
Abstract
ZnCrOx oxides coupled with zeolites (OXZEO) allow direct conversion of syngas into light olefins, while active sites in the composite oxides remain elusive. Herein, we find that ZnO particles physically mixed with ZnCr2O4 spinel particles can be well dispersed onto the spinel surfaces by treatment in syngas and through a reduction-evaporation-anchoring mechanism, forming monodispersed ZnOx species with uniform thickness or dimension on ZnCr2O4 up to a dispersion threshold ZnO loading of 16.0 wt% (ZnCr2O4@ZnOx). A linear correlation between CO conversion and surface ZnO loading clearly confirms that the ZnOx overlayer on ZnCr2O4 acts as the active structure for the syngas conversion, which can efficiently activate both H2 and CO. The obtained ZnCr2O4@ZnOx catalyst combined with SAPO-34 zeolite achieves excellent catalytic performance with 64% CO conversion and 75% light olefins selectivity among all hydrocarbons. Moreover, the ZnOx overlayer is effectively anchored on the ZnCr2O4 spinel, which inhibits Zn loss during the reaction and demonstrates high stability over 100 hours. Thus, a significant interface confinement effect is present between the spinel surface and the ZnOx overlayer, which helps to stabilize ZnOx active structure and enhance the catalytic performance.
Similar content being viewed by others

Introduction
Syngas conversion to value-added chemicals stands as a central industrial process in the efficient utilization of coal, natural gas, and biomass resources1,2,3,4. The oxide–zeolite (OXZEO) process, utilizing oxides and zeolites as bifunctional catalysts for direct syngas conversion, overcomes the limitations imposed by the Anderson–Schulz–Flory distribution inherent to the traditional Fischer–Tropsch synthesis process5,6,7,8. By optimizing the oxide and zeolite components, it is possible to selectively obtain high value-added chemical feedstocks9,10,11,12, including light olefins, gasoline, and aromatics10. Many oxide catalysts are reported for conversion of syngas to light olefins in the OXZEO process, including ZnO13, ZnCrOx5,14, ZnZrOx7, ZnO/ZrO215, ZnAlOx16,17, MnOx18, ZnGaOx19, and MnGaOx20. Among them, ZnCrOx exhibits excellent ability for the activation of CO/H2 and is demonstrated as one of the most active oxides10,21.
Although ZnCrOx is widely considered crucial for controlling overall activity6,22, the active phase remains a subject of ongoing debate10,21,23. Using X-ray diffraction (XRD) and high-resolution transmission electron microscopy (HRTEM), Jiao et al. confirmed that the ZnCrOx composite oxide possessed a typical spinel structure, which was regarded as the active phase6. Song et al. hypothesized that an amorphous ZnO overlayer on the ZnCr2O4 spinel surface acted as the active phase for syngas conversion to methanol22. Similarly, Gao et al. conjectured that Zn enrichment on the ZnCr2O4 spinel surface was key to syngas conversion to isobutanol24,25. Recently, Wang et al. discovered that ZnO–ZnCr2O4 interface exhibited low activation energy for syngas conversion to light olefins and was beneficial for the high catalytic performance26. Chen et al. conducted CO activation studies on ZnCr2O4 samples using scanning transmission electron microscopy (STEM) and electron energy loss spectroscopy (EELS), finding that ZnCr2O4 coated with a ZnO thin layer favors direct CO dissociation21. Despite significant progresses in understanding surface active phases, precise construction of the active structure and well control of structural evolution during reaction remain core challenges.
Surface oxygen vacancy (Ov) has been proposed to be active for both CO and H2 activation on ZnCrOx catalysts22,27. Jiao et al. found that the spinel ZnCr2O4(111) surface could be reduced by CO or H2, and the adsorption of CO and H was significantly enhanced with the increase in Ov concentration6. Lai et al. reported that two-coordinate O vacancy site served as the active site for CO activation and hydrogenation reaction, leading to formation of CHO, CH2O, CH2 and CH2CO intermediates28. Fu et al. demonstrated that reduced Cr-terminated ZnCr2O4(111) with a 50% Ov concentration exhibited higher activity for ketene (CH2CO) production compared to methanol and methane29. Despite these intensive studies, the active site of CO hydrogenation and the role of Ov/surface ZnO species on ZnCrOx catalysts still need further exploration, particularly for real catalysts.
The structural complexity of ZnCrOx composite oxide makes it challenging to experimentally identify active surface structures30, further obscuring the precise identification of CO and H2 reaction sites. Therefore, it is crucial to design real catalysts with well-defined surface and interface structures and understand their roles in the reaction. Accordingly, precise construction of high density of surface active sites allows to improve the catalytic performance. Herein, we utilize a vapor phase migration process to disperse ZnO onto the ZnCr2O4 spinel surface, effectively creating monodispersed ZnOx species on ZnCr2O4 surface. A reduction-evaporation-anchoring mechanism is established using various characterization methods. We have identified the monodispersed ZnOx species confined on ZnCr2O4 as the active sites for syngas conversion to light olefins. The active monodispersed ZnOx species can activate both H2 and CO, and further demonstrate excellent chemical and catalytic stability through the interface confinement effect of the oxide support.
Results
Syngas-induced ZnO dispersion onto ZnCr2O4 surface
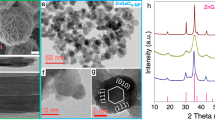
ZnCr2O4 was synthesized using traditional co-precipitation method, as detailed in the experimental procedures. XRD pattern and N2 adsorption–desorption analysis (Fig. S1) confirm the formation of a pure spinel phase with a mesoporous structure for the obtained powders31,32. The crystallite size calculated using the Scherrer equation based on ZnCr2O4(311) plane is 5.9 nm and the specific surface area determined by the Brunauer–Emmett–Teller (BET) method is 133.0 m2·g–1 (Table S1). Commercial ZnO and prepared ZnCr2O4 particles were physically mixed to obtain a ZnCr2O4-ZnO sample, containing 8.0 wt% ZnO. Diffraction peaks associated with wurtzite ZnO are clearly observed in XRD pattern from the mixture, as shown in Fig. 1a. ZnCr2O4-ZnO was then treated with a syngas (CO: H2 = 1: 2.5) flow at 450 °C and 0.1 MPa, which was investigated by in situ XRD (Fig. 1a). The results show that the ZnO(10\(\bar{1}\)0) diffraction peak gradually gets weakened during the process and eventually only the diffraction peaks of ZnCr2O4 spinel are evident. Inductively coupled plasma optical emission spectroscopy (ICP-OES) result (Table S2) reveals that Zn content does not change after the treatment. Thus, it is suggested that ZnO particles become highly dispersed after the syngas treatment.

a In situ XRD of ZnCr2O4-ZnO containing 8.0 wt% ZnO during syngas treatment at 450 oC for various time. The bottom black pattern is from the sample before the treatment and the top red pattern from the treated one for 6 h. b Quasi in situ XPS spectra of ZnCr2O4-ZnO, ZnCr2O4@ZnOx, fresh ZnCr2O4 (named ZnCr2O4-F), and treated ZnCr2O4 (named ZnCr2O4-T). c UV-Vis spectra of ZnCr2O4-ZnO, ZnCr2O4@ZnOx, ZnCr2O4-F, and ZnCr2O4-T. EDS mapping images of ZnCr2O4-ZnO (d) and ZnCr2O4@ZnOx (e). Scale bar: 400 nm.
Quasi in situ X-ray photoelectron spectroscopy (XPS) analysis (Fig. 1b) and Auger spectra of Zn LMM (Fig. S2) indicate that Zn species on the treated ZnCr2O4-ZnO sample surface remains at Zn2+ state33,34 and even exhibits a significant increase in the Zn 2p peak intensity. This further proves that ZnO species are dispersed onto the ZnCr2O4 surface (denoted as ZnCr2O4@ZnOx). Ultraviolet-visible (UV-Vis) spectra show that the typical absorption band at 370 nm from bulk ZnO structure35,36 cannot be observed in the ZnCr2O4@ZnOx sample (Fig. 1c), excluding the existence of three-dimensional ZnO aggregates. In contrast, no changes have been observed in the XPS (Fig. 1b) and UV-Vis (Fig. 1c) spectra after the syngas treatment of the pure ZnCr2O4 sample. Energy dispersive spectroscopy (EDS) mapping result (Fig. 1d) shows obvious separated ZnO aggregates in ZnCr2O4-ZnO. However, the spatial distribution of Zn and Cr elements in ZnCr2O4@ZnOx is very uniform. All the above results convincingly confirm that ZnO species in the oxide mixture dynamically migrate and then highly disperse onto the ZnCr2O4 surface after the syngas treatment.
Migration-dispersion mechanism and dispersion threshold
Gray deposits identified as Zn metal by XRD form on the inner wall of the quartz tube after the same syngas treatment of pure ZnO, suggesting that ZnO can be reduced and then Zn evaporates away from ZnO particles. When ZnCr2O4 sample is placed beneath ZnO and separated by an inert layer of quartz wool (ZnO | |ZnCr2O4), no Zn metal deposits on the quartz tube wall are observed, indicating that ZnCr2O4 may capture the evaporated Zn metal species (Fig. S3). To further study the dispersion mechanism in this syngas treatment process, we placed ZnCr2O4 particles beneath Zn metal powder and separated them by an inert layer of quartz wool (Zn | |ZnCr2O4). For the Zn | |ZnCr2O4 sample, H2 evolution is observed to occur predominantly above 300 °C when heated in an Ar flow, with a notable increase beyond 430 °C (Fig. 2a). However, no H2 generation is observed upon heating ZnO | |ZnCr2O4 in Ar. This indicates that Zn vapor from the Zn metal can migrate in the heated Ar flow and further react with surface OH on ZnCr2O4 to produce gaseous H2 and surface ZnOx species37. When the ZnO | |ZnCr2O4 sample is exposed to a CO flow, H2 generation is also observed at 400 °C (Fig. S4). The control experiments suggest that ZnO needs to be first reduced in reductive atmospheres, which then allows the further evaporation of Zn into the gas phase. Fourier transform infrared (FTIR) result (Fig. 2b, the whole spectra are shown in Fig. S5) confirms that surface OH groups on the treated ZnCr2O4 are strongly consumed after the migration and dispersion of ZnOx species37.

a H2 and H2O evolution upon temperature-programmed heating of ZnCr2O4 packed below Zn metal or ZnO, separated by an inert layer of quartz wool with the comparison of ZnCr2O4-ZnO (same as ZnO-ZnCr2O4) and ZnCr2O4. b FTIR spectra of OH groups on treated ZnCr2O4 and ZnCr2O4@ZnOx samples with various ZnO loadings. c Schematic illustrating formation mechanism of ZnCr2O4@ZnOx during syngas treatment. d Surface OH amount on ZnCr2O4@ZnOx as a function of ZnO loading. e Dispersion threshold of ZnO on ZnCr2O4 determined by XRD and XPS.
The ZnCr2O4-ZnO samples have been treated in various gases including Ar, O2, H2, and CO. The similar dispersion of ZnO particles happens in H2 and CO flows but not in O2 and Ar flows (Fig. S6). Therefore, ZnO particles should be first reduced to Zn metal in reductive gases e.g. CO, H2 and syngas, which then migrate through the gas phase and are further anchored by OH sites on ZnCr2O4 surface. Overall, a reduction-evaporation-anchoring mechanism is suggested for the dispersion of ZnO onto ZnCr2O4 upon the syngas treatment as shown in Fig. 2c.
D2 temperature programmed reaction (D2-TPR) experiments were conducted to determine surface OH38 of ZnCr2O4@ZnOx catalysts with different ZnO loadings (Fig. S7). The quantitative results shown in Fig. 2d indicate that the OH content gradually decreases with the increasing dispersed ZnO loading, which is consistent with the FTIR results (Fig. 2b). However, when ZnO loading exceeds 15% the OH content no longer changes with further increasing ZnO loading. Meanwhile, trace amounts of Zn are found to deposit onto the inner wall of the quartz tube (Fig. S8). It can be inferred that there is a limit to the amount of Zn which can be anchored and dispersed on the ZnCr2O4 surface.
ZnCr2O4@ZnOx catalysts prepared with different ZnO loadings were systematically studied using XRD and XPS (Fig. S9). With ZnO loading below 15.0 wt%, no XRD peaks corresponding to any ZnO phases are observed, confirming that all ZnO species are well dispersed on ZnCr2O4. Concurrently, Zn/Cr atomic ratio calculated from Zn 2p3/2 and Cr 2p XPS spectra increases linearly with the ZnO loading (Fig. 2e). Above 15.0 wt%, diffraction peaks of hexagonal ZnO appear (Fig. S9) and their intensity increases linearly with the ZnO loading. Meanwhile, ZnO particles are observed in EDS mapping images (Fig. S10). The plot of Zn/Cr atomic ratio versus ZnO loading shows a turning point at 16.0 wt%, which also corresponds to an intercept at x-axis of the fitted straight line of the plot of XRD AZnO/AZnCr2O4 (the peak area ratio of the ZnO(10\(\bar{1}\)0) vs. ZnCr2O4(400) planes) as a function of the ZnO loading. The XPS and XRD results suggest that the growth of ZnOx overlayer on ZnCr2O4 driven by the syngas treatment follows a characteristic monolayer-dispersion behavior39,40 at low ZnO loading and the dispersion threshold of ZnO is 16.0 wt% (1.9 mmol ZnO·100 m−2 ZnCr2O4).
Surface structure of the ZnCr2O4@ZnOx catalysts
HRTEM was used to identify dispersed ZnO species on the ZnCr2O4 spinel surface. As shown in Fig. 3a, ZnCr2O4@ZnOx-15, meaning the ZnCr2O4@ZnOx catalyst with a ZnO loading of 15.0 wt%, exhibits the main ZnCr2O4 spinel phase and 0.3 nm thick amorphous overlayer. The distribution of Zn and Cr elements in ZnCr2O4@ZnOx-15 was further analyzed using EELS with mapping and line scanning functions (Fig. 3b, c). Both Zn and Cr elements are uniformly distributed in the bulk region but the outermost amorphous layer is mainly composed of Zn element (Fig. 3b). For ZnCr2O4@ZnOx-30, ZnCr2O4 spinel particles are completely covered by ZnOx overlayer, and meanwhile isolated ZnO particles are also observed (Fig. 3d). The image data demonstrate that the ZnOx overlayer with a thickness close to that of a ZnO monolayer can fully cover the ZnCr2O4 spinel when the ZnO loading exceeds 16.0 wt% (Fig. 3d, e), which is consistent with the monolayer dispersion behavior of ZnO on the ZnCr2O4 spinel surface given in Fig. 2e. Accordingly, we propose that ZnOx species are highly dispersed on the ZnCr2O4 spinel surface with uniform thickness or dimension when ZnO loading is below the dispersion threshold, which is described as monodispersed ZnOx overlayer in this work. Above 16.0 wt% both a complete monodispersed ZnOx layer and additional ZnO particles form on the spinel surface.

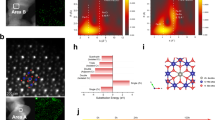
a HRTEM images of ZnCr2O4@ZnOx−15. Scale bar: 2 nm. b STEM-EELS mapping images of ZnCr2O4@ZnOx−15. Scale bar: 2 nm. c Elemental composition of Zn and Cr obtained from the line-scan along the yellow line in (b) by EELS. d STEM-EELS mapping images of ZnCr2O4@ZnOx−30. Scale bar: 20 nm. e Elemental composition of Zn and Cr obtained from the line-scan along the yellow line in (d) by EELS. f HS-LEIS spectra of ZnCr2O4@ZnOx catalysts.
High sensitivity-low energy ion scattering (HS-LEIS) technique, sensitive to the surface elements (escape depth of ~0.3 nm)41,42,43, was employed to study the outermost structure of ZnCr2O4@ZnOx. The results shown in Fig. 3f indicate that the surface of stoichiometric ZnCr2O4 spinel is almost entirely Cr exposed, which is consistent with the results of Wachs et al42. When a small amount of ZnO is dispersed onto ZnCr2O4, the surface Zn content on ZnCr2O4@ZnOx-5 increases while the surface Cr content decreases. The surfaces of ZnCr2O4@ZnOx-15 and ZnCr2O4@ZnOx-30 are primarily composed of Zn elements. In addition, the Cr/Zn element ratio on these three ZnCr2O4@ZnOx surfaces gradually increases with increasing He+ sputtering cycles (Fig. S11), particularly for ZnCr2O4@ZnOx-15 and ZnCr2O4@ZnOx-30. Apparently, the amorphous ZnOx overlayer at the outmost surface can be gradually etched away, leading to the progressive exposure of Cr from the spinel support, which is consistent with the EELS line scanning results (Fig. 3c, e).
Catalytic performance and active sites of ZnCr2O4@ZnOx catalysts
The ZnCr2O4@ZnOx catalysts coupled with SAPO-34 were evaluated for syngas conversion to light olefins at 4.0 MPa and 400 °C. Figure 4a shows that CO conversion on stoichiometric ZnCr2O4 spinel is 45%, with a selectivity of 57% for light olefins. The surface Cr species in ZnCr2O4 tend to promote excessive hydrogenation of light olefins presenting lower selectivity. Notably, the addition of 5.0 wt% ZnO species boosts CO conversion to 51% and elevate light olefin selectivity to 70%, suggesting that the monodispersed ZnOx species effectively enhance both selectivity and overall activity. In contrast, the same syngas treatment of pure ZnCr2O4 spinel does not affect its performance (Fig. S12). Further, the dispersed ZnOx overlayers over Cr2O3 and γ-Al2O3 show quite poor catalytic performance for the syngas conversion to light olefins (Fig. S13), indicating that the performance enhancement of ZnCr2O4@ZnOx is not due to any changes in ZnCr2O4 but rather results from the formation of the monodispersed ZnOx overlayer over ZnCr2O4 spinel. Interestingly, CO conversion of ZnCr2O4@ZnOx increases linearly with ZnO loading, reaching a maximum of 64% at a ZnO loading of 15.0 wt%. Beyond this optimal loading, there is a modest decline in CO conversion, which is predominantly attributed to the dilution effect caused by the presence of low-activity ZnO particles. The linear positive correlation between ZnO loading (<15.0 wt%) and CO conversion clearly indicates that the active site on ZnCr2O4@ZnOx should be related to the content of surface ZnOx species dispersed on the ZnCr2O4 spinel.

a Catalytic performance of ZnCr2O4 and ZnCr2O4@ZnOx catalysts coupled with SAPO-34. Reaction conditions: 0.20 g ZnCr2O4@ZnOx + 0.10 g SAPO-34, 68% H2 + 27% CO + 5% Ar, 4.0 MPa, 15 mL·min−1, 40–60 mesh, 400 °C. CO2 selectivity does not change with the ZnO loading and is in the range of 38–41% for each catalyst. b Relationship between surface Ov concentration, amount of adsorbed CO, and ZnO loading of ZnCr2O4@ZnOx catalysts. c Relationship between HD formation rate and ZnO loading of ZnCr2O4@ZnOx catalysts. The pretreated ZnCr2O4@ZnOx catalysts were exposed to a flow of 10% D2, 10% H2, and 80% Ar, and the formation of HD species was detected at 400 °C. d Scheme of the active sites on ZnCr2O4@ZnOx catalysts.
Oxygen vacancies on the catalyst surfaces were analyzed using O2-pulse experiments44,45. Specifically, the catalysts were treated in syngas at 450 °C and subsequently tested for oxygen adsorption capacity using pulse O2 gas at 120 °C (Fig. S14). The ZnCr2O4 spinel exhibits a significant oxygen adsorption capacity of 0.21 mmol O2 per gram of sample. Quasi in situ XPS results (Fig. S15) show no significant change in Cr 2p spectra of ZnCr2O4 following the O2 pulse, suggesting that the O2 consumption is mainly due to capture by Ov rather than oxidation of Cr3+ to Cr6+ on ZnCr2O4. Quantitative results show that the concentration of Ov on the ZnCr2O4 surface is 0.42 mmol·g–1, and it significantly decreases with the addition of monodispersed ZnOx species, demonstrating a negative correlation between surface Ov concentration and coverage of surface ZnOx species. Furthermore, Ov concentration of ZnCr2O4@ZnOx stays constant at 0.15 mmol·g–1 when ZnO loading exceeds 16.0 wt%.
To determine the influence of ZnO loading on CO adsorption capacity, the ZnCr2O4@ZnOx catalysts were investigated by CO temperature-programmed desorption (CO-TPD). The primary desorbed species, identified as CO and CO2 (Fig. S16), were quantified, as presented in Fig. 4b. Additionally, carbonate species from the adsorption of generated CO2 are observed in the infrared spectra46,47 (Fig. S17). Notably, a positive linear relationship is observed between the adsorbed CO amount and ZnO loading with ZnO loading below 16.0 wt%. However, once ZnO loading exceeds 16.0 wt%, further increase in ZnO loading results in a slight decrease in the adsorbed CO amount. This indicates that CO adsorption capacity of ZnCr2O4@ZnOx is closely related to the monodispersed ZnOx overlayer rather than the surface Ov.
To investigate H2 activation on ZnCr2O4 and ZnCr2O4@ZnOx catalysts, we conducted H2 − D2 exchange experiments. HD formation rate quantitatively reflects H2 activation ability of catalysts17. As shown in Fig. 4c stoichiometric ZnCr2O4 exhibits the highest HD formation rate, while the rate for ZnCr2O4@ZnOx decreases gradually as the ZnO loading increases. Once the dispersion threshold of 16.0 wt% is exceeded, HD formation rate for ZnCr2O4@ZnOx remains almost unchanged. This suggests that the exposed Cr species on the ZnCr2O4 surface have a stronger ability for HD formation under this reaction condition in comparison with monodispersed ZnOx overlayer on ZnCr2O4@ZnOx. Additionally, in contrast with heterolytic dissociation of D2 on bulk ZnCr2O4 and ZnO48,49,50, D2 dissociation by the monodispersed ZnOx overlayer tends to proceed via homolytic route, forming more stable Zn−D species at higher temperature (Fig. S18) and promoting hydrogenation of CO51,52. The negative correlation between CO conversion and Ov concentration but the positive correlation between CO conversion and dispersed ZnO loading imply that monodispersed ZnOx species rather than Ov from ZnCr2O4 spinel serve as the key active sites for the syngas conversion (Fig. 4d).
Interface confinement effect on the ZnOx overlayer
It is known that ZnO as an amphoteric oxide can be dissolved in (NH4)2CO3 solution53. XRD and ICP-OES analysis experiments were conducted on ZnCr2O4@ZnOx catalysts with ZnO loading above 16.0 wt% before and after leaching with an (NH4)2CO3 solution. After the leaching process, no ZnO diffraction peaks are detected in all XRD patterns (Fig. 5a). Moreover, ICP-OES results reveal that the leached catalysts all retain a ZnO amount close to 16.0 wt% (Fig. 5b). The results suggest that the ZnOx overlayer is strongly confined on the ZnCr2O4 spinel, which can withstand chemical leaching in the (NH4)2CO3 solution. In contrast, the excess ZnO particles are all removed by the leaching. Notably, ICP-OES analysis of the leaching filtrate shows an absence of Cr element and thus (NH4)2CO3 solution only selectively etches ZnO particles but not ZnCr2O4 support.

a XRD patterns of ZnCr2O4@ZnOx-30 catalysts before and after leaching with (NH4)2CO3 solution (named as ZnCr2O4@ZnOx-30-L). b ICP-OES results of ZnO content in ZnCr2O4@ZnOx before and after leaching with (NH4)2CO3 solution. c Catalytic performance of ZnCr2O4@ZnOx-30 and ZnCr2O4@ZnOx-30-L catalysts coupled with SAPO-34. Reaction conditions: 0.20 g ZnCr2O4@ZnOx + 0.10 g SAPO-34, 68% H2 + 27% CO + 5% Ar, 4.0 MPa, 25 mL·min−1, 40–60 mesh, and 400 °C. d Catalytic performance versus time on stream (TOS) of ZnCr2O4@ZnOx-15 and ZnCr2O4@ZnOx-30 catalysts coupled with SAPO-34. Reaction conditions: 0.20 g ZnCr2O4@ZnOx + 0.10 g SAPO-34, 68% H2 + 27% CO + 5% Ar, 4.0 MPa, 25 mL·min−1, 60–80 mesh, and 400 °C.
These ZnCr2O4@ZnOx catalysts, both post-leaching and untreated, were evaluated for syngas conversion to light olefins under conditions of 4.0 MPa and 400 °C (Fig. 5c). For the ZnCr2O4@ZnOx-30-L catalyst with the leaching treatment, both CO conversion and selectivity of light olefins are only slightly reduced compared to the ZnCr2O4@ZnOx-30 catalyst. This further substantiates that the active site on ZnCr2O4@ZnOx surface is the confined ZnOx overlayer but not those excess ZnO particles. Long-term reactions were conducted over ZnCr2O4@ZnOx-15 and ZnCr2O4@ZnOx-30 catalysts, as depicted in Fig. 5d. For both catalysts, CO conversion exhibits a slight decrease at the onset of the reaction and then remains stable (Fig. S19). For the ZnCr2O4@ZnOx-15 catalyst the selectivity of light olefins decreases from 79% to 76% after 100-hour reaction. In contrast, the light olefins selectivity of the ZnCr2O4@ZnOx-30 catalyst decreases from 77% to 67%. In this catalyst excess ZnO particles are negative for the long-term reaction since these unconfined bulk ZnO can migrate into the interior of SAPO-34 and lower the selectivity54. Overall, the monodispersed ZnOx species confined by the ZnCr2O4 surface exhibits high stability against chemical leaching in solution and Zn loss during syngas reaction, which catalyzes the syngas conversion effectively and presents confinement enhanced performance.
Discussion
Syngas treatment of the mixture of ZnO and ZrCr2O4 particles induces dispersion of ZnO and formation of ZnOx overlayer onto the ZnCr2O4 spinel. In this process, ZnO is reduced to Zn metal, which then migrates through gas phase and is re-oxidized by surface OH groups of ZnCr2O4, forming monodispersed ZnOx species with a dispersion threshold of 16.0 wt%. These ZnO species cover the ZnCr2O4 spinel surface in the form of ZnOx monolayer with a thickness of 0.3 nm as the ZnO loading is close to 16.0 wt%. The catalyst with high density of surface active sites can be well constructed through this unique reduction-evaporation-anchoring process and the roles of these surface sites can be clearly understood, which holds significant promise for preparation of well-defined oxide catalysts supported on oxides.
The formed ZnCr2O4@ZnOx combined with SAPO-34 zeolite achieves 64% CO conversion and 75% light olefins selectivity in syngas conversion to light olefins. A direct linear correlation between CO conversion and surface ZnO loading is clearly identified, confirming the active site of the monodispersed ZnOx species trapped on the ZnCr2O4 surface. The ZnOx overlayer is effectively confined on the ZnCr2O4 spinel, which inhibits excessive reduction of ZnO and loss of Zn during the reaction, demonstrating high stability over 100 hours. The mechanism of H2/CO activation at the active sites has been elucidated on the oxide catalysts. The monodispersed ZnOx overlayer contributes to the stabilization of H species and promotes the adsorption and activation of CO, thereby offering active sites for efficient syngas conversion. Our work emphasizes the importance of atomic level analysis of surface active sites in practical catalytic systems, identifying the reaction mechanism from a surface science study perspective. Moreover, the current understanding of active sites and reaction mechanisms can inspire further researches on syngas conversion over other composite oxide systems, offering crucial guidance for the rational design and optimization of efficient oxide catalysts.
Methods
Catalyst synthesis and treatment
ZnCr2O4 spinel oxide was synthesized by conventional co-precipitation method. Analytical reagent grade (99.7%) Zn(NO3)3·6H2O and Cr(NO3)3·9H2O were used as the precursors. 8.96 g Zn(NO3)3·6H2O and 24.01 g Cr(NO3)3·9H2O were dissolved into 60 mL distilled deionized water to get stable solutions. Afterward, an ammonium carbonate (4.6 mol·L–1) aqueous solution was dripped slowly into the mixed liquid until the pH value reached 8 under the condition of continuous stirring at 70 °C. After stirring for 3 h, the obtained mixtures were cooled down to room temperature, and the navy precipitate was washed three times with distilled deionized water. The precipitate was dried via vacuum drying at 70 °C for 12 h, which was followed by calcination at 500 °C in air for 1 h with a heating rate of 2 °C·min–1. The molar composition of ZnCr2O4 was determined by ICP-OES, and the Zn/Cr ratio of the obtained sample conformed to the stoichiometric ratio (Table S1). The wurtzite ZnO sample is commercially available (99.9%, Aladdin reagent (Shanghai) co. LTD) and used without further purification. The mixtures of ZnO and ZnCr2O4 with a specific content were put into the sample bottle and shaken for 6 h, and the obtained sample is noted as ZnCr2O4-ZnO-x, x means x wt% of ZnO in the mixture. The ZnCr2O4@ZnOx-x sample was gained from a treatment of ZnCr2O4-ZnO-x by syngas (68% H2 + 27% CO + 5% Ar) flow with a pressure of 0.1 MPa at 450 °C for 6 h.
SAPO-34 was synthesized by a hydrothermal method. In detail, 7.46 g pseudo boehmite (82 wt% Al2O3) was added into 54 mL H2O and stirred for 0.5 h. Then, 1.80 g silica sol (30 wt% SiO2), 13.84 g orthophosphoric acid (85 wt% H3PO4) and 18.22 g triethylamine were added to the mixture in turn and stirred separately for 0.5 h. Finally, 0.31 g SAPO-34 seeds (NKF-9, Nankai University Catalyst Co., Ltd.) were added, and the mixtures were placed in a Teflon-lined autoclave and heated in a rotating oven at 180 °C under autogenic pressure for 48 h. After crystallization, the samples were subjected to centrifugal separation, washing, and drying for over 12 h at 120 °C, and final calcination for 3 h at 600 °C. The sample was subjected to XRD, X‐ray fluorescence, N2 adsorption–desorption analysis, and NH3 temperature‐programmed desorption (Fig. S20 and Table S3). The results were consistent with the reported literature54.
The ZnO-containing catalyst is selectively leached in ammonium carbonate solution. In detail, 1.0 g oxide catalyst was added to 40 ml ammonium carbonate solution (1 mol·L−1) under the condition of continuous stirring at 40 °C. Next, the mixture was filtered and washed several times with pure water. The obtained precipitate was dried via vacuum drying at 70 °C for 12 h. The powder was treated with syngas under conditions before use, and noted as ZnCr2O4@ZnOx-x-L.
Catalyst characterization and syngas conversion reaction evaluation
X-ray diffraction (XRD) patterns were collected on an Empyrean diffractometer (Empyrean-100) using Cu Kα (λ = 1.5406 Å) radiation source and a scanning rate of 10 °·min–1. In situ XRD measurements were conducted on the same instrument, and the reaction cell was controlled using an XRK900 controller. The catalysts were heated in an Ar flow, and syngas (68% H2 + 27% CO + 5% Ar) was introduced at a flow rate of 30 mL·min–1 when the temperature was 450 °C. XRD patterns were recorded from 25 to 50 ° versus holding time.
The textural properties of the samples were measured by N2 adsorption–desorption on an automated gas sorption analyzer (Autosorb iQ2 and Quadrasorb evo) at liquid nitrogen temperature (−196 °C) to analyze the SAPO-34 zeolite and oxide catalyst. The specific surface area was calculated using the Brunauer-Emmett-Teller (BET) method, and the pore size distribution was derived from the adsorption isotherm using the Barrett-Joyner-Halenda (BJH) method. Prior to analysis, the samples were degassed under vacuum at 300 °C for 4 h.
The contents of Zn and/or Cr were determined by inductively coupled plasma optical emission spectrometry (ICP-OES).
Ex situ, quasi in situ X-ray photoelectron spectra (XPS) and X-ray-excited Auger spectra were acquired with a spectrometer equipped with a Mg Kα X-ray source operated at 200 W. The background pressure was in the range of 10−9 mbar. Ex situ treatments were performed in a high-pressure reactor (SPECS, HPC-20). All quasi in situ XPS spectra were collected under ultra-high vacuum (UHV, 10−9–10−10 mbar) at room temperature. To prevent contact with O2 and H2O in air, the catalysts treated with syngas were transferred to a glove box through an airtight reaction tube. Subsequently, the catalysts were transferred from the glovebox to the XPS system using a mobile transfer chamber. The sample was kept in Ar during the entire transfer process to ensure no exposure to air.
Ultraviolet-visible (UV-vis) spectra were recorded on a Lambda 950 (Perkin Elmer) operated in diffuse reflectance mode.
Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) element mapping images were acquired on a JSM-7900F instrument operated at 5 and 15 kV.
Ar temperature-programmed heating experiments were conducted on an AutoChem II 2920 equipped with a GSD 350 OmniStar mass spectrometer. The ZnCr2O4 particles were placed beneath Zn metal or ZnO powders, separated by an inert layer of quartz wool (Zn | |ZnCr2O4 or ZnO | |ZnCr2O4). Typically, the sample was pretreated with Ar at 300 °C for 1 h to remove H2O, then cooled to room temperature. The experiment was initiated to monitor the changes in the signals of H2 (m/z = 2) and H2O (m/z = 18) as the temperature was increased from 30 to 500 °C at a rate of 10 °C·min–1 in an Ar flow. Similar experiments were conducted for oxide samples (ZnO, ZnCr2O4, ZnO-ZnCr2O4, ZnO | |ZnCr2O4) in different reducing atmospheres, i.e., H2/Ar and CO/Ar, to monitor the changes in H2 (m/z = 2), H2O (m/z = 18), CO (m/z = 28), and CO2 (m/z = 44) during the temperature-programmed process.
D2 temperature-programmed reduction (D2-TPR) was conducted on a homemade reactor equipped with a GSD 350 OmniStar mass spectrometer. The sample (0.10 g) was loaded in a quartz tube and then pretreated with a 68% H2/Ar flow at 450 °C for 6 h, and then switched to an Ar flow for 1 h. The pretreated sample was cooled to room temperature under an Ar flow and then reduced by a 10% D2/Ar flow. The experiment was initiated to monitor changes in the signals of D2 (m/z = 4) as the temperature was increased from 30 to 600 °C at a rate of 10 °C·min–1.
High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images, annular bright field (ABF) images, and energy dispersive spectroscopy (EDS) elemental distribution of the catalysts were obtained using JEOL JEM-F200 and JEM-ARM300 microscopes. Electron energy loss spectroscopy (EELS) elemental distribution of the catalysts was acquired on a JEM-ARM300.
High-sensitivity low-energy ion scattering (HS-LEIS) spectra were obtained using an Ion-TOF Qtac100 instrument. To minimize the surface damage, helium was selected as the ion source with a kinetic energy of 3 keV, an ion flux of 6000 pA·m–2, and a spot size of 2 mm × 2 mm.
O2 pulse experiments were performed in a quartz reactor at 120 °C on an AutoChem II 2920. Typically, the sample was pretreated with a syngas flow at 450 °C for 6 h, followed by purging with Ar gas for 1 h. The sample was then cooled to 120 °C, and 5% O2/Ar gas was pulsed 40 times into the sample bed via the automatic six-way valve, with the thermal conductivity detector (TCD) signal monitoring the process. The amount of O2 consumption on the catalyst was quantified by measuring the molar amount of O2 species present in the quantitative loop.
CO temperature-programmed desorption (CO-TPD) experiments were conducted on an AutoChem II 2920 equipped with a GSD 350 OmniStar mass spectrometer. The ZnCr2O4@ZnOx sample was first pretreated under a syngas flow at 450 °C for 0.5 h to recover the reduced surface, followed by switching to Ar flow for another 0.5 h. After cooling to room temperature, it was then exposed to a 10% CO/Ar flow at 30 °C, followed by Ar flushing for 1 h, and the temperature was ramped from 30 to 700 °C at a rate of 10 °C·min–1. The desorbed CO (m/z = 28) and CO2 (m/z = 44) signals were detected as the temperature was increased.
H2–D2 exchange experiments were conducted on an AutoChem II 2920 equipped with a GSD 350 OmniStar mass spectrometer. The ZnCr2O4@ZnOx sample (0.01 g) mixed with the inert silica (0.09 g) was first pretreated under flowing 10% H2/Ar at 450 °C for 0.5 h, followed by an Ar flow for another 0.5 h and cooling to room temperature. After the pretreatment, a mixture flow of 10% D2, 10% H2 and 80% Ar was introduced. The experiment was initiated to monitor changes in the signals of HD (m/z = 3) as the temperature was increased from 30 to 400 °C, with the reaction maintained at 400 °C for 0.5 h.
Transmission Fourier transform infrared (FTIR) spectra were obtained using a transmission FTIR spectrometer (Bruker, VERTEX 70 v) under vacuum (base pressure <5 × 10–8 mbar). The IR spectra were recorded with 64 scans at a resolution of 4 cm–1 using a liquid-N2-cooled mercury cadmium telluride (MCT) detector. The sample with a mass of 10 mg was pretreated under vacuum at 300 °C to analyze the hydroxyl groups on the surface. And the sample was pretreated under vacuum condition at 480 °C to remove the surface impurities during the D2 activation experiments.
In situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) was measured on a Thermo Nicolet iS 10 equipped with an MCT detector at a spectral resolution of 4 cm−1 and an accumulation of 32 scans. Before the test, the powder samples were pretreated at 400 °C in helium flow (30 mL·min–1) for 30 min. Background spectra were recorded at different temperatures in a He flow (30 mL·min–1). 5% CO/He was fed into the sample cell at 30 °C for 10 min. Thereafter, 5% CO/He was introduced into the sample cell at 50 °C. Then, the sample was heated from 50 to 400 °C at a temperature interval of 50 °C in 5% CO/He. Sample spectra were obtained with the corresponding background spectra subtracted during the entire process.
Elemental analysis was carried out using a Zetium X‐ray fluorescence (XRF) spectrometer.
NH3 temperature‐programmed desorption (NH3‐TPD) experiment was carried out on a Micromeritics AutoChem II 2920 equipped with a TCD. The SAPO-34 zeolite was pretreated in flowing Ar at 550 °C for 1.5 h, followed by exposure to a 5% NH3/He mixture at 100 °C. After NH3 exposure, the sample was flushed with He and the temperature was ramped from 100 to 700 °C at a rate of 10 °C·min–1.
Catalytic reactions were performed in a fixed-bed stainless steel reactor furnished with a quartz lining with an inner diameter of 6 mm. Typically, a 0.30 g catalyst with oxide/SAPO-34 = 2/1 (mass ratio) was heated from room temperature to 400 °C in an Ar flow. Syngas containing CO and H₂ with 5% Ar as the internal standard for online gas chromatography (GC) analysis was used as the feed. Reaction conditions were H2/CO = 2.5, 4.0 MPa, 400 °C, 40–60 mesh, and 15 mL·min–1 unless otherwise stated. Data were collected after at least 6 h on stream. The products of syngas conversion to light olefins were analyzed by online GC (Agilent 8890B). It was equipped with a TCD and two flame ionization detectors (FID). HP-PLOT Q and 5 Å molecular sieve-packed columns were connected to the TCD, while HP-INNOwax and GS-Alumina capillary columns were connected to the FID. Oxygen-containing compounds and hydrocarbons up to C17 were analyzed by the FID, while CO, CO2, C2H4, and Ar were analyzed by the TCD. C2H4 was used as a reference bridge between the FID and TCD. CO conversion was calculated on a carbon atom basis using the following equation:
where COin and COout represent the concentrations of CO at the inlet and outlet, respectively.
CO2 selectivity (\({\mbox{Se}}{{\mbox{l}}}_{{\mbox{C}}{{\mbox{O}}}_{2}}\)) was calculated according to
where [CO2]out is the concentration of CO2 in the reactor outlet.
The selectivity of individual hydrocarbon CnHm (\({{\rm{Se}}}{{{\rm{l}}}}_{{{{\rm{C}}}}_{{{\rm{n}}}}{{{\rm{H}}}}_{{{\rm{m}}}}}\)) among hydrocarbons was obtained according to
The selectivity to oxygenate was below 0.5% C and therefore was not reported in the product selectivity. The carbon balance was around 100%.
Data availability
All data that support the findings in this paper are available within the article and its Supporting Information or are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Subramani, V. & Gangwal, S. K. A review of recent literature to search for an efficient catalytic process for the conversion of syngas to ethanol. Energy Fuels 22, 814–839 (2008).
Zhou, W. et al. New horizon in C1 chemistry: breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 48, 3193–3228 (2019).
Kasipandi, S. & Bae, J. W. Recent advances in direct synthesis of value-added aromatic chemicals from syngas by cascade reactions over bifunctional catalysts. Adv. Mater. 31, 1803390 (2019).
Wang, C. et al. Direct conversion of syngas to ethanol within zeolite crystals. Chem 6, 646–657 (2020).
Jiao, F. et al. Disentangling the activity-selectivity trade-off in catalytic conversion of syngas to light olefins. Science 380, 727–730 (2023).
Jiao, F. et al. Selective conversion of syngas to light olefins. Science 351, 1065–1068 (2016).
Cheng, K. et al. Direct and highly selective conversion of synthesis gas into lower olefins: design of a bifunctional catalyst combining methanol synthesis and carbon–carbon coupling. Angew. Chem. Int. Ed. 55, 4725–4728 (2016).
Torres Galvis, H. M. & de Jong, K. P. Catalysts for production of lower olefins from synthesis gas: a review. ACS Catal. 3, 2130–2149 (2013).
Cheng, K., Li, Y., Kang, J., Zhang, Q. & Wang, Y. Selectivity control by relay catalysis in CO and CO2 hydrogenation to multicarbon compounds. Acc. Chem. Res. 57, 714–725 (2024).
Pan, X., Jiao, F., Miao, D. & Bao, X. Oxide–zeolite-based composite catalyst concept that enables syngas chemistry beyond Fischer–Tropsch synthesis. Chem. Rev. 121, 6588–6609 (2021).
Chernyak, S. A., Corda, M., Dath, J.-P., Ordomsky, V. V. & Khodakov, A. Y. Light olefin synthesis from a diversity of renewable and fossil feedstocks: state-of the-art and outlook. Chem. Soc. Rev. 51, 7994–8044 (2022).
Mahmoudi, E. et al. Advances in catalysts for direct syngas conversion to light olefins: a review of mechanistic and performance insights. J. CO2 Util. 86, 102893 (2024).
Li, N. et al. Size effects of ZnO nanoparticles in bifunctional catalysts for selective syngas conversion. ACS Catal. 9, 960–966 (2019).
Su, J. et al. Syngas to light olefins conversion with high olefin/paraffin ratio using ZnCrOx/AlPO-18 bifunctional catalysts. Nat. Commun. 10, 1297 (2019).
Liu, Z. et al. Insights into effects of ZrO2 crystal phase on syngas-to-olefin conversion over ZnO/ZrO2 and SAPO-34 composite catalysts. Chin. J. Catal. 43, 877–884 (2022).
Su, J. et al. Unveiling the anti-trap effect for bridging intermediates on ZnAlOx/AlPO-18 bifunctional catalysts to boost syngas to olefin conversion. ACS Catal. 13, 2472–2481 (2023).
Liu, X. et al. Tandem catalysis for hydrogenation of CO and CO2 to lower olefins with bifunctional catalysts composed of spinel oxide and SAPO-34. ACS Catal. 10, 8303–8314 (2020).
Zhu, Y. et al. Role of manganese oxide in syngas conversion to light olefins. ACS Catal. 7, 2800–2804 (2017).
Li, N. et al. Steering the reaction pathway of syngas-to-light olefins with coordination unsaturated sites of ZnGaOx spinel. Nat. Commun. 13, 2742 (2022).
Bai, B. et al. Tuning the Crystal Phase to Form MnGaOx-Spinel for Highly Efficient Syngas to Light Olefins. Angew. Chem. Int. Ed. 62, e202217701 (2023).
Chen, Y. et al. Visualization of the active sites of zinc–chromium oxides and the CO/H2 activation mechanism in direct syngas conversion. J. Am. Chem. Soc. 146, 1887–1893 (2024).
Song, H. et al. Spinel-structured ZnCr2O4 with excess Zn is the active ZnO/Cr2O3 catalyst for high-temperature methanol synthesis. ACS Catal. 7, 7610–7622 (2017).
Del Piero, G., Trifiro, F. & Vaccari, A. Non-stoicheiometric Zn–Cr spinel as active phase in the catalytic synthesis of methanol. J. Chem. Soc., Chem. Commun. 656, 658 (1984).
Gao, X. et al. Binary ZnO/Zn–Cr nanospinel catalysts prepared by a hydrothermal method for isobutanol synthesis from syngas. Catal. Sci. Technol. 8, 2975–2986 (2018).
Wang, L. et al. The synergistic effect between ZnO and ZnCr2O4 on the catalytic performance for isobutanol synthesis from syngas. Fuel 253, 1570–1577 (2019).
Wang, X. et al. Synthesis gas conversion to lower olefins over ZnCr-SAPO-34 catalysts: role of ZnO−ZnCr2O4 interface. ChemCatChem 12, 4387–4395 (2020).
Gao, X. et al. Insight into the role of hydroxyl groups on the ZnCr catalyst for isobutanol synthesis from syngas. Appl. Catal. A. 547, 1–11 (2017).
Lai, Z. et al. Resolving the intricate mechanism and selectivity of syngas conversion on reduced ZnCr2Ox: a quantitative study from DFT and microkinetic simulations. ACS Catal. 11, 12977–12988 (2021).
Fu, X. & Xiao, J. Toward understanding and simplifying the reaction network of ketene production on ZnCr2O4 spinel catalysts. J. Phys. Chem. C. 125, 24902–24914 (2021).
Ma, S., Huang, S.-D. & Liu, Z.-P. Dynamic coordination of cations and catalytic selectivity on zinc–chromium oxide alloys during syngas conversion. Nat. Catal. 2, 671–677 (2019).
Guo, S. et al. Selective conversion of CO2 to trimethylbenzene and ethene by hydrogenation over a bifunctional ZnCrOx/H-ZSM-5 composite catalyst. ACS Catal. 14, 271–282 (2024).
Nie, Q. et al. In situ restructuring of ZnCr2O4 spinel catalyst alters elementary surface reaction kinetics and catalytic selectivity in CO hydrogenation reaction. ACS Catal. 14, 1922–1929 (2024).
Song, T. et al. Enhanced methanol synthesis over self-limited ZnO overlayers on Cu nanoparticles formed via gas-phase migration route. Angew. Chem. Int. Ed. 63, e202316888 (2024).
Winiarski, J., Tylus, W. & Szczygieł, B. EIS and XPS investigations on the corrosion mechanism of ternary Zn–Co–Mo alloy coatings in NaCl solution. Appl. Surf. Sci. 364, 455–466 (2016).
Haase, M., Weller, H. & Henglein, A. Photochemistry and radiation chemistry of colloidal semiconductors. 23. Electron storage on zinc oxide particles and size quantization. J. Phys. Chem. 92, 482–487 (1988).
Chen, J., Feng, Z., Ying, P. & Li, C. ZnO clusters encapsulated inside micropores of zeolites studied by UV raman and laser-induced luminescence spectroscopies. J. Phys. Chem. 108, 12669–12676 (2004).
Zhao, D. et al. In situ formation of ZnOx species for efficient propane dehydrogenation. Nature 599, 234–238 (2021).
Fan, Y., Wang, F., Li, R., Liu, C. & Fu, Q. Surface hydroxyl-determined migration and anchoring of silver on alumina in oxidative redispersion. ACS Catal. 13, 2277–2285 (2023).
Xie, Y.-C. & Tang, Y.-Q. Spontaneous monolayer dispersion of oxides and salts onto surfaces of supports: applications to heterogeneous catalysis. In Advances in Catalysis, Eley, D. D., ed. (Academic Press, 1990), pp. 1–43.
Feng, X. et al. Stable CuO/La2Sn2O7 catalysts for Soot combustion: study on the monolayer dispersion behavior of CuO over a La2Sn2O7 pyrochlore support. Chin. J. Catal. 42, 396–408 (2021).
ter Veen, H. R. J., Kim, T., Wachs, I. E. & Brongersma, H. H. Applications of high sensitivity-low energy ion scattering (HS-LEIS) in heterogeneous catalysis. Catal. Today 140, 197–201 (2009).
Wachs, I. E. & Routray, K. Catalysis science of bulk mixed oxides. ACS Catal. 2, 1235–1246 (2012).
He, J. et al. Subsurface A-site vacancy activates lattice oxygen in perovskite ferrites for methane anaerobic oxidation to syngas. Nat. Commun. 15, 5422 (2024).
Xie, Z. et al. Highly active nanosized anatase TiO2–x oxide catalysts in situ formed through reduction and Ostwald ripening processes for propane dehydrogenation. ACS Catal. 10, 14678–14693 (2020).
Otroshchenko, T. et al. ZrO2-based alternatives to conventional propane dehydrogenation catalysts: active sites, design, and performance. Angew. Chem. Int. Ed. 54, 15880–15883 (2015).
Noei, H., Wöll, C., Muhler, M. & Wang, Y. Activation of carbon dioxide on ZnO nanoparticles studied by vibrational spectroscopy. J. Phy. Chem. C. 115, 908–914 (2011).
Haruta, M. Spiers memorial lecture role of perimeter interfaces in catalysis by gold nanoparticles. Faraday Discuss. 152, 11–32 (2011).
Dong, A. et al. Modulating the formation and evolution of surface hydrogen species on ZnO through Cr addition. ACS Catal. 12, 6255–6264 (2022).
Eischens, R. P., Pliskin, W. A. & Low, M. J. D. The infrared spectrum of hydrogen chemisorbed on zinc oxide. J. Catal. 1, 180–191 (1962).
Feng, Z. et al. Asymmetric sites on the ZnZrOx catalyst for promoting formate formation and transformation in CO2 hydrogenation. J. Am. Chem. Soc. 145, 12663–12672 (2023).
Ling, Y. et al. Probing active species for CO hydrogenation over ZnCr2O4 catalysts. Chin. J. Catal. 43, 2017–2025 (2022).
Zhang, L. et al. Activation and surface reactions of CO and H2 on ZnO powders and nanoplates under CO hydrogenation reaction conditions. J. Energy Chem. 50, 351–357 (2020).
Jia, L. et al. Recovery of zinc resources from secondary zinc oxide via composite ammonia leaching: analysis of Zn leaching behavior. Chem. Eng. J. 472, 144930 (2023).
Ding, Y. et al. Effects of proximity-dependent metal migration on bifunctional composites catalyzed syngas to olefins. ACS Catal. 11, 9729–9737 (2021).
Acknowledgements
This work is supported by National Key R&D Program of China (2021YFA1502800, 2022YFA1504800, and 2022YFA1504500), National Natural Science Foundation of China (22332006, 22288201, and 22321002), Photon Science Center for Carbon Neutrality, and the Fundamental Research Funds for the Central Universities (20720220009). R.M. thanks support from the Dalian Innovation Support Plan for High Level Talents (2023RG002) and DICP (I202407, I202443). The authors thank the fruitful discussions with Feng Jiao and Xiulian Pan.
Author information
Authors and Affiliations
Contributions
X.F. performed material preparation, characterization, and catalytic activity tests. H.J. carried out the FTIR experiments. R.L. carried out the XPS experiments. M.L. carried out the STEM experiments. C.L. provided suggestions for the illustrations. X.D. provided suggestions for the characterization methods. M.C. provided HS-LEIS data support. X.F. wrote the original draft. H.J., L.L., Y.D., R.M., and X.W. were involved to discuss the results. X.B. and Q.F. conceived the idea, directed the project, and revised the paper. All authors discussed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks José Odriozola, Vitaly Ordomsky and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, X., Jia, H., Li, R. et al. ZnOx overlayer confined on ZnCr2O4 spinel for direct syngas conversion to light olefins. Nat Commun 16, 3711 (2025). https://doi.org/10.1038/s41467-025-58951-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58951-8
