
Abstract
Cells need to reprogram their metabolism to adapt to extracellular nutrient changes. The yeast histone acetyltransferase SAGA (Spt-Ada-Gcn5-acetyltransferase) has been reported to acetylate its subunit Ada3 and form homo-dimers to enhance its ability to acetylate nucleosomes and facilitate metabolic gene transcription. How cells transduce extracellular nutrient changes to SAGA structure and function changes remains unclear. Here, we found that SAGA is deacetylated by Rpd3L complex and uncover how its deacetylase activity is repressed by nutrient sensor protein kinase A (PKA). When sucrose is used as the sole carbon source, PKA catalytic subunit Tpk2 is activated, which phosphorylates Rpd3L catalytic subunit Rpd3 to inhibit its ability to deacetylate Ada3. Moreover, Tpk2 phosphorylates Rpd3L subunit Ash1, which specifically reduces the interaction between Rpd3L and SAGA. By phosphorylating both Rpd3 and Ash1, Tpk2 inhibits Rpd3L-mediated Ada3 deacetylation, which promotes SAGA dimerization, nucleosome acetylation and transcription of genes involved in sucrose utilization and tricarboxylate (TCA) cycle, resulting in metabolic shift from glycolysis to TCA cycle. Most importantly, PKA phosphorylates HDAC1, the Rpd3 homolog in mammals to repress its deacetylase activity, promote TCA cycle gene transcription and facilitate cell growth. Our work hence reveals a conserved role of PKA in regulating Rpd3/HDAC1 and metabolic adaptation.
Similar content being viewed by others


Introduction
To maintain proper cell homeostasis and growth, cells need to tune their metabolic output in response to nutritional changes or metabolic stress in the environment1,2. For example, the availability of nutrients in the tumor microenvironment is believed to fluctuate extensively during tumor development and progression3. Cancer cells couple nutrient changes to their metabolic outputs by regulating the expression of nutrient transporters and metabolic enzymes3. A better understanding of metabolic adaptation is critical for identifying potential anti-cancer targets4. Most epigenetic modifications are rapid, flexible, reversible, and can regulate metabolic gene transcription. The SAGA (Spt-Ada-Gcn5 acetyltransferase) complex is a transcriptional coactivator that activates the transcription of stress-responsive genes, i.e., SUC2 when sucrose is used as the sole carbon source5. SAGA is also a histone acetyltransferase (HAT) that regulates gene expression by acetylating nucleosomes6,7,8. We have previously reported an auto-regulatory mechanism of SAGA under metabolic stress conditions in Saccharomyces cerevisiae9. SAGA acetylates its subunit Ada3 at lysines 8, 14 and 182 and the acetylated Ada3 is bound by bromodomains within SAGA subunits Gcn5 and Spt7 to form SAGA homo-dimers9. Ada3 acetylation-dependent SAGA dimerization is enhanced when cells are grown under sucrose as the sole carbon source or phosphate starvation conditions9. Once dimerized, SAGA efficiently acetylates nucleosomes, facilitates gene transcription and promotes cell survival under stress conditions9. Hence, Ada3 acetylation-dependent SAGA dimerization is important for cells to respond to sucrose treatment, which provides a good model for studying the mechanism regulating metabolic adaptation. Further study showed that Ada3 is deacetylated by histone deacetylase Rpd39. The stress-induced increase of Ada3 acetylation could be primarily due to decreased Rpd3-mediated Ada3 deacetylation, as loss of Rpd3 makes Ada3 acetylation insensitive to sucrose treatment9. However, it remains unclear how cells sense extracellular sucrose signals and respond accordingly with alterations of these histone modifying enzymes.
Nutrient sensing and metabolic control are intimately linked4. Cells have evolved multiple signaling pathways to sense fluctuations in nutrient availability10. The extensively studied nutrient sensing molecules are AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR), and cyclic AMP (cAMP)-dependent protein kinase A (PKA)11. All these nutrient sensors are protein kinases, which transduce nutrient signals by phosphorylating their substrates. When energy supply is insufficient, AMP activates AMPK to reduce anabolism and promote catabolism to maintain energy homeostasis12. The yeast AMPK homolog is Snf1, which is activated under conditions of glucose limitation to maintain energy homeostasis and control responses to environmental stresses13. Cells sense the availability of amino acids and regulate protein synthesis via mTOR signaling pathway14. Activated mTOR phosphorylates p70-S6 kinase 1 and eukaryotic initiation factor 4E (4E-BP1), which promotes protein translation15. In Saccharomyces cerevisiae, TOR kinases assemble into two distinct complexes: TOR complex 1 (TORC1) and TOR complex 2 (TORC2). Only TORC1 is rapamycin sensitive and promotes anabolism and cell growth16. PKA is a tetramer composed of two catalytic subunits and two regulatory subunits. Upon binding of cAMP to PKA regulatory subunits, the PKA tetramer dissociates into free and active catalytic subunits, which exert their cellular functions by phosphorylating its target proteins17. The yeast PKA holoenzyme is composed of a regulatory subunit Bcy1 dimer and two catalytic subunits encoded by TPK1, TPK2 and TPK318. Upon binding of cAMP to Bcy1, the PKA tetramer dissociates into active Tpk catalytic subunits. These nutrient-sensing molecules can phosphorylate chromatin modifying enzymes to directly connect extracellular nutrient changes with chromatin modifications19. For example, AMPK phosphorylates DNA demethylase ten-eleven-translocation protein (TET2) at serine 99 (S99) to stabilize TET220. Increased glucose levels reduce AMPK-mediated phosphorylation of TET2-S99, which destabilizes TET2 and compromises its tumor suppressive function20.
As a histone deacetylase, Rpd3 has been reported to repress gene transcription by deacetylating histones21. In budding yeast, Rpd3 exists in three distinct complexes, Rpd3L, Rpd3S and Rpd3μ with different subunits and functions22. Rpd3L is recruited to gene promoters, where it deacetylates histones H3 and H4 to repress gene transcription21,23. We found that Rpd3L deacetylates non-histone proteins, such as histone variant H2A.Z and chromatin remodeling complex Ino80, to down-regulate autophagy-related gene expression24. In contrast, Rpd3S is recruited to gene bodies by RNA polymerase II25. Rpd3S represses cryptic transcription within gene bodies and maintains chromatin integrity26, while Rpd3μ is involved in resistance to oxidative stress27. Here, we identified Rpd3L as the primary Rpd3 complex that deacetylates Ada3 within SAGA and uncovered a new mechanism by which extracellular sucrose regulates the activity of Rpd3L and SAGA to stimulate an adaptive response. When cells are grown in sucrose as the sole carbon source, the PKA catalytic subunit Tpk2 is activated, which phosphorylates the catalytic subunit Rpd3 to directly inhibit its activity to deacetylate Ada3. Moreover, Tpk2 phosphorylates Rpd3L subunit Ash1 to disrupt the interaction between Rpd3L and SAGA, which further inhibits Rpd3L-mediated Ada3 deacetylation. Consequently, Tpk2-catalyzed Rpd3L phosphorylation promotes SAGA auto-acetylation and dimerization, which facilitates the transcription of enzymes involved in sucrose metabolism and TCA cycle. Most importantly, this phosphorylation-dependent repression of Rpd3 by Tpk2 is conserved in hepatocellular carcinoma cells to enhance TCA cycle and promote cell growth.
Results
Rpd3L complex deacetylates Ada3 and its deacetylase activity is repressed by sucrose treatment
Rpd3 exists in three distinct complexes, Rpd3L, Rpd3S and Rpd3μ with different subunits and functions in budding yeast22. To identify which Rpd3 complex(es) deacetylates Ada3 within SAGA complex, we examined the global levels of Ada3-K14 acetylation (Ada3-K14ac) and Ada3-K182 acetylation (Ada3-K182ac) in the deletion mutants of Rpd3L-, Rpd3S- and Rpd3μ-specific subunits24. Loss of Rpd3L-specific subunits (Ash1, Dep1, Cti6) increased global levels of Ada3-K14ac and Ada3-K182ac, whereas deletion of Rpd3S- or Rpd3μ-specific subunits had negligible effect (Fig. 1a). We also purified Rpd3 complex from WT (Wt Rpd3) and deletion mutant of Rpd3L-specific subunit Ash1 (Rpd3 ash1Δ) that contain TAP-tagged Rpd3 using tandem affinity purification (TAP)28. An in vitro deacetylase assay with purified Rpd3 complex and SAGA revealed that Wt Rpd3 complex reduced Ada3 acetylation much faster than Rpd3 ash1Δ complex (Fig. 1b), indicating that loss of Ash1 substantially reduced Rpd3-catalyzed Ada3 deacetylation. In contrast, loss of Rpd3S-specific subunit Rco1 had no effect on Rpd3-catalyzed Ada3 deacetylation (Supplementary Fig. 1a). Moreover, we purified Rpd3L complex and Rpd3S complex from TAP-tagged Rxt2 and TAP-tagged Rco1 cells28, respectively (Supplementary Fig. 1b). An in vitro deacetylase assay showed that Rpd3L reduced Ada3-K14ac and Ada3-K182ac to a lower level than Rpd3S (Fig. 1c), confirming that Ada3 within SAGA complex is primarily deacetylated by Rpd3L complex.

a Analysis of Ada3 acetylation in the deletion mutants of Rpd3L, Rpd3S, Rpd3μ by immunoblots with indicated antibodies. b Analysis of Ada3 deacetylase activity of Rpd3 complex from WT (Wt Rpd3 comp.) and ash1Δ mutant (Rpd3 ash1Δ comp.) using purified SAGA as the substrate. P-values refer to the statistical difference of Ada3-K14ac/Ada3 and Ada3-K182ac/Ada3 between Wt Rpd3 and Rpd3 ash1Δ mutant. c Rpd3L deacetylases Ada3 at a faster rate than Rpd3S using purified SAGA as the substrate. d Loss of Ash1 enhanced the interaction between Ada3-FLAG-containing SAGA and Ada3-Myc-containing SAGA as determined by Co-IP assay. e Analysis of Ada3 deacetylase activity of Rpd3L purified from cells grown in YP + 2% glucose (Rpd3L-Glu) and YP + 2% sucrose (Rpd3L-Suc), respectively. f WT and deletion mutants of Rpd3 complex were grown in YP + 2% sucrose and treated with 0.1% H2O2 or 5 M NaCl for 2 h. The survival percentage was calculated by counting the number of colonies formed on YPD agar. For (a–c, e, f), data represent means ± SE; n = 3 biological independent experiments; one-way ANOVA with Dunnett’s multiple comparison tests (a), two-way ANOVA with Šídák’s multiple comparisons tests (b, c, e), and two tailed unpaired t-tests (f) were used for statistical analysis. For (d), shown is the typical example of two biological independent experiments.
As Ada3 acetylation is required for SAGA dimerization9, we then examined the effect of Rpd3L on SAGA dimerization using the diploid strain (Ada3-FLAG/Ada3-Myc) that contains both Ada3-FLAG and Ada3-Myc. The Ada3-FLAG containing SAGA complex was specifically immunoprecipitated from the Ada3-FLAG/Ada3-Myc diploid strain by anti-FLAG beads. Myc-tagged Ada3 was co-IPed (Fig. 1d, lane 7) and loss of Rpd3L-specific subunit Ash1 enhanced the association between Ada3-FLAG and Ada3-Myc (Fig. 1d, lane 8 vs lane 7), which is consistent with increased Ada3 acetylation in ash1Δ mutant (Fig. 1a). In contrast, loss of Rpd3S-specific subunit Rco1 did not affect the interaction between Ada3-FLAG and Ada3-Myc (Supplementary Fig. 1c, lane 8 vs lane 7).
Ada3 acetylation is induced when cells are grown under stress conditions9. When cells are grown in medium supplemented with sucrose as the sole carbon source, the global level of Ada3 acetylation increases to activate the expression of genes that utilize sucrose, i.e., SUC29. We thus examined the activity of Rpd3L to deacetylate Ada3 when cells were grown in 2% glucose-containing medium and then switched to 2% sucrose-containing medium for 0–90 min. Sucrose treatment induced Ada3 acetylation in WT cells in a time-dependent manner, while Ada3 acetylation was relatively high and constant in ash1Δ mutant (Supplementary Fig. 1d). In contrast, sucrose treatment still induced Ada3 acetylation in rco1Δ mutant (Supplementary Fig. 1e). Moreover, we purified Rpd3L complex from cells when treated with 2% glucose (Rpd3L-Glu) and 2% sucrose (Rpd3L-Suc) for 0.5 h, respectively. An in vitro deacetylase assay revealed that Rpd3L-Glu had greater Ada3 deacetylase activity than Rpd3L-Suc (Fig. 1e), suggesting that the deacetylase activity of Rpd3L is repressed by sucrose treatment.
Ada3 acetylation has been reported to enhance cell resistance to stress conditions, such as oxidative stress caused by 0.1% H2O2 treatment and osmotic stress caused by 5 M NaCl treatment9. Loss of Rpd3L-specific subunits increased the percentage of surviving cells when treated with H2O2 and NaCl, while loss of Rpd3S-specific subunits or Rpd3μ-specific subunit had no effect on the percentage of surviving cells (Fig. 1f). These data suggest that Rpd3L deacetylates Ada3 to reduce SAGA dimerization and decrease cell resistance to stress conditions. Moreover, Rpd3L-mediated Ada3 deacetylation is repressed when cells are grown in sucrose-containing media.
Sucrose treatment inhibits Rpd3L-mediated Ada3 deacetylation via the Gpr1-Gpa2-Cyr1-Tpk2 pathway
To understand how Rpd3L-mediated Ada3 deacetylation is inhibited by sucrose treatment, we immunoprecipitated Rpd3 from Rpd3-FLAG cells and performed liquid chromatography-mass spectrometry (LC‒MS) analysis, which showed that Tpk2 was an Rpd3-interacting protein (Fig. 2a). LC‒MS analysis of immunoprecipitated Tpk2 also revealed the presence of Rpd3L subunits (Fig. 2a). The interaction between Rpd3 and Tpk2 was confirmed by co-immunoprecipitation (Co-IP) and reciprocal IP (Fig. 2b). Moreover, purified recombinant His-Tpk2 directly interacted with purified Rpd3L (Rxt2-CBP) complex (Fig. 2c).

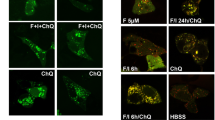
a IP-mass spectrometry analysis of proteins co-purified with Rpd3 and Tpk2. The endogenously expressed Rpd3-FLAG and Tpk2-FLAG were individually immunoprecipitated from cells with anti-FLAG beads. The identified unique peptides and sequence coverage were listed. b Tpk2 interacted with Rpd3 as determined by Co-IP and reciprocal IP. c Rpd3L interacted with purified recombinant His-Tpk2 as determined by in vitro Co-IP. d Loss of Tpk2 enhanced the interaction between endogenously expressed Ada3 and Rpd3. e Analysis of Ada3 acetylation in WT and tpk2Δ mutant when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. f Analysis of Ada3 deacetylase activity of Rpd3L purified from WT and tpk2Δ mutant. g The interaction between Tpk2 and Bcy1 was reduced when cells were shifted from YP + 2% glucose (Glu) to YP + 2% sucrose (Suc). h Diagram showing sucrose treatment activates Tpk2 via the Gpr1-Gpa2-cAMP pathway in budding yeast. i Analysis of intracellular cAMP levels in WT, gpr1∆ and gpa2∆ mutants when grown in YP, YP + 2% glucose (YP + Glu) or YP + 2% sucrose (YP + Suc) medium for 10 min. j, k Analysis of Ada3 acetylation in WT, gpr1∆ and gpa2∆ mutants when treated with YP + 2% glucose (YP + Glu) or YP + 2% sucrose (YP + Suc) for 0.5 h by immunoblots (j) and immunofluorescence (k). For Fig. k, cell nuclei was stained by DAPI (in blue). Ada3 acetylation was stained red with anti-Ada3-K14ac and anti-Ada3-K182ac antibodies. Anti-Ada3 antibody was used as a control. Shown are merged images. Scale bars, 5 μm. For (d–g, i, j), data represent means ± SE; n = 3 biological independent experiments; two tailed unpaired t-tests (d), one-way ANOVA with Tukey’s multiple comparison tests (g), two-way ANOVA with Tukey’s multiple comparisons tests (e, i, j) and Šídák’s multiple comparisons tests (f) were used for statistical analysis. For (b, c, k), shown is the typical example of at least two biological independent experiments.
Tpk2 is the catalytic subunit of protein kinase A (PKA). PKA is a tetrameric holoenzyme, which is composed of a regulatory subunit Bcy1 dimer and two catalytic subunits encoded by TPK1, TPK2 and TPK317,18. Loss of Tpk2 enhanced the interaction between Rpd3 and Ada3 (Fig. 2d, lane 3 vs lane 4). Moreover, loss of Tpk2 but not Tpk1 and Tpk3 abolished sucrose-induced Ada3-K14ac and Ada3-K182ac (Fig. 2e; Supplementary Fig. 2a). We also examined the effect of other potential nutrient-sensing molecules on sucrose-induced Ada3-K14ac and Ada3-K182ac, including TORC1, AMPK (Snf1), and Sch929,30. Inhibition of TORC1 by rapamycin (Rapa) or loss of Snf1 and Sch9 had negligible effects on sucrose-induced Ada3-K14ac and Ada3-K182ac (Supplementary Fig. 2b, c). These data indicate that Tpk2 is required for sucrose to induce Ada3 acetylation.
To examine the effect of Tpk2 on Rpd3L-mediated Ada3 deacetylation, we purified Rpd3L complex from WT (Wt Rpd3L) and tpk2Δ mutant (Rpd3L tpk2Δ) and then performed an in vitro deacetylase assay. Compared with Wt Rpd3L, Rpd3L tpk2Δ deacetylated Ada3 at a relatively faster rate as indicated by relatively lower Ada3-K14ac and Ada3-K182ac levels (Fig. 2f), suggesting that Tpk2 may inhibit Rpd3L-mediated Ada3 deacetylation.
PKA is activated by cyclic AMP (cAMP), which binds to its inhibitory subunit, Bcy1 and causes its dissociation17,18. Indeed, when cells were switched from glucose-containing medium to sucrose-containing medium, the intracellular cAMP was substantially increased (Supplementary Fig. 2d). With the increase of cAMP induced by sucrose treatment, the interaction between Bcy1 and Tpk2 was reduced but the interaction between Bcy1 and Tpk1/Tpk3 was unaffected (Fig. 2g; Supplementary Fig. 2e, f). The intracellular Tpk2 levels were greater than those of Tpk1 and Tpk3 (Supplementary Fig. 2g), which could explain why Tpk2 but not Tpk1 and Tpk3 are activated by sucrose. We then deleted the cAMP phosphodiesterases PDE1 and PDE2, which encode enzymes that hydrolyze cAMP and inactivate PKA31,32,33. As expected, the cAMP levels were significantly increased in pde1Δ and pde2Δ mutants (Supplementary Fig. 2d). Consequently, the global levels of Ada3-K14ac and Ada3-K182ac were elevated in pde1Δ and pde2Δ mutants and loss of Pde1 and Pde2 abrogated sucrose-induced Ada3-K14ac and Ada3-K182ac (Supplementary Fig. 2h).
cAMP is synthesized by adenylate cyclase (Cyr1), which can be stimulated by the small GTP-binding proteins Ras1 and Ras2 to form the Ras-cAMP-PKA signaling pathway34,35. However, sucrose treatment still induced Ada3-K14ac and Ada3-K182ac in ras1Δ and ras2Δ mutants (Supplementary Fig. 2i). The activity of Cyr1 can also be stimulated by glucose-sensing G-protein-coupled receptor (GPCR) system, which is composed of G-protein-coupled receptor Gpr1, G protein α subunit Gpa2, and its GTPase activating protein Rgs2, the so called Gpr1-Gpa2-Cyr1-Tpk2 pathway36 (Fig. 2h). Loss of Gpr1 and Gpa2 abrogated sucrose-induced increase of cAMP (Fig. 2i). Moreover, loss of Gpr1 and Gpa2 attenuated sucrose-induced Ada3-K14ac and Ada3-K182ac (Fig. 2j, k; Supplementary Fig. 2j). These results suggest that the Gpr1-Gpa2-Cyr1-Tpk2 pathway is required for sucrose to repress Rpd3L-mediated Ada3 deacetylation.
Tpk2 phosphorylates Rpd3 at serine 50 and serine 354
We next aimed to understand how Tpk2 represses Rpd3L’s activity to deacetylate Ada3. Tpk2 had no effect on Rpd3 expression and Rpd3L subunit composition (Supplementary Fig. 3a, b). As Tpk2 is a protein kinase, we examined whether Tpk2 could phosphorylate Rpd3L to inhibit its deacetylase activity. We purified Rpd3 complex from yeast cells when grown in glucose-containing medium and sucrose-containing medium, respectively, and then examined its phosphorylation status with pan-phosphoserine/threonine (Phos-Ser/Tyr) antibody. Immunoblot analysis showed that several Rpd3L subunits were phosphorylated, including Ash1, Cti6, Rpd3, Rxt2, Dep1, and Sds3 (Fig. 3a). We individually expressed and purified these proteins in E. coli and then performed an in vitro kinase assay with purified Tpk2-CBP. Tpk2-CBP can directly phosphorylate Rpd3 and Ash1 (Fig. 3b; Supplementary Fig. 3c–f). Bioinformatic analysis of Rpd3 sequence revealed five potential phosphorylation sites, S50, S123, S354, T394, and S408 (http://gps.biocuckoo.cn/). We thus individually mutated these residues to nonphosphorylatable alanine to construct Rpd3-S50A, Rpd3-S123A, Rpd3-S354A, Rpd3-T394A, and Rpd3-S408A mutants. Among these five mutants, the S50A and S354A mutations reduced Ada3-K14ac and Ada3-K182ac upon sucrose treatment (Supplementary Fig. 3g). To confirm that Tpk2 phosphorylates Rpd3 at S50 and S354, we developed antibodies against phospho-specific forms of these two sites, Rpd3-S50p and Rpd3-S354p with high specificity (Supplementary Fig. 3h). Immunoblot and immunofluorescence analysis with these two antibodies detected phosphorylated S50 and S354 in WT Rpd3 but not in Rpd3 double mutant on both S50 and S354 residues (Rpd3-S50A S354A, which we refer to hereafter as Rpd3-2SA) (Fig. 3c; Supplementary Fig. 3i).

a Rpd3 complex was purified from cells (Rpd3-TAP, Rpd3 tagged with protein A and CBP) when treated with YP + 2% glucose (Glu) and YP + 2% sucrose (Suc) for 0.5 h, respectively. After purification, the protein A tag of Rpd3-TAP was cleaved to produce Rpd3-CBP. The phosphorylation status of Rpd3 complex subunits were probed by immunoblots with pan-phosphoserine/threonine (Phos-Ser/Tyr) antibody. Rpd3-CBP was used as a loading control. b Purified Tpk2-CBP directly phosphorylated purified recombinant His-Rpd3 and His-Ash1 as determined by in vitro kinase assay. The phosphorylation was detected using pan-phos-Ser/Tyr antibody. c The intracellular Rpd3 was phosphorylated at S50 and S354. Rpd3 was immunoprecipitated from WT, Rpd3-S50A, Rpd3-S354A and Rpd3-2SA (Rpd3-S50A S354A) mutants. The phosphorylated Rpd3 was determined by immunoblots with anti-Rpd3-S50p and anti-Rpd3-S354p antibodies. d Analysis of Rpd3 phosphorylation in WT and tpk2∆ mutant. e Rpd3 was phosphorylated at S50 and S354 by purified Tpk2-CBP as determined by in vitro kinase assay. f–h Analysis of Rpd3 phosphorylation at S50 and S354 in WT, Rpd3-2SA and tpk2∆ mutants when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h as determined by immunofluorescence (f) and immunoblots (g, h). Scale bars, 5 μm. i Sucrose treatment enhanced Tpk2-catalyzed phosphorylation of Rpd3. Tpk2-CBP was purified from cells when treated with YP + 2% glucose (Glu) and YP + 2% sucrose (Suc), respectively. Tpk2 kinase activity was determined by in vitro kinase assay using purified His-Rpd3 as the substrate. For (f, i), data represent means ± SE; n = 3 biological independent experiments; two tailed unpaired t-tests (f) and two-way ANOVA with Šídák’s multiple comparisons tests (i) were used for statistical analysis. For (a–e, g, h), shown is the typical example of at least two biological independent experiments.
Loss of Tpk2 but not of Tpk1 or Tpk3 abolished the phosphorylation of Rpd3 at S50 and S354 (Fig. 3d; Supplementary Fig. 3j). An in vitro kinase assay with purified Tpk2-CBP and recombinant His-Rpd3 showed that Tpk2 phosphorylated His-Rpd3 at S50 and S354, while no phosphorylation was detected when His-Rpd3-2SA mutant was used as the substrate (Fig. 3e, lanes 5-7). These data indicate that Tpk2 directly phosphorylates Rpd3 at S50 and S354.
We then examined Tpk2-catalyzed Rpd3 phosphorylation in cells grown in media supplemented with 2% sucrose as the sole carbon source. Rpd3-S50 phosphorylation and Rpd3-S354 phosphorylation were increased by sucrose treatment in WT but not in Rpd3-2SA and tpk2Δ mutants (Fig. 3f–h). Deletion of GPA2 also abrogated sucrose-induced Rpd3 phosphorylation at S50 and S354 (Supplementary Fig. 3k). In addition, we purified Tpk2 from cells grown in glucose-containing and sucrose-containing medium, respectively. An in vitro kinase assay with purified recombinant His-Rpd3 revealed that Tpk2 purified from sucrose-treated cells (Tpk2-Suc) displayed greater activity to phosphorylate His-Rpd3 than Tpk2 purified from glucose-treated cells (Tpk2-Glu) (Fig. 3i), indicating that Tpk2 directly phosphorylates Rpd3 at S50 and S354 and its kinase activity is enhanced by sucrose treatment.
Tpk2 phosphorylates Rpd3 at S50 and S354 to inhibit its deacetylase activity
We next investigated the effect of Tpk2-mediated Rpd3 phosphorylation on its deacetylase activity. The global levels of Ada3-K14ac and Ada3-K182ac were reduced in Rpd3-2SA mutant and no increase was observed when cells were switched from glucose-containing to sucrose-containing medium (Fig. 4a). Immunofluorescence analysis revealed similar trend of Ada3-K14ac and Ada3-K182ac in Rpd3-2SA and tpk2Δ mutants (Fig. 4b). We also constructed Rpd3 phosphomimetic mutant, Rpd3-S50D S354D (Rpd3-2SD). The global levels of Ada3-K14ac and Ada3-K182ac were increased in Rpd3-2SD mutant and no increase was observed when cells were switched from glucose-containing medium to sucrose-containing medium (Fig. 4c; Supplementary Fig. 4a). Mutation of Rpd3-2SD rescued the reduced Ada3-K14ac and Ada3-K182ac in tpk2Δ mutant (Supplementary Fig. 4b, c).

a Analysis of Ada3 acetylation in WT and Rpd3-2SA mutant when treated with YP + 2% glucose (Glu) and YP + 2% sucrose (Suc) for 0.5 h. b Analysis of Ada3 acetylation in WT, Rpd3-2SA, and tpk2∆ mutants when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h by immunofluorescence. Scale bars, 5 μm. c Analysis of Ada3 acetylation in WT and Rpd3-2SD mutant when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. d Analysis of the effect of Tpk2-mediated Rpd3 phosphorylation on its activity to deacetylate Ada3. Rpd3-FLAG was immunoprecipitated from WT and Rpd3-2SA mutant by anti-FLAG beads. Wt Rpd3-FLAG and Rpd3-2SA-FLAG were phosphorylated by purified Tpk2-CBP for 1 h. Tpk2-CBP was then washed away and purified SAGA was added for in vitro deacetylase assay. e Analysis of the effect of Tpk2-catalyzed Rpd3 phosphorylation on His-Rpd3-mediated Ada3 deacetylation. The purified recombinant His-Rpd3 and His-Rpd3-2SA were phosphorylated by Tpk2-CBP as in Fig. 4d and then used for in vitro deacetylase assay with purified SAGA. f Analysis of the effect of Tpk2-catalyzed Rpd3 phosphorylation on Rpd3-catalyzed histone deacetylation. His-Rpd3 and His-Rpd3-2SA were phosphorylated by Tpk2-CBP and then used for in vitro histone deacetylation assay using nucleosomes as the substrate. For Fig. (a–f), data represent means ± SE; n = 3 biological independent experiments; two-way ANOVA with Tukey’s multiple comparisons tests (a–c) and Šídák’s multiple comparisons tests (d–f) were used for statistical analysis.
We then examined the effect of Tpk2-mediated Rpd3 phosphorylation on localization of Rpd3L complex by fluorescence microscopy of Rxt2-GFP cells. Mutation of Rpd3-2SA or Rpd3-2SD had no effect on nuclear localization of Rpd3L (Supplementary Fig. 4d). To examine the effect of Tpk2-mediated Rpd3 phosphorylation on Rpd3-catalyzed Ada3 deacetylation, we purified Rpd3 complex from yeast cells, which was treated with λ phosphatase to remove its phosphorylation and then used for an in vitro deacetylase assay. The dephosphorylated Rpd3 reduced Ada3-K14ac and Ada3-K182ac to lower levels than untreated Rpd3 (Supplementary Fig. 4e). To directly show that Tpk2 inhibits Rpd3-mediated Ada3 deacetylation by phosphorylating Rpd3, we immobilized the purified Rpd3-FLAG on anti-FLAG agarose beads, and then incubated it with purified Tpk2-CBP to phosphorylate Rpd3 (Fig. 4d). After phosphorylation, Tpk2 was then removed and the deacetylase activity of the bound Rpd3-FLAG was determined (Fig. 4d). An in vitro deacetylase assay revealed that the phosphorylated Rpd3 complex deacetylated Ada3 at a slower rate than unphosphorylated Rpd3 complex (Supplementary Fig. 4f). When purified Wt Rpd3-FLAG and Rpd3-2SA-FLAG were phosphorylated by Tpk2, Rpd3-2SA-FLAG reduced Ada3-K14ac and Ada3-K182ac to a lower level than Wt Rpd3-FLAG (Fig. 4d). We also purified recombinant His-Rpd3 and His-Rpd3-2SA from E. coli and performed an in vitro deacetylase assay. Tpk2-mediated phosphorylation of His-Rpd3 reduced its Ada3 deacetylase activity (Supplementary Fig. 4g). Tpk2-treated His-Rpd3-2SA displayed greater deacetylase activity than Tpk2-treated His-Rpd3 (Fig. 4e).
We next determined the effect of Tpk2-mediated phosphorylation of Rpd3 on its nucleosome deacetylase activity. Although there was no difference between the deacetylase activity of His-Rpd3 and His-Rpd3-2SA mutant before phosphorylation by Tpk2 (Supplementary Fig. 4h), Tpk2-treated His-Rpd3 displayed reduced nucleosome deacetylase activity than Tpk2-treated His-Rpd3-2SA mutant (Fig. 4f). All these data indicate that Tpk2 phosphorylates Rpd3 at S50 and S354 to inhibit its deacetylase activity.
Tpk2 phosphorylates Ash1 to reduce the interaction between Rpd3L and SAGA
As shown in Fig. 2d, loss of Tpk2 enhanced the interaction between Rpd3 and Ada3. The in vivo Co-IP assay indicated an interaction between Rpd3 and SAGA complex and this interaction was reduced by sucrose treatment (Fig. 5a, lane 3 vs lane 4). When Rpd3 was dephosphorylated by λ phosphatase, the interaction between Rpd3 and SAGA was enhanced (Supplementary Fig. 5a). When Rpd3 was pre-phosphorylated by Tpk2-CBP, this interaction was reduced (Supplementary Fig. 5b). An in vitro Co-IP revealed that there was a direct interaction between purified Rpd3L and SAGA (Fig. 5b). When Rpd3L was phosphorylated by Tpk2, the association between Rpd3L and SAGA was reduced (Fig. 5b, lane 5 vs lane 6). To determine whether Tpk2 reduces the interaction between Rpd3L and SAGA by phosphorylating Rpd3, we performed Co-IP with WT Rpd3 and Rpd3-2SA when cells were grown in glucose-containing medium and sucrose-containing medium, respectively. Surprisingly, sucrose treatment still reduced the interaction between Rpd3-2SA and Ada3 (Supplementary Fig. 5c). Consistently, mutation of Rpd3-2SA or Rpd3-2SD had no effect on co-localization of Rpd3L (Rxt2-GFP) and Ada3-tdTomato (Ada3 tagged with tdTomato) (Supplementary Fig. 5d), implying that Tpk2 inhibits the interaction between Rpd3L and SAGA independent on Rpd3 phosphorylation.

a Sucrose treatment reduced the interaction between Rpd3-FLAG and SAGA as determined by in vivo Co-IP. Rpd3-FLAG cells were treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. Rpd3-FLAG was immunoprecipitated with anti-FLAG beads and the co-IPed SAGA was detected with indicated antibodies. b Tpk2-catalyzed Rpd3L phosphorylation reduced its interaction with SAGA as determined by in vitro Co-IP. c The interaction between Rpd3-FLAG and Ada3 was reduced in sgf73∆, ubp8∆, cti6∆, rxt2∆, dep1∆, and ash1∆ mutants. d Summary for Co-IP results (c, Supplementary Fig. 5e–g) that determined the proteins required for Ada3-Rpd3 interaction. “+”: the subunit required for Ada3-Rpd3 interaction. “-”: the subunit not required for Ada3-Rpd3 interaction. “N/A”: as loss of Gcn5 reduced Ada3 level and loss of Spt7 compromised SAGA structure integrity, their contribution to the interaction between Ada3 and Rpd3 was unknown. e The purified recombinant Ubp8-FLAG directly interacted with purified recombinant His-Ash1 as determined by in vitro Co-IP. f The purified recombinant Ubp8-FLAG directly interacted with purified Rpd3L (Rxt2-CBP) as determined by in vitro Co-IP. g Ash1 and Ubp8 were required for sucrose to reduce the interaction between Rpd3 and Ada3. The endogenously expressed Rpd3-FLAG was immunoprecipitated from WT, ash1∆, and ubp8∆ mutants when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. h Ash1 and Ubp8 were required for sucrose to induce Ada3 acetylation as determined by immunofluorescence. Scale bars, 5 μm. i Analysis of Ada3 deacetylase activity of Rpd3L when Wt SAGA and SAGA ubp8∆ were used as the substrates. j Proposed model for Ubp8 and Ash1 act together to mediate the interaction between SAGA and Rpd3L and promote Rpd3-catalyzed Ada3 deacetylation. For (g, i), data represent means ± SE; n = 3 biological independent experiments; two-way ANOVA with Tukey’s multiple comparisons tests (g) and Šídák’s multiple comparisons tests (i) were used for statistical analysis. For (a–c, e, f, h) shown is the typical example of at least two biological independent experiments.
To elucidate how Tpk2 inhibits the interaction between Rpd3L and SAGA, we first characterized the subunits of Rpd3L and SAGA that mediate this interaction. We examined the interaction between Rpd3 and Ada3 in the deletion mutants of SAGA and Rpd3L by Co-IP. The histone H2B deubiquitylation (DUB) module in SAGA is required for the interaction between Rpd3 and Ada3 as loss of Sgf73 and Ubp8 reduced the Rpd3-Ada3 interaction (Fig. 5c, d). Other SAGA subunits, including Sgf29, Sgf11, Sus1, Ada1, Spt20, Spt8, and Spt3 had no effect on the interaction between Rpd3 and Ada3 (Supplementary Fig. 5e, f). As loss of Gcn5 reduced the global Ada3 protein level (Supplementary Fig. 5e) and loss of Spt7 has been reported to compromise SAGA structural integrity7, we cannot determine the direct effects of these proteins on Rpd3-Ada3 interaction. Deletion of Rpd3L subunits, including Cti6, Rxt2, Dep1, and Ash1 reduced the interaction between Rpd3 and Ada3 (Fig. 5c, d). Deletion of other Rpd3L subunits, including Sds3, Sin3, Rxt3, Pho23, Sap30, Ume6, and Ume1 had no effect on Rpd3-Ada3 interaction (Supplementary Fig. 5g). To determine the direct interaction between SAGA subunits (Sgf73, Ubp8) and Rpd3L subunits (Rxt2, Dep1, Ash1, Cti6), we individually expressed and purified these recombinant proteins in E. coli. By performing in vitro Co-IP with these purified proteins, we observed a direct interaction between purified recombinant Ubp8 and Ash1 but not other Rpd3L subunits (Fig. 5e; Supplementary Fig. 5h). The purified recombinant Sgf73 had no direct interaction with Rpd3L subunits (Supplementary Fig. 5i). The purified recombinant Ubp8 interacted with purified Rpd3L complex (Fig. 5f). Loss of Ash1 and Ubp8 reduced the interaction between Rpd3 and Ada3 and made this interaction insensitive to sucrose treatment (Fig. 5g). In agreement with the trend of interaction between Rpd3 and Ada3, the level of Ada3-K14ac and Ada3-K182ac was higher in ash1Δ and ubp8Δ mutants and less sensitive to sucrose treatment when compared with WT (Fig. 5h; Supplementary Fig. 1d, 5j). We also purified SAGA from WT (Wt SAGA) and ubp8Δ (SAGA ubp8Δ) mutant and then performed an in vitro deacetylase assay with purified Rpd3L. As expected, Rpd3L deacetylated Ada3 within Wt SAGA at a faster rate than Ada3 within SAGA ubp8Δ (Fig. 5i). As loss of Ash1 also reduced the activity of Rpd3 to deacetylate Ada3 within SAGA (Fig. 1b), these data indicate that Ash1 of Rpd3L directly interacts with Ubp8 of SAGA to bridge these two complexes together and promote Ada3 deacetylation by Rpd3 (Fig. 5j).
From Fig. 3b, we can tell that Tpk2 can phosphorylate both Rpd3 and Ash1 (Fig. 3b). As Rpd3 phosphorylation is not responsible for Tpk2 to reduce the interaction between Rpd3 and Ada3 (Supplementary Fig. 5c), we next examined the effect of Ash1 phosphorylation. We purified recombinant His-Ash1 and phosphorylated it with Tpk2-CBP. The phosphorylated Ash1 was incubated with purified SAGA and then immunoprecipitated with nickel beads. The results revealed that Tpk2-phosphorylated Ash1 had reduced interaction with SAGA when compared with unphosphorylated Ash1 (Fig. 6a, lane 2 vs lane 3). Loss of Tpk2 enhanced the intracellular interaction between Ash1 and Ubp8 without affecting Ash1 protein level (Fig. 6b; Supplementary Fig. 6a), suggesting that Tpk2-catalyzed Ash1 phosphorylation may inhibit the interaction between Ash1 and Ubp8.

a Tpk2-catalyzed Ash1 phosphorylation reduced the interaction between purified His-Ash1 and purified SAGA as determined by in vitro Co-IP. His-Ash1 was pre-phosphorylated by purified Tpk2-CBP and then incubated with purified SAGA for immunoprecipitation with nickel beads. b Loss of Tpk2 increased the interaction between Ubp8 and Ash1. c Ash1 was phosphorylated at S149 and S388. Ash1 was immunoprecipitated from WT, Ash1-S149A, Ash1-S388A and Ash1-2SA (Ash1-S149A S388A) mutants and its phosphorylation level was determined by immunoblots with phospho-PKA antibody. d The purified recombinant His-Ash1 was phosphorylated at S149 and S388 by purified Tpk2-CBP in vitro. e Analysis of Ash1 phosphorylation in WT, Ash1-2SA and Ash1-2SD mutants when cells were treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. f Analysis of the interaction between Ubp8 and Ash1 in WT, Ash1-2SA and Ash1-2SD mutants when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. g Mutation of Ash1-2SA enhanced the interaction between Rpd3L (Rxt2-CBP) and SAGA as determined by in vitro Co-IP. h Tpk2-catalyzed Ash1 phosphorylation reduced the interaction between Ubp8 and Ash1 as determined by in vitro Co-IP. i Immunoblot analysis of Ada3 acetylation in WT, Ash1-2SA and Ash1-2SD mutants when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. j Analysis of Ada3 acetylation in WT and Ash1-2SA mutant when cells were treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h by immunofluorescence. Scale bars, 5 μm. For (b, c, f–i), data represent means ± SE; n = 3 biological independent experiments; two tailed unpaired t-tests (b, g), one-way ANOVA with Tukey’s multiple comparisons tests (c), and two-way ANOVA with Tukey’s multiple comparisons tests (f, h, i) were used for statistical analysis. For (a, d, e, j), shown is the typical example of at least two biological independent experiments.
Analysis of Ash1 sequence revealed two potential phosphorylation sites, S149 and S388, which are located within the consensus PKA phosphorylation motif (146RLSS149, 385RKPS388). We thus mutated these two residues to construct Ash1-S149A, Ash1-S388A, Ash1-S149A S388A (Ash1 double mutant on both S149 and S388 residues, which we refer to hereafter as Ash1-2SA) mutants. Using anti-phospho-PKA antibody that recognizes phosphorylated serine/threonine within the consensus PKA motif, we found that mutation of Ash1-S149A or Ash1-S388A reduced Ash1 phosphorylation level, while double mutation of Ash1-S149A and Ash1-S388A totally abolished Ash1 phosphorylation signal (Fig. 6c). An in vitro kinase assay showed that Tpk2 directly phosphorylated recombinant His-Ash1 and simultaneous mutation of Ash1-S149A and S388A abolished Tpk2-mediated Ash1 phosphorylation (Fig. 6d, lane 6 vs lane 2). We then constructed yeast strains that expressed endogenous WT Ash1, Ash1-2SA and Ash1-S149D S388D (Ash1-2SD) mutants. Mutation of Ash1-2SA reduced the intracellular Ash1 phosphorylation (Fig. 6e). In vivo Co-IP showed that mutation of Ash1-2SA enhanced its intracellular interaction with Ubp8 and Ada3 within SAGA and mutation of Ash1-2SD reduced its interaction with Ubp8 and Ada3 within SAGA (Fig. 6f; Supplementary Fig. 6b). Moreover, the interaction between Ubp8 and Ash1-2SA was less sensitive to sucrose treatment (Fig. 6f), which is consistent with the changes of Ash1 phosphorylation (Fig. 6e). To further determine the effect of Ash1 phosphorylation on Rpd3L and SAGA interaction, we purified Rpd3L containing either WT Ash1 (Wt Rpd3L) or Ash1-2SA (Rpd3L Ash1-2SA) and examined their interaction with purified SAGA. Mutation of Ash1-2SA had no effect on the integrity of Rpd3L complex (Supplementary Fig. 6c). Compared with Wt Rpd3L, Rpd3L Ash1-2SA had increased interaction with SAGA (Fig. 6g, lane 3 vs lane 4). Tpk2-catalyzed His-Ash1 phosphorylation directly reduced the interaction between His-Ash1 and Ubp8-FLAG but not the His-Ash1-2SA and Ubp8-FLAG interaction (Fig. 6h).
We next examined the effect of Tpk2-catalyzed Ash1 phosphorylation on Rpd3L-catalyzed Ada3 deacetylation. An in vitro deacetylase assay with purified Rpd3L and Rpd3L Ash1-2SA revealed that Rpd3L Ash1-2SA deacetylated Ada3 at a faster rate than Rpd3L (Supplementary Fig. 6d). Consistently, the global levels of Ada3-K14ac and Ada3-K182ac were reduced in Ash1-2SA mutant and slightly increased in Ash1-2SD mutant (Fig. 6i). The reduced levels of Ada3-K14ac and Ada3-K182ac in Ash1-2SA mutant were also observed by immunofluorescence (Fig. 6j). Taken together, these data suggest that Tpk2 phosphorylates Ash1 at S149 and S388, which reduces the interaction between Rpd3L and SAGA and inhibits Rpd3L-mediated Ada3 deacetylation.
Tpk2-catalyzed Rpd3L phosphorylation promotes SAGA dimerization and histone acetylation
Ada3 acetylation has been reported to promote SAGA dimerization and enhance its ability to acetylate nucleosomes9. We then examined the effect of Tpk2-catalyzed Rpd3L phosphorylation on SAGA dimerization and histone acetyltransferase (HAT) activity. Loss of Tpk2 reduced the interaction between Ada3-FLAG and Ada3-Myc (Supplementary Fig. 7a), which is consistent with the trend of changes in Ada3 acetylation (Fig. 2e). In addition, mutation of Rpd3-2SA and Ash1-2SA decreased the interaction between Ada3-FLAG and Ada3-Myc (Fig. 7a, b), suggesting that Tpk2-mediated phosphorylation of Rpd3L promotes SAGA dimerization.

a, b Mutation of Rpd3-2SA (a) and Ash1-2SA (b) reduced the interaction between Ada3-FLAG-containing SAGA and Ada3-Myc-containing SAGA as determined by Co-IP and reciprocal IP. c Analysis of H3K9ac and H3K14ac in WT, Ash1-2SA, Rpd3-2SA and Rpd3-2SA/Ash1-2SA when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. d RT-qPCR analysis of SUC2 transcription in WT, Ash1-2SA, Rpd3-2SA and Rpd3-2SA/Ash1-2SA mutants when treated with YP + 2% glucose or YP + 2% sucrose for 0.5 h. e ChIP-qPCR analysis of H3K9ac/H3 and H3K14ac/H3 at SUC2 promoter regions (NUC1, NUC2, NUC3, NUC4) in WT and Rpd3-2SA mutant when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. f Venn diagrams showing the role of Ada3 acetylation and Rpd3 phosphorylation in sucrose-induced gene transcription (Fold change ≥ 1.5, fold change ≤ 0.75; P ≤ 0.05). g GSEA analysis of TCA cycle gene transcription in WT, Ada3-3KR, and Rpd3-2SA mutants when cells were treated with YP + 2% sucrose for 0.5 h. The RNA-seq data for Ada3-3KR were retrieved from GEO database GSE161887. One-side hypergeometric test was used for computing P-values. h RT-qPCR analysis of TCA cycle gene transcription in WT, Ash1-2SA and Rpd3-2SA mutants when treated with YP + 2% glucose (Glu) or YP + 2% sucrose (Suc) for 0.5 h. i ChIP-qPCR analysis of H3K9ac/H3 and H3K14ac/H3 at TCA cycle genes in WT and Rpd3-2SA mutant when treated with YP + 2% glucose (YP+Glu) or YP + 2% sucrose (YP+Suc) for 0.5 h. j, k LC-MS analysis of TCA cycle metabolites in WT, Ash1-2SA and Rpd3-2SA mutants when treated with YP + 2% glucose (YP+Glu) or YP + 2% sucrose (YP+Suc) for 0.5 h. For (a–e, h–k), data represent means ± SE; n = 3 biological independent experiments; two tailed unpaired t-tests (a, b) and two-way ANOVA with Tukey’s multiple comparisons tests (c–e, h–i, k) were used for statistical analysis.
We also examined the effect of Tpk2-mediated Rpd3L phosphorylation on histone acetylation. The global levels of H3K9ac and H3K14ac were gradually increased when cells were shifted from YP medium to YP + 2% glucose (YP + Glu) medium or to YP + 2% sucrose (YP + Suc) medium (Supplementary Fig. 7b). However, loss of Tpk2 reduced H3K9ac and H3K14ac and abrogated sucrose-induced H3K9ac and H3K14ac (Supplementary Fig. 7b). Similar results were observed for Rpd3-2SA and Ash1-2SA mutants (Supplementary Fig. 7c). Mutation of Ash1-2SA did not further reduce the global levels of H3K9ac and H3K14ac in Rpd3-2SA mutant (Fig. 7c). In contrast, the global levels of H3K9ac and H3K14ac were slightly higher in Rpd3-2SD and Ash1-2SD mutants than WT especially when grown in glucose-containing medium (Supplementary Fig. 7c).
Tpk2-catalyzed Rpd3L phosphorylation regulates the transcription of metabolic enzymes
We then investigated the effect of Tpk2-catalyzed Rpd3L phosphorylation on gene transcription. To utilize sucrose as a carbon source, yeast cells need to express the invertase enzyme Suc2, which hydrolyzes sucrose to glucose. SUC2 transcription is induced when cells are grown in sucrose-containing media37. Upon sucrose treatment, SUC2 was rapidly induced in WT but reduced in Ada3 acetylation mutant (Ada3-3KR) (Supplementary Fig. 7d), which is consistent with our reported study9. The reduced SUC2 transcription was also observed in Rpd3-2SA mutant and mutation of Rpd3-2SA in Ada3-3KR mutant did not further reduce SUC2 transcription (Supplementary Fig. 7d). Similarly, reduced SUC2 was observed in tpk2Δ and gpr1Δ mutants and loss of Tpk2 and Gpr1 in Ada3-3KR mutant did not further reduce SUC2 transcription (Supplementary Fig. 7d). Mutation of Ash1-2SA in Rpd3-2SA did not further reduce SUC2 transcription (Fig. 7d). These data suggest that Tpk2-mediated Rpd3L phosphorylation and Ada3 acetylation may act via the same pathway to promote SUC2 transcription. Consistent with the trend of SUC2 transcription, the enrichment of H3K9ac and H3K14ac at SUC2 gene promoter was significantly induced by sucrose treatment but was reduced in Rpd3-2SA and Ash1-2SA mutants (Fig. 7e; Supplementary Fig. 7e). These data suggest that Tpk2-mediated phosphorylation of Rpd3L promotes nucleosome acetylation to induce SUC2 transcription.
We then performed RNA-sequencing (RNA-seq) for WT and Rpd3-2SA mutant when treated with 2% glucose and 2% sucrose, respectively. By comparing the transcriptome data for Rpd3-2SA with the transcriptome data for Ada3-3KR mutant9, we found that sucrose treatment induced a total of 825 genes, among which the transcription of 773 genes was dependent on Ada3 acetylation and the transcription of 643 genes was dependent on Ada3 acetylation and Rpd3 phosphorylation (Fig. 7f). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that these 643 genes were significantly enriched in pathways, such as biosynthesis of secondary metabolites, biosynthesis of antibiotics, carbon metabolism, peroxisome, the tricarboxylate (TCA) cycle (Supplementary Fig. 7f). Gene set enrichment analysis (GSEA) revealed that TCA cycle genes were significantly reduced in both Ada3-3KR and Rpd3-2SA mutants upon sucrose treatment (Fig. 7g), which was confirmed by RT-qPCR for TCA cycle genes (CIT2, CIT3, IDP2 and KGD2) (Fig. 7h; Supplementary Fig. 7g). The enrichment of H3K9ac and H3K14ac at these gene promoters was significantly induced by sucrose treatment but was reduced in Rpd3-2SA and Ash1-2SA mutants (Fig. 7i; Supplementary Fig. 7h). Sucrose had no effect on histone acetylation at these gene promoters in Rpd3-2SA and Ash1-2SA mutants (Fig. 7i; Supplementary Fig. 7h). Consistent with the transcriptional changes of TCA cycle genes, metabolomic analysis using liquid chromatography-mass spectrometry (LC‒MS) revealed that sucrose treatment increased the amount of TCA cycle metabolites and reduced the amount of glycolytic metabolites (Fig. 7j). Mutation of Rpd3-2SA and Ash1-2SA alleviated sucrose-induced increase of TCA metabolites (Fig. 7j, k). These data suggest that Tpk2-mediated phosphorylation of Rpd3L promotes nucleosome acetylation to induce TCA cycle gene transcription and promote metabolic shift to the TCA cycle.
We next examined the effect of Tpk2-catalyzed Rpd3L phosphorylation on cell cycle progression and resistance to stress conditions. Flow cytometry analysis revealed that Rpd3-2SA mutant displayed slower cell cycle progression (Supplementary Fig. 8a). Rpd3-2SA and Ash1-2SA mutants also displayed slower cell growth in sucrose-containing medium (Supplementary Fig. 8b). We also examined the tolerance of WT, Rpd3-2SA and Ash1-2SA to oxidative stress and salt stress by treating cells with 0.1% H2O2 and 5 M NaCl, respectively. Both Rpd3-2SA and Ash1-2SA mutants displayed reduced survival after H2O2 and NaCl treatment (Supplementary Fig. 8c, d). We then examined the effect of these mutations on cell apoptosis. Chronological aging can be used to study the physiological scenario of apoptosis induction in yeast38,39. Compared to WT cells, Rpd3-2SA and Ash1-2SA mutants showed an early onset of cell death during chronological aging when grown in sucrose-containing medium (Supplementary Fig. 8e). Dihydroethidium (DHE) staining of Rpd3-2SA and Ash1-2SA mutants grown in YP + 2% sucrose medium for 3 days showed an increased accumulation of reactive oxygen species (ROS) (Supplementary Fig. 8f), which is an early step in the apoptotic process. TUNEL staining revealed that Rpd3-2SA and Ash1-2SA mutants had increased DNA fragmentation (Supplementary Fig. 8f). These results indicate that Tpk2-mediated Rpd3L phosphorylation improves cell survival under environmental stress conditions.
PKA-catalyzed HDAC1 phosphorylation inhibits its deacetylase activity and promotes the transcription of metabolic genes
We then investigated whether PKA could phosphorylate the homologue of Rpd3 in mammals. Mammalian PKA is activated by cAMP, which could be regulated in a G-protein-dependent manner40. The mammalian PKA is also a tetramer composed of a cAMP-binding regulatory subunit dimer and two catalytic subunits41. The mammalian PKA catalytic subunits are encoded by the two major genes PRKACA and PRKACB41. Knockdown of PRKACA and PRKACB with short hairpin RNA (shRNA) decreased the global levels of H3K9ac and H3K14ac in hepatocellular carcinoma HepG2 cells (Fig. 8a), similar to the reduction in H3K9ac and H3K14ac in yeast tpk2Δ mutant (Supplementary Fig. 7b). In mammals, there are four Rpd3 homologs, which belongs to class I HDACs (HDAC1, 2, 3, 8)42. We individually knocked down the expression of HDAC1, HDAC2, HDAC3, and HDAC8 by shRNA (Supplementary Fig. 9a). Knockdown of HDAC1 substantially increased H3K9ac and H3K14ac (Fig. 8b). Co-IP assays revealed that HDAC1 interacted with PRKACA and PRKACB (Fig. 8c), which was further confirmed by reciprocal IP (Fig. 8d). By analyzing the sequence of HDAC1, we found that its S434 residue is located within the consensus PKA phosphorylation motif (431RKXS434). We thus mutated HDAC1-S434 to nonphosphorylatable alanine (HDAC1-S434A). We knocked down the endogenous HDAC1 in HepG2 cells and then ectopically expressed Flag-HDAC1 and Flag-HDAC1-S434A, which were then immunoprecipitated with anti-FLAG beads. Analysis of immunoprecipitated HDAC1 with anti-phospho-PKA antibody revealed that mutation of HDAC1-S434A remarkably reduced HDAC1 phosphorylation level (Fig. 8e). In addition, mutation of HDAC1-S434A reduced the interaction between PRKACA and HDAC1 (Supplementary Fig. 9b). An in vitro kinase assay with purified Flag-PRKACA and recombinant His-HDAC1 revealed that Flag-PRKACA phosphorylated His-HDAC1 but not His-HDAC1-S434A mutant (Fig. 8f; Supplementary Fig. 9c).

a Immunoblot analysis of H3K9ac and H3K14ac in control, PRKACA knockdown and PRKACB knockdown HepG2 cells. b Immunoblot analysis of H3K9ac and H3K14ac in control, HDAC1 knockdown, HDAC2 knockdown, HDAC3 knockdown and HDAC8 knockdown HepG2 cells. c, d Co-IP and reciprocal IP showing HDAC1 interacted with PRKACA and PRKACB. e HDAC1 was phosphorylated at S434. Flag-HDAC1 and Flag-HDAC1-S434A were immunoprecipitated from HepG2 cells with anti-FLAG beads and the phosphorylation of HDAC1 was determined by phospho-PKA antibody. f An in vitro kinase assay showing purified Flag-PRKACA phosphorylated His-HDAC1 but not His-HDAC-S434A. g Ectopic expression of Flag-HDAC1-S434A in shHDAC1 HepG2 cells reduced global levels of H3K9ac and H3K14ac. h An in vitro deacetylase assay showing Flag-PRKACA-mediated HDAC1-S434 phosphorylation reduced the deacetylase activity of His-HDAC1 but not His-HDAC1-S434A. i Ectopic expression of HDAC1-S434A in shHDAC1 HepG2 cells reduced TCA cycle gene transcription. j Ectopic expression of HDAC1-S434A in shHDAC1 HepG2 cells reduced TCA cycle metabolites. k, l Ectopic expression of HDAC1-S434A in shHDAC1 HepG2 cells reduced cell growth (k) and colony formation (l). m–o Effect of PKA inhibitor H89 on HDAC1 phosphorylation, TCA cycle gene transcription, and TCA cycle metabolites in the liver of C57BL/6 J mice under fed and fasting conditions. Mice were injected with or without 5 mg/kg H89 followed by fed ad libitum or fasted for 12 h (5 mice per group). The live tissues were harvested. HDAC1 was immunoprecipitated with anti-HDAC1 antibody and its phosphorylation was probed with anti-phospho-PKA antibody. TCA cycle gene transcription and metabolites were analyzed by RT-qPCR and LC-MS, respectively. For (a, b, g–k, m, n), data represent means ± SE; n = 3 biological independent experiments; one-way ANOVA with Dunnett’s multiple comparisons tests were used for statistical analysis (a, b); two tailed unpaired t-tests were used for statistical analysis (g, i); two-way ANOVA with Šídák’s multiple comparisons tests (h, k) and Turkey’s multiple comparisons tests (m, n) were used for statistical analysis. For (o), n = 5 biological independent experiments. For (c–f, l), shown is the typical example of at least two biological independent experiments.
We next examined the effect of PKA-catalyzed HDAC1-S434 phosphorylation on its deacetylase activity. Ectopic expression of Flag-HDAC1-S434A reduced the global levels of H3K9ac and H3K14ac when compared with ectopic expression of Flag-HDAC1 (Fig. 8g). When cells were treated with PKA inhibitor H89, the global levels of H3K9ac and H3K14ac were reduced (Supplementary Fig. 9d). When cells were treated with PKA activator 8-Bromo-cAMP, the global levels of H3K9ac and H3K14ac were increased (Supplementary Fig. 9d). An in vitro deacetylase assay with purified recombinant His-HDAC1 and nucleosomes revealed that His-HDAC1 displayed higher activity to deacetylate nucleosomes and its activity was reduced after phosphorylation by PRKACA (Supplementary Fig. 9e). Moreover, we purified His-HDAC1 and His-HDAC1-S434A, which were then phosphorylated by Flag-PRKACA in vitro. His-HDAC1-S434A displayed higher deacetylase activity than His-HDAC1 (Fig. 8h), suggesting that PKA-catalyzed HDAC1-S434 phosphorylation repressed its deacetylase activity.
We also performed RNA-seq analysis of Flag-HDAC1 and Flag-HDAC1-S434A HepG2 cells treated with or without H89. H89 treatment reduced the transcription of 3106 genes, among which 2146 genes were repressed in Flag-HDAC1-S434A mutant (Supplementary Fig. 9f, g). We also performed Cleavage Under Targets & Tagmentation (CUT & Tag) for HDAC1 when HepG2 cells were treated with or without H89. H89 only slightly increased the occupancy of HDAC1 at chromatin (Supplementary Fig. 9h), suggesting that PKA-catalyzed HDAC1 phosphorylation has little to no effect on its binding to chromatin. HDAC1 directly bound 6837 genes, among which 475 genes were up-regulated by HDAC1-S434A and H89 treatment and 680 genes were down-regulated by HDAC1-S434A and H89 treatment (Supplementary Fig. 9i). KEGG analysis of 680 genes revealed that these genes were enriched in ribosome, pathway of neurodegeneration, Huntington disease, Amyotrophic lateral sclerosis, Alzheimer disease, Parkinson disease, prion disease, thermogenesis, oxidative phosphorylation, etc. (Supplementary Fig. 9j). KEGG analysis of 475 genes revealed that these genes were enriched in focal adhesion, parathyroid hormone synthesis, secretion and action, spinocerebellar ataxia, insulin resistance, acute myeloid leukemia (Supplementary Fig. 9j).
RNA-seq analysis revealed that genes involved in TCA cycle were repressed by ectopic expression of Flag-HDAC1-S434A, which was also confirmed by qRT-PCR (Fig. 8i, Supplementary Fig. 10a). Treatment with H89 reduced the expression of these TCA cycle genes in Flag-HDAC1 cells but not in Flag-HDAC1-S434A cells (Supplementary Fig. 10b). In contrast, treatment with 8-Bromo-cAMP increased the expression of these TCA cycle genes in Flag-HDAC1 cells and marginally induced TCA cycle gene expression in Flag-HDAC1-S434A cells (Supplementary Fig. 10c). CUT & Tag analysis of HDAC1 revealed that HDAC1 directly bound at these TCA cycle genes and H89 treatment slightly increased the binding of HDAC1 at these genes (Supplementary Fig. 10d). H3K9ac was increased at these gene promoters by 8-Bromo-cAMP treatment in Flag-HDAC1 (Supplementary Fig. 10e). Mutation of HDAC1-S434A reduced H3K9ac at these gene promoters and abolished the inducing effect of 8-Bromo-cAMP (Supplementary Fig. 10e). Consistent with gene regulation, mutation of HDAC1-S434A reduced the amount of TCA cycle metabolites (Fig. 8j; Supplementary Fig. 10f). Both growth curves and colony formation assays showed that ectopic expression of Flag-HDAC1-S434A slowed cell growth compared with ectopic expression of Flag-HDAC1 (Fig. 8k, l; Supplementary Fig. 10g).
We then investigated the physiological significance of PKA-HDAC1 pathway in regulating TCA cycle in mammalian system. C57BL/6 J mice were fasted for 12 h, which has been reported to induce PKA43. HDAC1 was immunoprecipitated from the liver tissues and immunoblots revealed that HDAC1 phosphorylation was induced after fasting and this phosphorylation was abolished upon treatment with PKA inhibitor H89 (Fig. 8m). Consistently, the expression of TCA cycle genes as well as the intracellular amount of TCA cycle metabolites were increased under fasting condition and this inducing effect was abolished by H89 treatment (Fig. 8n, o; Supplementary Fig. 11).
Taken together, these data suggest that PKA phosphorylates HDAC1 to repress its deacetylase activity, promote the TCA cycle and maintain normal cell growth.
Discussion
Cells undergo metabolic adaptations to reinforce their ability to survive and proliferate when they encounter fluctuations in external nutritional changes. Understanding how cells couple nutrient-sensing to metabolic adaptations would help identify potential anti-cancer targets and develop anti-cancer strategies4. Chromatin modifying enzymes allow cells to tune their adaptive metabolism by regulating metabolic gene expression. How cells transduce extracellular nutrient changes into specific nuclear chromatin modifications remains largely unknown19. The current study demonstrated an intricate link between nutrient/energy sensing, chromatin modifications, and metabolism adaptation. We found that PKA catalytic subunit Tpk2 is activated when cells are grown under stress conditions with sucrose serving as the carbon source. Activated Tpk2 then phosphorylates Rpd3 at serine 50 and serine 354, which represses its ability to deacetylate Ada3. Moreover, Tpk2 phosphorylates Rpd3L subunit Ash1 serine 149 and serine 388, which reduces the interaction between SAGA and Rpd3L. By phosphorylating both Rpd3 and Ash1 within Rpd3L, Tpk2 inhibits Rpd3L-mediated Ada3 deacetylation under sucrose treatment, which then promotes SAGA dimerization, nucleosome H3 acetylation as well as the transcription of genes involved in sucrose metabolism and TCA cycle. Most importantly, PKA phosphorylates HDAC1, the Rpd3 homolog in mammals, and inhibits its deacetylase activity to promote TCA cycle gene expression and cell growth, suggesting that this mechanism is conserved from yeast to mammals. Overall, our work reveals a conserved mechanism for regulation of Rpd3L activity by PKA and provides insights into how cells sense and respond to environmental nutritional changes.
Although the PKA catalytic subunits are encoded by Tpk1, Tpk2 and Tpk3, only Tpk2 phosphorylates Rpd3L to inhibit its activity. The different effects of Tpk isoforms on Rpd3L are consistent with the findings of Ptacek et al. who reported that three Tpk isoforms have distinct substrate preferences and most substrates are recognized by only one of the Tpks44. Tpk1, but not Tpk2 or Tpk3 regulates non-homologous end joining double-stranded break repair by phosphorylating Nej145. Tpk2 activates pseudohyphal growth, while Tpk3 inhibits filamentation and Tpk1 has no effect46. Tpk1 and Tpk2 have distinct functions in regulating iron uptake and respiration47. To understand the specific regulation of Rpd3L by Tpk2, we examined the expression of PKA subunits (Tpk1, Tpk2, Tpk3) when cells were treated with 2% glucose and 2% sucrose, respectively. Although the expression of Tpk1/2/3 was not altered when cells were treated by sucrose, the expression of Tpk2 was higher than Tpk1 and Tpk3 (Supplementary Fig. 2g). We also examined the effect of sucrose treatment on the interaction between Bcy1 and Tpk1/2/3. Our data showed that Tpk2 but not Tpk1 and Tpk3 dissociated with Bcy1 upon sucrose treatment (Fig. 2g; Supplementary Fig. 2e, f). It is possible that the relatively high expression of Tpk2 and the relatively rapid dissociation of Tpk2 from Bcy1 in response to sucrose determine the specific regulation of Rpd3L by Tpk2.
We identified a new pathway by which yeast cells to sense and respond to extracellular sucrose treatment. Tpk2 phosphorylates Rpd3L to enhance SAGA auto-acetylation and dimerization, which facilitates the transcription of SUC2 and TCA cycle genes to help cells survive under the stress conditions. Tpk2 is activated by two main pathways. The glycolytic metabolite, fructose-1,6-biphosphate (FBP) activates small GTP-binding proteins, Ras1 and Ras2 through the guanine exchange factor, Cdc2534. Ras then stimulates adenylate cyclase (Cyr1) to synthesize cAMP, which then activates PKA by binding to Bcy1 regulatory subunit to release the catalytic subunits36. We found that sucrose induced Ada3 acetylation in ras1Δ and ras2Δ mutants (Supplementary Fig. 2i), suggesting that sucrose activates Tpk2 not via the Ras-cAMP pathway. This is conceivable as the intracellular levels of glucose and FBP were reduced when cells were shifted from glucose-containing medium to sucrose-containing medium (Fig. 7j). This would lead to decreased Ras1/2 activity. In addition to the Ras-cAMP-PKA pathway, Cyr1 can be stimulated by GPCR system, which is composed of G-protein-coupled receptor Gpr1, G protein α subunit Gpa2, and its GTPase activating protein Rgs211. Gpr1 acts as the receptor protein for detection of glucose and sucrose, while Gpa2 acts as a stimulatory G protein on adenylate cyclase48. Here, we found that sucrose treatment activates Tpk2 via the Gpr1-Gpa2-Cyr1-Tpk2 pathway. Compared with the Ras-cAMP-PKA pathway, the Gpr1-Gpa2-Cyr1 pathway depends on the recognition of extracellular nutrients by GPCR protein Gpr1 but not by glycolysis rate. Gpr1 is a nutrient sensor with much higher affinity for sucrose than for glucose, which may allow yeast cells to survive on low concentrations of sucrose released from plants48. Moreover, sucrose has been reported to induce filamentation via Gpr1 more efficiently than glucose49. When yeast cells are grown in sucrose as the sole carbon source, cells need to secrete periplasmic invertase to hydrolyze sucrose to glucose in order to survive. Under this stress situation, the Gpr1-Gpa2-Cyr1 pathway is probably much more rapid and efficient than the Ras-cAMP pathway in activating Tpk2 and repressing Rpd3L.
Although sucrose-induced SUC2 transcription was reduced in Ada3-3KR, Rpd3-2SA, tpk2Δ, and gpr1Δ mutants, sucrose can still induce SUC2 transcription in these mutants (Fig. 7d; supplementary Fig. 7d). This may be caused by the fact that SUC2 transcription is not only controlled by histone acetylation but also regulated by other proteins. For example, when cells are grown in high glucose medium, the Ssn6-Tup1 complex is recruited to SUC2 promoter to repress SUC2 transcription via its interaction with Mig150,51,52. When cells are grown in low glucose, Mig1 is phosphorylated and inactivated by Snf1/Snf4 serine-threonine protein kinase complex, which then derepresses SUC252. These pathways work together to regulate SUC2 transcription in response to extracellular carbon source changes.
Chromatin modifying enzymes can be phosphorylated by these nutrient sensors, which thus directly connect nutrient sensing with chromatin modifications19. We have previously reported that Tpk2 phosphorylates histone demethylase Jhd2, which regulates its nuclear translocation, binding to chromatin and protein stability, which eventually reduce its activity to demethylate H3K432. Here, we found that Tpk2 phosphorylates Rpd3L subunits, Rpd3 and Ash1. We noted that Tpk2 does not affect the protein levels of both Rpd3 and Ash1, which is quite different with Jhd2. Instead, Tpk2-catalyzed Rpd3 phosphorylation impairs the deacetylase activity of Rpd3 and this inhibition is not limited to Ada3 deacetylation but also nucleosome H3 deacetylation. Tpk2-catalyzed Ash1 phosphorylation reduces the interaction between Rpd3L and SAGA, which further diminished Ada3 deacetylation within SAGA. Whereas Rpd3L deacetylates histones at promoter regions, Rpd3S targets transcribed regions to suppress intragenic transcription initiation21,23. We found that it is Rpd3L not Rpd3S that deacetylates Ada3 within SAGA. This specificity is mediated by the direct interaction between Rpd3L specific subunit Ash1 and SAGA subunit Ubp8. By phosphorylating Rpd3 and Ash1, Tpk2 connects extracellular sucrose with repression of Rpd3L and auto-acetylation of SAGA. Although we cannot rule out that Rpd3L may regulate gene expression by directly deacetylating histone H3 at gene promoter, our data with Ada3-3KR Rpd3-2SA mutant revealed that Tpk2-catalyzed Rpd3L phosphorylation may primarily promote gene expression by repressing Ada3 deacetylation and enhancing SAGA-catalyzed histone acetylation.
The phosphorylation of Rpd3 by Tpk2 is conserved in mammals. PKA has been reported to phosphorylate HDAC4, which belongs to class IIa HDACs, to enhance its nuclear influx and promote its ability to repress the transcription factor myocyte enhancer factor 2 (MEF2)53,54. Here, we found that PKA phosphorylates class I HDACs, HDAC1, the Rpd3 homolog in mammals. Similar to Tpk2-catalyzed phosphorylation of Rpd3, PKA-mediated HDAC1 phosphorylation reduces its deacetylase activity, which increases histone acetylation and promotes TCA cycle. Hence, we identified a new mechanism to regulate HDAC1 activity by post-translational modifications. ATP-citrate lyase (ACLY) is the enzyme providing acetyl-CoA for histone acetylation and has been demonstrated to localize in the nucleus in mammalian cells55. We also examined the effect of treatment with PKA activator 8-Bromo-cAMP on ACLY localization in nuclei of HepG2 cells. It revealed that 8-Bromo-cAMP treatment slightly but significantly increased the nuclear localization of ACLY in both HDAC1-Flag and HDAC1-S434A-Flag mutant (Supplementary Fig. 12a). This modest yet significant increase in nuclear ACLY likely provides acetyl-CoA for histone acetylation. 8-Bromo-cAMP treatment increased histone acetylation in Flag-HDAC1 cells and mutation of HDAC1-S434A significantly reduced 8-Bromo-cAMP-induced increase of histone acetylation (Supplementary Fig. 12b), suggesting that PKA induced histone acetylation primarily by repressing HDAC1 activity. HDAC1 is an important factor that regulate cell growth and survival. For example, HDAC1 promotes apoptosis through inhibition of extracellular signal-regulated kinases 1/2 (ERK1/2) in mouse hepatocytes56. We found that loss of HDAC1 phosphorylation site enhances its deacetylase activity and renders cells grow slowly. It has been reported that PKA promotes hepatocellular carcinoma (HCC) invasion and metastasis by phosphorylating CIP457. Our data suggest that PKA could promote tumorigenesis by phosphorylating and inhibiting HDAC1. Nonetheless, it remains to be explored whether the homolog of Ash1 is phosphorylated by PKA.
Together, we identified a molecular signaling pathway that directly connects extracellular nutrient changes to histone modifications and metabolic gene transcription, which is conserved in mammalian cells. We also uncovered a regulatory mechanism for histone deacetylase Rpd3/HDAC1, which could be exploited for future cancer treatment.
Methods
Yeast strains
All yeast strains used in this study are listed in Supplementary Table 1. The gene deletion mutants and genomic integration of C-terminal epitope tags were constructed by homologous recombination of PCR fragments. The endogenously expressed Rpd3-2SA (Rpd3-S50A S354A) mutant was constructed by homologous recombination of PCR fragments as follows: The Saccharomyces cerevisiae Rpd3 was cloned and inserted into plasmid pRS316 to generate pRS316-Rpd3. The pRS316-Rpd3-S50A S354A plasmid was obtained by site-directed mutagenesis and used as the template to amplify the Rpd3-S50A S354A fragment with a selectable marker URA3 and 60 nt homologous sequence to endogenous RPD3 flanking sequence. The PCR fragment was then integrated into a RPD3 knockout strain through homologous recombination. Successful transformants were screened to obtain the Rpd3-S50A S354A mutant. The endogenously expressed Ash1-2SA (Ash1-S149A S388A) mutant was constructed using the similar approach except that the Ash1-S149A S388A fragment with a selectable marker HIS3 and 60 nt homologous sequence to endogenous ASH1 flanking sequence was amplified by PCR and then integrated into a ASH1 knockout strain through homologous recombination. All yeast strains used in this study were verified by colony PCR, DNA sequencing, quantitative reverse transcription PCR (RT-qPCR), and/or immunoblots.
Cell culture
HEK293T cells (STCC10301P) and HepG2 cells (STCC10114P) were obtained from the Servicebio Company. HepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were reauthenticated by short tandem repeat analysis after resuscitation in our laboratory. For cAMP treatment, the stable analog 8-Bromo-cAMP (HY-12306A, MCE) was used.
Animals
All animal experiments were approved by the Animal Care and Use Committee of Hubei University (NO. 20240116). Wild-type C57BL/6 J male mice were obtained from Henan Skobes Biotechnology Co., Ltd, and housed in a specific pathogen-free (SPF) facility on a 12-hour light/dark cycle with ad libitum access to food and water. Animals were fed with a normal chow diet. Animals used for experiments were all healthy prior to experiments. For PKA inhibitor treatment, animals were injected with or without 5 mg/kg H89 (HY-15979, MCE) at 9 pm, followed by fed ad libitum or fasted for 12 h (6 mice per group, including 3 male and 3 female mice). 5 mice were randomly chosen and sacrificed to harvest the liver tissues were for immunoblot, RT-qPCR and metabolite analysis.
Plasmids and transfection
The pHAGE plasmids that ectopically express HDAC1 and PRKACA were constructed by standard molecular biology techniques. Point mutations were generated by site-directed mutagenesis. Cells were infected with lentivirus to stably overexpress genes of interest58. In brief, HEK-293T cells were cultured to 70–80% confluence and then transfected with 2 μg of pHAGE (expression vector), 1 μg of psPAX2 and 1 μg of pMD2.G plasmids with Neofect reagent (TF20121201, Neofect). After 48 h, the supernatant-containing lentivirus was collected. HepG2 cells were infected with lentivirus and 5 μg/mL polybrene by centrifugation at 3,000 × g for 1.5 h. After 24 h, cells were selected using 2 μg/mL puromycin.
To construct stable knockdown cell lines, shRNA hairpins were cloned and inserted into the lentiviral vector pLKO.158. A control hairpin that targeted GFP was cloned and inserted into the pLKO.1 vector and used as a negative control. HEK293T cells were transfected with pLKO.1 vectors and lentiviral packaging vectors. 48 h later, the supernatant-containing lentivirus was collected. HepG2 cells were transfected with lentivirus and selection was performed under 2 μg/mL puromycin. The knockdown efficiency was examined by RT-qPCR and/or immunoblots. The shRNA sequences are listed in Supplementary Table 2.
Cell growth and treatment
To examine the effect of glucose and sucrose on histone modifications, yeast cells were grown in YPD (Yeast Extract Peptone Dextrose) medium until OD600 of 0.7–1.0. Cells were then collected, washed and resuspended in YP medium followed by the addition of 2% glucose or 2% sucrose for 0.5-1 h.
Cell counting, CCK-8 and colony formation assays
For cell counting assay, cells were seeded into 12-well plates at 5 × 104 cells/well. Cells were collected at different time points and counted with cell counter (HSCORE, China). For CCK-8 assay, cells were seeded into 96-well plates at 2 × 103 cells/well. After 48 h, cell proliferation rate was determined by Cell Counting Kit (CCK-8, Dojindo, Japan) according to the manufacturer’s instructions. For colony formation assay, cells were seeded into 6-well plates at 2 × 103 cells/well and then cultured for 14 days. The colonies were stained with 0.1% (w/v) crystal violet (Solarbio).
Protein expression and purification
The recombinant His-tagged proteins were expressed in BL21(DE3) cells with 0.5 mM IPTG induction at 16 °C for 16 h. Cells were harvested by centrifugation at 2350 × g for 10 min, resuspended with phosphate buffered saline (PBS) and then subjected to sonication on ice. The cell lysate was centrifuged at 15,000 × g at 4 °C for 1 h. The supernatant was then incubated with Ni-NTA Agarose (Abclonal) at 4 °C for 3 h. The beads were sequentially washed with 3 mL PBS, 10 mL PBS buffer plus 20 mM imidazole and then eluted with PBS buffer plus 200 mM imidazole. The eluted fractions were polled and dialyzed in 2 L PBS for 4 times at 4 °C and concentrated using Amicon Ultra-0.5 ml Centrifugal Filters (Millipore).
The recombinant FLAG-tagged proteins were expressed in BL21(DE3) cells and induced with 0.5 mM IPTG at 18 °C for 16 h. Cells were harvested and sonicated in PBS on ice. The cleared cell lysate was then incubated with anti-FLAG IP Resin (GenScript, L00425) at 4 °C for 4 h. The beads were washed with 5 ml E100 buffer (25 mM HEPES-KOH pH7.6, 100 mM KCl, 2 mM MgCl2, 1 mM EDTA, 10% glycerol, 0.02% NP40), eluted with E100 buffer plus 0.25 mg/ml FLAG peptides and then concentrated using Amicon Ultra-0.5 ml Centrifugal Filters (Millipore).
Tandem affinity purification
Rpd3 complex and SAGA complex were purified by TAP purification from yeast cells as described previously9. The purified complex was analyzed by SDS-PAGE followed by silver staining or Coomassie blue staining.
Immunoblots
Cells were grown in 5 mL YPD (Yeast Extract-Peptone-Dextrose) or selective medium until OD600 of 0.7-1.0. Cells were harvested and lysed in the lysis buffer (2 M NaOH, 8% 2-mercaptoethanol). After centrifugation, the protein pellet was resuspended in 150 μL of 2×SDS-sample buffer. Protein samples were separated by 8-15% SDS-PAGE and transferred to PVDF membrane. The blots were probed by primary antibodies followed by incubation with horseradish peroxidase-labelled IgG secondary antibodies. The protein bands were visualized using the ECL Chemiluminescence Detection Kit (Bio-Rad, 170-5061) and quantified with ImageJ software (v.1.8.0).
Antibodies
Antibody against H4 (1:2,000; ab10158) was purchased from Abcam; anti-FLAG (1:5,000; AE024) was purchased from Abclonal; antibodies against H3 (1:5,000; 9715S), H3K9ac (1:5,000; 9649S), Myc (1:3,000; 2276S), H3K14ac (1:1,000; 7627S), phospho-PKA substrate (1:2000; 100G7E), HDAC1 (1:3,000; 34589S) were purchased from Cell Signaling Technology; antibody against CBP (1:1,000; Abs130593) was purchased from Absin; anti-GAPDH (1:5,000; 10494-1), beta-actin (1:10000; 20536-1-AP), 6×His (1:5000; HRP-66005) were purchased from Proteintech; antibodies against PRKACA (1:3000; A21869), PRKACB (1:3000; A22721) and ACLY (1:2000; A15251) were purchased from Abclonal; antibodies against Rpd3 phosphorylated at S50 (anti-Rpd3-S50p, 1:2000) and S354 (anti-Rpd3-S354p, 1:1000), Ada3 acetylated at K14 (anti-Ada3K14ac, 1:2000) and K182 (anti-Ada3K182ac, 1:2000) were custom made by Abclonal. The specificity of custom-made antibodies was confirmed by immunoblot analysis with the cell extract or IP of corresponding mutants. The custom-made antibodies against Ada3 (1:1,000), Gcn5 (1:2,000) and Spt3 (1:2,000) were kind gifts from J. L. Workman (Stowers Institute for Medical Research).
Microscopy
Microscopy analysis was performed as described previously59. In brief, yeast cells were grown to an OD600 of 1.0 and fixed with 1/10 volume 37% formaldehyde for 0.5 h. After fixation, cells were centrifuged at 500 × g for 5 min and resuspended in 1 mL cold PBS. Cells were washed three times with PBS and then resuspended in 0.5 mL of 1.2 M sorbitol phosphate citrate buffer (SPC) containing 0.01% zymolyase 20 T at 30 °C for 1 h. After cold SPC buffer wash, the spheroplasts were added on the poly-L-lysine-coated slides, blocked with 1% bovine serum albumin (BSA) in PBS at 30 °C for 1 h, and then incubated with primary antibodies at 4 °C overnight. Cells were washed with PBS for three times and incubated with Alexa Fluor 594-conjugated goat anti-rabbit IgG (Abclonal) or Alexa Fluor 488-conjugated goat anti-mouse IgG (Abclonal) at 4 °C for 5 h. After washing with PBS, cells were treated with DAPI (Solarbio) for 10 min at room temperature. Cells were then washed with PBS and visualized with a Zeiss LSM710 microscope using ZEN Imaging Software (Zeiss). The merged color images were generated by Fiji software.
Co-immunoprecipitation (Co-IP)
Yeast whole cell extracts were prepared by glass bead vortex, centrifuged at 23,447 × g for 15 min. The supernatant was incubated with anti-FLAG M2 agarose, IgG Sepharose (GE Healthcare) for 5 h at 4 °C. The beads were washed three times with IP washing buffer (25 mM HEPES-KOH, pH7.5, 0.1% NP40, 10% glycerol, 1 mM PMSF, 350 mM KCl, 2 mM MgCl2, 1 mM EDTA, 10% glycerol, 150 mM NaCl, 0.02% NP40) and boiled in SDS sample buffer for 10 min. Supernatants from the boiled beads were subjected to SDS-PAGE and immunoblots.
In vitro kinase assay
Purified Rpd3 or Ash1 was mixed with or without 200 ng TAP-purified Tpk2 (Tpk2-CBP) in reaction buffer (100 mM Tris-HCl pH7.4, 100 mM MgCl2, 10 mM ATP, 0.2 μM PMSF, 5% glycerol) at 30 °C for 0– 45 min. Equal aliquot was taken at different time points and the reaction was quenched by adding 2×SDS-PAGE loading buffer followed by boiling at 95 °C for 10 min. After centrifugation, the supernatant was resolved on 10% SDS-PAGE and analyzed by immunoblots.
In vitro deacetylase assay
An in vitro deacetylase assay was performed using purified Rpd3 complex and SAGA complex as previously described in ref. 24. Briefly, 10 μg of SAGA was incubated with 0.5 mM acetyl-CoA for 1 h and immobilized on calmodulin beads to remove unreacted acetyl-CoA. The pre-acetylated SAGA was then incubated with 2 μg of purified Rpd3 in 50 μL of deacetylation buffer (20 mM Tris pH7.5, 150 mM NaCl, 10% glycerol) at 30 °C for 0−90 min. The reaction products were resolved on 10% SDS-PAGE followed by immunoblots with anti-Ada3, anti-Ada3-K14ac and anti-Ada3-K182ac antibodies.
Recombinant His-Rpd3 and His-Rpd3-2SA were used for in vitro deacetylase assays. 20 μg of His-Rpd3 and His-Rpd3-2SA were incubated with 5 μg of Tpk2-CBP in reaction buffer (100 mM Tris-HCl pH7.4, 100 mM MgCl2, 10 mM ATP, 0.2 μM PMSF, 5% glycerol) for 1 h. Tpk2-treated His-Rpd3 and His-Rpd3-2SA were incubated with 10 μg pre-acetylated SAGA or nucleosomes in 50 μL deacetylase reaction buffer (20 mM Tris pH7.5, 150 mM NaCl, 10% glycerol) at 30 °C for 0−90 min.
Immunoprecipitation-mass spectrometry analysis
Yeast cells were grown in 2 L of YPD to an OD600 of 1.0, washed with PBS and resuspended in IP buffer (40 mM HEPES-KOH pH 7.5, 150 mM NaCl, 10% glycerol). Cells were lysed with glass beads using a Biospec bead beater. After centrifugation at 26,900 g at 4 °C for 1.5 h, the supernatant was incubated with anti-FLAG M2 affinity gel (Sigma) at 4 °C for 4 h. The immunoprecipitated proteins were digested overnight with sequencing-grade trypsin (Promega) at 37 °C. The supernatant peptides were desalted using C18 columns (Thermo Fisher) and lyophilized. The dried peptides were reconstituted in 0.1% FA, loaded onto an Acclaim PepMap 100 C18 LC column (Thermo Fisher) utilizing a Thermo Easy nLC 1000 LC system (Thermo Fisher) connected to Q Exactive HF mass spectrometer (Thermo Fisher) and analyzed as previously described32.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assays were performed as previously described60. Yeast cells were grown in 200 mL of YPD media at 30 °C until an OD600 of 1.0. Cells were then collected, washed, and resuspended in YP + 2% sucrose or YP + 2% glucose at 30 °C for 0.5 h. The crosslinking was performed in 1% formaldehyde for 10 min and quenched by adding 2.5 M glycine for 5 min. Cells were then harvested, washed with PBS buffer and lysed by vortexing with glass beads in FA-SDS lysis buffer (40 mM HEPES-KOH pH7.5, 1 mM EDTA pH8.0, 1% Triton X-100, 0.1% Nadeoxycholate, 0.1% SDS, 1 mM PMSF, protease inhibitor cocktail). The chromatin was sonicated to ~200–500 bp fragments and incubated with antibodies (H3, H3K9ac or H3K14ac) pre-bound to Protein G Dynabeads (Invitrogen). Beads were washed sequentially with PBS containing with 0.5% BSA, FA buffer containing 1 M NaCl, FA buffer with 0.5 M NaCl, TEL buffer (10 mM Tris pH8.0, 1 mM EDTA, 0.25 M LiCl, 1% NP40, 1% sodium deoxycholate) and TE buffer. The eluted DNA/protein complexes were treated with 20 µg proteinase K (Roche) at 55 °C for 1 h and the cross-link was reversed at 65 °C overnight. The DNA was treated by RNase (Roche), purified with ethanol precipitation and quantified by qPCR with the primers listed in Supplementary Table 3.
For ChIP assays in HepG2 cells, Flag-HDAC1 and Flag-HDAC1-S434A HepG2 cells were treated with or without 10 μM 8-Bromo-cAMP (HY-12306A, MCE) for 24 h. HepG2 cells were fixed with 1% formaldehyde to cross-link proteins to DNA, followed by quenching with 0.125 M glycine. The cells were then harvested, washed, and lysed in lysis buffer (50 mM Tris pH 8.0, 5 mM EDTA, 1% SDS, 1 mM PMSF, and a protease inhibitor cocktail) followed by the above procedures.
Quantitative reverse-transcription PCR (RT-qPCR)
Total RNA was isolated from exponentially growing yeast cells by standard phenol-chloroform extraction procedures, treated with DNase I (RNase-free) (Takara, 2270 A) and quantified by Nanodrop 2000 (Thermo Scientific). The integrity of RNA was assessed by agarose gel electrophoresis. A total of 0.5 μg RNA was used for reverse transcription PCR with Reverse Transcriptase Kit (M-MLV) (ZOMANBIO). qPCR was carried out on a Bio-Rad real-time PCR machine with iTaq™ Universal SYBR® Green Supermix (Bio-Rad, 1725121). The primers used for RT-qPCR are described in Supplementary Table 3. We used the 2^(-ΔΔCt) method to determine the quantity of relative transcription level. Flag-HDAC1 and Flag-HDAC1-S434A HepG2 cells treated with 10 μM H89 (HY-15979, MCE) or 10 μM 8-Bromo-cAMP (HY-12306A, MCE) for 24 h. Total RNA is extracted with Trizol reagent RNAiso Plus (Takara).
RNA sequencing (RNA-seq)
Total RNA was isolated from exponentially growing yeast cells by standard phenol-chloroform extraction procedures and the quality of RNA was examined using Agilent Bioanalyzer61. Total RNA was extracted from Flag-HDAC1 and Flag-HDAC1-S434A HepG2 cells treated with or without 10 μM H89 for 24 h by Trizol reagent RNAiso Plus (Takara). Library construction, sequencing and bioinformatics analysis were performed by GeneRead Biotechnology Co., Ltd (Wuhan, China). There were three biological replicates. The differential expression levels of the aligned sequences were calculated using significant thresholds as indicated in figure legends. The differentially expressed genes were further used for KEGG pathway analysis. A one-sided hypergeometric test was used for computing P values.
cAMP concentration measurement
Metabolite samples were prepared as described previously32,62. Briefly, cells were grown in 6 mL of YPD medium at 28 °C until an OD600 of 0.7–1.0. Cells were washed and treated with YP medium for 2 h. Afterwards, cells were treated with YP + 2% glucose and YP + 2% sucrose, respectively. Quenching was performed by adding 4 mL buffer 1 (60% methanol with 10 mM tricine) at −80 °C for 5 min. Cells were then centrifuged and resuspended in 400 μL buffer 2 (75% ethanol plus 0.5 mM triazine). Cells were lysed by incubating at −80 °C for 3 min followed by cooling on ice bath for 5 min. Cell suspension was centrifuged at 20,000 × g for 10 min. The supernatant was collected and the cAMP concentration was determined with a cAMP ELISA kit (Shanghai Huding Biotechnology Co., Ltd). At least three biological replicates per sample were used for cAMP quantification.
LC‒MS analysis of metabolites
The intracellular concentration of metabolites was measured by liquid chromatograph-mass spectrometry (LC‒MS). Cells were collected and resuspended with 1 mL of 80% methanol, treated in liquid nitrogen for 5 min, then placed on ice for 5 min until the samples thawed, and then treated with an ultrasonic breaker for 5 min. The samples were centrifuged at 23,447 g at 4 °C for 15 min. The supernatant was collected and the metabolite concentration was measured by Q Exactive™ Hybrid Quadrupole Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific). The raw data were normalized and processed by MetaboAnalyst (v.6.0).
CUT & Tag experiment and analysis
CUT & Tag experiment was performed by CUT & Tag Pro kit (Vazyme, TD904). HepG2 cells treated with DMSO or H89 were harvested, mixed with ConA beads and incubated overnight with anti-HDAC1 antibody (Cell Signaling, 34589) and rabbit IgG (Abclonal, A19711). A secondary antibody (Vazyme, Ab207) was added and incubated at room temperature with gentle rotation for 1 h. The samples were then treated with pA/G-Tnp Pro and TTBL at 37 °C for 1 h. The supernatant was mixed with DNA Extract Beads Pro to dissolve DNA and used for PCR amplification. VAHTS DNA Clean Beads were added to purify DNA, which was then subjected to next-generation sequencing.
For CUT & Tag data analysis, the raw sequenced reads were filtrated by Fastqc (v.0.11.9) and Trim Galore (v.2.11) with quality control of Q25 and 6 base pairs removed at 5’ end of each paired reads. Clean reads were mapped to human genome version hg19 using bowtie2 0.12.9 and default settings. Samtools (version v1.11) was used for format conversion and peaks were called using macs2 callpeak with parameter -g hs -p 0.01 --nomodel. Peaks annotation was performed by R package ChIPseeker (version v1.42.0). Tracks was smoothed by deepTools software and visualized by IGV software with a reference genome of hg19. The heatmap and metagene plots were analyzed using deepTools plotHeatmap and deepTools plotProfile.
Quantification and statistical analysis
For quantification of the immunoblot data, Image J (v.1.8.0) software was used to measure the relative intensity of each band. Unless otherwise indicated, data are presented as mean ± SEM (standard error of the mean), calculated using GraphPad Prism 10 (version 10.1.2). A P-value of < 0.05 was considered statistically significant. Specific P-values and the statistical tests used are detailed in each figure legend. For comparisons between two groups, unpaired two-tailed t-tests were performed. For comparisons among multiple groups, one-way ANOVA was utilized, followed by Dunnett’s multiple comparison tests or Tukey’s multiple comparisons tests as appropriate. For analysis involving two factors, two-way ANOVA was conducted, followed by Tukey’s multiple comparisons tests or Sidak’s multiple comparisons test to identify specific group differences.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are included in the manuscript and its supplementary files. The RNA-seq data for WT and Rpd3-2SA generated in this study have been deposited in the NCBI under accession number GSE260970. [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE260970]. The RNA-seq data for WT and Ada3-3KR are available in the GEO database under accession number GSE161887. [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161887]. The RNA-Seq accession number for the Flag-HDAC1 and Flag-HDAC1-S434A treated with or without H89 is GSE284216.
[https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE284216].
The CUT &Tag accession number for the HDAC1 treated with or without H89 is GSE285017. [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE285017]. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD058634. [https://www.ebi.ac.uk/pride/archive/projects/PXD058634]. Source data are provided with this paper.
References
Wellen, K. E. & Thompson, C. B. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell 40, 323–332 (2010).
Li, X., Egervari, G., Wang, Y., Berger, S. L. & Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 19, 563–578 (2018).
Lobel, G. P., Jiang, Y. & Simon, M. C. Tumor microenvironmental nutrients, cellular responses, and cancer. Cell Chem. Biol. 30, 1015–1032 (2023).
Torrence, M. E. & Manning, B. D. Nutrient Sensing in Cancer. Annu Rev. Cancer Biol. 2, 251–269 (2018).
Huisinga, K. L. & Pugh, B. F. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 13, 573–585 (2004).
Govind, C. K., Zhang, F., Qiu, H., Hofmeyer, K. & Hinnebusch, A. G. Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol. Cell 25, 31–42 (2007).
Grant, P. A. et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 11, 1640–1650 (1997).
Li, S. & Shogren-Knaak, M. A. The Gcn5 bromodomain of the SAGA complex facilitates cooperative and cross-tail acetylation of nucleosomes. J. Biol. Chem. 284, 9411–9417 (2009).
Huang, J. et al. Acetylation-dependent SAGA complex dimerization promotes nucleosome acetylation and gene transcription. Nat. Struct. Mol. Biol. 29, 261–273 (2022).
Chantranupong, L., Wolfson, R. L. & Sabatini, D. M. Nutrient-sensing mechanisms across evolution. Cell 161, 67–83 (2015).
Zaman, S., Lippman, S. I., Zhao, X. & Broach, J. R. How Saccharomyces responds to nutrients. Annu Rev. Genet 42, 27–81 (2008).
Hardie, D. G., Ross, F. A. & Hawley, S. A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012).
Coccetti, P., Nicastro, R. & Tripodi, F. Conventional and emerging roles of the energy sensor Snf1/AMPK in Saccharomyces cerevisiae. Micro. Cell 5, 482–494 (2018).
Efeyan, A., Comb, W. C. & Sabatini, D. M. Nutrient-sensing mechanisms and pathways. Nature 517, 302–310 (2015).
Garami, A. et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 (2003).
Loewith, R. et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 (2002).
Toda, T. et al. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol. Cell Biol. 7, 1371–1377 (1987).
Toda, T., Cameron, S., Sass, P., Zoller, M. & Wigler, M. Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 50, 277–287 (1987).
Yu, X. & Li, S. Specific regulation of epigenome landscape by metabolic enzymes and metabolites. Biol. Rev. Camb. Philos. Soc. 99, 878–900 (2024).
Wu, D. et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641 (2018).
Kadosh, D. & Struhl, K. Targeted recruitment of the Sin3-Rpd3 histone deacetylase complex generates a highly localized domain of repressed chromatin in vivo. Mol. Cell Biol. 18, 5121–5127 (1998).
McDaniel, S. L. & Strahl, B. D. Stress-free with Rpd3: a unique chromatin complex mediates the response to oxidative stress. Mol. Cell Biol. 33, 3726–3727 (2013).
Kadosh, D. & Struhl, K. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89, 365–371 (1997).
Li, X. et al. The TORC1 activates Rpd3L complex to deacetylate Ino80 and H2A.Z and repress autophagy. Sci. Adv. 9, eade8312 (2023).
Govind, C. K. et al. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell 39, 234–246 (2010).
Wagner, E. J. & Carpenter, P. B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 (2012).
Baker, L. A. et al. The yeast Snt2 protein coordinates the transcriptional response to hydrogen peroxide-mediated oxidative stress. Mol. Cell Biol. 33, 3735–3748 (2013).
Carrozza, M. J. et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 (2005).
Powers, R. W. et al. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184 (2006).
Pedruzzi, I. et al. TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol. Cell 12, 1607–1613 (2003).
Nikawa, J., Sass, P. & Wigler, M. Cloning and characterization of the low-affinity cyclic AMP phosphodiesterase gene of Saccharomyces cerevisiae. Mol. Cell Biol. 7, 3629–3636 (1987).
Yu, Q. et al. Phosphorylation of Jhd2 by the Ras-cAMP-PKA(Tpk2) pathway regulates histone modifications and autophagy. Nat. Commun. 13, 5675 (2022).
Sass, P., Field, J., Nikawa, J., Toda, T. & Wigler, M. Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 83, 9303–9307 (1986).
Peeters, K. et al. Fructose-1,6-bisphosphate couples glycolytic flux to activation of Ras. Nat. Commun. 8, 922 (2017).
Helmlinger, D. & Tora, L. Sharing the SAGA. Trends Biochem Sci. 42, 850–861 (2017).
Tamaki, H. Glucose-stimulated cAMP-protein kinase A pathway in yeast Saccharomyces cerevisiae. J. Biosci. Bioeng. 104, 245–250 (2007).
Sarokin, L. & Carlson, M. Upstream region of the SUC2 gene confers regulated expression to a heterologous gene in Saccharomyces cerevisiae. Mol. Cell Biol. 5, 2521–2526 (1985).
Carmona-Gutierrez, D. et al. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 17, 763–773 (2010).
Walter, D., Matter, A. & Fahrenkrog, B. Loss of histone H3 methylation at lysine 4 triggers apoptosis in Saccharomyces cerevisiae. PLoS Genet 10, e1004095 (2014).
Zhang, H., Kong, Q., Wang, J., Jiang, Y. & Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 9, 32 (2020).
Taylor, S. S. et al. PKA Cbeta: a forgotten catalytic subunit of cAMP-dependent protein kinase opens new windows for PKA signaling and disease pathologies. Biochem J. 478, 2101–2119 (2021).
Yang, X. J. & Seto, E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 (2008).
Sakai, M. et al. The GCN5-CITED2-PKA signalling module controls hepatic glucose metabolism through a cAMP-induced substrate switch. Nat. Commun. 7, 13147 (2016).
Ptacek, J. et al. Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 (2005).
Jessulat, M. et al. The conserved Tpk1 regulates non-homologous end joining double-strand break repair by phosphorylation of Nej1, a homolog of the human XLF. Nucleic Acids Res 49, 8145–8160 (2021).
Robertson, L. S. & Fink, G. R. The three yeast A kinases have specific signaling functions in pseudohyphal growth. Proc. Natl Acad. Sci. USA 95, 13783–13787 (1998).
Robertson, L. S., Causton, H. C., Young, R. A. & Fink, G. R. The yeast A kinases differentially regulate iron uptake and respiratory function. Proc. Natl Acad. Sci. USA 97, 5984–5988 (2000).
Lemaire, K., Van de Velde, S., Van Dijck, P. & Thevelein, J. M. Glucose and sucrose act as agonist and mannose as antagonist ligands of the G protein-coupled receptor Gpr1 in the yeast Saccharomyces cerevisiae. Mol. Cell 16, 293–299 (2004).
Van de Velde, S. & Thevelein, J. M. Cyclic AMP-protein kinase A and Snf1 signaling mechanisms underlie the superior potency of sucrose for induction of filamentation in Saccharomyces cerevisiae. Eukaryot. Cell 7, 286–293 (2008).
Varanasi, U. S., Klis, M., Mikesell, P. B. & Trumbly, R. J. The Cyc8 (Ssn6)-Tup1 corepressor complex is composed of one Cyc8 and four Tup1 subunits. Mol. Cell Biol. 16, 6707–6714 (1996).
Tzamarias, D. & Struhl, K. Functional dissection of the yeast Cyc8-Tup1 transcriptional co-repressor complex. Nature 369, 758–761 (1994).
Treitel, M. A. & Carlson, M. Repression by SSN6-TUP1 is directed by MIG1, a repressor/activator protein. Proc. Natl Acad. Sci. USA 92, 3132–3136 (1995).
Liu, Y. & Schneider, M. F. Opposing HDAC4 nuclear fluxes due to phosphorylation by beta-adrenergic activated protein kinase A or by activity or Epac activated CaMKII in skeletal muscle fibres. J. Physiol. 591, 3605–3623 (2013).
Doddi, S. K., Kummari, G., M, V. J. & Kalle, A. M. Protein kinase A mediates novel serine-584 phosphorylation of HDAC4. Biochem Cell Biol. 97, 526–535 (2019).
Wellen, K. E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Lei, W. W. et al. Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis. 1, e44 (2010).
Tonucci, F. M. et al. Identification of a CIP4 PKA phosphorylation site involved in the regulation of cancer cell invasiveness and metastasis. Cancer Lett. 461, 65–77 (2019).
Wu, Y. et al. Phosphoglycerate dehydrogenase activates PKM2 to phosphorylate histone H3T11 and attenuate cellular senescence. Nat. Commun. 14, 1323 (2023).
Zhang, S. et al. Metabolic regulation of telomere silencing by SESAME complex-catalyzed H3T11 phosphorylation. Nat. Commun. 12, 594 (2021).
Zhang, X. et al. Glc7/PP1 dephosphorylates histone H3T11 to regulate autophagy and telomere silencing in response to nutrient availability. Cell Discov. 9, 71 (2023).
Chen, W. et al. The SESAME complex regulates cell senescence through the generation of acetyl-CoA. Nat. Metab. 3, 983–1000 (2021).
Yu, Q. et al. Regulation of SESAME-mediated H3T11 phosphorylation by glycolytic enzymes and metabolites. PLoS One 12, e0175576 (2017).
Acknowledgements
We thank Li lab members for suggestions on this project. We especially thank J.L. Workman (Stowers Institute for Medical Research) for reagents. We thank Prof. F. Yu (Hubei University) for assistance with LC‒MS. This work was supported by funding from Natural Science Foundation of Hubei Province (2021CFA013, 2023BCA005 to S. L.) National Natural Science Foundation of China (32400458 to Q.Y.), Postdoctoral Fellowship Program of CPSF (GZC20230749 to Q.Y.), China Postdoctoral Science Foundation (2024M750849 to Q.Y.), Postdoctor Project of Hubei Province (2004HBBHCXA003 to Q.Y.).
Author information
Authors and Affiliations
Contributions
Conceptualization, X.Y. and S.L.; Experiments were performed by W.D., Q.Y., R.M., Z.Z., L.H., Y.Q.; data analysis was performed by W.D., Q.Y., Z.Z., R.M., L.H., F.H.; Mass spec analysis was performed by M.W., F.G.; Writing-original Draft, X.Y. and S.L.; Writing-Review and Editing, X.Y. and S.L.; Supervision, X.Y. and S.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fabrice Caudron, Maurizio Crestani and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dai, W., Yu, Q., Ma, R. et al. PKA plays a conserved role in regulating gene expression and metabolic adaptation by phosphorylating Rpd3/HDAC1. Nat Commun 16, 4030 (2025). https://doi.org/10.1038/s41467-025-59064-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59064-y
