
Abstract
Low-bandgap (LBG) mixed tin-lead (Sn−Pb) perovskite solar cells (PSCs) suffer from inferior performance due to their high defect density. Conventionally, ethylenediammonium diiodide (EDADI) is used as a surface passivator to reduce defects and improve device photovoltaic performance, but it introduces severe hysteresis caused by excessive mobilized ions at the top interface. Here, we report a mobile ion suppressing strategy of using hydrazine monohydrochloride (HM) as a bulk passivator to anchor the free ions in LBG perovskites. The protonated hydrazine (N2H5+) of HM formed hydrogen bonds with iodine (I–) ions, while the chloride (Cl–) ions occupied the I– vacancies, collectively impeding the migration of I– and thus mitigating the ion movement-induced hysteresis that arose from EDADI usage. The synergistic strategy of HM doping and EDADI post-treatment significantly suppresses the oxidation of Sn2+, decreases trap density, and inhibits rapid crystallization of perovskite. Consequently, we achieved a champion efficiency of 23.21% for LBG PSCs. Integrating these cells with wide-bandgap PSCs into all-perovskite tandem solar cells yields a high efficiency of 28.55% (certified 28.31%) with negligible hysteresis.
Similar content being viewed by others
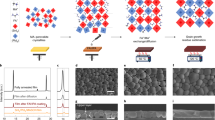

Introduction
Mixed tin-lead (Sn−Pb) perovskite solar cells (PSCs) have been extensively investigated as the bottom subcells of all-perovskite tandem solar cells (TSCs). Nevertheless, the vulnerability of Sn2+ oxidation poses a challenge, as it triggers the formation of defects and increases non-radiative recombination losses1,2,3. Additionally, the uncontrolled crystallization process of the Sn-based perovskite results in inhomogeneous film surface and compromised carrier diffusion lengths4. Research has demonstrated that hydrazine and its derivatives can effectively inhibit Sn oxidation and control crystallization5,6,7. However, earlier efforts have not adequately investigated functions beyond the reducing properties of hydrazine derivatives.
Additionally, surface defect passivation strategies, particularly at the perovskite surface, have emerged as a prominent approach due to the high defect density encountered8,9. However, despite notable progresses, the utilization of ethylenediamine (EDA) for surface defect passivation10, or the combined use of ethylenediammonium diiodide (EDADI) and glycine hydrochloride (GlyHCl) to modify top and bottom interfaces11, also leave unresolved ion migration issues. Specifically, the iodine-rich EDADI always introduced excessive I– ions onto the perovskite surface, leading to undesirable ion migration and device failure. In Pb-based PSCs, halogen bonding has been employed to capture and confine Ix–, inhibiting ion migration and thereby enhancing thermal-photostability12. However, the exploration of ion migration in Sn−Pb PSCs and all-perovskite TSCs remains inadequate, highlighting the urgence for further research in this field.
In this work, we develop a unique approach that incorporates hydrazine monohydrochloride (HM) as a bulk passivator and EDADI as a surface anchoring agent, representing a dual-pronged strategy for synergistic passivation of bulk and interface defects. Specifically, the protonated hydrazine (N2H5+) moiety within HM engages in hydrogen bonds with I– ions13, while the chlorine (Cl–) effectively occupies I– vacancies14. This combination elevates the activation energy required for ion migration, diminishing its occurrence. Moreover, this synergistic approach significantly inhibited the oxidation of Sn2+, mitigated non-radiative recombination, and promoted well-controlled crystallization. Consequently, the best-performing LBG PSC exhibits a power conversion efficiency (PCE) of 23.21%, with an open-circuit voltage (VOC) of 0.900 V, and significantly improved stability. We further fabricated all-perovskite TSCs based on the optimized devices, and achieved an impressive PCE of 28.55% with negligible hysteresis. The unencapsulated TSC sustained 90% of its initial PCE following 700 h operation under 1-sun illumination.
Results and discussion
Mechanism of inhibiting ion migration in Sn−Pb perovskite
We fabricated the LBG Sn−Pb PSCs with an inverted structure (ITO/PEDOT:PSS/perovskite/C60/BCP/Cu), where the absorber is (FASnI3)0.6(MAPbI3)0.4 (FA = formamidinium, MA = methylammonium)15,16. HM was employed as a bulk passivator, while EDADI as a surface anchoring agent. Devices with HM doping, EDADI post-treatment, and both were designated as HM, EDADI, and EDADI + HM, respectively.
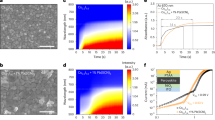
EDA2+ molecules can effectively bind with residual anions on the film surface, forming a stable interface. Therefore, EDADI as a passivator in LBG PSCs alleviates the interface defects17,18. As evidenced in Fig. 1a, b, the merely application of EDADI significantly enhances the VOC and fill factor (FF) of LBG PSCs under reverse voltage scan. However, excessive EDADI also leads to an increase in hysteresis (Supplementary Tables 1 and 2). The hysteresis index, defined in Supplementary Eq. (1), monotonically increased after more times EDADI post-treatment or higher EDADI concentrations (Fig. 1c and Supplementary Fig. 1). We attribute this to the vulnerability of ion migration due to the presence of excessive I– ions introduced by EDADI. In addition, we also revealed the positive correlation between hysteresis and the concentration of mobile ions from the perspective of comprehensive optoelectronic simulation. As shown in Supplementary Fig. 2, higher ion concentrations result in more pronounced hysteresis effect. According to the simulation results, increasing the durations of EDADI post-treatment raises the ion concentration from 2.0 × 1015 to 7.6 × 1015 cm–3. Therefore, we calculated the activation energy (EA) of ion mobilization in perovskite films, as it serves as an indicator of the ease of ion migration. A lower EA reflects easier ion mobilization, which in turn intensifies the hysteresis effect. Figure 1d shows the reduction of EA upon more spin-coating cycles of EDADI treatment19,20.

a J−V curves under reverse (forward) voltage scan of PSCs with different spin-coating cycles of EDADI post-treatment. Statistics of b VOC and c hysteresis index of PSCs with different durations of EDADI post-treatment. Each parameter was calculated from 10 individual devices. d The EA of devices with different durations of EDADI post-treatment. e, f 3D ToF-SIMS profiles of EDADI + HM LBG perovskite film deposited on PEDOT:PSS. g Schematic mechanisms of inhibiting ion migration of Sn–Pb perovskite films with HM doping. h EA of the control, EDADI, and EDADI + HM devices. i J−V curves of HM and EDADI + HM PSCs under reverse (forward) voltage scan.
Free ions usually exhibit a significantly slower velocity compared to charge carriers, resulting in a delayed response to changes in the electric field21. We then experimentally and theoretically investigated the dependence of hysteresis on the scan rates, as shown in Supplementary Figs. 3–5 and Supplementary Table 3. The results reveal a significant positive correlation between the hysteresis and scan rate at relatively low scan speeds (<0.1 V/s), while reversed back with extremely faster scan rates (>1 V/s). This is caused by the mismatch between the ion migration speed and the electric field change velocity. At low scan rates (<0.1 V/s), ions have sufficient time to migrate and redistribute, creating a balanced spatial electric field (Supplementary Fig. 5a). When the scan rate increases up to above 1 V/s, ion freezing leads to a stable electric field distribution (Supplementary Fig. 5c)22. While for the scan rate range between 0.1 and 1 V/s, a pronounced mismatch emerges between ion migration and changes in the electric field, where the rapid fluctuations in the electric field prevent certain low-mobility ions from responding promptly. This would result in the non-uniform ion spatial distribution, which hinders carrier transport and enhances the hysteresis effect (Supplementary Fig. 5b)23.
We then performed the time-of-flight secondary-ion mass spectrometry (ToF-SIMS) for perovskite film with HM and EDADI to dig out their distribution, as illustrated in Fig. 1e, f and Supplementary Figs. 6–8. The samples deposited on PEDOT:PSS and the profile shows that EDA2+ cations introduced via post-treatment are primarily located at the top surface of perovskite film. Here, N2H5+ ions, as the representative marker of HM, are present at both interfaces and in the film bulk, indicating that HM is distributed throughout the film. Moreover, the free Cl– ions primarily serve to fill iodine vacancies at the top and buried interfaces, which was further confirmed by the presence of Cl– in ToF-SIMS and X-ray photoelectron spectroscopy (XPS) results (Supplementary Figs. 8 and 9). This will effectively help mitigate hysteresis issue caused by EDADI.
We thoroughly explored the action mechanism of HM in perovskites, as shown in Fig. 1g. The N2H5+ in HM anchors free I– ions through hydrogen bonding13. The reaction process is shown in Supplementary Eqs. (2) and (3). This approach is complemented by Cl– filling iodine vacancies, as evidenced by slightly peak shift to larger angle in XRD patterns, shown in Supplementary Fig. 10. The (110) peak shifted to larger angles with increasing HM concentration, indicating that trace amounts of Cl– have been incorporated into the perovskite lattice. Additionally, a slight increase in the bandgap is observed, attributed to lattice shrinkage caused by the substitution of I– by Cl– 14. Therefore, the addition of HM significantly elevates the EA from 0.00434 eV for EDADI treatment to 0.05786 eV (Fig. 1h), over one order of magnitude. Furthermore, our XPS results reveal that the introduction of HM shifts the I 3d peak towards higher binding energy (Supplementary Fig. 11), indicating the formation of coordination bonds between protonated hydrazine with I– ions, thereby increasing the energy required for iodide ion migration23,24. These results unequivocally demonstrate that HM effectively mitigates the ion migration induced by EDADI. We then investigated the individual roles of chlorine and hydrazine by using lead chloride (PbCl2) and hydrazine monohydroiodide (HM-I) as additives. As shown in Supplementary Figs. 12–18 and Supplementary Table 4, the N2H5+ component interacts with I– through hydrogen bonding, which reduces ion-induced instability, prevents Sn2+ oxidation, and passivates defects in the perovskite. Meanwhile, Cl– promotes grain growth, passivates crystal defects, occupies I vacancies, and increases the EA for ion migration, effectively reducing hysteresis and enhancing device efficiency.
We conducted comprehensive evaluations of devices with and without EDADI and HM treatments, yielding the optimal J−V curves depicted in Fig. 1a, i. The corresponding photovoltaic parameters are listed in Supplementary Table 5. The hysteresis index for the control device is 1.28%. Post-processing with EDADI led to an improvement in device performance, while resulted in an increase in hysteresis index to 4.66%. Interestingly, the merely HM doping reduced the hysteresis index from 1.28% to 0.13%. Moreover, when EDADI and HM were employed simultaneously, the device performance increased with the hysteresis index dropping from 4.66% to 0.38%.
The control device attains the PCE of 20.20 (19.94)% under reverse (forward) voltage scan. The EDADI device gets a PCE of 21.68 (20.67)%, with a VOC of 0.884 (0.870) V, an FF of 80.2 (78.0)%. The HM device realizes a PCE of 21.86 (21.81)%. The best-performing PSC is achieved with both EDADI post-treatment and HM doping, delivering a PCE of 23.21 (23.12)% under reverse (forward) voltage scan. Furthermore, our findings indicate the EDADI post-treatment slightly compromises stability due to the excessive mobilized ions, while the introduction of HM mitigates this issue without sacrificing the efficiency (Supplementary Fig. 19).
Characterizations of Sn−Pb perovskite films
The introduction of EDADI and HM have gentle effects on the optical properties of Sn−Pb perovskite films due to the minimal concentration, given a bandgap of around 1.25 eV (Supplementary Fig. 20). However, it significantly affects the surface morphology and crystallinity of Sn−Pb perovskites. Film treated with EDADI exhibits smoother surface with clear grain boundaries compared to the control sample, which is likely caused by the washing effect of the iso-propenal (IPA) solution11,25. As shown in Supplementary Fig. 21, the control film has a very rough surface with blurred grain boundaries due to the amorphous feature, while after the IPA treatments, the unstable amorphous phase has been washed out26. Moreover, we compare the photovoltaic parameters of control, IPA-treated, and EDADI-treated LBG PSCs as shown in the Supplementary Figs. 22 and 23. Our findings indicate that employing IPA washing can negatively impact device performance, which can be attributed to the corrosive effect of IPA and the increased interfacial resistance between perovskite layer and electron transport layer27.
Moreover, perovskite films doped with HM displayed increased grain size and smooth surface, indicating the sufficient regulation on the crystallization process of LBG films (Fig. 2a−d and Supplementary Fig. 24).The surface roughness of the perovskite films was reduced upon EDADI surface treatment and HM doping, leading to flatter surface favorable for C60 deposition and thereby promoted electron extraction (Supplementary Fig. 25)28,29. All films display prominent diffraction peaks at (110) and (220) (Supplementary Fig. 26). The post-treatment of EDADI and the doping of HM individually enhanced the intensity of (110) peak. When they acted synergistically, the diffraction peaks were further enhanced. As shown in Fig. 2e, the half-width at half-maximum (FWHM) value of (110) peak gradually reduced for the control, EDADI, HM, and EDADI + HM films, indicating an improvement in the crystallinity of the perovskite30,31.

Top-view SEM images of a control, b EDADI, c HM, and d EDADI + HM perovskite films. e FWHM values of (110) peak from XRD results. Sn 3d5/2 XPS spectra of f control and g EDADI + HM perovskite films. h FTIR spectra of HM before and after mixing with SnI2 and FASnI3. i 1H NMR spectra of FAI and FAI + HM. j Schematic of energy levels of mixed Sn−Pb perovskite films with and without HM.
We further investigated the perovskite growth dynamics upon HM addition with two-step annealing process at 65 °C for 3 min and 100 °C for another 8 min (Supplementary Figs. 27–29). At 65 °C annealing, HM film shows similar surface morphology compared to control, however, its (110) peak intensity is much higher than that of the control, indicating better crystal quality. When further heated to 100 °C for a longer time, the HM film shows significantly enlarged grain size than the control sample, indicating the significantly enhanced crystallinity, which was further confirmed by the enhanced peak intensities from XRD results (Supplementary Fig. 29). Notably, the HM incorporation slowed down the grain growth kinetics as evidenced by the prolonged time for films to reach the maximized peak intensity. The control sample showed the highest peak intensity at 1 minute upon 100 °C annealing, while the HM sample presented the best crystallinity at 5 min. This confirms that the introduction of HM has a positive effect on improving the film quality due to the delayed crystallization and then reduced bulk defect density6,32.
We further delved deeper into the roles of EDADI and HM in suppressing the oxidation of Sn2+ in Sn−Pb perovskite films. The oxidation of Sn2+ to Sn4+ can trigger a charge imbalance, leading to the creation of lattice vacancies or defects33,34. As shown in Fig. 2f, g and Supplementary Fig. 30, control film has a large ratio of Sn4+ accounted for 19.43%, indicating a substantial presence of Sn4+ 35. However, the Sn4+ percentage of EDADI film and HM film dropped to 9.76% and 6.05%, respectively. Remarkably, the Sn4+ concentration of EDADI + HM film further dropped to 4.81%. This reduction can be partially related to the ability of EDADI on passivating uncoordinated Sn, forming a protective barrier10. Additionally, the hydrazine groups in HM exhibit strong reducing properties36,37, releasing electrons during crystallization, which reduces Sn4+ to Sn2+ (detailed in Supplementary Eqs. (4) and (5)). Moreover, the Sn 3d5/2 XPS peak shifts from 485.9 eV to lower binding energies for treated film. This shift indicates an increased electron density around the Sn atom, improving its coordination environment, and repairing dangling states. The nitrogen in N-H donates lone electron pairs to Sn2+, enhancing the electron cloud and in turn causing the Sn 3d5/2 peak shift. Additionally, stronger coordination interactions prevent oxidation of Sn2+, improving the chemical stability of Sn−Pb perovskite38. As shown in Supplementary Fig. 31, the control solution with an initial yellow color turned orange and deep red after 16 h exposure in air36. Conversely, due to the strong interaction between HM and SnI2, the oxidization of Sn2+ to Sn4+ was effectively inhibited. Even after 72 h, the color of HM-presented solution remained unchanged.
We employed Flourier transform infrared spectroscopy (FTIR) to scrutinize the interaction between HM and perovskites. As illustrated in Fig. 2h, the peak at 3442 cm–1, representing the N-H bond vibration in HM molecules, shifted by 14 and 27 cm–1 when mixed with SnI2 and FASnI3, respectively. This suggests a substantial weakening of the N-H bonds in HM upon the addition of SnI2 and FAI39, potentially due to the formation of coordination bonds between HM and Sn2+ and hydrogen bonding interactions with FAI. Moreover, the appearance of a C=N vibration bond at 1720 cm−1 found in the HM+FASnI3 film originates from the FA+ cation40.
Furthermore, we conducted a series of nuclear magnetic resonance spectroscopy (NMR) measurements to explore the hydrogen bonding interactions between the perovskite and HM additives. Figure 2i shows the 1H-NMR measurements of FAI and FAI + HM in deuterated dimethyl sulfoxide (DMSO-d6) solution. For pure FAI, 1H peaks at 8.82 ppm indicate N-H proton signal, while mixing with HM causes the peak splitting from one to two distinct peaks, indicating the presence of hydrogen bonds between N-H groups of FA+ and the N2H5+ group of HM41. We then measured liquid-state 119Sn-NMR spectra, as shown in Supplementary Fig. 32. The SnI2 signal shifted from −700 to −625 ppm, suggesting increased electron density surrounding Sn atoms due to the reduction of HM42. Moreover, in comparison to the control film, both EDADI and HM caused a shift of the Pb 4f7/2 peaks towards lower binding energies through XPS measurements (Supplementary Fig. 33). This shift is attributed to the interaction of HM and EDADI with Pb2+, mitigating undesirable Pb defects43,44.
To assess how HM affects the electronic properties of LBG perovskite films, we conducted ultraviolet photoelectron spectroscopy (UPS) measurements. Supplementary Fig. 34 shows that adding HM increases the secondary electron cutoff energy (Ecut-off) from 16.34 to 16.60 eV, giving the work function changing from −4.88 eV to −4.62 eV for control and HM samples, respectively45,46. Considering the optical bandgap of 1.25 eV, we determined that the conduction band minimum (CBM) values are −4.19 eV and −4.10 eV, respectively (Fig. 2j). The Fermi level rises closer to the CBM, which correlates with reduced Sn2+ oxidation and lower background hole concentration47,48. Moreover, the film surface displays weak n-type doping, benefiting the carrier transport.
Optoelectronic properties of perovskite films and devices
Through photoluminescence (PL) mapping, the optimized perovskite films exhibit brighter and more uniform PL, indicating increased radiative recombination with suppressed defects (Fig. 3a)49. We further evaluated the carrier dynamics using steady-state PL spectra and transient PL (TRPL, Fig. 3b, c). The EDADI + HM film shows the highest PL peak intensity and significantly prolonged carrier lifetime, which helps reduce non-radiative recombination, defect density, and allows more efficient carrier transport and collection50,51,52. Photoluminescence quantum yield (PLQY) was performed to quantitatively evaluate the passivation effect of HM on perovskite. The PLQY increased from 1.87% for control film to 2.23% for HM-treated film (Fig. 3d), indicating effective reduction of defects and non-radiative recombination. The quasi-Fermi level splitting (QFLS) was further estimated following the protocol reported in our previous work49, which increased by 16.10 meV upon HM treatment. These findings confirm the effective passivation capability of HM53.

a PL mapping images, b PL spectra, c TRPL spectra of different perovskite films. d PLQY values of perovskite films with and without HM treatment. e Statistics of carrier mobility of different perovskite films. f VOC versus light intensity, and g EIS plots of different LBG PSCs.
Using time-resolved microwave conductivity (TRMC) measurements, we gained insights into the electrical performance of perovskite films (Fig. 3e). The optimized EDADI + HM sample demonstrated improved carrier mobility, indicating fewer trap states on the film surface and reduced non-radiative recombination. Higher carrier mobility means faster and more efficient transfer of photoexcited carriers to the electrodes54.
We performed space charge limited current (SCLC) measurements to evaluate the trap density (Supplementary Fig. 35). After EDADI and HM addition, we observed a significant decrease in the trap field limited voltage (VTFL). Specifically, VTFL decreased from 0.333 V for the control device to 0.245 V for the synergistic treatment device, giving a reduced trap density from 1.53 ×1015 to 1.13 ×1015 cm–3. The lowest VTFL of EDADI + HM film indicates the highest quality of Sn−Pb perovskite films55.
As shown in Fig. 3f and Supplementary Fig. 36, we investigated the light intensity dependence of VOC and short-circuit current density (JSC) for different PSCs. VOC has a linear relationship to the logarithm of light intensity. The control device had an ideal factor of 1.65, while the HM, EDADI, and EDADI + HM devices present the values of 1.27, 1.26, and 1.25, respectively56,57, suggesting a reduction in Shockley-Read-Hall (SRH) recombination and improved diode quality after EDADI post-treatment and HM doping58. Additionally, JSC exhibited a linear relationship with the light intensity. The EDADI + HM device has the highest α value of 0.980, indicating the lowest space charge accumulation16. This is consistent with the increased recombination resistance and reduced dark current density as shown in Fig. 3g and Supplementary Fig. 37, respectively, indicating a reduced shunting risk due to the suppression of non-radiative recombination59,60.
Performance of LBG PSCs and all-perovskite TSCs
We fabricated LBG PSCs utilizing Sn−Pb perovskite absorbers with EDADI post-treatment and HM doping, where the optimal concentration of HM as an additive is 4 mg/mL (Supplementary Fig. 38). As shown in Fig. 4a, the devices subjected to EDADI post-treatment and HM doping exhibit the best performance. The external quantum efficiency (EQE) spectra of these devices are illustrated in Fig. 4b. The EQE-integrated JSC values over the entire solar spectrum for the control, EDADI, HM, and EDADI + HM devices are 30.46, 30.56, 30.89, and 31.27 mA/cm2, respectively, which closely match the JSC values obtained from the J–V measurements. We conducted statistical analysis on the photovoltaic parameters, as shown in Fig. 4c and Supplementary Fig. 39. The photovoltaic performance is significantly improved after EDADI and HM synergistic effect, and the HM addition could efficiently reduce the hysteresis caused by EDADI treatment. The averaged PCE increases from 20.02 ± 0.6% for control devices to 22.60 ± 0.7% for EDADI + HM devices, indicating high reproducibility (Supplementary Fig. 40). The modification by HM and EDADI significantly improves the stability of single-junction LBG PSCs, the unencapsulated LBG PSC retained 90% of its initial efficiency after being stored in an N2-filled glovebox for 2000 h (Supplementary Fig. 41).

a J−V curves, b EQE spectra, and c statistics of PCE of different LBG PSCs. d Cross-sectional SEM image of the all-perovskite TSC. e J–V curves of different all-perovskite TSCs with and without optimization. f EQE spectra of all-perovskite TSCs with HM + EDADI processing. g PCE evolution of TSC through MPP tracking. The unencapsulated TSC was measured under 100 mW/cm2 illumination with a LED light source in a glovebox.
By integrating the optimized LBG PSCs with the 1.77 eV wide-bandgap (WBG) FA0.8Cs0.2Pb(I0.6Br0.4)3 top subcells, we fabricated two-terminal all-perovskite TSCs with a structure of glass/ITO/4PADCB/WBG perovskite/C60/SnO2/IZO/PEDOT:PSS/LBG perovskite/C60/BCP/Cu49. The PCE of our single-junction WBG PSC is 19.03 (18.98)% under reverse (forward) scan, with an impressive VOC of 1.339 V, and the EQE-integrated JSC is 17.51 mA/cm2 (Supplementary Fig. 42 and Supplementary Table 6). Figure 4d shows the cross-sectional SEM image of the TSC, the absorber thicknesses of WBG and LBG subcells are around 400 and 900 nm, respectively.
As illustrated in Fig. 4e, the control TSC shows a PCE of 25.05% with a VOC of 2.020 V. After EDADI treatments, it achieves a PCE of 26.37 (24.79)% under reverse (forward) voltage scan, accompanied by a VOC of 2.090 (2.056) V, a JSC of 15.56 (15.52) mA/cm2, and an FF of 81.1 (77.7)%, indicating a huge hysteresis. The HM + EDADI treated TSC achieves the best-performing PCE of 28.55 (28.34)% under reverse (forward) voltage scan, accompanied by a VOC of 2.131 (2.130) V, a JSC of 16.05 (16.01) mA/cm2, and an FF of 83.5 (83.1)%. The hysteresis index of control, EDADI, and HM + EDADI is 0.27%, 5.99% and 0.73%, respectively (Supplementary Table 7). This indicates that, despite the EDADI can significantly increase the VOC and PCE, however, it sacrifices the overall performance and stability of TSCs due to the huge hysteresis. When EDADI treatments are combined with HM, it can address the hysteresis issues in TSCs, giving the best performance of 28.55% and improved stability due to the mitigation of the ion migration. Furthermore, the EQE-integrated JSC values for the respective top and bottom subcells in the EDADI + HM tandem device is 15.80 and 15.86 mA/cm2, demonstrating a well-matched current density between the two subcells (Fig. 4f). We obtained an independently certified efficiency of 28.31% (27.82%) under reverse (forward) voltage scan for our 2-T all-perovskite tandem cell (Supplementary Fig. 43). We continuously monitored the operational stability of EDADI and EDADI + HM based TSCs (Fig. 4g). The unencapsulated TSC with EDADI processing retained 90% of its initial PCE for approximately 500 hours of maximum power point (MPP) tracking, while the EDADI + HM device can still retain 90% of its initial PCE for 700 hours of MPP tracking, giving the good stability among the first class of reported61,62.
Discussion
We reported a synergistic strategy combining EDADI post-treatment and HM addition to address the issues of high defect density and hysteresis in LBG Sn–Pb PSCs, which are caused by unintentional Sn2+ oxidation and ion migration. Although EDADI treatments partially improve the VOC, they also introduce excessive migrated ions, leading to severe hysteresis. Through the addition of HM, the protonated N2H5+ forms hydrogen bonds with mobilized I–, and the Cl– fills the I– vacancies, effectively preventing ion migration and mitigating the hysteresis caused by EDADI. This synergistic method significantly inhibits Sn2+ oxidation, reduces trap density, and modulates the rapid crystallization of Sn−Pb perovskite films. Consequently, we achieved a high PCE of 23.21% for LBG PSCs. Furthermore, we obtained a remarkable PCE of 28.55% in all-perovskite TSCs with negligible hysteresis. Our method provides an effective route to address the adverse hysteresis issues caused by EDADI treatment.
Methods
Materials
All materials were obtained from commercial suppliers and used without any purification. Indium doped tin oxide (ITO) glass substrate (15 Ω/square) were purchased from Shangyang and Advanced election technology Co., diethyl ether was purchased from Alfa Aesar. Tin iodide (SnI2) and methylammonium bromide (MABr) were purchased from Advanced election technology Co., Ltd. Leadthiocyanate (Pb(SCN)2, 99.5%), tin floride (SnF2, 99%), N,N-dimethylmethanamide (DMF, anhydrous), dimethyl sulfoxide (DMSO, anhydrous), iso-propanol (IPA) were purchased from Sigma Aldrich. Poly (3,4-ethylenedioxythiophene): polystyrene sulfonate (PEDOT:PSS, CLEVIOS™ PVP AI 4083) was purchased from Heraeus, LLC. Formamidinium iodide (FAI) was purchased from Greatcell company (Australia). Bathocuproine (BCP) was purchased from Jilin OLED Company (China). Lead bromide (PbBr2), lead iodide (PbI2), methylammonium iodide (MAI), cesium iodide (CsI), ethylenediammonium diiodide (EDADI), (4-(7H-dibenzo[c,g]carbazol-7-yl)butyl) phosphonic acid (4PADCB) were purchased from Xi’an polymer light company. Hydrazine monohydrochloride (HM) was purchased from Aladdin company. The ceramic 2-inch indium zinc oxide (IZO) target was purchased from Shijiazhuang Huake metal material technology Co., Ltd. The gold (Au) and copper (Cu) were purchased from Kairui Xincai. All chemicals used in this work were commercially available and were used as received.
Preparation of precursors
The LBG Sn−Pb (FASnI3)0.6(MAPbI3)0.4 perovskite precursor. The (FASnI3)0.6(MAPbI3)0.4 precursor solution was acquired by mixing stoichiometric amounts of FASnI3 and MAPbI3 perovskite precursor. The FASnI3 precursor solution was prepared by dissolving 372 mg of SnI2 and 172 mg of FAI with 5 mol% (7.8 mg) of SnF2 in 626 μL mixed solution of DMF and DMSO (DMF:DMSO = 2:1). The MAPbI3 precursor solution was prepared by dissolving 461 mg PbI2 and 159 mg MAI with 3.5 mol% (13.8 mg) Pb(SCN)2 dissolved in 565 μL DMF and 71 μL DMSO. For HM bulk doping precursor, 4 mg (6 mol%, compared to the total amount of perovskite) of HM was added into the perovskite precursor. The EDADI post-treatment solution was prepared by dissolving 1 mg EDADI in 1 mL IPA. The WBG FA0.8Cs0.2Pb(I0.6Br0.4)3 perovskite precursor was prepared by dissolving 0.4 mmol FAI, 0.4 FABr mmol, 0.2 mmol CsI, 0.6 mmol PbI2 and 0.4 mmol PbBr2 in 0.833 mL mixed solvents of DMF and DMSO with a volume ratio of 3:1. All of the perovskite precursor and post treatment precursor were stirred at 50 °C for 1 h and filtered through a 0.22 μm PTFE filter before using.
Fabrication of PSCs
LBG PSCs
The ITO substrates underwent a 20-minute UV-ozone treatment, after which a 20 nm PEDOT layer was applied via spin-coating. This was followed by a 20-minute annealing process at 150 °C in ambient conditions. The substrates were then transferred into a N2-filled glovebox for the deposition of perovskite films. (FASnI3)0.6(MAPbI3)0.4 perovskite absorber layers were deposited on the PEDOT:PSS substrates at 4000 rpm for 60 s. During the spin-coating, 800 μL diethyl ether was dripped on the substrates. The as-deposited film was first annealed at 65 °C for 3 min, followed by 100 °C for 7 min. Then, 100 µL of the EDADI solution was applied to the annealed perovskite films and spin-coated at 4000 rpm for 20 s. Immediately after, the films were annealed at 100 °C for 5 min and then placed into a high vacuum chamber for subsequent deposition of functional layers. A 20 nm layer of C60 and a 5 nm layer of BCP were sequentially evaporated onto the perovskite, followed by the deposition of a 100 nm Cu with an active area of 0.09 cm2.
WBG PSCs
The 4PADCB solution, dissolved in ethanol at a concentration of 0.5 mg/mL, was filtered prior to deposition. An 80 μL aliquot of this solution was then dropped onto the ITO substrate and spin-coated at 3000 rpm for 30 s, followed by annealing at 100 °C for 10 min. The WBG perovskite precursor was applied to the HTLs and rapidly spin-coated using a fast deposition-crystallization method, with diethyl ether serving as the antisolvent. The resulting perovskite film was then annealed at 60 °C for 2 min and subsequently at 100 °C for 10 min. EDADI treatment was performed on the WBG perovskite films similarly to the LBG perovskite films. Sequential evaporation of 20 nm C60 and 5 nm BCP layers onto the perovskite surface followed. Finally, a 100 nm Cu was deposited, achieving an active area of 0.09 cm2.
Fabrication of 2-T all-perovskite TSCs
ITO substrates were cleaned using detergent, water, and organic solvents with the assistance of an ultrasonic bath. After cleaning, the substrates underwent a 20-minute UV-ozone treatment and were then transferred to a glovebox for the deposition of 4PADCB. The 4PADCB layer was annealed at 100 °C for 10 min, followed by the deposition of a WBG perovskite film. For 2-T tandem cells, a 30 nm layer of C60 was deposited by thermal evaporation in a vacuum chamber. Subsequently, the samples were transferred to an ALD system (Ensure Scientific) for the deposition of SnO2. IZO layer was then deposited on the SnO2 using a sputtering method. After depositing the ALD SnO2/IZO interconnection layer, PEDOT was spin-coated onto the samples and post-annealed at 100 °C for 10 min. The prepared subcells were moved into a glovebox for the deposition of the LBG perovskite layer and functional layers. Finally, the 2T TSCs were encapsulated with cover glass and ultraviolet-curable epoxy.
Film and device characterizations
J−V curves of PSCs and TSCs were measured with a Keithley 2400 sourcemeter. The devices were exposed to standard AM 1.5 G 100 mW/cm2 illumination using a solar simulator (SS-F5-3A, Enlitech). For the light intensity dependence test, the light intensity was varied between 1 and 100 mW/cm2 by adjusting the source power. EQE measurements were performed using an EQE system (QE-R, Enlitech) under near-dark conditions. For TSCs, bias illumination was provided by a 150 W halogen lamp with optical filters of 850 nm and 550 nm to measure the spectral response of the WBG and LBG subcells, respectively. The MPP tracking measurement was performed using a multicolor LED solar simulator (Tianjin Meitong Intelligent Technology Co., Ltd.) with a light intensity equivalent to 100 mW/cm2. No temperature control unit was used during the test. The dark J–V curves were measured using a semiconductor device parameter analyzer (B1500A) with a scanning range from −0.5 to 1.2 V for LBG PSCs. Activation energy of ion mobilization was performed using INSTEC temperature control probe station (Shanghai Hengshang Precision Instrument Co., Ltd.) and a semiconductor device parameter analyzer (B1500A). Electrochemical impedance spectroscopy (EIS) measurements were carried out using an electrochemical workstation (CIMPS, Zennium Zahner) under dark conditions. Nyquist plots of the impedance spectra were obtained in the frequency range of 1 MHz to 1 Hz with a bias voltage of 0.8 V and an amplitude of 5 mV. The top-view and cross-section structures of perovskite films and PSCs were characterized with field emission SEM instruments (Hitachi S-4800 and ZEISS Sigma 300). FTIR spectra were acquired using a NICOLET iS50 FTIR spectrometer. The crystallinity and crystal structure of the perovskite layers were analyzed using an Ultima III X-ray Diffractometer with a Ni-filtered Cu Kα X-ray source (Rigaku Corp.). Absorbance spectra were obtained using a UV-vis spectrophotometer (PerkinElmer Lambda 1050S).
For steady-state PL and TRPL measurements, perovskite films were fabricated on glass substrates followed by a layer of polymethylmethacrylate (PMMA). PL measurements were conducted in ambient air at room temperature, with the samples illuminated from the film side. A 532 nm continuous-wave laser (beam diameter ≈ 90 μm) at 40 mW/cm2 was used as the excitation source for steady-state PL measurement. The PL signal was detected using a Symphony-II CCD detector (from Horiba) after passing through a 300 g/mm grating monochromator, with an integration time of 0.5 s. For TRPL measurements, the Sn–Pb perovskite films were excited by a pulsed laser with a wavelength of 650 nm and an excitation frequency of 0.1 MHz, the TRPL spectra were fitted using double exponential.
PLQY of the corresponding film was measured by Edinburgh FLS1000 with a 532 nm laser to photoexcite the samples placed in an integrating sphere, and were provided technical support by Shiyanjia Lab (www.shiyanjia.com). UPS measurements were taken using a photoelectron spectrometer (ESCALAB 250Xi, Thermo Fisher Scientific). According to Einstein’s photo-emission law and calculating from the formula EF photon energy (21.22 eV) and Ecutoff. XPS spectra of various perovskite films were performed with a photoelectron spectrometer (ESCALAB 250Xi, Thermal Fisher Scientific), and were provided technical support by “Ceshigo Research service” (www.ceshigo.com). For time-resolved microwave conductivity (TRMC) measurements, the samples were pumped with a tunable OPO laser, which has a pulse width of ≈ 5 ns and repetition rate of 10 Hz. A FieldFox Handheld Microwave Analyzer (Keysight, N9915A) was used as microwave source and detector and the source power was set to be 1 mW. ToF-SIMS analysis was conducted using a ToF-SIMS6 instrument from IONTOF GmbH. Samples were analyzed in dual-beam profiling mode, and all profiles were performed in non-interlaced mode. Spectral data were acquired in high mass-resolution mode using a 30 keV Bi3+ primary ion beam over a 50 × 50 μm2 area at the center of the sputter crater. The sputter ion was a 1 keV Ar+ ion source with an electron impact ion source. The sputtering area was 200 × 200 μm2 at 0.3 pA, with a Bi liquid metal area at 120 nA. During sputtering, the mass spectra within the same layer remained consistent, and different layers exhibited well-defined profiles, indicating no beam damage was observed. 119Sn-NMR characterization was performed using Bruker AVII 400 MHz or AVIII 500 MHz spectrometers equipped with TBO (400 MHz) and BBO (500 MHz) room-temperature probe heads, respectively. 1H-NMR analyses were conducted using standardized acquisition protocols from Bruker Topspin software (version 2.1 for AV400 and 3.0 for AV500 systems), maintaining consistency across measurements.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the main data are available in the main text, the Supplementary Information, and the Source Data file. All other data of this study are available from the corresponding authors on request. Source data are provided with this paper.
References
Nakamura, T. et al. Sn(IV)-free tin perovskite films realized by in situ Sn(0) nanoparticle treatment of the precursor solution. Nat. Commun. 11, 3008 (2020).
Chen, Q. et al. Unveiling Roles of Tin Fluoride Additives in High‐Efficiency Low‐Bandgap Mixed Tin–Lead Perovskite Solar Cells. Adv. Energy Mater. 11, 2101045 (2021).
Savill, K. J., Ulatowski, A. M. & Herz, L. M. Optoelectronic Properties of Tin–Lead Halide Perovskites. Adv. Mater. Interfaces 6, 2413–2426 (2021).
Liu, H. et al. Modulated Crystallization and Reduced VOC Deficit of Mixed Lead–Tin Perovskite Solar Cells with Antioxidant Caffeic Acid. ACS Energy Lett. 6, 2907–2916 (2021).
Dai, X. et al. Efficient monolithic all-perovskite tandem solar modules with small cell-to-module derate. Nat. Energy 7, 923–931 (2022).
Zhang, W. et al. Component Distribution Regulation in Sn‐Pb Perovskite Solar Cells through Selective Molecular Interaction. Adv. Mater. 35, 2303674 (2023).
Bai, Y. et al. Decoupling light- and oxygen-induced degradation mechanisms of Sn–Pb perovskites in all perovskite tandem solar cells. Energy Environ. Sci. 17, 8557–8569 (2024).
Zhang, L. et al. Surface Defect Passivation of Pb–Sn‐Alloyed Perovskite Film by 1,3‐Propanediammonium Iodide toward High‐Performance Photovoltaic Devices. Sol. RRL 5, 2100299 (2021).
Hu, S. et al. Synergistic Surface Modification of Tin–Lead Perovskite Solar Cells. Adv. Mater. 35, 2208320 (2023).
Kapil, G. et al. Tin‐Lead Perovskite Fabricated via Ethylenediamine Interlayer Guides to the Solar Cell Efficiency of 21.74%. Adv. Energy Mater. 11, 2101069 (2021).
Hu, S. et al. Optimized carrier extraction at interfaces for 23.6% efficient tin-lead perovskite solar cells. Energy Environ. Sci. 15, 2096–2107 (2022).
Ren, X. et al. Mobile iodides capture for highly photolysis- and reverse-bias-stable perovskite solar cells. Nat. Mater. 23, 810–817 (2024).
Tsarev, S. et al. Hydrazinium-assisted stabilisation of methylammonium tin iodide for lead-free perovskite solar cells. J. Mater. Chem. A. 6, 21389–21395 (2018).
Zhao, D. et al. Efficient two-terminal all-perovskite tandem solar cells enabled by high-quality low-bandgap absorber layers. Nat. Energy 3, 1093–1100 (2018).
Wang, C. et al. A universal close-space annealing strategy towards high-quality perovskite absorbers enabling efficient all-perovskite tandem solar cells. Nat. Energy 7, 744–753 (2022).
Ma, T. et al. Hole Transport Layer‐Free Low‐Bandgap Perovskite Solar Cells for Efficient All‐Perovskite Tandems. Adv. Mater. 36, 2308240 (2023).
Zhou, J. et al. Mixed tin-lead perovskites with balanced crystallization and oxidation barrier for all-perovskite tandem solar cells. Nat. Commun. 15, 2324 (2024).
Zhou, W. et al. A Soldering Flux Tackles Complex Defects Chemistry in Sn–Pb Perovskite Solar Cells. Adv. Mater. 36, 2405807 (2024).
Ighodalo, K. O. et al. Negligible Ion Migration in Tin‐Based and Tin‐Doped Perovskites. Angew. Chem. Int. Ed. 62, e202213932 (2022).
Wang, Y. et al. Ordered Perovskite Structure with Functional Units for High Performance and Stable Solar Cells. Adv. Mater. 36, 2401416 (2024).
Tress, W. Metal Halide Perovskites as Mixed Electronic–Ionic Conductors: Challenges and Opportunities—From Hysteresis to Memristivity. J. Phys. Chem. Lett. 8, 3106–3114 (2017).
Wang, Z. S. et al. Device deficiency and degradation diagnosis model of Perovskite solar cells through hysteresis analysis. Nat. Commun. 15, 9647 (2024).
Ai, Z. et al. Physics, Simulation, and Experiment of Perovskite Solar Cells with Addressing Hysteresis Effect. Sol. RRL 6, 2200606 (2022).
Singh, S., Siliavka, E., Löffler, M. & Vaynzof, Y. Impact of Buried Interface Texture on Compositional Stratification and Ion Migration in Perovskite Solar Cells. Adv. Funct. Mater. 34, 2402655 (2024).
Fu, S. et al. Polishing the Lead‐Poor Surface for Efficient Inverted CsPbI3 Perovskite Solar Cells. Adv. Mater. 34, 2205066 (2022).
Yoo, J. J. et al. An interface stabilized perovskite solar cell with high stabilized efficiency and low voltage loss. Energy Environ. Sci. 12, 2192–2199 (2019).
Chen, D. et al. Performance enhancement of perovskite solar cells via material quality improvement assisted by MAI/IPA solution post-treatment. Dalton Trans. 48, 5292–5298 (2019).
Wang, Q. et al. Reductive Sn2+ Compensator for Efficient and Stable Sn–Pb Mixed Perovskite Solar Cells. Adv. Sci. 11, 2400962 (2024).
He, R. et al. Pure 2D Perovskite Formation by Interfacial Engineering Yields a High Open‐Circuit Voltage beyond 1.28 V for 1.77‐eV Wide‐Bandgap Perovskite Solar Cells. Adv. Sci. 9, 2203210 (2022).
Duan, C. et al. Scalable fabrication of wide-bandgap perovskites using green solvents for tandem solar cells. Nat. Energy 10, 318–328 (2025).
Liu, Z. et al. All-perovskite tandem solar cells achieving >29% efficiency with improved (100) orientation in wide-bandgap perovskites. Nat. Mater. 24, 252–259 (2025).
Zhou, X. et al. Additive engineering with 2,8-dibromo-dibenzothiophene-S,S-dioxide enabled tin-based perovskite solar cells with 14.98% power conversion efficiency. Energy Environ. Sci. 17, 2837–2844 (2024).
Song, J. et al. Synergistic Effect of Sodium Cyanoborohydride in Pb‐Sn Perovskite Solar Cells. Adv. Funct. Mater. 33, 2304201 (2023).
Lee, S. et al. Buried interface modulation via PEDOT:PSS ionic exchange for the Sn-Pb mixed perovskite based solar cells. Chem. Eng. J. 479, 147587 (2024).
Zou, S. et al. Efficient Environment‐friendly Lead‐free Tin Perovskite Solar Cells Enabled by Incorporating 4‐Fluorobenzylammonium Iodide Additives. Energy Environ. Mater. 6, e12465 (2023).
HAPascual, J. et al. Fluoride Chemistry in Tin Halide Perovskites. Angew. Chem. Int. Ed. 60, 21583–21591 (2021).
Cao, J. et al. High‐Performance Tin–Lead Mixed‐Perovskite Solar Cells with Vertical Compositional Gradient. Adv. Mater. 34, 2107729 (2021).
Sun, Q. et al. Surface charge transfer doping of narrow-bandgap Sn–Pb perovskites for high-performance tandem solar cells. Energy Environ. Sci. 17, 2512–2520 (2024).
Liu, J. et al. Alkyl Chains Tune Molecular Orientations to Enable Dual Passivation in Inverted Perovskite Solar Cells. Angew. Chem. Int. Ed. 63, e202403610 (2024).
Hills-Kimball, K., Nagaoka, Y., Cao, C., Chaykovsky, E. & Chen, O. Synthesis of formamidinium lead halide perovskite nanocrystals through solid–liquid–solid cation exchange. J. Mater. Chem. C. 5, 5680–5684 (2017).
Sun, X. et al. Multifunctional Modification of the Buried Interface in Mixed Tin‐Lead Perovskite Solar Cells. Angew. Chem. Int. Ed. 63, e202409330 (2024).
Agustin, D. & Ehses, M. 119Sn NMR spectroscopic and structural properties of transition metal complexes with terminal stannylene ligands. C. R. Chim. 12, 1189–1227 (2009).
Wang, J. et al. Highly efficient all-inorganic perovskite solar cells with suppressed non-radiative recombination by a Lewis base. Nat. Commun. 11, 177 (2020).
Zheng, L. et al. Reducing the Surface Reactivity of Alkyl Ammonium Passivation Molecules Enables Highly Efficient Perovskite Solar Cells. Adv. Energy Mater. 13, 2301066 (2023).
Li, S. et al. Flexible semitransparent perovskite solar cells with gradient energy levels enable efficient tandems with Cu(In,Ga)Se2. Nano Energy 78, 105378 (2020).
Ke, K., Kondamareddy, K. K., Gao, F., Zhang, X. & Yuan, X. Fabrication of Highly Efficient and Stable Hole‐Transport Material‐Free Perovskite Solar Cells through Morphology and Interface Engineering: Full Ambient Process. Energy Technol. 7, 1900446 (2019).
Brinkmann, K. O. et al. Perovskite–organic tandem solar cells with indium oxide interconnect. Nature 604, 280–286 (2022).
Zhou, S. et al. Aspartate all-in-one doping strategy enables efficient all-perovskite tandems. Nature 624, 69–73 (2023).
He, R. et al. Improving interface quality for 1-cm2 all-perovskite tandem solar cells. Nature 618, 80–86 (2023).
Shao, W. et al. Modulation of nucleation and crystallization in PbI2 films promoting preferential perovskite orientation growth for efficient solar cells. Energy Environ. Sci. 16, 252–264 (2023).
Zhu, J. et al. A donor–acceptor-type hole-selective contact reducing non-radiative recombination losses in both subcells towards efficient all-perovskite tandems. Nat. Energy 8, 714–724 (2023).
Huang, Y. et al. Substitution of Ethylammonium Halides Enabling Lead-Free Tin-Based Perovskite Solar Cells with Enhanced Efficiency and Stability. ACS Appl. Mater 15, 15775–15784 (2023).
Wang, J. et al. Bimolecular Crystallization Modulation Boosts the Efficiency and Stability of Methylammonium‐Free Tin–Lead Perovskite and All‐Perovskite Tandem Solar Cells. Adv. Energy Mater. 14, 2402171 (2024).
Luo, J. et al. Improved Carrier Management via a Multifunctional Modifier for High‐Quality Low‐Bandgap Sn–Pb Perovskites and Efficient All‐Perovskite Tandem Solar Cells. Adv. Mater. 35, 2300352 (2023).
Zhao, Y. et al. Reduced 0.418 V VOC-deficit of 1.73 eV wide-bandgap perovskite solar cells assisted by dual chlorides for efficient all-perovskite tandems. Energy Environ. Sci. 16, 2080–2089 (2023).
Chen, J. et al. Managing Lead Leakage in Efficient Perovskite Solar Cells with Phosphate Interlayers. Adv. Mater. Interfaces 9, 2200570 (2022).
Wan, Z. et al. Suppression of Nonradiative Recombination by Vacuum‐Assisted Process for Efficient Lead‐Free Tin Perovskite Solar Cells. Adv. Mater. Interfaces 8, 2100135 (2021).
Sun, H. et al. Scalable Solution‐Processed Hybrid Electron Transport Layers for Efficient All‐Perovskite Tandem Solar Modules. Adv. Mater. 36, 2308706 (2023).
Dey, K. et al. Substitution of lead with tin suppresses ionic transport in halide perovskite optoelectronics. Energy Environ. Sci. 17, 760–769 (2024).
Wu, D. et al. Efficient Flexible Perovskite Solar Cells with Reduced Hysteresis Employing Cobalt Nitrate Treated SnO2. Sol. RRL 6, 2200210 (2022).
Zhou, Y. et al. Defect-less Formamidinium Sn-Pb Perovskite Grown on Fluorinated Substrate with Top-down Crystallization Control for Efficient and Stable Photovoltaic. Energy Environ. Sci. 17, 2845–2855 (2024).
Zhao, J. et al. Suppressed Defects by Functional Thermally Cross‐Linked Fullerene for High‐Efficiency Tin–Lead Perovskite Solar Cells. Adv. Mater. 36, 2406246 (2024).
Acknowledgements
The work was financially supported by the National Key R&D Program of China (No. 2022YFB4200303 to D.Z. and C.W.), National Natural Science Foundation of China (Nos. 62005188 to C.W., 62174112 and 52461160298 to D.Z., 62120106001 to X.L.), Natural Science Foundation of Jiangsu Province (No. BK20190825 to C.W.), Natural Science Research Project of Jiangsu Higher Education Institutions (No. 22KJA480003 to C.W.), and Tang Scholar to C.W., Postdoctoral Fellowship Program of CPSF (No. GZB20240516 to T.M.).
Author information
Authors and Affiliations
Contributions
C.W., Y.L., and T.M. proposed the research idea and directed the overall project. Y.L. and T.M. wrote the manuscript. Y.L. fabricated low-bandgap (LBG) perovskite solar cells (PSCs) and all-perovskite tandem cells, and performed film and device characterizations. Yue.Z., Z.W., and Chen.C. assisted in the fabrication of wide-bandgap (WBG) subcells for all-perovskite tandem cells. Z.Y. and Y.B. supported the simulation experiments. Yuhang.Z. and T.J. assisted in the XPS measurements. All authors discussed the results and participated in revising the manuscript. Z.Y., D.Z., Cong.C., X.L., and C.W. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Ma, T., Wang, C. et al. Synergistic immobilization of ions in mixed tin-lead and all-perovskite tandem solar cells. Nat Commun 16, 3477 (2025). https://doi.org/10.1038/s41467-025-58810-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58810-6
