
Abstract
Lignin represents the most abundant biomass resource, which contains aromatic units. Lignin refinery is a promising, sustainable alternative for the production of aromatic chemicals. However, depolymerization and transformation of lignin into aryl halides, which are indispensable chemicals in both the academic and industrial communities, remain challenging. Here, we report a simple and mild method for the depolymerization and halogenation of lignin, leading to the production of useful aryl halides. Notably, hydrogen bond activation for halogenation reagents is essential for substantially increasing the reactivity, resulting a highly efficient cleavage of C–C bonds in lignin. This method is highly selective for breaking C(sp2)–C(sp3) bonds of lignin linkages, enabling the application of precise depolymerization and halogenation reactions from lignin models to native lignin from various wood resources, which provides a sustainable and efficient access to various synthetically useful aryl halides.
Similar content being viewed by others
Introduction
Aryl halides are indispensable chemicals in both the academic and industrial communities. Owing to their unique reactivity patterns, aryl halides have played crucial roles in the development of modern chemical synthesis1. The carbon–halogen bonds in these chemicals have versatile and robust applications in synthetic chemistry and materials manufacture2. For example, aryl halides are widely used as starting materials for the synthesis of fused benzene-based advanced materials such as conjugated polymers3,4,5, organic optoelectronic materials6,7,8, and organic semiconductors9,10,11. Moreover, aryl halides represent the most significant precursors in cross-coupling12,13,14,15 and nucleophilic aromatic substitution reactions or the preparation of Grignard reagents16, which are widely used in the synthesis of pharmaceuticals, agrochemicals, and synthetic building blocks17,18,19,20. Currently, aryl halides are produced predominantly via the halogenation of aromatics12,21,22,23, which are produced from petroleum and coal through catalytic reforming and steam cracking processes (Fig. 1a)24,25,26. However, these fossil resource-dependent routes involve complicated and multiple procedures, high energy consumptio,n and environmental pollution27,28. Thus, the development of efficient, environmentally friendly, and sustainable processes for chemical production from renewable biomass has attracted much attention.

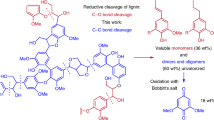
a Traditional production of aryl halides. b Conversion of lignin into aromatic compounds. c This work: producing aryl halides from lignin.
Lignin, which is one of the main components of lignocellulosic biomass, represents the most abundant resource featuring aromatic structures, and is the most promising sustainable alternative for the production of functionalized aromatic compounds29,30,31,32. Numerous efforts have been made to valorize lignin into value-added aromatic chemicals via the depolymerization of lignin linkages under oxidative33,34,35,36,37, reductive38,39,40, and redox-neutral conditions41,42. Indeed, simple oxygen-containing aromatic products can be efficiently obtained, which can be used as useful intermediates for further diverse functionalization (Fig. 1b). Furthermore, various methodologies have been developed for the one-pot depolymerization and functionalization of lignin linkages to yield heteroatom-containing (N, S, and Si) aromatic molecules, which could increase the value of lignin valorization43. However, there have been no documented examples of the direct preparation of useful aryl halides from lignin, which is a fascinating and promising pathway. Owing to the complex three-dimensional structure and low reactivity of real lignin, most current methods for preparing aromatics from lignin are limited to lignin models, which are largely insufficient for practical applications with real lignin. In addition, increasing the reactivity and selectivity of halogenation is challenging due to the complex lignin structure. Thus, developing a highly efficient catalytic method, that can both depolymerize real lignin and halogenate the formed fragments to produce aryl halides is highly important and challenging.
Here, we describe depolymerization and halogenation of native lignin for the production of polysubstituted aryl bromides and chlorides (Fig. 1c). The reaction is initiated by the electrophilic attack of the phenyl rings of lignin by bromine or chorine cations from halogenation reagents, and the hydrogen bond activation of halogenation reagents by 2,2,2-trifluoroethanol (TFE) is key to their success. This work provides an efficient route for lignin valorization that involves the one-pot depolymerization and halogenation of native lignin under simple and mild conditions with readily/commercially available chlorine and bromine sources and selective and efficient C(sp2)–C(sp3) bond cleavage to produce useful lignin-derived aryl halides for diverse synthetic transformations.
Results
Screening conditions for depolymerization and halogenation
We hypothesized that the electron-rich aromatic motifs of lignin which are constructed of syringyl (S), guaiacyl (G), and para-hydroxy-phenyl (H) units, provide the possibility for electrophilic attack by electrophiles such as bromine and chorine cations. Subsequent bond cleavage could promote the depolymerization of lignin and afford halogenated monomers (Fig. 2a). To survey the aforementioned hypothesis, we initially employed N-bromosuccinimide (NBS), which is a commercially available and commonly used electrophilic bromination reagent, as the cut-off and functionalization tool to address lignin β-O-4 linkages (Fig. 2b). Treating lignin model 1a with NBS at room temperature in normal solvents such as acetone, 1,4-dioxane (Fig. 2b, entries 1 and 2), ethyl acetate, and tetrahydrofuran did not disrupt the bonds, and no target products were obtained (Supplementary Table 1). This could be attributable to the inefficient electrophilicity of the Br+ of NBS in the reaction system, leading to unsuccessful electrophilic attack of the phenyl rings. To increase the electrophilicity of Br+, TFE was introduced into the reaction as a hydrogen bond activator44. As expected, when 0.2 mL of TFE in 1.8 mL of acetone was added, the depolymerization successfully yielded dibrominated products 2a and 3a (Fig. 2b, entry 3). Increasing the dosage of TFE further promoted the reaction (Fig. 2b, entries 4 and 5). Notably, yields of 99% for 2a and 61% for 3a were obtained (Fig. 2b, entry 6) when TFE was used as the sole solvent, indicating efficient cleavage of the C(sp2)–C(sp3) bond with excellent selectivity. Other bromination reagents, such as N-bromophthalimide (NBP) and 1,3-dibromo-5,5-dimethylhydantoin (DBDMH), also performed well in providing brominated products with slight decreases of yields (Fig. 2b, entries 7 and 8). Moreover, dibromoisocyanuric acid (DBI) showed poor reactivity for this reaction, and only a 5% yield of 2a was obtained (Fig. 2b, entry 9). Notably, in entries 3-9, phenol product 4a was observed in low yields (7-16%), which was derived from the decomposition of 3a. Compared with strong acids such as Brønsted or Lewis acids, which exhibit a strong ability to active hydrogen bonding, TFE is a mild reagent that has excellent system tolerance and is an excellent hydrogen bond donor. In addition, TFE has a good dissolving capacity due to the effect of florin atoms, which can contribute to the dissolution of the native lignin.

a Initial considerations for the depolymerization and halogenation of lignin. b Optimization of the conditions for depolymerization and bromination. Reaction conditions: 1a (0.2 mmol), [Br+] reagent (0.9 mmol), and solvent (2 mL) at room temperature for 12 h. c Optimization of conditions for depolymerization and chlorination. Reaction conditions: 1a (0.2 mmol), [Cl+] reagent (0.3 mmol), and solvent (2 mL) at room temperature (r.t., ~25 °C) for 12 h. 8a 3,4-dichlorinated product. 9a 2,3,4,5-tetrachlorinated product. 10a 4,5-dichloro-2-methoxyphenol. 11a 3,4,5-trichloro-2-methoxyphenol. 12a 2,3,4,5-tetrachloro-6-methoxyphenol. The yields of 2a and 5a-9a were determined by 1H nuclear magnetic resonance (NMR) spectra with CH2Br2 as an internal standard, the yields of 3a, 4a, and 10a-12a were detected via GC-MS analysis of the unpurified reaction mixture with n-octadecane and n-dodecane as an internal standard, respectively.
The depolymerization and chlorination reactions of 1a were also investigated, and the results are shown in Fig. 2c. The chlorination reagents (1.5 equiv.) were screened using TFE as the solvent at room temperature. N-Chlorosuccinimide (NCS), 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), and sodium dichloroisocyanurate (Na-DCC) showed poor reactivity for 1a depolymerization and very low yields of dichlorinated product 5a were obtained (Fig. 2c, entries 1-3). When trichloroisocyanuric acid (TCCA), which is a versatile and efficient reagent that is used in chlorination reactions due to its excellent performance for the release of active chlorine cations, was employed in the reaction, the cleavage of 1a produced 5a and 8a in 48% and 32% yields, respectively (Fig. 2c, entry 4). Moreover, trichlorinated product 6a was found in the reaction, which was probably derived from the overchlorination of primary depolymerization products. In common with bromination, phenol products 10a-12a with different extent of chlorination were also observed (Fig. 2c, entries 5-8), which were derived from the decomposition of 8a or 9a. Owing to the strong electrophilic effect of TCCA, several reaction conditions were screened to control the degree of chlorination. The use of a mixed solvent of TFE/methyl acetate (1/1, v/v) slightly increased the yield of 5a (58%) (Fig. 2c, entry 5). Increasing the amount of TCCA to 2.5 equiv. promoted the formation of the trichlorinated product 6a (Fig. 2c, entry 6). When the same TCCA dosage as that used in entry 6 was used, complete chlorination occurred in the sole solvent TFE, providing the tetrachlorinated product 7a and a mixture of di-/tetrachlorinated products 8a/9a in 78% and 24 % yields, respectively (Fig. 2c, entry 7). A further increase in TCCA (4 equiv.) afforded the tetra-chlorinated product 7a in a 92% yield. Thus, the depolymerization and chlorination of lignin β-O-4 models could be controlled by changing the reaction conditions. Notably, both Br+ and Cl+ exhibited high selectivity for C(sp2)–C(sp3) bond cleavage in the lignin β-O-4 models.
Depolymerization and halogenation of lignin β-O-4 models
Under optimal reaction conditions, the scope of lignin β-O-4 models was initially investigated for depolymerization and halogenation. As the methoxyl substituent can considerably change the properties of lignin models, which can directly affect the halogenation process, we investigated the reactivity of β-O-4 models containing syringyl (S), guaiacyl (G), and p-hydroxy-phenyl (H) units (Fig. 3). For the depolymerization and bromination reactions, substrates with a G-type (left) and a methoxyl substituent (ortho-position, right) were successfully depolymerized and brominated (1a and 1c), yielding dibrominated products 2a, 3a, 3c, and 4a in good to excellent yields. Substrates 1b and 1d also performed well and afforded 2a in high yields. Notably, the right parts of 1b and 1d could be transformed into mono-brominated products 3b, 3d, and 4b, which might be attributable to the low reactivity of bromo-substituted phenyl rings for further bromination. For the H-type models, 1e and 1f could smoothly afford mono-brominated products (2b, 3e, and 4c) and dibrominated product (3c and 4a). The use of electron-rich S-type models 1g and 1h as substrates led to tribrominated product 2c and corresponding mono-brominated products (3b and 4b) and dibrominated product (3a and 4a). Thus, for this method, the degree of bromination could be controlled by the electronic properties of the lignin models. For depolymerization and chlorination reactions, various models exhibit high reactivity for depolymerization. The remaining part (G-type) of 1a-1d could be completely transformed into tetra-chlorinated product 7a in good to excellent yields (62-92%). However, owing to the strong electrophilic ability of Cl+, mixed products from the right part of the 1a-1d were found to have different degrees of chlorination (Supplementary Figs. 4-7). This method was also adapted for lignin model 1i containing a hydroxyl group, affording corresponding halogenated products in moderate yields.

aReaction conditions: 1 (0.2 mmol), NBS (0.9 mmol), TFE (2 mL), room temperature, and 12 h. bReaction conditions: 1 (0.2 mmol), TCCA (0.8 mmol), TFE (2 mL), room temperature, and 12 h. Isolated yields. 4b 4-bromophenol. 4c: 3-bromo-4-methoxyphenol.
Mechanistic investigations
To provide insights into the mechanism of this depolymerization and halogenation reaction, the activation effect of TFE was initially examined (Fig. 4a). The 1H and 13C NMR shifts of NBS with the addition of TFE were studied. The 1H NMR signal of the methylene group in NBS shifted to a lower field in the presence of TFE, indicating that the electron density of the methylene group decreased. In addition, the 13C NMR signal of the carbonyl group in NBS shifted to a lower field with the addition of TFE, revealing that the electron density of the carbonyl group decreased. A natural population analysis (NPA) for the electron density of NBS reveals that more positive charge distributes on the Br atom of TFE-activated NBS, which can explain the higher electrophilic reactivity (Supplementary Fig. 28). Next, the reactivity of lignin linkages with different electron properties was tested to investigate the behavior of bond cleavage by the electrophilic attack of halogen cations (Fig. 4b). Substrates with no substituent on the phenyl rings (1j and 1k) or with a para-fluoro substituent (1l) exhibited inert reactivity, indicating that an electron-rich aromatic ring is crucial for the reaction. The reaction was also ineffective in oxidizing lignin models 1 m containing a Cα=O group (Fig. 4c). Methyl-protected lignin model 1n was used under standard reaction conditions, and depolymerization yielded brominated product 2b in a 56% yield, indicating that the hydroxyl group at the α position of the substrate is not necessary in this reaction (Fig. 4d). Notably, acetal product 3f was obtained in a 45% yield, which was formed via the capture of carbocation intermediate by TFE. This result could prove the cleavage of C(sp2)–C(sp3) bond and the formation of carbocation intermediate. Moreover, the formation of polysubstituted aryl halides was confirmed by the reaction of mono-substituted 3,4-dimethoxy-1-chlorobenzene (13) and 3,4-dimethoxy-1-bromobenzene (14) under standard conditions (Fig. 4e). The reactivity of polybromination reactions was explored using mono-/di-/trimethoxyl-substituted bromobenzene (2a, 2b, and 15) as substrates under standard bromination reaction conditions (Fig. 4f). The result shows that 2a and 2b exhibited inert reactivities for further bromination, while trimethoxyl-substituted substrate 15 successfully produced tribrominated product 2c. Notably, the result of bromination reactions for substrates with S-, G-, and H-type shows that the directing effect of methoxy group play an essential role for selective bromination, yielding brominated products with excellent para-selectivity (Fig. 3). Hence, the electronic property of substrates with S-, G-, and H-type directly affects the degree of overbromination and the directing effect of methoxyl group controls the site selectivity of bromination. Halogen cation reagents (N–X, where X = Cl, Br, or I) can produce nitrogen and halogen radicals45, possibly triggering a radical-involving pathway. In this work, the addition of the free-radical scavenger 2,2,6,6-tetramethylpiperidinooxy (TEMPO) did not quench the depolymerization and halogenation reaction, indicating that a radical pathway can be excluded (Fig. 4g). To explore more details, reaction of 1b under standard bromination conditions was detected within 5 min. Dibromination of substrate 1b could be observed via the detection of 1o and monosubstituted product 14 was also formed via the C(sp2)–C(sp3) bond cleavage of 1b (Fig. 4h). This result indicates that the polyhalogenation and the carbon-carbon bond cleavage occurred simultaneously. Piecing together the above details and the reported relative mechanism46, a possible reaction mechanism is proposed in Fig. 4i. According to this mechanism, the halogenation reagent is initially activated by TFE via hydrogen bond to generate a halogen cation, which subsequently performs a selective electrophilic attack on lignin model 1a to afford intermediate 16. Notably, a simultaneous electrophilic substitution of halogen cation to benzene rings also proceeds. The C–C bond cleavage of intermediate 16 yields polyhalogenated product 18 and cation intermediate 17. Further halogenation of compound 18 produces the final polyhalogenated product 2a, 6a or 7a. The cation intermediate 17 is transformed into aldehyde 19, which subsequently undergoes halogenation to yield polyhalogenated product 3a, 8a or 9a.

a 1H and 13C NMR spectra study for halogenation reagent activation. b and c Reactivity of lignin linkages for research on bond cleavage. d Reaction of methyl protected lignin model for detection of intermediate. e Formation of polysubstituted aryl halides. f Interaction of electronic property of aromatics with reactivity of bromination. g Radical trapping experiments. e Proposed mechanism. h Bond cleavage and bromination research. i Proposed mechanism.
Depolymerization and halogenation of native lignin
After the lignin models were tested, we explored the application of this depolymerization and halogenation strategy in native lignin. Dioxasolv larch lignin was treated with TCCA as the cut-off and chlorination reagent in TFE at room temperature (25 °C), and a 0.311 wt% yield of tetrachlorinated product 12a was successfully obtained (Fig. 5a). Increasing the reaction temperature to enhance the depolymerization process was further studied and the yield of monomer 12a increased to 0.533 wt% at 100 °C. Considering the solubility of native lignin, mixed solvents were investigated to further increase the reaction efficiency. When the reaction was conducted in a mixed solvent of TFE and 1,4-dioxane (1:1, v/v), a 1.864 wt% yield of 12a was obtained. Methyl acetate (MA) and dimethyl sulfoxide (DMSO) were also effective in this process when used as part of the mixed solvent, and decreased yields were obtained (0.309 and 0.12 wt%). Notably, increasing the dosage of TCCA significantly increased the yield of the target tetrachlorinated product 12a, resulting in a 2.691 wt% yield when 4 mmol of TCCA was used in the reaction. The depolymerization and bromination of dioxasolv larch lignin were investigated under these conditions. As shown in Fig. 5b, increasing the reaction temperature had a negative effect on the reaction of lignin with NBS, and the highest yield of 0.177 wt% for dibrominated product 4a was obtained. Unfortunately, further screening of the solvent, dosage of NBS, or species of Br+ reagent failed to increase the yield of the depolymerization and bromination process.

a Screening conditions for depolymerization and chlorination. b Screening conditions for depolymerization and bromination. Larch lignin (50 mg) was used. c Standard process for separation and purification. d Depolymerization and halogenation of various lignins. aReaction conditions: lignin (100 mg), NBS (6 mmol), TFE (10 mL), room temperature (25 °C), and 72 h. bReaction conditions: lignin (100 mg), TCCA (8 mmol), TFE/1,4-dioxane (1:1, 10 mL), 100 °C, and 72 h. Products were analysed by GC-MS.
Under the optimized conditions and directed by the standard process for separation and purification (Fig. 5c), various lignins from softwoods (larch, Scots pine, Korean pine, and Chinese fir) and hard woods (poplar, birch, and beechwood) were used to test the developed method for the production of aryl halides. From the perspective of structural analysis, G-type units are the main components of lignin from soft woods, whereas in hard woods, both S- and G-type units exist. First, the reaction of lignin with NBS was investigated (Fig. 5d). Lignin from soft woods such as larch, Scots pine, and Korean pine exhibited good depolymerization performance and 38.4-47.2 wt% lignin was degraded, yielding 0.237-0.329 wt% brominated product 4a. Chinese fir lignin showed lower reactivity for this transformation, affording 0.15 wt% target product with 73.2 wt% insoluble fraction. Compared with softwood-derived lignin, for lignin from hardwoods such as poplar, birch, and beechwood, a similar degree of degradation was achieved (40.4-47.3 wt%). The total yields of di- and tribrominated products 4a and 20 were 0.452, 0.319, and 0.339 wt%, respectively. For depolymerization and chlorination reactions, both softwood- and hardwood-derived lignin exhibited higher degradation efficiencies than depolymerization and bromination of lignin did, and only 1.6-10.7 wt% of the insoluble fraction was recovered; this might be attributed to the higher reactivity of Cl+, which has a stronger interaction with lignin than Br+ does. Lignin from softwoods produced tetrachlorinated product 12a in 0.914-2.497 wt% yields. Moreover, lignin derived from hardwoods of poplar, birch, and beechwood generated tetra- and trichlorinated products 12a and 21 in total yields of 1.821, 1.04, and 1.398 wt%, respectively. In addition, the gram scale experiments for depolymerization and halogenation of larch lignin were performed and showed higher efficiency than that of 100 mg scale. The result shows that 6.2 mg/g (0.62 wt%) and 35.7 mg/g (3.57 wt%) of dibrominated product 4a and tetrachlorinated product 12a were obtained using 1 g larch lignin as substrate, respectively, exhibiting potential practicability for producing aryl halides from lignin. Notably, the solvent and the residues (succinimide and cyanuric acid) from NBS and TCCA could be recollected by rotary evaporation and extraction, respectively (Supplementary Figs. 29 and 30).
The depolymerization and halogenation processes were also investigated via heteronuclear single quantum coherence spectroscopy (HSQC) NMR and gel permeation chromatography (GPC) experiments. The β-O-4, β-β, and β-5 linkages could be clearly identified by HSQC spectrum of dioxasolv birch lignin (Fig. 6a). The HSQC analysis of the soluble fraction after depolymerization and halogenation suggested a significant degree of depolymerization, as almost all the signals of Cα–H, Cβ–H, and Cγ–H of these three linkages were diminished (Fig. 6a). In dioxasolv larch lignin, β-O-4 and β-5 linkages could be observed via HSQC analysis (Fig. 6b). The disappearance of the Aα, Aβ, Aγ, Cα, Cβ, and Cγ signals revealed by HSQC spectra also confirmed a significant depolymerization process using our method (Fig. 6b). Moreover, compared to the original lignin from Birch and larch, GPC analysis of the soluble fraction after the reaction indicated a notable decrease in the molecular weight (MW) of the depolymerized lignin, and lower-MW oligomers and polymers could also be detected (Supplementary Figs. 19-26).

a Birch lignin before and after depolymerization and halogenation. b Larch lignin before and after depolymerization and halogenation.
Synthetic application of lignin-derived aryl halides
As mentioned in the introduction, aryl halides are useful precursors and building blocks for chemical production, and further diverse synthetic transformations of lignin-derived dibromo- (Fig. 7a) or tetrachloro-substituted (Fig. 7b) aromatics were investigated to demonstrate their utility. First, the demethylation of dibrominated product 4a from lignin could form 4,5-dibromobenzene-1,2-diol 22, which could further afford heterocycle dioxine 23 and polymer precursor phenazine 24 via the transformation of the phenolic hydroxyl group. Then, cascade cyclization reactions of methylated product 2a could generate heterocyclic motifs 25-28. A two-step Heck reaction of 2a with methyl acrylate could afford 1,2-phenylene product 29, which is the starting material for the synthesis of the bioactive natural products scocycamide47. Further diarylation and diamination of 2a could also be realized through Suzuki (30) and Buchwald-Hartwig (31) cross-coupling reactions. Notably, dialkynylated, diborated, and dicyanated products 32-34, which are widely used as precursors for the synthesis of organic semiconductors48, fused aromatics49, and conjugated metal-organic frameworks (MOFs)50, could be synthesized from dibromo product 2a. Moreover, tetrachlorinated product 12a could be converted into 3,4,5,6-tetrachlorobenzene-1,2-diol 35, which can be further used for catalyst synthesis51. The simple oxidation of 35 could produce tetrachloro-o-benzoquinone 36, which has been widely applied for the preparation of metal ion sensors52, organic semiconductors53, Lewis acid catalysts54, and chirality sensors55. These results demonstrate the robust utility and synthetic applications of aryl halides from lignin.

a Synthetic transformation of dibrominated product 4a. b Synthetic transformation of tetrachlorinated product 12a. Compounds 4a and 12a were obtained from commercial sources. DCM: dichloromethane. DMF: N,N-dimethylformamide. THF: tetrahydrofuran. Ts: p-toluenesulfonyl. DMEDA: N,N’-dimethyl-1,2-ethanediamine. DIEA: N,N-diisopropylethylamine. TEA: triethylamine. DMA: N,N-dimethylaniline. IPr·HCl: 1,3-bis (2,6-diisopropylphenyl) imidazolium chloride. dppf: 1,1’-bis(diphenylphosphino)ferrocene. B2(pin)2: bis(pinacolato)diboron.
Discussion
In summary, we have developed an efficient and mild method for the depolymerization and halogenation of lignin into useful aryl halide compounds. Various wood-derived lignins can be converted into polybrominated and polychlorinated aromatics, which are useful precursors for diversiform synthetic applications in the preparation of functional molecules. The hydrogen bond activation of halogenation reagents by TFE is crucial for enhancing the reactivity of halonium ions (Br+ and Cl+). The electrophilic attack of halonium ions on the aromatic ring of lignin initiates the breakage of lignin linkages and further facilitates halogenation. This work represents a significant advancement in the development of simple and efficient protocols for lignin depolymerization and the production of value-added aromatics from lignin.
Methods
General procedure for depolymerization and bromination of lignin
A 25 mL sealed tube was charged with extracted native lignin (100 mg), NBS (6.0 mmol, 1.0678 g), and TFE (10 mL) under air atmosphere. The reaction was stirred for 72 h at room temperature. After the reaction is complete, the mixture was centrifuged with ethyl acetate (20 mL × 3) to separate the solid and liquid phases. The solid-phase fraction was stirred in 100 mL of water for 3 h to remove residues from the bromination reagent, and the insoluble fraction was collected by Brinell funnel filtration before drying and weighing. The liquid phase was concentrated in a vacuum and diluted with 30 mL of ethyl acetate, then extracted with water (50 mL × 3) to remove bromination reagent residues from the solution. The organic phase was collected and dried with anhydrous sodium sulfate, then concentrated in a vacuum to quantify the product by GC-MS.
General procedure for depolymerization and chlorination of lignin
A 25 mL sealed tube was charged with extracted native lignin (100 mg), TCCA (8.0 mmol, 1.8592 g) and TFE/1,4-dioxane (1/1, 10 mL) under air atmosphere. The reaction was stirred for 72 h at 100 °C. After the reaction is complete, the mixture is centrifuged with ethyl acetate (20 mL × 3) to separate the solid and liquid phases. The solid-phase fraction was stirred in 100 mL of water for 3 h to remove residues from the chlorination reagent, and the insoluble fraction was collected by Brinell funnel filtration before drying and weighing. The liquid phase was concentrated in a vacuum and diluted with 30 mL of ethyl acetate, then extracted with water (50 mL × 3) to remove chlorination reagent residues from the solution. The organic phase was collected and dried with anhydrous sodium sulfate, then concentrated in a vacuum to quantify the product by GC-MS.
Data availability
Data generated in this study are provided in the main text and Supplementary Information files. The general information, optimization of reaction conditions, experimental procedures, and characterization of all compounds are provided in the Supplementary Information. All other data supporting the findings of this study are available within the paper and its Supplementary Information, or from the corresponding author upon request. Source data are provided with this paper.
References
Organic Bromine and Iodine Compounds. In Handbook of Environmental Chemistry, (ed Neilson, A. H.) (Springer, Heidelberg, Berlin, 2003).
Ruiz-Castillo, P. & Buchwald, S. L. Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 116, 12564–12649 (2016).
Bunz, U. H. F. & Freudenberg, J. N-heteroacenes and N-heteroarenes as N-nanocarbon segments. Acc. Chem. Res. 52, 1575–1587 (2019).
Bunz, U. H. F., Seehafer, K., Bender, M. & Porz, M. Poly(aryleneethynylene)s (PAE) as paradigmatic sensor cores. Chem. Soc. Rev. 44, 4322–4336 (2015).
Freudenberg, J., Jänsch, D., Hinkel, F. & Bunz, U. H. F. Immobilization strategies for organic semiconducting conjugated polymers. Chem. Rev. 118, 5598–5689 (2018).
Lee, K.-W. et al. Organic optoelectronic materials: a rising star of bioimaging and phototherapy. Adv. Mater. 36, 2306492 (2024).
Zhang, C. & Zhu, X. Thieno[3,4-b]thiophene-based novel small-molecule optoelectronic materials. Acc. Chem. Res. 50, 1342–1350 (2017).
Liu, Y., Li, C., Ren, Z., Yan, S. & Rryce, M. R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 3, 18020 (2018).
Ding, L. et al. Polymer semiconductors: synthesis, processing, and applications. Chem. Rev. 123, 7421–7497 (2023).
Tang, M. L. & Bao, Z. Halogenated materials as organic semiconductors. Chem. Mater. 23, 446–455 (2011).
Wang, J., Xue, P., Jiang, Y., Huo, Y. & Zhan, X. The principles, design and applications of fused-ring electron acceptors. Nat. Rev. Chem. 6, 614–634 (2022).
Petrone, D. A., Ye, J. & Lautens, M. Modern transition-metal-catalyzed carbon-halogen bond formation. Chem. Rev. 116, 8003–8104 (2016).
Yoffe, D. et al. Bromine Compounds. In Ullmann’s Encyclopedia of Industrial Chemistry 7th edn, (ed Ley, C.) (John Wiley-VCH, 2013).
Larock, R. C. & Zhang L. Aromatic halogenation. In Comprehensive Organic Transformations: A Guide to Functional Group Preparations 3rd edn, (ed Larock, R. C.) (John Wiley & Sons, Inc., 2018).
de Meijere, A., Brase, S. & Oestreich M. (eds) Metal Catalyzed Cross-Coupling Reactions and More 1st edn, (Wiley-VCH, 2013).
Garst, J. F. & Ungváry, F. In Grignard Reagents: New Developments, (ed Richey, H. G.) (Wiley-VCH, 2000)
Smith, B. R., Eastman, C. M. & Njardarson, J. T. Beyond C, H, O, and N! Analysis of the elemental composition of U.S. FDA approved drug architectures. J. Med. Chem. 57, 9764–9773 (2014).
Hernandes, M. Z., Cavalcanti, S. M., Moreira, D. R., de Azevedo Junior, W. F. & Leite, A. C. Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr. Drug Targets 11, 303–314 (2010).
Kambe, N., Iwasakia, T. & Terao, J. Pd-catalyzed cross-coupling reactions of alkyl halides. Chem. Soc. Rev. 40, 4937–4947 (2011).
Wan, X., Li, C., Zhang, M. & Chen, Y. Acceptor-donor-acceptor type molecules for high performance organic photovoltaics-chemistry and mechanism. Chem. Soc. Rev. 49, 2828–2842 (2020).
Saikia, I., Borah, A. J. & Phukan, P. Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev. 116, 6837–7042 (2016).
Cavallo, G. et al. The halogen bond. Chem. Rev. 116, 2478–2601 (2016).
Song, S. et al. DMSO-catalysed late-stage chlorination of (hetero)arenes. Nat. Catal. 3, 107–115 (2020).
Davis, R. J. & Derouane, E. G. A non-porous supported-platinum catalyst for aromatization of n-hexane. Nature 349, 313–315 (1991).
Squires, A. M. Chemicals from coal. Science 191, 689–700 (1976).
Mochida, I., Okuma, O. & Yoon, S.-H. Chemicals from direct coal liquefaction. Chem. Rev. 114, 1637–1672 (2014).
Chow, J., Kopp, R. J. & Portney, P. R. Energy resources and global development. Science 302, 1528–1531 (2003).
Huber, G. W., Iborra, S. & Corma, A. Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem. Rev. 106, 4044–4098 (2006).
Corma, A., Iborra, S. & Velty, A. Chemical routes for the trans-formation of biomass into chemicals. Chem. Rev. 107, 2411–2502 (2007).
Zakzeski, J., Bruijnincx, P. C. A., Jongerius, A. L. & Weckhuysen, B. M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 110, 3552–3599 (2010).
Meng, Q. et al. Sustainable production of benzene from lignin. Nat. Commun. 12, 4534 (2021).
Shao, Y. et al. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 8, 16104 (2017).
Liu, M. & Dyson, P. J. Direct conversion of lignin to functionalized diaryl ethers via oxidative cross-coupling. Nat. Commun. 14, 2830 (2023).
Subbotina, E. et al. Oxidative cleavage of C–C bonds in lignin. Nat. Chem. 13, 1118–1125 (2021).
Rahimi, A., Ulbrich, A., Coon, J. & Shannon, S. S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 515, 249–252 (2014).
Tong, X., Zheng, J., Xue, S. & Guo, S. High-entropy materials as the catalysts for valorization of biomass and biomass-derived platform compounds. ACS Sustain. Chem. Eng. 11, 10203–10218 (2023).
Lim, S. H. et al. Effects of alkoxy groups on arene rings of lignin β-O-4 model compounds on the efficiencies of single electron transfer- promoted photochemical and enzymatic C–C bond cleavage reactions. J. Org. Chem. 78, 9431–9443 (2013).
Shuai, L. et al. Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization. Science 354, 329–333 (2016).
Li, N. et al. Selective lignin arylation for biomass fractionation and benign bisphenols. Nature 630, 381–386 (2024).
Liu, M., Sun, Z. & Elangovan, S. Bioactive molecules from lignin via homogeneous and heterogeneous catalytic pathways. Trends Chem. 5, 713–716 (2023).
Dong, L. et al. Sustainable production of dopamine hydrochloride from softwood lignin. Nat. Commun. 14, 4996 (2023).
Wu, X. et al. Solar energy-driven lignin-first approach to full utilization of lignocellulosic biomass under mild conditions. Nat. Catal. 1, 772–780 (2018).
Li, H., Bunrit, A., Li, N. & Wang, F. Heteroatom-participated lignin cleavage to functionalized aromatics. Chem. Soc. Rev. 49, 3748–3763 (2020).
Tang, R.-J., Milcent, T. & Crousse, B. Regioselective halogenation of arenes and heterocycles in hexafluoroisopropanol. J. Org. Chem. 83, 930–938 (2018).
Jiang, M., Wei, Y. & Shi, M. Acetoxylation and hydroxylation of diarylmethylenecycloalkanes via radical approach. J. Org. Chem. 75, 2528 (2010).
Shibata, A. et al. Dehydroxymethyl bromination of alkoxybenzyl alcohols by using a hypervalent iodine reagent and lithium bromide. Synlett 29, 2275–2278 (2018).
Wang, J. X. et al. Scocycamides, a pair of macrocyclic dicaffeoylspermidines with butyrylcholinesterase inhibition and antioxidation activity from the roots of Scopolia tangutica. Org. Lett. 22, 8240–8244 (2020).
Maier, S., Hippchen, N., Rominger, F., Freudenberg, J. & Bunz, U. H. F. Cyclodimers and cyclotrimers of 2,3-bisalkynylated anthracenes, phenazines and diazatetracenes. Chem. Eur. J. 27, 16320–16324 (2021).
Shimizu, M., Tomioka, Y., Nagao, I., kadowaki, T. & Hiyama, T. Palladium-catalyzed annulation of 1,2-diborylalkenes and -arenes with 1-bromo-2-[(Z)-2-bromoethenyl]arenes: a modular approach to multisubstituted naphthalenes and fused phenanthrenes. Chem. Asian J. 7, 1644–1651 (2012).
Meng, Z., Luo, J., Li, W. & Mirica, K. A. Hierarchical tuning of the performance of electrochemical carbon dioxide reduction using conductive two-dimensional metallophthalocyanine based metal-organic frameworks. J. Am. Chem. Soc. 142, 21656–21669 (2020).
Maki, T., Ishihara, K. & Yamamoto, H. 4,5,6,7-Tetrachlorobenzo[d][1,3,2]dioxaborol-2-ol as an effective catalyst for the amide condensation of sterically demanding carboxylic acids. Org. Lett. 8, 1431–1434 (2006).
Bryant, J. J. et al. Alkynylated phenazines: synthesis, characterization, and metal-binding properties of their bis-triazolyl cycloadducts. J. Org. Chem. 77, 7479–7486 (2012).
Xu, X. et al. Synthesis, solution-processed thin film transistors and solid solutions of silylethynylated diazatetracenes. Chem. Commun. 50, 12828–12831 (2014).
Roth, D., Stirn, J., Stephan, D. W. & Greb, L. Lewis superacidic catecholato phosphonium ions: phosphorus-ligand cooperative C-H bond activation. J. Am. Chem. Soc. 143, 15845–15851 (2021).
Formen, J. S. S. K. & Wolf, C. Chiroptical switching and quantitative chirality sensing with (pseudo)halogenated quinones. Angew. Chem. Int. Ed. 60, 27031–27038 (2021).
Acknowledgements
This work was supported by the Natural Science Funding of Heilong Jiang province for Excellent Young Scholar (YQ2022C004, B.T.), the National Natural Science Foundation of China (22378056, B.T.) and the Fundamental Research Funds for the Central Universities (2572022BB01, B.T., 2572023CT06, S.Liu).
Author information
Authors and Affiliations
Contributions
B.T., S.Liu., and Z.C. conceived the work and designed the experiments. Y.Liu. designed and performed the laboratory experiments. Y.Liu., Y.Li. Z.H., and S.W. collected the data. Y.Liu., Y.Li. Z.H. S.W. C.M. W.L., and S.Li. analyzed the data and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fengxia Sun, Xingchen Yan, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Li, Y., He, Z. et al. Producing aryl halides from lignin. Nat Commun 16, 3673 (2025). https://doi.org/10.1038/s41467-025-59054-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59054-0
