
Abstract
The conjugation of ubiquitin (Ub) or ubiquitin-like proteins (UBL) to target proteins is a crucial post-translational modification that typically involves nucleophilic attack by a lysine on a charged E2 enzyme (E2~Ub/UBL), forming an oxyanion intermediate. Stabilizing this intermediate through an oxyanion hole is vital for progression of the reaction. Still, the mechanism of oxyanion stabilization in E2 enzymes remains unclear, although an asparagine residue in the conserved HPN motif of E2 enzymes was suggested to stabilize the oxyanion intermediate. Here, we study the E2 enzyme UFC1, which presents a TAK rather than an HPN motif. Crystal structures of UFC1 mutants, including one that mimics the oxyanion intermediate, combined with in vitro activity assays, suggest that UFC1 utilizes two distinct types of oxyanion holes, one that stabilizes the oxyanion intermediate during trans-ufmylation mediated by the E3 ligase, and another that stabilizes cis-driven auto-ufmylation. Our findings indicate that oxyanion stabilization is influenced by multiple factors, including C-alpha hydrogen bonding, and is adaptable, enabling different modes of action.
Similar content being viewed by others
Introduction
The covalent attachment of ubiquitin (Ub) or ubiquitin-like proteins (UBLs) to target proteins is involved in numerous cellular processes, including proliferation, survival, and programmed cell death1,2,3,4. In humans, approximately a dozen UBLs are conjugated to target proteins via a three-enzyme cascade, similar to Ub5. Initially, an E1 enzyme activates Ub/UBL by hydrolyzing ATP, forming a thioester bond through its active site Cys6. Ub/UBL is then transferred to an E2 enzyme via a trans-thiolation reaction, forming a thioester bond with the E2 active site Cys7. In the last step, an E3 ligase delivers the substrate and mediates the transfer of Ub/UBL to a target protein8. In humans, some 800 different E3 ligases are responsible for substrate specificity. These E3 ligases can be classified into two groups based on their mechanism of action9. One group comprises the HECT and RBR families of E3 ligases, which utilize a two-step mechanism10. Here, Ub/UBL initially forms a thioester bond with a Cys in the E3 ligase and only then is Ub/UBL transferred to the target protein. The other group, mainly comprising E3 ligases belonging to the RING family, transfers Ub/UBL directly from the E2 enzyme to the target protein11. In this group, the E3 ligase typically brings a lysine residue from the target protein close to the E2~Ub/UBL thioester, thereby enabling the lysine to carry out a nucleophilic attack. This results in an uncharged E2 and Ub/UBL covalently attached to that target protein lysine via an isopeptide bond.
Before the formation of this isopeptide bond, a tetrahedral intermediate is formed5. In this intermediate, the carbonyl group of Ub/UBL, which is linked to the sulfur of the E2 Cys, no longer presents a double bond to the oxygen, instead forming a single bond, resulting in a negatively charged oxygen atom known as an oxyanion (Supplementary Fig. 1a). Stabilization of this oxyanion, facilitated by a structure known as the oxyanion hole, is essential for progression of the reaction and subsequent product formation. In 2003, Wu et al. suggested that an Asn residue within a highly conserved HPN motif in E2 enzymes plays a role in oxyanion stabilization by forming a hydrogen bond between the Asn side chain and the oxyanion12. Subsequently, it was proposed that this Asn serves a structural role in stabilizing a loop adjacent to the active site through hydrogen bonding13, which is necessary for deprotonating the attacking lysine14. In parallel, mutagenesis analysis of this Asn raised doubts regarding its role in oxyanion stabilization. As such, the mechanism of oxyanion stabilization in E2 enzymes remains unsolved14,15.
UFC1 is the E2 enzyme for the UBL ubiquitin-fold modifier 1 (UFM1)16. Initially, UFM1 is activated by the E1 enzyme UBA5, forming a thioester bond with the active site Cys of UBA516,17. The activated UFM1 is then transferred to the active site Cys of UFC1 in a trans-thiolation reaction18. Finally, with the assistance of the E3 enzyme complex comprising UFL1 and DDRGK1, UFM1 is transferred to target proteins19. Unlike E2 enzymes possessing an HPN motif, UFC1 does not contain this motif, instead harboring a TAK motif20. Like the HPN motif, the TAK motif is crucial for UFC1 activity, as revealed by mutagenesis19. However, unlike the side chain of the Asn residue of the HPN motif, the side chain of the corresponding residue in the TAK motif, i.e. K108, does not participate in hydrogen bonding. The absence of hydrogen bonding is due to UFC1 lacking a loop adjacent to the active site18. This loop is stabilized by the Asn residue of the HPN motif in other E2 enzymes13 (Supplementary Fig. 1b). This difference prompted us to investigate whether the TAK motif could be exploited to elucidate the mechanism of oxyanion stabilization, without the structural constraints presented by the HPN motif.
In this work, structural and biochemical characterization of UFC1 mutants, including one that mimics the oxyanion intermediate, allowed us to identify a critical hydrogen bond between the T106 side chain and the backbone nitrogen of K108 within the TAK motif. This interaction restricts the movement of the K108 C-alpha, enabling its involvement in C-alpha hydrogen bonding with the oxyanion, thereby contributing to oxyanion stabilization. Interestingly, our findings reveal that UFC1 possesses an additional TAK-independent oxyanion hole, which facilitates auto-ufmylation via a cis mechanism. Collectively, our study highlights the complexity and adaptability of the oxyanion hole in E2 conjugating enzymes.
Results
The UFC1 T106-K108 hydrogen bond is essential for ufmylation
UFC1 contains its active site cysteine at position 116 and a conserved TAK motif spanning residues 106–108. A mutation in this motif, T106I, has been linked to infantile encephalopathy with progressive microcephaly21. Recently, Peter et al. reported that this mutation hinders UFM1 ligation, thus prompting inquiries into the underlying mechanism19. To further investigate this point, we solved the crystal structure of this mutant to 1.35 Å resolution (Supplementary Fig. 2 and Supplementary Table 1a). The asymmetric unit comprises a single molecule of UFC1 T106I. Super-positioning the C-alpha atoms onto WT UFC1 (PDB 2z6o) yielded an RMSD of 0.266 Å, indicating no discernible differences in the structures (Fig. 1a). The Tm of T106I is 3 °C lower than that of the WT (as measured by a thermal shift assay; Supplementary Fig. 1c). However, this mutant does not differ from the WT protein in terms of solubility, unlike the H77A mutant, which affects the HPN motif of Ubc13 and severely impairs protein solubility12. Overall, our structural data suggest that the T106I mutation does not introduce significant structural changes.

a Super-positioning of UFC1 T106I (orange) onto WT UFC1 (PDB 2z6o) (blue) yields C-alpha atoms with an RMSD of 0.266 Å. Side chains of C116 and T/I106 are shown in stick representation. b A representative western blot showing the effect of UFC1 T106I, T106V, and T106A mutants on in vitro ufmylation (see Supplementary Fig. 1g for protein loading control). c The band corresponding to the ufmylated product (~40 kDa) was quantified for each mutant and normalized against the product formed in WT protein (data represented as mean ± SD, n = 5 independent experiments). Differences were significant for T106I, T106V (with p < 0.0001) and T106A (with p < 0.01, all calculated using two‐tailed unpaired Student’s t‐tests). d Western blot showing the effect of UFC1 T106I, T106V, T106A, and T106L mutants on ufmylation of substrate (RPL26) (see Supplementary Fig. 1h for protein loading control). Ufmylation reactions were performed in the presence of the 60S ribosomes and the above mutants. Data is representative of three independent experiments. e Superposition of the crystal structure of UFC1 T106A (red) with the WT protein (PDB 2z6o) (blue). The C-alpha of A106 is moved 1.8 Å, relative to the position of C-alpha of T106 in the WT protein. f The crystal structure of the T106A mutant shows that the side chain of E149 adopts two alternative conformations, one of which forms a hydrogen bond with the amide backbone of K108. Source data are provided as a Source Data file.
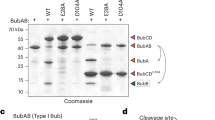
Considering that Ile possesses a larger side chain than Thr, we investigated whether this accounts for the defect in ufmylation seen with the T106I mutant. Accordingly, we mutated T106 to Val, which has a similar size as Thr but cannot participate in hydrogen bonding. We solved the crystal structure of the T106V mutant to 2.0 Å resolution (Supplementary Table 1a and Supplementary Figs. 1d, 2) and found no structural changes, as compared to the WT or T106I proteins (C-alpha RMSDs of 0.332 and 0.349 Å, respectively). Moreover, we did not detect any effect on the thermal stability of the protein (Supplementary Fig. 1c). We next tested the effect of this mutation on ufmylation. For this, we performed an in vitro assay, which probes changes in the ufmylation pattern due to the presence of UFL1 fused to DDRGK122. In contrast to WT UFC1, both the T106I and T106V mutants generated ufmylation patterns reminiscent of that observed in the absence of the DDRGK1-UFL1 fusion protein (Fig. 1b). Specifically, an additional band of ~40 kDa was observed in the presence of WT UFC1 (given the similar sizes of UFC1-(UFM1)2 and DDRGK1-UFL1-UFM1, we consider this band to represent both species; see Supplementary Fig. 1e, f). This band was very weak when the assay was conducted with the T106I mutant and absent using the T106V mutant (Fig. 1b, c). Moreover, the profiles generated by both mutants lacked the smear above the 53 kDa band observed with the WT protein. In parallel, we tested the ability of these mutants to ufmylate RPL26 in an in vitro assay using purified 60S ribosomes. As expected, both mutants prevented RPL26 ufmylation (Fig. 1d). It is of note, no defects in the charging of the UFC1 mutants by UBA5 were seen in the in vitro charging assay (Supplementary Fig. 1i, k, m), indicating that the defect affects the transfer of UFM1 from UFC1 and not the charging process. Overall, the results suggest that the defect associated with the T106I mutant is not due to the larger size of Ile, as compared to Thr.
Recently, Peter et al. reported that ufmylation activity of the UFC1 T106A mutant was hardly affected, relative to that of the WT protein19. This prompted us to assess whether the small size of Ala, as compared to Ile, permits structural changes that would allow the regaining of catalytic activity. We, therefore, solved the crystal structure of UFC1 T106A to 1.21 Å resolution (Supplementary Table 1b and Supplementary Fig. 2). The crystal structure indeed showed changes in the loop harboring A106, as compared to the same loop in the WT protein. Specifically, the C-alpha atom of A106 moved 1.8 Å, relative to the position of the C-alpha atom of T106 in WT UFC1 (Fig. 1e). In parallel, an in vitro ufmylation assay indicated that the T106A mutation damaged ufmylation activity, although considerably less than seen with the T106I/V mutants (Fig. 1b, c). Similarly, a very weak band corresponding to ufmylated RPL26 was detected in the presence of UFC1 T106A (Fig. 1d). Interestingly, T106A substitution increased the thermal stability of the protein by 4 °C (Supplementary Fig. 1c). At the structural level, the side chain of E149 in the T106A mutant adopts two alternative conformations, one forming a hydrogen bond with the amide backbone of K108 (Fig. 1f), which replaces the hydrogen bond formed by the T106 side chain in the WT protein (Fig. 2a). This raises the possibility that the activity of the T106A mutant is linked to the partial recovery of the hydrogen bond with K108, which is absent in the T106I/V mutants.

a UFC1 T106 forms two hydrogen bonds, one with the backbone amide of K108 and one with the side chain of E149. b Super-positioning the crystal structures of UFC1 E149D (yellow) onto WT UFC1 (blue). The loop harboring T106 (residues 103–112) adopts different conformation in the mutant. c The crystal structure of UFC1 E149D shows that the distance between the side chains of T106 and D149 is 7.0 Å, which is too far for hydrogen bonding (indicated by a red dotted line). The N-zeta atom of K108 forms hydrogen bond with the side chain of D149. K108 retains the hydrogen bond with T106, as in the WT. d A representative western blot showing the effect of UFC1 E149I and E149D mutants on in vitro ufmylation (see Supplementary Fig. 3a for protein loading control). e The band corresponding to the ufmylated product (~40 kDa) was quantified for each mutant and normalized against the product obtained in WT protein (data represented as mean ± SD, n = 5 independent experiments). The difference is significant for UFC1 E149D (two‐tailed unpaired Student’s t‐test, p < 0.01) but not for UFC1 E149I. f A representative western blot showing the effect of UFC1 E149I and E149D mutants on RPL26 ufmylation (see Supplementary Fig. 3b for protein loading control). g The band corresponding to the ufmylated RPL26 (RPL26-UFM1) was quantified for each mutant and normalized against RPL26-UFM1 obtained with WT protein (data represented as mean ± SD, n = 3 independent experiments). The difference is significant for UFC1 E149D (two‐tailed unpaired Student’s t‐test, p < 0.05) but not for UFC1 E149I. h Crystal structure for UFC1 T106S. The T106S mutant forms a hydrogen bond with the backbone N of K108. i A representative western blot showing the effect of the UFC1 T106S and T106C mutants on in vitro ufmylation (see Supplementary Fig. 3d for protein loading control). j The band corresponding to the ufmylated product (~40 kDa) was quantified for each mutant and normalized against the product formed in WT protein (data represented as mean ± SD, n = 5 independent experiments). The difference is significant for the T106C (p < 0.0001) but not for the T106S mutant in two‐tailed unpaired Student’s t‐test. k Western blot showing the effect of UFC1 T106S and T106C on RPL26 ufmylation (see Supplementary Fig. 3e for protein loading control). Data is representative of two independent experiments. Source data are provided as a Source Data file.
UFC1 T106 forms two hydrogen bonds, one with the backbone amide of K108 and one with the side chain of E149 (Fig. 2a). To elucidate the structural and functional roles of the hydrogen bond between T106 and E149, we generated the E149D mutant. This mutant has a shorter side chain than Glu yet retains the negative charge. The thermal stability of this mutant was reduced by 7.5 °C (Supplementary Fig. 1c). We determined the crystal structure of this mutant to 1.96 Å resolution (Supplementary Table 1b and Supplementary Fig. 2). Super-positioning the C-alpha atoms of the E149D mutant onto the WT protein yielded an RMSD of 0.729 Å, suggesting no major changes. However, local structural changes were clearly observed in the loop harboring T106 (residues 103–112). Specifically, the C-alpha atoms of T106 and K108 had moved 3.4 and 3.0 Å, respectively, relative to their positions in the WT protein (Fig. 2b). Accordingly, the distance between the T106 and D149 side chains is 7.0 Å, a length that is not amenable for hydrogen bonding (Fig. 2c). However, the position of the K108 side chain in the E149D mutant permits hydrogen bonding between its N-zeta atom and the side chain of D149 that is not present in the WT protein (Fig. 2c). Importantly, the above structural changes did not prevent hydrogen bond formation between the T106 side chain and the amide group of K108, a bond found in the WT protein. The ufmylation activity of the E149D mutant showed a reduction in the intensity of the 40 kDa band and the disappearance of the upper smear (Fig. 2d, e). Accordingly, this mutation diminished RPL26 ufmylation (Fig. 2f, g), although no defect in UFC1 charging was observed (Supplementary Fig. 1j, m). This suggests that the presence of a small amino acid at position 149 (i.e., Asp vs. Glu) leads to conformational changes that harm ufmylation, despite retaining the hydrogen bond between T106 and the amide group of K108.
Since the structural changes described above could be due to the smaller size of Asp, relative to Glu, we mutated E149 to an Ile, which has a large side chain, yet cannot form hydrogen bonds. This change reduced the thermal stability of the protein by 2.5 °C, which is less than the effect of the E149D substitution (Supplementary Fig. 1c). We solved the UFC1 E149I crystal structure to 2.03 Å resolution (Supplementary Table 1b and Supplementary Fig. 2). Super-positioning the C-alpha atoms from this mutant onto the WT protein yielded an RMSD of 0.281 Å, reflecting the absence of conformational changes in the loop harboring T106 (Supplementary Fig. 3c). Interestingly, the mutation did not affect the level of ufmylated RPL26 (Fig. 2f, g). Moreover, in the in vitro ufmylation assay, we found an increase in the intensity of the 40 kDa band and the upper smear associated with the mutant, as compared to the WT protein (Fig. 2d, e). Overall, our results show that the hydrogen bond between T106 and E149 is not essential for protein activity and can be replaced by a bulky residue, such as Ile, which is not amenable to hydrogen bonding. This implies that the defect in UFC1 T106I, which cannot form a hydrogen bond with E149, is unlikely to be due to the absence of this bond.
To determine the importance of the hydrogen bond that T106 forms with the backbone nitrogen of K108, we mutated T106 to Ser, which has the ability to form hydrogen bonds. This substitution increased the thermal stability of the protein by 4 °C (Supplementary Fig. 1c). We solved the crystal structure of UFC1 T106S to 1.11 Å resolution (Supplementary Table 1a and Supplementary Fig. 2). Super-positioning the C-alpha atoms in this variant onto WT UFC1 yielded an RMSD of 0.274 Å, suggesting no structural differences. Accordingly, as in the WT protein, UFC1 T106S forms a hydrogen bond with the backbone amide of K108 (Fig. 2h). In contrast to the T106I/V variants considered above, UFC1 T106S showed ufmylation activity that was equivalent to that of the WT protein (Fig. 2i, j), as well as RPL26 ufmylation (Fig. 2k). To further support the claim that this hydrogen bond is essential for activity, we next substituted T106 with a Cys. Although Cys and Ser differ by only one atom, namely, sulfur instead of oxygen, respectively, the former rarely acts as an acceptor in hydrogen bonding, unlike the latter. As with Ser substitution, the presence of a Cys increased the protein thermal stability by 3 °C (Supplementary Fig. 1c). We solved the crystal structure of UFC1 T106C to 1.43 Å resolution (Supplementary Table 1a and Supplementary Fig. 2). When the C-alpha atoms were super-posed onto the WT protein, an RMSD of 0.276 Å was obtained, reflecting no structural changes. However, in contrast to the T106S variant, the T106C change significantly reduced ufmylation activity and prevented RPL26 ufmylation (Fig. 2i–k). Overall, our data suggest that the hydrogen bond between T106 and K108 is critical for UFC1 function.
The T106-K108 hydrogen bond restricts movement of the K108 C-alpha
To understand why the hydrogen bond between the T106 side chain and the amide of K108 is important for activity, we asked whether it affects K108 mobility. We thus normalized the B-factor of the K108 C-alpha (see Methods for calculation). In WT UFC1 (PDB 2z6p) or the T106S mutant, which behaves as does the WT protein and retains the hydrogen bond, the B-factors of the K108 C-alpha are very similar (1.74 and 1.67 standard deviations (SDs) above average, respectively). However, in those UFC1 mutants (T106V/L/C) that showed reduced activity and lacked the hydrogen bond in question, the B-factor values of the K108 C-alpha are significantly higher (2.58, 2.33 and 2.66 SD above average, respectively) (Fig. 3a). Since the T106I mutant crystallized in a different P 21 21 21 space group than did the other mutants (P 41 21 2), we excluded it from the B-factor calculation to avoid crystal packing effects. Taken together, our results suggest that the hydrogen bond linking T106 with the K108 amide restricts the movement of the K108 C-alpha.

a B-factor analysis of the K108 C-alpha in UFC1 WT and mutants. b A representative western blot showing the effect of the UFC1 T106I/W145H double mutant on in vitro ufmylation, in comparison to the T106I or W145H mutants (see Supplementary Fig. 4h for protein loading control). c The band corresponding to the ufmylated product (~40 kDa) was quantified for each mutant and normalized to the product formed in WT protein (data represented as mean ± SD, n = 4 independent experiments). The difference was significant for the T106I/W145H double mutant (p < 0.0001, two-tailed unpaired Student’s t-test) when compared with the T106I mutant. The difference was significant for the W145H mutant when compared with WT (p < 0.01, two-tailed unpaired Student’s t-test). d Super-position of UBCH5B charged with Ub (PDB 3a33) onto the structure of UFC1. UFC1 active site C116 is super-imposed onto S95 of UBCH5B (the presence of a serine instead of a cysteine at this position allows the formation of an oxy-ester bond with Ub (white)). The C-terminal carboxyl of Ub faces the C-alpha of UFC1 K108, allowing C-alpha hydrogen bonding. e Mimicry of the oxyanion intermediate through a Cys to Glu replacement. The side chain of Glu places a negatively charged oxygen at a distance from the Glu C-alpha that resembles the distance between the C-alpha of the active site Cys and the negatively charged oxygen of the Ub/UBL carbonyl during the nucleophilic attack. f Crystal structure of UFC1 C116E showing the hydrogen bond between the Glu side chain and the C-alpha of K108; and the distance between the Glu side and the backbone amide of M109. g B factor analysis of M109 C-alpha in the indicated mutants. The presence of E116 reduces the B-factor of M109 C-alpha. h Crystal structure of UFC1 T106I/C116E showing the distance between K108 C-alpha and E116 side chain oxygen. i B-factor analysis of the K108 C-alpha in the indicated UFC1 mutants.
Restriction of K108 C-alpha movement is required for UFC1 activity
To further support our hypothesis that the defect in the UFC1 T106I mutant is at least partially due to the unrestricted movement of the K108 C-alpha, we explored the impacts of additional mutations outside the 106 position that might influence K108 mobility. Similar to the hydrogen bond with the amide of K108, the hydrogen bond with the carbonyl of K108 could also influence C-alpha mobility. In UFC1, the side chain of W145 forms a hydrogen bond with this carbonyl (Supplementary Fig. 4a). We, therefore, asked how replacing W145 with Phe would affect C-alpha mobility and UFM1 activity. We solved the crystal structure of UFC1 W145F at 1.79 Å resolution (Supplementary Table 1c and Supplementary Fig. 2). As expected, the B-factor of the K108 C-alpha in this mutant is very high, i.e., 3.2 SDs above average (Fig. 3a). Hence, similar to the T106I mutant, the W145F replacement significantly impaired UFC1 activity (Supplementary Fig. 4b, c, e). This further supports the idea that K108 C-alpha mobility impacts activity.
We next investigated whether we could restore the activity of UFC1 T106I by restricting K108 C-alpha movement. To that end, we substituted W145 with His, which can form a stronger hydrogen bond with the carbonyl of K108. As shown in Fig. 3b, c, we observed increased activity with the T106I/W145H double mutant, as compared to the single T106I substitution. Specifically, we observed enhancement of the 40 kDa band in the double mutant, as compared to the single T106I mutant. Interestingly, W145H substitution alone significantly impaired UFM1 activity (Fig. 3b, c). To investigate this unexpected observation, we solved the crystal structure of UFC1 W145H to 2.02 Å resolution (see Supplementary Table 1c and Supplementary Fig. 2). Structural analysis showed that the distance between the H145 side chain and the carbonyl of K108 is 4 Å, as compared to 2.96 Å in the WT protein with W145, thereby weakening the hydrogen bonding (Supplementary Fig. 4g). Our attempts to obtain structural data for the T106I/W145H double mutant were unsuccessful, leaving the question of how this mutation improves ufmylation activity in the context of the T106I mutation unresolved. One possibility is that, in the context of T106I, H145 forms a hydrogen bond with the carbonyl of K108, thereby stabilizing it. On the other hand, we cannot rule out the possibility that the defect in W145H arises from the difference in hydrophobicity between tryptophan and histidine, with the latter being significantly less hydrophobic. However, the presence of the T106I mutation, which increases hydrophobicity, counteracts the effect of W145H, thus restoring activity.
Stabilization of the oxyanion involves K108 C-alpha hydrogen bonding
UFC1 K108, a constituent of the TAK motif, overlaps with the proposed oxyanion-stabilizing Asn of the HPN motif in other E2 enzymes, suggesting a comparable role in UFC119. This led us to investigate whether K108 could contribute to oxyanion stabilization. Accordingly, we considered whether the TAK motif could be positioned at a distance conducive to hydrogen bonding with the C-terminal carbonyl of UFM1 when the latter is linked to the active site Cys of UFC1. Presently, the only crystal structures of UFM1 linked to UFC1 available include an isopeptide bond (PDB 8bzr)23, which is considerably longer than the native thioester bond, and, therefore, cannot be used to address our goal. As such, we superimposed the structure of UFC1 onto the structure of the E2 enzyme UBCH5B charged with Ub via an oxyester bond (PDB 3a33)24. In the model, the C-alpha of K108 is positioned 3.4 Å away from the C-terminal carbonyl of Ub. The nature of the C-alpha carbon in proteins polarizes the hydrogen, making it suitable to act as a donor in hydrogen bonding25. In UFC1, after adding the hydrogen to the K108 C-alpha, the distance to the C-terminal carbonyl of Ub becomes 2.4 Å with an angle of 159°, consistent with the known characteristics of hydrogen bonding26 (Fig. 3d).
To date, structural studies of the mechanisms of oxyanion stabilization in E2 enzymes required their charging with Ub/UBL via a thio- or oxy-ester bond. This enables determination of the position of the Ub/UBL carbonyl, relative to the E2 enzyme, thus facilitating the study of residues contributing to the stabilization mechanism. However, using such structures introduces caveats. These include the inability to account for the negative charge of the oxyanion and the susceptibility to crystal packing effects, which can affect the position of the Ub/UBL globular domain and subsequently influence the conformation of the C-terminus27. Given these considerations, we took an alternative approach, namely, one that involves a mimic of the oxyanion intermediate. As depicted in Fig. 3e, the side chain of Glu places a negatively charged oxygen at a distance from the Glu C-alpha similar to that between the C-alpha of the active site Cys and the oxygen of the Ub/UBL carbonyl. With this in mind, we purified and solved the crystal structure of UFC1 C116E to 1.92 Å resolution (Supplementary Table 1d and Supplementary Fig. 2). In the structure, the oxygen of the Glu side chain approaches the C-alpha of K108 with a distance of 3.76 Å, or a distance of 2.8 Å from the C-alpha hydrogen, indicating potential hydrogen bonding (Fig. 3f). Of note, super-positioning UFC1 C116E onto the structure of UBCH5B~Ub clearly showed how the Glu side chain mimics the C-terminal Gly of Ub attached to the active site Cys (Supplementary Fig. 4i). In addition to the C-alpha hydrogen bond, our structure suggests that the amide of M109, located 4 Å from the oxygen of the Glu side chain, also plays a role in oxyanion stabilization by forming hydrogen bonds (Fig. 3f). To support the involvement of M109 in oxyanion stabilization, we asked whether the B-factor of M109 decreases in UFC1 presenting the C116E mutation due to hydrogen bonding. Indeed, B-factor analysis reveals that in the structure of UFC1 T106S with the oxyanion-mimetic mutation C116E, there is a reduction in the B-factor of the M109 C-alpha and the backbone nitrogen, as compared to the structure with only the T106S mutation (Fig. 3g). This finding supports the possibility that hydrogen bonding involving the amide of M109 also contributes to oxyanion stabilization.
To address whether T106I substitution prevented the formation of a C-alpha hydrogen bond with the oxyanion, we solved the crystal structure of UFC1 T106I/C116E to 1.77 Å resolution (Supplementary Table 1d and Supplementary Fig. 2). Super-positioning of this double mutant onto the structures of UFC1 T106I, UFC1 C116E, or WT UFC1 revealed no discernible structural changes, with the C-alpha atoms yielding RMSD values of 0.314, 0.116, and 0.309 Å, respectively. Similar to the C116E single mutant, we noted that in the double mutant, the distance between the K108 C-alpha and the side chain oxygen of E116 is 3.5 Å, suggesting the feasibility of a hydrogen bond (Fig. 3h). However, the B-factor of the K108 C-alpha in the double mutant was significantly higher than in the single mutant (Fig. 3i). Specifically, while the B-factor of K108 C-alpha is 1.26 SDs above average in the C116E mutant, it is 2.55 SDs above average in the T106I/C116E double mutant. This difference is due to the absence of the hydrogen bond between T106I and the amide of K108, which could have stabilized the movement of the C-alpha atom. Indeed, in the crystal structure of UFC1 T106S/C116E (Supplementary Table 1d and Supplementary Fig. 2), where the hydrogen bond between the S106 side chain and the backbone amide of K108 is retained, the measured B-factor for K108 C-alpha was 1.25 SDs above average, similar to the value obtained for the C116E single mutant (Supplementary Table 2). Overall, our findings indicate that both UFC1 K108, via C-alpha hydrogen bonding, and the hydrogen bond mediated by the amide of M109 play a role in forming the oxyanion hole. However, this oxyanion stabilization is disrupted in UFC1 variants with mutations at T106 that prevent hydrogen bonding with the K108 amide.
While our data suggest that the K108 C-alpha is part of the oxyanion hole, it remains unclear whether the side chain is also required. To address this, we tested the activity of the UFC1 K108M mutant, which lacks the positive charge but retains a similar size, and the K108R mutant, which preserves the positive charge. As shown in Fig. 4a, these mutations disrupted ufmylation activity, including the intensity of the band corresponding to mono-ufmylated UFC1, suggesting that K108 is critical for activity. However, since K108 can also serve as a ufmylation site, we tested the ability of these mutants to ufmylate RPL26 using purified 60S ribosomes. As shown in Fig. 4b, these mutants were unable to ufmylate RPL26, confirming that the side chain of K108 is critical for activity.

a Western blot showing the effect of K108 mutants of UFC1 (K108A, K108M, K108R) on in vitro ufmylation assay (see Supplementary Fig. 5a for protein loading control). b Western blot showing the effect of K108A, K108M, and K108R mutants on RPL26 ufmylation. Ufmylation reactions were performed in the presence of the 60S ribosomes and the above mutants. (Supplementary Fig. 5b for protein loading control). Data in a, b are representative of three independent experiments. Source data are provided as a Source Data file. c B-factor analysis for the C-alpha atom in position 108 for WT UFC1 (2z6p) and the indicated mutants. d The UBC9 cleft next to the active site is shown in the structure of UBC9-SUMO2-RANGAP1 (PDB 5d2m). This cleft facilitates the entry of the lysine from one end and the C-terminus of SUMO2 from the other. UBC9 is depicted in yellow using a surface representation. K524 of RANGAP1 is colored green and is linked to the C-terminal glycine of SUMO2 (gray). e Superposition of UFC1 (blue) and UBC9 (yellow) from the structure, depicted in panel d, shows that the side chain of K108 lines the top of the cleft. In UBC9, this region is formed by a loop that is absent in UFC1. Therefore, the K108 side chain is critical for generating a proper cavity to accommodate the attacking lysine and the C-terminus of UFM1. f As in panel e, but for simplicity, UBC9 is not shown. UFC1 is displayed in surface representation, allowing better visualization of the cleft adjacent to the active site and the role of K108 in shaping it. g and h Superposition of WT UFC1 with the UFC1 mutants K108R (g) and K108M (h). The residue at position 108 is shown in the stick representation. In both mutants, the residue enters the cleft and narrows its size. i Superposition of UFC1 K108A/C116E (shown in green) with WT UFC1. K108 of WT UFC1 is shown in stick representation colored blue. The short side chain of A108 prevents the formation of the top of the cleft.
Next, to investigate the defect in these mutants, we solved the crystal structures of UFC1 K108R and UFC1 K108M at 1.90 and 2.05 Å resolution, respectively (Supplementary Table 1c and Supplementary Fig. 2). Superpositioning of these structures onto the wild-type UFC1 structure (PDB 2z6o) yielded RMSDs of 0.2967 and 0.2870 Å, respectively, indicating no significant conformational changes caused by the mutations. However, B-factor analysis revealed that the C-alpha of R/M108 displayed high values, suggesting that these mutations prevent the C-alpha at position 108 from participating in hydrogen bonding with the oxyanion (Fig. 4c). Since the hydrogen bond between T106 and the amide of M/R108 is preserved in the structures of these mutants, we hypothesized that the high B-factor for the C-alpha at position 108 is due to the side chain of Met or Arg, which could impact the C-alpha B-factor. To cancel the potential effect of the side chain on the C-alpha B-factor, we tested the activity of the UFC1 K108A mutant, which has only a methyl group at the side chain. Similar to the other mutations at position 108, this mutation prevented activity (Fig. 4a, b). Then, to confirm that this mutation does not affect the C-alpha B-factor, we solved the crystal structure of UFC1 C116E/K108A to 1.88 Å resolution (Supplementary Table 1d and Supplementary Fig. 2). Indeed, the C-alpha B-factor of A108 was less than the value obtained for K108 in the WT protein (Fig. 4c), suggesting that stabilizing the C-alpha at position 108 alone is insufficient for activity and that the K108 side chain is needed for activity.
To further explore the importance of the K108 side chain, we asked whether it is required for orienting the attacking lysine and the UFM1 tail for a productive reaction. To this end, we superimposed UFC1 onto the structure of UBC9,SUMO2-RANGAP1 with the SUMO E3 ligase ZNF451 (PDB 5d2m)28. In this structure, the lysine of RANGAP1 is linked to the C-terminal glycine of SUMO2 via an isopeptide bond, with this linkage being located next to the active site cysteine of UBC9. In the structure, UBC9 possesses a cleft next to the active site that allows the entrance of the lysine from one end and the C-terminus of SUMO2 from the other end (Fig. 4d).
The top of the cleft is formed by residues located on a loop that is absent in UFC1. However, the cleft is maintained in UFC1 because the K108 side chain compensates for the missing loop and lines the top of the cleft (Fig. 4e, f). This suggests that the side chain of K108 plays a role in forming the cleft, enabling the substrate lysine to attack and form an isopeptide bond. Indeed, superpositioning of the UFC1 K108R and K108M mutants, in which activity is prevented, clearly shows that the side chains of these residues enter the cleft and narrow their size (Fig. 4g, h). In contrast, in the UFC1 K108A/C116E structure, the top of the cleft is missing (Fig. 4i), potentially allowing greater flexibility of the UFM1 tail, thereby preventing the reaction. Overall, our results suggest that the K108 side chain plays a critical role in forming the cleft adjacent to the active site, which is necessary for holding the attacking Lys and the UFM1 C-terminal tail in position for a productive reaction.
UFC1 possesses an additional oxyanion hole that only enables intramolecular transfer
While our data suggest that UFC1 T106I exhibits a defect in the oxyanion stabilization mechanism, we, nonetheless, observed robust mono-ufmylation of UFC1 T106I (Fig. 1b). This prompted us to search for an additional mechanism of oxyanion stabilization, one potentially independent of the TAK motif. Previously, Hamilton et al. determined the structure of Ubc1 bound with Ub (Ubc1~Ub) using NMR29. In this structure, the position of the Ub carbonyl is unsuitable for oxyanion stabilization via Asn 79 of the HPN motif. Instead, the carbonyl is directed towards the backbones of Asp 90 and Leu 89 (Supplementary Fig. 6a). Specifically, the carbonyl of Ub is positioned at a distance of 3 Å from both the L89 amide and the C-alpha hydrogen, enabling hydrogen bonds. Additionally, the amide of D90 is located at a distance of 2.6 Å from the Ub carbonyl, which can further contribute to oxyanion stabilization. Super-positioning this structure onto that of UFC1 C116E clearly revealed distinct positioning of the carbonyl, which is no longer within a distance conducive to hydrogen bonding with K108 C-alpha (Fig. 5a). In this superposition, the carbonyl group of Ub is oriented toward UFC1 L117 and T118 (the equivalent residues of L89 and D90 in UBC1), enabling hydrogen bonding with these residues (Fig. 5a). This supports the presence of an additional oxyanion hole that is TAK-independent and which remains unaffected in the T106I mutant.

a Super-positioning UFC1 C116E (blue) onto the structure of Ubc1 charged with Ub (pink) (PDB 1fxt). For simplicity, Ubc1 is omitted, with only its active site C88, and the linked Ub is shown. The Ub G76 carbonyl adopts a different conformation, relative to the UFC1 E116 side chain and is oriented toward the L117 and T118 C-alpha atoms. b A representative western blot showing the levels of mono-ufmylated WT UFC1 and UFC1 T106I in an in vitro ufmylation assay without E3 (See Supplementary Fig. 6b for protein loading controls). c The band corresponding to mono-ufmylated UFC1 was quantified for the T106I mutant and was normalized against mono-ufmylated UFC1 formed in WT protein (data represented as mean ± SD, n = 4 independent experiments). The difference was significant (p < 0.001, one‐tailed unpaired Student’s t‐test). d and e. A western blot showing the effect of WT UFC1 and the T106I mutant on in vitro ufmylation of UFC1 C116A. Blots are shown with anti-FLAG antibody as UFM1 has a FLAG-tag (d) and with anti-His antibody against Trx-UFC1 C116A that has a His-tag (e) (for protein loading controls see Supplementary Fig. 6c, d respectively). f Western blot of an in vitro ufmylation assay in the absence of E3, comparing the levels of mono-ufmylated UFC1 in the indicated mutants (see Supplementary Fig. 6e for protein loading controls). g Western blot of an in vitro ufmylation assay in the presence and absence of DDRGK1-UFL1. Only UFC1 WT and K122R in the presence of DDRGK1-UFL1 show an additional band of ~40 kDa (see Supplementary Fig. 6f for protein loading controls). Data in d, g and e, f are representative of two and three independent experiments, respectively. Source data are provided as a Source Data file.
Interestingly, mono-ufmylation of UFC1 T106I or the WT protein can also occur without the E3 fusion protein (Fig. 5b). Moreover, in the absence of the E3 fusion protein, the T106I mutant showed more mono-ufmylation than did the WT protein (Fig. 5c). E3 ligase-independent ufmylation raises the possibility of intramolecular nucleophilic attack wherein UFM1 is transferred to a UFC1 lysine residue via a cis mechanism. To support the possibility of a cis mechanism, we first excluded the possibility of a trans mechanism. Specifically, we wanted to show that the T106I mutation prevents ufmylation via a trans mechanism. Accordingly, we conducted a ufmylation assay using catalytically inactive UFC1 C116A, which can only undergo ufmylation through a trans mechanism. Specifically, we investigated whether the latter can be ufmylated by WT UFC1 or UFC1 T106I. As illustrated in Fig. 5d, e, both WT UFC1 and the T106I mutant failed to ufmylate UFC1 C116A in the absence of E3 fusion protein. However, in the presence of the E3 ligase, only WT UFC1 facilitated ufmylation of UFC1 C116A. This suggests that charged UFC1 T106I is not susceptible to a trans nucleophilic attack, and cannot transfer UFM1 to target proteins. Overall, our data argue that UFC1 possesses one oxyanion hole that relies on K108 C-alpha hydrogen bonding, which is essential for trans-ufmylation, and another that is K108-independent and suitable for intramolecular transfer.
To further investigate the TAK-independent oxyanion, we examined which UFC1 lysine residues undergo ufmylation via this oxyanion hole. UFC1 contains 14 lysine residues, but two—K108 and K122—are located near the active site cysteine (C116), at distances of 8 and 6 Å, respectively. Initially, we tested ufmylation of UFC1 carrying the K108R, K122R, or both substitutions in the absence of E3, so as to focus on cis-ufmylation. The combination of both mutations significantly reduced ufmylation, suggesting that these residues are the primary sites for UFC1 cis-ufmylation (Fig. 5f). Next, we asked whether the presence of the T106I mutation alters the ufmylation site. To investigate this, we tested the above mutations in the context of the T106I mutation. Interestingly, a comparison of the K108R single mutant with the K108R/T106I double mutant revealed mono-ufmylated UFC1 levels to be increased in the presence of the double mutant (Fig. 5f). Additionally, in the presence of the T106I mutation, UFC1 carrying the K108R and K122R substitutions showed increased mono-ufmylation, as compared to the K108R/K122R double mutant without T106I. This suggests that, in the presence of T106I, cis-ufmylation is enhanced on another lysine residue of UFC1, probably K114, located in the active site cysteine loop.
Recently, Millrine et al. reported that UFC1 undergoes ufmylation on Lys122, which serves as a mechanism to regulate UFC1 activity30. Since our data show that in the presence of T106I there is more cis-ufmylation, we asked whether this could account for the defect in UFC1 T106I. Mutations at the K108 site, which prevent it from serving as a ufmylation site, also abolish trans-ufmylation activity. Therefore, mutating K108 eliminates activity entirely. However, whether mutation at the K122 site has a similar effect on trans-ufmylation is unclear. As shown in Fig. 5g, in contrast to the K108R mutation, the K122R mutation did not disrupt the ufmylation pattern observed in our in vitro assays with E3, suggesting that K122 is not essential for activity. This allowed us to test whether the T106I mutant regains trans-activity in the presence of K122R, as it cannot undergo ufmylation at that site. Similar to the T106I single mutant, the double mutant did not show ufmylation activity beyond -ufmylation of UFC1 (Fig. 5g). This suggests that the defect in T106I is not due to increased ufmylation of K122, but is rather likely due to the presence of a non-functional oxyanion hole.
An HPN-dependent oxyanion hole requires conformational changes
Interestingly, although E2 enzymes other than UFC1 contain an HPN motif, and not a TAK motif, a comparable hydrogen bond as described above, is observed31. In the HPN motif, the histidine side chain forms a hydrogen bond with the asparagine backbone, similar to the hydrogen bond between UFC1 T106 and K108 (Fig. 6a). This led us to address whether C-alpha-oxyanion stabilization also applies to E2 enzymes possessing the HPN motif. To test this possibility, we used the structures of E2 enzymes charged with Ub/UBL via a thio- or oxy-ester bond. Specifically, we asked whether the carbonyl group of Ub/UBL is positioned at a distance conducive to hydrogen bonding with the Asn C-alpha of the HPN motif. In the structure of UBCH5B linked with Ub, the distance of the C-alpha of Asn77 to the Ub carbonyl is 4.1 Å, while in the structure of Ubc13~Ub, the equivalent distance is 5.9 Å (Supplementary Fig. 7a, b). Interestingly, in the latter structure, the distance of the Ub carbonyl to the proposed oxyanion stabilizer, namely, the side chain of Asn, is 4.9 Å, a distance also too far for hydrogen bonding27. While the structures described above do not directly demonstrate this mode of stabilization, we cannot rule out the possibility that small conformational changes may enable it to occur upon Lys attack. It is well established that interaction with an E3 ligase restricts the highly dynamic E2-Ub/Ub complex, promoting a productive closed conformation that positions the oxyanion intermediate near the stabilizing Asn7. Indeed, in the structure of the SUMO E3 ZNF451 together with UBC9-SUMO2-RANGAP1, one that captures the configuration after isopeptide bond formation, the distance between the SUMO C-terminal carboxyl and the Asn75 C-alpha is 3.5 Å28.

a Super-positioning of His and Asn in the HPN motif of different E2 enzymes, specifically, UBC9 (PDB 1a3s), E2-25K (PDB 2bep), UBC7 (PDB 2cyx), Rad6 (PDB 2yb6), and E2S (PDB 1zdn), shows a conserved hydrogen bond between the side chain of His and the backbone nitrogen of Asn. b–d Super-positioning of the crystal structures of UBC9 C93E, Ubc13 C87E, and UBCH5B C85E onto the corresponding WT structure of each of these E2 mutants charged with Ub/UBL (PDBs; 5d2m, 2gmi and 3a33; Ub/UBL is omitted from the figure for simplicity). The conformation of UFC1 E116 (side chain shown in stick representation (blue), with van der Waals surface) super-imposed onto the corresponding Glu in each E2 enzyme. e Western blot showing the effect of UFC1 (HPN) on ufmylation of ribosomal protein RPL26. Ufmylation reactions were performed in the presence of the 60S ribosomes and UFC1 (HPN) (see Supplementary Fig. 7d for protein loading controls). f Western blot showing the effect of UFC1 (HPN) on in vitro ufmylation. UFC1 (HPN) in the presence of DDRGK1-UFL1 did not generate ufmylated product (~40 kDa) (See Supplementary Fig. 7e for protein loading controls). g Western blot showing the effect of UFC1 (HPN) on the ufmylation of UFC1 C116A in trans. See Supplementary Fig. 7f for the protein loading controls. Data in e, f are representative of two independent experiments. Data in g is performed once. Source data are provided as a Source Data file. h Superposition of UBC9 with UFC1 (HPN). For simplicity, only the HPN motif of UBC9 is shown (yellow), along with the helix corresponding to the long helix in UFC1 (blue). While Pro and Asn in the HPN motif of UFC1 (HPN) adopt similar positions as in the UBC9 HPN motif, the His in UFC1 (HPN) adopts a different orientation compared to the His in the UBC9 HPN motif. This difference is likely due to the longer helix in UFC1 that prevents His from reaching the right position.
To further investigate the role of C-alpha hydrogen bonding in oxyanion stabilization within E2 enzymes possessing the HPN motif, we employed our approach of mimicking the oxyanion intermediate through a Cys-to-Glu mutation in the active site. As such, we purified and solved the crystal structure of the UBC9 C93E, Ubc13 C87E, and UBCH5B C85E E2 enzymes (Supplementary Table 1e and Supplementary Fig. 2). Super-positioning each of these E2 enzyme mutants onto the apo WT structure revealed no distinct structural changes (C-alpha atoms RMSDs of 0.438, 0.289, and 0.308 Å, respectively), indicating that the changes introduced did not affect the overall structure of the enzyme. Surprisingly, in all three structures, the side chain of the Glu adopted a conformation unsuitable for hydrogen bonding with the Asn C-alpha (Fig. 6b–d).
We next asked why the Glu side chain in the structures of the E2 enzymes possessing the HPN motif adopts a conformation that does not allow C-alpha hydrogen bonding (Fig. 6b–d). This prompted us to explore whether these E2 enzymes undergo conformational changes upon charging, thereby allowing the carbonyl to position correctly. To test this possibility, we super-imposed the structure of UFC1 C116E onto the three E2 enzymes described above and tested whether the conformation of the Glu was accessible in these E2 structures, as in UFC1 (Fig. 6b–d). Such analysis revealed that the loop adjacent to the active site, present in these E2 enzymes but absent in UFC1, hinders the orientation of the Glu side chain necessary for forming the aforementioned C-alpha hydrogen bond. While we found clashes with the Glu in apo UBCH5B and apo Ubc13, repulsions between D127 and the Glu may prevent the desired conformation in UBC9 (Fig. 6b–d). However, when we super-imposed the UFC1 C116E structure onto the E2 enzyme structures charged with Ub/SUMO, the orientation of the loop no longer affected the position of the Glu, such that hydrogen bonding with Asn C-alpha was feasible. Overall, our data align with the concept that oxyanion stabilization by C-alpha hydrogen bonding can also occur in E2 enzymes with the HPN motif, although a structural rearrangement is required.
We next explored whether the TAK motif in UFC1 could be replaced with the HPN motif. As such, we purified UFC1 containing HPN at positions 106–108 (referred to as UFC1(HPN)). Initially, we tested the ability of this mutant to ufmylate RPL26. As shown in Fig. 6e, UFC1(HPN) failed to ufmylate RPL26 in an in vitro assay. However, a reduced level of mono-ufmylated UFC1(HPN) was observed (Fig. 6f), suggesting that the TAK-independent oxyanion hole retains some level of functionality in UFC1(HPN). We next tested whether UFC1(HPN) could ufmylate the UFC1 C116A mutant, which can only occur in trans. As expected, while UFC1 WT successfully ufmylated UFC1 C116A, UFC1(HPN) failed (Fig. 6g). This suggests that UFC1 (HPN) is not suitable for trans-ufmylation, despite supporting weak cis-ufmylation. Of note, UFC1 (HPN) showed a defect in UFM1-charging (Supplementary Fig. 7c), which, in turn, could reduce the level of cis-ufmylation. In addition, it showed no charging with Ub, suggesting that UFC1 possessing the HPN motif cannot act with the ubiquitin machinery (Supplementary Fig. 7g).
Next, to further understand the defects in UFC1 (HPN), we solved its crystal structure to 1.54 Å resolution (Supplementary Table 1e and Supplementary Fig. 2). Interestingly, the electron density for the HPN motif was poor, suggesting this region as being highly mobile. Accordingly, B-factor normalization for the C-alpha atoms of these residues was more than 4.5 SDs. above average. This contrasts with the HPN motif in other E2 enzymes, where a low B-factor is measured. In the UFC1 (HPN) structure, the H106 side chain is oriented away from N108, in a position that is not suitable for hydrogen bonding with the backbone nitrogen of N108 (Fig. 6h). This suggests that the HPN motif cannot serve its role in UFC1 since the key hydrogen bond between H106 and K108 is not retained.
To explore whether the structure of UFC1 prevents His106 from adopting the conformation needed for hydrogen bonding with Asn, we super-imposed the structure of the HPN motif from UBC9 onto our structure (Fig. 6h). This superposition revealed that the position of the His side chain in UBC9 clashes with the long helix of UFC1. In UBC9, this helix is shorter and turns into a loop, which is missing in UFC1. Overall, our data suggest that the HPN motif introduced into UFC1 cannot function properly due to the longer helix that prevents the His side chain from achieving the right conformation.
Discussion
A substantial challenge in studying enzyme mechanisms often arises when the motif or domain under investigation plays more than one role. Specifically, if mutational analysis interferes with both roles, it becomes difficult to investigate a specific function without the other being affected. This is true for the highly conserved HPN motif in E2 conjugating enzymes, which has been suggested to play both structural and catalytic roles12,13. This dual functionality presents a challenge when exploring if and how the HPN motif contributes to oxyanion stabilization. In this study, we overcame this challenge by focusing on the E2 enzyme UFC1, which allowed us to explore catalytic roles without dealing with structural considerations. We attributed this to two main factors, namely, the presence of a TAK motif instead of an HPN motif, allowing mutagenesis analysis without perturbing structural integrity, and the absence of a loop next to the active site normally found in E2 enzymes18. These attributes allowed us to propose that the TAK motif in UFC1 contributes to the stabilization of the oxyanion intermediate, in part, through C-alpha hydrogen bonding. By comparing the TAK motif to the HPN motif, which was previously proposed to stabilize the oxyanion via the Asn side chain, we found that the HPN structure can also support C-alpha hydrogen bonding, suggesting this mode of stabilization to be more general.
Oxyanion stabilization via conventional hydrogen bonding has been extensively studied32. In these cases, the oxyanion hole is mainly formed by backbone amides that donate hydrogen bonds to the negatively charged oxygen of the intermediate32. Over the last decade, appreciation of the involvement of C-alpha hydrogen bonds in protein structures has significantly increased33. In proteins, the C-alpha carbon is connected to two electron-withdrawing groups, i.e., the amide and carbonyl groups, which activate the C-alpha, enabling it to act as a hydrogen donor34. Here, we proposed that this mode of hydrogen bonding also contributes to the stabilization of the oxyanion. Specifically, the distance of K108 C-alpha from the oxyanion intermediate, combined with its restricted movement, makes it suitable to serve as a hydrogen donor in C-alpha hydrogen bonding. The function of the oxyanion hole is to stabilize an intermediate during the reaction, but not the final product, where the oxygen is no longer negatively charged, thus allowing product release. In this context, having C-alpha hydrogen bonds, which are considered weaker than canonical hydrogen bonds, can be advantageous. Specifically, the relative weakness of the C-alpha as a hydrogen donor is compensated by the strong acceptor properties of the oxyanion, which exists only as an intermediate. Consequently, interactions with the product, where the oxygen is neutral and only polar, are less favorable for this type of C-alpha hydrogen bonding.
Oxyanion holes are formed by two or three hydrogen bond donors oriented towards a central oxygen atom35. This suggests that, in addition to the proposed C-alpha hydrogen bond, other hydrogen bonds are involved. Our structural analysis of UFC1 suggests that hydrogen bonding with the backbone amide of M109 can also contribute to oxyanion stabilization. Another component of the oxyanion hole can be proposed from a recent structure mimicking the oxyanion intermediate during the trans-thiolation reaction between an E2 enzyme and a HECT E3 enzyme. In this structure, the Arg residue in the Ub C-terminal tail is located near the oxyanion, allowing for hydrogen bonding36. Interestingly, this Arg is conserved in UFM1 (R81), and mutation resulting in R81C replacement leads to infantile encephalopathy, similar to the phenotype of the UFC1 T106I mutant21, suggesting that R81 may also contribute to oxyanion stabilization. At the same time, it remains unclear whether the K108 side chain is involved in oxyanion stabilization. Although our data suggest that the K108 side chain is essential for UFC1 activity, its solvation and the lack of movement toward E116 in the structure of the UFC1 oxyanion-mimetic mutant weaken the likelihood of its involvement in oxyanion stabilization.
One possibility is that the side chain of K108 assists in orienting the attacking lysine, making it essential for catalytic activity. Similar to the shallow cleft in the active sites of other E2 enzymes that accommodates the C-terminus of Ub/UBL and positions the attacking lysine, the cleft identified in the active site of UFC1—lined by the backbone of Leu117–Asp119 and the side chain of Lys108—may accommodate the C-terminal tail of UFM1 and position the incoming lysine. The cleft is narrowed in UFC1 T106I and lost its topology in the K108M and K108R mutants, hampering their activities. Although the arginine replacement retains the positively charged long side chain similarly to lysine, the specific orientation of the K108 side chain, essential for designing the cleft, is not seen with Arg. Also, the possible angle strain due to the side chain orientation of Arg/Met108 is attributed to the increased B factor of C-alpha in these replacements. All of these points suggest that the lysine side chain may assist in positioning the attacking lysine. However, further research is needed to fully understand the role of the K108 side chain in catalysis.
A significant contribution to our finding of how UFC1 stabilizes the oxyanion came from the genetic mutation that generates UFC1 T106I21. This mutation, together with others, led us to identify a hydrogen bond between the side chain of T106 and the backbone amide of K108 that is essential for UFC1 functionality. This hydrogen bond, in turn, restricts the movement of the K108 C-alpha, enabling the formation of a hydrogen bond with the oxyanion. Therefore, the defect associated with the T106I mutant can, at least in part, be attributed to the highly mobile K108 C-alpha, which is unable to effectively stabilize the oxyanion. Given this realization, we propose that small molecules that restrict the movement of K108 C-alpha could overcome the defect in the T106I mutant and serve as potential therapeutic agents.
The E2-Ub/UBL complex adopts multiple conformations, whereas interaction with an E3 ligase locks it into a closed conformation suitable for conjugation37,38. In the absence of E3, some E2 proteins undergo modification at specific lysine residues, often serving an auto-inhibitory function39. This highlights the need for more than one oxyanion hole in E2 proteins, given the dynamic nature of the E2-UB/UBL conjugate. Specifically, the varying orientations of the carbonyl oxygen in the thioester complex and the potential positional differences in the approach of the attacking lysine further support this requirement. The genetic mutation that yields UFC1 T106I assisted us in discovering an additional oxyanion hole in UFC1. Specifically, the presence of mono-ufmylated UFC1 T106I raises the possibility of an additional oxyanion hole that operates independently of the TAK motif. This oxyanion hole is formed by the backbone of residues L117 and T118. Additionally, there is an alpha-helix (His120–Val129) in proximity to this proposed oxyanion hole, with the positive dipole end of this helix possibly also contributing to the oxyanion hole structure. This TAK-independent oxyanion hole is suitable for a cis mechanism, enabling intramolecular ufmylation of UFC1, primarily at K122 and K108.
The presence of two oxyanion holes raises questions about how their activity is regulated and what conditions favor one mode over the other. The NMR structure of UFC1 (PDB 2k07) shows that in some models, the active site Cys 116 is closer to the TAK-dependent oxyanion, while in others, the TAK-independent oxyanion approaches the active site Cys. This supports the dynamic nature of this region to allow its functioning with both oxyanion holes. Our data suggest that the presence of the E3 complex decreases the activity of the TAK-independent oxyanion hole that functions in cis, as evidenced by the reduced levels of ufmylated UFC1 T106I in the presence of DDRGK1-UFL1. Conversely, trans-ufmylation via the TAK-dependent oxyanion hole occurs only in the presence of the E3 complex. This suggests that one mode of regulation could involve the level of E3 complex that is available for charged UFC1, with higher levels disfavoring cis-ufmylation.
UFC1 T106I showed higher levels of mono-ufmylation, as compared to the WT (Fig. 5b). This suggests that the TAK-independent oxyanion hole is enhanced in this mutant. However, the primary defect in UFC1 T106I activity is not due to mono-ufmylation but rather because of the loss of a functional TAK-dependent oxyanion.
Previously, Liu et al. proposed an induced-fit model in which UFC1 requires a highly flexible active site to accommodate the C-terminal tail of UFM140. This raises the possibility that non-functional UFC1 mutants (i.e., T106I/V/L) increase the flexibility of the TAK-independent oxyanion hole, thereby enhancing its potency for cis-ufmylation. Indeed, prior to oxyanion formation, the flexibilities of residues L117 and T118 are higher in the non-functional T106V/L mutants, as compared to the functional T106S mutant (Supplementary Table 2), supporting the induced-fit model. Simultaneously, the B-factor of the C-alpha atoms of residues 105–113 (excluding K108) comprising the TAK-dependent oxyanion hole, is lower in the non-functional mutants than in the functional T106S mutant. This reduction in flexibility of the loop harboring the TAK-dependent oxyanion hole diminishes its activity, rendering it less effective. Moreover, since the primary ufmylation sites of the TAK-independent oxyanion hole are K108 and K122, their lower B-factors in the functional mutant, as compared to the non-functional mutant, further reduce their ability to reach and attack the thioester. This, in turn, can result in decreased cis-ufmylation in the WT enzyme.
An intriguing question is how mono-ufmylation of UFC1 affects activity. Our data suggest that mono-ufmylation occurs mainly at K108 or K122, but not simultaneously at both sites. Due to the proximity of these lysine residues to the active site, ufmylation at either site would prevent UFC1 from being charged, thereby blocking its activity and conferring auto-inhibition. Indeed, ufmylation of UFC1 has been reported in cells and proposed as a mechanism for regulating UFC1 activity. In that case, UFC1 undergoes ufmylation at K122, which can be reversed by the UFM1-specific protease UFSP130. Such an auto-inhibitory mechanism is a common feature of E2 enzymes. Auto-ubiquitination at a highly conserved lysine located at the (n + 5) position downstream of the active site cysteine is well studied in other E2 proteins39. Liess et al. reported the intramolecular transfer of ubiquitin to Lys100 from ubiquitin-loaded Cys95 in UBE2S proteins39. Auto-ubiquitination at this lysine prevents further loading of Ub at the E2 active site cysteine in the next cycle of ubiquitination.
Our data further suggest that E2 enzymes with an HPN motif also possess two oxyanion holes, one of which is HPN-dependent, involving hydrogen bonding with the main chain C-alpha atom and with the amide nitrogen of Asn side chain. This oxyanion hole requires conformational changes in the loop near the active site, enabling the terminal carbonyl of Ub/UBL to correctly orient so as to act with this oxyanion hole. Structure determination of E2 proteins with the active site Cys mutated to Glu, an oxyanion mimetic, revealed that the Glu side chain does not align with the HPN oxyanion hole. Instead, it interacts with the backbone of residues at n + 1 and n + 2 positions from the active site cysteine, forming an alternate oxyanion hole, independent of the HPN motif. Interestingly, in these E2s, the Glu side chain adopts a conformation similar to that of the ubiquitin terminal carbonyl linked to UBC1 (PDB 1fxt), allowing it to interact with the HPN-independent oxyanion hole.
Overall, our data suggest that one oxyanion hole is HPN-dependent and requires conformational changes in the loop near the active site, while the other does not rely on such changes. The data also suggest that similar to cis-ufmylation of UFC1 K122 located N + 6 residues downstream of the active site cysteine, ubiquitination at the N + 5 site in E2 enzymes bearing an HPN motif can occur in cis mode via the HPN-independent oxyanion hole. Further studies are needed to explore this mode of oxyanion stabilization, particularly to clarify how the presence of E3 affects ubiquitination via the HPN-independent oxyanion hole.
While our data suggest that the HPN motif can participate in C-alpha hydrogen bonding and function similarly to the TAK motif in oxyanion hole stabilization, our swapping experiment showed that a chimeric UFC1 possessing the HPN motif cannot function in trans-ufmylation. Our structural analysis suggests that the longer helix in UFC1, which is shorter in other E2s and turns into a loop, prevents the His residue from adopting the correct conformation required for hydrogen bonding with the amide of the Asn. Therefore, it cannot stabilize the latter for C-alpha hydrogen bonding with the oxyanion. This implies that in the context of UFC1, the HPN motif is not functional. It is unclear whether the longer helix in UFC1 is the only structural element preventing the HPN motif from functioning properly. The lack of a loop adjacent to the active site in UFC1, along with the role of Lys in the TAK motif for orienting the attacking Lys and the C-terminus of UFM1, further suggests that the HPN motif cannot recapitulate the function of the TAK motif.
Before attacking the thioester bond, the lysine residue needs to undergo deprotonation so as to enhance its nucleophilicity. Yunus and Lima suggested a desolvation mechanism that suppresses the pKa of the attacking lysine and assists in deprotonation41. The desolvation takes place via the replacement of the water-mediated or hydrogen-bonding interactions between the lysine and the bulk solvent, with interactions with side chains of active site residues that line the catalytic groove that holds the lysine in an optimal position. Similarly, the residues lining the identified cleft in UFC1 that possibly accommodate the attacking lysine may suppress the pKa of lysine and assist in deprotonation. Moreover, studies on E2 enzymes have proposed that acidic residues in the loop adjacent to the active site facilitate this process41,42. However, this loop is absent in UFC1, leaving the active site completely solvated. One possibility is that the DDRGK1–UFL1 complex that approaches the active site of UFC1 may provide a microenvironment that desolvates the active site and suppresses the pKa of the lysine. Interestingly, unlike ubiquitin or other UBLs, UFM1 contains an aspartic acid residue at position 80 (four residues upstream of the terminal glycine). This raises the possibility that the proximity of Asp80 to the active site assists in the deprotonation of the attacking lysine. To date, it remains unclear whether the mechanism of lysine deprotonation is influenced by which oxyanion hole is utilized during the reaction.
Taken together, the data presented here provide insights into the complexity of oxyanion intermediate stabilization in E2 enzymes. Through detailed studies of UFC1 and analysis of related findings in other E2 enzymes, we propose that these enzymes possess two structurally distinct oxyanion holes that rely not only on canonical hydrogen bonds but also on C-alpha hydrogen bonds, which are essential for their function. One oxyanion hole facilitates cis-modification of the E2 itself, while the other supports substrate modification in a trans mode (Fig. 7). Overall, our work suggests that oxyanion stabilization in E2 enzymes is a complex process involving different oxyanion holes, each relying on distinct structural components, including C-alpha hydrogen bonding.

In the cis mechanism, a charged UFC1 undergoes self-ubiquitination, where a Lys residue on the surface of UFC1 attacks the thioester bond. This generates an oxyanion, which is stabilized by a dedicated oxyanion hole composed of hydrogen bonding via the backbone of L117 and T118. In the case of trans-ufmylation, where UFM1 is transferred to a Lys residue on a substrate, a different oxyanion hole takes on this role. This oxyanion hole involves hydrogen bonding with the backbone of K108 and M109. Specifically, K108 contributes C-alpha hydrogen bonding with the oxyanion.
Methods
Cloning
Sequences encoding human UBA5, UFM1, DDRGK1207–314-UFL11–200 (DDRGK1-UFL1), DDRGK187–314-UFL11–200 (DDRGK1ext-UFL1), Ubc13, UBCH5B, and UBC9 were cloned into plasmid pET15b, while that of UFC1 was cloned into plasmid pET32a17,22. The UFC1, Ubc13, UBCH5B, and UBC9 point mutants were generated by PCR and Gibson assembly (Gibson assembly master mix, New England Biolabs). All constructs were verified by DNA sequencing. All primer sequences used in this study are listed in Supplementary Table 3.
Protein expression and purification
Wild type and mutant UFC1 variants were expressed and purified as previously described17. The fusion construct DDRGK1-UFL1 or DDRGK1ext-UFL1 was used as the E3 ligase for in vitro ufmylation22. Ubc13, UBCH5B, UBC9 mutants, and all other proteins were expressed in the Escherichia coli T7 express strain (New England Biolabs). The transformed cells were grown in 2xYT and induced with 0.3 mM isopropyl-β-thio-galactoside (IPTG) overnight at 16 °C. The induced cells were harvested by centrifugation at 8000 × g for 15 min. Pellets were resuspended in lysis buffer (50 mM NaH2PO4, pH 8.0, 400 mM NaCl, 10 mM imidazole, and 5 mM β-mercaptoethanol), supplemented with 1 mM phenyl-methyl sulphonyl fluoride (PMSF) and DNase. The cells were disrupted using a microfluidizer (Microfluidics). The lysate was cleared by centrifugation at 29,000 × g for 45 min, with the supernatant being applied to 5 ml His-Trap columns (GE Healthcare). The protein was eluted with a linear imidazole gradient of 15–300 mM in 30-column volumes. Fractions containing the purified protein were pooled and dialyzed against dialysis buffer (25 mM NaH2PO4, pH 8.0, 300 mM NaCl, and 5 mM β-mercaptoethanol) in the presence of TEV protease overnight at 4 °C. Cleaved protein was then subjected to a second round of His-Trap chromatography and the flow-through containing the cleaved protein was collected. Further purification was achieved using HiLoad 16/600 Superdex 75 pg or Superdex 200 pg size-exclusion chromatography columns (GE Healthcare) according to the size of the protein. The purified proteins were concentrated, flash-frozen in liquid N2, and stored at −80 °C.
Purification of 60S ribosomal subunit
60S ribosomal subunit was purified from HEK293T cells grown to ∼80% confluency in T75 flasks with medium containing high-glucose DMEM supplemented with 10% (v/v) FBS, penicillin (100 units),streptomycin (0.1 mg/ml) and 2 mM l-glutamine. The cells were collected in ice-cold PBS and were pelleted by centrifugation at 500 g for 5 min. Next, the cell pellet was resuspended in lysis buffer (containing 25 mM Tris pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1% Triton X-100, 1 mM DTT, RNAsin (60 U), EDTA-free 1× protease inhibitor cocktail) followed by incubation on ice for 10 min. After lysis, nuclei, and mitochondria were removed by two consecutive centrifugations at 800 × g for 5 min followed by centrifugation at 8000 × g for 5 min. Then the supernatant was centrifuged at 17,000 × g for 10 min and the supernatant was collected again. The collected supernatant was layered directly onto a linear 10–30% sucrose gradient containing 25 mM Tris pH 7.5, 150 mM KCl, 15 mM MgCl2, and 1 mM DTT. The 80S, 60S, and 40S were separated by centrifugation at 210,000 × g (38,000 rpm) for 3 h at 4 °C using a SW41 Ti rotor (Beckman Coulter). Gradients were fractionated into 0.75 ml fractions using the BioComp fractionating system. The fractions containing 60S ribosomal subunits were collected and exchanged into buffer containing 20 mM HEPES pH 7.5, 100 mM KCl, 5 mM MgCl2, 2 mM DTT and stored at −80 °C.
Differential scanning fluorimetry
Differential scanning fluorimetry was performed with a StepOnePlus real-time PCR system (Applied Biosystems, Life Technologies) using 96-well plates (Applied Biosystems, Life Technologies) covered with a clear adhesive seal. Assays were performed in a final volume of 25 μL containing 1 μM UFC1 (WT or mutant variant) and 7.5X SYPRO Orange (Invitrogen, Life Technologies) mixed in buffer containing 20 mM Tris–HCl, pH 7.5, 150 mM NaCl. Measurements were recorded at an excitation wavelength of 465 nm and an emission wavelength of 580 nm over a temperature rise of 1% from 25 to 90 °C. Each experiment was carried out at least in triplicate and the average was obtained at each temperature. Data were then plotted using GraphPad Prism 6.0 software and fitted using Boltzmann sigmoidal curve fit.
In vitro UFC1 charging assay
E1 (UBA5 or UBA1) (1 μM), UFM1 or Ub (10 μM), and E2 (UFC1 or UbCH5c) (5 μM) were combined in a buffer containing 50 mM Bis–Tris (pH 6.5) or HEPES (pH 8.0), 100 mM NaCl and 10 mM MgCl2. Protein concentrations were the same in all reactions involving point mutants and truncations unless otherwise specified. Reactions were initiated by the addition of ATP (5 mM) and ran for a specified time at 30 °C. Before activation, an aliquot of the reaction mix was collected and taken as the control at time 0. The reactions were quenched with SDS sample buffer without β-mercaptoethanol, separated by 12% Bis–Tris non-reducing PAGE, and visualized by Coomassie brilliant blue R staining.
In vitro ufmylation assay
UBA5 (0.5 μM), FLAG-UFM1 (6 μM), UFC1 WT or mutants (3 μM), and DDRGK1-UFL1 or DDRGK1ext-UFL1 (3 μM) were incubated in a buffer containing HEPES (50 mM, pH 8.0), NaCl (100 mM) and MgCl2 (10 mM). Protein concentrations were the same in all reactions involving point mutants unless otherwise specified. Reactions were initiated by the addition of ATP (5 mM) and ran for 45 min at 30 °C. The negative control sample was incubated without ATP. After incubation, reactions were stopped by adding an SDS-sample buffer containing β-mercaptoethanol. The samples, along with the control, were then separated by 12% Bis–Tris PAGE followed by immunoblot with anti-FLAG (Merck, F1804) to detect ufmylated products (as UFM1 has a FLAG tag).
To detect trans-ufmylation on UFC1, 3 μM of His-TRX-UFC1 C116A protein was added in a reaction containing UBA5 (0.5 μM), FLAG-UFM1 (6 μM), UFC1 WT or T106I (3 μM) and DDRGK1-UFL1 (3 μM). The negative control samples were without DDRGK1-UFL1. Reactions were initiated by the addition of ATP (5 mM) and ran for 45 min at 30 °C. After incubation, reactions were stopped by adding an SDS-sample buffer containing β-mercaptoethanol. The samples were then separated by 12% Bis-Tris PAGE followed by immunoblot with anti-His (Abcam, ab18184) antibody to detect ufmylated UFC1C116A.
For ribosome ufmylation assay 1.5 nM purified 60S was mixed with UBA5 (0.5 μM), FLAG-UFM1 (6 μM), UFC1 WT or mutants (3 μM), and DDRGK1-UFL1 (3 μM) in a buffer containing HEPES (50 mM, pH 8.0), NaCl (100 mM) and MgCl2 (10 mM). Reactions were initiated by the addition of ATP (5 mM) and ran for 1 h at 30 °C. The negative control samples were incubated either without ribosomes or without ATP. After incubation, reactions were stopped by adding SDS-sample buffer containing β-mercaptoethanol. The samples, along with the controls, were then separated by 12% Bis-Tris PAGE followed by immunoblot with anti-RPL26 (Abcam, ab59567) antibody to detect ufmylated RPL26.
Crystallization
Crystals of all UFC1 mutants were grown at 20 °C using the hanging drop vapor diffusion method. UFC1 T106I was crystallized in a solution containing 1.5 M ammonium sulfate and 0.1 M Na-HEPES. UFC1 E149I was crystallized in a solution containing 0.1 M Bis-Tris, pH 5.5, 2.0 M ammonium sulfate. UFC1 E149D was crystallized in a solution containing 2% (v/v) tacsimate, pH 7.0, 0.1 M HEPES, pH 7.5, 20% PEG 3350. UFC1 T106V was crystallized in a solution containing 1.5 M ammonium sulfate, 0.1 M Tris–HCl, pH 8.0. UFC1 T106C was crystallized in a solution containing 1 M ammonium Citrate tribasic, pH 7.0, 0.1 M Bis–Tris propane, pH 7.0. UFC1 T106L was crystallized in a solution containing 1.4 M sodium potassium monobasic monohydrate/potassium phosphate dibasic, pH 9.0. UFC1 T106A was crystallized in a solution containing 0.1 M NaCl, 0.1 M Bis–Tris, pH 6.5, and 1.5 M ammonium sulfate. UFC1 T106S was crystallized in a solution containing 1.4 M sodium malonate dibasic monohydrate, pH 6.0. UFC1 W145F was crystallized in a solution containing 1.5 M ammonium sulfate; and 0.1 M Bis–Tris propane pH 7.0. UFC1 C116E was crystallized in a solution containing 0.1 M Bis–Tris, pH 5.5, and 1.8 M ammonium sulfate. UFC1 W145H crystals were obtained from a solution containing 0.2 M Lithium sulfate monohydrate, 0.1 M HEPES pH 7.5, 25% w/v PEG 3350. Crystals of UFC1 K108R were obtained in a solution containing 0.1 M Bis–Tris pH 5.5, and 2 M Ammonium Sulfate. UFC1 K108M crystals are obtained in a solution containing 0.15 M Ammonium sulfate, 0.1 M Sodium HEPES pH 7.0, 20% PEG 4000. Crystals of UFC1 T106I/C116E were obtained in a solution containing 0.1 M Bis–Tris, pH 5.5, and 2 M ammonium sulfate. UFC1 T106S/C116E was crystallized in a solution containing 2 M ammonium sulfate, 0.1 M Tris–HCl, pH 8.0. Yeast Ubc13 C87E was crystallized in a solution containing 0.1 M sodium cacodylate, pH 6.5, 25% PEG 4000. UFC1 K108A/C116E was crystallized in a solution containing 0.1 M Tris pH 8.0, and 2 M Ammonium sulfate. Crystals of the chimeric UFC1 (HPN) were obtained in 0.1 M sodium citrate tribasic dihydrate pH 5.5, 20% v/v 2-propanol, 20% PEG 4000. Human UBC9 C93E crystals were obtained in a solution containing 2% tacsimate, pH 5.0, 0.1 M sodium citrate, and 16% PEG 3350. Crystals of human UBCH5B C85E were obtained in a solution containing 10% PEG 200, 0.1 M Bis–Tris propane, pH 9, and 15% PEG 8000. Crystals appeared after 24–48 h and subsequently grew to the required size in 4 days. The crystals were soaked in the corresponding reservoir solution with or without 22% glycerol and flash-frozen in liquid nitrogen.
X-ray data collection, processing and structure determination
Diffraction data for the crystals used in this study were collected as detailed in Tables 1a–e, and then processed using autoproc43 or XDS as applicable44. The phases were determined by the molecular replacement method implemented in the PhaserMR module of CCP4 using the wild-type UFC1 structure (PDB 2z6o) as a template for all UFC1 mutants45. Phases were obtained for human UBC9 C93E, yeast Ubc13 C87E, and human UBCH5B C85E using the appropriate native structures (PDB 1u9a, 1jbb, and 3tgd, respectively), available in the Protein data bank (https://www.rcsb.org/). The positions affected in the mutants were promptly shown as a negative difference Fourier map masked over the appropriate positions, which were changed to the mutant residue using COOT46. The MR model was subsequently refined using the REFMAC module in CCP447. The refined model was inspected and remodeled appropriately using COOT. Water molecules were subsequently added and refined to obtain the final model with acceptable Rwork/Rfree values.
B-factor analysis
B-factor values of C-alpha atoms in UFC1 WT (2z6p) and mutants, determined in space group P 41 21 2, were extracted and normalized using the method described in Smith et al.48. WT UFC1 (PDB 2z6o) and the UFC1 T106I mutant, which were determined in a P 21 21 21 space group, were excluded to remove any anomalies in the calculation that may arise because of differences in crystal packings in different space groups. UFC1 T106S compensated for the exclusion of WT UFC1, while UFC1 T106V compensated for the exclusion of UFC1 T106I. A coordinate file in PDB file format was generated, listing only the main chain C-alpha atom of all the residues. Then, the atoms with a B-factor exceeding a cut-off threshold value as defined were considered outliers and omitted using a median-based method. Finally, the normalized B-factor was calculated as described.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates and structure factors were deposited in the RCSB PDB with the accession codes: 9GMM (Human UFC1 T106I); 9GLH (Human UFC1 T106S); 9GLI (Human UFC1 T106C); 9GMN (Human UFC1 T106V); 9GLJ (Human UFC1 T106A); 9GLK (Human UFC1 E149I); 9GLL (Human UFC1 T106L); 9GLM (Human UFC1 W145F); 9GLN (Human UFC1 C116E); 9GLO (Human UFC1 C116E/T106S); 9GLP (Human UFC1 C116E/T106I); 9GN8 (Human UFC1 E149D); 9I9M (Chimeric UFC1 HPN); 9I9N (Human UFC1 C116E/K108A); 9I9O (Human UFC1 K108M); 9I9P (Human UFC1 W145H); 9IA8 (Human UFC1 K108R); 9GLR (UBC9 C93E); 9GLS (Human UBCH5B C85E); 9GLT (Yeast UBC13 C87E). Previously published crystal structures used in this study are available from the RCSB PDB under the accession codes: 2Z6O (Human UFC1); 2Z6P (Human UFC1 M109MSe); 1U9A (Human UBC9); 1JBB (Yeast Ubc13); 1J7D (Human Mms2-Ubc13); 3TGD (Human UbcH5b); 3A33 (Human UbcH5b~UB conjugate); 5D2M (Human SUMO2-RANGAP1, UBC9 and ZNF451);1FXT [https://doi.org/10.2210/pdb1FXT/pdb] (Yeast Ubc1~Ub);1A3S [https://doi.org/10.2210/pdb1A3S/pdb] (Human UBC9); 2BEP (Ubiquitin conjugating enzyme E2-25kDa); 2CYX (Human UB2G2); 2YB6 (Human Rad6); 1ZDN (Human E2S); 2GMI (Mms2/Ubc13~Ub); 5BNB (Human Ube2S~Ubiquitin); 2K07 [https://doi.org/10.2210/pdb2K07/pdb] (Human UFC1). Source data are provided with this paper.
References
Herrmann, J., Lerman, L. O. & Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 100, 1276–1291 (2007).
Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 458, 422–429 (2009).
Kerscher, O., Felberbaum, R. & Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180 (2006).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012).
Cappadocia, L. & Lima, C. D. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem. Rev. https://doi.org/10.1021/acs.chemrev.6b00737 (2017).
Schulman, B. A. & Harper, J. W. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 (2009).
Stewart, M. D., Ritterhoff, T., Klevit, R. E. & Brzovic, P. S. E2 enzymes: more than just middle men. Cell Res. 26, 423–440 (2016).
Scheffner, M., Nuber, U. & Huibregtse, J. M. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373, 81–83 (1995).
Berndsen, C. E. & Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307 (2014).
Lorenz, S. Structural mechanisms of HECT-type ubiquitin ligases. Biol. Chem. 399, 127–145 (2018).
Metzger, M. B., Pruneda, J. N., Klevit, R. E. & Weissman, A. M. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60 (2014).
Wu, P. Y. et al. A conserved catalytic residue in the ubiquitin-conjugating enzyme family. EMBO J. 22, 5241–5250 (2003).
Berndsen, C. E., Wiener, R., Yu, I. W., Ringel, A. E. & Wolberger, C. A conserved asparagine has a structural role in ubiquitin-conjugating enzymes. Nat. Chem. Biol. 9, 154–156 (2013).
Wilson, R. H., Zamfir, S. & Sumner, I. Molecular dynamics simulations reveal a new role for a conserved active site asparagine in a ubiquitin-conjugating enzyme. J. Mol. Graph. Model. 76, 403–411 (2017).
Jones, W. M., Davis, A. G., Wilson, R. H., Elliott, K. L. & Sumner, I. A conserved asparagine in a ubiquitin-conjugating enzyme positions the substrate for nucleophilic attack. J. Comput Chem. 40, 1969–1977 (2019).
Komatsu, M. et al. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 23, 1977–1986 (2004).
Oweis, W. et al. Trans-binding mechanism of ubiquitin-like protein activation revealed by a UBA5–UFM1 complex. Cell Rep. 16, 3113–3120 (2016).
Kumar, M. et al. Structural basis for UFM1 transfer from UBA5 to UFC1. Nat. Commun. 12, 5708 (2021).
Peter, J. J. et al. A non-canonical scaffold-type E3 ligase complex mediates protein UFMylation. EMBO J. 41, e111015 (2022).
Burroughs, A. M., Jaffee, M., Iyer, L. M. & Aravind, L. Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J. Struct. Biol. 162, 205–218 (2008).
Nahorski, M. S. et al. Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 141, 1934–1945 (2018).
Banerjee, S. et al. Structural study of UFL1–UFC1 interaction uncovers the role of UFL1 N-terminal helix in ufmylation. EMBO Rep. 24, e56920 (2023).
Makhlouf, L. et al. The UFM1 E3 ligase recognizes and releases 60S ribosomes from ER translocons. Nature 627, 437–444 (2024).
Sakata, E. et al. Crystal structure of UbcH5b∼Ubiquitin intermediate: insight into the formation of the self-assembled E2∼Ub conjugates. Structure 18, 138–147 (2010).
Scheiner, S., Kar, T. & Gu, Y. Strength of the Calpha H.O hydrogen bond of amino acid residues. J. Biol. Chem. 276, 9832–9837 (2001).
Itoh, Y. et al. N(+)–C–H…O Hydrogen bonds in protein–ligand complexes. Sci. Rep. 9, 767 (2019).
Eddins, M. J., Carlile, C. M., Gomez, K. M., Pickart, C. M. & Wolberger, C. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat. Struct. Mol. Biol. 13, 915–920 (2006).
Cappadocia, L., Pichler, A. & Lima, C. D. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat. Struct. Mol. Biol. 22, 968–975 (2015).
Hamilton, K. S. et al. Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure 9, 897–904 (2001).
Millrine, D. et al. Human UFSP1 is an active protease that regulates UFM1 maturation and UFMylation. Cell Rep. 40, 111168 (2022).
Cook, B. W. & Shaw, G. S. Architecture of the catalytic HPN motif is conserved in all E2 conjugating enzymes. Biochem. J. 445, 167–174 (2012).
Simón, L. & Goodman, J. M. Enzyme catalysis by hydrogen bonds: the balance between transition state binding and substrate binding in oxyanion holes. J. Org. Chem. 75, 1831–1840 (2010).
Derewenda, Z. S. C–H groups as donors in hydrogen bonds: a historical overview and occurrence in proteins and nucleic acids. Int. J. Mol. Sci. 24, 13165 (2023).
Scheiner, S., Kar, T. & Gu, Y. L. Strength of the CH••O hydrogen bond of amino acid residues. J. Biol. Chem. 276, 9832–9837 (2001).
Menard, R. & Storer, A. C. Oxyanion hole interactions in serine and cysteine proteases. Biol. Chem. Hoppe Seyler 373, 393–400 (1992).
Kochanczyk, T. et al. Structural basis for transthiolation intermediates in the ubiquitin pathway. Nature (2024).
Branigan, E., Carlos Penedo, J. & Hay, R. T. Ubiquitin transfer by a RING E3 ligase occurs from a closed E2~ubiquitin conformation. Nat. Commun. 11, 2846 (2020).
Page, R. C., Pruneda, J. N., Amick, J., Klevit, R. E. & Misra, S. Structural insights into the conformation and oligomerization of E2∼Ubiquitin conjugates. Biochemistry 51, 4175–4187 (2012).
Liess, A. K. L. et al. Autoinhibition mechanism of the ubiquitin-conjugating enzyme UBE2S by autoubiquitination. Structure 27, 1195–1210.e1197 (2019).
Liu, G. et al. NMR and X-RAY structures of human E2-like ubiquitin-fold modifier conjugating enzyme 1 (UFC1) reveal structural and functional conservation in the metazoan UFM1-UBA5-UFC1 ubiquination pathway. J. Struct. Funct. Genom. 10, 127–136 (2009).
Yunus, A. A. & Lima, C. D. Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat. Struct. Mol. Biol. 13, 491–499 (2006).
Buetow, L., Gabrielsen, M. & Huang, D. T. Single-turnover RING/U-Box E3-mediated lysine discharge assays. Methods Mol. Biol. 1844, 19–31 (2018).
Vonrhein, C. et al. Advanced exploitation of unmerged reflection data during processing and refinement with autoPROC and BUSTER. Acta Crystallogr. Sect. D Struct. Biol. 80, 148–158 (2024).
Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 (2011).
Smith, D. K., Radivojac, P., Obradovic, Z., Dunker, A. K. & Zhu, G. Improved amino acid flexibility parameters. Protein Sci. 12, 1060–1072 (2003).
Acknowledgements
We thank the staff of ESRF beamlines ID30B, ID23-2, and ID23-1, and Diamond Light Source Beamline I03 for the help with data collection. This work was supported, in whole or in part, by the Israel Science Foundation, founded by the Israel Academy of Science and Humanities (grant number 491/2021 to R.W.) and by the Israel Cancer Research Fund (award ID 21‐113‐PG to R.W.).
Author information
Authors and Affiliations
Contributions
M.K., S.B., and R.W. designed the experiments. S.B. and E.C.K. performed the biochemical experiments. S.B., M.B.M., and E.C.K. carried out the cloning. M.K. and S.B. performed the protein purification. M.K. and S.B. grew the crystals and collected the crystallographic data with assistance from S.T., M.D., and R.W. M.K., M.D., M.N.I., and R.W. determined the crystal structures. M.K., S.B., and R.W. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Helge Magnussen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kumar, M., Banerjee, S., Cohen-Kfir, E. et al. UFC1 reveals the multifactorial and plastic nature of oxyanion holes in E2 conjugating enzymes. Nat Commun 16, 3912 (2025). https://doi.org/10.1038/s41467-025-58826-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58826-y
This article is cited by
-
Evaluating evidence for UFMylation client diversity
Nature Reviews Molecular Cell Biology (2026)
