sodium-Ion Storage Behaviors of Layered Oxide Cathodes. Adv. Funct. Mater. 32, 2203424 (2022).
Xiong, B., Zhao, W., Chen, L. & Shi, J. One‐Step synthesis of W2C@N,P‐C nanocatalysts for efficient hydrogen electrooxidation across the whole pH range. Adv. Funct. Mater. 29, 1902505 (2019).
Ma, Y. et al. Highly efficient hydrogen evolution triggered by a multi-interfacial Ni/WC hybrid electrocatalyst. Energ Environ Sci 11, 2114–2123 (2018).
Yan, H. et al. Small-sized and high-dispersed WN from [SiO4(W3O9)4]4− clusters loading on GO-derived graphene as promising carriers for methanol electro-oxidation. Energy Environ. Sci. 7, 1939–1949 (2014).
Chaco´N, C. et al. Synthesis and characterization of WO3 polymorphs: monoclinic, orthorhombic and hexagonal structures. J Mater Sci: Mater Electron 26, 5526–5531 (2015).
Wang, Z. et al. Atomically dispersed Co2–N6 and Fe–N4 costructures boost oxygen reduction reaction in both alkaline and acidic media. Adv. Mater. 33, 2104718 (2021).
Ma, T. et al. Crystalline lattice‐confined atomic Pt in metal carbides to match electronic structures and hydrogen evolution behaviors of platinum. Adv. Mater. 34, 2206368 (2022).
Yu, X. et al. Strengthening the hydrogen spillover effect via the phase transformation of W18O49 for boosted hydrogen oxidation reaction. ACS Catal 13, 2834–2846 (2023).
Sarma, P. V. et al. Nanostructured tungsten oxysulfide as an efficient electrocatalyst for hydrogen evolution reaction. ACS Catal 10, 6753–6762 (2020).
Wu, Z. et al. Microwave synthesis of Pt Clusters on Black TiO2 with abundant oxygen vacancies for efficient acidic electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 62, e202300406 (2023).
Jin, H. et al. Mesoporous Pt@Pt-skin Pt3Ni core-shell framework nanowire electrocatalyst for efficient oxygen reduction. Nat. Commun. 14, 1518 (2023).
Wang, L. et al. Stabilizing low‐valence single atoms by constructing metalloid tungsten carbide supports for efficient hydrogen oxidation and evolution. Angew. Chem. Int. Ed. 62, e202311937 (2023).
He, J., Fu, G., Zhang, J., Xu, P. & Sun, J. Multistage electron distribution engineering of Iridium oxide by codoping W and Sn for enhanced acidic water oxidation electrocatalysis. Small 18, 2203365 (2022).
Jia, Z. et al. Fully-exposed Pt-Fe cluster for efficient preferential oxidation of CO towards hydrogen purification. Nat. Commun. 13, 6798 (2022).
Dong, Y. et al. Pb‐Modified Ultrathin RuCu Nanoflowers for Active, Stable, and CO‐resistant Alkaline Electrocatalytic Hydrogen Oxidation. Angew. Chem. Int. Ed. 62, e202311722 (2023).
Sheng, W., Gasteiger, H. A. & Shao-Horn, A. C. Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs Alkaline Electrolytes. J. Electrochem. Soc. 157, B1529 (2010).
Wang, C., Yang, S., Chang, H., Peng, Y. & Li, J. Dispersion of tungsten oxide on SCR performance of V2O5WO3/TiO2: Acidity, surface species and catalytic activity. Chem. Eng. J. 225, 520–527 (2013).
Peng, L. et al. Dual synergetic catalytic effects boost hydrogen electric oxidation performance of Pd/W18O49. Nano Res 14, 2441–2450 (2021).
Sung, S. & Hoffmann, R. How carbon monoxide bonds to metal surfaces. J. Am. Chem. Soc. 107, 578–584 (1985).
Tong, M. et al. Operando Cooperated Catalytic Mechanism of Atomically Dispersed Cu@N4 and Zn@N4 for Promoting Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 60, 14005–14012 (2021).
Gorczyca, A. et al. Monitoring morphology and hydrogen coverage of nanometric Pt/γ‐Al2O3 Particles by in situ HERFD–XANES and quantum simulations. Angew. Chem. Int. Ed. 53, 12426–12429 (2014).
Yang, H. et al. Metastable-phase platinum oxide for clarifying the Pt–O active site for the hydrogen evolution reaction. Energy Environ Sci 16, 574–583 (2023).
Su, B. et al. Hydroxyl-Bonded Ru on metallic TiN surface catalyzing CO2 reduction with H2O by infrared light. J. Am. Chem. Soc. 145, 27415–27423 (2023).
Wu, Z.-Z. et al. Identification of Cu (100)/Cu (111) interfaces as superior active sites for CO dimerization during CO2 electroreduction. J. Am. Chem. Soc. 144, 259–269 (2021).
Mo, S. et al. Non‐Interacting Ni and Fe dual‐atom pair sites in N‐doped carbon catalysts for efficient concentrating solar‐driven photothermal CO2 reduction. Angew. Chem. Int. Ed. 62, e202313868 (2023).
Wu, Y. et al. Dynamic formation of Brønsted acid sites over supported WOx/Pt on SiO2 inverse catalysts─spectroscopy, probe chemistry, and calculations. ACS Catal 13, 7371–7382 (2023).
Ma, W. et al. Ru–W pair sites enabling the ensemble catalysis for efficient hydrogen evolution. Adv. Sci. 10, 2303110 (2023).
Nakka, L., Molinari, J. E. & Wachs, I. E. Surface and bulk aspects of mixed oxide catalytic nanoparticles: oxidation and dehydration of CH3OH by polyoxometallates. J. Am. Chem. Soc. 131, 15544–15554 (2009).
Nørskov, J. K., Abild-Pedersen, F., Studt, F. & Bligaard, T. Density functional theory in surface chemistry and catalysis. PNAS 108, 937–943 (2011).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238–240 (1995).
Wang, X. et al. Embedding oxophilic rare-earth single atom in platinum nanoclusters for efficient hydrogen electro-oxidation. Nat. Commun. 14, 3767 (2023).
Men, Y. et al. Oxygen-Inserted top-surface layers of Ni for boosting alkaline hydrogen oxidation electrocatalysis. J. Am. Chem. Soc. 144, 12661–12672 (2022).
Acknowledgements
We gratefully acknowledge the support of this research by the National Key R&D Program of China (2023YFA1507204), the National Natural Science Foundation of China (U20A20250, 22179034), and the Natural Science Foundation of Heilongjiang Province (ZD2023B002).
Author information
Authors and Affiliations
Contributions
D.S. conceived the idea. L.W. and H.G.F. co-supervised the project. D.S. designed the experiments and analyzed the data. D.S. and Z.J.L. performed electrochemical experiments and analyzed the electrochemical data. D.S. and Y.X. conducted DFT calculations. D.S prepared catalysts, took and analyzed TEM images. Y.C.W. contributed to implementation of H2-O2 fuel cell measurements. F.F.S. and B.B.M took and analyzed the XAFS spectra. D.S. fit the XANES and EXAFS spectra. All authors discussed the results and implications at all stages. The manuscript was written through contributions of all authors. D.S and F.F.S contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jin-Song Hu, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, D., Sun, F., Liang, Z. et al. Oxygen spillover on supported Pt-cluster for anti-CO-poisoning hydrogen oxidation. Nat Commun 16, 3883 (2025). https://doi.org/10.1038/s41467-025-58735-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58735-0
