
Abstract
Ferroptosis, a form of cell death instigated by iron-dependent lipid peroxidation reactions (LPO), is emerging as a promising therapeutic target for cancer. While the mechanisms governing LPO induction and suppression have gradually been unveiled, questions persist regarding the specific cellular location of LPO and the utilization of iron in driving cell death. A comprehensive understanding of these aspects holds significant potential for advancing therapeutic applications in disease management. Here, we show lysosomal LPO in the initiation of ferroptosis, leveraging the hidden abilities of fluorescent detection probes. Intra-lysosomal LPO triggers iron leakage, fostering cell-wide LPO by augmenting lysosomal membrane permeabilization (LMP). Conversely, cell lines with low susceptibility to ferroptosis do not exhibit LMP. This deficiency is rectified by the concurrent administration of chloroquine, leading to LMP induction and subsequent cell death. These findings underscore enhancing LMP induction efficacy as a strategic approach to surmount resistance to therapies in cancer.
Similar content being viewed by others

Introduction
Inhibition of glutathione peroxidase 4 (GPX4) is known to induce cell death triggered by oxidized lipids1. This cell death is named ferroptosis because of the important role of iron in oxidized lipids generation2. Ferroptosis induction and inhibition have garnered attention as a novel therapeutic approach for cancer treatment and neurodegenerative diseases3,4,5,6,7. However, some cancer cells have been found to be less susceptible to ferroptosis, which may limit cancer therapy.
The identification and understanding of ferroptosis-inducing factors and their inhibitors have progressed, and the occurrence of pores formation in the plasma membrane (PM) during the final step of cell death has been documented8. Nevertheless, the origins of highly reactive lipid peroxides responsible for ferroptosis remain subjects of ongoing debate. For example, the use of alkyne-tagged ferrostatin or deuterated polyunsaturated fatty acids (PUFA), along with Raman microscopy, suggested the ER as a site of oxidized lipid formation9,10. On the other hand, it has been reported that ROS generated in lysosomes contribute to the induction of ferroptosis11. Identifying oxidized lipid formation and iron distribution is a pivotal aspect of understanding the ferroptosis induction mechanism, with potential implications for cancer therapies and explaining variations in ferroptosis susceptibility among cancer cell lines.
Here, we show the site of lipid radical generation and Fe2+ release mechanism for ferroptosis induction, proposing an antitumor effect on cancer cells with low susceptibility to ferroptosis beyond the additional inhibition of enzymes that reduce oxidized lipids.
Results
Optimal use of LPO detection probes identified important organelle targets for cell death
We evaluated the characteristics of the commonly used LPO detection probes (i.e., NBD-Pen12, C11-BODIPY13 and Liperfluo14) to act as antioxidants (Supplementary Fig. 1). If these probes act as ferroptosis inhibitors and prevent cell death, some probe molecules may directly capture the molecules that induce ferroptosis. Thus, we investigated whether these LPO detection probes exhibit ferroptosis-inhibitory effects by focusing on non-small cell lung cancer (NSCLC) cell lines (e.g., Calu-1). Notably, C11-BODIPY inhibited RSL3-induced ferroptosis at a concentration of 10 µM, while Liperfluo showed no inhibition up to 10 µM (Fig. 1a). Conversely, NBD-Pen completely abrogated RSL3-induced ferroptosis at 2 µM. Furthermore, NBD-Pen demonstrated the ability to suppress ferroptosis induced by other inducers (ML162 and erastin) in addition to an RSL3-treated NSCLC cell line (H661) and a fibrosarcoma cell line (HT1080) at lower concentrations than C11-BODIPY (Supplementary Fig. 2a). Remarkably, the fluorescence intensity of NBD-Pen increased as detected by flow cytometry upon RSL3 treatment, and this increase was effectively suppressed by adding OH-Pen12, a compound with a significant inhibitory effect on ferroptosis (Supplementary Fig. 2b, c). In contrast, the increased fluorescence intensity of C11-BODIPY in flow cytometry upon RSL3 treatment remained unaffected by OH-Pen. Moreover, when each probe was used at the concentration that did not exhibit any inhibitory effect on ferroptosis induction, C11-BODIPY displayed a fluorescence shift, while NBD-Pen did not (Supplementary Fig. 2d). These findings strongly suggested that NBD-Pen is capable of capturing lipid peroxidation, a critical trigger of ferroptotic cell death.

a Viability of Calu-1 cells treated with RSL3 (0.1 µM) and each probe simultaneously for 24 h. b MS peak areas of NBD-Pen radical adducts. Calu-1 cells were treated with RSL3 (0.1 µM) ± liproxstatin-1 (Lip-1, 1 µM) for 2.5 h and administered NBD-Pen (10 µM) for 20 min. c Scheme of NBD-Pen fluorescence imaging. Cells were co-treated with RSL3 and NBD-Pen for 3 h. NBD-Pen, 2 µM; RSL3, 0.1 µM; Lip-1, 1 µM. d Live-cell fluorescence imaging with NBD-Pen in Calu-1 cells. e Quantification of the NBD-Pen puncta per cell. f Viability of Calu-1 cells treated with RSL3 (0.1 µM) + Lyso-NBD-Pen (more polar diastereomer) or NBD-Pen for 24 h. g Co-localization of Lyso-NBD-Pen (more polar diastereomer) with lysosomal marker. Cells were treated with RSL3 (0.1 µM) ± Lip-1 (1 µM) and Lyso-NBD-Pen (0.1 µM) simultaneously for 3 h and then labeled with LysoTracker Red DND-99 and Hoechst 33342 for 20 min. Data are mean ± s.d. (a, b, f), median ±95% CI (e). Sample size: (a, b, f) n = 3 independent experiments, (e) n = 39 (Control), 35 (RSL3), 49 (RSL3 + Lip-1) number of cells present in three different images from each sample. Three independent experiments were performed with similar results (e). Statistical analysis: p values were calculated by one-way ANOVA with Tukey–Kramer test (a, b, e) or by two-way ANOVA with Sidak’s multiple comparison test (f). Scale bar: 20 µm (d, g). Source data are provided as a Source Data file.
We subsequently explored radical species as the inducing ferroptotic cell death molecules. In our previous study, we developed a method for detecting lipid-derived radicals based on their reaction with NBD-Pen and carbon-centered radicals, combined with LC-MS/MS analysis15. Using this method, we successfully detected six truncated alkyl-type radical species, including •C5H11 and •C8H15, in RSL3-treated Calu-1 cells (Fig. 1b). Importantly, the levels of these radical species were significantly reduced in the presence of the liproxstatin-1 (Lip-1) which prevents LPO production.
Our results provided insights into the essential organelle involved in ferroptosis induction when using cell imaging with NBD-Pen. Specifically, fluorescence puncta were observed in some cancer cells (i.e., Calu-1, H661, and HT1080) treated with ferroptosis inducers, including RSL3, ML162, and erastin, and these puncta were effectively eliminated by Lip-1 or OH-Pen (Fig. 1c–e and Supplementary Fig. 3a–h). In contrast, fluorescence puncta derived from C11-BODIPY were not detected (Supplementary Fig. 3i). In cells treated with tert-butyl hydroperoxide (TBHP), which induces LPO throughout the cell, NBD-Pen was widely distributed intracellularly, ruling out the possibility that the NBD-Pen puncta depend on the probe’s biased distribution (Supplementary Fig. 3j). Consistent with this, subcellular localization studies with NBD-Pen-NOMe12, an “always-on” type of NBD-Pen probe, revealed a distribution throughout the entire cell that remained unaltered upon RSL3 treatment (Supplementary Fig. 3k, l). It was observed that NBD-Pen puncta co-localized with the LysoTracker but not with markers for the ER, PM, or mitochondria, regardless of the ferroptosis inducer or cell type used in this study (Supplementary Fig. 4a–c). Moreover, NBD-Pen puncta co-localized with a fluorescent protein-fused lysosome marker (LAMP1) and parts of an early endosome marker (Rab5) and a late endosome marker (Rab7) (Supplementary Fig. 4d, e). These results indicate that NBD-Pen detects LPO induced in the endosomal/lysosomal compartment.
To further investigate whether lysosomal LPO is responsible for ferroptosis induction, we synthesized a lysosome-targeting probe based on NBD-Pen with morpholine (a lysosomal targeting group)16, named 2,2,6-trimethyl-4-(2-morpholinoethyl)(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino-6-pentylpiperidine-1-oxyl (Lyso-NBD-Pen) (more polar diastereomer 1, less polar diastereomer 2) (Supplementary Fig. 5a). Importantly, Lyso-NBD-Pen (more polar diastereomer) effectively suppressed ferroptosis induced by RSL3, ML162, and erastin at lower concentrations than NBD-Pen (Fig. 1f and Supplementary Fig. 5b, c). Therefore, the following experiments used Lyso-NBD-Pen (more polar diastereomer). Lyso-NBD-Pen showed better inhibitory effects on ferroptosis in H661, IA-LM and HT1080 cells (Supplementary Fig. 5d). In particular, Lyso-NBD-Pen exhibited specific localization within the lysosome, supported by the result that Lyso-NBD-Pen-NOMe (3) is more abundant in lysosomes than NBD-Pen-NOMe (Fig. 1g and Supplementary Fig. 5e–h). Furthermore, Lyso-NBD-Pen puncta were not observed with Lip-1 treatment, suggesting that Lyso-NBD-Pen has an inhibitory effect on ferroptosis by acting as a radical trapping antioxidant (Fig. 1g). Our studies demonstrated that the reactivity of Lyso-NBD-Pen towards lipid radicals generated by 2,2’-azobis(2-aminodipropane) dihydrochloride (AAPH, an LPO initiator) in liposomes was nearly identical to that of NBD-Pen (Supplementary Fig. 5i). These findings provide compelling evidence that the lysosomal lipid peroxidation are critical factors responsible for ferroptosis induction under the experimental conditions examined in this study.
Lysosomal lipid peroxidation induces lysosomal membrane permeabilization
The next question that emerged was how lysosomal LPO induces ferroptosis in these cells. Some molecular dynamics simulations and experiments have shown that the oxidation of lipid membranes increases their permeability17,18,19. Thus, our attention was directed towards LMP in particular, a phenomenon associated with the disruption of lysosomal function characterized by an increase in lysosomal pH and the leakage of lysosomal contents20. Notably, imaging and flow cytometric analysis with acridine orange as a pH sensor revealed that treatment with RSL3 for 3 h increased lysosomal pH, which was effectively prevented by treatment with lysosomal LPO inhibitors, including Lip-1, NBD-Pen, and Lyso-NBD-Pen (Lyso-NP) (Fig. 2a and Supplementary Fig. 6a). Additionally, lower concentrations of RSL3 did not induce cell death and did not increase lysosomal pH (Supplementary Fig. 6b, c).

a, b Live-cell fluorescence imaging with acridine orange or fluorescent dextran in Calu-1 cells treated with RSL3 (0.1 µM) ± inhibitors for 3 h. c Time-dependent change of lactate dehydrogenase (LDH) released from Calu-1 cells treated with RSL3 (0.1 µM) ± Lip-1. d Lysosomal Fe2+ detected by Lyso-RhoNox. Calu-1 cells were pretreated with deferoxamine (DFO, 50 µM) for 1 h and then labeled with Lyso-RhoNox (1 µM) and Hoechst 33342 for 30 min. Relative intensity per cell is presented. e Intracellular Fe2+ detected by FerroOrange. Calu-1 cells were treated with RSL3 (0.1 µM) ± inhibitors for 3 h and then labeled with FerroOrange (1 µM) and Hoechst 33342 for 30 min. Relative intensity per cell is shown. NBD-Pen fluorescence analysis after LMP in Calu-1 cells treated with RSL3 (0.1 µM) ± Lip-1. Scheme (f), live-cell fluorescence imaging (g), and co-staining with ER-Tracker or CellMask Orange Plasma Membrane Stain at 3 and 5 h (h). i Death of Calu-1 cells treated with RSL3 (0.1 µM) ± inhibitors for 24 h. Inhibitors were added either at the same time as RSL3 (0 h) or 1–6 h later. Inhibitors: Lip-1 (1 µM); NBD-Pen (0.3 µM); Lyso-NBD-Pen (Lyso-NP, more polar diastereomer, 0.1 µM). Data are mean ± s.d. (c, d, e, i). Sample size: (c, i) n = 3 independent experiments, (d) n = 21 number of cells present in three different images from each sample, (e) n = 27 (Control), 36 (RSL3), 28 (RSL3 + Lip-1), 31 (RSL3 + NBD-Pen), 34 (RSL3 + Lyso-NBD-Pen) number of cells present in three different images from each sample. Three independent experiments were performed with similar results (d, e). Statistical analysis: p values were calculated by two-way ANOVA with Sidak’s multiple comparison test (c), by the two-tailed unpaired t test (d) or by one-way ANOVA with Tukey–Kramer test (e). Scale bar, (a, h) 10 µm; (b, d, e, g) 20 µm. Source data are provided as a Source Data file.
To better understand the effect of RSL3 on LMP, we monitored the release of FITC-dextran (40 kDa), which accumulates in lysosomes through endocytosis. Although FITC-dextran remained within lysosomes after 3 h of RSL3 treatment, it diffused into the cytosol after 4 h (Fig. 2b). Additionally, cathepsin B, which is typically confined to the lysosomal lumen, was found to be distributed not only within lysosomes but also in the cytosol in RSL3-treated cells, whereas it co-localized with the lysosomal marker LAMP1 in control cells and cells treated with lysosomal LPO inhibitors (e.g., Lip-1, NBD-Pen, and Lyso-NBD-Pen) (Supplementary Fig. 6d). Consistent with this observation, the cytosolic level of cathepsin B protein increased with RSL3 treatment (Supplementary Fig. 6e). These results strongly suggested that lysosomal LPO induces LMP in RSL3-treated Calu-1 cells. Notably, the release of lactate dehydrogenase (LDH) was not observed within 3 h after RSL3 treatment (Fig. 2c). In addition, LMP was observed in erastin-treated Calu-1 cells and RSL3-treated H661 cells before LDH release (Supplementary Fig. 7a, b). LMP was not observed in Calu-1 cells treated with staurosporine (STS), a typical apoptosis inducer (Supplementary Fig. 7c–e). These suggest that LMP precedes PM disintegration in ferroptotic cells, but not in cells with STS-induced apoptosis.
The cathepsin B inhibitor CA-074Me has previously been demonstrated to inhibit erastin-induced ferroptosis in pancreatic cancer cells21. However, in our study, CA-074Me did not suppress RSL3- or erastin-induced ferroptosis in Calu-1, H661 and HT1080 cells (Supplementary Fig. 8a), showing that cathepsin B is not involved in ferroptosis, at least in these three cell lines. Given that iron is abundant in lysosomes, primarily due to cellular uptake via transferrin receptor- or CD44-mediated endocytotic pathway and ferritinophagy (a form of autophagy specific to ferritin)22,23,24, we hypothesized that LMP might lead to the diffusion of lysosomal iron into the cytosol and trigger cell death. In fact, Lyso-RhoNox25, a lysosomal ferrous iron-selective probe, showed fluorescence emission in Calu-1 cells, which was decreased by pretreatment with deferoxamine (DFO, an iron chelator) (Fig. 2d). Moreover, in Calu-1 cells treated with RSL3 for 3 h, fluorescence intensity of the subcellular iron-selective probe FerroOrange was enhanced throughout the cells (Fig. 2e). It was effectively inhibited by Lip-1, NBD-Pen, and Lyso-NBD-Pen treatment. These findings suggest that lysosomal Fe2+ is diffused throughout the cell upon RSL3 treatment.
Given that Fe2+ promotes lipid peroxidation through the Fenton reaction26, it is plausible that lysosomal iron release accelerates lipid peroxidation throughout the ferroptotic cell. To assess lipid peroxidation after LMP induction, Calu-1 cells were stained with NBD-Pen 3 h after RSL3 treatment (Fig. 2f). Under these conditions, NBD-Pen fluorescence was observed throughout a wide intracellular range in RSL3-treated cells (Fig. 2g), which was in stark contrast to the image shown in Fig. 1d, where RSL3 and NBD-Pen were simultaneously applied. Our results indicated that LPO in other organelles besides lysosome is rarely induced upon simultaneous treatment of NBD-Pen due to the lysosomal LPO and LMP inhibition, suggesting that LPO elsewhere than in lysosome is induced following lysosomal LPO and LMP. A similar distribution of NBD-Pen fluorescence intensity throughout the cells was observed in erastin-treated Calu-1 cells (Supplementary Fig. 8b). Notably, this fluorescence co-localized with the ER-Tracker after 3 h of RSL3 treatment and subsequently co-stained well with CellMask at 5 h (Fig. 2h).
To clarify the contribution of lysosomal Fe2+ leakage to cell death, we investigated whether inactivation of leaked lysosomal Fe2+ suppresses ferroptosis. Treatment with FerroOrange (which oxidizes ferrous iron27) or deferiprone (DFP, an iron chelator that can penetrate cells) after LMP suppressed ferroptosis (Supplementary Fig. 8c), revealing that increased cytoplasmic Fe2+ is responsible for cell death. Consistent with this, FerroOrange or DFP inhibited LPO enhancement throughout the cell under these conditions (Supplementary Fig. 8d,e). Furthermore, analysis of NBD-Pen radical adducts showed that truncated alkyl radicals are predominantly generated at a wide range of cellular sites following LMP (Supplementary Fig. 8f). Given that these truncated alkyl radicals are presumed to result from β-scission of alkoxy radicals generated by the reaction of lipid hydroperoxides with ferrous iron15,28,29, this result indicates that Fe2+ is essential for LPO expansion from the lysosome to other organelles. In line with this, DFP treatment following LMP reduced radical production (Supplementary Fig. 8f). These results support our hypothesis that leaked lysosomal Fe2+ plays a causal role in LPO spread to the entire cell during ferroptosis.
A recent study by von Krusenstiern et al. showed that exogenous fatty acids, such as oleic acid (OA), incorporate primarily into ER phospholipids and alter susceptibility to ferroptosis10. This suggests that the ER membrane is a crucial site of lipid peroxidation. Accordingly, we investigated the effect of exogenous OA (used as an anti-ferroptotic fatty acid) on lysosomal LPO and LMP. Consistent with that previous report, OA pretreatment inhibited RSL3-induced ferroptosis and lipid peroxidation in the ER (Supplementary Fig. 9a,b). However, RSL3-induced lysosomal lipid peroxidation and LMP remained unaffected in OA-pretreated cells (Supplementary Fig. 9c,d). These results rule out the possibility of lysosomal LPO and LMP resulting from LPO in the ER.
To further investigate the importance of LMP in ferroptotic cell death, we evaluated the cell death inhibitory effect of NBD-Pen and Lyso-NBD-Pen when they were administered after LMP had occurred. Cell death was entirely prevented when these compounds were added within 2 h of RSL3 treatment. However, the inhibitory effect rapidly diminished upon administration more than 3 h after RSL3 treatment (Fig. 2i and Supplementary Fig. 9e). These findings collectively demonstrate that lysosomal lipid peroxidation-mediated LMP is a crucial mechanism in ferroptotic cell death.
Effect of lysosomotropic agents on cell lines with low susceptibility to ferroptosis
The vulnerability to ferroptosis depends on the cell state and cell types6,23. For instance, susceptibility to ferroptosis differs significantly within NSCLC cells in an epithelial state. HARA, H460, A549, KNS62, and PC9 cells are low susceptibility to RSL3-induced cell death than Calu-1 cells (Fig. 3a). Consequently, proposing ferroptosis-based therapy for NSCLC cells has limitations. To shed light on the response to RSL3 of cells with low susceptibility to ferroptosis, we conducted cell imaging experiments using NBD-Pen. NBD-Pen puncta were observed even when RSL3 concentrations were insufficient to induce cell death. However, treatment with Lip-1 completely inhibited the increased fluorescence intensity (Fig. 3b, c and Supplementary Fig. 10a, b). In particular, lysosomal pH did not increase in RSL3-treated cells under these conditions (Fig. 3d and Supplementary Fig. 10c). These findings suggest that RSL3 induces lysosomal LPO but does not trigger LMP in cells with low susceptibility to ferroptosis. Notably, this mechanism also occurred in a neuroblastoma cell line (SH-SY5Y) (Supplementary Fig. 10a–c).

a Viability of non-small cell lung cancer (NSCLC) cells treated with RSL3 for 24 h. b Live-cell fluorescence imaging in PC9 cells treated with NBD-Pen (2 µM) after 6 h of treatment with RSL3 (1 µM) ± Lip-1 (1 µM). c Quantification of the NBD-Pen puncta per cell of PC9. d Flow cytometric analysis of acridine orange red fluorescence in PC9 cells treated with RSL3 (1 µM) for 6 h. e Viability of PC9 cells treated with RSL3 + Chloroquine (CQ, 100 µM), ammonium chloride (NH4Cl, 120 mM), and methylamine (80 mM) for 24 h. Viability was normalized to untreated conditions or treatment with lysosomotropic agent alone as a control. f Viability of PC9 cells treated with RSL3 + CQ (100 µM) ± inhibitors (1 µM Lip-1, 50 µM DFO, 2 µM NBD-Pen, 10 µM Z-VAD-FMK, 5 µM Nec-1) for 24 h. g Intracellular Fe2+ detected by FerroOrange. PC9 cells were treated with RSL3 (1 µM) ± CQ (100 µM) ± Lip-1 (1 µM) for 3 h and then labeled with FerroOrange (1 µM) and Hoechst 33342 for 30 min. Relative intensity per cell is presented. h Live-cell fluorescence imaging of FITC-dextran (40 kDa) in PC9 cells treated with RSL3 (1 µM) + CQ (100 µM) ± Lip-1 (1 µM) for 6 h. Data are mean ± s.d. (a, e, f, g), median ± 95% CI (c). Sample size: (a, e, f) n = 3 independent experiments, (c) n = 35 (Control), 27 (RSL3), 35 (RSL3 + Lip-1) number of cells present in three different images from each sample, (g) n = 29 (Control), 34 (RSL3), 34 (CQ), 33 (RSL3 + CQ), 37 (RSL3 + CQ + Lip-1) number of cells present in three different images from each sample. Three independent experiments were performed with similar results (c, g). Statistical analysis: p values were calculated by one-way ANOVA with Tukey–Kramer test (c, f, g). Scale bar: 20 µm (b, g), 10 µm (h). Source data are provided as a Source Data file.
We hypothesized that facilitating LMP in low-susceptible cells could induce ferroptosis at lower RSL3 concentrations. Lysosomotropic agents, such as chloroquine (CQ), ammonium chloride (NH4Cl), and methylamine, accumulate in lysosomes, leading to lysosomal swelling and pH elevation30,31,32. A previous report indicated that enlarged lysosomes are more susceptible to membrane rupture33. Therefore, we employed a combination treatment of lysosomotropic agents and ferroptosis inducers to induce LMP, resulting in ferroptotic cell death. Consistent with prior reports, CQ, NH4Cl, and methylamine induced lysosomal swelling in PC9 cells, while the V-ATPase inhibitor bafilomycin A1 (BafA1) did not (Supplementary Fig. 10d). In PC9 cells, lysosomotropic agents enhanced ferroptosis induction with lower doses of GPX4 inhibitors (RSL3, ML162) (Fig. 3e and Supplementary Fig. 11a–c). In contrast, BafA1 did not enhance RSL3-induced ferroptosis, ruling out the possibility that the inhibition of lysosomal degradation function is responsible for sensitizing cells to ferroptosis (Supplementary Fig. 11d). The cell death accelerated by lysosomotropic agents was effectively suppressed by Lip-1, DFO, and NBD-Pen, but not by the apoptosis inhibitor Z-VAD-FMK or the necroptosis inhibitor Necrostatin-1 (Nec-1) (Fig. 3f and Supplementary Fig. 11e). Notably, STS- and H2O2-induced cell death was not augmented by CQ (Supplementary Fig. 11f). Other NSCLC cell lines and SH-SY5Y with low susceptibility to ferroptosis also displayed increased sensitivity to RSL3-induced ferroptosis when treated with CQ (Supplementary Fig. 11g). Collectively, these findings demonstrate that lysosomotropic agents promote ferroptotic cell death induction in NSCLC and other cancer type with low susceptibility to ferroptosis via GPX4 inhibition.
Although it is conceivable that CQ blocks lysosomal iron uptake by inhibiting lysosomal function, lysosomal iron did not decrease significantly, at least after 6 h of CQ treatment (Supplementary Fig. 12a, b). Notably, co-treatment with RSL3 and CQ elevated the cytoplasmic Fe2+ level (Fig. 3g). In support of these results, FITC-dextran remained within lysosomes in RSL3-treated cells, but it diffused into the cytoplasm in CQ co-treated cells (Fig. 3h). These findings suggest that co-treatment with CQ facilitates lysosomal iron leakage throughout the cell. Inactivation of lysosomal Fe2+ upon pre-treatment with Lyso-RhoNox or cytoplasmic iron upon post-treatment with FerroOrange or deferiprone suppressed CQ-enhanced ferroptosis (Supplementary Fig. 12c–e), supporting the assertion that lysosomal iron and its leakage are responsible for the induction of ferroptosis amplified by CQ co-treatment. Consistent with these results, the levels of various LPO products that are hallmarks of ferroptosis were increased by co-treatment with RSL3 and CQ (Supplementary Fig. 12f). Therefore, we revealed that combining CQ with RSL3 promotes iron diffusion from lysosomes and increases LPO levels throughout the cell, resulting in cell death in cells with low susceptibility to ferroptosis.
Chloroquine enhances GPX4 KO H460 tumor suppression in vivo
Lastly, we examined the possibility of CQ treatment as a therapeutic approach to ferroptosis-low-susceptible cells. GPX4-knockout (GPX4KO) cells were adopted to examine the effect of CQ in ferroptosis-low-susceptible cells on in vivo tumor development. To confirm that CQ also induces cell death due to GPX4 deficiency, we generated GPX4KO H460 cells that can survive after withdrawal of Lip-1 (Supplementary Fig. 13a, b). CQ induced cell death and LMP in GPX4KO cells, but not in wild-type cells (Supplementary Fig. 13c, d). Interestingly, CQ treatment produced a pronounced reduction in tumor growth of GPX4KO H460 cells, without affecting body weight (Fig. 4a and Supplementary Fig. 13e, f). Additionally, tumor weight displayed a significant decrease in the group subjected to CQ treatment on tumor of GPX4KO cells, coupled with accumulation of the lipid peroxidation product 4-HNE (Fig. 4b, c and Supplementary Fig. 13g). These results collectively suggest that concurrent CQ treatment, in conjunction with GPX4 inhibition, may effectively overcome low susceptibility to ferroptosis.

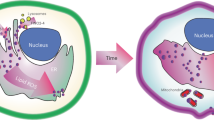
a In vivo growth of subcutaneous GPX4KO H460 xenografts in Balb/c-nu mice. Five days after transplantation, mice were treated randomly with vehicle or CQ (25 mg/kg intraperitoneally daily). The distribution of tumor volume is shown. b Tumor weight at the end of the experiment. Data for panel (a, b) are mean ± s.d., n = 9 (vehicle), n = 10 (CQ) independent experiments, p values were calculated by two-way ANOVA with Sidak’s multiple comparison test (a) or by the two-tailed unpaired t test (b). c 4-HNE accumulation in the group subjected to CQ treatment on tumor of GPX4KO cells. Representative 4-HNE immunostaining images of four different tumor samples and the relative intensity normalized to vehicle group of WT H460 tumor are shown. Scale bar, 40 µm. Data are mean ± s.d., n = 4 independent experiments, p values were calculated by the two-tailed unpaired t test. d Model illustrating the mechanism of cell death initiated by lysosomal LPO. Purple closed circles represent lipid radicals. Created in BioRender. Yuma, S. (2025) https://BioRender.com/c82c186. Source data are provided as a Source Data file.
Discussion
In this study, we have demonstrated that lysosomal LPO triggers LMP during ferroptosis, and Fe2+ diffuses throughout the cell to induce LPO (Fig. 4d). Interestingly, NBD-Pen inhibited ferroptosis induction most effectively. This could be considered to be based on the reacting molecular species, the second-order rate constant, and the intracellular organelle distribution of probes. Previously, lipid peroxides were detected during ferroptosis as phospholipid-derived hydroperoxides and their end products34,35,36. Here, we detected lipid-derived alkyl radicals such as •C5H11 radical (Fig. 1b). These alkyl radicals, which are causative molecules during the LPO chain reaction, may be produced via Fe2+-induced β-scission reaction in lipid hydroperoxide15,28,29. Additionally, the second-order reaction rate constant between nitroxide (reaction molecule of NBD-Pen) and carbon-centered radicals such as lipid-derived alkyl radicals is generally 108 to 109 M−1s−1 (that of C11-BODIPY and ROO• is 104 M−1s−1)37,38, and this high secondary reaction rate constant could effectively suppress the chain reaction during LPO. Moreover, when TBHP, which induces LPO across the cell, was applied simultaneously with NBD-Pen or NBD-Pen-NOMe (the always-on form of NBD-Pen) was co-treated with RSL3 for 3 h, these probes were distributed throughout the cell (Supplementary Fig. 3j,l). This cell-wide distribution further supports the assertion that lysosomes are the essential site in the early stages of ferroptosis induction. Furthermore, the use of Lyso-NBD-Pen made the analysis more straightforward. Otherwise, under our conditions, C11-BODIPY could not detect LPO in lysosomes (Supplementary Fig. 3i). The results suggest that the detection probe can function as an inhibitor by reacting with the molecule causing the effect, rather than just being used for imaging. Furthermore, since the detection probe is present throughout the cell, it provides strong evidence for identifying a molecule within the lipid radical-generating organelle as the one responsible for ferroptosis.
LPO is detected in various organelles such as ER, mitochondria and plasma membrane during ferroptosis induction10,34,39. However, it had remained unclear at which organelle LPO is initiated and triggers cell death. Notably, the lysosome plays an important role in cellular iron homeostasis22 and this lysosomal function has been reported to be involved in reactive oxygen species (ROS) generation and ferroptosis11. Furthermore, salinomycin induces the sequestration of iron in lysosomes, leading to lysosomal ROS generation, LMP and the resulting ferroptotic cell death, highlighting that lysosomal iron as the initial trigger of salinomycin-induced ferroptosis6. In this manner, lysosomal iron and ROS have long been pointed out as triggers of ferroptosis, and a recent study reported LPO starts in lysosomes upon ferroptosis induction40. However, how lysosomal iron induces LPO throughout the cell and ultimately results in plasma membrane damage are still debated. In this study, we characterized that LPO initiates in lysosomes during GPX4 inhibition-induced ferroptosis and that lysosomal LPO is critical for cell death (Fig. 1d). Interestingly, our results suggested that lysosomal iron leaked by lysosomal LPO-mediated LMP contributes to the spread of LPO to organelles other than the lysosome (Supplementary Fig. 8c–f). A recent study by Rodrigues et al. focusing on lysosomal iron showed that LPO is initiated in the lysosome, which propagates to the other proximal organelle membranes via a radical chain reaction40. Presumably, the expansion of LPO from lysosomes to the whole cell is caused by both membrane-to-membrane propagation and leaked lysosomal iron. In light of the above, our results provide one mechanism by which lysosomal iron enhances cell-wide LPO and further emphasize the key roles of iron and LPO in the lysosome for ferroptosis, along with the recent study using a different approach40.
Our results revealed that lysosomal LPO induces LMP during GPX4 inhibition-induced ferroptosis. Recently, several papers have shown that LMP is induced in a ROS-dependent manner upon stimulation with the ferroptotic cell death inducer FIN56 or disulfiram41,42. In addition, Mai et al. pointed out that LMP is presumably the result of Fe-mediated lysosomal ROS production and lysosomal membrane oxidation through the analysis of salinomycin-treated cells6. We have shown that lipid radicals were generated in lysosomes under the experimental conditions in this study (Fig. 1d,g). Previous molecular dynamics simulations predicted that oxidation of the lipid bilayer membrane causes the hydrophobic tail to fold toward the aqueous phase, leading to the loss of membrane stability and increased membrane permeability17,18,43. It is quite possible that lipid-derived alkyl radicals and its products are the causal molecules that induce LMP, as evidenced by the generation of lipid-derived alkyl radicals in lysosomes using NBD-Pen and Lyso-NBD-Pen. Furthermore, regarding the causal relationship between ferroptosis and LMP induced by GPX4 inhibition, we explicate that LMP is essential for LPO expanding during ferroptosis progression. Importantly, facilitating LMP upon treatment with lysosomotropic agents sensitizes low-susceptible cells to ferroptosis (Fig. 3e), highlighting the importance of LMP in ferroptosis.
To date, cell types in which it is challenging to induce ferroptosis have been suggested. To overcome this ferroptosis resistance, the inhibition of GPX4, FSP1, and other enzymes that suppress LPO across cell lines has been the primary strategy44,45,46. In this study, we successfully exploited the fact that LPO occurs in lysosomes even in cancer cells with low sensitivity to ferroptosis; additional LMP induction led to ferroptotic cell death in cultured cells and animal tumors. The next challenge is to explore proteins or molecules involved in LMP suppression in cancer cells with low susceptibility to ferroptosis, which should help to elucidate regulatory mechanisms of LMP.
In summary, this study has demonstrated the accumulation of LPO in lysosomes and LMP contribute to ferroptosis induction through the successful use of LPO-detecting fluorescent probes. Furthermore, LMP induction can overcome cell lines with low susceptibility to ferroptosis. These findings suggest the future potential for cancer therapy that induces ferroptosis beyond targeting the inhibition of enzymes that suppress LPO.
Methods
Ethical statement
This research complies with all relevant ethical regulations. All procedures and animal care were approved by the Committee on Ethics of Animal Experiments, Graduate School of Pharmaceutical Sciences, Kyushu University, and conducted in accordance with the Guidelines for Animal Experiments of the Graduate School of Pharmaceutical Sciences, Kyushu University.
Chemicals and reagents
NBD-Pen, NBD-Pen-NOMe, and OH-Pen were prepared as previously reported7. The synthetic method for Lyso-NBD-Pen and Lyso-NBD-Pen-NOMe can be found in the Supplementary Note 1. The reagents that were used in this study are as follows: RSL3 (Sigma-Aldrich), erastin (Cayman Chemical), ML162 (Sigma-Aldrich), liproxstatin-1 (Sigma-Aldrich), BODIPY 581/591 C11 (Invitrogen), Liperfluo (Dojindo), tert-butyl hydroperoxide (Tokyo Chemical Industry), acridine orange (Nacalai Tesque), FerroOrange (Dojindo), Lyso-RhoNox (synthesized at Prof. Hideko Nagasawa’s laboratory), FITC-dextran (40 kDa) (Sigma-Aldrich), hydrogen peroxide (FUJIFILM Wako Pure Chemical), staurosporine (Cayman Chemical), CA-074Me (Peptide Institute), oleic acid (FUJIFILM Wako Pure Chemical), chloroquine (Nacalai Tesque), ammonium chloride (Sigma-Aldrich), methylamine (Tokyo Chemical Industry), bafilomycin A1 (Sigma-Aldrich), deferiprone (Tokyo Chemical Industry), deferoxamine mesylate (Abcam), Z-VAD-FMK (Cayman Chemical), and necrostatin-1 (Cayman Chemical).
Cell culture
Calu-1 (HTB-54), H661 (HTB-183), H460 (HTB-177) and SH-SY5Y (CRL-2266) were purchased from the ATCC; A549 (86012804) and PC9 (90071810) were from ECACC; HARA (JCRB1080.0), KNS62 (IFO50358) and HT1080 (IFO50354) were from the Japanese Collection of Research Bioresources (JCRB); and IA-LM (RCB0554) was from RIKEN Bioresource Center (BRC). All cell line identities were confirmed by the commercial source on each provider on website (ATCC, ECACC, JCRB, BRC). First, H661, H460, PC9, HARA, KNS62, and IA-LM were cultured in RPMI1640 (Nacalai Tesque), Calu-1 was cultured in McCoy’s 5 A Medium (Gibco), and A549 was cultured in DMEM (4.5 g/L glucose) (Nacalai Tesque). SH-SY5Y was cultured in DMEM/Hamʼs F-12 (Nacalai Tesque). HT1080 was cultured in MEM (Nacalai Tesque) supplemented with 1 mM sodium pyruvate solution (Sigma) and 1% MEM non-essential amino acid solution (Nacalai Tesque). All growth media were supplemented with 10% FBS and 100 U/mL penicillin and 100 µg/mL streptomycin (Nacalai Tesque). All cell lines were grown at 37 °C in a humid atmosphere with 5% CO2.
Cell viability assay
Cells (1 × 104 cells/well) were seeded in a 96-well plate and cultured for 24 h. Cell viability was assessed by MTT assay (Nacalai Tesque) after drug treatment for 24 h. Briefly, pretreatment with oleic acid (20 µM) was performed for 24 h, followed by RSL3 (0.1 µM). MTT solution was added to each well, followed by incubation for 1 h at a final concentration of 5 mg/mL in 5% CO2 at 37 °C. The supernatant was removed and 100 µL of DMSO was added to each well. Absorbance at 570 nm was measured using an EnSpire Multimode Plate Reader (Perkin Elmer) and EnSpire Manager v4 (Perkin Elmer). The relative cell viability (%) was calculated by setting the absorbance of the control to 100%. In cases where lysosomotropic agent and cell death inducer were simultaneously added, treatment with lysosomotropic agent alone was used as a control.
LDH release assay
Cells (1 × 104 cells/well) were seeded in a 96-well plate and cultured for 24 h. The Cytotoxicity LDH Assay Kit-WST (Dojindo) was used in accordance with the manufacturer’s protocol to measure cell death. After drug treatment, 100 µL of supernatant was transferred to a new 96-well plate, and assay buffer was added. Then, the mixture was incubated at room temperature for 30 min, followed by addition of a stop solution. Absorbance was measured at 490 nm using an EnSpire Multimode Plate Reader (Perkin Elmer) and EnSpire Manager v4 (Perkin Elmer). The LDH release rate was calculated using the absorbance of the lysis buffer group as 100%.
Flow cytometric analysis
Cells (3 × 105 cells/well) were seeded in a six-well plate and cultured for 24 h. Then, they were stained with either NBD-Pen or C11-BODIPY to detect lipid peroxidation. To conduct an LMP assay, the cells were stained with 10 µM acridine orange (Nacalai Tesque) and incubated for 30 min in 5% CO2 at 37 °C. The cells were rinsed with PBS and detached using accutase (Nacalai Tesque). Next, they were washed with 1 mL of HBSS and resuspended in 0.5 mL of HBSS. Afterwards, the cells were filtered through a cell strainer and analyzed using a flow cytometer (FACS Verse, Becton Dickinson) with a 488 nm laser for excitation and BD FACSuite Software v1.0.6 (Becton Dickinson). Fluorescence intensity was measured using the FITC detector with wavelengths at 511–543 nm for NBD-Pen and C11-BODIPY or the PerCP-Cy5.5 detector with wavelengths at 673–727 nm for acridine orange red fluorescence. At least 10,000 events were assayed per sample. Data were analyzed using FlowJo v10 software (Becton Dickinson).
Live-cell confocal imaging
Cells (3 × 104 cells/compartment) were seeded in four-well CELL View glass-bottomed dishes (Greiner Bio-One) and incubated for 24 h. After treatment and staining, the cells were washed three times with PBS. For the FITC-dextran release assay, the cells were seeded in four-well CELL View glass-bottomed dishes (Greiner Bio-One) at 2 × 104 cells per compartment and incubated for 24 h. The cells were pretreated with 1 mg/ml FITC-dextran for 24 h. They were treated after washing three times with PBS. Images were acquired using a confocal laser microscope (LSM780; Carl Zeiss Microscopy GmbH) with a 63× objective lens and ZEN 2011 v14 (Carl Zeiss). The following wavelengths were used for excitation and emission, respectively: Hoechst33342, λex =405 nm, λem = 410–476 nm; NBD-Pen, C11-BODIPY (oxidized), FITC-dextran, λex = 488 nm, λem = 516–559 nm; LysoTracker, ER-Tracker, MitoTracker, CellMask, C11-BODIPY (reduced), FerroOrange, Lyso-RhoNox, λex = 561 nm, λem = 595–637 nm; and acridine orange, λex =488 nm, λem (green fluorescence) = 515–560 nm, λem (red fluorescence) = 600–710 nm. The images were analyzed using ZEN 3.4 software (Carl Zeiss Microscopy GmbH). The images shown in the figure are representative of at least three independent experiments. Quantification of puncta or mean fluorescence intensity per cell was performed using Fiji (Image J) v1.54 software (NIH). Cell outlines were manually marked as regions of interest (ROI), and each ROI was measured. Relative intensity was calculated using the average intensity of the control group as 1 for ferrous probe imaging. Three different images were analyzed for each group and the total number of cells in the image was defined as n. Colocalization rate was quantified by Coloc2 in ImageJ with Pearson’s correlation coefficient.
Fluorescent staining of live cells
To evaluate co-localization with organelle markers, cells were treated with 0.2 µM LysoTracker Red DND-99 (Invitrogen), 0.5 µM ER-Tracker Red (Invitrogen), 2.5 µg/mL CellMask Orange Plasma Membrane Stain (Invitrogen), and 0.15 µM MitoTracker Red CM-H2Xros (Invitrogen) and incubated for 20 min in 5% CO2 at 37 °C. For nuclear staining, cells were treated with 1 µg/mL Hoechst 33342 (Nacalai Tesque) along with other probes. For live-cell imaging with acridine orange, cells were stained with 10 µM acridine orange (Nacalai Tesque) and incubated for 30 min in 5% CO2 at 37 °C. For ferrous iron imaging, cells were stained with 1 µM FerroOrange (Dojindo) or 1 µM Lyso-RhoNox in HBSS and incubated for 30 min in 5% CO2 at 37 °C.
Immunofluorescence imaging
Cells (3 × 104 cells/compartment) were seeded in four-well CELL View glass-bottomed dishes (Greiner Bio-One) and incubated for 24 h. After treatment with drugs, the cells were washed three times with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. They were then permeabilized and blocked with 5% normal goat serum (Jackson ImmunoResearch) in PBST (PBS with 0.3% Triton X-100) for 30 min at room temperature. Next, the cells were stained with mouse anti-Cathepsin B antibody (Santa Cruz Biotechnology, sc-365558) 1:200 and rabbit anti-LAMP1 antibody (Cell Signaling Technology, 9091) 1:200 for 3 h at room temperature. The cells were then washed three times with PBST and stained with Cy3 AffiniPure Goat Anti-Mouse IgG (Jackson ImmunoResearch, 115-165-146) 1:200, Alexa Fluor 488 AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch, 111-545-003) 1:200, along with 1 µg/mL Hoechst 33342 (Nacalai Tesque) at room temperature for 1 h. The cells were subsequently washed three times with PBST. Images were acquired using a confocal laser microscope (LSM780; Carl Zeiss Microscopy GmbH) with a 63× objective lens and ZEN 2011 v14 (Carl Zeiss). The following were used as excitation and emission wavelengths, respectively: Hoechst33342, λex =405 nm, λem = 410–476 nm; Alexa Fluor 488, λex = 488 nm, λem = 516–559 nm; and Cy3, λex = 561 nm, λem = 595–637 nm. For quantification of lysosome size, the size of LAMP1 puncta per cell was averaged using Fiji (Image J) v1.54 software (NIH). Relative size was calculated using the average size of the control group as 1. Three different images were analyzed for each group and the total number of cells in the image was defined as n.
Western blotting
Cells were washed and incubated in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% deoxycholic acid, 1 mM benzylsulfonyl fluoride, 1 mM sodium orthovanadate, protease inhibitor cocktail, pH 7.5) on ice for 30 min. The solution was collected with a scraper and centrifuged at 10,000 × g for 15 min. Protein concentration was determined by the Bradford assay using the Bio-Rad Protein Assay Kit (Bio-Rad). Protein concentrations were normalized and diluted with 5× SDS sample buffer (50% glycerol, 0.05% Bromophenol blue, 150 mM Tris–HCl, 5% SDS, 25% 2-mercaptoethanol, pH 6.8). Protein was resolved on 10% or 15% polyacrylamide gel using SDS-PAGE and transferred onto PVDF membranes (Merck Millipore). Membranes were blocked in Blocking One (Nacalai Tesque) at room temperature for 30 min, and incubated overnight at 4 °C with primary antibodies in Blocking One. They were then washed three times with TBS-T and incubated for 1 h at room temperature with secondary antibodies. Next, membranes were washed three times with TBS-T and reacted with WSE-7120 EzWestLumi Plus (ATTO). ChemiDoc MP and Image Lab v5.2.1 (Bio-Rad) were used for imaging. The antibodies used included mouse anti-Cathepsin B antibody (Santa Cruz Biotechnology, sc-365558) 1:1000; mouse anti-GAPDH antibody (MBL, M171-3) 1:3000; rabbit anti-LAMP1 antibody (Cell Signaling Technology, 9091) 1:1000; rabbit anti-Calreticulin antibody (Cell Signaling Technology, 12238) 1:1000; rabbit anti-VDAC antibody (Cell Signaling Technology, 4661) 1:1000; rabbit anti-GPX4 antibody (Abcam, ab125066) 1:1000; goat anti-mouse IgG & IgM antibody, HRP conjugate (Merck Millipore, AP130P) 1:3000; and goat anti-rabbit IgG HRP antibody (R&D Systems, HAF008) 1:3000.
Overexpression plasmid construct and generation of cell lines
For subcloning, desired DNA sequences of LAMP1-RFP (Origene, RC100048), mRFP-Rab5 (addgene, 14437) and mCh-Rab7A (addgene, 61804) were amplified using KOD -Plus- Neo (TOYOBO) or Q5® Hot Start High-Fidelity 2X Master Mix (New England Biolabs) and PCR products were purified using FastGene™ Gel/PCR Extraction Kit (Fast Gene). Ligation reaction of PCR products with digested pLJM1 vectors was performed using NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs). The plasmid was confirmed by sequencing and used for lentiviral production. After 72 h of infection with Calu-1 cells, selection was performed with 5 µg/mL puromycin (Nacalai Tesque).
Lentiviral production
Lenti-X 293 T (TaKaRa Bio) cells (3 × 105 cells/well) were seeded in 12-well plates and cultured overnight. To 200 µL of OptiMEM (Gibco) were added 0.8 µg of cloned lentiviral plasmid, 0.6 µg of packaging plasmid (psPAX2, Addgene, 12260), and 0.4 µg of envelope plasmid (pMD2.G, Addgene, 12259). Next, 10.8 µg of PEI was added and incubated at room temperature for 10 min. The solution was added dropwise to Lenti-X 293 T cells cultured in 300 µL of medium and incubated for 24 h in 5% CO2 at 37 °C. The transfection medium was replaced with 500 µL of fresh medium and cells were incubated for 48 h. Viral supernatant was collected and used for lentiviral transduction.
ICP-MS analysis for iron in lysosome-enriched fraction
For lysosome purification, the well-established magnetic bead technique Lyso-IP47 was used. To generate cells stably expressing T192-mRFP-3×HA, PC9 cells were infected with lentivirus produced by transfection of Lenti-X 293 T cells with pLJM1-Tmem192-mRFP-3×HA (addgene, 134631). The cells were seeded in a 15 cm dish at 4 × 106 cells and cultured for 24 h. After CQ treatment, the cells were washed with PBS and detached using a scraper. The pelleted cells were resuspended in 1 ml of KPBS and gently homogenized with 20 strokes of a Dounce homogenizer. Then, the homogenate was centrifuged at 1000 × g for 2 min. The supernatant was incubated with 100 µL of anti-HA magnetic beads (Thermo Scientific, 88837) for 20 min. The beads with bound lysosomes were washed three times with KPBS on a DynaMag Spin magnet. The immunoprecipitates were eluted using lysis buffer for 10 min and quantified using protein assay BCA kit (Nacalai Tesque).
The lysosome fraction samples were treated with 62% nitric acid, followed by centrifugation at 20,000 × g for 10 min. Ultrapure water was added to the supernatant and nitric acid was diluted down to a final concentration of 1%. The samples were filtered and analyzed with an ICP-MS (Agilent 7900). The iron concentration of the sample was calculated from the calibration curve prepared with iron standard solution (Nacalai Tesque). Iron concentration in samples was normalized against protein concentration. Three biological replicates were prepared for each sample and each sample was measured once.
Generation of GPX4-knockout cell lines using CRISPR/Cas9
The single guide RNA (sgRNA) sequence targeting GPX4 was 5′-CTTGGCGGAAAACTCGTGCA-3′, referring to a previous study3. sgRNA was cloned into BsmbI-digested lentiCRISPR v2-Blast vectors (Addgene, 83480). Ligation reaction of sgRNA with digested vectors was performed using Ligation High Ver. 2 (TOYOBO). The reaction mixture was transformed into competent DH5α cells (TOYOBO) and plasmids were isolated using the Mini Plus Plasmid DNA Extraction System (VIOGENE). The plasmid was confirmed by sequencing and used for lentiviral production.
H460 cells (2.5 × 104 cells/well) were seeded in a 12-well plate and incubated overnight. Cell culture medium was replaced with 400 µL of fresh medium supplemented with 12.5 µg/mL polybrene and 100 µL of viral supernatant. After incubation for 24 h, medium was replaced with fresh medium containing 0.05 µM Lip-1 and incubated for 48 h. Selection was performed with 5 µg/mL blasticidin (Nacalai Tesque). After selection for 3 days, the single-cell clone was isolated, and the knockout clone was validated by western blotting. GPX4-knockout H460 cells were maintained in medium with or without Lip-1.
Lipid peroxidation assay in liposome
For liposome preparation, 200 mg of egg phosphatidylcholine (egg PC, NOF) and 8 mg of dihexadecyl phosphate (DCP, Sigma-Aldrich) were dissolved in chloroform/methanol (4:1, v/v). Solvent was evaporated for 30 min and vacuum-dried for 10 min to form a lipid film at the bottom of the flask. The film was hydrated with 10 mL of phosphate buffer (10 mM, pH 7.4). The liposome suspension was then extruded 10 times using a mini extruder (Avanti Polar Lipids) equipped with a 100 nm polycarbonate membrane (Cytiva). Then, liposome suspension (containing 5 mg/mL egg PC) with 2,2’-azobis(2-aminodipropane) dihydrochloride (AAPH, FUJIFILM Wako Pure Chemical) were incubated at 37 °C for 1 h. After the addition of 5 µM NBD-Pen or Lyso-NBD-Pen, fluorescence intensity was measured every 2 min using an EnSpire Multimode Plate Reader (Perkin Elmer) and EnSpire Manager v4 (Perkin Elmer) at 37 °C. Excitation and emission wavelengths were λex = 470 nm and λem =530 nm. Fluorescence intensities were normalized to the average fluorescence intensity of the first measurement for each probe set as 1.
Apoptosis assay
Calu-1 cells (3 × 105 cells/well) were seeded in a six-well plate and cultured for 24 h. After treating the cells with 1 µM STS for 6 h, they were washed with PBS. A scraper was used to detach cells, which were then washed with 1 mL of HBSS. To detect apoptosis, the Annexin V-FITC Apoptosis Detection Kit (Nacalai Tesque) was used, in accordance with the manufacturer’s protocol. Cells were passed through a cell strainer and assayed with a flow cytometer (FACS Verse, Becton Dickinson) with a 488 nm laser for excitation and BD FACSuite Software v1.0.6 (Becton Dickinson). Fluorescence intensity was measured using the FITC detector with wavelengths at 511–543 nm for Annexin V-FITC and the PerCP-Cy5.5 detector with wavelengths at 673–727 nm for propidium iodide. At least 10,000 events were assayed per sample. Data were analyzed using FlowJo v10 software (Becton Dickinson).
Isolation of cytosol fraction
Calu-1 cells were seeded in a 10 cm dish at 2 × 106 cells and cultured for 24 h. After drug treatment, the cells were washed with PBS and detached using a scraper. Next, the cells were centrifuged at 800 × g for 5 min and the supernatant was removed. The cells were then resuspended in 100 µL of fraction buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, pH 7.4) containing protease inhibitor cocktail and homogenized in an ice-cold 1-mL homogenizer. After centrifugation at 800 g for 5 min, the supernatant was transferred and ultracentrifuged at 100,000 × g for 1 h. The supernatant was then used as the cytosolic fraction.
LC-MS/MS analysis for lipid radicals and NBD-Pen adducts
Calu-1 cells were seeded in a 10 cm dish at 2 × 106 cells and cultured for 24 h. The cells were treated with RSL3 (0.1 µM) for 2.5 h and administered NBD-Pen (10 µM) for 20 min. The cells were then washed with cold PBS and detached using a scraper. After centrifugation at 10,000 g for 10 min and removal of the supernatant, the cells were resuspended in 200 µL of ice-cold methanol including 100 µM dibutylhydroxytoluene (BHT) and EDTA. The cell suspension was homogenized by Sonifier 250D (Branson) and centrifuged at 10,000 × g for 10 min. The supernatant was used as the sample. Three biological replicates were prepared for each sample and each sample was measured once. For the analysis of NBD-Pen and lipid radical adducts, LC-MS/MS was performed using an LCMS-8060 (Shimadzu). The mass spectrometer was equipped with an electron spray ionization source. The LC conditions were as follows: injection volume, 5 µL; autosampler temperature, 4 °C; column, InertSustain C18 column (2.1 × 150 mm, particle size of 3 µm, GL Sciences); column temperature, 40 °C; mobile phase, 5 mM ammonium formate in acetonitrile/H2O (2:1, v/v) (A) and 5 mM ammonium acetate in isopropanol/H2O (95:5, v/v) (B); flow rate, 0.4 mL/min; and gradient, 0%–100% B, 0–20 min; 100% B, 20–25 min. MS analysis was performed in negative-ion mode and MRM transitions for individual radical adducts are described in Supplementary Table 1. The LC-MS/MS analysis was performed using LabSolutions v5 (Shimazu).
LC-MS/MS analysis for lipid peroxidation products
A total of 2 × 106 PC9 cells were seeded in a 10 cm dish and cultured for 24 h. The cells were treated with RSL3 (1 µM) and CQ (100 µM) for 6 h. The cells were then washed with cold PBS and detached using a scraper. After centrifugation at 10,000 × g for 10 min and removal of the supernatant, the cells were resuspended in 200 µL of ice-cold methanol including 100 µM dibutylhydroxytoluene (BHT) and EDTA. The cell suspension was homogenized by Sonifier 250D (Branson) and centrifuged at 10,000 × g for 10 min. The supernatant was used as the sample. Three biological replicates were prepared for each sample and each sample was measured once. LC-MS/MS was performed using an LCMS-8060 mass spectrometer (Shimadzu) equipped with an electron spray ionization source. The LC conditions were as follows: injection volume, 5 µL; autosampler temperature, 4 °C; column, InertSustain C18 column (2.1 × 150 mm, 3 μm particle size; GL Sciences); column temperature, 40 °C; mobile phase, 5 mM ammonium formate in acetonitrile:H2O = 2:1 (A) and isopropanol:methanol = 95:5 (B); flow rate, 0.4 mL/min; and gradient elution, 0–22.5 min 0–100% B, 22.5–27.5 min 100% B. MS analysis was performed in positive-ion mode and MRM transitions for individual lipid peroxidation products are described in Supplementary Table 2. The LC-MS/MS analysis was performed using LabSolutions v5 (Shimazu). MS peak areas were normalized by LysoPC [18:1(d7)] as an internal standard. The normalized MS peak areas were log10-transformed and auto-scaled using MetaboAnalyst 6.048. The heatmap was generated using GraphPad Prism v9 (GraphPad Software).
Animal experiment
Four-week-old male BALB/c-nu mice were purchased from Jackson Laboratory Japan. All mice were housed in a light-controlled room (light/dark cycle of 12 h/ 12 h) at 24 ± 1 °C and 60% ± 10% humidity, and the animals had free access to water and CLEA Rodent Diet CE-2 (Clea Japan). For the xenograft tumor experiment, wild-type or GPX4-knockout H460 cells (8 × 106 cells in 100 µl of PBS) were injected subcutaneously into the right flank of 6-week-old male BALB/c-nu mice. After the tumor reached approximately 100 mm3 in size, the mice were randomized and treated with vehicle or chloroquine (25 mg/kg intraperitoneally) in PBS once daily. Tumor was measured using a caliper daily, and tumor volume was calculated according to the following formula: length × width2 × 1/2. Endpoints apply when tumors reach 1000 mm3 in volume, 1.5 cm in diameter, or 10% of body weight, none of which were exceeded in this study. Dissected tissues were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek) and frozen in liquid nitrogen. Frozen tissues were sectioned at a thickness of 10 μm using a cryostat for immunohistochemistry analysis. The tissue sections were fixed with 4% paraformaldehyde in PBS for 10 min and then incubated with PBST (PBS with 0.3% Triton X-100) for 30 min at room temperature. They were blocked with 5% normal donkey serum (Jackson ImmunoResearch) in PBST for 30 min at room temperature. Subsequently, sections were incubated with primary antibody (mouse anti-HNE-histidine monoclonal antibody, a gift from Dr. Uchida, University of Tokyo) 1:100 at 4 °C overnight. After washing, sections were incubated with secondary antibody (Donkey Anti-Mouse IgG H&L Alexa Fluor 647, Abcam, ab150107) 1:400 for 30 min at room temperature. Sections were mounted using Vibrance Antifade Mounting Medium with DAPI (Vector Laboratories) and analyzed using a confocal laser microscope (LSM780; Carl Zeiss Microscopy GmbH) with a 40× objective lens and ZEN 2011 v14 (Carl Zeiss). The following were used as excitation and emission wavelengths, respectively: DAPI, λex = 405 nm, λem = 410–476 nm; and Alexa Fluor 647, λex = 633 nm, λem = 638–746 nm. Quantification of mean fluorescence intensity was performed using Fiji (Image J) v1.54 software (NIH).
Statistical analysis
Data are expressed as the mean ± standard deviation or median ± 95% confidence interval for each group. Statistical significance was evaluated using two-tailed unpaired t test and one-way or two-way analysis of variance (ANOVA), followed by Sidak’s or Tukey’s multiple comparison tests. p < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism v9 (GraphPad Software).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIf-mediated cell death. Cell Metab. 8, 237–248 (2008).
Dixon, S. J. et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Stockwell, B. R. et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Mai, T. T. et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 9, 1025–1033 (2017).
Rodriguez, R., Schreiber, S. L. & Conrad, M. Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol. Cell 82, 728–740 (2022).
Riegman, M. et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 22, 1042–1048 (2020).
Gaschler, M. M. et al. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 13, 1013–1020 (2018).
von Krusenstiern, A. N. et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat. Chem. Biol. 19, 719–730 (2023).
Torii, S. et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 473, 769–777 (2016).
Yamada, K. I. et al. Fluorescence probes to detect lipid-derived radicals. Nat. Chem. Biol. 12, 608–613 (2016).
Drummen, G. P. C., Van Liebergen, L. C. M., Op den Kamp, J. A. F. & Post, J. A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe: (Micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med 33, 473–490 (2002).
Yamanaka, K. et al. A novel fluorescent probe with high sensitivity and selective detection of lipid hydroperoxides in cells. RSC Adv. 2, 7894–7900 (2012).
Matsuoka, Y., Izumi, Y., Takahashi, M., Bamba, T. & Yamada, K. I. Method for structural determination of lipid-derived radicals. Anal. Chem. 92, 6993–7002 (2020).
Lin, J., Yang, K. & New, E. J. Strategies for organelle targeting of fluorescent probes. Org. Biomol. Chem. 19, 9339–9357 (2021).
Wong-Ekkabut, J. et al. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 93, 4225–4236 (2007).
Wiczew, D., Szulc, N. & Tarek, M. Molecular dynamics simulations of the effects of lipid oxidation on the permeability of cell membranes. Bioelectrochemistry 141, 107869 (2021).
Freitas, F. P. et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature 626, 401–410 (2024).
Wang, F., Gómez-Sintes, R. & Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931 (2018).
Gao, H. et al. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res Commun. 503, 1550–1556 (2018).
Rizzollo, F., More, S., Vangheluwe, P. & Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem Sci. 46, 960–975 (2021).
Müller, S. et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 12, 929–938 (2020).
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W. & Kimmelman, A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 508, 105–109 (2014).
Hirayama, T., Miki, A. & Nagasawa, H. Organelle-specific analysis of labile Fe(ii) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 11, 111–117 (2019).
Winterbourn, C. C. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 82–83, 969–974 (1995).
Hirayama, T., Niwa, M., Hirosawa, S. & Nagasawa, H. High-throughput screening for the discovery of iron homeostasis modulators using an extremely sensitive fluorescent probe. ACS Sens 5, 2950–2958 (2020).
Qian, S. Y., Guo, Q. & Mason, R. P. Identification of spin trapped carbon-centered radicals in soybean lipoxygenase-dependent peroxidations of ω-3 polyunsaturated fatty acids by LC/ESR, LC/MS, and tandem MS. Free Radic. Biol. Med 35, 33–44 (2003).
Qian, S. Y., Yue, G. H., Tomer, K. B. & Mason, R. P. Identification of all classes of spin-trapped carbon-centered radicals in soybean lipoxygenase-dependent lipid peroxidations of ω-6 polyunsaturated fatty acids via LC/ESR, LC/MS, and tandem MS. Free Radic. Biol. Med 34, 1017–1028 (2003).
Ohkuma, S. & Poole, B. Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J. Cell Biol. 90, 656–664 (1981).
Kondratskyi, A. et al. Ferroquine, the next generation antimalarial drug, has antitumor activity. Sci. Rep. 7, 15896 (2017).
Villamil Giraldo, A. M., Appelqvist, H., Ederth, T. & Öllinger, K. Lysosomotropic agents: Impact on lysosomal membrane permeabilization and cell death. Biochem Soc. Trans. 42, 1460–1464 (2014).
Ono, K., Kim, S. O. & Han, J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol. Cell Biol. 23, 665–676 (2003).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Sun, W. Y. et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 17, 465–476 (2021).
Feng, H. & Stockwell, B. R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 16, e2006203 (2018).
Bagryanskaya, E. G. & Marque, S. R. A. Scavenging of organic C-centered radicals by nitroxides. Chem. Rev. 114, 5011–5056 (2014).
Yoshida, Y., Shimakawa, S., Itoh, N. & Niki, E. Action of DCFH and BODIPY as a probe for radical oxidation in hydrophilic and lipophilic domain. Free Radic. Res 37, 861–872 (2003).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363.e3 (2019).
Rodriguez, R. et al. Activation of lysosomal iron triggers ferroptosis in cancer. Preprint at https://doi.org/10.21203/rs.3.rs-4165774/v1 (2024).
Zhang, X., Guo, Y., Li, H. & Han, L. FIN56, a novel ferroptosis inducer, triggers lysosomal membrane permeabilization in a TFEB-dependent manner in glioblastoma. J. Cancer 12, 6610–6619 (2021).
Qiu, C. et al. Disulfiram, a ferroptosis inducer, triggers lysosomal membrane permeabilization by up-regulating ros in glioblastoma. Onco Targets Ther. 13, 10631–10640 (2020).
Agmon, E., Solon, J., Bassereau, P. & Stockwell, B. R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 8, 5155 (2018).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Soula, M. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360 (2020).
Chen, C., Sidransky, E. & Chen, Y. Lyso-IP: Uncovering pathogenic mechanisms of lysosomal dysfunction. Biomolecules 12, 616 (2022).
Pang, Z. et al. MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res. 52, W398–W406 (2024).
Acknowledgements
This work was supported in part by an AMED-CREST grant (JP22gm0910013 to K.Y.), JSPS KAKENHI grants (23H05481, 22H05572, 20H00493, and 18K19405 to K.Y., and 22KJ2465 to Y.S.), and Takeda Science Foundation (to K.Y.). This work was also supported by Platform Project for Supporting Drug Discovery and Life Science Research of AMED. We appreciate the technical support provided by Research Support Center of the Graduate School of Medical Sciences at Kyushu University. We also thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
Y.S., D.K., E.K., and K.T. performed and analyzed the cell experiments. Y.S. and N.K. performed and analyzed the animal experiments. T.U., T.K., S.M., and S.K. synthesized and analyzed the NBD-Pen analog probes. M.T., T.H., and H.N. synthesized and analyzed the iron probes. Y.S., D.K., K.M., Y.M., K.U., and M.J. contributed scientific insights and analyzed the results. Y.S. and M.J. wrote parts of the manuscript. K.Y. designed the experiments, conceived and supervised the study, and wrote and edited the manuscript. All authors have read and agreed to the contents of the paper.
Corresponding author
Ethics declarations
Competing interests
K.Y. is a co-founder and shareholder of the FELIQS Corporation. All other authors declare no conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Donat Kögel, Raphaël Rodriguez and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saimoto, Y., Kusakabe, D., Morimoto, K. et al. Lysosomal lipid peroxidation contributes to ferroptosis induction via lysosomal membrane permeabilization. Nat Commun 16, 3554 (2025). https://doi.org/10.1038/s41467-025-58909-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58909-w
This article is cited by
-
Fatty acid uptake mediated by FABP4 promotes the formation of CD8+T cell senescence through lipid peroxidation in the adipocyte-rich microenvironment of Ovarian Cancer
Oncogenesis (2026)
-
Lymph node environment drives FSP1 targetability in metastasizing melanoma
Nature (2026)
-
SARS-CoV-2 spike triggers TLR7-dependent endolysosome dysfunction and senescence in human astrocytes
Journal of Neuroinflammation (2025)
-
From “lysosomal addiction” to targeted therapies: exploiting novel windows in colorectal cancer
European Journal of Medical Research (2025)
-
Live-cell imaging with fluorogenic radical-trapping antioxidant probes reveals the onset and progression of ferroptosis
Nature Chemistry (2025)
