
Abstract
Hydrogels are extensively utilized in stem cell-based tissue regeneration, providing a supportive environment that facilitates cell survival, differentiation, and integration with surrounding tissues. However, designing hydrogels for regenerating hard tissues like bone presents significant challenges. Here, we introduce macroporous hydrogels with spatiotemporally programmed mechanical properties for stem cell-driven bone regeneration. Using liquid-liquid phase separation and interfacial supramolecular self-assembly of protein fibres, the macroporous structure of hydrogels provide ample space to prevent contact inhibition during proliferation. The rigid protein fibre-coated pore shell provides sustained mechanical cues for guiding osteodifferentiation and protecting against mechanical loads. Temporally, the hydrogel exhibits tunable degradation rates that can synchronize with new tissue deposition to some extent. By integrating localized mechanical heterogeneity, macroporous structures, surface chemistry, and regenerative degradability, we demonstrate the efficacy of these stem cell-encapsulated hydrogels in rabbit and porcine models. This marks a substantial advancement in tailoring the mechanical properties of hydrogels for stem cell-assisted tissue regeneration.
Similar content being viewed by others

Introduction
Stem cells offer immense promise for tissue engineering, given their capacity to differentiate into various cell types representative of different tissues and organs1,2,3,4,5,6,7. However, the clinical application of stem cell-based therapies for tissue engineering encounters several hurdles. Foremost among these challenges is ensuring the successful engraftment and viability of implanted cells8,9. A significant portion of transplanted cells succumbs within the initial days post-implantation due to inadequate support for cell adhesion, proliferation, and functionality within the host environment10,11,12. Another obstacle lies in steering the differentiation of stem cells toward the desired lineage13,14,15,16. To circumvent these challenges, biomaterial scaffolds have been widely explored to improve the survival of implanted stem cells and guide their differentiation17,18,19,20,21,22,23,24,25,26,27,28,29. Hydrogels, in particular, have garnered attention for their ability to furnish a three-dimensional milieu that closely emulates the extracellular matrix (ECM) of native tissues30,31,32,33,34.
The mechanical properties of hydrogels, such as elasticity35,36, viscoelasticity37,38, stress relaxation39,40, and heterogeneity41,42, have been demonstrated to be important physical factors that influence the survival and subsequent differentiation of implanted stem cells. Current hydrogels often fail to provide a suitable combination of mechanical cues that support the growth and differentiation of implanted stem cells. For instance, in bone regeneration, achieving a high matrix elasticity is essential for promoting mechanotransduction and guiding stem cells towards osteoblastic or chondroblastic differentiation43,44,45,46,47,48. Yet, conventional methods to enhance matrix elasticity, such as biomineralization or increased crosslinking density49,50,51, frequently hinder cell behaviours like growth, spreading, and migration. Additionally, these hydrogels must degrade to accommodate cell proliferation and new matrix deposition52,53,54. However, matrix degradation inevitably compromises mechanical integrity, hindering sustained mechanical signalling crucial for new bone tissue formation. Furthermore, excessively stiff mechanical environments can disrupt the early inflammatory cascade regulation, impeding scar-free healing initiation55. Certain approaches, such as employing macroporous hydrogels43,56,57,58,59 or dynamically crosslinked and degradable ultrastiff hydrogels60,61,62,63,64, have shown promise in addressing some of these challenges and delivering favourable outcomes. Nevertheless, it remains challenging to develop a biomaterial capable of degrading over time while providing a continuous, appropriate mechanical stimulus to support stem cell differentiation towards osteoblasts and facilitate bone regeneration.
In this study, we develop shell-hardened macroporous hydrogels with distinctive mechanical properties and adjustable degradability to facilitate stem cell-mediated bone regeneration. Our approach involves creating macroporous structures using a soft-templating technique based on liquid-liquid phase separation, with the pore shell reinforced through interfacial self-assembly of rigid protein fibres. The macropore structure provides ample space to prevent contact inhibition during stem cell proliferation. Simultaneously, the rigid pore shell offers sustained mechanical cues to guide stem cell osteodifferentiation and protects cells from damage under mechanical stress. Moreover, the soft hydrogel matrix exhibits tunable degradation, which may align with the rate of new tissue formation to some degree. As a proof of concept, we showcase the application of stem cell-encapsulated hydrogels for bone regeneration in rabbit and porcine models. This research signifies a crucial advancement in engineering mechanically heterogeneous hydrogels for stem cell-assisted tissue regeneration.
Results
Design and fabrication of shell-hardened macroporous hydrogels
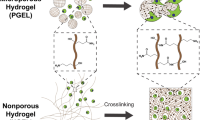
The design of shell-hardened macroporous hydrogels is depicted in Fig. 1. These hydrogels are fabricated using a soft-templating technique, involving liquid-liquid phase separation between polyethylene glycol (PEG) and dextran. By adjusting the concentrations of dextran and PEG, the liquid‒liquid phase separation can be controlled, with PEG serving as the percolating phase and dextran forming nonpercolating droplets. The dextran phase acts as a soft template for macropore formation (Fig. 1a). Polymerizable monomers, such as acrylamide, can be present in the PEG phase, facilitating hydrogel formation via free radical polymerization. The addition of preassembled lysozyme nanofibers (protein fibres) into the phase-separated system leads to the self-assembly of protein fibres at the interface, ultimately forming a stable protein fibrous shell (Fig. 1a). To enable covalent attachment of protein nanofibers to the hydrogel matrix, lysozyme is modified with acryl groups (right of Fig. 1b). Additionally, a cyclo(Arg-Gly-Asp-D-Phe-Lys-acrylate) (acrylated RGD) ligand is also introduced to facilitate cell attachment (left of Fig. 1b). Two types of PEG crosslinkers are utilized: 4-armed PEG acrylate (PEG-ACLT) containing degradable ester bonds and 4-armed PEG acrylamide (PEG-ACA) lacking degradable bonds (Supplementary Fig. 1). By altering the ratio of these crosslinkers, the degradation rate of the hydrogels can be modulated.

a Schematic depicting the fabrication and structure of shell-hardened macroporous hydrogels. A mixture of polyethylene glycol (PEG) and dextran solution induces liquid‒liquid phase separation, serving as a soft template for macroporous hydrogel formation. Protein fibres within the precursors assemble at the interface of dextran nanodrops, forming a stable protein fibrous shell. b Detailed illustrations highlighting the shell-hardened macropore and cell encapsulation. The shells are anchored to the hydrogel matrix through covalent linkages. Cells are encapsulated and adhere to the interface of macropores with the assistance of RGD peptide.
Following the formation of the phase-separated system, components containing acryl groups—including PEG-ACLT, acrylated RGD, acrylated lysozyme nanofiber, and acrylamide—were copolymerized under blue light (405 nm) illumination using lithium phenyl-2,4,6-trimethylbenzoylphosphinate (Lap) as the photoinitiator (Supplementary Fig. 2). This process resulted in the formation of hydrogels featuring shell-hardened macroporous structures. To determine the optimal PEG-to-dextran ratio for macropore diameters suitable for cell encapsulation and culture, we first examined the diameters of shell-free macropores. The average macropore diameters in hydrogel precursors ranged from 6.7 ± 1.5 to 24.2 ± 6.2 μm, depending on the concentrations of PEG and dextran (Supplementary Fig. 3). Upon hydrogel swelling, the pore sizes expanded to 14.9 ± 3.3 to 49.2 ± 8.8 μm at equilibrium (Supplementary Fig. 4). Based on these results, PEG concentrations of 80 mg mL-1 and dextran concentrations of 50 mg mL-1 were chosen for hydrogel preparation, as the diameter of ~50 μm was considered suitable for cell encapsulation and culture.
Cell-encapsulated hydrogels were prepared by directly adding cell suspensions to the dextran phase before phase separation and polymerization. We proposed that the shells and hydrogel matrix might possess distinct mechanical properties owing to the presence of rigid lysozyme nanofibers on the pore shells. These shells obstruct the encapsulated cells’ ability to sense the elasticity of the hydrogel matrix, resulting in mechanotransduction-induced differentiation of stem cells primarily being triggered by mechanical signals from the shells rather than the hydrogel matrix (Fig. 1b). Consequently, stem cell differentiation is mainly influenced by the mechanical signals from the stiff nanofiber shells, regardless of variations in hydrogel matrix elasticity due to degradation. This ensures consistent osteodifferentiation of encapsulated stem cells within the hydrogel matrix. Hereafter, shell-hardened macroporous hydrogels are referred to as SP hydrogels. For comparison, hydrogels containing macropores without shells (referred to as NP hydrogels) and hydrogels without macropores (referred to as NN hydrogels) were also prepared as controls. Live/dead staining showed that the viabilities of mouse embryonic osteoblast cells (MC3T3-E1) on all hydrogels were above 97-98% after 24 h, indicating in vitro biocompatibility (Supplementary Fig. 5). Notably, although dextran can be removed via dialysis (Supplementary Fig. 6), this step may affect for eventual translation. To minimize its impact, cell-encapsulated hydrogels can be immersed in a cell culture medium, with removal expedited by increasing the frequency of medium changes.
Hydrogel structures
In the left panel of Fig. 2a, nanodrops were observed in the precursors of NP and SP hydrogels, resulting from the phase-phase separation of PEG and dextran solutions, which were absent in NN hydrogels. The nanodrops in the precursor of SP hydrogels exhibited a more uniform diameter compared to those in the precursor of NP hydrogels, attributed to the stabilizing effect of the lysozyme nanofibers. Fluorescent rings were detected at the periphery of the nanodrops in the precursor of SP hydrogels prepared with fluorescein isothiocyanate (FITC)-labelled lysozyme, confirming the presence of lysozyme nanofiber coatings on the nanodrop structures (right panel of Fig. 2a). After gelation and swelling, the NN hydrogel appeared transparent while the NP and SP hydrogels were semi-transparent, indicating the presence of micron-sized structures (Fig. 2b).

a Micrographs (left) and fluorescent micrographs (right) for the precursors of NN, NP and SP hydrogels. FITC-labelled lysozyme was used in the precursor of SP hydrogels. Each experiment was repeated 3 times independently with similar results. b Optical image of NN, NP and SP hydrogels. c FESEM images of NN, NP and SP hydrogels. Each experiment was repeated 3 times independently with similar results. 3D reconstruction of the macroporous structures in NN (d), NP (e) and SP (f) hydrogels using LCFM. The scanning area was 200 μm × 318 μm × 318 μm. FITC-labelled PEG was used in NN and NP hydrogels to visualize the hydrogel matrix, while FITC-labelled lysozyme was utilized in SP hydrogels to visualize the shells of the macropores. g Enlarged fluorescent micrograph (left) showing the precursors of SP hydrogels, with the corresponding intensity profile (right) along the red line in the left panel. The green shading indicates the ring thickness. h Summary of shell thickness measurements from three independent fabrication batches, determined using fluorescence labelling-assisted analysis in g (n = 43 points for each batch). The p values for the batch 1 and batch 2, batch 2 and batch 3, and batch 1 and batch 3 are 0.4381, 0.0623 and 0.2484, respectively. i Enlarged top view of the 3D reconstruction of macroporous structures in SP hydrogels (left), along with a summary of pore sizes (right) from three different fabrication batches, based on 3D reconstruction analysis (n = 43 points for each batch). The p values for the batch 1 and batch 2, batch 2 and batch 3, and batch 1 and batch 3 are 0.1861, 0.2583 and 0.8003, respectively. Statistical significance was assessed using Student’s t-test. NS: not significant.
Field emission scanning electron microscopy (FESEM) images further revealed the presence of typical microporous structures in NN hydrogels, while macroporous structures were observed in NP and SP hydrogels (Fig. 2c). The macroporous structures, occupying space within the PEG/acrylamide polymer network, contributed to a higher crosslinking density in matrix of NP and SP hydrogels compared to NN hydrogels. Nonetheless, when factoring in the volume occupied by the macropores, the overall crosslinking densities among all hydrogels appeared to be uniform at the macroscopic scale. This was substantiated by the comparable swelling ratios and water contents observed across all hydrogels (Supplementary Fig. 7).
Fluorescent labelling-assisted 3D reconstruction based on laser confocal fluorescence microscopy (LCFM) was further employed to characterize the macroporous structures (Fig. 2d–f). In NP hydrogels, macroporous structures lacking shells were predominantly formed by the PEG phase, as evidenced by FITC-labelled PEG (Fig. 2e). Conversely, macropores within SP hydrogels were enveloped by green shells, as indicated by FITC-labelled lysozyme (Fig. 2f). Given that the diameters of the macropores (>50 μm) exceeded the typical size of cells (~5–15 μm) at the presence of lysozyme shells, SP hydrogels were deemed highly suitable for cell encapsulation and culture. Notably, both the NN hydrogels prepared with FITC-labelled PEG and those prepared in the presence of FITC-labelled lysozyme both showed uniform fluorescence, suggesting that the macroporous structures cannot be attributed to the assembly of lysozyme fibrils in the PEG phase (Fig. 2d and Supplementary Fig. 8). To verify the reproducibility of shell-hardened macroporous hydrogels, we have evaluated the shell thickness and pore size of SP hydrogels across different fabrication batches (Fig. 3g–i). The consistent shell thickness and pore size with no significant differences suggested the good reproducibility.

a Schematic depiction of the AFM-based nanoindentation on the cross profile of SP hydrogels. The cantilever tip compresses and retracts from the hydrogel surface to obtain force-displacement curves. b Typical micrograph (left) and force‒displacement curve (right) of AFM-based nanoindentation. Young’s modulus was calculated by fitting the approaching traces using the Hertz model. Each experiment was repeated 3 times independently with similar results. Histograms displaying the distribution of Young’s modulus of the matrix (M) and macropore (P) in SP (c) and NP (d) hydrogels determined by AFM-based nanoindentation in PBS (10 mM, pH = 7.4). e Degradation rates of SP hydrogels at various total concentrations of PEG-ACLT and PEG-ACA (10, 40 and 80 mg mL−1). The concentration of PEG-ACA was maintained at 10 mg mL-1 while the concentration of PEG-ACLT varied from 0 to 70 mg mL-1. Values represent the mean and the standard deviation (n = 6 independent samples). The p values for the comparisons between the 10 and 40 mg mL-1 groups, 40 and 80 mg mL-1 groups, and 10 and 80 mg mL-1 groups are 0.0143, 0.0277, and 0.0020, respectively. f Degradation rates of SP hydrogels at various mass ratios of PEG-ACLT to PEG-ACA (1:3, 2:2 and 3:1). The total mass of PEG-ACLT and PEG-ACA was maintained at 40 mg mL-1. Values represent the mean and the standard deviation (n = 6 independent samples). The p values for the comparisons between the 1:3 and 2:2 groups, 2:2 and 3:1 groups, and 1:3 and 3:1 groups are 0.1862, 0.00003, and 0.00002, respectively. g Normalized Young’s modulus of the matrix (M) and macropore (P) in SP-low hydrogels over 42 days in SBF at 37 °C. Values represent the mean and the standard deviation (n = 3 independent samples). The p value is 0.00005. Statistical significance in e-g was assessed using Student’s t-test. *: p < 0.05; **p < 0.01; ***p < 0.001; NS: not significant.
Mechanical and Degradation Properties
Subsequently, we explored the localized mechanical strength of SP hydrogels at the microscopic level. Hydrogels with different matrix elasticity were prepared by substituting PEG-ACLT with hydroxyl-terminated 4-armed polyethylene glycol (PEG-OH, 20 K). The NN, NP and SP hydrogels at different concentrations of PEG-ACLT (high: CPEG-ACA = 10 mg mL-1 and CPEG-ACLT = 70 mg mL-1; low: CPEG-ACA = 10 mg mL-1 and CPEG-OH = 70 mg mL-1) were named NN-high, NN-low, NP-high, NP-low, SP-high, and SP-low hydrogels, respectively. The macropore structures of SP hydrogels with low matrix elasticity (SP-low hydrogel, Supplementary Fig. 9) closely resembled those with high matrix elasticity (SP-high hydrogel, Fig. 2c, f), since the mass concentrations of PEG (PEG-ACLT/PEG-ACA or PEG-OH/PEG-ACA) and dextran remained constant.
We assessed the stiffness of shelled macropores and hydrogel matrices in aqueous environments (PBS, 10 mM, pH=7.4) through AFM-based nanoindentation (Fig. 3a, b). The Young’s modulus of both macropores and the hydrogel matrix were calculated by fitting the approaching traces of force-displacement curves using the Hertz model (right of Fig. 3b). As shown in Fig. 3c, the Young’s modulus of shelled macropores (P) in SP-low and SP-high hydrogels were ~ 28.7 ± 3.4 and 163.2 ± 29.3 kPa, considerably higher than the 12.3 ± 2.0 and 63.3 ± 7.0 kPa of matrix (M). In contrast, no significant stiffness difference was observed between the macropore and matrix in NP-low hydrogels (~11.6 ± 2.5 kPa and 11.8 ± 2.7 kPa, left of Fig. 3d) and NP-high hydrogels (~59.9 ± 16.8 kPa and 57.8 ± 10.9 kPa, right of Fig. 3d). These findings underscored the successful fabrication of shell-hardened macroporous hydrogels, promising for spatial cell differentiation regulation.
We further conducted in vitro degradation measurements on SP hydrogels with varying crosslinking densities and different ratios of PEG-ACLT to PEG-ACA (Fig. 3e, f)65. When exposed to simulated body fluid (SBF) buffer at 37 °C for 48 days, SP hydrogels with different concentrations of PEG crosslinking displayed distinct degradation rates, demonstrating the tunable biodegradability of SP hydrogels (Fig. 3e and Supplementary Fig. 10a). Notably, when the ratio of ester-containing PEG (PEG-ACLT) exceeded 50%, the degradation rate was significantly accelerated (Fig. 3f and Supplementary Fig. 10b). The tunable degradation rates enabled us to pinpoint an appropriate hydrogel formulation that may match the deposition rates of the matrix for in vivo stem cell-assisted regeneration66,67. Furthermore, we assessed the variation in the Young’s modulus of the shelled macropores and the matrix of SP-low hydrogels during in vitro degradation to confirm the consistency of mechanical cues from the shell-hardened macropores. The Young’s modulus of the shelled macropores decreased at a much slower rate compared to that of the matrix, with a slight reduction of 20% over 42 days (Fig. 3g and Supplementary Fig. 11). In contrast, the Young’s modulus of the matrix decreased significantly for more than 70% over 42 days. This indicates that the rigid nanofiber shells can provide consistent mechanical cues to the encapsulated cells, even as the matrix undergoes significant softening.
Protection of cells from mechanical damage
We then investigated whether the stiff lysozyme shell could shield cells inside the macropores from mechanical damage under intensive mechanical loads on the hydrogels. MC3T3-E1 cells were encapsulated in the shelled macropores of hydrogels, as confirmed by FESEM imaging (Fig. 4a). Hydrogels containing cells were subjected to cyclic compressions at a frequency of 0.5 Hz (strains: 0%, 30%, and 60%) to evaluate cytoprotection (Fig. 4b, c)68. The initial cell viabilities were ~81% and 57% in SP-low and SP-high hydrogels after encapsulation (Supplementary Fig. 12). It is worth noting that initial cell viabilities in SP-high hydrogels were lower than in other hydrogels, as cells trapped in the matrix may be damaged by stress from hydrogel swelling.

a FESEM images show encapsulated cells in shelled macropores (left), while the illustration depicts cell protection during continuous compression-relaxation cycles using SP hydrogels (right). Each experiment was repeated 3 times independently with similar results. Optical images (b) and strain signals (c) of continuous compression-relaxation applied to hydrogels. MC3T3-E1 cells in hydrogels identified by live/dead staining (green/red) and against ROS staining (blue) after 1500 compression-relaxation cycles at low (d) and high (e) matrix elasticities. A space of 1272 μm × 1272 μm × 400 μm in each sample was scanned layer by layer, and the images were projected onto the z-axis to show the staining. Cell viabilities (f) and normalized ROS intensities (g) of MC3T3-E1 cells encapsulated in different hydrogels after 1500 cycles of compression and relaxation. Values represent the mean and the standard deviation (n = 3 independent experiments). The p-values for NN and SP hydrogels at low concentration under strains of 0%, 30%, and 60% are 0.3005, 0.0127, and 0.0118, respectively, in (f), and 0.0039, 0.0041, and 0.0203, respectively, in g. At high concentration, the p-values for NN and SP hydrogels under strains of 0%, 30%, and 60% are 0.0037, 0.0002, and 0.00001, respectively, in (f), and 0.0012, 0.0439, and 0.0285, respectively, in (g). Statistical significance was determined by two-tailed t-test. NS: P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
After 1500 cycles of compression and relaxation, live/dead staining revealed that cells encapsulated in SP-low hydrogels exhibited 10% and 15% higher viability than those in NN-low hydrogels under moderate and excessive compression, respectively (Fig. 4d, f). Moreover, cells in SP-low hydrogels generated 49% and 42% fewer reactive oxygen species (ROS) than cells in NN-low hydrogels (Fig. 4d, g). Differences in viability and ROS production were more pronounced in hydrogels with higher concentrations of PEG-ACLT (Fig. 4e–g). Cell viability in SP-high hydrogels was 9 and 7 times higher than in NN-high hydrogels (<4%) under moderate and excessive compression, respectively. Notably, cell viability in NN-high hydrogels was significantly lower than in NN-low hydrogels due to mechanical injury caused by the swelling of rigid hydrogel networks. More importantly, cell viability remained almost constant with increasing strain in SP-high hydrogels, indicating that the shelled macropore structures effectively protected cells from mechanical stress, even under significant deformations. The divergent trends of cell viability and corresponding ROS levels suggested that cell damage primarily resulted from mechanical injury during compression cycles. Note that the lysozyme nanofibers in SP hydrogels were fully denatured, shifting from an α-helix to a β-sheet structure69. Thus, the decrease in ROS in SP hydrogels results from the mechanical protection of the hard shell, not from ROS scavenging by the deactivated lysozyme. Additionally, the shelled macropore structures did not restrict cell proliferation based on the assessment of the total DNA content within the hydrogel (Supplementary Fig. 13); in contrast, cells tended to form clusters inside macropores, and some may have migrated out of the macropores to the matrix because the lysozyme nanofiber shells were primarily assembled by weak physical interactions (Supplementary Fig. 12a). These results demonstrate that SP hydrogels can effectively safeguard encapsulated cells from excessive mechanical stress under dynamic loading without compromising cell proliferation.
Rigid Shells Offer Consistent Mechanical Cues for Osteodifferentiation
We then investigated whether shell-stiffened SP hydrogels could consistently provide mechanical cues to guide stem cell differentiation toward osteoblasts, using SP-low, NP-low, SP-high, and NP-high hydrogels as model systems. After 5 and 10 days of cell culture, we assessed the differentiation of MC3T3-E1 cells encapsulated within these hydrogels (Fig. 5), with cells cultured on tissue culture wells serving as a control group. Immunostaining of molecular markers (OCN and Runx2) showed no upregulation of osteogenic differentiation-related genes in NP-low hydrogels, suggesting that low hydrogel matrix elasticity does not induce cell differentiation (Fig. 5a and Supplementary Figs. 14–17). In contrast, significant upregulation of OCN and Runx2 was observed in SP-low hydrogels, suggesting the initiation of osteoblast differentiation. This indicates that the stiffened macropore shell of SP-low hydrogels can promote osteogenic differentiation even with low matrix stiffness. Moreover, OCN and Runx2 upregulation was also observed in both NP-high and SP-high hydrogels, attributed to the high elasticity of their hydrogel matrices, regardless of the presence of shelled macropores (Fig. 5a and Supplementary Fig. 14–17). The quantification of nuclear Runx2 and OCN intensity across different replicates further confirmed the findings observed in the immunostaining images (Supplementary Fig. 18).

a MC3T3-E1 cells in different hydrogels identified by immunostaining of molecular markers (OCN or Runx2, shown in red) and F-actin (phalloidin, shown in green) after 5 and 10 days of culture. MC3T3-E1 cells on the cell culture wells served as the control group. OCN and Runx2, specific markers for osteogenic differentiation, were upregulated in SP-low, NP-high and SP-high hydrogels. Cell nuclei were stained with DAPI (blue). A space of 318 μm × 318 μm × 100 μm in each sample was scanned layer by layer, and the images were projected onto the z-axis to show immunostaining. b Heatmap illustrating mRNA expression levels corresponding to osteogenesis genes (Runx2, OCN, Col I and ALP) for cells in different hydrogels or cell culture wells (Control) after 5 and 10 days of culture. The intensity represents the expression relative to the control group. c Summary of relative mRNA expression levels of osteogenesis-related genes (Runx2, OCN, Col I, and ALP) in cells cultured in different hydrogels or cell culture wells (Control) after 10 days. The expression levels of these genes were normalized to those of the control group. Values represent the mean ± standard deviation (n = 6 independent experiments). The p-values for the comparisons between the control group and the NP-low, SP-low, NP-high, and SP-high groups are as follows: for Runx2, 0.2938, 0.0048, 0.0012, and 0.0098, respectively; for OCN, 0.0068, 0.0025, 0.0019, and 0.0007, respectively; for Col I, 0.5670, 0.0130, 0.0003, and 0.0020, respectively; and for ALP, 0.0127, 0.0028, 0.00001, and 0.0003, respectively. Statistical significance between different groups and the control group was assessed using two-tailed Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001; NS: not significant.
Interestingly, dispersed staining of OCN and Runx2 was observed around the encapsulated cells, showing a spherical distribution in the NP-high and SP-high hydrogels (Fig. 5a and Supplementary Figs. 14–17), probably due to a small amount of release from the cells which requires further investigation beyond immunostaining70,71,72,73,74. In contrast, minimal staining of OCN and Runx2 was detected around cells in SP-low hydrogels. This is likely attributed to the highly crosslinked hydrogel matrix in the NP-high and SP-high hydrogels, which restricts the diffusion of these factors. We calculated that the mesh size of the NP-high and SP-high hydrogels was ~ 3–4 nm, smaller than the size of OCN and Runx2 molecules. The inhibition of the macromolecule diffusion was further demonstrated using a model fluorescent protein encapsulated within the macropores (Supplementary Fig. 19). A critical aspect of stem cell-mediated regeneration is the secretion of growth factors and cytokines that modulate the local environment, recruit other cells to the injury site, and reduce inflammation, thereby facilitating the healing process75,76,77,78. We showed that SP-low hydrogels are more effective in dispersing the stem cell secretion compared to macroporous hydrogels with densely crosslinked external matrices (NP-high and SP-high hydrogels), which also enhances bone regeneration.
Moreover, the effectiveness of SP-low hydrogels in promoting bone regeneration was underscored by F-actin staining. F-actin is pivotal in transducing mechanical signals into biochemical signals that enhance osteoblast differentiation and functionality79,80,81,82. In SP-low hydrogels, F-actin was organized into long, dense fibres under significant tension, with higher intensity and area coverage observed (Fig. 5a and Supplementary Figs. 14–17 and 20). Conversely, F-actin fibres in SP-high and NP-high hydrogels appeared notably shorter and declined over time, suggesting a less favourable local mechanical environment83. Given that the mechanotransduction of cells is significantly influenced by the mechanical microenvironment, we anticipate that the stiffened macropore shell would promote the activation of YAP/TAZ, the mechanically sensitive transcriptional coactivators, which is expected to further regulate cell differentiation41.
The differentiation of MC3T3-E1 cells was further characterized by real-time quantitative reverse transcription polymerase chain reaction (qRT‒PCR) analysis of four well-established markers (Runx2, OCN, Col I, and ALP, Supplementary Table 1). Consistently, the expression levels of these marker mRNAs in NP-high, SP-high, and SP-low hydrogels were significantly higher than those in NP-low hydrogels and cell culture wells (Fig. 5b, c and supplementary Fig. 21). Moreover, neither lysozyme nor lysozyme nanofibers affected MC3T3-E1 cell differentiation (Supplementary Fig. 22), suggesting that shell stiffness may contribute to differentiation. However, aside from mechanical effects, other factors—such as differential capture of ECM proteins or growth factors—may also influence differentiation due to differences in surface chemistry. We acknowledge that cell differentiation likely results from a combination of mechanical and chemical cues, with the stiff shells providing a consistent mechanical influence.
Bone regeneration using Rabbit Models
Given that SP hydrogels can shield cells from mechanical harm and steer stem cell differentiation towards osteoblasts, we anticipated that they can be used for transplanted stem cell-assisted bone regeneration. To circumvent immune rejection in rabbit models, we opted for rabbit bone mesenchymal stem cells (rBMSCs) instead of mouse-derived MC3T3-E1 cells. SP hydrogels demonstrated no adverse effects on rBMSC viability (Supplementary Fig. 23). The in vitro differentiation of rBMSCs in NP and SP hydrogels was similar to that of MC3T3-E1 cells (Supplementary Fig. 24, 25 and Supplementary Table 2). Markers indicative of early and late stages of osteogenic differentiation, such as ALP activity and mineralized nodule formation, were notably heightened in cells within SP-low hydrogels after 14 days of culture (Supplementary Fig. 26), suggesting robust osteogenic differentiation of rBMSCs. Furthermore, ALP and Alizarin red S staining revealed increased cell density and spreading in SP-low hydrogels, implying that lower matrix stiffness facilitates cell migration and proliferation.
Subsequently, we explored bone regeneration using rBMSC-encapsulated hydrogels in a rabbit model. NN, NP, and SP hydrogels with encapsulated rBMSCs were directly implanted into freshly created bone defects in the femoral condyle of New Zealand white rabbits (Fig. 6a). No growth factors or hyaluronic acid were utilized in these experiments. Bone regeneration was evaluated after the implantation for 12 weeks, with a blank group serving as the control. The bone defects were repaired to varying degrees by cell-encapsulated hydrogels (Fig. 6b and Supplementary Fig. 27). Microcomputed tomography (micro-CT) imaging analysis (Fig. 6c) showed that bone defects in the SP-low hydrogel group were nearly completely filled with bony tissues and covered with continuous cortical bone. Conversely, other groups displayed evident cavities or intermittent cortical bones, albeit with partial defect repair. The control group exhibited a sizable cavity within the defect area, indicative of the poorest bone regeneration. Moreover, the SP-low hydrogel group demonstrated the highest values of bone volume/total volume (BV/TV), trabecular thickness (Tb. Th), and trabecular number (Tb. N) (Fig. 6d), suggesting significant osteogenesis in the hydrogel filling region. The other groups exhibited higher trabecular separation (Tb. Sp), indicating notable bone resorption. It is worth noting that the bone regeneration observed in the NP-low hydrogel groups was notably inferior to that in the SP-low groups, underscoring the essential role of the macropore shell in bone regeneration.

a Schematic depicting the surgical procedure for creating bone defects and filling them with hydrogel in New Zealand white rabbits. A defect (diameter ~5 mm) was created in the femoral condyle, followed by implantation of rBMSC-encapsulated hydrogel into the defect. Scale bar: 5 mm. b Overall views of the bone defect regions for the SP-low hydrogel and control groups 12 weeks post-implantation. The blank group served as the control. Scale bar: 5 mm. c Bone regeneration assessed through micro-CT analysis for different groups at 12 weeks post-implantation. Scale bar: 2.5 mm. d Quantitative micro-CT analysis for different groups, including bone volume/total volume (BV/TV), trabecular thickness (Tb. Th), trabecular number (Tb. N), and trabecular separation (Tb. Sp). Values represent the mean and the standard deviation (n = 5 independent samples). The p-values for the comparisons of BV/TV between the SP-low group and the control, NP-low, NP-high, and SP-high groups are 0.000001, 0.0001, 0.0024, and 0.0141, respectively. For Tb. Th, the p values are 0.0000009, 0.00002, 0.0058, and 0.0273, respectively. For Tb. N, the p values are 0.000002, 0.000004, 0.0010, and 0.0008, respectively. For Tb. Sp, the p values are 0.00000007, 0.0009, 0.0185, and 0.0480, respectively. Statistical significance between different groups and the SP-low group was assessed using two-tailed Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001; NS: not significant. e, Representative histological images showing H&E staining, Masson’s trichrome staining, Trap staining, and immunofluorescence staining of Col I, Col X and IBSP (shown in red) for regenerated bones at 12 weeks post-implantation. Cell nuclei were counterstained with DAPI (blue) in immunostaining. The arrows indicate the specific areas of interest.
Histological assessments further validated the superior bone regeneration observed in the SP-low hydrogel group (Fig. 6e). H&E staining reveals uniform and seamlessly regenerated bone tissues that completely fill the defects in this group, accompanied by the formation of a fully developed bone cortex. In contrast, irregularly regenerated tissues, gaps, or discontinuous bone cortex were observed in the NP-high and SP-high hydrogel groups, indicating inadequate bone regeneration. Masson’s trichrome staining illustrated extensive cortical bone regeneration in the SP-low hydrogel group, contrasting with minimal or discontinuous cortical bone regeneration in the other groups. Additionally, minimal Trap staining in the SP-low hydrogel groups suggested nearly complete bone regeneration. The heightened level of immunofluorescent staining for type I (Col I-IF), type X collagen (Col X-IF) and bone sialoprotein (IBSP-IF) in the SP-low hydrogel group further indicated enhanced bone regeneration compared to other groups (Fig. 6e). The quantification of the histological analysis across different replicates confirmed the observation from the representative images (Supplementary Fig. 28).
Moreover, the micro-CT analysis and histological results from the SP-low group showed the greatest similarity to the uninjured tissue, further supporting the superior bone regeneration observed in the SP-low hydrogel group (Supplementary Fig. 29). Notably, SP-low hydrogels without encapsulated cells exhibited poor bone regeneration, indicating that the bone-promoting effects were driven by the combined action of the hydrogel and BMSCs, rather than by the hydrogel alone (Supplementary Fig. 30). These results emphasized the critical role of the decoupled shell and matrix elasticity in macroporous hydrogels for successful bone regeneration. The rigid shell induced directional differentiation of stem cells, while the low restriction of the hydrogel matrix promoted cell proliferation, migration and new matrix deposition, synergistically contributing to the enhanced bone regeneration observed in the SP-low hydrogel group.
Bone regeneration using porcine models
Bone regeneration in large animal models can be more challenging due to the larger size and scale of defects, and the slower bone turnover and metabolic rates characteristic of larger animals. Following successful bone regeneration outcomes in rabbit models, we extended our investigations to a porcine model, using hydrogels encapsulating porcine bone mesenchymal stem cells (pBMSCs) (Fig. 7a). Given that hydrogels with high rigidity and densely crosslinked external matrices, such as NP-high and SP-high hydrogels, did not yield favourable regeneration outcomes in rabbit models, we mainly focused on hydrogels characterized by low external matrix rigidity. Similar to the results in rabbit models, the group of pBMSCs-encapsulated SP-low hydrogel (SP-low-cell) demonstrated nearly complete repair of bone defects, forming consecutive cortical bone and bony tissues, as confirmed by micro-CT imaging analysis (Fig. 7b and Supplementary Fig. 31). Conversely, noticeable cavities and injuries persisted in other groups, despite partial defect repair. The SP-low-cell hydrogel group attained the highest scores for bone volume/total volume (BV/TV), trabecular thickness (Tb. Th), and trabecular number (Tb. N), while registering the lowest scores for trabecular separation (Tb. Sp) (Fig. 7c). In contrast, the groups of SP-low hydrogel without encapsulated cell (SP-low) and NP-low hydrogel with encapsulated cell (NP-low-cell) showed inferior bone regeneration, highlighting the critical role of the implanted cells and macropore shell in facilitating effective bone regeneration. The enhanced bone regeneration in the SP-low-cell hydrogel group was further validated through histological evaluations, including H&E staining, safranin O staining, and immunofluorescent staining of collagen types I (Col I-IF), X (Col X-IF) and IBSP (IBSP-IF) (Fig. 7d and Supplementary Fig. 32). The SP-low group also exhibited micro-CT and histological results comparable to those of uninjured bones in the porcine model, similar to the findings observed in the rabbit model (Supplementary Fig. 33).

a Schematic illustrating the surgical procedure for creating bone defects and filling them with hydrogel in male Bama mini pigs. A defect (diameter ~12 mm) was made in the femoral condyle, followed by implantation of hydrogels into the defect. b Assessment of bone regeneration through micro-CT analysis for different groups at 12 weeks post-implantation. The blank group served as the control. c Quantitative micro-CT analysis for different groups, including bone volume/total volume (BV/TV), trabecular thickness (Tb. Th), trabecular number (Tb. N), and trabecular separation (Tb. Sp). Values represent the mean and the standard deviation (n = 6 independent samples). The p-values for the comparisons of BV/TV between the SP-low-cell groups and the control, SP-low, and NP-low-cell groups are 0.0001, 0.0001, and 0.000003, respectively. For Tb. Th, the p values are 0.00004, 0.00006, and 0.0002, respectively. For Tb. N, the p values are 0.0002, 0.0004, and 0.0003, respectively. For Tb. Sp, the p values are 0.000005, 0.000002, and 0.000005, respectively. Statistical significance between different groups and the SP-low group was assessed using two-tailed Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001; NS: not significant. d Representative histological images showing H&E staining, Safranin O staining, and immunofluorescence staining of Col I, Col X and IBSP (shown in red) for regenerated bones at 12 weeks post-implantation. Cell nuclei were counterstained with DAPI (blue) in immunostaining. The arrows indicate the specific areas of interest.
In vivo biocompatibility and degradation
Finally, we assessed the biocompatibility and degradation of SP hydrogels through dorsal subcutaneous implantation in a rat model. Histological evaluations and assessments of inflammatory factors conducted 24 hours post-implantation revealed only mild inflammation with no significant tissue damage (Supplementary Fig. 34). Blood chemistry and cell indicators remained stable, indicating a mild systemic response (Supplementary Fig. 35). Further haematological analysis and evaluation of major organs after prolonged exposure to SP hydrogels for 42 days yielded similar results with no pathological changes observed (Supplementary Figs. 36, 37). Additionally, we investigated the in vivo degradability of the hydrogels. The SP-low hydrogel exhibited gradual degradation over 42 days, with cells and granulation tissue infiltrating the divided hydrogels (Supplementary Fig. 38). In contrast, the SP-high hydrogel showed significantly slower biodegradation, leaving over 50% residue (Supplementary Fig. 39). This slow degradation rate aligns with the observed inferior bone regeneration in vivo. Noting that the major regeneration of bone tissues typically begins between half to one month after the creation of defects and persisted for 6-12 weeks66,67,84,85,86, the tunable biodegradation rate of SP-low hydrogels may potentially synchronize with the pace of new tissue regeneration.
Discussion
Bone tissue regeneration remains a great challenge for bioengineers87,88. Bones possess a complex hierarchical structure with a combination of organic and inorganic components, making it difficult to replicate the precise architecture and properties required for successful regeneration89. Furthermore, bones endure diverse mechanical loads within the body, necessitating regenerated tissue to exhibit requisite strength, stiffness, and resilience to withstand these forces without excessive deformation or breakage90,91. In addition, regenerated bone must seamlessly integrate with the surrounding native bone tissue to restore mechanical stability and functionality. Achieving this level of integration can be challenging, particularly in cases where significant bone loss has occurred92.
Stem cells have the capacity to differentiate into various cell types, including osteoblasts responsible for bone formation. Harnessing this potential, stem cell-assisted bone regeneration has proven to enhance the healing process and facilitate more effective tissue repair93. The integration of stem cells with advanced hydrogel techniques enables the development of complex, functional bone tissue constructs, suitable for treating larger bone defects or replacing damaged tissue when conventional treatment modalities fall short44,94,95. However, traditional hydrogels often fail to provide the right combination of mechanical cues for optimal stem cell growth and differentiation. For example, mineralized hydrogels, formed by the deposition of inorganic salts, lack degradability and cannot be remodelled by implanted cells96. Additionally, rigid hydrogels, despite supporting the osteodifferentiation of stem cells, often impede cell proliferation and migration66. Although incorporating degradable units97 or void-forming mechanisms43 can promote cell growth, the dramatic decrease in rigidity upon degradation undermines their ability to provide consistent, sustainable mechanical signals for bone regeneration. Moreover, aligning degradation rates with the pace of new tissue regeneration also poses a challenge.
In this study, we developed shell-hardened macroporous hydrogels with unique mechanical properties and adjustable degradability to support stem cell-mediated bone regeneration. They offer a macropore structure that prevents contact inhibition during cell proliferation, a rigid pore shell for sustained mechanical cues and protection from mechanical load, and a soft hydrogel matrix with tunable degradation. These characteristics facilitate gradual degradation while providing continuous mechanical signals for stem cell differentiation and bone regeneration, addressing the limitations of other hydrogel strategies. Investigating the clinical applications of shell-hardened macroporous hydrogels for stem cell-mediated tissue regeneration will be our subsequent pursuit.
Methods
Ethical Statement
All animal studies were carried out in compliance with the regulations and guidelines of the Ethics Committee of Drum Tower Hospital affiliated to the Medical School of Nanjing University and adhered to the Institutional Animal Care and Use Committee (IACUC) guidelines (approval number: IACUC-D2310006).
Materials
4-armed PEG acrylate (PEG-ACLT), 4-armed PEG acrylamide (PEG-ACA), and 4-armed PEG oxhydryl (4-armed PEG-OH) were purchased from SinoPEG, Inc. (Xiamen, China). Acrylated RGD peptide (Cyclo(Arg-Gly-Asp-D-Phe-Lys-acrylate)) was purchased from Xi’an Ruixi Biological Technology (Xi’an, China). The MC3T3-E1 cell line was kindly provided by the Cell Bank of the Chinese Academy of Sciences (Chinese Academy of Sciences, Shanghai, China). The rBMSC line was purchased from OriCell® (Guangzhou, China). The cell culture medium α-MEM (cat: 310-010-CL) was purchased from Wisent (Nanjing, China). The simulated body fluid (SBF) was purchased from Biobomei (Hefei, China). Other reagents included cell lysis buffer (cat: P0013, Beyotime, China), bovine serum albumin (BSA, cat: ST2254, Beyotime, China), Tween 20 (cat: CT371, U-CyTech, USA), Triton X-100 (cat: 0694, Amresco, USA), primary antibodies, including Runx2 rabbit monoclonal antibody (cat: AF2593, Beyotime, China), anti-osteocalcin antibody (OCN, cat: sc-365797, Santa Cruz, USA), anti-bone sialoprotein antibody (IBSP, cat: ab52128, Abcam, UK), mouse monoclonal alkaline phosphatase antibody (ALP, cat: NB600-540, Novus, USA), rabbit polyclonal collagen I antibody (col I, cat: NB600-408, Novus, USA), 4’,6-diamidino-2-phenylindole (DAPI, cat: MBD0015, Sigma‒Aldrich, China), Alexa fluor 647-labelled goat anti-rabbit IgG (H + L, cat: A0468, Beyotime, China), FITC AffiniPure goat anti-mouse IgG (H + L, cat: E031210, Earthox, USA); a calcein-AM and propidium iodide (PI) double staining kit (cat: C542-3000T, Dojindo, Japan), an alkaline phosphatase (ALP) colour development kit (cat: C3206-1, Beyotime, China), an alkaline phosphatase assay kit (Beyotime, China), a DNeasy blood & tissue kit (cat: 69506, QIAGEN, Germany), an alizarin red s staining kit (cat: A5533, Sigma‒Aldrich, China), TRIzol™ reagent (cat: 11596026, Invitrogen™, USA), DEPC water (DNase, RNase free) (cat: R0021, Beyotime, China), DNase and RNase-free pipette tips, tubes and ultraclear caps in strips of 8 (cat: AB1183, Thermo Scientific™, USA), a PrimeScript™ RT Reagent Kit (cat: RR037A, TaKaRa, Japan), PowerUp™ SYBR™ Green Master Mix (cat: A25742, Applied Biosystems™, USA), a SYBR Premix Ex TaqTM II kit (DRR081A, Takara Bio Inc), forward and reverse primers (GenScript, China), and a real-time PCR system (cat: A30299, Applied Biosystems™, USA) for the quantitative real-time polymerase chain reaction (qRT‒PCR) analysis of gene transcripts. All other reagents, unless otherwise stated, were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (China). All reagents were used without further purification.
Preparation of the self-assembled lysozyme nanofiber
Before the preparation of self-assembled lysozyme nanofibers, lysozyme was first modified using methacrylic anhydride. Typically, lysozyme solutions at a concentration of 1.4 mM and methacrylic anhydride solutions at a concentration of 28 mM were prepared using ddH2O. After adjusting the pH of the methacrylic anhydride solution to 6.0, the lysozyme solution was added dropwise into the methacrylic anhydride solution to achieve a final methacrylic anhydride concentration of 14 mM. The mixture was stirred for 12 hours at 50 °C and then dialyzed to remove any unreacted methacrylic anhydride. The resulting products were lyophilized and stored at 4 °C.
To prepare the self-assembled lysozyme nanofibers, the methacrylic anhydride-modified lysozyme was dissolved in a 10 mM NaCl solution with the pH adjusted to 2.0 using hydrochloric acid. The resulting solutions were filtered with PVDF (0.22 µm, Millipore) and incubated at 60 °C for 60 hours to obtain self-assembled lysozyme nanofibers. Then, the buffer of the solution was changed into PBS (10 mM, pH = 7.4) and stored at 4 °C for the next step.
Preparation of NN, NP and SP hydrogels
The preparation of NN, NP, and SP hydrogels followed similar procedures with varying compositions. For NN-high hydrogel preparation, 4-armed PEG-ACLT (20 K), 4-armed PEG-ACA (20 K), acrylamide, acrylate-RGD peptide and photoinitiator (lithium phenyl-2,4,6-trimethylbenzoylphosphinate, Lap) were dissolved in PBS (10 mM, pH = 7.4) to concentrations of 70, 10, 360, 5.0 and 0.5 mg mL-1, respectively. The mixture was then filtered with PVDF (0.22 µm, Millipore) and illuminated under blue light (405 nm, 253 mw cm-2) for 3 minutes to complete gelation. After dialyzing in PBS (10 mM, pH = 7.4) for 24 hours to achieve swelling equilibrium, the resulting hydrogels were stored at 4 °C.
For NN-low hydrogel preparation, 4-armed PEG-ACA (20 K), acrylamide, acrylate-RGD peptide, and Lap were dissolved in PBS (10 mM, pH = 7.4) to concentrations of 10, 45, 5.0, and 0.5 mg mL-1, respectively. The solution was then filtered through PVDF (0.22 µm, Millipore) before undergoing the same gelation process.
For NP-high hydrogel preparation, 4-armed PEG-ACLT (20 K), 4-armed PEG-ACA (20 K), acrylamide, acrylate-RGD peptide, and Lap were dissolved in PBS (10 mM, pH = 7.4) to concentrations of 140, 20, 720, 5 and 1.0 mg mL-1, respectively. Dextran and acrylate-RGD peptide were dissolved in PBS (10 mM, pH = 7.4) to concentrations of 100 and 5 mg mL-1, respectively. The solutions were filtered with PVDF (0.22 µm, Millipore) and mixed at a 1:1 volume ratio. The mixture was stored at room temperature for 1 minute, and then illuminated under blue light (405 nm, 253 mw cm-2) for 3 minutes to complete gelation. After dialyzing in PBS (10 mM, pH = 7.4) for 24 hours, the resulting hydrogels were stored at 4 °C.
For NP-low hydrogel preparation, 4-armed PEG-OH (20 K), 4-armed PEG-ACA (20 K), acrylamide, acrylate-RGD peptide and Lap were dissolved in PBS (10 mM, pH = 7.4) to concentrations of 140, 20, 90, 5 and 1.0 mg mL-1, respectively. The solution was then filtered through PVDF (0.22 µm, Millipore) and mixed with the dextran solution at a 1:1 volume ratio before undergoing the same gelation process.
For SP-high hydrogel preparation, 4-armed PEG-ACLT (20 K), 4-armed PEG-ACA (20 K), acrylamide, acrylate-RGD peptide, and Lap were dissolved into PBS (10 mM, pH = 7.4) containing lysozyme nanofibers (20 mg mL-1) to concentrations of 140, 20, 720, 5 and 1.0 mg mL-1, respectively. Dextran and acrylate-RGD peptide were dissolved in PBS (10 mM, pH=7.4) containing lysozyme nanofibers (20 mg mL-1) to concentrations of 100 and 5 mg mL-1, respectively. Then, the solutions were filtered with PVDF (0.22 µm, Millipore) and mixed at a 1:1 volume ratio. The mixture was stored at room temperature for 1 minute, and then illuminated under blue light (405 nm, 253 mw cm-2) for 3 minutes to complete gelation. After dialyzing in PBS (10 mM, pH=7.4) for 24 hours, the resulting hydrogels were stored at 4 °C.
For SP-low hydrogel preparation, 4-armed PEG-OH (20 K), 4-armed PEG-ACA (20 K), acrylamide, acrylate-RGD peptide, and Lap were dissolved into PBS (10 mM, pH = 7.4) containing lysozyme nanofibers (20 mg mL-1) to concentrations of 140, 20, 90, 5.0 and 1.0 mg mL-1, respectively. The solution was then filtered through PVDF (0.22 µm, Millipore) and mixed with the dextran solution at a 1:1 volume ratio before undergoing the same gelation process.
Swelling ratio and water content determination
To determine the swelling ratios of hydrogels, the volume of the hydrogel after illumination was recorded as V1. Then, the hydrogels were swollen in ddH2O for more than 24 hours, and the volume was measured again and recorded as V2. The swelling ratio was calculated as V2/V1 × 100%. To determine the water content, the wet weight of hydrogel after swelling in ddH2O was recorded as W1, and the dry weight of the hydrogel after lyophilization was recorded as W2. The water content was calculated as (W1-W2)/W1 × 100%.
Evaluation of dextran removal
During the dextran removal test, SP hydrogels were prepared with 2% dextran replaced by FITC-labelled dextran (FITC-dextran). Typically, 200 µl of the SP hydrogel was prepared as described above and immersed in 8 mL PBS (10 mM, pH = 7.4) for 5 hours at room temperature (22 °C). Fluorescence spectroscopy of the leachates was recorded using a fluorescence spectrophotometer (FP-6500, JASCO, Japan). The FITC-dextran concentrations in the leachates were determined according to the calibration curves of FITC-dextran concentrations and fluorescence intensity at 518 nm. Then, the leachates were removed, and 8 mL of fresh PBS was added again. The dextran removal operation was repeated three times, and the accumulated FITC-dextran removal percentage was calculated as the removal percentage of dextran.
Field emission scanning electron microscopy (FESEM) imaging
For the observation of the microstructures, hydrogels were dialyzed in ddH2O for 24 h and then lyophilized. After coating with Pt, the samples were imaged using a field emission scanning electron microscope (Gemini 500, Zeiss, Germany) at 15 kV. Additionally, for observing cell encapsulation, hydrogels containing encapsulated cells were incubated at 37 °C and 5% CO2 for 2 days. Then, hydrogels were washed three times with PBS (10 mM, pH = 7.4) and fixed with an electron microscope fixative solution (2.5% glutaraldehyde) overnight. Subsequently, the hydrogels were cut into pieces and dehydrated sequentially with ethanol. Finally, the hydrogels were lyophilized and coated with Pt before imaging using FESEM (Gemini 500, Zeiss, Germany) at 15 kV.
3D reconstruction of microstructures of hydrogels
3D reconstruction of the microstructures of NN, NP and SP hydrogels was conducted through fluorescent labelling and 3D scanning using a laser confocal fluorescence microscope (Olympus FV3000, Japan). To enable fluorescent labeling, 4-armed PEG-ACLT with one end labelled with fluorescein isothiocyanate (denoted FITC-PEG) was prepared, and fluorescence-labelled lysozyme nanofibers were prepared using FITC-labelled lysozyme. For NN hydrogels prepared with FITC-labelled PEG, 3% of the PEG was replaced with FITC-PEG. For NN hydrogels containing FITC-labelled lysozyme, lysozyme nanofibers with 3% replaced by FITC-labelled lysozyme nanofibers were added to the precursor at a concentration of 20 mg mL-1. For NP hydrogels prepared with FITC-labelled PEG, 3% of the PEG was replaced with FITC-PEG. For SP hydrogels prepared with FITC-labelled PEG, 3% of the lysozyme nanofibers was replaced with FITC-labelled lysozyme nanofibers during hydrogel preparation. Subsequently, the hydrogels were scanned using a laser confocal fluorescence microscope (Olympus FV3000, Japan) with a scanning space dimension of 200 μm × 318 μm × 318 μm and a step size of 2 μm. Reconstruction was accomplished using the commercial software (FV31S-SW, version: 2.4.1.198) provided by Olympus.
AFM-based nanoindentation
For the AFM nanoindentation of dried hydrogels in air, the hydrogel samples were cut to expose the cross section and dried. Then, the samples were scanned at room temperature using a NanoWizard IV (JPK, Germany) in QI mode (conditions: Z length: 0.8 μm; extend and retract speed: 30 μm s-1; Z resolution: 80,000 Hz; maximum loading force: 150 nN; pixel number: 256 × 256). Silica cantilevers (PPP-NCHR with a half-open angle of the pyramidal face of θ < 25°, tip radius: 7–10 nm, spring constant 42 N m-1) were used in the experiments in air. Typically, the cantilever extended to the hydrogel surface and retracted at a constant speed of 30 µm s-1, during which the force and displacement of the cantilever were recorded. The Young’s modulus and stiffness were calculated by fitting the extended curve with the Hertz model (1).
where F corresponds to the force, \(h\) corresponds to the depth of the sample pressed by the cantilever tip, \(R\) is the radius of the tip, \(E\) is the Young’s modulus of the crystals and \(v\) is the Poisson ratio. We chose \(v\) = 0.4 in this calculation. Typically, 5-8 such regions were randomly selected on each sample to make the elasticity histogram, and at least three cantilevers were used in the experiments to exclude the tip-to-tip dependency. All the data were analyzed, and two-dimensional maps were reconstructed using the commercial software (JPK data processing 7.0.46) provided by JPK.
For the AFM nanoindentation of hydrogels in PBS, the cantilever of SNL-10 (B type, half-open angle of the pyramidal face of θ~22.5°, tip radius: ~2 nm, spring constant: 0.12 N m-1) was used. Typically, the band-like hydrogel was fixed on a glass dish, and PBS was added to cover the hydrogel. Then, the cantilever was moved to the surface of the hydrogel and held on the different areas of the hydrogel surface (matrix and macropore). The QI mode (conditions: Z length: 5 μm; extend and retract speed: 30.0 μm s-1; Z resolution: 29200 Hz; maximum loading force: 1.8 nN; pixel number: 40 × 40) was used to obtain the force‒displacement curves. Since the stiffness of wet hydrogels was much weaker than that of dry hydrogels, resulting in a higher press depth of the cantilever, Young’s modulus was calculated by fitting the extended curve with the Hertz/Sneddon model (2).
where F is the stress of the cantilever, \(h\) is the depth of the hydrogel pressed by the cantilever tip, \(\alpha\) is the half angle of the tip, \(E\) is the Young’s modulus of the hydrogel and \(v\) is the Poisson ratio. We chose \(v\) = 0.5 in this calculation.
Tensile and compressive tests
The mechanical properties of hydrogels at the macroscopic level were evaluated using tensile/compressive stress‒strain measurements. All experiments were performed using a tensile-compressive tester (Instron-5944 with a 2 kN sensor) in air at room temperature (22 °C). A humidifier was applied to keep the hydrogel wet during the measurements. In the tension-crack and compression-crack tests, the strain rates of stretching and compressing were both maintained at 2 mm min-1. The Young’s modulus for tension and compression comprised the approximate linear fitting values at a strain of 20%.
In vitro biocompatibility and biodegradation evaluation
To evaluate the cytotoxicity in vitro, different hydrogels were prepared in the wells of 48-well plates and allowed to reach swelling equilibrium in PBS (10 mM, pH = 7.4). Then, MC3T3-E1 cells and rBMSCs were seeded into the hydrogel-covered wells at a density of 105 per well. After 48 h of cell culture, each well was washed three times using PBS. Then, a mixture of calcein AM and propidium iodide (PI) dye solution was added to each well and incubated at 37 °C for 15 minutes. After washing three times with PBS, live/dead staining images were obtained using a laser confocal fluorescence microscope (Olympus FV3000, Japan).
To evaluate biodegradation in vitro, hydrogels at the same volume (50 μL) were prepared, and the original dry weight of each hydrogel after washing with ddH2O was defined as W0. Then, a piece of hydrogel (50 μL) was soaked in 0.5 mL of simulated body fluid (SBF). The sample was incubated at 37 °C. At a certain time (t), the hydrogel was removed from the incubation medium and washed with ddH2O three times. The sample was lyophilized, and the weight was defined as Wt. The remaining mass at time t was determined as \(\varepsilon=\frac{{W}_{t}}{{W}_{0}}\times 100\%\).
3D cell culture and protection in hydrogels
During the fabrication of the hydrogel with cells encapsulated inside, the solution containing acrylamide, PEG, acrylate-RGD peptide, and Lap was prepared using PBS (10 mM, pH = 7.4) containing lysozyme nanofibers. Cells were harvested from cell culture plates and mixed into the dextran/acrylate-RGD/lysozyme nanofiber solution at a density of 2 × 106 mL-1. After mixing these two solutions at a 1:1 volume ratio, gelation was completed in 3 minutes under blue light illumination. As a result, acrylamide was retained in the PEG phase as much as possible and polymerized into non-cytotoxic polyacrylamide as quickly as possible, thereby reducing the concentration of free acrylamide in the hydrogel. After gelation, the dialysis of the hydrogel with cells encapsulated inside was achieved by immersing the hydrogel into cell culture medium (α-MEM) at 37 °C and 5% CO2, with the medium being replaced three times in the first 12 hours to remove any remaining free acrylamide and dextran. These procedures minimized the cytotoxicity of acrylamide and ensured the maintenance of cell viability during dialysis. During subsequent cell culture, the medium was replaced daily.
To evaluate cell proliferation in hydrogels, cells were encapsulated in hydrogels according to the aforementioned procedure. After 12 hours of dextran removal in the medium, hydrogels were allowed to swell for another 12 hours. Subsequently, live/dead staining of cells was conducted at different time points (0, 5, and 10 days). Fluorescent images were then captured using a laser confocal fluorescence microscope (Olympus FV3000, Japan). Typically, each sample was scanned layer by layer, and the images were projected onto the z-axis to show the staining. Cell viabilities and live cell intensities were calculated based on the live/dead staining images.
To test cell protection in hydrogels, cells were cultured in hydrogels for 48 hours. Then, the hydrogels underwent cyclic compression (0.5 Hz) at different strains in cell culture medium. After 1500 cycles of compression, the cell viability and ROS production were investigated using live/dead staining and ROS staining. Then, fluorescent images were obtained using a laser confocal fluorescence microscope (Olympus FV3000, Japan). Typically, a region of 1272 μm × 1272 μm × 400 μm in the hydrogel was scanned layer by layer at a step size of 2 μm, and the images were projected onto the z-axis. Cell viabilities were determined by analyzing the images using ImageJ.
Cell proliferation assessment based on DNA quantification
To determine the cell expansion fold, hydrogels encapsulating cells were cultured for varying durations and then lyophilized. The lyophilized samples were thoroughly ground into a fine powder using a mortar and pestle in the presence of liquid nitrogen. The resulting powders were rehydrated in PBS solution, and DNA was extracted using the DNeasy Blood & Tissue Kit (QIAGEN) and quantified with a Nanodrop spectrophotometer (Thermo Fisher). A standard curve correlating total DNA yield with cell numbers was established by diluting a cell suspension into gradient concentrations and measuring the DNA content in each corresponding solution. Finally, the cell numbers in SP hydrogels were calculated based on this standard curve.
Cell differentiation inside hydrogels
Differentiation of MC3T3-E1 and RBMSCs was evaluated using immunostaining and qRT–PCR analyses. For the fluorescent staining analysis of nuclei, cytoskeleton and OCN (or Runx2) in MC3T3-E1 cells, hydrogels containing cells were fixed in 4% (vol/vol) paraformaldehyde for 30 min and then treated with 0.25% Triton X-100 for 15 min. After blocking with 1% (wt/vol) BSA in PBST (0.1 wt% Tween-20 in PBS) for 1 hour to minimize nonspecific binding, primary antibodies against OCN (or Runx2), diluted with antibody dilution buffer, were added to the fixed hydrogels and incubated overnight at 4 °C. Then, the primary antibody solution was decanted, and the sample was immediately washed with PBS 3 times for 5 minutes each time. After rinsing with PBST 3 times, secondary antibodies (Alexa Fluor 647-labelled goat anti-rabbit IgG) were added to the dish, and the cells were incubated for 60 min at room temperature. The secondary antibody was decanted, and the cells were washed with PBST 3 times for 10 minutes each. Subsequently, phalloidin (phalloidin dilution 1:1000) in PBST with 1% BSA was added to the dish, and the cells were incubated at 4 °C overnight before being washed with PBST 3 times for 10 minutes each time. At last, DAPI (1 µg mL-1, 1:500) in PBST with 1% BSA was added to the dish, and the cells were incubated for 60 min at room temperature, followed by washing with PBST 3 times for 10 minutes each. Immunofluorescence images were obtained using a laser confocal fluorescence microscope (Olympus FV3000, Japan). A space of 318 μm × 318 μm × 100 μm or 636 μm × 636 μm × 400 μm in the hydrogel was scanned layer by layer at a step size of 1 μm, and the images were projected onto the z-axis to show the staining.
For the immunostaining analysis of nuclei, ALP and Col I of RBMSCs, hydrogels containing cells were fixed in 4% (vol/vol) paraformaldehyde for 30 minutes and then treated with 0.25% Triton X-100 for 15 minutes. After blocking with 1% (wt/vol) BSA in PBST (0.1 wt% Tween-20 in PBS) for 1 hour to minimize nonspecific binding, primary antibodies against ALP and Col I, diluted with antibody dilution buffer, were added to the fixed hydrogels and incubated overnight at 4 °C. Then, the primary antibody solution was decanted, and the sample was immediately washed with PBS 3 times for 5 minutes each time. After rinsing with PBST 3 times, secondary antibodies (FITC AffiniPure goat anti-mouse IgG; Alexa Fluor 647-labelled goat anti-rabbit IgG) were added to the dish, and the cells were incubated for 60 min at room temperature. The secondary antibody was decanted, and the cells were washed with PBST 3 times for 10 minutes each. DAPI in PBS was added to the dish, and the cells were incubated for 5 minutes at room temperature before being washed with PBST 3 times for 10 minutes each. Immunofluorescence images were obtained using a laser confocal fluorescence microscope (Olympus FV3000, Japan). A space of 636 μm × 636 μm × 400 μm in the hydrogel was scanned layer by layer at a step size of 2 μm, and the images were projected onto the z-axis to show the staining.
For the qRT–PCR analysis, hydrogels with cells were crushed down to extract total RNA using TRIzol reagent. Subsequently, 1000 ng of total RNA was reverse-transcribed into cDNA with 5 × HiScript III qRT SuperMix following the manufacturer’s instructions. Real-time PCR was conducted using the SYBR Premix Ex TaqTM II kit on the LightCycler 96 system (Roche, Mannheim, Germany). The primers used for quantitative RT-PCRs are summarized in Supplementary Tables 1 and 2. The PCR involved incubation at 95 °C for 2 min, denaturation at 94 °C for 15 s, annealing at 58 °C for 15 s, and polymerization at 72 °C for 60 s for 40 cycles, followed by a final extension at 72 °C for 5 minutes. All CT values were analyzed quantitatively and normalized to their respective GAPDH values. The relative levels were calculated using the 2 − ΔΔCt method.
Protein engineering
The gene for the fusion fluorescent protein (SpyCatcher-green fluorescent protein-SpyCatcher, denoted as SC-GFP-SC) was custom synthesized and codon-optimized for expression in Escherichia Coli (GenScript, China). They were designed to include 5′ BamHI, 3′ BglII, and KpnI restriction sites. The genes encoding the chimeras used in this work were assembled in the pUC57 vector (Novagen, USA) using standard molecular biology techniques, with the same sticky ends generated by BamHI and BglII. Subsequently, the genes were excised from pUC57 and inserted into the pET30-a vector. The resulting plasmid was sequenced to confirm the correct protein sequences. The full amino acid sequence is provided in Supplementary Table 3.
Diffusion of SC-GFP-SC in hydrogels
The diffusion behaviour of the model fluorescent protein (SC-GFP-SC) within hydrogels was investigated to simulate protein diffusion within hydrogel macropores. Typically, SC-GFP-SC was incorporated into a solution containing dextran and acrylate-RGD peptide at a concentration of 1 mg mL-1 during precursor preparation. Subsequently, hydrogels were formed and allowed to swell in PBS (10 mM, pH=7.4) for 24 hours at room temperature. Subsequently, the hydrogels were incubated in cell culture medium (α-MEM) at 37 °C for 2 days and then examined using a laser confocal fluorescence microscope (Olympus FV3000, Japan). A scanning area of 400 μm × 636 μm × 636 μm was systematically scanned layer by layer with a step size of 2.5 μm. 3D reconstruction was accomplished using commercial software (FV31S-SW, version: 2.4.1.198) provided by Olympus.
ALP activity and mineralization determination
For alkaline phosphatase (ALP) staining, hydrogels with cells cultured inside for 14 days were washed three times with PBS and fixed with 4% paraformaldehyde for 30 min. After being washed three times with PBS again, hydrogels were stained with the ALP staining kit following the manufacturer’s instructions. The stained hydrogels were immediately washed with PBS 3 times for 5 minutes each. Images of staining were obtained using a fluorescence microscope (IX81, OLYMPUS, Japan). For the quantitative evaluation of ALP activity, hydrogels with cells cultured inside for 14 days were washed three times with PBS and crushed. The samples were then coincubated with p-nitrophenyl phosphate solution from the Alkaline Phosphatase Assay Kit (Beyotime, China) at 37 °C for 20 min, followed by the addition of the stop solution. The absorbance at 405 nm of the supernatant was recorded using an ultraviolet spectrophotometer (V550, Jasco, Japan).
For alizarin red S staining, hydrogels with cells cultured inside for 14 days were washed three times with PBS and fixed with 4% paraformaldehyde for 30 min. After being washed three times with ddH2O, hydrogels were stained with alizarin red S (0.2%, pH=5.3) solution for 5 min. The stained hydrogels were then washed with ddH2O 3 times for 10 min each time. Images of staining were obtained using a fluorescence microscope (IX81, OLYMPUS, Japan). For the quantitative evaluation of mineralization, alizarin red S was quantified by measuring absorbance at 562 nm after the stained cells were desorbed with 10% (w/v) cetylpyridinium chloride.
Bone regeneration in vivo
For the bone regeneration in the rabbit model, a total of 25 female New Zealand white rabbits with a bodyweight of 2.5 kg were enroled and randomly divided into 5 groups (control, NP-low hydrogel, SP-low hydrogel, NP-high hydrogel and SP-high hydrogel) with 5 rabbits per group. General anaesthesia was induced with a 0.2 mL kg-1 intramuscular injection of xylazine hydrochloride (Jilin Huamu Animal Health Products Co., Ltd., Jilin, China). Through a lateral parapatellar incision, we exposed the lateral femoral condyle and created a defect measuring 3.5 mm in depth and 5 mm in diameter. These defects were filled with rBMSC-encapsulated hydrogels, while defects in the control groups remained untreated. All the hydrogels used for implantation were from the same batch. Cefuroxime sodium was administered intramuscularly for 3 days post-operation to prevent infection. After 12 weeks, all animals were euthanized, and their femurs were harvested for bone regeneration assessment.
For the bone regeneration in the porcine model, a total of 12 male Bama mini pigs (9-month-old male Bama mini pig, bodyweight ~40 kg) were enroled and randomly divided into 4 groups (control, NP-low-cell hydrogel, SP-low hydrogel and SP-low-cell hydrogel) with 3 pigs per group. Anaesthesia was induced using Zoletil (Virbac S.A., France), and surgery was performed on both hind legs of each pig. Through a lateral parapatellar incision, we exposed the lateral femoral condyle and created a defect measuring 10 mm in depth and 12 mm in diameter, which was then filled with hydrogels. Control groups underwent the same surgical procedure without hydrogels. All hydrogels used for implantation were from the same batch. Cefuroxime sodium was administered intramuscularly for 3 days post-operation to prevent infection. After 12 weeks, all animals were euthanized, and their femurs were harvested for bone regeneration evaluation.
Micro-CT & 3D reconstruction
The harvested femurs were scanned by a micro-CT system (XM, Hiscan, China) with a 25 μm resolution at 90 kV and 89 μA. The 3D models of the harvested femurs were reconstructed with MIMICS 19.0 (Materialise, Leuven, Belgium). Micro-CT scanned images were imported into the software and segmented in accordance with grayscale values. Analyses of the bone tissue volume (BV/TV), trabecular thickness (Tb. Th), trabecular number (Tb. N), and trabecular separation (Tb. Sp) were used to evaluate the bone regeneration.
In vivo biocompatibility and biodegradation evaluation
For the in vivo biocompatibility evaluation, band-shaped hydrogels were implanted subcutaneously into male Sprague Dawley rats through a 1.5-cm skin incision at the centre of the back. The incision was then closed using interrupted sutures (3-0 Vicryl Plus, JNJ). After 24 hours, the rats were sacrificed, and target subcutaneous regions were obtained and fixed in 10% formalin for 48 hours before histological analysis (H&E staining) and immunofluorescence staining (TNF-α and IL-1β). Blood samples were collected for systemic toxicity tests.
For the in vivo biodegradation test, SP hydrogels were implanted subcutaneously as described above. After 7, 14, 28 and 42 days, the rats were sacrificed, and target tissues and blood were collected for histological analysis (H&E staining) and systemic toxicity tests (haematological examination and blood biochemistry).
Histological evaluation
For the histological analysis of in vivo biocompatibility and biodegradation evaluations, the harvested samples were fixed in 4% (vol/vol) paraformaldehyde for 24 h. The samples were then embedded in paraffin and sectioned into 6 μm thick slices. These sections underwent staining with hematoxylin & eosin (H&E), while the expression levels of TNF-α and IL-β were detected through immunofluorescence staining. For the histological analysis of in vivo bone regeneration, the harvested samples were fixed in 4% (vol/vol) paraformaldehyde for 24 h and then decalcified with 10% formic acid for 35 days. Following decalcification, the samples were embedded in paraffin and cut into 6 μm thick sections for subsequent histological, immunohistochemical, and immunofluorescence staining. All histological images were obtained using Pannoramic MIDI (3DHISTECH Ltd., Budapest, Hungary) and browsed with CaseViewer software (3DHISTECH Ltd., Budapest, Hungary).
Statistical analysis
Statistical analysis and exponential curve fitting were performed using IGOR Pro 6.37 software (WaveMetrics Inc., OR, USA) and SPSS 19.0 software (SPSS Inc., IL, USA). Data were presented as means ± standard deviation. Statistical significance was determined using Student’s t-test with significance set at a P-value < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the Supplementary Information. Data is available from the authors on request. Source data are provided with this paper.
References
Zakrzewski, W., Dobrzyński, M., Szymonowicz, M. & Rybak, Z. Stem cells: past, present, and future. Stem Cell Res. Ther. 10, 68 (2019).
Cossu, G. et al. Lancet Commission: Stem cells and regenerative medicine. Lancet 391, 883–910 (2018).
Fitzsimmons, R. E. B., Mazurek, M. S., Soos, A. & Simmons, C. A. Mesenchymal stromal/stem cells in regenerative medicine and tissue engineering. Stem Cells Int. 2018, 8031718 (2018).
Yin, X. et al. Engineering stem cell organoids. Cell Stem Cell 18, 25–38 (2016).
Chan, C. K. F. et al. Identification of the human skeletal stem cell. Cell 175, 43–56.e21 (2018).
Laurenti, E. & Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426 (2018).
Robinton, D. A. & Daley, G. Q. The promise of induced pluripotent stem cells in research and therapy. Nature 481, 295–305 (2012).
Mitrousis, N., Fokina, A. & Shoichet, M. S. Biomaterials for cell transplantation. Nat. Rev. Mater. 3, 441–456 (2018).
Müller-Ehmsen, J. et al. Effective engraftment but poor mid-term persistence of mononuclear and mesenchymal bone marrow cells in acute and chronic rat myocardial infarction. J. Mol. Cell. Cardiol. 41, 876–884 (2006).
Kaur, G. & Cai, C. Current progress in the rejuvenation of aging stem/progenitor cells for improving the therapeutic effectiveness of myocardial repair. Stem Cells Int. 2018, 9308301 (2018).
Li, X. et al. Improve the viability of transplanted neural cells with appropriate sized neurospheres coated with mesenchymal stem cells. Med. Hypotheses 79, 274–277 (2012).
Hu, X. et al. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 135, 799–808 (2008).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Bio. 15, 786–801 (2014).
Watt, F. M. & Huck, W. T. S. Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Bio. 14, 467–473 (2013).
Hynes, R. O. The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 (2009).
Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006).
Han, S. B., Kim, J. K., Lee, G. & Kim, D. H. Mechanical properties of materials for stem cell differentiation. Adv. Biosyst. 4, e2000247 (2020).
Hogrebe, N. J., Augsornworawat, P., Maxwell, K. G., Velazco-Cruz, L. & Millman, J. R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 38, 460–470 (2020).
Jang, J. et al. 3D printed complex tissue construct using stem cell-laden decellularized extracellular matrix bioinks for cardiac repair. Biomaterials 112, 264–274 (2017).
Huang, Q. et al. Hydrogel scaffolds for differentiation of adipose-derived stem cells. Chem. Soc. Rev. 46, 6255–6275 (2017).
Nikolaev, M. et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature 585, 574–578 (2020).
Gaharwar, A. K., Singh, I. & Khademhosseini, A. Engineered biomaterials for in situ tissue regeneration. Nat. Rev. Mater. 5, 686–705 (2020).
Huebsch, N. & Mooney, D. J. Inspiration and application in the evolution of biomaterials. Nature 462, 426–432 (2009).
Lutolf, M. P., Gilbert, P. M. & Blau, H. M. Designing materials to direct stem-cell fate. Nature 462, 433–441 (2009).
Huang, G. et al. Functional and biomimetic materials for engineering of the three-dimensional cell microenvironment. Chem. Rev. 117, 12764–12850 (2017).
Trappmann, B. et al. Extracellular-matrix tethering regulates stem-cell fate. Nat. Mater. 11, 642–649 (2012).
Wang, X. et al. Chromatin reprogramming and bone regeneration in vitro and in vivo via the microtopography-induced constriction of cell nuclei. Nat. Biomed. Eng. 7, 1514–1529 (2023).
Álvarez, Z. et al. Bioactive scaffolds with enhanced supramolecular motion promote recovery from spinal cord injury. Science 374, 848–856 (2021).
Li, R. et al. Synthetic presentation of noncanonical Wnt5a motif promotes mechanosensing-dependent differentiation of stem cells and regeneration. Sci. Adv. 5, eaaw3896 (2019).
Cao, H., Duan, L., Zhang, Y., Cao, J. & Zhang, K. Current hydrogel advances in physicochemical and biological response-driven biomedical application diversity. Signal Transduct. Target. Ther. 6, 426 (2021).
Luo, Y. & Shoichet, M. S. A photolabile hydrogel for guided three-dimensional cell growth and migration. Nat. Mater. 3, 249–253 (2004).
Tibbitt, M. W. & Anseth, K. S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 103, 655–663 (2009).
Prince, E. & Kumacheva, E. Design and applications of man-made biomimetic fibrillar hydrogels. Nat. Rev. Mater. 4, 99–115 (2019).
Daly, A. C., Riley, L., Segura, T. & Burdick, J. A. Hydrogel microparticles for biomedical applications. Nat. Rev. Mater. 5, 20–43 (2020).
Vining, K. H. & Mooney, D. J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 18, 728–742 (2017).
Discher, D. E., Janmey, P. & Wang, Y. L. Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143 (2005).
Chaudhuri, O., Cooper-White, J., Janmey, P. A., Mooney, D. J. & Shenoy, V. B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 584, 535–546 (2020).
Agarwal, P. et al. A dysfunctional TRPV4–GSK3β pathway prevents osteoarthritic chondrocytes from sensing changes in extracellular matrix viscoelasticity. Nat. Biomed. Eng. 5, 1472–1484 (2021).
Chaudhuri, O. et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 15, 326–334 (2016).
Adebowale, K. et al. Enhanced substrate stress relaxation promotes filopodia-mediated cell migration. Nat. Mater. 20, 1290–1299 (2021).
Xue, B. et al. Engineering hydrogels with homogeneous mechanical properties for controlling stem cell lineage specification. Proc. Natl Acad. Sci. USA 118, e2110961118 (2021).
Daly, A. C., Davidson, M. D. & Burdick, J. A. 3D bioprinting of high cell-density heterogeneous tissue models through spheroid fusion within self-healing hydrogels. Nat. Commun. 12, 753 (2021).
Huebsch, N. et al. Matrix elasticity of void-forming hydrogels controls transplanted-stem-cell-mediated bone formation. Nat. Mater. 14, 1269–1277 (2015).
Bai, X. et al. Bioactive hydrogels for bone regeneration. Bioact. Mater. 3, 401–417 (2018).
Goto, R. et al. Gelatin Methacryloyl-Riboflavin (GelMA-RF) Hydrogels for Bone Regeneration. Int. J. Mol. Sci. 22, 1635 (2021).
Li, L. et al. Biofabrication of a biomimetic supramolecular-polymer double network hydrogel for cartilage regeneration. Mater. Des. 189, 108492 (2020).
Sun, W. et al. Polymer-Supramolecular Polymer Double-Network Hydrogel. Adv. Funct. Mater. 26, 9044–9052 (2016).
Chatterjee, K. et al. The effect of 3D hydrogel scaffold modulus on osteoblast differentiation and mineralization revealed by combinatorial screening. Biomaterials 31, 5051–5062 (2010).
Kim, S. Y. & Park, J.-S. Biomineralized hyaluronic acid/poly(vinylphosphonic acid) hydrogel for bone tissue regeneration. J. Appl. Polym. Sci. 131, 41194 (2014).
Xu, B. et al. A mineralized high strength and tough hydrogel for skull bone regeneration. Adv. Funct. Mater. 27, 1604327 (2017).
Fang, J. et al. A strong, tough, and osteoconductive hydroxyapatite mineralized polyacrylamide/dextran hydrogel for bone tissue regeneration. Acta Biomater. 88, 503–513 (2019).
Simmons, C. A., Alsberg, E., Hsiong, S., Kim, W. J. & Mooney, D. J. Dual growth factor delivery and controlled scaffold degradation enhance in vivo bone formation by transplanted bone marrow stromal cells. Bone 35, 562–569 (2004).
Khetan, S. et al. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat. Mater. 12, 458–465 (2013).
Wade, R. J., Bassin, E. J., Rodell, C. B. & Burdick, J. A. Protease-degradable electrospun fibrous hydrogels. Nat. Commun. 6, 6639 (2015).
Duda, G. N. et al. The decisive early phase of bone regeneration. Nat. Rev. Rheumatol. 19, 78–95 (2023).
Dinu, M. V. & Dragan, E. S. in Hydrogels: Recent Advances (eds Vijay Kumar Thakur & Manju Kumari Thakur) 51–85 (Springer Singapore, 2018).
Broguiere, N. et al. Macroporous hydrogels derived from aqueous dynamic phase separation. Biomaterials 200, 56–65 (2019).
Cui, Z.-K. et al. Microporous methacrylated glycol chitosan-montmorillonite nanocomposite hydrogel for bone tissue engineering. Nat. Commun. 10, 3523 (2019).
Yang, X. et al. Coacervation-mediated cytocompatible formation of supramolecular hydrogels with self-evolving macropores for 3D multicellular spheroid culture. Adv. Mater. 35, 2300636 (2023).
Stevens, K. R., Miller, J. S., Blakely, B. L., Chen, C. S. & Bhatia, S. N. Degradable hydrogels derived from PEG-diacrylamide for hepatic tissue engineering. J. Biomed. Mater. Res. A 103, 3331–3338 (2015).
Ozcelik, B. in Biosynthetic Polymers for Medical Applications (eds L. Poole-Warren, P. Martens, & R. Green) 173–188 (Woodhead Publishing, 2016).
Li, Y., Xue, B. & Cao, Y. 100th anniversary of macromolecular science viewpoint: synthetic protein hydrogels. ACS Macro Lett. 9, 512–524 (2020).
Yang, B. et al. Enhanced mechanosensing of cells in synthetic 3D matrix with controlled biophysical dynamics. Nat. Commun. 12, 3514 (2021).
Wang, H. Y. & Heilshorn, S. C. Adaptable Hydrogel Networks with Reversible Linkages for Tissue Engineering. Adv. Mater. 27, 3717–3736 (2015).
Xu, X. et al. Bioadhesive hydrogels demonstrating pH-independent and ultrafast gelation promote gastric ulcer healing in pigs. Sci. Transl. Med. 12, eaba8014 (2020).
Fu, L. et al. Cartilage-like protein hydrogels engineered via entanglement. Nature 618, 740–747 (2023).
Einhorn, T. A. & Gerstenfeld, L. C. Fracture healing: mechanisms and interventions. Nat. Rev. Rheumatol. 11, 45–54 (2015).
Chen, X. et al. Conformational manipulation of scale-up prepared single-chain polymeric nanogels for multiscale regulation of cells. Nat. Commun. 10, 2705 (2019).
Cao, A., Hu, D. & Lai, L. Formation of amyloid fibrils from fully reduced hen egg white lysozyme. Protein Sci. 13, 319–324 (2004).
Kim, J. W. Transcription factors crossing the cell membrane. Nat. Rev. Mol. Cell Bio. 25, 86–86 (2024).
Gholami Farashah, M. S. et al. Bone marrow mesenchymal stem cell’s exosomes as key nanoparticles in osteogenesis and bone regeneration: specific capacity based on cell type. Mol. Biol. Rep. 49, 12203–12218 (2022).
Lu, G. et al. An instantly fixable and self-adaptive scaffold for skull regeneration by autologous stem cell recruitment and angiogenesis. Nat. Commun. 13, 2499 (2022).
Policastro, G. M. et al. OGP functionalized Phenylalanine-based Poly(ester urea) for enhancing osteoinductive potential of human mesenchymal stem cells. Biomacromolecules 16, 1358–1371 (2015).
Lee, S.-H. et al. Runx2 protein stabilizes Hypoxia-inducible Factor-1α through competition with von Hippel-Lindau Protein (pVHL) and stimulates angiogenesis in growth plate hypertrophic Chondrocytes*. J. Biol. Chem. 287, 14760–14771 (2012).
Trounson, A. & McDonald, C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell 17, 11–22 (2015).
Borlongan, C. V. Age of PISCES: stem-cell clinical trials in stroke. Lancet 388, 736–738 (2016).
Xia, H. et al. Tissue repair and regeneration with endogenous stem cells. Nat. Rev. Mater. 3, 174–193 (2018).
Forbes, S. J. & Rosenthal, N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat. Med. 20, 857–869 (2014).
Khan, A. U., Qu, R., Fan, T., Ouyang, J. & Dai, J. A glance on the role of actin in osteogenic and adipogenic differentiation of mesenchymal stem cells. Stem Cell Res. Ther. 11, 283 (2020).
Fan, Y. L., Zhao, H. C., Li, B., Zhao, Z. L. & Feng, X. Q. Mechanical roles of F-Actin in the differentiation of stem cells: a review. ACS Biomater. Sci. Eng. 5, 3788–3801 (2019).
Pocaterra, A. et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. J. Hepatol. 71, 130–142 (2019).
Harris, A. R., Jreij, P. & Fletcher, D. A. Mechanotransduction by the Actin Cytoskeleton: Converting mechanical stimuli into biochemical signals. Annu. Rev. Biophys. 47, 617–631 (2018).
Yin, S. et al. Structurally and mechanically tuned macroporous hydrogels for scalable mesenchymal stem cell–extracellular matrix spheroid production. P. Natl Acad. Sci. USA 121, e2404210121 (2024).
Robin, M. et al. Mineralized collagen plywood contributes to bone autograft performance. Nature 636, 100–107 (2024).
Shuai, Y. et al. Oriented cortical-bone-like silk protein lamellae effectively repair large segmental bone defects in pigs. Adv. Mater. 37, 2414543 (2025).
Yang, F. et al. Full-thickness osteochondral defect repair using a biodegradable bilayered scaffold of porous zinc and chondroitin sulfate hydrogel. Bioact. Mater. 32, 400–414 (2024).
Salhotra, A., Shah, H. N., Levi, B. & Longaker, M. T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Bio. 21, 696–711 (2020).
Kanczler, J. M. et al. in Principles of Tissue Engineering (Fifth Edition) (eds Robert Lanza, Robert Langer, Joseph P. Vacanti, & Anthony Atala) 917-935 (Academic Press, 2020).
Zhu, G. et al. Bone physiological microenvironment and healing mechanism: Basis for future bone-tissue engineering scaffolds. Bioact. Mater. 6, 4110–4140 (2021).
Ma, C., Du, T., Niu, X. & Fan, Y. Biomechanics and mechanobiology of the bone matrix. Bone Res. 10, 59 (2022).
Wang, L., You, X., Zhang, L., Zhang, C. & Zou, W. Mechanical regulation of bone remodeling. Bone Res. 10, 16 (2022).
Koons, G. L., Diba, M. & Mikos, A. G. Materials design for bone-tissue engineering. Nat. Rev. Mater. 5, 584–603 (2020).
Grayson, W. L. et al. Stromal cells and stem cells in clinical bone regeneration. Nat. Rev. Endocrinol. 11, 140–150 (2015).
Xue, X., Hu, Y., Deng, Y. & Su, J. Recent advances in design of functional biocompatible hydrogels for bone tissue engineering. Adv. Funct. Mater. 31, 2009432 (2021).
Hasani-Sadrabadi, M. M. et al. An engineered cell-laden adhesive hydrogel promotes craniofacial bone tissue regeneration in rats. Sci. Transl. Med. 12, eaay6853 (2020).
Wu, X. et al. Mineralized hydrogels induce bone regeneration in critical size cranial defects. Adv. Healthc. Mater. 10, e2001101 (2021).
Lutolf, M. P. et al. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat. Biotechnol. 21, 513–518 (2003).
Acknowledgements
This work is supported mainly by the National Natural Science Foundation of China (Grant nos. T2322010 (B.X.) and T2225016 (Y.C.)), the National Key R&D Programme of China (Grant no. 2020YFA0908100 (Y.C.), 2023YFC3605802 (Y-R.L.), 2024YFA0919300 (Y.C. and B.X.)), the Natural Science Foundation of Jiangsu Province (Grant no. BK20220120 (B.X.)), the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (Nos. JNL-2025008B (X.G.), JNL-2025009B (C.J.) and JNL-2025010B (Y.C.)), the Shandong Provincial Laboratory Project (No. SYS202202 (C.J.)), the Fundamental Research Funds for the Central Universities (Grant no. 020414380232 (B.X.) and 020514380274 (Y.C.)) and NJU-Skoltech Collaborative Research Projects provided by International Joint Research Centre for STEM (B.X.).
Author information
Authors and Affiliations
Contributions
Y.C., W.W., Q.J. and B.X. conceived the idea and designed the study. B.X., Z.X. and L.L. performed the experiments and analysed the results. K.G., H.W. and Y-R.L. prepared some of the hydrogel samples. J.M., C.X., J.J. and J.X. performed some of in vivo analyses. B.X., Y.C., W.W. and Q.J. wrote and refined the paper. C.J. and X.G. designed some of in vivo validations. Y.C., W.W., Q.J. and M.Q. supervised the project. All the authors discussed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xue, B., Xu, Z., Li, L. et al. Hydrogels with programmed spatiotemporal mechanical cues for stem cell-assisted bone regeneration. Nat Commun 16, 3633 (2025). https://doi.org/10.1038/s41467-025-59016-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59016-6
This article is cited by
-
Shell-reinforced macroporous hydrogels for bone repair
Nature Reviews Materials (2025)
