
Abstract
Electrocatalytic upcycling of polyethylene terephthalate (PET) plastics offers a promising and sustainable route that not only addresses serious waste pollution but also produces high value-added chemicals. Despite some important achievements, their activity and selectivity have been slower than needed. In this work, pulsed electrocatalysis is employed to engineer chemisorption properties on a lamellar mesoporous PdCu (LM-PdCu) catalyst, which delivers high activity and stability for selective electrosynthesis of high value-added glycolic acid (GA) from PET upcycling under ambient conditions. LM-PdCu is synthesized by in situ nucleation and attachment strategy along assembled lamellar templates, whose stacked morphology and lamellar mesoporous structure kinetically accelerate selective desorption of GA and expose fresh active sites of metal catalysts for continuous electrocatalysis at pulsed mode. This strategy thus delivers GA Faraday efficiency of >92% in wide potential windows, yield rate of reaching 0.475 mmol cm–2 h–1, and cycling stability of exceeding 20 cycles for electrocatalytic PET upcycling. Moreover, pulsed electrocatalysis discloses good electrocatalytic performance for scaled-up GA electrosynthesis from real bottle waste plastics. This work presents a sustainable route for selective electrosynthesis of value-added chemicals through upcycling of various waste feedstocks.
Similar content being viewed by others


Introduction
Being recognized as one of most consumed plastics, polyethylene terephthalate (PET) has revolutionized human life over the past one century. However, exponentially increasing with an annual output of exceeding 70 million tons has resulted in severe waste pollution of PET plastics1. Currently, a large number of PET waste plastics ends directly up in our earth’s curst by physical landfills, with <20% of waste plastics being upcycled into various value-added chemicals and/or fuels2,3. Traditionally, thermal catalysis has presented a popular route for PET upcycling to produce value-added chemicals and address waste plastic pollution4,5,6,7,8. Controversially, strict conditions of thermal catalysis, especially performed under high temperature (>200 °C) and high pressure (>20 atm), consume significant energy resources and thus disfavor sustainable development9,10. In this regard, it is of great significance to search for environment-friendly and sustainable routes for producing value-added chemicals from PET upcycling11,12,13.
Electrocatalysis, powered by renewable electricity, is becoming an alternative yet highly sustainable route for direct PET upcycling to produce fine chemicals under ambient conditions14,15,16,17,18,19,20. By alkaline hydrolysis of PET plastics and further separation of terephthalic acid (TPA), PET-derived ethylene glycol (EG) monomers can be electrocatalytically oxidized into various value-added products relied strongly on multiple C1 and C2 reaction pathways21,22,23,24. Traditionally, EG is favorably electrooxidized into CO2, involving 10 electrons (10e–) and releasing higher electrical energy, in fuel cell electrocatalysis through a C1 pathway25,26. In sharp comparison, selective conversion of EG to partially oxidized C2 products, for example specific value-added glycolic acid (GA), through a C2 pathway is more chemically significant and economically viable27. Currently, some important electrocatalysts with different nanostructures and elemental compositions have been developed to optimize chemisorption properties and further promote selective EG-to-GA electrocatalysis28,29,30,31,32,33. Despite some considerable efforts, GA electrosynthesis from PET-derived EG still suffered from poor selectivity and low yield as well as rapid catalysts deactivation, all of which disabled their development for practical large-scale application34,35. Therefore, there is an urgent need to develop new yet highly reliable routes that can achieve high selectivity, yield rate, and stability for selective GA electrosynthesis from PET upcycling in a sustainable and scaled-up manner.
In this work, pulsed electrocatalysis (PE) was employed for selective oxidation of PET-derived EG to GA with superior yield and stability, with a lamellar mesoporous palladium-copper (LM-PdCu) as electrocatalyst. In comparison to traditional current-potential electrocatalysis (CE), alternating periods of high oxidation potential and low open-circuit potential (OCP) at PE mode kinetically accelerated desorption of product and poisoning intermediates from catalyst surfaces and thus exposed fresh metal sites for further electrocatalysis36,37. High GA Faradaic efficiency (FE) of >92% and yield rate of 0.475 mmol cm–2 h–1 as well as good cycling stability (20 cycles) were achieved at PE model. Meanwhile, this route has been generally extended to practical upcycling system, holding high electrocatalytic performance with a yield of 64% for robust, scaled-up electrosynthesis of GA from real bottle waste plastics. This work thus provides important insights in engineering electrocatalytic modes for selective electrosynthesis of high value-added chemicals from various waste feedstocks in a reliable yet sustainable manner.
Results
LM-PdCu electrocatalyst with stacked morphology and lamellar mesoporous structure was synthesized by a simple solution-phase route with (NH4)2PdCl4 and Cu(NO3)2 as metal precursors, cetrimonium bromide (CTAB) as mesopore-forming template, 1,3,5-trimethylbenzene (TMB) as structure-controlled agent, and NaBH4 as reducing agent. Typically, predominant (NH4)2PdCl4 and Cu(NO3)2 were first mixed in the solution containing pre-dissolved CTAB and TMB. Then, freshly prepared NaBH4 was injected into above solution and further reacted at 90 °C for 1.5 h. Finally, mesopore-forming template and structure-controlled agent were removed by H2O/ethanol for several times to expose clean metal sites in mesopores. Similarly, monometallic LM-Pd and other bimetallic LM-PdM alloys were prepared accordingly with the same procedures but using different metal precursors.
LM-PdCu electrocatalyst and corresponding counterparts were thoroughly revealed by various advanced characterization techniques. Low-magnification transmission electron microscopy (TEM) image showed that LM-PdCu was homogeneously dispersed with nearly one-dimensional (1D) stacked morphology (Fig. 1a). The average length and width of LM-PdCu were estimated as 192 nm and 117 nm by being measured from more than 200 samples (Supplementary Fig. S1). TEM image further disclosed high degree of uniformity of products with 2D lamellar mesoporous structure (Fig. 1b). Different to traditional spherical mesopores, LM-PdCu were constructed by interlayer stackings, forming stacked morphology and lamellar mesoporous structure. Enlarged TEM image of single LM-PdCu further revealed open nanochannels of lamellar mesoporous structure (Fig. 1c), which were also confirmed by corresponding equalized rainbow color mode image (Fig. 1d). Above characterizations clearly demonstrated that LM-PdCu was morphologically stacked and structurally lamellar (Fig. 1e).

a, b Low-magnification TEM images, c enlarged TEM image and d corresponding equalized rainbow color mode image(The color scale is a grayscale value, with high grayscale values being blue and low grayscale values being red.), e schematic illustration, f HAADF-STEM EDS mapping images, g high-magnification and h high-resolution TEM images of LM-PdCu. i TEM image of LM-Pd. j HAADF-STEM image and corresponding EDS mapping images of LM-PdAg.
Atomic structure of LM-PdCu was further characterized. Spatial distribution of Pd and Cu in the product was first revealed by high-angle annular dark-field scanning TEM (HAADF-STEM) and corresponding energy dispersive X-ray spectroscopy (EDS) mapping images (Fig. 1f). Two metal elements, Pd and Cu, were uniformly distributed in whole sample, having atomic ratio of 76:24 (Supplementary Fig. S2), proving they were compositionally alloyed rather than phase-separated. High-magnification TEM image recorded from side view of LM-PdCu further implied lamellar mesostructures with alternative interlayered metal frameworks and 2D mesopores (Fig. 1g). The average framework thickness and mesopore size were calculated as 3.8 nm and 3.1 nm, respectively. High-resolution TEM image further showed a clear lattice fringe with a d-spacing distance of 0.221 nm, which corresponded to the (111) plane of a face-centered cubic (fcc) PdCu alloys (Fig. 1h). In addition to the result from powder X-ray diffraction (PXRD) pattern, our result definitely confirmed successful synthesis of well-alloyed LM-PdCu. Moreover, X-ray photoelectron spectroscopy (XPS) showed the strong electronic interaction between Pd and Cu in LM-PdCu, corresponding to the better oxidation resistance of Pd for electrocatalysis (Supplementary Fig. S3). More importantly, this strategy can be generally extended to various lamellar mesoporous structures with different metal compositions. Not only monometallic LM-Pd but also bimetallic LM-PdM (M = Ag, Ni, Co, and Fe) were successfully prepared with same stacked morphology and lamellar mesoporous structure as well as well-alloyed composition (Fig. 1i, j and Supplementary Figs. S4–S6). Considering potential applications induced by different metal compositions, this study would provide an opportunity for materials discovery and application exploration.
On the basis of synthetic parameters presented above, we believed the utilization of CTAB as mesopore-forming template and TMB as structure-controlled agent as well as NaBH4 as reducing agent played concurrent roles in precise synthesis ofLM-PdCu electrocatalyst with stacked morphology and lamellar mesoporous structure. To explore their important effects, all synthetic parameters were investigated systemically. First, we found, in the absence of CTAB, the product was metal nanoparticles without any mesopores. After the addition of CTAB, the structure first evolved into spherical nanoparticles with ring-like lamellar mesopores. With further increase of CTAB concentrations, perfect LM-PdCu with 2D lamellar mesoporous structure and 1D stacked morphology was formed accordingly (Supplementary Fig. S7). Then, TMB as concurrent structure-controlled agent was investigated (Supplementary Fig. S8). In the absence of TMB, non-porous products were obtained. In comparison, 1D stacked morphology was formed immediately after the addition of 0.36 mmol TMB. However, small amount of TMB disabled the formation of perfect lamellar mesoporous structure. With further addition of TMB, 2D lamellar mesoporous structure was produced accordingly and became wider with a width of >200 nm in the higher TMB concentrations. The results clearly demonstrated the concurrent roles of CTAB as mesopore-forming surfactant and TMB as structure-controlling agent for the formation of LM-PdCu. Besides, we explored the effect of reducing agents for adjusting its reduction kinetics38. Other reducing agents with weaker reduction ability, for example N2H4·H2O and ascorbic acid (AA), formed a stack of nanoparticles, instead of 2D lamellar mesopores (Supplementary Fig. S9), indicating the importance of NaBH4 as reducing agent that can adjust the reduction kinetics for the formation of LM-PdCu.
To gain more insights on formation mechanism of LM-PdCu, we also performed time-dependent structure evolution characterizations by detailed TEM studies. Upon the addition of NaBH4 to reaction solution (30 s), some crystalline nanoparticles with an average size of 5.2 nm were immediately formed (Fig. 2a). As the nucleation proceeded for 5 min, small nanoparticles evolved into lamellar stacked structures with discrete metal aggregates (Fig. 2b). With a longer reaction time of 20 min, discrete metals further attached into bigger crystals (Fig. 2c). Finally, perfect stacked metal frameworks were obtained after being reacted for 1.5 h (Fig. 2d). On the basis of above results, a nucleation and attachment mechanism along lamellar assembled template was proposed for the formation of LM-PdCu (Fig. 2d). In an aqueous solution, CTAB and TMB co-assembled and further stabilized with (NH4)2PdCl4 and Cu(NO3)2 by electrostatic and coordinated interactions (with quaternary head of CTAB) into ordered lamellar structures, in which CTAC and TMB were used as mesopore-forming surfactant and structure-controlling agent, respectively39,40. After the injection of strong reducing agent (NaBH4), PdCu alloys were immediately nucleated into small nanoparticles and further attached into discrete aggregates along lamellar assembles. With further attachment of discrete nanoparticles, continuous 2D metal frameworks with stacked morphology and lamellar mesoporous structure were produced accordingly. Surface-clean metal frameworks of LM-PdCu were finally obtained by further removing CTAB and TMB.

Low-magnification and high-magnification TEM images as well as simulated structures of LM-PdCu synthesized with different reaction times of a–c 30 s, d–f 5 min, g–i 20 min, and j–l 1.5 h. m A schematic illustration for proposed formation mechanism of LM-PdCu.
Physicochemical properties of lamellar mesoporosity and bimetallic alloys rendered LM-PdCu promising electrocatalyst for PET upcycling41,42. Meanwhile, monometallic LM-Pd and bimetallic nanoparticle PdCu (NP-PdCu) were also investigated as controls for sharp comparisons. Before electrocatalysis, waste PET plastic was hydrolyzed into PET hydrolysate (PETH) in KOH, which consisted of terephthalic acid (TPA) and ethylene glycol (EG)43,44,45. After that, electrochemical upcycling of PETH was probed in a three-electrode system to explore the EG oxidation reaction (EGOR) at anode. In the process, EG was first electrooxidized to glycolaldehyde (GAD) by losing two electrons (2e−) and then electrooxidized to high value-added glycolic acid (GA) by further losing 2e−. After the separation and purification of GA, isolated KOH can be recycled for further PET hydrolysis. Meanwhile, at cathode, H2O was electroreduced into H2 accordingly (Fig. 3a).

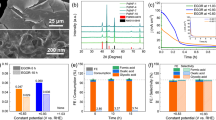
a A schematic diagram illustrating the reaction pathways for selective GA electrosynthesis from PET upcycling at anode. b LSV curves of LM-PdCu, LM-Pd and NP-PdCu collected in 1 M KOH and PETH at a sweep rate of 5 mV s−1 (Tested in PETH containing 0.10 M EG). c FE values and yield rates of GA over different electrocatalysts at CE model (The duration of the constant potential electrocatalysis is 3600 s, and the error bars are the standard deviation of three replicate tests). d In situ Raman spectra over LM-PdCu collected at different potentials (Raman signals were acquired at the beginning of the test at 200 s). e A schematic illustration for GA electrosynthesis from PET upcycling at CE mode.
Linear sweep voltammetry (LSV) curves were first collected and further compared in N2-saturated 1.0 M KOH with or without PETH containing 0.10 M EG (Fig. 3b). On the LM-PdCu catalyst, an anodic potential of 1.70 V (vs. reversible hydrogen electrode (RHE) hereafter) was required at a current density of 10 mA cm−2 in KOH, which drove the oxygen evolution reaction (OER). In comparison, the potential shifted negatively to 0.76 V in PETH under the same conditions, indicating that the EGOR in PETH occurred at a low potential. Differently, the higher potentials to drive EGOR electrocatalysis were required for LM-Pd (0.86 V) and NP-PdCu (0.84 V), indicating their lower activities for EGOR electrocatalysis in PETH. FE and yield rates of GA were then collected in the optimized potential window from 0.8 to 1.1 V. As summarized in Fig. 3c and Supplementary Fig. S10, LM-PdCu disclosed the highest GA FE of 96.8% and yield rate of 0.129 mmol cm–2 h–1 at 0.95 V. In comparison, the highest FE and yield rate were 92.5% and 0.113 mmol cm–2 h–1 for LM-Pd at 0.90 V and 91.2% and 0.079 mmol cm–2 h–1 for NP-PdCu at 0.90 V, respectively. Electrochemical impedance spectroscopy (EIS) also showed that electrochemical oxidation of catalyst surface at higher potentials led to an increase in the charge transfer resistance (Rct) at the electrode-electrolyte interface (Supplementary Fig. S11)36,46, suggesting that the introduction of Cu enhanced the antioxidant capacity of Pd, which allowed LM-PdCu to remain stable at the operating potential. Meanwhile, LM-PdCu electrocatalyst hold the highest mass activity and electrochemical active surface area (Supplementary Figs. S12–S14). Moreover, the fastest electrocatalytic kinetics, including the lowest apparent activation energy (Ea) and Tafel slope values, and the best electron transfer resistance, were also achieved by LM-PdCu (Supplementary Figs. S15–S17). In addition, in comparison to other LM-PdM, LM-PdCu disclosed the best EGOR performance in PETH (Supplementary Fig. S18).
In general, noble metal electrocatalysts can be quickly deactivated by poisoning with C–O/C–H intermediates during EGOR electrocatalysis47. Although LM-PdCu electrocatalyst hold the strongest OH adsorption capacity (Supplementary Fig. S19)48,49, its exposed active sites were still occupied by C–O/C–H intermediates, as being confirmed by the disappearance of adsorption and desorption peaks of OH– in EG solution (Supplementary Fig. S20). This would result in a huge waste of noble metal sites and cause the deactivation of electrocatalysts. To further prove our hypothesis, in situ Raman spectroscopy of LM-PdCu was further performed during EGOR electrocatalysis in PETH under different potentials. As shown in Fig. 3d, with the increase of potentials, the adsorption signals of EG became weaker for 200 s of electrocatalysis50. However, Raman signals for Pd-CO at 2122 cm–1 appeared at higher potentials of >1.5 V, suggesting that poisoning by COad occurred only at high potentials (Supplementary Fig. S21)51,52. In addition, the Pd-O signal at 522 cm–1 was gradually evident at >1.3 V53, demonstrating that Pd oxidation occurred at higher potentials (Supplementary Fig. S22). More active sites were occupied by oxidized intermediates and/or products at high potentials, definitely indicating LM-PdCu can be deactivated during EGOR electrocatalysis. Similarly, open-circuit potential (OCP) tests with different test times showed that adsorption capacity of EG in PETH decreased rapidly at CE mode (Supplementary Fig. S23)54. We thus concluded, on fresh catalyst surface, EG can be progressively electrooxidized to GA, but the desorption of GA and/or other intermediates (for example GAD) was slower due to serious poisoning of metal sites55,56. With continued electrocatalysis, the reaction intermediates and products occupied most active sites of noble metals, making difficult for EG to be further adsorbed, which thus deactivated the electrocatalyst for selective EGOR electrocatalysis in PETH at CE mode (Fig. 3e).
Pulsed electrocatalysis (PE) has recently been recognized as an effective route to redistribute surface microenvironment of noble metal electrocatalysts and accelerate removal of poisoning intermediates, thus enhancing electrocatalytic activity and selectivity as well as stability in various reactions57,58,59. Therefore, we performed and compared EGOR electrocatalysis from PETH in the potential square wave outputs at PE and CE modes (Fig. 4a). In CE mode, a high oxidation potential was continuously output. In PE mode, differently, the alternating periods of high oxidation potential and low OCP were output repeatably. When being pulsed from high potential to low OCP, a large reversed charging current was generated in a very short time for charging the surface double–layer of electrocatalyst60,61. Meanwhile, four output forms of square waves were designed with different high potential output times, all of which exhibited high reversed performance (Supplementary Fig. S24). We further compared chronoamperometric performance at CE and PE modes. As presented in Fig. 4b, current density rapidly decreased as the reaction proceeded at CE mode. In comparison, PE mode retained perfectly current density (Supplementary Fig. S25). At PE mode, oxidated intermediates and products (GA) were promptly desorbed from metal surface of electrocatalyst, resulting in pretty clean sites for further GA electrosynthesis from PETH. We also compared electrocatalytic EGOR performance in PETH under various pulsed pattens with different output times. The best FE and yield rate of GA from PETH were achieved when being pulsed at high potential for 1.0 s and low potential for 0.5 s (Supplementary Fig. S26).

a Output potentials and b corresponding chronoamperometric curves in CE and PE modes. c Current retention rates of LM-PdCu after being electrocatalyzed under different potentials at CE and PE modes. d Yield rates (Only high potential durations were considered when calculating the yield rate.) and e FE values of GA over LM-PdCu for EGOR electrocatalysis from PETH at CE and PE modes. f Concentrations of EG and GA during electrocatalysis collected with different input charges in PETH. g Consecutive recycling stability of LM-PdCu for selective GA electrosynthesis from PET upcycling. (All the error bars are the standard deviation of three replicate tests.).
Electrocatalytic performance of LM-PdCu was then explored and further compared at CE and PE modes for GA electrosynthesis from PETH (Fig. 4c). After being performed at CE mode, the stability gradually deteriorated with the increase of input potentials, having only 1.8% of current retention rate at 1.1 V for 3600 s. At PE mode, however, the stability became much better even at high potentials, retaining 42.8% of current retention rate at 1.1 V, after being electrochemically pulsed for 5400 s. Moreover, the increase of PE potentials facilitated the desorption of toxic intermediates/products, thus holding better performance in the higher potentials (Supplementary Fig. S27). Typically, PE mode accelerated the desorption of toxic intermediates and/or products on catalyst surface and thus enhanced catalyst resistance to toxification during EGOR electrocatalysis in PETH. Electrocatalytic EGOR performance in PETH was then evaluated at PE mode. As poisoning species can be removed quickly from catalyst surface at PE mode, GA yield rate did not reach the peak at 0.95 V (at CE mode). As presented in Fig. 4d, GA yield rate gradually increased with the increase of input potentials, reaching 0.475 mmol cm–2 h–1 at 1.1 V, which was 10.4 times higher than that at CE mode. More importantly, FE values of GA were high as >92% in all potentials tested (Fig. 4e), followed by a relatively low FE of formic acid (Supplementary Fig. S28). It was mostly because electrosynthesized GA was timely removed from catalyst surface and thus prevented GA being over-oxidized to other C2 and/or C1 products. We also collected concentrations of reactant (EG) and product (GA) during the electrocatalysis. With an input charge of 900C, EG concentrations in the PETH gradually decreased, followed by the increase of GA concentrations (Fig. 4f). EG conversion and GA selectivity finally reached as 97.1% and 95.4%, respectively (Supplementary Fig. S29), indicating EG in PETH was almost electrooxidized into value-added GA at PE mode. Moreover, 1H nuclear magnetic resonance (NMR) spectra showed that, with the increase of input charge, EG was gradually consumed and further electrooxidized into GA, while TPA do not engage in the reaction (Supplementary Figure S30).
Not only superior activity and selectivity but also high stability was achieved by LM-PdCu for GA electrosynthesis from PETH at PE mode. As shown in Fig. 4g, after being continued PE for even 20 cycles, both activity and selectivity were retained perfectly with high GA FE of 92.7 % and yield rate of 0.464 mmol cm–2 h–1. More impressively, the performance can be recovered, even being performed for 70 h in the flow cell (Figure S31). Meanwhile, TEM images showed high distribution and uniformity of LM-PdCu after the stability tests (Figures S32), although metal Cu was slightly dealloyed into the electrode (Figures S33-S34 and Table S1). Meanwhile, when compared with activity and selectivity in EGOR electrocatalysis, LM-PdCu also behave well for GA electrosynthesis (Table S2). More impressively, a good yield rate of 3.72 mmol h−1 cm−2 was achieved in 1.0 M pure EG that was generally reported for EGOR electrocatalysis (Figure S35). Meanwhile, LM-PdCu was electrochemically stable for EGOR electrocatalysis even in a high potential of 1.1 V, when comparing with previously reported electrocatalysts Table S3). These results definitely highlighted high potential of LM-PdCu as a high-performance electrocatalyst for selective GA electrosynthesis from PET upcycling.
In order to further explore how reacted species were adsorbed and desorbed on catalyst surface during electrocatalysis, we performed in situ Raman spectra at CE and PE modes. As shown in Fig. 5a, there were three main Raman signals at 875 cm–1, 1083 cm–1, and 1464 cm–1, which can be exactly assigned to νs (C-C-O), γ (C-O-H), and δ (CH2) of GA on catalyst35,62. After being electrocatalyzed at CE mode for 5 min, all Raman signals increased dramatically, indicating that a large amount of GA and counterpart intermediates were adsorbed on catalyst surface. After switching to PE mode directly, by contrast, the Raman signals gradually weakened with the reaction time being prolonged to 3 min. Obviously, adsorbed GA and other intermediates were desorbed from catalyst surface at PE mode. Similarly, 2D in situ Raman spectra showed that oxidated species were rapidly adsorbed on catalyst surface at the high potential of 1.05 V, resulting in a serious poisoning of metal active sites during electrocatalysis at CE mode (Fig. 5b). However, after being switched to low OCP of 0.50 V, δ (CH2) signals of GA gradually became weaker. The result further confirmed that switching to PE mode desorbed poisoning GA and/or intermediates and thus realized the site clean of metal catalyst surface for further electrocatalysis.

a in situ Raman spectra at CE and PE modes collected with different times. b 2D in situ Raman spectra at PE mode switched from high potential to low OCP during electrocatalysis. c in situ Raman spectra at PE mode under continuous switching electrocatalysis between 1.05 V and 0.50 V. d 2D in situ Raman spectra at PE mode with three continuous pulsed cycles (Tested the duration of each cycle for 18 s). e OCP tests after 30 min of electrocatalysis at CE and PE modes. f A schematic diagram illustrating electrocatalytic mechanism of LM-PdCu for selective GA electrosynthesis from PETH at PE mode.
Dynamic adsorption and desorption of reacted species and products during GA electrosynthesis at PE mode were further required by in situ Raman spectroscopy. As presented in Fig. 5c, adsorption signals of GA and intermediates were immediately generated on catalyst surface at high potentials based on electrocatalytic oxidation of EG, all of which can be then desorbed by cleaning step after continuously switching high potential to OCP. More interestingly, 2D in situ Raman spectra visualized dynamic processes of selective electrooxidation of EG and rapid desorption of GA on catalyst surface at three continuous PE cycles (Fig. 5d). We also carried out OCP tests after 30 min of electrocatalysis at CE and PE modes. In comparison to CE mode, PE mode disclosed much better EG adsorption capacity, further highlighting that removal of poisoning species exposed active metal sites for further electrocatalysis (Fig. 5e). On the basis of above observations, we proposed a reliable electrocatalytic mechanism for GA electrosynthesis from PETH at PE mode (Fig. 5f). There were two dynamic yet continuous steps at PE mode, including selective electrosynthesis of GA at high potentials and oxidation removal of GA at low potentials. Clean metal sites of catalyst surfaces facilitated continuous electrooxidation of EG and fast desorption of GA, which accelerated electrocatalytic kinetics and thus achieved high performance for robust GA electrosynthesis from PET upcycling at PE mode. Meanwhile, electrocatalytic oxidation reactions were performed with different substrates (alcohols). Similarly, better performance was achieved in the PE mode, further highlighting its practical application for selective electrosynthesis of value-added products from alcohols (Supplementary Fig. S36).
Considering high performance in GA electrosynthesis from PET upcycling, we finally demonstrated its practical scaled-up application at PE mode from real bottle waste plastics. As presented in Fig. 6a, bottle waste plastics were first hydrolyzed in a KOH solution to obtain PETH. Then, 5.1 g of TPA was separated from PETH by direct filtration. After being diluted, EG was electrocatalytically oxidized into glycolate at PE mode. After being further separated and acidified, 1.53 g of high-purity yet value-added GA was produced accordingly. Impressively, a high yield of 64% for GA electrosynthesis from upcycling of bottle waste plastics was achieved. LSV curves collected from bottle waste plastics and PET in KOH and PETH were almost overlapped, indicating same electrocatalytic performance, which thus confirmed high performance of LM-PdCu for practical GA electrosynthesis from PET bottle plastics at PE mode (Fig. 6b). Moreover, 1H NMR spectra and PXRD patterns of products (GA) verified high purity of GA produced from waste bottle plastics at PE mode (Fig. 6c, d). The results definitely highlighted high efficiency of LM-PdCu electrocatalyst for robust GA electrosynthesis from real waste plastics at PE mode. In addition, the cost-effectiveness analysis exhibited high potential of our strategy for considerable economic benefits (Supplementary Fig. S37).

a Synthetic routes of GA from electrocatalytic upcycling of PET wastes. b LSV curves for PETH waste at a sweep rate of 5 mV s−1. c 1H NMR spectra of reaction solutions in the separation and purification processes. d PXRD patterns for regenerated GA from upcycling of PET wastes.
Discussion
We developed a electrocatalyst of LM-PdCu for selective pulsed electrosynthesis of value-added GA for upcycling of PET waste plastics. LM-PdCu electrocatalyst was prepared by in situ nucleation and further attachment growth of metal frameworks along assembled lamellar structure of CTAB and TMB. Different to traditional mesoporous metals, LM-PdCu featured stacked morphology and lamellar mesoporous structure as well as bimetallic alloy composition, which kinetically accelerated transport of poisoning reactants and thus enhanced electrocatalytic performance. For selective GA electrosynthesis from PET upcycling, LM-PdCu disclosed superior performance, including high FE of 92% and yield rate of 0.475 mmol cm–2 h–1 as well as high cycling stability of reaching 20 cycles at PE model. Mechanism studies revealed that, compared with CE mode, PE mode accelerated oxidation removal of poisoning intermediates and thus cleaned metal sites for continuous electrocatalysis. More impressively, LM-PdCu was electrocatalytically efficient at PE mode in electrocatalytic upcycling of practical bottle waste plastics, achieving 64% of yield for GA electrosynthesis. Our work highlighted the importance of electrocatalytic modes in cleaning metal sites of catalyst surfaces, providing an effective strategy to promote electrocatalytic upcycling of waste plastics for fine electrosynthesis of high value-added chemicals in a sustainable manner.
Methods
Synthesis of LM-PdCu and other counterparts
LM-PdCu was synthesized by a simple solution-phase route with (NH4)2PdCl4 and Cu(NO3)2 as metal precursors, CTAB as mesopore-forming template, TMB as structure-controlled agent, and NaBH4 as reducing agent. Typically, 200 mg of CTAB was dissolved in 40 mL of water at room temperature. Then, 200 μL of TMB was added into above solution. After the mixture was stirred for 20 min, 500 μL of aqueous solution containing 12.5 mg of (NH4)2PdCl4 and 250 μL of aqueous solution containing 6.7 mg of Cu(NO3)2 were added. After being further stirred for 30 min, 1.5 mL of fresh solution containing 12.6 mg of NaBH4 was injected to above solution. After being reacted for 1.5 h, the product (LM-PdCu) was cleaned with H2O and ethanol for several times. Similarly, LM-Pd was prepared with the same procedures but with (NH4)2PdCl4 as sole metal precursor. Besides, other LM-PdM were prepared with the same procedures but with (NH4)2PdCl4 and corresponding metal salts as metal precursors.
Hydrolysis of PET plastics into PETH
PETH was made by immersing 16.0 g of PET powder in 200 mL of 4.0 M KOH and reacted for 48 h at 90 °C. The concentration of KOH in the hydrolysate was then diluted to 1.0 M and used directly as the electrolyte (containing 1.0 M KOH with 0.10 M EG, pH 14.0 ± 0.02). Similarly, bottle-based PETH was prepared by soaking 6.0 g of PET bottle powder in 100 mL of 10 M KOH and further reacted for 48 h at 90 °C. Stored all electrolytes at room temperature with dry ventilation and the electrolyte was remade every two weeks.
Preparation of electrocatalyst ink
The electrocatalyst ink was prepared by mixing 5.0 mg of electrocatalyst and 2.5 mg of Vulcan XC-72 carbon with a mixed solution of 0.20 mL of water, 0.70 mL of ethanol, and 0.10 mL of Nafion. After being sonicated thoroughly, 100 μL of electrocatalyst ink was dropped onto carbon paper (geometric area of working electrode is 0.5*1.0 cm2; catalyst loading is 1.0 mg cm−2), followed by natural drying within a culture dish with a cover. A compact film was finally formed and used as the working electrode in the following electrocatalysis process. All working electrodes were treated identically. Before the electrochemical test, all working electrodes were cleaned by continuous cyclic voltammetry scanning in 1.0 M KOH in the potential range of -0.7 ~ +0.4 V vs. Ag/AgCl (Saturated KCl).
Electrocatalytic measurement
All electrochemical measurements were carried out using a CHI 660E electrochemical workstation (Chenhua, Shanghai) in a single electrolytic cell with a test temperature of 25 °C. A standard three-electrode system, in which Ag/AgCl (with saturated KCl as the filling solution) and carbon rod were used as the reference and counter electrodes. All electrochemical potentials were initially measured versus Ag/AgCl and subsequently adjusted to the RHE scale by the following equation: ERHE = EAg/AgCl + (0.197 + 0.0591 × pH), with final values uniformly expressed as E vs. RHE. The Ag/AgCl electrode to be calibrated was immersed in 3.5% NaCl solution at the same time as the standard calomel electrode for calibration. Data collection was performed by the CHI 660E electrochemical workstation, with explicit acknowledgment that the electrochemical data obtained were used directly without post-processing or algorithmic manipulation. Before electrocatalytic tests, linear sweep voltammetry (LSV) curves were performed until that the polarization curves achieved the steady-state ones at a rate of 10 mV s−1 from 0.30 V to 1.40 V. The PE was conducted by using muti-potential steps technique. EIS measurements were performed in the frequency range of 100 kHz to 0.01 MHz with an alternating current voltage of 5.0 mV. The performance of PETH oxidation reaction was evaluated in a single-cell system without membrane. The test time was measured by the electrochemical workstation.
The experimental setup utilized a custom-designed flow cell configuration operating in a membrane-free electrolytic system. The electrolyte consisted of 1.0 M KOH solution saturated with N2 gas. A carbon rod served as the counter electrode, while the working electrode comprised pre-fabricated LM-PdCu material with a defined geometric surface area of 1.0 cm² (1.0 × 1.0 cm). The anolyte solution, containing 0.10 M ethylene glycol (EG) in 1.0 M KOH base electrolyte, was maintained at a constant volume of 500 mL under continuous magnetic stirring at 400 revolutions per minute46. Electrolyte circulation was achieved through a closed-loop peristaltic pumping system operating at 8 mL min-1. All reported electrode potentials were recorded without implementation of iR compensation measures.
In situ Raman spectroscopy
In situ Raman measurements were carried out on a Renishaw (inVia-Qontor) reflex Raman microscope under an excitation of 532 nm laser light with 50 mW57. A homemade cell and CHI660E electrochemical workstation were used. The cell was made of Teflon and equipped with a quartz glass plate to protect the objective. The WE (carbon paper coated with catalyst) was submerged in the electrolyte and kept perpendicular to the laser. Ag/AgCl was acted as the RE. A platinum wire embraced around the cell wall was served as the CE.
Data availability
All data for this study are available within the article and supplementary files or from the corresponding author upon reasonable request. Supplementary data generated for this study are provided in the Supplementary Information file. Source data of main text and Supplementary Information are provided with this paper. Source data are provided with this paper.
References
Tournier, V. et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580, 216–219 (2020).
Rahimi, A. & García, J. M. Chemical recycling of waste plastics for new materials production. Nat. Rev. Chem. 1, 0046 (2017).
Liu, F. et al. Concerted and selective electrooxidation of polyethylene-terephthalate-derived alcohol to glycolic acid at an industry-level current density over a Pd−Ni(OH)2 catalyst. Angew. Chem. Int. Ed. 62, e202300094 (2023).
Liu, Y. et al. Solar thermal catalysis for sustainable and efficient polyester upcycling. Matter 5, 1305–1317 (2022).
Wu, Y. et al. Catalytic degradation of polyethylene terephthalate using a phase-transitional zirconium-based metal-organic framework. Angew. Chem. Int. Ed. 61, e202117528 (2022).
Li, Y. et al. Catalytic transformation of PET and CO2 into high-value chemicals. Angew. Chem. Int. Ed. 61, e202117205 (2022).
Gao, Z., Ma, B., Chen, S., Tian, J. & Zhao, C. Converting waste PET plastics into automobile fuels and antifreeze components. Nat. Commun. 13, 3343 (2022).
Cao, R. et al. Co-upcycling of polyvinyl chloride and polyesters. Nat. Sustain 6, 1685–1692 (2023).
Jing, Y. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a Ru/Nb2O5 Catalyst. Angew. Chem. Int. Ed. 60, 5527–5535 (2021).
Rorrer, N. A. et al. Combining reclaimed PET with bio-based monomers enables plastics upcycling. Joule 3, 1006–1027 (2019).
Ellis, L. D. et al. Chemical and biological catalysis for plastics recycling and upcycling. Nat. Catal. 4, 539–556 (2021).
Lu, H. et al. Machine learning-aided engineering of hydrolases for PET depolymerization. Nature 604, 662–667 (2022).
Chen, J. et al. Toward carbon neutrality: Selective conversion of waste plastics into value-added chemicals. Matter 6, 3322–3347 (2023).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 12, 4679 (2021).
Sun, L., Lv, H., Xiao, J. & Liu, B. Enzymatic mesoporous metal nanocavities for concurrent electrocatalysis of nitrate to ammonia coupled with polyethylene terephthalate upcycling. Adv. Mater. 36, 2402767 (2024).
Yan, Y. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144–6155 (2023).
Shi, K., Si, D., Teng, X., Chen, L. & Shi, J. Pd/NiMoO4/NF electrocatalysts for the efficient and ultra-stable synthesis and electrolyte-assisted extraction of glycolate. Nat. Commun. 15, 2899 (2024).
Shi, Q. et al. Electrocatalytic upgrading of plastic and biomass-derived polyols to formamide under ambient conditions. Angew. Chem. Int. Ed. 63, e202407580 (2024).
Li, Q. et al. Layered (AlO)2OH·VO3 composite superstructures for ultralong lifespan aqueous zinc-ion batteries. J. Colloid Interface Sci. 663, 697–706 (2024).
Guo, X. et al. Confining Iodine into Metal‐Organic Framework Derived Metal‐Nitrogen‐Carbon for Long‐Life Aqueous Zinc‐Iodine Batteries. Adv. Mater. 36, 2408317 (2024).
Behera, S., Dinda, S., Saha, R. & Mondal, B. Quantitative electrocatalytic upcycling of polyethylene terephthalate plastic and its oligomer with a cobalt-based one-dimensional coordination polymer having open metal sites along with coproduction of hydrogen. ACS Catal. 13, 469–474 (2022).
Xin, L., Zhang, Z., Qi, J., Chadderdon, D. & Li, W. Electrocatalytic oxidation of ethylene glycol (EG) on supported Pt and Au catalysts in alkaline media: Reaction pathway investigation in three-electrode cell and fuel cell reactors. Appl. Catal. B Environ. 125, 85–94 (2012).
Kang, H. et al. Mining the carbon intermediates in plastic waste upcycling for constructing C–S bond. J. Am. Chem. Soc. 146, 18639–18649 (2024).
Li, J. & Duan, H. Recent progress in energy-saving hydrogen production by coupling with value-added anodic reactions. Chem 10, 1–32 (2024).
Qin, Y. et al. Extraordinary p–d hybridization interaction in heterostructural Pd−PdSe nanosheets boosts C−C bond cleavage of ethylene glycol electrooxidation. Angew. Chem. Int. Ed. 61, e202200899 (2022).
Lv, H. et al. Ir-doped CuPd single-crystalline mesoporous nanotetrahedrons for ethylene glycol oxidation electrocatalysis: Enhanced selective cleavage of C−C bond. Angew. Chem. Int. Ed. 63, e202400281 (2024).
Liu, K. et al. Energy-saving hydrogen production by seawater splitting coupled with PET plastic upcycling. Adv. Energy Mater. 14, 2304065 (2024).
Chen, J. et al. Unveiling synergy of strain and ligand effects in metallic aerogel for electrocatalytic polyethylene terephthalate upcycling. Proc. Natl Acad. Sci. USA 121, e2318853121 (2024).
Mao, Q. et al. In situ reconstruction of partially hydroxylated porous Rh metallene for ethylene glycol-assisted seawater splitting. Adv. Funct. Mater. 32, 2201081 (2022).
Lai, J. et al. Efficient bifunctional polyalcohol oxidation and oxygen reduction electrocatalysts enabled by ultrathin PtPdM (M = Ni, Fe, Co) nanosheets. Adv. Energy Mater. 9, 1800684 (2019).
Du, R. et al. Unveiling reductant chemistry in fabricating noble metal aerogels for superior oxygen evolution and ethanol oxidation. Nat. Commun. 11, 1590 (2020).
Si, D., Xiong, B., Chen, L. & Shi, J. Highly selective and efficient electrocatalytic synthesis of glycolic acid in coupling with hydrogen evolution. Chem. Catal. 1, 941–955 (2021).
Song, M. et al. Corrosion engineering of part-per-million single atom Pt1/Ni(OH)2 electrocatalyst for PET upcycling at ampere-level current density. Adv. Mater. 36, 2403234 (2024).
Xu, H. et al. Facile construction of fascinating trimetallic PdAuAg nanocages with exceptional ethylene glycol and glycerol oxidation activity. Nanoscale 9, 17004–17012 (2017).
Du, M. et al. Electrochemical production of glycolate fuelled by polyethylene terephthalate plastics with improved techno-economics. Small 19, 2303693 (2023).
Chen, W. et al. Pulse potential mediated selectivity for the electrocatalytic oxidation of glycerol to glyceric acid. Nat. Commun. 15, 2420 (2024).
Li, Z. et al. Alcohols electrooxidation coupled with H2 production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Yao, L., Yan, Y. & Lee, J.-M. Synthesis of porous Pd nanostructure and its application in enzyme-free sensor of hydrogen peroxide. ACS Sustain. Chem. Eng. 5, 1248–1252 (2017).
Sun, L. et al. Noble-metal-based hollow mesoporous nanoparticles: synthesis strategies and applications. Adv. Mater. 34, 2201954 (2022).
Zhang, G. et al. MXene‐mediated interfacial growth of 2D‐2D heterostructured nanomaterials as cathodes for Zn‐based aqueous batteries. Angew. Chem. Int. Ed. 63, e202401903 (2024).
Zhou, Y., Shen, Y. & Xi, J. Seed-mediated synthesis of PtxAuy@Ag electrocatalysts for the selective oxidation of glycerol. Appl. Catal. B Environ. 245, 604–612 (2019).
Chen, J., Jiang, M., Zhang, F., Wang, L. & Yang, J. Interstitial boron atoms in Pd aerogel selectively switch the pathway for glycolic acid synthesis from waste plastics. Adv. Mater. 36, 2401867 (2024).
Zhang, H. et al. Electrocatalytic upcycling of polyethylene terephthalate plastic to formic acid coupled with energy-saving hydrogen production over hierarchical Pd-doped NiTe nanoarrays. Appl. Catal. B Environ. 340, 123236 (2024).
Wang, J. et al. Electrocatalytic valorization of poly(ethylene terephthalate) plastic and CO2 for simultaneous production of formic acid. ACS Catal. 12, 6722–6728 (2022).
Yue, H., Zhao, Y., Ma, X. & Gong, J. Ethylene glycol: properties, synthesis, and applications. Chem. Soc. Rev. 41, 4218–4244 (2012).
Zhao, G. et al. Potential cycling boosts the electrochemical conversion of polyethylene terephthalate-derived alcohol into valuable chemicals. Nat. Commun. 15, 8463 (2024).
Wang, H. et al. Electrocatalysis of ethylene glycol oxidation on bare and Bi-modified Pd concave nanocubes in alkaline solution: An interfacial infrared spectroscopic investigation. ACS Catal. 7, 2033–2041 (2017).
Huang, W. et al. Promoting effect of Ni(OH)2 on palladium nanocrystals leads to greatly improved operation durability for electrocatalytic ethanol oxidation in alkaline solution. Adv. Mater. 29, 1703057 (2017).
Zhou, M. et al. Synthesis of Pd3Sn and PdCuSn nanorods with L12 phase for highly efficient electrocatalytic ethanol oxidation. Adv. Mater. 34, 2106115 (2021).
Cai, R. et al. Engineering Cu(I)/Cu(0) interfaces for efficient ethanol production from CO2 electroreduction. Chem 10, 211–233 (2024).
Hao, J. et al. Suppression of structural heterogeneity in high‐entropy intermetallics for electrocatalytic upgrading of waste plastics. Angew. Chem. Int. Ed. 64, e202419369 (2024).
Wei, D.-Y. et al. In situ Raman observation of oxygen activation and reaction at platinum–ceria interfaces during CO oxidation. J. Am. Chem. Soc. 143, 15635–15643 (2021).
Han, S., Wang, C., Wang, Y., Yu, Y. & Zhang, B. Electrosynthesis of nitrate via the oxidation of nitrogen on tensile‐strained palladium porous nanosheets. Angew. Chem. Int. Ed. 60, 4474–4478 (2021).
Lu, Y. et al. Tuning the selective adsorption site of biomass on Co3O4 by Ir single atoms for electrosynthesis. Adv. Mater. 33, 2007056 (2021).
Gao, F. et al. Shape-control of one-dimensional PtNi nanostructures as efficient electrocatalysts for alcohol electrooxidation. Nanoscale 11, 4831–4836 (2019).
Figueiredo, M. C., Trieu, V., Eiden, S. & Koper, M. T. M. Spectro-electrochemical examination of the formation of dimethyl carbonate from CO and methanol at different electrode materials. J. Am. Chem. Soc. 139, 14693–14698 (2017).
Huang, Y. et al. Pulsed electroreduction of low-concentration nitrate to ammonia. Nat. Commun. 14, 7368 (2023).
Hu, Q. et al. Pulsed co-electrolysis of carbon dioxide and nitrate for sustainable urea synthesis. Nat. Sustain. 7, 442–451 (2024).
Timoshenko, J. et al. Steering the structure and selectivity of CO2 electroreduction catalysts by potential pulses. Nat. Catal. 5, 259–267 (2022).
Casebolt, R., Levine, K., Suntivich, J. & Hanrath, T. Pulse check: Potential opportunities in pulsed electrochemical CO2 reduction. Joule 5, 1987–2026 (2021).
Ding, Y. et al. Pulsed electrocatalysis enables an efficient 2-electron oxygen reduction reaction for H2O2 production. J. Mater. Chem. A 9, 15948–15954 (2021).
Wang, H. et al. Copper-doped nickel oxyhydroxide for efficient electrocatalytic ethanol oxidation. Chin. J. Catal. 43, 1478–1484 (2022).
Acknowledgements
The authors kindly thank the financial supports from the National Key Research and Development Program of China (2023YFC3905002), the Natural Science Foundation of Sichuan Province (2025ZNSFSC0903) and the Fundamental Research Funds for the Central Universities. The authors would like to thank Dr. Feng Yang for her assistance of TEM imaging and Dr. Yanhong Liu for her assistance of Raman tests.
Author information
Authors and Affiliations
Contributions
B.L. conceived and supervised the project. S.H. and L.S. conducted the experiments, performed the structural characterizations, and analyzed the data. D.F. performed the electron microscopy imaging. All the authors discussed the results and cowrote the paper.
Corresponding authors
Ethics declarations
Competing interests
B.L., S.H., and L.S. are the inventors on a Chinese patent application related to the pulsed electrosynthesis of glycolic acid through polyethylene terephthalate upcycling submitted by Sichuan University (application number CN202411557324.5). The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, S., Sun, L., Fan, D. et al. Pulsed electrosynthesis of glycolic acid through polyethylene terephthalate upcycling over a mesoporous PdCu catalyst. Nat Commun 16, 3426 (2025). https://doi.org/10.1038/s41467-025-58813-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58813-3
