
Abstract
Orchestrated hormonal interactions in response to feeding and fasting play a pivotal role in regulating glucose homeostasis. Here, we show that in obesity, the production of follistatin-like 3 (FSTL3), an endogenous inhibitor of Activin B, in adipose tissue is increased in both mice and humans. The knockdown of FSTL3 improves insulin sensitivity and glucose tolerance in diabetic obese db/db mice. Notably, the overexpression of Activin B, a member of the TGFβ superfamily that is induced in liver sinusoidal endothelial cells by fasting, exerts multiple metabolically beneficial effects, including improvement of insulin sensitivity, suppression of hepatic glucose production, and enhancement of glucose-stimulated insulin secretion, all of which are attenuated by the overexpression of FSTL3. Activin B increases insulin sensitivity and reduces fat by inducing fibroblast growth factor 21 (FGF21) while suppressing glucagon action in the liver by increasing phosphodiesterase 4 B (PDE4B), leading to hepatic glucagon resistance and resultant hyperglucagonemia. Activin B-induced hyperglucagonemia enhances glucose-stimulated insulin secretion by stimulating glucagon-like peptide-1 (GLP-1) receptor in pancreatic β-cells. Thus, enhancing the action of Activin B which improves multiple components of the pathogenesis of diabetes may be a promising strategy for diabetes treatment.
Similar content being viewed by others
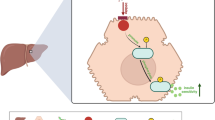

Introduction
In response to environmental changes, such as fasting and refeeding, the liver undergoes rapid adaptations that are crucial for maintaining metabolic homeostasis1. Dysregulation of these processes is a key feature observed in obesity and type 2 diabetes2. Several humoral factors involved in adaptive responses to nutritional cues have potent antidiabetic effects and have been applied as diabetes therapeutics3. Among them, glucagon-like peptide-1 (GLP-1) or GLP-1/gastric inhibitory peptide (GIP) receptor dual agonists have been widely used. Recently, GLP-1/GIP/glucagon receptor triple agonists have been developed and shown to have strong antidiabetic and anti-obesity effects4,5. Glucagon is a fasting-induced hormone that increases gluconeogenesis in the liver. Suppression of glucagon has been attempted as a treatment for diabetes mellitus. However, glucagon is also known to contribute to the improvement of glucose metabolism by promoting lipolysis in adipocytes and suppressing appetite in the central nervous system. More importantly, glucagon serves as an insulin secretagogue and has recently attracted attention as a novel drug for obesity and diabetes6,7,8.
Hepatocytes are the dominant cell type in the liver, and their roles in metabolic adaptations to environmental nutritional status have been emphasized in previous studies1. Several hepatokines regulated by fasting and feeding, such as fibroblast growth factor 21 (FGF21), play key roles in regulating glucose homeostasis9,10,11,12,13,14. However, the precise mechanisms by which the liver appropriately responds to fasting and feeding remain not fully understood. The liver is composed of various cell types other than hepatocytes, among which, liver sinusoidal endothelial cells (LSECs) comprise the largest proportion of liver non-parenchymal cells. Recent studies have highlighted the essential roles of LSEC in a wide range of liver functions15,16, although their significance in metabolic adaptations remains largely unexplored.
Activin, a member of transforming growth factor-β (TGF-β) superfamily cytokines, is a hetero or homodimer of four different Inhibin β subunits and thus comprises several isoforms, including Activin A (Inhibin βA/Inhibin βA) and Activin B (Inhibin βB/Inhibin βB)—the two major isoforms of activin. These two activins are abundantly expressed in the gonadal tissues and are classically known as regulators of developmental and reproductive processes17. However, studies have revealed their expression in other organs and their previously unknown roles in immune responses and fibrosis17,18,19. Reports have also suggested possible roles in metabolism and energy expenditure20,21,22,23, while the mechanisms of action or precise roles in hepatic metabolism are still not fully understood.
In this study, we found that the expression of Activin B, a TGF β superfamily protein, was induced in LSECs in the fasting state. Administration or overexpression of Activin B in the liver markedly decreased glucose levels in diabetic mice. The glucose-lowering effects of Activin B are mediated by at least two independent mechanisms; one is Fgf21-dependent enhancement of whole-body insulin sensitivity, and the other is the suppression of hepatic glucagon action leading to inhibition of gluconeogenesis and the resultant increase in circulating glucagon levels, which enhances glucose-stimulated insulin secretion. Furthermore, the glucose-lowering effects of Activin B were inhibited by its endogenous inhibitor, follistatin-like protein 3 (FSTL3), whose expression in white adipose tissue is increased by obesity in mice and humans. These data suggest a previously unknown mechanism through which Activin B regulates glucose metabolism, thereby providing a promising target for the treatment of diabetes.
Results
FSTL3 was increased in obesity and negatively modulated glucose homeostasis
Adipokines have been shown to be involved in the regulation of glucose metabolism, and changes in their expression play pivotal roles in the development of diabetes and related diseases in obesity. Thus, we screened for the adipokines whose expression starts changing at the very early stage of obesity and thereby attempted to identify those involved in the regulation of glucose metabolism. We have analyzed the gene expression profiles in subcutaneous white adipose tissue (WAT) of 176 Japanese subjects undergoing gastric surgery. We performed expression analysis along with the clinical data, and extracted 28 genes whose expression increased with increasing body mass index (BMI), as candidate adipokines potentially involved in glucose metabolism (Supplementary Fig. 1a). Among these candidates, we focused follistatin-like 3 (FSTL3), whose expression was significantly correlated with BMI (r = 0.44, P < 0.001, Supplementary Fig. 1b). FSTL3 has been suggested to be involved in glucose metabolism24,25,26, while their precise mechanisms of action remain to be elucidated. We also confirmed in an independent cohort that the FSTL3 expression both in visceral and subcutaneous adipose tissues were strongly correlated with BMI (Fig. 1a, b). Furthermore, the glucose infusion rate (GIR) during the steady state of a euglycemic-hyperinsulinemic clamp study, an indicator of whole-body insulin sensitivity showed a strong negative correlation with the expression levels of FSTL3 in visceral WAT (r = −0.74, P < 0.001, Fig. 1c) and subcutaneous WAT (r = −0.71, P < 0.001, Fig. 1d). Importantly, correlations between FSTL3 expression and BMI and GIR were observed in both men and women. These data indicate that the expression levels of FSTL3 in WAT correlate with obesity and whole-body insulin sensitivity in humans.

a, b Relative expression levels of FSTL3 in visceral (a) or subcutaneous fat (b) of subjects. n = 188 (male n = 86; female n = 102). c, d Negative correlation of FSTL3 mRNA expression in visceral (c) or subcutaneous fat (d) with whole-body insulin sensitivity. The glucose infusion rate (GIR) was determined by hyperinsulinemic-euglycemic clamp studies. e, f Blood glucose levels of DIO mice received Ad-LacZ or Ad-Fstl3 during GTT (e, n = 7 mice per group) or ITT (f, Ad-LacZ; n = 6 mice, Ad-Fstl3; n = 7 mice). g Blood glucose levels of male db/db mice treated with a control antisense oligonucleotide (ASO) or Fstl3-targeting ASO (described in “Materials and method”) during GTT. h Relative expression levels of Fstl3 in the adipose tissues of mice used in (g). n = 8 mice per group. Data were shown as mean ± SEM. Statistical significance was determined by Spearman’s rank correlation test (a–d); unpaired two-tailed Student’s t-test (e, g, h); two-way ANOVA with Šídák’s multiple comparison test (e, f) as compared to the respective control group. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data File.
While FSTL3 is strongly expressed in the reproductive organs26,27,28, it is also expressed in a broad range of tissues, including white adipose tissues27,29. Consistent with previous reports30, northern blot analysis detected Fstl3 mRNA in the white adipose tissue of normal lean mice, although to a lesser extent than that in the testes and kidneys (Supplementary Fig. 1c). Notably, however, the expression of Fstl3 was greatly increased in obesity-diabetic db/db mice, especially in subcutaneous and epididymal WAT, compared with that in lean control mice, amounting to levels similar to those in the testis (Supplementary Fig. 1c). Quantitative expression analysis revealed that Fstl3 expression levels in the epididymal WAT of normal lean mice were higher than those in the subcutaneous WAT (Supplementary Fig. 1d, e). In both diet-induced obese (DIO) and db/db mice, Fstl3 expression in both subcutaneous and epididymal WAT was markedly increased compared with that in lean control mice (Supplementary Fig. 1d, e). Increased expression of the Fstl3 gene was also observed in white adipose tissue from female db/db mice (Supplementary Fig. 1f). Importantly, plasma levels of FSTL3 were barely detectable in lean wild-type mice but were elevated in DIO or db/db mouse models (Supplementary Fig. 1g, h). Notably, the expression levels of another follistatin family member, Fst, in white adipose tissue did not correlate with BMI in humans (Supplementary Fig. 1i, j), and were not altered in the subcutaneous and epididymal WAT of DIO and db/db mice compared with their respective controls (Supplementary Fig. 1k, l). Furthermore, adenovirus-mediated gene transfer of Fstl3 into normal C57BL6 mice resulted in increased ectopic hepatic expression of the Fstl3 gene and circulating FSTL3 protein levels, which in turn impaired glucose tolerance and insulin sensitivity, as demonstrated by the glucose tolerance test (GTT) and insulin tolerance test (ITT) results (Fig. 1e, f and Supplementary Fig. 1m, n), suggesting a functional link between FSTL3 and systemic glucose metabolism. Conversely, treatment with an antisense oligonucleotide targeting Fstl3 decreased its expression in adipose tissues and improved glucose tolerance in db/db mice (Fig. 1g, h). Consistent with the human data, these findings suggest that FSTL3 negatively regulates glucose metabolism under obese conditions.
Overexpression of Activin B in the liver improved systemic glucose tolerance via simultaneously suppressing gluconeogenesis and insulin secretion
Although our data suggest that Fstl3 contributes to glucose metabolism, its precise mechanism of action remains to be elucidated. It is known that FSTL3 directly binds to Activins and inhibits their biological activities mediated by their respective receptors31,32.
First, we investigated the effects of Activin A and Activin B on systemic glucose metabolism. Adenovirus-mediated overexpression of INHBA (Ad-INHBA) or INHBB (Ad-INHBB), the human homologs of mouse Inhba or Inhbb, respectively, which share 96% amino acid sequence identity, resulted in lower glucose levels during GTT and ITT (Fig. 2a–c and Supplementary Fig. 2a–c). Treatment with Ad-INHBB alone was associated with higher insulin levels during GTT in DIO mice compared to the control virus treatment (Fig. 2b). In contrast, FSTL3 overexpression ameliorated Activin B-mediated glucose tolerance and insulin sensitivity in obese mice (Fig. 2d–f). These data suggest a beneficial role for activin signaling in systemic glucose homeostasis.

a, b Blood glucose (a) and plasma insulin levels (b) of DIO mice treated with Ad-LacZ or Ad-INHBB during GTT. c Blood glucose levels during ITT. n = 8 per group. d–f Blood glucose (d) and plasma insulin levels (e) of DIO mice treated with Ad-INHBB with or without Ad-FSTL3 during GTT. f Blood glucose levels during ITT. Ad-LacZ; n = 8, Ad-INHBB; n = 7, Ad-INHBB + Ad-FSTL3; n = 8. g Relative expression levels of Inhibin β subunits (Inhba, Inhbb, Inhbc, and Inhbe) in liver. n = 5 per group. h–k Relative expression levels of Inhbb gene in the liver of ad libitum fed, fasted, or refed mice (h, n = 5), fractionated liver of wild-type mice (i, n = 5), isolated CD11b+ cells and CD146+ cells (LSECs) from the liver of ad libitum fed (white bars) or fasted (black bars) mice (j, n = 3), or in liver tissues of conditional Inhbb knockout mice (k, the sample size (n) is indicated within the graph.). l–s Metabolic phenotyping in DIO mice treated with AAV-TBG-EGFP or AAV-TBG-INHBB mice. l, m Body weight changes (l) and ambient blood glucose levels (m). EGFP; n = 14, INHBB; n = 18. n–p Blood glucose levels during GTT (n, n = 8) or ITT (o, EGFP; n = 8, INHBB; n = 6) and plasma insulin levels during GTT in (n). q Whole-body energy expenditure measured in a metabolic cage. n = 8 mice per group. r Relative expression levels of genes in BAT. GFP; n = 6, INHBB; n = 8. s Representative images of H&E staining of BAT. Data were shown as mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t-test (a, g, n, r); two-way ANOVA with Šídák’s (a–c, l–n, o–q), or Dunnett’s (d–f) multiple comparison test; one-way ANOVA Sidak’s multiple comparison test (h) as compared to the respective control group. *p < 0.05, **p < 0.01, ***p < 0.001. #p < 0.05, ##p < 0.01, ###p < 0.001 versus Ad-INHBB treated mice in (d–f). Source data are provided as a Source Data File.
Furthermore, we found that hepatic expression of Inhbb, which encodes a subunit of Activin B, was markedly induced in the fasted state compared to the feeding state, and this induction was more pronounced than that of other inhibinβ subunits (Inhba, Inhbc, and Inhbe) (Fig. 2g). The experiment with the fasting and refeeding challenge in mice demonstrated that Inhbb expression decreased in the liver 6 h post refeeding (Fig. 2h). We also found that expression of Inhbb was induced by ketogenic diet (Supplementary Fig. 2d). Thus, Activin B, but not Activin A may be involved in the physiological regulation of glucose metabolism. Therefore, we explored the role of Activin B in glucose metabolism.
To further explore the physiological responses of the expression of Inhbb in the liver, we fractionated the liver cells into subpopulations and examined the expression levels of Inhbb in each fraction. Higher expression levels of Inhbb were detected in the non-parenchymal cell fraction and, more finely, in the CD146 positive cell fraction, known as liver sinusoidal endothelial cells (LSECs) (Fig. 2i, j). Importantly, Inhbb expression was barely detectable in the CD11b-positive fraction or the CD11b and CD146 double-negative fraction, indicating that Inhbb-expressing cells were not present in these fractions, that is, in liver non-parenchymal cells other than LSECs. (Supplementary Fig. 2e). Indeed, the disruption of Inhbb gene expression was observed in the liver tissue of Inhbb-Lyve1 KO and Inhbb-Tie2 KO mice, which were generated by crossing InhbbFlox mice and mice expressing Cre recombinase specifically in endothelial cells, Lyve1-Cre, and Tie2-Cre driver mice, respectively, whereas crossing InhbbFlox mice and Alb-Cre transgenic mice did not affect the expression of Inhbb in the liver (Fig. 2k and Supplementary Fig. 2f). Taken together, these results clearly demonstrate that LSECs is the only cell population in the liver that express the Inhbb gene, and that its gene product, Activin B, may be an “LSECkine”.
We next investigated the long-term effect of Activin B using adeno-associated virus (AAV) gene transfer in DIO mice. Endogenous Inhbb expression in the liver tissue was limited in LSECs. However, a method of exogenous gene transfer specifically for LSECs has not been established; thus, we adopted an alternative method to produce Activin B in hepatocytes, following a recent, clinically approved gene therapy for hemophilia using a virus vector to produce coagulation factor VIII, which is originally expressed in LSECs, in hepatocytes33. As expected, expression of INHBB was detected predominantly in the hepatocytes (Supplementary Fig. 2g). Notably, adenovirus-mediated expression of INHBA or INHBB genes was also detected mostly in the hepatocyte fraction (Supplementary Fig. 2h).
AAV-mediated sustained Activin B overproduction resulted in a significant decrease in ambient blood glucose levels and body weight of DIO mice than in the control mice (Fig. 2l, m), with no significant difference detected in food intake between the two groups (Supplementary Fig. 2i). DIO mice administered with INHBB-harboring AAV showed lower blood glucose levels during the GTT and ITT than did those administered with the control AAV (Fig. 2n, o), suggesting an improvement in insulin sensitivity under Activin B overexpression. In contrast, plasma insulin levels during the GTT were markedly increased in mice overexpressing Activin B than in control mice, despite their enhanced insulin sensitivity (Fig. 2p). Furthermore, comprehensive metabolic cage studies using indirect calorimetry revealed that whole-body energy expenditure was significantly higher in DIO mice treated with AAV-TBG-INHBB than in control mice (Fig. 2q). Notably, the increase in energy expenditure was not due to changes in food intake or physical activity (Supplementary Fig. 2i, j).
Activin B is known to activate a common signaling pathway with TGFβ, an important factor in fibrosis, and its involvement has been reported in a mouse model of carbon tetrachloride-induced liver fibrosis34. In our DIO model, AAV-mediated Activin B overproduction showed an upward trend in the expression of inflammation and fibrotic marker gene expression in the liver, with no statistically significant difference (Supplementary Fig. 2k).
Collectively, these data indicated that the overproduction of Activin B improved not only adiposity but also whole-body glucose tolerance, possibly by simultaneously affecting insulin sensitivity and glucose-stimulated insulin secretion (GSIS) capacity, in obese mice.
Activin B induced FGF21 from hepatocytes, which partly mediated the metabolic effects of Activin B
We sought to elucidate the mechanism by which Activin B enhances insulin sensitivity in mice. Ample evidence has demonstrated that FGF21 is a hepatokine induced by fasting and plays an important role in the regulation of glucose homeostasis9,10,11,12,13,14. Here, the hepatic expression and plasma levels of FGF21 protein were significantly higher in mice overexpressing Activin B (Fig. 3a, b), whereas the hepatic expression of Fgf21 in the fasted state was significantly decreased in Inhbb-Lyve1 KO mice than in the respective controls (Fig. 3c, d). Moreover, the administration of recombinant Activin B increased Fgf21 expression in a dose-dependent manner in cultured hepatocytes (Fig. 3e), with an increase in its promoter activity (Supplementary Fig. 3a). These findings collectively indicate that Activin B induced FGF21 in the liver, plausibly by regulating its gene expression.

a, b Relative expression levels of Fgf21 in liver (a, n = 8) and plasma FGF21 concentration (b, n = 6) of mice treated with Ad-LacZ or Ad-INHBB. c, d Relative expression levels of Fgf21 (c) and Inhbb (d) in the liver. n = 5 mice per group. e Dose-dependent effects of recombinant Activin B protein on Fgf21 expression. n = 3. f–k Relative expression levels of Fgf21 in the liver (f), body weight changes (g), body fat (h), blood glucose levels during ITT (i), blood glucose levels (j), and plasma insulin levels (k) during GTT of wild-type or Fgf21 deficient mice treated with indicated AAV. (WT, EGFP; n = 7, WT, INHBB; n = 7, Fgf21 KO, EGFP; n = 6, Fgf21 KO, INHBB; n = 7). Data were shown as mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t-test (a, b); two-way ANOVA with Tukey’s (c, d, g, i–k) or Šídák’s (f, h, j) multiple comparisons test; one-way ANOVA with Dunnett’s multiple comparisons test (e) as compared to the respective control group. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data File.
Mechanistically, the activin signaling cascade is initiated by the binding of activin to the activin receptor type II or IIB (ActRII/IIB). This results in the recruitment and phosphorylation of the type I receptor (activin receptor-like kinase [ALK]), leading to the formation of the Smad2/3 and Smad4 complex. Fgf21 induction by Activin B was suppressed by blocking ALK4/7 and subsequent Smad2 phosphorylation but not by an inhibitor specific to ALK2/3/6 (Supplementary Fig. 3b, c). ALK4 knockdown inhibited Activin B-induced Fgf21 expression (Supplementary Fig. 3d), whereas a constitutively active mutant of ALK4 significantly induced Fgf21 expression and Smad2 phosphorylation (Supplementary Fig. 3e, f). Notably, previous reports have identified PPARα as the inducer of hepatic Fgf21 upon fasting, but silencing hepatic Ppara by adenovirus-mediated shRNA delivery (Ad-shPpara) did not affect the Activin B-mediated hepatic Fgf21 induction (Supplementary Fig. 3g, h). Collectively, these findings indicate that Activin B induces FGF21 expression in the liver by activating the ALK4-Smad2 pathway.
We hypothesized that FGF21 induced by Activin B could mediate the metabolic effects of Activin B. To test this hypothesis, we overexpressed Activin B in wild-type or Fgf21 knockout (KO) mice fed a high-fat diet. We found that the enhancement of insulin sensitivity, as well as the reduction in body weight and body fat by Activin B overexpression, was attenuated in Fgf21 KO mice compared with wild-type mice, while the changes in glucose tolerance and GSIS by Activin B overexpression remained comparable in the two genotypes of mice (Fig. 3f–k). We also administered recombinant Activin B protein with or without the concomitant administration of an adenovirus harboring a shRNA targeting Fgf21. Consistent with the results of the overexpression experiments, the mice administered recombinant Activin B showed enhanced insulin sensitivity, as validated by lower glucose levels during ITT, compared with the control mice, while concomitant knockdown of hepatic Fgf21 expression abolished the recombinant Activin B-mediated insulin-sensitizing effects in the ITT (Supplementary Fig. 3i, j). Collectively, these data indicate that activin B enhances insulin sensitivity and suppresses adiposity in obese mice via FGF21 induction. However, some metabolic effects such as improved glucose tolerance and insulin secretion induced by Activin B overproduction—were observed even in the absence of FGF21, suggesting the possibility of an FGF21-independent mechanism.
Activin B suppressed the gluconeogenic gene expression in hepatocytes
In further attempts to elucidate the mechanisms of the metabolic effects of Activin B other than FGF21-mediated improvement of insulin sensitivity, the enhanced insulin secretion capacity by Activin B, as revealed by our analyses, could make it difficult to precisely assess the changes occurring in the insulin target tissues under overexpression of Activin B. To mitigate this limitation, we next adopted streptozotocin (STZ)-induced insulin-deficient mice. Ad-INHBB-mediated overexpression of Activin B in STZ-induced insulin-deficient mouse livers significantly reduced blood glucose levels during GTT compared to Ad-LacZ control administration, without changes in plasma insulin levels (Fig. 4a, b). Analyses of the livers of insulin-deficient mice treated with Ad-INHBB showed that the mRNA levels of the key gluconeogenic genes, G6pc and Pck1 (Fig. 4c), as well as the protein levels of PEPCK (Fig. 4d), were significantly decreased, suggesting that overexpression of Activin B led to the suppression of hepatic gluconeogenesis. In the pyruvate tolerance test (PTT), a test to assess the potential of hepatic glucose production, Ad-INHBB-mediated overexpression of Activin B significantly lowered glucose levels compared with those in the controls, both in STZ-treated mice (Fig. 4e) and in normal mice (Supplementary Fig. 4a), along with reduced hepatic Pck1 expression (Supplementary Fig. 4b), but without activation of Akt in the liver (Supplementary Fig. 4c). Conversely, fasting and pyruvate-stimulated glucose levels in STZ-treated Inhbb-Lyve1 KO mice were significantly higher than those in controls (Fig. 4f, g). In addition, in Fgf21 deficient STZ mice, Ad-INHBB treatment lowered the blood glucose levels during PTT (Supplementary Fig. 4d). Collectively, these data indicate that Activin B suppresses hepatic glucose production independent of increased insulin secretion or FGF21 induction. An in vitro experiment using primary hepatocytes showed that stimulation with recombinant Activin B significantly reduced glucose production induced by pyruvate and glucagon (Fig. 4h). Recombinant Activin B suppressed Pck1 mRNA levels, indicating modulation of Pck1 expression by Activin B (Fig. 4i).

a, b Blood glucose (a) and plasma insulin levels (b) of STZ mice treated with Ad-LacZ or Ad-INHBB during GTT. Ad-LacZ; n = 8, Ad-INHBB; n = 7. c Relative expression levels of genes regulating hepatic gluconeogenesis of STZ mice treated with Ad-LacZ or Ad-INHBB. Ad-LacZ; n = 6, Ad-INHBB; n = 4. d PEPCK protein levels in the liver from 4 independent animals. e Blood glucose levels of STZ mice during PTT. n = 5 mice per group. f, g Fasting blood glucose levels (f) or blood glucose levels during PTT (g) in STZ-treated Inhbb-Lyve1 KO or control mice. Inhbb-Flox; n = 6, Inhbb-Lyve1 KO; n = 9. h Glucose production in primary hepatocytes treated with recombinant Activin B. n = 4. i–k Relative expression levels of Pck1 treated with recombinant Activin B with or without Forskolin and Dexamethasone (Frk/Dex) (i, n = 3), subtype-specific ALK inhibitors (j, n = 3), or treated with adenovirus encoding constitutive active mutant of ALK2 (Ad-ALK2(QD)) (k, n = 3). Data were presented as means ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t-test (a, f, g); two-way ANOVA (g) or with Šídák’s (a, c, j), Dunnett’s (e), or Tukey’s (i) multiple comparison test, one-way ANOVA with Dunnett’s (e) or Šídák’s (h, k) multiple comparison test as compared to the respective control group. *p < 0.05, **p < 0.01, ***p < 0.001. #p < 0.05 versus normal mice in (e). Source data are provided as a Source Data File.
We further investigated the mechanism by which Activin B regulates gluconeogenic gene expression in hepatocytes. Previous studies have suggested that Activin B can activate not only the canonical ALK4-Smad2 pathway, but also a non-canonical pathway, which comprises ALK2/3-Smad1/5/835,36,37. Our assessments in cultured cells confirmed that Activin A and Activin B comparatively activated the canonical pathway, estimated by inducing the phosphorylation of Smad2 and Smad3, while greater Smad1/5/8 phosphorylation was observed in Activin B stimulation (Supplementary Fig. 4e), which was recapitulated in the livers of mice that received Ad-INHBB (Supplementary Fig. 4f). The reporter assays demonstrated that Activin B activated both Smad2/3-and Smad1/5/8-dependent transactivation, whereas Activin A activated only Smad2/3-dependent transactivation (Supplementary Fig. 4g, h). These findings suggest that Activin B stimulates both the canonical ALK4-Smad2/3 and non-canonical ALK2/3-Smad1/5/8 pathways in hepatocytes. However, the ALK2/3/6 inhibitor (LDN-193189), but not the ALK4/7 inhibitor (SB525334), abrogated Activin B-induced suppression of Pck1 expression (Fig. 4j and Supplementary Fig. 4i). Additionally, the expression of a constitutively active mutant of ALK2 (ALK2 QD) significantly attenuated forskolin/dexamethasone-induced Pck1 expression (Fig. 4k). Collectively, these results suggest that Activin B suppresses Pck1 expression via a non-canonical ALK2-dependent signaling pathway.
Hepatic glucagon signaling was suppressed by Activin B
To further assess the mechanism of Activin B-mediated suppression of hepatic gluconeogenesis, we performed a glucagon challenge test and found that the increase in blood glucose levels after glucagon injection was significantly lower in Ad-INHBB-treated mice than in control mice (Fig. 5a). Immunoblotting analysis using antibodies recognizing phosphorylated protein kinase A substrates showed that glucagon induces activation of PKA signaling as well as induction of cAMP production in the liver, which were clearly reduced in Ad-INHBB-treated mice compared with the controls (Fig. 5b, c).

a–c Blood glucose (a, n = 6 mice per group), hepatic glucagon signaling (b), and cAMP contents (c, the sample size (n) is indicated within the graph) in response to glucagon in mice treated with Ad-LacZ or Ad-INHBB. d–f Blood glucose (d, n = 6 mice per group), hepatic glucagon signaling (e), and cAMP contents (f, the sample size (n) is indicated within the graph) in response to glucagon in Inhbb-Lyve1 KO mice. g, h Plasma glucagon levels in fasted adenovirus treated wild-type mice (g, n = 6 mice per group) or Inhbb-Lyve1 KO mice (h, n = 6 mice per group). i, j Relative expression levels of genes encoding phosphdiesterase four subtypes in liver of mice treated with Ad-LacZ or Ad-INHBB (i, n = 3 mice per group) or Inhbb-Lyve1 KO mice (j, n = 3 mice per group). k–m Changes in hepatic cAMP contents (k, the sample size (n) is indicated within the graph) or blood glucose (l, the sample size (n) is indicated within the graph) in response to glucagon challenge and plasma glucagon levels (m, n = 5 mice per group) in mice treated with Ad-LacZ or Ad-INHBB with or without PDE4 inhibitor (PDE4i). Data were shown as mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t-test (a, d, g–j); two-way ANOVA (a, d) or with Šídák’s (c, f), or Tukey’s (k–m) multiple comparison test as compared to the respective control group. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data File.
Of note, there were no significant changes in glucagon receptor or PKA protein levels or, interestingly, in phosphorylation of CREB, which is known to be phosphorylated by PKA in a glucagon-dependent manner (Fig. 5b). These findings suggested that glucagon-stimulated cAMP production is attenuated in the livers of Ad-INHBB-treated mice, leading to a downregulation of PKA activity. In contrast, Inhbb-Lyve1 KO mice showed significantly higher gluconeogenic capacity, hepatic cAMP production, and phosphorylation of PKA substrates in response to acute glucagon stimulation compared to control mice (Fig. 5d–f), indicating that Inhbb-Lyve1 KO mice are hypersensitive to glucagon. It has been proposed that glucagon levels are influenced by hepatic glucagon demand, known as the “Liver-α-cell axis” concept38. To investigate this, we measured plasma glucagon levels using a sandwich ELISA assay. We found that fasting glucagon levels were significantly elevated in Ad-INHBB-treated mice and reduced in Inhbb-Lyve1 KO mice (Fig. 5g, h), suggesting that Activin B signaling suppresses hepatic glucagon action. Even in the STZ-induced insulin-deficient mice, overproduction of Activin B tended to increase plasma glucagon levels, albeit without statistical significance—likely because insulin deficiency alone had already elevated glucagon levels. Nevertheless, Activin B overproduction significantly suppressed glucagon-induced glucose elevation and reduced hepatic cAMP levels (Supplementary Fig. 5a–c). Taken together, these data indicate that Activin B induces hepatic glucagon resistance in hepatocytes independently of insulin action.
We further explored the molecular mechanisms by which Activin B regulates glucagon actions in the liver. We found that the hepatic expression of the cAMP-degrading phosphodiesterase genes Pde4a and Pde4b was significantly increased in Ad-INHBB-treated mice (Fig. 5i), whereas hepatic Pde4b expression was decreased in Inhbb-Lyve1 KO mice (Fig. 5j) than in the respective control mice. Treatment of mice with roflumilast, a selective PDE4 inhibitor, abrogated Ad-INHBB-mediated suppression of glucagon-induced cAMP and glucose production, along with hyperglucagonemia (Fig. 5k–m). Taken together, these data indicate that Activin B inhibits hepatic glucagon activity by suppressing cAMP production, leading to hyperglucagonemia.
Activin B enhanced GSIS via glucagon-mediated GLP-1 receptor activation
Next, we investigated the mechanisms by which Activin B overexpression enhances insulin secretion. To evaluate the effect of Activin B on insulin secretion in detail, we performed an in vivo GSIS test with Activin B overexpression39. Overproduction of Activin B significantly enhanced glucose-stimulated insulin secretion in both the first and second phases and increased the plasma C-peptide concentration compared with the control virus treatment (Fig. 6a, b). In contrast, in Inhbb-Lyve1 KO mice, insulin responses in the in vivo GSIS test were significantly compromised compared with those in control mice (Fig. 6c, d). Similarly, impaired insulin secretion was observed in Inhbb-Tie2 KO mice (Supplementary Fig. 6a). These data indicate that LSEC-derived Activin B was required for proper insulin secretion in response to glucose in mice.

a, b Plasma levels of insulin (a) and c-peptide (b) in response to bolus glucose challenge (in vivo GSIS test). n = 6 mice per group. c, d Plasma levels of insulin (c) and c-peptide (d) of Inhbb-Lyve1 KO mice during in vivo GSIS test (Inhbb-Flox; n = 7, Inhbb-Lyve1 KO; n = 9). e Effects of Activin B on glucose-stimulated insulin secretion (GSIS). Isolated islets were cultured with or without Activin B and stimulated with high glucose (22.2 mM l-glucose). Data were presented as secreted insulin concentration normalized to insulin content. n = 6. f Pancreatic insulin contents. n = 5. g–i Plasma levels of glucagon (g), active GLP-1 (h), and active GIP (i) in mice received Ad-INHBB mice during in vivo GSIS test. n = 6 mice per group. j, k Plasma levels of glucagon in fasting (j) or plasma insulin during in vivo GSIS test (k) in Ad-INHBB received mice pretreated with or without Pramlintide. The sample size (n) is indicated within the graph. l, m Plasma insulin levels in Ad-INHBB received mice pretreated with or without PDE4 inhibitor (l, n = 5 mice per group) or GLP-1R blocker Ex9–39 (m, the sample size (n) is indicated within the graph). Data were shown as mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t-test (a–d, f); two-way ANOVA (g–i, m) or with Šídák’s (a–d) or Tukey’s (e, j–n) multiple comparison test as compared to the respective control group. *p < 0.05, **p < 0.01, ***p < 0.001. Source data are provided as a Source Data File.
In contrast, stimulation of freshly isolated islets with recombinant Activin B did not change insulin secretion under low- and high-glucose conditions (Fig. 6e). Although some reports claim that activins can directly modulate insulin secretion23,40,41,42, our data suggest that Activin B itself does not possess a direct insulinotropic effect. We also did not detect any changes in pancreatic insulin contents from mice received Ad-INHBB compared to control (Fig. 6f) as well as in the gene expressions related to β-cell functions, with Ins1 and Ins2 gene expressions being even decreased in the islets of mice treated with Ad-INHBB compared to the controls (Supplementary Fig. 6b). Additionary, we did not detect significant differences in the amount of pancreatic insulin and insulin secretion in freshly isolated islets from Inhbb-Lyve1 KO mice or Ad-INHBB-treated mice compared to the respective control mice (Supplementary Fig. 6c–e). These results suggest that Activin B does not directly act on β-cells to stimulate insulin secretion, nor does it increase insulin secretory capacity.
Although GLP-1 and GIP levels were not altered in mice treated with Ad-INHBB (Fig. 6g–i), Ad-INHBB increased the rapid surge of glucagon following the glucose challenge. Thus, we tested the effect of increased glucagon levels by Activin B on insulin secretion, as glucagon is now thought to be an insulin secretagogue43,44,45,46,47. Suppression of glucagon levels—achieved either by pretreatment with pramlintide, a synthetic amylin analog and potent inhibitor of glucagon secretion (Fig. 6j), or by a PDE4 inhibitor, which increased hepatic glucagon sensitivity (Fig. 5k–m)— largely abrogated the enhanced GSIS response observed following Ad-INHBB administration compared to control treatment (Fig. 6k, l). Moreover, pretreatment of mice with Ex9–39, a potent and selective GLP-1R inhibitor, almost completely abolished the enhanced GSIS response induced by Ad-INHBB (Fig. 6m, n). These data indicate that Activin B downregulates hepatic glucagon sensitivity, thereby increasing the secretion of glucagon, which binds to and activates GLP-1R, leading to enhanced glucose-stimulated insulin secretion.
Taken together, these data indicate that the glucose-lowering effect of Activin B is mediated through at least two independent mechanisms: one, FGF21-dependent enhancement of systemic insulin sensitivity, and the other, inhibition of gluconeogenesis by suppression of hepatic glucagon signaling and consequent hyperglucagonemia-mediated increase in insulin secretion (Supplementary Fig. 6f).
Discussion
In this study, we characterized Activin B as an LSECkine encoded by the fasting-responsive Inhbb gene, which has a beneficial effect on glucose metabolism. We found that Activin B ameliorated glucose intolerance in DIO mice by affecting multiple, mutually-independent processes involved in diabetic pathogenesis. Activin B increased FGF21 production, thereby improving peripheral insulin sensitivity. Additionally, Activin B suppressed gluconeogenesis by inhibiting hepatic glucagon action independently of FGF21, leading to hyperglucagonemia, which enhanced glucose-stimulated insulin secretion from pancreatic β cells through activation of GLP-1 receptor (Supplementary Fig. 6f).
Hepatic expression of Inhbb is attributed to LSECs, which are highly specialized endothelial cells forming a structurally and functionally unique capillary network that vascularizes the liver15,16. Although LSECs are known to secrete hemostatic factors, here, we report that LSECs could have metabolic regulatory functions through the production of “LSECkines” in response to fasting-feeding cycles. It has been established that in the hepatic regenerative process and the inflammatory reactions, growth factors and cytokines secreted from cells composed of the liver sinusoid, including Kupffer cells and stellate cells, stimulate hepatocytes and exert their functions. In this study, we demonstrated that stimulating Activin B signaling in hepatocytes affects metabolic procedures in the liver, suggesting that LSECkines could also closely communicate with hepatocytes and exert their regulatory function in metabolism. As gene transfer methods targeting LSECs have not yet been established, in this study, the biological effects of Activin B secreted by overproduction in hepatocytes were evaluated using viral vectors which are already applied for clinical use. The lack of complete mimicry of the physiological production mechanism is a potential limitation in this study.
Activin B increases Fgf21 expression in the liver through the canonical activin signaling ALK4-Smad2 dependent pathway. The Activin B-mediated improvement in insulin sensitivity largely depend on this increase in FGF21, as it was almost abolished in Fgf21 knockout mice. Furthermore, the expression of Fgf21 in the liver of mice with disrupted Inhbb was decreased by half compared to that in wild-type mice, underscoring the significance of FGF21 induction among the multiple metabolic functions of Activin B. While previous studies have shown that PPARα pathway controls expression of Fgf21 upon fasting, our data indicate that Activin B regulates Fgf21 expression independently of PPARα, suggesting a novel mechanism of FGF21 induction under fasting, which is through communication from LSECs to hepatocytes via Activin B. In the physiological fasting state, Activin B-induced suppression of hepatic glucagon actions and subsequent glucagon release could, together with the induction of FGF21, impact the energy metabolism of tissues other than hepatocytes, leading to the rewiring of whole-body fuel metabolism. In contrast, Activin B potentiates insulin secretion as well as insulin actions, facilitating body adaptation in the postprandial state. However, these possibilities need to be addressed in future studies.
Activin B increases insulin secretion despite enhancing insulin sensitivity. Although previous studies have reported that activin signaling suppresses insulin secretion23,42, we did not detect any direct effect of Activin B on insulin or glucagon secretion, at least under our subacute experimental conditions using isolated islets. In contrast, the overexpression of Activin B causes hyperglucagonemia, whereas the knockout of Activin B in endothelial cells results in hypoglucagonemia. It is known that hyperglucagonemia and pancreatic α-cell hyperplasia are induced in mice lacking liver-specific glucagon receptor48. The amount of glucagon supply from α-cells is thought to be regulated by hepatic glucagon demand, and the concept of “liver-α cell axis” has been proposed38. In this study, overexpression of Activin B suppressed hepatic glucagon activity along with reduced cAMP levels, whereas Inhbb-Lyve1 KO mice exhibited increased hepatic glucagon sensitivity. These findings suggest that Activin B, a fasting-responsive LSECkine, may regulate hepatic glucagon sensitivity. Furthermore, glucagon has been proposed to enhance insulin secretion by activating GLP-1 receptor signaling43,45,46,49. Enhancement of the GSIS response has also been observed in mice with impaired hepatic glucagon signaling50,51, which may have been mediated by hyperglucagonemia. The hyper-GSIS response observed in Ad-INHBB-treated mice was largely abolished by the inhibition of glucagon secretion and by a GLP-1 receptor-selective blocker. In contrast, the attenuated GSIS observed in Inhbb-Lyve1 KO mice was restored by glucagon, suggesting that glucagon levels play a critical role in modulating physiological GSIS response, and that hyperglucagonemia resulting from hepatic glucagon resistance may contribute to enhanced insulin responsiveness. Regarding the mechanism linking Activin B and glucagon, we found that Activin B suppressed intracellular cAMP levels by increasing the expression of PDE4, a key enzyme involved in cAMP degradation.
While the expression of Activin B does not seem to be altered under obese or diabetic conditions, the expression of its endogenous antagonist, FSTL3, is increased in the adipose tissue of mice and humans with obesity, which may contribute to the development of glucose metabolism disorders. The inhibition of FSTL3 in diabetic obese mice significantly improved glucose tolerance and insulin sensitivity.
In diabetes mellitus, several pathological disorders coincide, such as insulin resistance in peripheral insulin target tissues, increased glucose production from the liver, and impaired insulin secretion from pancreatic β cells. Treatment of patients with diabetes is usually initiated with a single medication, such as metformin, in combination with lifestyle modifications. In cases of insufficient blood glucose control, the treatment must be intensified to multidrug therapy, which potentially causes poor drug adherence, leading to the development of diabetic complications and poor prognosis. The problem is attributed to the fact that current therapeutic options lack medication capable of simultaneously fixing multiple components of the pathogenesis of diabetes at the same time. Here, we found that Activin B ameliorated glucose intolerance in diabetes by improving multiple components of diabetes pathogenesis, enhancing insulin sensitivity via FGF21 production, and suppressing hepatic glucagon signaling associated with glucose-induced insulin secretion. Therefore, therapeutic strategies that enhance the action of Activin B, such as the suppression of FSTL3 with neutralizing antibodies or the development of long-acting Activin B, are expected to improve all pathological disorders of diabetes, thereby achieving sustained glycemic control and preventing diabetic complications.
In conclusion, Activin B produced in the liver in the fasting state contributes to the maintenance of glucose homeostasis by increasing insulin sensitivity, suppressing gluconeogenesis, and promoting glucose-stimulated insulin secretion. Under obese conditions, increased FSTL3 antagonizes Activin B, thereby causing hyperglycemia. Thus, enhancing Activin B activity may represent a promising strategy for the treatment of diabetes.
Methods
This research complies with all relevant ethical regulations. Human studies were conducted in accordance with the Declaration of Helsinki and were approved by the Human Ethics Committee of Tokyo University (G0951) or the ethics committee of the University of Leipzig (363-10-13122010 and 017-12-230112). All subjects gave written informed consent before taking part in the study. All animal studies were reviewed and approved for the accepted standards of humane animal care under protocols approved by the Institutional Animal Care and Use Committee of the National Center for Global Health and Medicine (A045-2024) and the Animal Care Committee of the University of Tokyo (#21077).
Human tissue samples
To identify candidate adipokines with gene expression alterations associated with obesity, we conducted a comprehensive gene expression analysis on human adipose tissue. The study included 176 subjects (men n = 125; women n = 51) who were inpatients at the Department of Gastrointestinal Surgery, Tokyo University Hospital, and were undergoing surgery for gastric cancer. Adipose tissue samples were immediately frozen in liquid nitrogen after removal. Total RNA was extracted from adipose tissue samples using the Qiagen RNeasy Lipid Tissue Mini Kit (Venlo, Netherlands) and subsequently processed for microarray analysis. Extracted RNA was converted into labeled cDNA and hybridized onto the SurePrint G3 Human GE Microarray (Agilent Technologies, Santa Clara, CA, USA). Hybridization signals were quantified using an Agilent Technologies Scanner G2505B (Santa Clara, CA, USA). Data analysis was conducted using the Limma package (3.54.2) implemented in R (4.2.3) with Bonferroni correction.
Paired samples of visceral and subcutaneous adipose tissue were obtained from 188 participants (men n = 86; women n = 102) of European ancestry who underwent abdominal surgery as described in detail elsewhere52. The age of the subjects ranged from 16 to 86 years, and all had a BMI between 20.8 and 54.1 kg m−2. Liver tissue samples were obtained from 66 study participants (men n = 37; women n = 29) with a wide range of BMI (22.7–45.6 kg m−2) who underwent open abdominal surgery for Roux-en-Y bypass, sleeve gastrectomy, elective cholecystectomy or explorative laparotomy53. Participants were recruited in the context of the Leipzig Obesity BioBank to donate liver biopsies in addition to visceral and subcutaneous abdominal adipose tissue samples. There was no additional objective or invention related to the liver biopsy. Patients with liver cirrhosis and any malignancies (including hepatocellular carcinoma) were excluded from the biobank. Biopsies were taken in the context of standard medical care. Liver and adipose tissue samples were immediately frozen in liquid nitrogen. The mRNA purification, cDNA synthesis, and qPCR analyses were performed as described below. The relative expression levels were normalized to the value of 18S rRNA in each sample. Additional methods of phenotypic characterization have been described previously52,53.
Animal studies
Mice were housed in a temperature and humidity-controlled, specific pathogen-free animal facility at 22 ± 1 °C, under a 12 h light/dark cycle, and had free access to pelleted food and sterile water unless otherwise indicated. All mice used in the experiments for this study were male, C57BL6 background, aged between 6 and 24 weeks old, unless otherwise noted. C57BL/6 J and db/db (BKS.Cg-m +/+ Leprdb/Jcl) mice were obtained from CLEA Japan (Tokyo, Japan) and were maintained on standard laboratory chow (CE-2; CLEA Japan). Inhbb-Flox mice were generated at the National Center for Global Health and Medicine (NCGM, Tokyo, Japan) and crossed with Lyve1-Cre mice (JAX #012601). For diet-induced obesity (DIO) studies, mice were fed a high-fat diet (HFD, 60% kcal fat, D12492, Research Diet) for a minimum of 12 weeks to induce insulin resistance. Insulin deficiency was induced by intraperitoneal injection of STZ (100 mg/kg, twice every other day; Wako, Tokyo, Japan) into male C57BL/6J mice and the development of hyperglycemia was subsequently confirmed.
Intraperitoneal glucose tolerance tests (ipGTT), insulin tolerance tests (ITT), pyruvate tolerance tests (PTT), and glucagon challenge tests were performed following standard protocols. For the ipGTTs, mice were administered 20% d-glucose (2 g/kg, Otsuka Pharmaceuticals) via intraperitoneal injection after overnight fasting. For the ITTs, mice received intraperitoneal injections of human insulin (Humulin R; Eli Lilly, Indianapolis, IN). For the PTTs or glucagon challenge test, mice were administered 15% sodium pyruvate (1.5 g/kg, Wako Pure Chemicals) or glucagon G Novo (1 mg/kg, Novo Nordisk Pharma Ltd.) via intraperitoneal injection after an overnight fast. To investigate the insulin secretion response to glucose, in vivo GSIS tests were performed by intraperitoneally injecting 30% d-glucose (2 g/kg) after an overnight fast, and the plasma insulin concentration was determined.
For indirect calorimetry, animals were monitored individually in a metabolic cage system (ARCO-2000, ARCO SYSTEMS Ins., Kashiwa, Japan) with free access to a normal chow diet and drinking water. Oxygen consumption (VO2) and carbon dioxide production (VCO2) rates were recorded, and energy expenditure (EE) was calculated. The activity of animals was measured using beam break counts, which were recorded by infrared light sources and optical sensors. The total number of beam breaks per mouse was interpreted as physical activity. Body mass composition of non-anesthetized mice was assessed using the time-domain NMR minispec (Live Mice Analyzer system, Model LF90II, Bruker Optik). For blood chemistry analysis, tail vein blood was collected to measure glucose and hormone levels. Blood glucose was assessed using Glutest Neo Alpha or Glutest Mint (Sanwa Kagaku), and plasma hormone levels were measured with ELISA kits listed in the Supplementary Information.
Recombinant virus
Recombinant adenoviruses expressing InhibinβB (Ad-INHBB) and Fstl3 (Ad-Fstl3) were generated using the Adenovirus Expression Vector Kit (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions.
Recombinant adenoviruses for expression of short hairpin RNA targeting Inhbb (Ad-shInhbb) or Fgf21 (Ad-shFgf21) were generated using the BLOCK-iT adenoviral RNAi expression system (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Briefly, double-stranded oligonucleotides encoding shRNAs were generated, annealed, and cloned into a pENTR/U6 entry vector (Invitrogen). Sequences targeting murine Inhbb and Fgf21 were 5′-(1647) GCCTGTACTTCTTCGTCTCTA-3′ and 5′-(514) TCTCTATGGATCGCCTCACTT-3′, respectively. The linearized plasmid was subsequently transfected into HEK293A cells to generate viruses. As a negative control, a recombinant adenoviral vector expressing shRNA directed against LacZ was generated. The recombinant viruses were amplified in HEK293A cells, purified by cesium chloride density centrifugation, and dialyzed against PBS containing 10% glycerol prior to animal injection. Male mice received the purified adenoviruses (1 × 109 PFU) via tail vein injection and were sacrificed within 7 days after injection.
For a longer induction of Activin B, DIO mice were injected with AAV encoding EGFP or INHBB via the tail vein at a titer of 3 × 1011 vp/animal.
Blockade of FSTL3 using antisense oligonucleotide
To estimate the effect of Fstl3 knockdown, 8-week-old male db/db mice were dosed with antisense oligonucleotide or control oligonucleotide solutions (i.p., 50 mg/kg/wk) twice per week for 4 weeks. The antisense oligonucleotide used in this study had a uniform phosphorothioate backbone and a 20-base chimeric design with a 2′-O-(methoxy)ethyl modification on the first 5 and the last 5 bases. 5′-mG*mG*mG*mU*mU*G*G*A*A*G*G*T*A*C*T*G*G*G*C*mA*mG*mG*mG*mC-3′ Oligonucleotides with inversed sequence were used as control.
Administration of recombinant Activin B protein
Mice were injected with phosphate-buffered saline (PBS) or recombinant Activin B (i.p., 0.5 mg/kg) three times every other day for 1 week.
Fractionation of mouse liver and LSEC isolation
Hepatocytes and non-parenchymal cell (NPC) suspensions were obtained by digesting freshly removed livers from male C57BL6/J mice (9–12 weeks old) using Liver Perfusion Kit (Miltenyi Biotec, #130-128-030) or Mouse Liver Dissociation Kit (Miltenyi Biotec, #130-105-807) with a gentleMACS Dissociator. Hepatocytes were pelleted from the cell suspension by additional low-speed centrifugation (50×g, 2 min, 4 °C) and NPC-containing supernatants were collected. After filtering through a 70-μm cell strainer, the cells were collected by centrifugation and resuspended. After trapping the Kupffer cells from the NPC fraction using CD11b MicroBeads UltraPure (Miltenyi Biotec), the remaining cell suspensions were labeled with CD146 (LSEC) MicroBeads (Miltenyi Biotec) and applied onto LS magnetic-activated cell sorting (MACS) columns (Miltenyi Biotec), which were placed within the magnetic field of an MACS separator and washed three times with MACS buffer (Miltenyi Biotec). The flowthrough was collected as CD11b-CD146- double-negative cell fractions, and the CD146+ LSECs were eluted from the column.
Quantitative PCR analysis
Total RNA from tissues or fractionated cells were extracted using TRIzol reagent (Thermo Fisher) and an RNA Extraction kit (Favorgen Biotech Corporation). cDNA was generated from total RNA using high-capacity cDNA reverse transcription kit (Applied Biosystems) and quantitative PCR analysis using TaqMan assays (Applied Biosystems) or PrimeTime qPCR assays (Integrated DNA Technologies) were performed. The probes used are listed in Supplementary Information. Relative expression levels were normalized to the value of cyclophilin in each sample.
Immunoblot analysis
For protein extraction from isolated tissues, frozen tissues were homogenized in ice-cold tissue lysis buffer (25 mM Tris-HCl, 100 mM NaF, 50 mM Na4P2O7, 10 mM EGTA, 10 mM EDTA, 10 mM Na3VO4, pH 7.4, and 1% Nonidet P-40 supplemented with a protease inhibitor cocktail). Cells were washed with ice-cold PBS and lysed in ice-cold cell lysis buffer (25 mM Tris-HCl, 100 mM NaF, 10 mM Na4P2O7, 10 mM EGTA, 10 mM EDTA, 10 mM Na3VO4, pH 7.4, and 1% Nonidet P-40 supplemented with a protease inhibitor cocktail). Lysates were centrifuged at 17,000×g for 20 min at 4 °C, and the supernatant was recovered. The protein concentration was determined using the Bradford method (Bio-Rad) and adjusted to 2 mg/mL. Samples for immunoblotting were prepared by heating at 95 °C with Leammli buffer for 5 min. Equal amounts of lysate samples were resolved by SDS-PAGE and electroblotted onto polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Turbo system (Bio-Rad). After blocking in TBST containing 1% BSA, the membrane was incubated with primary antibody (1:1000, unless otherwise stated) at 4 °C overnight. After washing, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody and visualized using an electrochemiluminescence (ECL) substrate.
Reporter gene assays
HepG2 cells (ATCC, HB-8065) were seeded onto 24-well plates and transfected with Smad2/3-responsive SRE-Luc (from Dr. Bert Vogelstein, Addgene #16495)54, Smad1/5/8-responsive BRE-Luc (from Dr. Martine Roussel and Dr. Peter ten Dijke, Addgene #45126)55, or the indicated expression plasmids using Lipofectamine 3000 (Invitrogen). After incubation, as described in the figure legend, cells were harvested, and luciferase activity was measured using the dual-luciferase reporter assay system (Promega) following the manufacturer’s protocol.
Statistical analysis
All statistical analyses were performed using Prism 9 software (9.5.1, GraphPad Software). Differences between groups were analyzed for significance using the unpaired Student’s t-test, one-way analysis of variance (ANOVA) with Dunnett’s, Bonferroni’s, or Tukey’s multiple comparisons test, or two-way ANOVA with Sidak’s, Dunnett’s, or Tukey’s multiple comparisons test, according to the comparison scheme. P < 0.05 was considered statistically significant at p < 0.05. Statistical analysis for correlation in human data were performed using Spearman’s rank correlation test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings from this study are provided in the Article, Supplementary Information, and Source Data file. The microarray data generated in this study have been deposited in the GEO database under accession code GSE283367. Source data are provided with this paper.
References
Kubota, N. et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 8, 49–64 (2008).
Efeyan, A., Comb, W. C. & Sabatini, D. M. Nutrient-sensing mechanisms and pathways. Nature 517, 302–310 (2015).
Shah, N., Abdalla, M. A., Deshmukh, H. & Sathyapalan, T. Therapeutics for type-2 diabetes mellitus: a glance at the recent inclusions and novel agents under development for use in clinical practice. Ther. Adv. Endocrinol. Metab. 12, 20420188211042145 (2021).
Nogueiras, R., Nauck, M. A. & Tschop, M. H. Gut hormone co-agonists for the treatment of obesity: from bench to bedside. Nat. Metab. 5, 933–944 (2023).
Jastreboff, A. M. & Kushner, R. F. New frontiers in obesity treatment: GLP-1 and nascent nutrient-stimulated hormone-based therapeutics. Annu. Rev. Med. 74, 125–139 (2023).
Caruso, I. et al. Glucagon in type 2 diabetes: friend or foe? Diabetes Metab. Res. Rev. 39, e3609 (2023).
Haedersdal, S., Andersen, A., Knop, F. K. & Vilsboll, T. Revisiting the role of glucagon in health, diabetes mellitus and other metabolic diseases. Nat. Rev. Endocrinol. 19, 321–335 (2023).
Capozzi, M. E., D’Alessio, D. A. & Campbell, J. E. The past, present, and future physiology and pharmacology of glucagon. Cell Metab. 34, 1654–1674 (2022).
Inagaki, T. et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 (2007).
Pal, D. et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 18, 1279–1285 (2012).
Oike, Y. et al. Angiopoietin-related growth factor antagonizes obesity and insulin resistance. Nat. Med. 11, 400–408 (2005).
El Ouaamari, A. et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab. 23, 194–205 (2016).
Misu, H. et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 12, 483–495 (2010).
Iroz, A., Couty, J. P. & Postic, C. Hepatokines: unlocking the multi-organ network in metabolic diseases. Diabetologia 58, 1699–1703 (2015).
Gracia-Sancho, J., Caparros, E., Fernandez-Iglesias, A. & Frances, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 18, 411–431 (2021).
Poisson, J. et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J. Hepatol. 66, 212–227 (2017).
Wijayarathna, R. & de Kretser, D. M. Activins in reproductive biology and beyond. Hum. Reprod. Update https://doi.org/10.1093/humupd/dmv058 (2016).
Sjoholm, K. et al. The expression of inhibin beta B is high in human adipocytes, reduced by weight loss, and correlates to factors implicated in metabolic disease. Biochem. Biophys. Res. Commun. 344, 1308–1314 (2006).
Namwanje, M. & Brown, C. W. Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a021881 (2016).
Hashimoto, O. et al. Activin E controls energy homeostasis in both brown and white adipose tissues as a hepatokine. Cell Rep. 25, 1193–1203 (2018).
Adam, R. C. et al. Activin E-ACVR1C cross talk controls energy storage via suppression of adipose lipolysis in mice. Proc. Natl Acad. Sci. USA 120, e2309967120 (2023).
Li, L. et al. Activin signaling: effects on body composition and mitochondrial energy metabolism. Endocrinology 150, 3521–3529 (2009).
Bertolino, P. et al. Activin B receptor ALK7 is a negative regulator of pancreatic beta-cell function. Proc. Natl Acad. Sci. USA 105, 7246–7251 (2008).
Tao, R. et al. Inactivating hepatic follistatin alleviates hyperglycemia. Nat. Med. 24, 1058–1069 (2018).
Brown, M. L. et al. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity 19, 1940–1949 (2011).
Mukherjee, A. et al. FSTL3 deletion reveals roles for TGF-beta family ligands in glucose and fat homeostasis in adults. Proc. Natl Acad. Sci. USA 104, 1348–1353 (2007).
Brandt, C. et al. Obesity and low-grade inflammation increase plasma follistatin-like 3 in humans. Mediators Inflamm. 2014, 364209 (2014).
Walton, K. L., Makanji, Y. & Harrison, C. A. New insights into the mechanisms of activin action and inhibition. Mol. Cell. Endocrinol. 359, 2–12 (2012).
Bolton, J. et al. Molecular biomarkers for weight control in obese individuals subjected to a multiphase dietary intervention. J. Clin. Endocrinol. Metab. 102, 2751–2761 (2017).
Tortoriello, D. V., Sidis, Y., Holtzman, D. A., Holmes, W. E. & Schneyer, A. L. Human follistatin-related protein: a structural homologue of follistatin with nuclear localization. Endocrinology 142, 3426–3434 (2001).
Sidis, Y. et al. Follistatin-related protein and follistatin differentially neutralize endogenous vs. exogenous activin. Endocrinology 143, 1613–1624 (2002).
Sidis, Y. et al. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147, 3586–3597 (2006).
Ozelo, M. C. et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N. Engl. J. Med. 386, 1013–1025 (2022).
Wang, Y. et al. Activin B promotes the initiation and progression of liver fibrosis. Hepatol. Commun. 6, 2812–2826 (2022).
Kanamori, Y. et al. Regulation of hepcidin expression by inflammation-induced activin B. Sci. Rep. 6, 38702 (2016).
Canali, S. et al. Activin B induces noncanonical SMAD1/5/8 signaling via BMP type I receptors in hepatocytes: evidence for a role in hepcidin induction by inflammation in male mice. Endocrinology 157, 1146–1162 (2016).
Besson-Fournier, C. et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 120, 431–439 (2012).
Galsgaard, K. D. et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Endocrinol. Metab. 314, E93–E103 (2018).
Kaneko, K. et al. Class IA phosphatidylinositol 3-kinase in pancreatic beta cells controls insulin secretion by multiple mechanisms. Cell Metab. 12, 619–632 (2010).
Tsuchida, K. et al. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol. Cell Endocrinol. 220, 59–65 (2004).
Bonomi, L. et al. Activin B regulates islet composition and islet mass but not whole body glucose homeostasis or insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 303, E587–E596 (2012).
Wu, H. et al. Differential regulation of mouse pancreatic islet insulin secretion and Smad proteins by activin ligands. Diabetologia 57, 148–156 (2014).
Capozzi, M. E. et al. Glucagon lowers glycemia when beta-cells are active. JCI Insight https://doi.org/10.1172/jci.insight.129954 (2019).
Cabrera, O. et al. Intra-islet glucagon confers beta-cell glucose competence for first-phase insulin secretion and favors GLP-1R stimulation by exogenous glucagon. J. Biol. Chem. 298, 101484 (2022).
Zhu, L. et al. Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight https://doi.org/10.1172/jci.insight.127994 (2019).
Capozzi, M. E. et al. β cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight https://doi.org/10.1172/jci.insight.126742 (2019).
Svendsen, B. et al. Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25, 1127–1134 e1122 (2018).
Longuet, C. et al. Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes 62, 1196–1205 (2013).
Farahani, R. A. et al. The effect of glucagon-like peptide 1 receptor blockade on glucagon-induced stimulation of insulin secretion. Diabetes 72, 449–454 (2023).
Gu, W. et al. Glucagon receptor antagonist-mediated improvements in glycemic control are dependent on functional pancreatic GLP-1 receptor. Am. J. Physiol. Endocrinol. Metab. 299, E624–E632 (2010).
Sorensen, H. et al. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 55, 3463–3469 (2006).
Berndt, J. et al. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes 54, 2911–2916 (2005).
Cansby, E. et al. GCKIII kinases control hepatocellular lipid homeostasis via shared mode of action. J. Lipid Res. 65, 100669 (2024).
Zawel, L. et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1, 611–617 (1998).
Korchynskyi, O. & ten Dijke, P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 277, 4883–4891 (2002).
Acknowledgements
We thank F. Takahashi, R. Honma, and Y. Masaki for their technical assistance and all the members of the Kadowaki and Ueki laboratories for discussions and suggestions about this project. Activin B and Smad constructs were kindly provided by Kunihiro Tsuchida (Fujita Health University) and Tetsuro Izumi (Gunma University), respectively. We also thank Tadashi Okamura, Kenta Nakano Tetsuya Arai, Miwa Tamura-Nakano and Chinatsu Oyama (NCGM) for their technical support on Inhbb-Flox mice generation and histological analysis. This work was supported by Practical Research Project for Life-Style related Diseases including Cardiovascular Diseases and Diabetes Mellitus from Japan Agency for Medical Research and Development (AMED, 19ek0210127h0001, to K.U.), Grants-in-Aid for Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (#24249053, to K.U.), a grant from the Ministry of Health, Labor and Welfare (to K.U.), a grant for the Research Program on Hepatitis from AMED (JP18fk0210040, JP19fk0210040, JP20fk0210040, JP21fk0210090, JP22fk0210090, JP23fk0210090, and 24fk0210149 to K.U.), a Grant-in-Aid for Scientific Research (C) from MEXT (18K08503, to Y.O.), Grant-in-Aid for Young Scientists from MEXT (18K15075, to N.K.), and grants for Front Runner of Future Diabetes Research by the Japan Foundation for Applied Enzymology (to N.K.). The human studies in Leipzig were supported by a grant from Deutsche Forschungsgemeinschaft the SFB 1052 “Obesity mechanisms” (B01).
Author information
Authors and Affiliations
Contributions
N.K. designed the study, performed the experiments, and wrote the manuscript. Y.O., A.I., K.H., M.H., K.S., M.M., T.S., K.Y., Y.S., T.Y., H.A., M.B., and M.A. performed the experiments and contributed to the discussion. N.I. and K.K. performed the experiments and provided research materials. T.K. contributed to the discussion. K.U. designed the study, contributed to the discussion, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
Y.O., M.B., T.K., and K.U. are co-inventors of the patents related to the FSTL3 and Activin B (JP 2012-517332, JP 2012-517350, US 13/699,876, and EP 15163160.3), details of each are given below. The remaining authors declare no competing interests. JP 2012-517332 (Registration No. 5422799). Patent applicant: The University of Tokyo and SEKISUI MEDICAL CO., LTD. Inventors: Takashi KADOWAKI, Kohjiro UEKI, Yukiko OKAZAKI, Matthias BLUHER, and Sumiko OZAWA. Status: Registered. Patentee: Kohjiro UEKI and Takashi KADOWAKI. Relevant aspect: This patent covers the relationship between the expression of FSTL3 and BMI. JP 2012-517350 (Registration No. 5286602). Patent applicant: The University of Tokyo and SEKISUI MEDICAL CO., LTD. Inventors: Takashi KADOWAKI, Kohjiro UEKI, Yukiko OKAZAKI, Matthias BLUHER, and Sumiko OZAWA. Status: Registered. Patentee: Kohjiro UEKI and Takashi KADOWAKI. Relevant aspect: This patent covers the role of Activin B and FSTL3 in the regulation of insulin sensitivity. US 13/699,876 (Registration No. 8,895,011). Patent applicant: The University of Tokyo and SEKISUI MEDICAL CO., LTD. Inventors: Takashi KADOWAKI, Kohjiro UEKI, Yukiko OKAZAKI, Matthias BLUHER, and Sumiko OZAWA. Status: Registered. Patentee: Kohjiro UEKI and Takashi KADOWAKI. Relevant aspect: This patent covers the role of Activin B and FSTL3 in the regulation of insulin sensitivity. EP 15163160.3 (Registration No. 2921562). Patent applicant: The University of Tokyo and SEKISUI MEDICAL CO., LTD. Inventors: Takashi KADOWAKI, Kohjiro UEKI, Yukiko OKAZAKI, Matthias BLUHER, and Sumiko OZAWA. Status: Registered in Germany, France, and the United Kingdom. Patentee: Kohjiro UEKI and Takashi KADOWAKI. Relevant aspect: This patent covers the relationship between the expression of FSTL3 and BMI.
Peer review
Peer review information
Nature Communications thanks Enis Kostallari and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kobayashi, N., Okazaki, Y., Iwane, A. et al. Activin B improves glucose metabolism via induction of Fgf21 and hepatic glucagon resistance. Nat Commun 16, 3678 (2025). https://doi.org/10.1038/s41467-025-58836-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58836-w
