
Abstract
The direct synthesis of ammonia from nitrate (NO3–) reduction in acid is a promising approach for industrialization. However, the difficulty arises from the intense competition with the inevitable hydrogen evolution reaction, which is favoured due to the overwhelming protons (H+). Here, we systematically explore and rationally optimize the microenvironment using multivariate covalent organic frameworks (COFs) as catalyst adlayers to promote the nitrate-to-ammonia conversion in acid. With the application of tailored positive electrostatic potential generated over the multivariate COFs, both the mass transfer of NO3– and H+ are regulated via appropriate electrostatic interactions, thus realizing the priority of NO3RR with respect to HER or NO3–-to-NO2–. As a result, an NH3 yield rate of 11.01 mmol h–1 mg–1 and a corresponding Faradaic efficiency of 91.0% are attained, and solid NH4Cl with a high purity of 96.2% is directly collected in acid; therefore, this method provides a practical approach for economically valorising wastewater into valuable ammonia.
Similar content being viewed by others
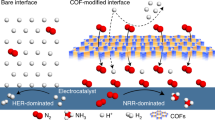
Introduction
Ammonia (NH3) is an essential feedstock in the fertiliser, textile, and pharmaceutical industries and is considered an ideal energy storage medium and carbon-neutral hydrogen carrier because of its high energy density1,2,3. To date, industrial-scale NH3 synthesis is dominated by the energy-intensive Haber-Bosch (HB) process under harsh conditions (400–500 °C and 150–300 atm)4,5. Alternatively, the electrocatalytic nitrogen reduction reaction (NRR) has attracted tremendous attention because of its mild conditions and the utilisation of renewable electricity6,7,8. However, the high dissociation energy of the N≡N bond and low solubility of nitrogen in aqueous systems usually lead to an NH3 yield rate that is two orders of magnitude lower than that of the HB process, hindering the practical application of the NRR9,10. In contrast to nitrogen, nitrate (NO3−) has a relatively lower dissociation energy of the N=O bond and higher solubility11,12. Furthermore, NO3− is one of the most common water-soluble contaminants that is widely present in agriculture and industry and causes serious environmental problems and human diseases13. Thus, the electrocatalytic nitrate reduction reaction (NO3RR) represents a more efficient and more economic route for industrial-scale NH3 synthesis.
Extensive efforts have been dedicated to the development of NO3RR systems with neutral or alkaline electrolytes14,15,16,17. Although relatively good performance can be achieved with state-of-the-art electrocatalysts, a serious limitation occurs in industrial practice. Industrial wastes originating from sectors such as metallurgy, chemical fibre, and electroplating are usually acidic18,19. To accommodate the required working conditions, these nitrate-containing wastewaters must undergo a pre-neutralisation step in treatment plants20. Moreover, considering the possible volatilisation of the product ammonia from neutral/alkaline electrolytes, an acid adsorption unit is also needed for ammonia capture21,22. All of these factors lead to additional increases in cost and energy consumption for industrialisation. In this context, the development of direct nitrate reduction in acid is urgently needed. Unfortunately, few systems are able to achieve high selectivity under acidic conditions, and the Faradaic efficiencies are generally less than 30%23.
The difficulty occurs from the intense competition with the inevitable hydrogen evolution reaction (HER), which is favoured due to the presence of overwhelming protons (H+) in acid24,25,26. In the heterogeneous catalysis field, several corresponding strategies for inhibiting this undesired side reaction have been proposed. For example, recent studies have shown that the optimisation of the local microenvironment by a catalyst adlayer has great potential to suppress the competing HER by reducing the proton amount27,28. Furthermore, microenvironment engineering is also capable of increasing the local reactant concentration and further improving product selectivity29,30. However, the proton accessibility cannot be too low since the NO3RR is a proton-coupled electron transfer reaction, and an insufficient proton supply would, in turn, impose restrictions on the intermediate hydrogenation and thus lead to byproducts, such as nitrite (NO2−), that are not fully reduced31,32. In this context, the development of a specific catalyst adlayer that is able to regulate the interfacial concentrations of electroactive substances in a targeted manner and achieve the optimum nitrate/proton ratio to increase NO3RR is needed.
Recent advances in materials science provide opportunities to address this pressing need, and a potential candidate is covalent organic frameworks (COFs)33. The atomic integration of building blocks into periodic structures enables precise manipulation of the spatial arrangement of the desired functional groups within the predetermined COF structure and thus achieves microenvironment modulation34,35,36. Here, a well-designed multivariate COF with a tailored electrostatic potential (ESP) is employed as an adlayer over the electrocatalyst to directly promote NO3RR in a strong acid (pH = 1 ± 0.2). The coated COFs with well-controlled cationic sites over the framework generate appropriate repulsive electrostatic forces towards H+, achieving moderate proton accessibility to the reaction interface that efficiently suppresses the electron-stealing HER while satisfying the hydrogenation process to prevent the predesorption of undesired NO2−. Moreover, the presence of cationic sites could also induce attractive electrostatic forces towards NO3– via electrostatic interactions, thus achieving localised enrichment of NO3– and prioritising the chemisorption competition with H+. Equipped with a multivariate COF adlayer, the system with the optimal NO3–/H+ ratio at the reaction interface delivers an NH3 yield rate of 11.01 mmol h–1 mg–1 and a corresponding Faradaic efficiency of 91.0% for acidic NO3–-to-NH3 conversion. By constructing a set of tailor-made devices, a complete conversion process for the primary production of high-purity solid NH4Cl (96.2%) in acid was demonstrated, indicating the potential of this method for direct industrial practice.
Results
Rational design of multivariate COFs and theoretical analysis
Multivariate COFs consisting of 2,4,6-triformylphloroglucinol (TP) as a fixed node and biphenyl diamines, including benzidine (BD) and ethidium bromide (EB), as tailored linkers were selected as proof-of-concept models (Fig. 1a). The two-component COFs synthesised by TP and pure BD or EB are denoted as BCOF or ECOF, respectively, whereas the three-component COFs synthesised by TP and the BD/EB mixture are denoted as BECOF. This facile bottom-up multicomponent design enables the sophisticated regulation of the charge distribution by simply varying the feed amounts of the two amine species with or without charge. Specifically, four kinds of assemblies can be obtained by the aldimine condensation of TP with BD and EB monomers, including TPBD3, TPBD2EB1, TPBD1EB2, and TPEB3 (Supplementary Fig. 1). The cationic site is the intrinsic N+ ionic site in EB, exists in the viologen units and is paired with the halogen (Br–)37. ESP is defined as the amount of energy needed per unit of electric charge to move the charge from a reference point to a specific point in an electric field. It is created by a system of nuclei and electrons and has a specific value at each point in the space of the system; thus, ESP is a very popular real space function in revealing possible electrostatic interactions between a molecule and environmental molecules38,39. Therefore, ESP calculations were carried out first on these four structures using density functional theory (DFT) to determine their intrinsic properties. The ESP maps and corresponding distribution ranges are displayed in Fig. 1b, c. When BD without charge is used as the only linker, the ESP map of TPBD3 shows an electrically neutral state and its ESP distribution ranges from –40 to 21 kcal mol–1. With an increasing EB ratio, a positive charge appears in the corresponding area because of the presence of cationic sites. A greater amount of EB correlates to a greater positive charge over the entire network, and a more positive shift in the ESP distribution occurs from 2 to 85 kcal mol–1 for TPBD2EB1, 34 to 106 kcal mol–1 for TPBD1EB2, and 71 to 158 kcal mol–1 for TPEB3. Since multivariate COFs would be used in an electrochemical environment, the external electric field is also included in the calculation process. Upon comparison, the ESP results with and without applied electrode potential slightly differ, and the overall trend remains unchanged (Supplementary Figs. 2 and 3). Therefore, multivariate COFs with specific ESPs are expected to function effectively under electrochemical conditions. Considering the electrophilicity of protons and the nucleophilicity of nitrate (Supplementary Figs. 4–6), the positive charge property has great potential for regulating the mass transfer of electroactive substances via electrostatic interactions.

a Synthetic scheme of multivariate COFs. Aromatic rings are coloured to assist in the differentiation of the origin of the different moieties (orange, 2,4,6-triformylphloroglucinol, TP; green, benzidine, BD; azure, ethidium bromide, EB). b Electrostatic potential (ESP) maps and (c) corresponding distribution ranges of TPBD3, TPBD2EB1, TPBD1EB2, and TPEB3. d Electrostatic interactions of H+ and NO3– with BCOF, BECOF, and ECOF.
The electrostatic interactions of H+ and NO3– with multivariate COFs were subsequently evaluated by molecular dynamics (MD) simulations (Supplementary Fig. 7)40. According to the MD trajectories (Fig. 1d), the electrostatic interaction generated by the near-neutral BCOF is relatively weak, with neither H+ nor NO3–. In contrast, BECOF and ECOF, which have different degrees of positive charge over the framework, could generate repulsive electrostatic forces towards H+ and attractive electrostatic forces towards NO3–. Notably, the electrostatic interactions between the ECOF made entirely of TPEB3 and the target molecules are stronger than those of BECOF. Specifically, ECOF exhibits stronger repulsion to H+ and stronger attraction to NO3–. However, an excessively high NO3–/H+ ratio is not necessarily beneficial for the NO3RR since it is a proton-coupled electron transfer reaction. The utilisation of multivariate COFs as catalyst adlayers is likely capable of achieving rational regulation of the surrounding mass transfer and thus optimising the interfacial microenvironment for a target reaction.
Synthesis and characterisation of multivariate COFs
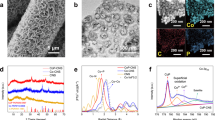
Inspired by the computational results, BCOF, BECOF, and ECOF were experimentally synthesised via a solvothermal condensation reaction of TP and amine monomers with different contents. The reactions were conducted in a sealed Pyrex tube at 120 °C for 3 days in a mixture of mesitylene and 1,4-dioxane, followed by suction filtration to collect the precipitates. The BCOF powder appeared orange, and an increase in EB addition resulted in more reddish-coloured products (Supplementary Fig. 8). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF STEM) was used to reveal the ordered internal structures of the as-synthesised COFs (Fig. 2a, b and Supplementary Figs. 9 and 10). A periodic structure is observed for all samples and clearly indicates their high crystallinity. For BECOF, the corresponding elemental mapping images show a uniform distribution of C, N, O, and Br throughout the framework (Fig. 2c). The crystallinities were further examined using X-ray diffraction (XRD) on a series of as-synthesised COFs (Fig. 2d). All of the XRD patterns exhibit identical characteristic peaks, and among these, BECOF has a relatively weak diffraction signal, which is attributed to the irregular sequence of BD and EB units in BECOF.

a, b Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF STEM) images and (c) corresponding elemental mappings of the synthesised BECOF. Scale bars, a 5 nm; b 1 nm; c 50 nm. d X-ray diffraction (XRD) patterns, e Fourier transform infrared (FTIR) spectra, f 13C solid-state nuclear magnetic resonance (SSNMR) spectra, g X-ray photoelectron spectroscopy (XPS) survey spectra, and (h) high-resolution N 1s XPS spectra of BCOF, BECOF, and ECOF.
The Fourier transform infrared (FTIR) spectra of different COFs and the initial monomers are shown in Fig. 2e. The characteristic absorbance bands at 3313 and 3201 cm−1 (amine νN−H stretches) observed in BD and EB and the characteristic absorbance bands at 2893 cm−1 (aldehyde νO=C–H stretch) and 1641 cm−1 (aldehyde νO=C stretch) observed in TP are absent in the spectrum of BECOF; this result indicates the successful condensation of the reactants37,41. Moreover, a strong peak at 1586 cm−1 (νC=C stretch) appears for all COFs, demonstrating the oxidation of the phenol hydroxyl group to ketone. The atomic-level construction of the multivariate COF was further confirmed by 13C solid-state nuclear magnetic resonance (SSNMR) spectroscopy (Fig. 2f). The 13C SSNMR spectrum of BCOF reveals broad multi-peaks that are attributed to the carbonyl carbon (185 ppm), enamine carbon (107 ppm), exocyclic carbon‒carbon double bond (147 ppm) and carbon signals in the benzene ring (137, 127, 120, and 115 ppm)42. In addition to its homologous structure to BCOF, the 13C SSNMR spectrum of ECOF shows distinct NMR shifts assigned to the ethidium carbon in the range of 130–150 ppm37. Moreover, the well-resolved peak associated with the ethyl carbon in EB is observed at 46 and 15 ppm and shows that ethidium bromide is retained in ECOF37. For BECOF, its 13C SSNMR spectrum inherits the characteristics of both BCOF and ECOF; this result also indicates the successful synthesis of multivariate COFs.
The element compositions and chemical natures of different COFs were subsequently investigated using X-ray photoelectron spectroscopy (XPS). Only C 1s, N 1s, and O 1s peaks are observed in the survey spectrum of BCOF, whereas additional Br 3d peaks are shown in both BECOF and ECOF, with Br contents of 1.39 at.% for BECOF and 2.02 at.% for ECOF (Fig. 2g). The high-resolution N 1s spectra were deconvoluted for further analysis. In contrast to BCOF, which shows only one peak, the high-resolution N 1s spectra of BECOF and ECOF exhibit two peaks corresponding to the amino N atoms (399.9 eV) and quaternary ammonium N atoms (401.2 eV) (Fig. 2h)43. The peak intensity of quaternary ammonium clearly increases with an increasing proportion of EB in the reactants; these results indicate the successful incorporation of EB species, namely, the cation sites. A greater incorporation of EB correlates to a greater presence of the cation sites, demonstrating the regulability of the ESP of multivariate COFs and, thus, its potential in regulating the interfacial microenvironment via electrostatic interactions. In addition to ESP, other characteristics, such as specific surface area, pore structure, and hydrophobicity, were controlled to be as close as possible (Supplementary Figs. 11 and 12); thus, the electrostatic interaction within the multivariate COFs is the primary variable. Considering the acidic application conditions, the acid tolerance of BECOF was also evaluated using XRD and FTIR. BECOF clearly maintains its initial framework and crystalline structure after treatment with 0.05 M H2SO4 + 0.1 M KNO3 for 3 days, as confirmed by the retained characteristic peaks in the corresponding pattern (Supplementary Fig. 13).
Experimental verifications of regulated mass transfer
To explore whether the multivariate COF with a tailored ESP could indeed regulate the mass transfer of NO3– and H+ in a targeted manner, a series of carefully designed experiments were conducted. In situ FTIR spectroscopy without an applied potential in a 0.1 M nitrate solution was initially utilised to evaluate the BECOF-triggered enrichment effect towards NO3– (Fig. 3a and Supplementary Fig. 14a). During in situ characterisation, unpolarised IR radiation sequentially passes through the CaF2 window and a thin-layer solution (approximately 10 μm) and is then reflected by the sample surface44. Consequently, both adsorbed species on the sample surface and dissolved substances in the thin-layer solution can be detected45. For the blank substrate, the in situ FTIR spectra as a function of time exhibit only one peak at 1660 cm−1, and this peak is attributed to the H‒O‒H bending mode of the interfacial water molecules, whereas no nitrate-related variation is observed (Fig. 3b)46. In contrast, upon modification with BECOF, the FTIR spectra show two new peaks at 1227 and 1490 cm−1; these peaks are attributed to the absorption of NO3– 47,48,49 and does not appear with the absence of nitrate in the solution (Supplementary Fig. 15). Moreover, these peaks gradually intensify as time increases from 0 to 10 min; these results indicate the enrichment of NO3– induced by the presence of BECOF. In situ Raman characterisation was further carried out under the same experimental conditions (Fig. 3c and Supplementary Fig. 14b). An evident peak located at 1347 cm−1 attributing to the D band of the carbon substrate is always present. If enriched and captured by Raman spectroscopy, the NO3– ions would exhibit a characteristic peak at 1047 cm−1 50. Similar to the in situ FTIR characterisation results, no peak representing NO3– was observed in the in situ Raman spectra of the blank substrate. With the coating of COFs, all three samples show the signal from the NO3– ions, but different samples experience different degrees of intensity increase as a function of time. Owing to the weak electrostatic interaction with NO3–, as indicated by the MD trajectories, the BCOF is able to enrich NO3– at a relatively slow pace, and the corresponding peak appears after approximately 45 min. In regard to cationic BECOF and ECOF, a strong attractive electrostatic force towards NO3– is generated such that it takes only 20 min for BECOF and 5 min for ECOF to exhibit the enriched NO3– peak. The in situ FTIR and in situ Raman characterisations effectively match with each other; these results clearly verify the enrichment effect towards NO3– triggered by the COFs with tailored cation sites.

a Configuration of the tailor-made cell and schematic illustration of the detection range for in situ FTIR characterisation without an applied potential. Blue spheres, N; red spheres, O; white spheres, H. b In situ FTIR spectra of the Au substrate loaded with or without BECOF in 0.1 M nitrate solution. c In situ Raman spectra of the blank, BCOF-coated, BECOF-coated, and ECOF-coated substrates without an applied potential in 0.1 M nitrate solution. d Schematic of the H+ diffusion test. e pH changes versus time when glass microfibre (GF), BCOF/GF, BECOF/GF, and ECOF/GF were used as the membranes.
The H+ diffusion test was subsequently conducted using a custom-made dual-chamber diffusion cell (Fig. 3d)51. Each half-cell contained the same volume of 0.05 M H2SO4 or deionized water and was separated by multivariate COF-loaded glass microfibre (GF) membranes. The H+ in 0.05 M H2SO4 continued to spread into the other chamber of the cell via free diffusion, and the pH within it was detected as a function of time using a high-precision pH metre. In the case of pristine GF, the pH of the deionized water side decreases quickly at the beginning of the experiment, from 7.0 to 1.9 in 5 min, and finally reaches 1.3 after 30 min. The modification of BCOF slows the H+ diffusion rate to some extent, probably due to physical sieving, but the final pH after 30 min still decreases to 2.1. As expected, when cationic BECOF/GF or ECOF/GF is used, the penetration of H+ is significantly restrained, as is the pH drop versus time, with values of 4.2 for BECOF/GF and 5.3 for ECOF/GF after 30 min, which can be attributed to the repulsive electrostatic force towards H+ generated by the positive ESP of the cation sites. Notably, ECOF with a more positive ESP would block H+ diffusion to the maximum extent, which in turn may be detrimental to the actual NO3RR since the undersupply of H+ would impose restrictions on the hydrogenation process. The above results strongly confirm the theoretical conjecture that the application of multivariate COFs with tailored ESPs as catalyst adlayers could achieve rational regulation of the mass transfer of NO3– and H+, thus constructing an ideal interfacial microenvironment for the NO3RR.
Acidic NO3RR performance of the multivariate COF-modified catalyst
To validate the proposed microenvironment design, electrochemical NO3RR experiments were systematically executed in an H-type electrolytic cell with 0.05 M H2SO4 + 0.1 M KNO3 (pH = 1 ± 0.2) as the electrolyte. Palladium-copper (PdCu) alloy nanocrystals have been reported to be able to facilitate ammonia electrosynthesis from nitrate16,52 and were thus synthesised and used as catalysts for subsequent electrochemical measurements (Supplementary Fig. 16). The PdCu catalysts were first deposited onto carbon paper, followed by drop-casting of the multivariate COFs to obtain BCOF/PdCu, BECOF/PdCu, and ECOF/PdCu as the working electrodes. Scanning electron microscopy revealed a homogeneous BECOF adlayer over the carbon fibre-supported PdCu catalyst (Supplementary Fig. 17). As indicated by further FTIR characterisation, there is no new characteristic peak in BECOF/PdCu compared with that of the bare BECOF alone (Supplementary Fig. 18), demonstrating that the BECOF and PdCu in BECOF/PdCu are simple physical mixtures. Linear sweep voltammetry (LSV) analysis was first performed on the PdCu, BCOF/PdCu, BECOF/PdCu, and ECOF/PdCu electrodes in 0.05 M H2SO4 without nitrate to evaluate the HER performance (Fig. 4a). Evidently, bare PdCu shows the strongest HER activity with the highest current density, which is suppressed to varying degrees with the modification of different COF adlayers. The BCOF adlayer can only block the access of a small number of H+ ions via physical sieving, leading to a slightly inhibited HER. However, BECOF and ECOF, with much more positive ESPs, generate strong repulsive electrostatic forces towards H+ and achieve much greater suppression of the HER. Compared with the experimental conditions above, the increase in current density with the addition of 0.1 M KNO3 demonstrates the activity towards the NO3RR for all the electrodes. A minimal increase in the current density is observed over PdCu, and the modification of the COF adlayers helps further increase the NO3RR activity, which can be attributed to efficient regulation of the interfacial microenvironment.

a Linear sweep voltammetry (LSV) curves of different electrodes in Ar-saturated 0.05 M H2SO4 without (dashed line) and with (solid line) 0.1 M KNO3 (mass loading was 0.2 mg cm–2 and reaction temperature was 25 °C) at a scan rate of 5 mV s–1. b NH3 yield rates, c corresponding Faradaic efficiencies, d H2 Faradaic efficiencies, and (e) NO2– yield rates of PdCu, BCOF/PdCu, BECOF/PdCu, and ECOF/PdCu at each given potential. The electrochemical cell resistance was 4.4 ± 0.3 Ω, and all the potentials were not iR-corrected. The error bars correspond to the standard deviations of measurements of three separately prepared samples under the same conditions. f Schematic illustration for improving NO3RR performance by using multivariate COFs with tailored ESPs. COFs are coloured to assist in the differentiation of the origins of the different moieties (orange vertices, TPs; green linkers, BDs; blue linkers, EBs). Blue spheres, N; red spheres, O; white spheres, H.
The NO3RR activities of all the samples were subsequently quantified by chronoamperometric measurements in 0.05 M H2SO4 + 0.1 M KNO3 at different potentials (Supplementary Fig. 19), and all the possible liquid products, including NH3, N2H4, and NO2−, were detected via a UV‒vis spectrophotometer and quantified according to the corresponding calibration curves (Supplementary Figs. 20–22). The possible byproduct N2H4 was determined spectrophotometrically using the Watt and Chrisp method, and negligible substances were detected in the proposed system (Supplementary Fig. 23). Upon calculation, the ammonia yield rates (Fig. 4b), the corresponding Faradaic efficiencies (Fig. 4c), and the NO3RR-to-NH3 partial current densities (Supplementary Fig. 24) of PdCu, BCOF/PdCu, BECOF/PdCu, and ECOF/PdCu are displayed, which all show a volcanic tendency with the applied potentials. Specifically, pristine PdCu has an NH3 yield rate of 3.59 mmol h–1 mg–1 and a low Faradaic efficiency of 29.8% at −1.3 V versus the reversible hydrogen electrode (RHE). With the intervention of the interfacial microenvironment triggered by the presence of multivariate COFs, the NO3RR performance gradually improved with increasing positive ESP, from an NH3 yield rate of 4.94 mmol h–1 mg–1 and a corresponding Faradaic efficiency of 43.1% for BCOF/PdCu to the maximised NH3 yield rate of 11.01 mmol h–1 mg–1 and a corresponding Faradaic efficiency of 91.0% for BECOF/PdCu. Interestingly, with further increases in the positive ESP, a decrease in the NO3RR performance is observed for ECOF/PdCu, with an NH3 yield rate of 8.18 mmol h–1 mg–1 and a Faradaic efficiency of 69.5% at −1.3 V versus RHE.
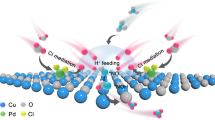
To obtain more insights into the interfacial reaction mechanism, the competing HER and NO3–-to-NO2– conversion processes were analysed. The H2 selectivity of all samples steadily increases as the potential becomes more negative but varies according to the interface regulation, and the selectivity decreases with increasing positive ESP (Faradaic efficiencies of 91.7% for PdCu, 77.9% for BCOF/PdCu, 46.7% for BECOF/PdCu, and 42.8% for ECOF/PdCu, respectively, at −1.5 V versus RHE) (Fig. 4d); these results show that the mass transfer of protons is effectively regulated upon surface modification with multivariate COFs. The NO3–-to-NO2– performance shows the opposite trend compared with that of the HER because of the trade-off relationship (Fig. 4e). Apparently, for pristine PdCu, the unrestricted protons are more inclined to participate in the HER process, and the repulsion of surface proton sources effectively suppresses the HER to some extent. However, an excessive lack of protons also restricts the subsequent hydrogenation of the nitrogenous intermediates, leading to the detection of byproducts (NO2–) that are not fully reduced; NO2– yield rates are 0.02, 0.06, 0.41, and 1.73 mmol h–1 mg–1 for PdCu, BCOF/PdCu, BECOF/PdCu, and ECOF/PdCu, respectively, at −1.1 V versus RHE.
The underlying mechanism is illustrated schematically in Fig. 4f. In acidic media, an overwhelming excess of protons exists at the interface of the bare electrocatalyst, the limited nitrate randomly moves in the bulk electrolyte, and only a small number of nitrate ions can access the catalyst surface. Under these circumstances, the active sites are more likely to be preferentially occupied by protons, thus contributing to the competing HER. With the modification of a near-neutral BCOF, interfacial H+ diffusion could be inhibited to a certain extent via physical sieving. Moreover, interfacial NO3– is also attracted and enriched through the weak electrostatic interaction generated by the BCOF adlayer, resulting in an increase in the interfacial NO3–/H+ ratio and, thus, a relative improvement in NO3–-to-NH3 conversion. Unfortunately, this level of regulation has a very limited effect; thus, the extent of improvement is modest. To confirm this deduction, the thickness of the BCOF layer on PdCu was rationally tuned by halving and doubling the loading to obtain 0.5BCOF/PdCu and 2BCOF/PdCu, and their electrocatalytic NO3RR activities were measured and compared with those of BCOF/PdCu and PdCu (Supplementary Fig. 25). Evidently, increasing the BCOF loading from 0.5BCOF/PdCu to BCOF/PdCu enhances the NO3RR performance, whereas further increasing the BCOF loading to 2BCOF/PdCu leads to a decrease in the NH3 yield rate; these results demonstrate that excess surface coating could over-shield the interface. The most favourable chemical microenvironment for the NO3RR on PdCu is achieved when BECOF is present as the adlayer. With the application of a tailored positive ESP, both the mass transfer of NO3– and H+ are regulated via appropriate electrostatic interactions, attaining the optimal NO3–/H+ ratio and, thus, the dominant NO3RR in the electrochemical process. The influence of BECOF loading was also explored, and the obtained performance exhibited a volcano-like dependence on the thickness of the BECOF adlayer (Supplementary Fig. 26). In the case of ECOF/PdCu, which has the most positive ESP, the optimal balance of the microenvironment may be broken, resulting in an excess of NO3– and an insufficient supply of H+; this subsequently hinders the hydrogenation process of the NO3RR (Supplementary Fig. 27). The controlled experiments indicate that a more positive ESP, namely a higher NO3–/H+ ratio, is not always better; these results highlight the critical role played by rational control of the interfacial microenvironment.
For real-time observation of the facilitated NO3RR process in the BECOF/PdCu system, operando measurements under working conditions, including operando Raman and FTIR spectroscopy (Supplementary Fig. 28), were employed. Figure 5a, b show the operando Raman spectra results collected for BECOF/PdCu and PdCu under potentiostatic operation for 1 h, respectively. For BECOF/PdCu, two new peaks located at 1150 and 1523 cm–1 corresponding to *NH2 and *NH, respectively53,54, appear and increase in intensity as the reaction progresses. On the other hand, no NO3RR-related intermediates are captured on PdCu. For operando attenuated total reflection-FTIR (ATR-FTIR) characterisation, in addition to the H‒O‒H bending mode of the interfacial H2O molecules, the downwards peaks attributed to NO3– negatively increase as a function of time because of its continuous consumption over BECOF/PdCu during electrolysis (Fig. 5c)48. Correspondingly, two peaks representing *NO2 and *NH2 intermediates appear, and their intensities also gradually increase55. In contrast, for bare PdCu, only the vibration band of deoxidation intermediates (*NO2) is observed, whereas the consumption of NO3– and the hydrogenation intermediate are barely detected (Fig. 5d). The combination of operando Raman and ATR-FTIR characterisation under working conditions strongly indicates the enhancement of the nitrate reduction process triggered by the BECOF modification.

Operando Raman spectra of (a) BECOF/PdCu and (b) PdCu collected at −1.3 V versus RHE as a function of time. Operando FTIR spectra of (c) BECOF/PdCu and (d) PdCu collected at −1.3 V versus RHE as a function of time. e 1H NMR spectra of standard samples and the NO3RR products using 14NO3– and 15NO3– as the isotopic nitrate source in 0.05 M H2SO4 electrolyte. f Comparison of the calculated NH3 yield rates and Faradaic efficiencies at −1.3 V versus RHE using different quantification methods. The potential was not iR-corrected. g Schematic of the ammonia product synthesis process from nitrate-containing influent to NH4Cl(s). h XRD analysis results of generated and commercial NH4Cl solid products.
To verify that the produced ammonia was from electrocatalytic nitrate reduction instead of other nitrogenous pollutants, a series of control experiments were performed56. Electrochemical measurements of bare BECOF/BCOF/ECOF-coated carbon paper indicate that none of these samples leads to the production of ammonia and that only PdCu serves as an electrocatalyst and works thermodynamically (Supplementary Fig. 29). The 1H nuclear magnetic resonance (NMR) analyses confirmed that negligible NH3 was detected in the absence of NO3–, whereas a typical double-peak feature with a coupling constant of 73.1 Hz representing 15NH4+ without any triplet peak related to 14NH4+ was observed when K15NO3 was used as the reactant (Fig. 5e). Upon further quantification using 1H NMR spectroscopy (Supplementary Figs. 30 and 31), the corresponding NH3 yield rates and Faradaic efficiencies are determined to be 11.14 mmol h–1 mg–1 and 91.1% for the 14NO3RR and 10.73 mmol h–1 mg–1 and 93.1% for the 15NO3RR, respectively; these data are in agreement with those from the colorimetric tests (Fig. 5f and Supplementary Table 1). The above results strongly indicate that the detected NH3 in the electrolyte is clearly from the NO3RR process rather than from environmental contamination. The superior NO3RR performances of the BECOF/PdCu system are considerably greater than those of the extensively reported systems tested under acidic conditions and even the majority of those tested in neutral or alkaline electrolytes reported to date (Supplementary Table 2).
Practical ammonia product synthesis
In light of the impressive acidic NO3RR performance of the BECOF/PdCu system, high-purity ammonia products were continuously collected from a set of tailor-made devices to demonstrate its practical application potential (Fig. 5g)57. Due to the advantages of acid systems, no air stripping or acid adsorption unit is needed for ammonia capture to avoid possible ammonia volatilisation from neutral/alkaline electrolytes, and the ammonia produced from electrocatalysis can be directly collected via evaporation58. During operation, long-term electrolysis at −1.3 V versus RHE was conducted in the H-type electrolytic cell equipped with the BECOF/PdCu cathode, and the system could work robustly over consecutive electrolysis cycles for up to 20 h, and negligible changes were observed in the NH3 yield rate and Faradaic efficiency (Supplementary Fig. 32). After further ion exchange between SO42– and Cl– as well as rotary evaporation for purification and collection, the ammonia in the collected solution could be converted into high-purity NH4Cl powder, as shown in the photograph of the collected product. The powder product is highly consistent with standard NH4Cl (JCPDS NO. 07-0007), as confirmed by the XRD pattern (Fig. 5h), and the product purity was determined to reach up to 96.2% from the UV‒vis absorption results (Supplementary Fig. 33); thus, this NH4Cl sheds light on the potential as a feedstock for fertiliser, textile, pharmaceutical industries, and so on. The post-test BECOF is also thoroughly characterised. The results indicate that the honeycomb-like porous morphology, crystallinity, and chemical structure of BECOF remain unchanged after long-term electrolysis in acid (Supplementary Figs. 34–36), confirming its robustness for practical applications. Overall, we demonstrated a complete process in which nitrate-containing feed is directly converted into actual ammonia products using our proposed system with BECOF modification. Moreover, since the concentrations of nitrate in various waters are different, the impact of the nitrate concentration on the NO3RR of BECOF/PdCu was further evaluated in electrolytes with different nitrate concentrations (Supplementary Fig. 37). The high performance of BECOF/PdCu was effectively maintained over a wide range; these results further indicate its promising application potential for the NO3RR.
Discussion
In summary, we have demonstrated that multivariate COFs with tailored ESPs enable rational control of the catalyst microenvironment and, thus, efficient ammonia electrosynthesis from the direct nitrate reduction in strongly acidic environments. With well-controlled cationic sites over the COF skeleton, both repulsive electrostatic forces towards H+ and attractive electrostatic forces towards NO3– are generated via electrostatic interactions. In particular, by sophisticated regulation of the charge distribution, H+ access is restrained to a certain extent to efficiently suppress the competing HER and maintain an adequate supply for hydrogenation of NO3RR-related intermediates. At the same time, localised enrichment of NO3– triggered by COFs with tailored cation sites is also achieved, and a favourable local environment for the NO3RR is attained. The multivariate COFs with the optimal ESP enable a maximised NH3 yield rate of 11.01 mmol h–1 mg–1 and a corresponding Faradaic efficiency of 91.0%; these values, surpass not only the extensively reported systems tested under acidic conditions but also the majority of those tested in neutral or alkaline electrolytes reported to date. A set of tailor-made devices without air stripping or acid adsorption units further achieves the direct production of high-purity NH4Cl in acid. This study can be used to facilitate sustainable ammonia electrosynthesis under acidic conditions and has the potential to directly convert nitrate-containing wastewater into practical feedstocks for various industries.
Methods
Electrostatic potential computational method
All quantum chemical calculations were performed with Gaussian 16. Molecular geometry was optimised at the B3LYP-D3/6-31G* level of theory, electronic structure calculations based on optimised structures are conducted at the B3LYP-D3/6-311G** level of theory. The ESP analysis was performed with the help of Multiwfn59. The specific ESP value, V(R), created at the distance R by the charge Q was calculated as follows39:
V(R) is a scalar quantity and indicates the potential of Q for interacting at a distance R with any other charge Qi. The ESP is mapped to the molecular van der Waals surface defined by electron density isosurface corresponding to ρ = 0.001e Bohr–3 as suggested by Bader60. The ESP coloured molecular surface is visualised by VMD software61. When considering the influence of applied electrode potential, the electric field corresponding to −1.3 V versus RHE was included in the calculation process62.
Electrostatic interaction evaluation method
The electrostatic interactions of H+ and NO3– with multivariate COFs were evaluated by MD simulations, which were carried out with Gromacs version 2019.663. For system construction, multivariate COFs were modelled as six rings with different charges, which were AA-stacked at a spacing of 0.34 nm. Multivariate COFs were placed in the centre of the simulation box, and then 60 H+, 60 NO3–, and 6600 water molecules were added to fully fill the simulation boxes, which were 6.0 nm in x-y dimensions and 6.4 nm in z dimension. Multivariate COFs, H+, and NO3– are modelled by the General Amber Force Field (GAFF)64, with RESP partial charges generated by the Multiwfn programme adopted to describe the interatomic electrostatic interactions65. TIP3P model was adopted for water, which is compatible with GAFF66. The Coulombic potential is used for the interatomic electrostatic interactions67:
where q is the partial charge of atoms i and j, εr is the actual permittivity, ε0 is the relative permittivity in vacuum, and rij is the distance between atoms i and j.
Reagents
2,4,6-Triformylphloroglucinol (98%) was purchased from Jilin Chinese Academy of Sciences Yanshen Technology Co., Ltd, China. Biphenyl diamine (98%), mesitylene (99%), 1,4-dioxane (99%), tetrahydrofuran (THF, 99.9%), methanol (99.9%), potassium nitrate (KNO3, 99.9%), potassium nitrate-15N (K15NO3, 99%), acetic acid (99.7%), palladium(II) acetylacetonate [Pd(acac)2, 99.9%], copper(II) acetylacetonate [Cu(acac)2, 97%], oleylamine (OAM, 80–90%), ammonium bromide (NH4Br, 99%), ascorbic acid (AA, 99%), ethanol (99.5%), and cyclohexane (99.5%) were purchased from Shanghai Aladdin Biochemical Technology. Ethidium bromide (99%) and Nafion solution (5 wt.%) were purchased from Shanghai Macklin Biochemical Co., Ltd. Sulphuric acid (H2SO4, 95–98%) was purchased from Sinopharm Chemical Reagent Co., Ltd. All chemical reagents were used without further purification.
Synthesis of BECOF
BD (5.15 mg, 0.028 mmol), EB (21.68 mg, 0.055 mmol), and TP (11.6 mg, 0.055 mmol) were dissolved in 6 mL of mesitylene/1,4-dioxane (1:1, v/v) mixture in a 25 mL Pyrex tube under sonication of 30 min. Then, 0.1 mL of glacial acetic acid (6 M, aqueous) was added, and the Pyrex tube was sealed after degassing by the three freeze-pump-thaw cycles. The reaction was maintained under 120 °C for 72 h. After cooling to room temperature, the produced precipitate was collected and washed successively with methanol and THF. The obtained jacinth powder was dried at 100 °C under vacuum for 24 h to obtain BECOF.
Synthesis of BCOF
BD (15.27 mg, 0.083 mmol) and TP (11.6 mg, 0.055 mmol) were dissolved in 6 mL of mesitylene/1,4-dioxane (1:1, v/v) mixture in a 25 mL Pyrex tube under sonication of 30 min. Then, 0.1 mL of glacial acetic acid (6 M, aqueous) was added, and the Pyrex tube was sealed after degassing by the three freeze-pump-thaw cycles. The reaction was maintained under 120 °C for 72 h. After cooling to room temperature, the produced precipitate was collected and washed successively with methanol and THF. The obtained orange powder was dried at 100 °C under vacuum for 24 h to obtain BCOF.
Synthesis of ECOF
EB (32.72 mg, 0.083 mmol) and TP (11.6 mg, 0.055 mmol) were dissolved in 6 mL of mesitylene/1,4-dioxane (1:1, v/v) mixture in a 25 mL Pyrex tube under sonication of 30 min. Then, 0.1 mL of glacial acetic acid (6 M, aqueous) was added, and the Pyrex tube was sealed after degassing by the three freeze-pump-thaw cycles. The reaction was maintained under 120 °C for 72 h. After cooling to room temperature, the produced precipitate was collected and washed successively with methanol and THF. The obtained deep-red powder was dried at 100 °C under vacuum for 24 h to obtain ECOF.
Physical characterisations
The AC HAADF STEM images of multivariate COFs were recorded using a Titan Themis Cubed G2 300. The crystallinity of multivariate COFs was characterised by XRD (D8 Advance, Bruker). FTIR spectra were collected using the Tensor 27 (Bruker, Germany) with pure KBr pellets. SSNMR spectra were collected using the AvanceIII/Wb-400 (Bruker, Germany). Surface elemental analysis was performed on XPS (Kratos Axis Ultra Dld, Japan). N2-sorption analysis was performed with a surface area and porosimetry instrument (Micromeritics, ASAP 2460, USA). The specific surface area and pore parameters were investigated by the Brunauer-Emmett-Teller equation and the Barrett-Joyner-Halenda method. Contact angles were conducted using water droplets via Attension Theta (JY-82B Kruss DSA). The pH values were collected using an FB20 ST310 (OHAUS, USA). The morphology of the PdCu catalyst was studied with a field emission transmission electron microscope (FETEM, FEI Tecnai G2 F20 S-TWIN TMP). The structure of the PdCu catalyst was characterised by XRD and XPS. The morphology of BECOF/PdCu electrode was studied with a scanning electron microscope (SEM, SU8010).
In situ FTIR and Raman characterisations
The in situ characterisations without applied potential were conducted using different tailor-made cells. In situ FTIR experiments were conducted using external reflection mode on a Nicolet iS50 FT-IR spectrometer equipped with an MCT detector cooled with liquid nitrogen. Before the test, the Au substrate was first polished with several grades of alumina powder and then cleaned in deionized water in an ultrasonic bath for 10 min. The Au substrate loaded with or without BECOF was placed in close contact with the CaF2 window plate to form a thin liquid layer with a thickness of approximately 10 μm. 0.05 M H2SO4 with or without 0.1 M KNO3 was selected as the solution and saturated with Ar before testing. The spectrum that recorded with the substrate loaded with or without BECOF in the solution prior to the in situ characterisation was captured as the reference spectrum, which was then subtracted from all the sample spectra collected in in situ FTIR characterisation as a function of time. The recording frequency of FTIR spectra was 1 min for the same position.
In situ Raman experiments were conducted on a Horiba Jobin Yvon HR evolution instrument (France) with a 532 nm laser source (Ventus LP 532) and a 50 × objective. The carbon paper coated with different COFs was used as the electrode. Raman frequency was calibrated by a Si wafer during each experiment. 0.05 M H2SO4 combined with 0.1 M KNO3 was selected as solution and saturated with Ar before testing. The recording frequency of Raman spectra was 10 min for the same position.
Synthesis of PdCu nanocrystals
NH4Br (25 mg) and AA (15 mg) was added into OAM (5 mL) in a 20 mL vial. After sonication for about 15 min, Pd(acac)2 (10 mg) and Cu(acac)2 (5 mg) were added into the vial. In order to obtain a homogeneous solution, the mixture was sonicated below 25 °C for 30 min. After sealing, the vial was heated to 150 °C at 2 °C min−1 and then kept for 3 h in an oil bath. The cooled product was collected by centrifugation and washed three times with an ethanol/cyclohexane mixture (9:1, v/v). Finally, the final product was dried at 60 °C under a vacuum oven overnight.
Cathode preparation
The carbon paper was used as the substrate of the working electrode. The as-synthesised PdCu nanocrystals was dispersed into ethanol and Nafion solution and sonicated for 1 h to get a homogeneous catalyst ink. The catalyst ink was pipetted onto carbon paper (0.5 × 0.5 cm2) with a mass loading of 0.2 mg cm–2 to obtain a PdCu electrode. The BCOF, BECOF, or ECOF was dispersed into ethanol solution, and sonicated for 1 h to get a homogeneous adlayer ink. Then, the solution was pipetted onto the PdCu electrode and dried naturally to obtain the BCOF/PdCu, BECOF/PdCu, ECOF/PdCu electrode with an adlayer loading of 0.2 mg cm–2. Notably, only PdCu acted as the active component and its loading mass was used for quantitative analysis.
Electrochemical NO3RR measurements
Electrochemical NO3RR measurements were performed in a gas-tight H-type cell, which has an ion exchange membrane (Nafion 117) to separate the cathode and anode chambers. The measured electrochemical cell resistance was 4.4 ± 0.3 Ω. Before use, the Nafion membrane was boiled in a 5% H2O2 aqueous solution at 80 °C for 1 h and subjected to multiple rinses. Both the cathode chamber and anode chamber contained 20 mL of 0.05 M H2SO4 + 0.1 M KNO3 electrolyte (pH = 1 ± 0.2). Herein, quantitative H2SO4 was added into a certain amount of deionized water, followed by weighing and dissolving of the required quantity of KNO3. The solution was transferred into a flask and diluted to the mark with deionized water. This solution was used immediately as the electrolyte and was purged with high-purity argon to remove impurities prior to the experiment. A Pt wire was used as the counter electrode, and an Ag/AgCl electrode (saturated KCl solution) was used as the reference electrode. The reference electrode was calibrated prior to the electrochemical measurements. It was immersed in a 3.5 M potassium chloride standard solution, and its potential was calibrated by comparing the measured potential with the theoretical potential at room temperature. The LSV and chronoamperometric measurements were conducted using a CHI660E electrochemical workstation (Shanghai Chenhua Instrument Co.) under constant stirring of 400 rpm. The current density was normalised to the geometric electrode area (0.25 cm2). Before the measurement, the electrolyte was saturated by Ar gas through purging for 30 min, and a flow of Ar was maintained throughout the entire electrochemical measurement with a gas flow rate of 10 sccm. The potentiostatic tests were tested at different potentials, including –1.1, –1.2, –1.3, –1.4, and –1.5 V versus RHE for 1 h. All potentials were not iR-corrected and were adjusted to the RHE reference scale using the formula E(versus RHE) = E(versus Ag/AgCl) + 0.0591 × pH + 0.197. After the electrochemical reduction reaction, the electrolyte was collected and analysed by colorimetric and NMR methods.
NO3RR product analysis
NH3 was determined by the indophenol blue method29. Specifically, 1 M NaOH solution containing 5 wt.% sodium citrate and 5 wt.% salicylic acid solution was prepared as solution A. 0.05 M sodium hypochlorite solution and 1 wt.% sodium nitroferricyanide were prepared as solutions B and C, respectively. For NH3 quantification, 0.1 mL of the electrolyte after the potentiostatic test was removed from the cathode chamber and diluted to 20 mL. Then, 2 mL of the above diluted solution was successively mixed with 2 mL of solution A, 1 mL of solution B, and 0.2 mL of solution C. After standing in the dark for 2 h, the absorption spectrum of the resulting solution was measured using an ultraviolet-visible spectrophotometer (UV-2700i, Shimadzu), and the absorption intensity at a wavelength of 655 nm was recorded. The concentration-absorbance curve was calibrated using standard ammonium chloride solutions in 0.05 M H2SO4 at a series of given concentrations.
N2H4 was determined by the Watt and Chrisp method29. Specifically, the chromogenic reagent (solution D) was prepared by dissolving 5.99 g of para-(dimethylamino) benzaldehyde in the solution containing 300 mL of ethanol and 30 mL of concentrated HCl. For N2H4 quantification, 5 mL of the electrolyte after the potentiostatic test was removed from the cathode chamber, and 5 mL of solution D was added. After stirring in the dark for 10 min, the absorption spectrum of the resulting solution was measured, and the absorption intensity at a wavelength of 455 nm was recorded. The concentration-absorbance curve was calibrated using standard hydrazine monohydrate in 0.05 M H2SO4 solutions at a series of given concentrations.
NO2– was determined by the Greiss spectrophotometric method29. Specifically, 0.5 g of sulforaphane was dissolved in the mixture containing 90 mL of H2O and 5 mL of acetic acid. Then, 5 mg N-(1-naphthyl)-ethylenediamine dihydrochloride was added, and the solution was fixed to 100 mL to obtain the chromogenic reagent (solution E). For NO2– quantification, 0.1 mL of the electrolyte after the potentiostatic test was removed from the cathode chamber and diluted to 20 mL. Then, 1 mL of the above diluted solution was mixed with 4 mL of solution E. After standing in the dark for 15 min, the absorption spectrum of the resulting solution was measured, and the absorption intensity at a wavelength of 540 nm was recorded. The concentration-absorbance curve was calibrated using a standard potassium nitrite solution in 0.05 M H2SO4 at a series of given concentrations.
Determination of hydrogen evolution
The gaseous hydrogen was monitored by a gas chromatography (GC-2014, SHIMADZU, Japan) equipped with a thermal conductivity detector. During the test, the cathode chamber was sealed and directly connected to the on-line GC instrument. Nitrogen was used as the carrier gas, and the tail gas was analysed by manual sampling under different potentials, including –1.1, –1.2, –1.3, –1.4, and –1.5 V versus RHE.
Faradaic efficiency and the yield rate
The Faradaic efficiency and yield rate of NH3 were calculated as follows:
where F is the Faraday constant, t is the electrolysis time, m is the loading mass of the catalysts, Q is the total charge, V is the electrolyte volume, and c(NH3) is the measured ammonia concentration.
The Faradaic efficiency of H2 was calculated as below:
where Vj is the volume percentage obtained from the GC analysis of H2, P is the atmospheric pressure (101.325 KPa), R is the gas constant (8.314 J mol−1 K−1), V is the volume of sampling loop (1 cm3), T is the temperature in Kelvin (298.15 K), Itotal is the recording current, and tH is the time required to fill the sampling loop.
The yield rate of NO2– was calculated using the following formula:
where c(NO2–) is the measured nitrite concentration, V is the electrolyte volume, t is the electrolysis time, m is the loading mass of the catalysts.
Operando Raman and FTIR measurements
The operando Raman and FTIR measurements were conducted using different tailor-made cells. The operando Raman experiments were conducted on a Horiba Jobin Yvon HR evolution instrument (France) with a 532 nm laser source (Ventus LP 532) and a 50 × objective. Raman frequency was calibrated by a Si wafer during each experiment. 0.05 M H2SO4 combined with 0.1 M KNO3 was selected as an electrolyte and saturated with Ar before testing. The chronoamperometric measurements were conducted at –1.3 V versus RHE using a CHI660E electrochemical workstation (Shanghai Chenhua Instrument Co.). To eliminate the influence of evolved gas bubbles, the electrolyte circulation was constantly maintained by using a peristaltic pump. The recording frequency of Raman spectra was 10 min for the same position.
Electrochemical operando ATR-FTIR measurements were conducted using the internal reflection mode to minimise absorption interference of IR light on a Nicolet iS50 FT-IR spectrometer equipped with an MCT detector cooled with liquid nitrogen. The silicon crystal was loaded successively with PdCu and BECOF, which was then assembled into a tailor-made spectral-electrochemical cell containing a Pt wire as a counter electrode and an Ag/AgCl electrode as a reference electrode. 0.05 M H2SO4 combined with 0.1 M KNO3 was selected as an electrolyte and saturated with Ar before testing. The chronoamperometric measurements were conducted at –1.3 V versus RHE using a CHI660E electrochemical workstation (Shanghai Chenhua Instrument Co.). The spectrum that recorded with the electrode prior to the operando characterisation was captured as the reference spectrum, which was then subtracted from all the sample spectra collected in operando ATR-FTIR characterisation as a function of time. The recording frequency of ATR-FTIR spectra was 100 s for the same position.
K15NO3 isotopic labelling experiment
K15NO3 and K14NO3 were used as the nitrate source in the labelling experiment. The electrolyte was collected after operating for 1 h at –1.3 V versus RHE. For quantitative 1H NMR measurements, various concentrations of ammonium chloride were used to prepare the standard curves. The electrolyte was collected after electrolysis at −1.3 V versus RHE. Then, the solution diluted to the detection range and was mixed with 0.1 mL of dimethylsulfoxide-d6. Maleic acid was used as the internal standard. The produced ammonia was quantified by using 1H NMR spectroscopy (Agilent 600 MHz).
Ammonia products collection
Continuous NO3RR coupled with the collection of high-purity ammonia products (NH4Cl) was accomplished in a set of tailor-made devices. A gas-tight H-type cell was chosen as the subject of the chronoamperometric measurements. Both the cathode chamber and anode chamber contained 20 mL of 0.05 M H2SO4 + 0.1 M KNO3 electrolyte. The electrolyte was collected every 1 h for ammonia quantification, and the chronoamperometric measurements were then carried out at the same conditions using the fresh electrolyte. It is worth noting that to avoid the effects of sulphuric acid, SO42– was removed using a barium chloride solution before initiating the rotary evaporator. To obtain the NH4Cl product, the purified electrolyte, after ion exchange was dried using a rotary evaporator and further dried overnight at 80 °C in a vacuum. The final NH4Cl solid was analysed by XRD and quantified by UV–vis absorption spectra.
Data availability
All data supporting the findings of this study are available within the article and the Supplementary Information file. Source data are provided in this paper.
References
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 568, 536–540 (2019).
Liu, S. et al. Awakening (220) as one more active facet of PtMo alloy via single-atom doping to boost ammonia electrooxidation in direct ammonia fuel cell. Adv. Funct. Mater. 33, 2306204 (2023).
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209–2244 (2009).
Soloveichik, G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nat. Catal. 2, 377–380 (2019).
Cheng, Q. et al. High-entropy alloys for accessing hydrogen economy via sustainable production of fuels and direct application in fuel cells. Rare Met. 42, 3553–3569 (2023).
Chen, J. G. et al. Beyond fossil fuel–driven nitrogen transformations. Science 360, eaar6611 (2018).
He, Y. et al. Donor-site-acceptor covalent organic frameworks enable spontaneous nitrogen dissociation for boosted photoelectrochemical ammonia synthesis. Adv. Funct. Mater. 34, 2315548 (2024).
He, Y. et al. A superaerophilic gas diffusion electrode enabling facilitated nitrogen feeding through hierarchical micro/nano channels for efficient ambient synthesis of ammonia. Chem. Eng. J. 454, 140106 (2023).
Daiyan, R. et al. Nitrate reduction to ammonium: from CuO defect engineering to waste NOx-to-NH3 economic feasibility. Energy Environ. Sci. 14, 3588–3598 (2021).
He, Y. et al. Advanced in situ characterization techniques for direct observation of gas-involved electrochemical reactions. Energy Environ. Mater. 6, e12552 (2023).
Liu, D. et al. Recent advances in electrocatalysts for efficient nitrate reduction to ammonia. Adv. Funct. Mater. 33, 2303480 (2023).
Zhao, X. et al. Comprehensive understanding of the thriving electrocatalytic nitrate/nitrite reduction to ammonia under ambient conditions. J. Energy Chem. 92, 459–483 (2024).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402–414 (2023).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Wu, Z.-Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
Wang, Z. et al. Interfacial defect engineering triggered by single atom doping for highly efficient electrocatalytic nitrate reduction to ammonia. ACS Mater. Lett. 5, 1018–1026 (2023).
Lv, Y. et al. Highly efficient electrochemical nitrate reduction to ammonia in strong acid conditions with Fe2M‐trinuclear‐cluster metal-organic frameworks. Angew. Chem. Int. Ed. 62, e202305246 (2023).
Ba, J. et al. Red carbon mediated formation of Cu2O clusters dispersed on the oxocarbon framework by Fehling’s route and their use for the nitrate electroreduction in acidic conditions. Adv. Mater. 36, 2400396 (2024).
Hou, H. et al. Fabrication of a sulfur/steel slag-based filter and its application in the denitrification of nitrate-containing wastewater. Water 16, 690 (2024).
Zhang, R. et al. Electrochemical nitrate reduction in acid enables high-efficiency ammonia synthesis and high-voltage pollutes-based fuel cells. Nat. Commun. 14, 8036 (2023).
Liao, W. et al. Sustainable conversion of alkaline nitrate to ammonia at activities greater than 2 A cm−2. Nat. Commun. 15, 1264 (2024).
Lv, Y. et al. Atomically precise integration of multiple functional motifs in catalytic metal-organic frameworks for highly efficient nitrate electroreduction. J. Am. Chem. Soc. Au 2, 2765–2777 (2022).
Chen, F.-Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
He, Y. et al. Deciphering engineering principle of three-phase interface for advanced gas-involved electrochemical reactions. J. Energy Chem. 80, 302–323 (2023).
Wu, H. et al. Electrocatalyst microenvironment engineering for enhanced product selectivity in carbon dioxide and nitrogen reduction reactions. ACS Catal. 13, 5375–5396 (2023).
Liu, S. et al. Molecular imprinting technology enables proactive capture of nitrogen for boosted ammonia synthesis under ambient conditions. Adv. Mater. 35, 2303703 (2023).
Cheng, Q. et al. Eliminating concentration polarization with cationic covalent organic polymer to promote effective overpotential of nitrogen fixation. Angew. Chem. Int. Ed. 62, e202308262 (2023).
Wang, M. et al. Salting-out effect promoting highly efficient ambient ammonia synthesis. Nat. Commun. 12, 3198 (2021).
Cheng, Q. et al. Li+-ion bound crown ether functionalization enables dual promotion of dynamics and thermodynamics for ambient ammonia synthesis. J. Energy Chem. 85, 191–197 (2023).
Abbott, D. F. et al. Understanding the synergy between Fe and Mo sites in the nitrate reduction reaction on a bio‐inspired bimetallic MXene electrocatalyst. Angew. Chem. Int. Ed. 62, e202313746 (2023).
Zhang, G. et al. Tandem electrocatalytic nitrate reduction to ammonia on MBenes. Angew. Chem. Int. Ed. 62, e202300054 (2023).
Zhao, X., Pachfule, P. & Thomas, A. Covalent organic frameworks (COFs) for electrochemical applications. Chem. Soc. Rev. 50, 6871–6913 (2021).
Qian, Y. & Jiang, H.-L. Structural regulation of covalent organic frameworks for catalysis. Acc. Chem. Res. 57, 1214–1226 (2024).
Jiao, L., Wang, J. & Jiang, H.-L. Microenvironment modulation in metal-organic framework-based catalysis. Acc. Mater. Res. 2, 327–339 (2021).
Wen, L. et al. Electronic state and microenvironment modulation of metal nanoparticles stabilized by MOFs for boosting electrocatalytic nitrogen reduction. Adv. Mater. 35, 2210669 (2023).
Ma, H. et al. Cationic covalent organic frameworks: a simple platform of anionic exchange for porosity tuning and proton conduction. J. Am. Chem. Soc. 138, 5897–5903 (2016).
Lu, T. & Chen, Q. Van der Waals potential: an important complement to molecular electrostatic potential in studying intermolecular interactions. J. Mol. Model. 26, 315 (2020).
Murray, J. S. & Politzer, P. The electrostatic potential: an overview. WIRES Comput. Mol. Sci. 1, 153–163 (2011).
Zeng, L. et al. Molecular dynamics simulations of electrochemical interfaces. J. Chem. Phys. 159, 091001 (2023).
Sasmal, H. S. et al. Covalent self-assembly in two dimensions: connecting covalent organic framework nanospheres into crystalline and porous thin films. J. Am. Chem. Soc. 141, 20371–20379 (2019).
Yang, H. et al. Highly water-selective membranes based on hollow covalent organic frameworks with fast transport pathways. J. Membr. Sci. 565, 331–341 (2018).
Hou, L. et al. Understanding the ion transport behaviour across nanofluidic membranes in response to the charge variations. Adv. Funct. Mater. 31, 2009970 (2021).
Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Zhou, Z.-Y., Wang, Q., Lin, J.-L., Tian, N. & Sun, S.-G. In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 55, 7995–7999 (2010).
Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study on the nitrogen electrochemical reduction reaction on gold and platinum surfaces. J. Am. Chem. Soc. 140, 1496–1501 (2018).
Zhou, B. et al. Cu1-Fe dual sites for superior neutral ammonia electrosynthesis from nitrate. Angew. Chem. Int. Ed. 63, e202406046 (2024).
Zhou, B. et al. Renewable energy driven electroreduction nitrate to ammonia and in-situ ammonia recovery via a flow-through coupled device. Water Res. 242, 120256 (2023).
Yang, J., Li, L., Xiao, C. & Xie, Y. Dual-plasmon resonance coupling promoting directional photosynthesis of nitrate from air. Angew. Chem. Int. Ed. 62, e202311911 (2023).
Butcher, D. P. Jr & Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: Effect of oxide and Cl−. Nano Energy 29, 457–465 (2016).
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Wang, Y. et al. Wide-pH-range adaptable ammonia electrosynthesis from nitrate on Cu-Pd interfaces. Sci. China Chem. 66, 913–922 (2023).
Philip, D., Eapen, A. & Aruldhas, G. Vibrational and surface enhanced Raman scattering spectra of sulfamic acid. J. Solid State Chem. 116, 217–223 (1995).
Ziegler, L. & Hudson, B. Resonance rovibronic Raman scattering of ammonia. J. Phys. Chem. 88, 1110–1116 (1984).
Zhang, N. et al. Lewis acid Fe-V pairs promote nitrate electroreduction to ammonia. Adv. Funct. Mater. 33, 2211537 (2023).
Hodgetts, R. Y. et al. Refining universal procedures for ammonium quantification via rapid 1H NMR analysis for dinitrogen reduction studies. ACS Energy Lett. 5, 736–741 (2020).
Dai, J. et al. Spin polarized Fe1-Ti pairs for highly efficient electroreduction nitrate to ammonia. Nat. Commun. 15, 88 (2024).
Liu, W. et al. Efficient ammonia synthesis from the air using tandem non-thermal plasma and electrocatalysis at ambient conditions. Nat. Commun. 15, 3524 (2024).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Bader, R. F., Carroll, M. T., Cheeseman, J. R. & Chang, C. Properties of atoms in molecules: atomic volumes. J. Am. Chem. Soc. 109, 7968–7979 (1987).
Humphrey, W., Dalke, A. & Schulten, K. V. M. D. visual molecular dynamics. J. Mol. Graph. Model. 14, 33–38 (1996).
Gurav, N. D., Gejji, S. P., Bartolotti, L. J. & Pathak, R. K. Encaged molecules in external electric fields: A molecular “tug-of-war”. J. Chem. Phys. 145, 074302 (2016).
Spoel, D. V. D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Bayly, C. I., Cieplak, P., Cornell, W. & Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 (1993).
Yao, Q. et al. Supercrystal engineering of atomically precise gold nanoparticles promoted by surface dynamics. Nat. Chem. 15, 230–239 (2023).
Sajid, H. Effect of interlayer slipping on the geometric, thermal and adsorption properties of 2D covalent organic frameworks: a comprehensive review based on computational modelling studies. Phys. Chem. Chem. Phys. 26, 8577–8603 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China [No. U21A20332 to C.L. Yan, No. 52202275 to M.F. Wang, No. 52401284 to S.S. Liu] and the Natural Science Foundation of Jiangsu Province [No. BK20240957 to S.S. Liu]. We acknowledge support from the Collaborative Innovation Centre of Suzhou Nano Science and Technology.
Author information
Authors and Affiliations
Contributions
S.S. Liu, C.L. Yan and J.M. Lu conceived and designed this work. Q.Y. Cheng, S.S. Liu and Y.F. Huan prepared and characterised the material and performed the experiments. H.Q. Ji conducted the theoretical calculations. M.F. Wang, Y.Z. He and T. Qian discussed the results and commented on the manuscript. Q.Y. Cheng wrote the paper, and S.S. Liu, C.L. Yan, and J.M. Lu revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, Q., Liu, S., He, Y. et al. Multivariate covalent organic frameworks with tailored electrostatic potential promote nitrate electroreduction to ammonia in acid. Nat Commun 16, 3717 (2025). https://doi.org/10.1038/s41467-025-59052-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59052-2
