
Abstract
Enzymes that catalyze long-range electron transfer (ET) reactions often function as higher order complexes that possess two structurally symmetrical halves. The functional advantages for such an architecture remain a mystery. Using cryoelectron microscopy we capture snapshots of the nitrogenase-like dark-operative protochlorophyllide oxidoreductase (DPOR) during substrate binding and turnover. DPOR catalyzes reduction of the C17 = C18 double bond in protochlorophyllide during the dark chlorophyll biosynthetic pathway. DPOR is composed of electron donor (L-protein) and acceptor (NB-protein) component proteins that transiently form a complex in the presence of ATP to facilitate ET. NB-protein is an α2β2 heterotetramer with two structurally identical halves. However, our structures reveal that NB-protein becomes functionally asymmetric upon substrate binding. Asymmetry results in allosteric inhibition of L-protein engagement and ET in one half. Residues that form a conduit for ET are aligned in one half while misaligned in the other. An ATP hydrolysis-coupled conformational switch is triggered once ET is accomplished in one half. These structural changes are then relayed to the other half through a di-nuclear copper center at the tetrameric interface of the NB-protein and leads to activation of ET and substrate reduction. These findings provide a mechanistic blueprint for regulation of long-range electron transfer reactions.
Similar content being viewed by others

Introduction
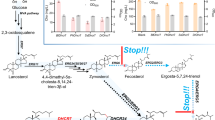
Photosynthetic organisms employ two distinct approaches for reducing protochlorophyllide (Pchlide; substrate) to chlorophyllide (Chlide; product), a key intermediate in the chlorophyll/bacteriochlorophyll biosynthetic pathway1. In angiosperms, this reductive step is catalyzed by the light-dependent Pchlide oxidoreductase (LPOR)2. In photosynthetic bacteria, cyanobacteria, green algae, and gymnosperms, this reaction is catalyzed in the dark by the ATP-dependent dark-operative protochlorophyllide oxidoreductase (DPOR)3. While these enzymes are not structurally related, they both catalyze the 2-electron reduction of the C17 = C18 double bond in Pchlide using differing mechanisms (Fig. 1A)4. DPOR shares structural homology with nitrogenase, the enzyme that catalyzes the reduction of dinitrogen to ammonia5,6, and thus is classified as a nitrogenase-like enzyme. Nitrogenase, DPOR, and other nitrogenase-like enzymes are composed of electron donor and electron acceptor component proteins7,8,9. In DPOR, the homodimeric BchL protein (L-protein) is the electron donor with an ATPase site per subunit and one [4Fe-4S]L cluster coordinated at the dimer interface (Fig. 1B)10. The electron acceptor BchN-BchB protein (NB-protein) is an α2β2 heterotetramer with two structurally identical halves (Fig. 1B)11. Each half possesses a [4Fe-4S]NB cluster and an active site for Pchlide binding and reduction. ATP binding to the L-protein promotes transient association of the component proteins, resulting in one electron being transferred to the substrate (Pchlide)12. Two such rounds of electron transfer (ET) are required to reduce Pchlide to Chlide.

A Schematic of the substrate reduction reaction catalyzed by DPOR. Two rounds of electron transfer are required to reduce the C17 = C18 double bond (shown in red) of Pchlide to form Chlide. B Crystal structure of P. marinus DPOR (PDB: 2YNM) stabilized in the presence of ADP-AlF3 depicting symmetrical binding of two L-protein homodimers to an NB-protein heterotetramer. The [4Fe-4S]L and [4Fe-4S]NB clusters, Pchlide, and ADP molecules are highlighted. C A 2.7 Å cryo-EM map of R. sphaeroides NB-protein. The subunits are colored light blue (BchN), dark blue (BchN’), light purple (BchB), and dark purple (BchB’), respectively. D Representative structure of the NB-protein showing the position of [4Fe-4S]NB cluster and the di-Cu site at the interface. E Carved electron density for the two helices coordinating the di-Cu site and the His-404 and Met-408 residues that interact with the Cu ions (colored orange) at the NB-protein tetramer interface. Water molecules are colored cyan. F Inductively coupled plasma mass spectrometry analysis of metal content shows a stoichiometry of 2 Cu ions bound per NB-protein. The Cu content for the H404A-M408A NB-protein mutant was near the level of detection. Mean values from two independent measurements are shown. G CW X-band EPR spectra of 500 µM NB-protein and of a 150 µM CuSO4 standard, as displayed on a g-scale. (i) CuSO4; (ii) 10-times magnified spectrum of anaerobic NB-protein; (iii) as-acquired spectrum of NB-protein. Conditions: T = 15 K, MW frequency, 9.37 GHz; Mod. Amp., 10 G; spectra cavity-background subtracted. H Pchlide reduction activity of NB-protein and the BchN-BchBH404A-M408A double mutant were assessed by adding L-protein and Pchlide (pre-bound to the respective NB-protein) in the presence of ATP-Mg2+. Alanine substitution of both His-404 and Met-408 residues that coordinate the di-Cu site abolishes the substrate reduction activity of DPOR. The apparent rate for Chlide formation under this single-turnover condition for wild-type NB-protein is 0.32 ± 0.02 µM.min−1. Std. Error from n = 3 experiments are plotted.
Structures of Rhodobacter capsulatus NB-protein in the absence/presence of Pchlide have been solved using X-ray crystallography11,13. Similarly, a structure of the Prochlorococcus marinus NB-protein bound to L-protein (DPOR complex) in a 1:2 stoichiometry was trapped in the presence of ADP-AlF3 (PDB: 2YNM; Fig. 1B)7. The general mechanistic assumption has been that ET events in the two halves are uncoupled. However, in both DPOR and nitrogenase, extensive allosteric communication exists between the two halves with events in one half regulating ET in the other14,15,16,17. Recent cryo-EM work on nitrogenase, performed under turnover conditions, further shows that the electron donor (Fe-protein) binds to only one half of the acceptor component complex (MoFe-protein), thereby introducing structural and functional asymmetry18. Turnover is defined as the activity of these proteins in the presence of ATP and substrate. In DPOR, we showed that the two halves of the NB-protein function in an asymmetric manner initiated upon Pchlide binding17. Thus, long-range allosteric communication between the NB-protein halves must exist to coordinate asymmetry, ensure proper delivery of the electron, and to distinguish between substrate (Pchlide) and product (Chlide) that differ by a single double bond at the C17 = C18 position (Fig. 1A). There are numerous steps in the DPOR catalytic cycle including Pchlide binding to both active sites in the NB-protein, engagement of ATP-bound L-protein to both halves of the NB-protein, ET, ATP hydrolysis (ADP & Pi dissociation), L-protein dissociation, and two repetitions of this cycle1,12.
Here, we used cryo-EM to capture the choreography of these events and establish how the two halves communicate through structural changes to function in an asymmetric manner. We uncovered a di-nuclear copper center (di-Cu site) at the center of the NB-protein tetrameric interface that is integral to propagating long-range allosteric communication in DPOR.
Results
Cryo-EM structure of NB-protein reveals the presence of a di-nuclear copper center at the tetramer interface
We solved a 2.7 Å cryo-EM structure of Rhodobacter sphaeroides (now reclassified as Cereibacter sphaeroides19) NB-protein in the absence of Pchlide (Fig. 1C, Supplementary Fig. 1, and Supplementary Table 1). Overall, the arrangement of the BchN and BchB subunits are similar to the previously published R. capsulatus NB-protein structure with one [4Fe-4S]NB cluster per half coordinated by 3 Cys and 1 Asp residues11. For clarity, the following nomenclature is used to distinguish the BchN and BchB subunits in the two halves: BchN, BchB, BchN′, and BchB′. One major difference in our structure is the presence of strong additional density at the tetrameric interface between the two halves (Fig. 1D). His-404 and Met-408 from both BchB and BchB′ protrude into this density and coordinate two Cu ions (Fig. 1E). The presence of two Cu ions per NB-protein complex was confirmed by inductively-coupled plasma mass spectrometry (ICP-MS) (Fig. 1F). However, the Cu in the NB-protein is EPR silent (Fig. 1G). As established by comparison with a 15 K X-band EPR spectra of CuSO4 as reference, there is no signature for mononuclear Cu(II) in the NB-protein, and no half-field transition associated with the S = 1 state of a Cu dimer. Instead, only a weak signal with g// ~ 2.11, g\(\perp\) ~ 2.01 is observed upon background subtraction, which we assign as the reduction of a small fraction of the EPR-silent [4Fe-4S]2+ cluster into an EPR-active [4Fe-4S]1+ cluster20,21,22,23. Thus, the Cu ions are either positioned as a dinuclear Cu(II) center (di-Cu site) that gives rise to a S = 0 spin state at 15 K, or reduced to Cu(I) by the excess dithionite in the buffer. In agreement with this interpretation, the additional density in the structure fits well to two Cu ions coordinated by the two His-404 and Met-408 residues along with two water molecules (Fig. 1E). To test whether the di-Cu site is important for substrate reduction, we substituted His-404 and Met-408 with Ala (DPORH404A-M408A) and measured Chlide formation in the presence of L-protein and ATP. ICP-MS analysis shows that no Cu ions are bound to the mutant NB-protein (Fig. 1F), and the mutant is catalytically inactive for Pchlide reduction (Fig. 1H). These findings suggest that the di-Cu site plays an essential role in DPOR activity.
Cryo-EM structure of the Pchlide-bound NB-protein reveals a path of allosteric communication that is routed through the di-Cu site
To better resolve the role of the di-Cu site in DPOR function, we solved a 3.0 Å cryo-EM structure of the NB-protein bound to Pchlide (Fig. 2A, Supplementary Fig. 2, and Supplementary Table 1). Overall, the structure determined using cryo-EM resembles the X-ray structure of Pchlide-bound NB-protein from R. capsulatus (PDB: 3AEQ & 3AEK) in terms of arrangement of the BchN and BchB subunits within the tetramer11. However, there are several key differences. Cryo-EM allows us to sample and average a wide array of conformational states. However, both heterogeneity and conformational flexibility contribute to local resolution. In our data, we believe that conformational flexibility is the major factor that dictates local resolution. A comparison of the local resolution differences between the top and bottom halves of the apo NB-protein, and when bound to Pchlide, reveals an intrinsic asymmetry between the BchN-BchB and BchN′-BchB′ halves. (Fig. 2B, C). Please note that the resolutions of the apo NB-protein (2.7 Å) and the Pchlide-bound NB-protein structures (3.0 Å) are different. Therefore, this analysis should be used as a coarse-grained overview of structural changes. In addition, the degree of movement and the heterogeneity of the sample would also contribute to the observed differences in local resolution. Thus, the following interpretations are only pertinent with respect to differences between the two halves of the NB-protein within each structure.

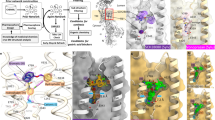
A A 3.0 Å cryo-EM map of R. sphaeroides NB-protein bound to Pchlide. B Local resolution analysis of the NB-protein-Pchlide complex is denoted as a heatmap. The scale of the heatmap represents regions of the structure that are higher (red) versus lower (blue) resolution. Asymmetry in the NB-protein is visible through more dynamic changes in one half of the complex. i.e., the lower half is more dynamic/flexible. An intrinsic path of dynamic changes can be traced from one half of the NB-protein to the other displaying a potential path of communication that traverses through the di-Cu site. C Local resolution map for the apo NB-protein displays lesser dynamic changes compared to the Pchlide-bound NB-protein complex in panel (B). One half is slightly more dynamic compared to the other, suggesting that asymmetry might be an inherent property that is enhanced upon Pchlide binding. B-factors for individual amino acids are plotted for the BchN/BchN′ (top panel) or the BchB/BchB′ (bottom panel) subunits from each half in the (D) Pchlide-bound BchNB and (E) Apo-BchNB structures. A higher degree of changes is observed in the BchN subunit compared to BchB.
A plot of the B-factor changes across the individual amino acids in each structure reveals that the changes are more prominent in the BchN/BchN′ subunit compared to BchB/BchB′ (Fig. 2D, E). Moreover, these changes are enhanced in the Pchlide-bound BchNB structure, suggesting that asymmetry is further selectively imprinted by Pchlide binding. The differences are also noticeable when the root mean square deviation (RMSD) values between the two halves in the structures are compared (Supplementary Fig. 3). In both the Pchlide-bound and apo-NB-protein structures, there is a noticeable ~ 0.6 Å difference between the two halves, suggesting an intrinsic asymmetry within the NB-protein. The conformational movements are marginally higher in the Pchlide-bound NB-protein structure compared to the apo-NB structure. Interestingly, in both structures, a path of dynamic changes can be traced from one half to the other and routes through the di-Cu site (Fig. 2B, C). To further probe these conformational changes, we reassessed the Pchlide-bound NB-protein structure using C2 symmetry with no relaxation. In this case, asymmetric dynamic changes between the two halves will be averaged. While the resolution of the structure marginally changed from 3.0 Å to 2.9 Å, the path of dynamic changes passing through the di-Cu site was still prominent (Supplementary Fig. 3C). Collectively, these coarse-grain observations raise an intriguing possibility for the presence of an allosteric communication path between the two halves in the NB-protein. We recently uncovered evidence for such allosteric communication in DPOR using a half-reactive NB-protein where perturbation of substrate sensing within one half completely abolished ET and substrate reduction in the entire complex17.
Cryo-EM structure of the DPOR complex under single-turnover conditions reveals asymmetric and sequential complex formation between the donor and acceptor component proteins
In DPOR and nitrogenase, we showed that ET was asymmetric and sequential14,15,17. Recent cryo-EM studies of nitrogenase under turnover conditions revealed that this asymmetry extends to binding of only one donor component (Fe-protein) to the acceptor component protein (MoFe-protein)18. To test whether the intrinsic asymmetry in the NB-protein resulted in asymmetric binding of the L-protein, and to better understand the role of the di-Cu site, we solved the structure of DPOR under single-turnover conditions (Fig. 3A, B, Supplementary Fig. 4, and Supplementary Table S2). Single-turnover conditions were achieved by first pre-forming the Pchlide-NB-protein complex and removing unbound Pchlide using size-exclusion chromatography. The Pchlide-bound NB-protein was then mixed with an excess of L-protein (8 fold excess over NB-protein) and 3 mM ATP-Mg2+ for 5 minutes, applied onto the grids, frozen, and imaged. The time point was chosen based on DPOR activity assays that were performed under identical single-turnover conditions (Fig. 1H). The ATPase rate of DPOR is very slow (kcat ~ 0.08 s-1)24, thus an ATP-regeneration system was not required to maintain levels of ATP in the reaction. The 2D classification analysis of the dataset shows the presence of unbound L-protein homodimers, NB-protein tetramers, and NB-protein tetramers bound to one L-protein dimer (Fig. 3A and Supplementary Fig. 4). We were able to solve a 3.68 Å structure of the one L-protein bound NB-protein complex during turnover (Fig. 3B).

A Representative subset of the 2D classes from the single-turnover cryo-EM analysis at the t = 5 min time point shows the presence of free L-protein, free NB-protein, and only one L-bound NB-protein complex. B A 3.7 Å cryo-EM map of DPOR solved under single turnover conditions in the presence of ATP. During turnover, at t = 5 min post addition of ATP, only one L-protein is bound to the NB-protein. The two BchL subunits (L1 and L2) are shown in dark and light green, respectively. C 2D classes of DPOR in the presence of ADP-AlF3 show free L-protein, free NB protein, and only two L-bound NB-protein complexes. D A 3.7 Å cryo-EM map of DPOR in the presence of ADP-AlF3 (transition state) shows two L-proteins bound to the NB-protein. E 2D-classes from a single-turnover cryo-EM dataset at t = 30 min timepoint show the presence of free L-protein, free NB-protein, one L-bound NB-protein complex, and (F) a few classes that show hazy density for an additional L-protein bound to NB-protein. The bottom panel shows the same 2D class images with higher contrast for visualization. The white arrows denote the position of L-proteins in the DPOR complex.
Interestingly, in the 5 min dataset, we observed no evidence for the hetero-octameric (BchL)2-(BchN-BchB)2-(BchL)2 complex as captured for Prochlorococcus marinus DPOR in the presence of ADP-AlF3 (PDB: 2YNM; Fig. 1B)7. To better understand this discrepancy, we solved a 3.7 Å cryo-EM structure of R. sphaeroides DPOR in the presence of ADP-AlF3 (Fig. 3C & D, Supplementary Fig. 5 and Supplementary Table 2). Under these conditions, 2D classification shows the presence of free L-protein and NB-protein molecules in addition to complexes where NB-protein is bound to two L-proteins (Fig. 3C, Supplementary Fig. 5, and Supplementary Table S2). Surprisingly, we did not observe 2D classes that show evidence for the one L-protein-bound NB-protein complex. One explanation for the difference in oligomeric states between the turnover (ATP) versus transition-state (ADP-AlF3) complexes is that asymmetry during normal catalysis results in asymmetric substrate reduction activity between the two halves. In other words, there is a defined order of events dictated by asymmetry within the Pchlide-bound NB-protein.
The ability to capture the two L-bound states with the ATP-analog suggests that this transition state must exist at some point during turnover. We reasoned that assessing the single-turnover DPOR complex after thirty minutes of incubation might help trap the state where NB-protein might be bound to two L-proteins, as the kinetics of substrate reduction by DPOR is quite slow, as reflected by the timescale of Pchlide reduction under the cryo-EM conditions (Fig. 1H). Cryo-EM data were collected on a 30-min incubated DPOR turnover sample and 2D classification reveals unbound L-protein, NB-protein, and predominantly single L-protein bound NB-protein tetramer complexes (Fig. 3E). In this dataset, we do observe a few 2D classes where density for one L-protein bound to the NB-protein tetramer is well defined, but hazy density for what appears to be a second L-protein is also observed (Fig. 3F). Unfortunately, the lack of sufficient particles for this class along with significant motion precluded us from obtaining meaningful structural information. These findings resolve the differences in complex stoichiometry between the turnover and transition-state structures of DPOR. The data also establish that L-protein binding to NB-protein during turnover is asymmetric and sequential. The asymmetric properties with respect to binding of L-protein are similar to observations for nitrogenase during turnover, where only one Fe-protein is bound to the MoFe-protein18.
Pchlide molecules bound in the active site of the NB-protein are non-planar during turnover
In our structures, Pchlide is bound to both active sites in the NB-protein. However, they are not strictly planar as observed in the crystal structures7,11. In the Pchlide-bound NB-protein structure, there is a small difference in the conformation of Pchlide molecules bound in each active site (Fig. 4A and B). These differences are minimized or lost when the structure is solved using C2 symmetry (Fig. 4C & D). Therefore, the differences in the Pchlide densities when C2 symmetry is not imposed suggest that the conformations sampled within the two active sites are not similar at any given instance. Thus, asymmetry in the NB-protein is likely enhanced/propagated upon Pchlide binding. In addition, the conformation of the Pchlide molecules bound to the two active sites in the turnover and the transition-state complexes shows striking differences (Fig. 4E–H). In these structures, the electron density for the Pchlide molecules do not fit well to planar conformations. In addition, the shape of the Pchlide densitities in each half of the NB-protein differ. While the best-fitting models for Pchlide are represented in each structure (Fig. 4), the ensemble of conformations for Pchlide captured in cryo-EM precludes us from obtaining higher resolution structural information to resolve bond angles or calculate the degree of bending. Nevertheless, based on comparisons of the averaged electron densitities within the two active sites in each structure, we reason that the Pchlide conformations are different. This is especially prominent during turnover, suggesting that the active site likely probes and differentiates between substrate (Pchlide) versus product (Chlide) by sensing the differences in bendability. Since Pchlide and Chlide only differ by a single double-bond (Fig. 1A), local changes in energetics within the active site through bending/puckering likely drives binding of Pchlide and dissociation of Chlide. The structural asymmetry observed in the NB-protein upon binding of Pchlide also suggests that asymmetry is introduced or enhanced upon substrate binding in agreement with our previous biochemical observations17.

A, B Carved electron density for the Pchlide molecules bound to each active site in the structure of the Pchlide-bound NB-protein cryo-EM structure. The Pchlide molecules are non-planar, and (C, D) when analyzed using C2 symmetry, these changes average out. E–H Carved electron density for the Pchlide molecules in the single-turnover and transition-state DPOR structures are shown. Asymmetric bending/puckering of the Pchilde molecules are visible during both conditions. The Pchlide molecules are colored pink and purple to denote the two active sites in the NB-protein tetramer. The green sphere in the middle of each Pchlide molecule is Mg2+.
Conventionally, in crystal structures of other chlorophyll-associated enzymes, chlorophyll or precursor molecules such as Pchlide are observed as planar rings within the active sites25. Thus, the aromatic rings are reductively construed as flat and rigid molecules. However, many in-solution and computational studies of tetrapyrrole molecules such as porphyrins, have described the existence of various shapes such as saddles, ruffles, domes, and waves26,27,28. The energetics to bend a Pchlide molecule are likely not driven by minimal local movements alone within the active site. We tested this idea through evaluation of the stability of the Pchlide-bent conformation using a combination of DFT and QM/MM calculations. We used the Pchlide molecules from our DPOR structure obtained under single-turnover conditions, which were further modified to include an axial water ligand to the Mg2+ ion, consistent with Pchlide structure analogues in the literature29,30,31,32,33. Both the unconstrained DFT and the protein-constrained QM/MM calculations resulted in rapid unfolding of the starting constrained ring within its binding pocket leading to significant stabilization of the model by ~ 246 kcal/mol (243 and 248 kcal/mol for QM and QM/MM measurements, respectively; Supplementary Fig. 6). This indicates that the bending of Pchlide observed in the structures cannot be induced through local changes alone. Both the DFT and QM/MM predictions do suggest that the Mg2+ ion is pulled out of the Pchlide plane by ~ 0.5 Å which is consistent with a distortion of the planar ring (Supplementary Fig. 6). Thus, long-range, and collective motions across the entire DPOR complex likely contribute to the distortion of Pchlide.
Asymmetry in the NB-protein is propagated through helix remodeling, Cu coordination, and Pchlide sensing in trans
The Cu ions in our NB-protein structures are coordinated by Met-404 and His-408 residues that are positioned in a long helix (residues 398–420) in the BchB subunit (Fig. 1E). The helix from one BchB subunit is positioned such that it interacts in trans with Pchlide bound in the other half of the NB-protein. Met-404 and His-408 are well conserved in Rhodobacter (Supplementary Fig. 7), thus how this helix mediates substrate binding and reduction remains to be established. Interestingly, in the X-ray structures, this helix is unbent under apo conditions, but is bent in the Pchlide-bound structure11 (Fig. 5A). Therefore, the bending of this helix was proposed as a mechanism for substrate binding in R. capsulatus DPOR11. In contrast, this helix is intact and does not bend in our cryo-EM structures in the absence or presence of Pchlide (Fig. 5B). In our turnover DPOR structure, we do not observe bending of this helix (Fig. 5C). However, in our transition-state DPOR structure, we observe additional density around Pchlide that when resolved shows two alternate conformations that depicts an average of the straight and bent states (Fig. 5D, and Supplementary Fig. 8). Residues 410–415 reside at the end of this helix and His-413 is poised to interact with Mg2+ in Pchlide. How these interactions contribute to Pchlide binding and reduction are not clear. His coordination of Mg2+ in chlorophyll molecules has been captured in other photosynthetic proteins34. The two conformations captured in this region, and their movement during turnover, suggest that such interactions occurring in trans are an integral part of the asymmetric substrate reduction mechanism of DPOR.

A In the crystal structures of R. capsulatus NB-protein, distortion of the two helices is observed upon Pchlide binding and thus proposed as a feature utilized for substrate binding. B In our cryo-EM structures of the NB-protein, we do not observe such structural changes within these helices in the absence or presence of Pchlide, or during turnover in the DPOR complex. C, D In our cryo-EM transition-state DPOR structure, two alternate conformations are observed where the helices show a transition to the distorted form. His-413 is poised to interact with the Mg2+ in Pchlide when the helix is distorted.
The density around the Cu ions vary in our cryo-EM structures, suggesting that movements in the helix could be enhanced, coordinated, and/or regulated by the di-Cu site. In the apo structure, Met-408 adopts two conformations, but becomes ordered in the Pchlide-bound, turnover, and transition-state structures (Supplementary Fig. 9). In the previously solved X-ray structures of NB-protein, there are no reports of bound Cu (Supplementary Fig. 10A-C)7,11,13. For some of these structures, NB-proteins were endogenously purified from Rhodobacter capsulatus11. Thus, we reassessed the deposited electron density maps of NB-protein bound to Pchlide (PDB: 3AEQ/3AEK) and indeed find additional density at the tetramer interface. As deposited, water molecules are modeled into the additional density (Supplementary Fig. 10B). We model two Cu molecules within this density with excellent coordination by two His residues (Supplementary Fig. 10D). Surprisingly, in the apo structure (PDB:3AER), this additional density is missing (Supplementary Fig. 10A). Crystallization conditions for the apo structure contained 0.2 M ammonium chloride11 and ammonium salts are known to interact with and precipitate Cu35. Thus, over the course of crystallization, the Cu ions were likely sequestered away by ammonium salts. We propose that the di-Cu site coordinates communication within DPOR by relaying information through the two helices. The ability to sense Pchlide/Chlide in trans could also be used as a mechanism to keep the electron transfer reactions in register with respect to L-protein binding and to maintain asymmetry within the DPOR complex. The path of asymmetric dynamic changes that are routed through the di-Cu site in the Pchlide-bound NB-protein structure (Fig. 2B) supports such a model.
Conformational changes during turnover reveal a path of allosteric communication in DPOR
To test whether the observed structural changes in DPOR are maintained and propagated through turnover, we assessed the dynamic changes in our turnover and transition-state cryo-EM structures (Fig. 6). To understand the differences in complex formation during turnover (ATP-Mg2+) versus under transition-state (ADP-AlF3) conditions, we compared the differences in the local resolution between the two halves of the NB-protein within each structure (Fig. 6A and B). While the resolutions of the two structures are relatively similar (3.68 versus 3.7 Å), the following interpretations might be more appropriate if compared with respect to differences between the two halves of the NB-protein within each structure. In the turnover data, asymmetric dynamic changes are pronounced in one half compared to the other, and L-protein binding induces relative stability to one half of the NB-protein. In both these structures, analysis of the B-factor changes within the NB-protein reveals significant differences between BchN and BchN′ compared to the BchB/BchB′ subunits (Fig. 6C and D). Such asymmetric changes are predominantly smaller in the presence of ADP-AlF3. Nevertheless, the path of allosteric changes is strikingly visible in both structures and the route through the di-Cu site at the NB-protein interface (Fig. 6A and B).

Local resolution maps for the structures of DPOR under (A) single-turnover and (B) transition state are shown. Both structures show robust dynamic changes. Stark asymmetrical and dynamic changes are observed in the turnover complex. The asymmetry, while present, is considerably muted in the transition-state complex. Under both conditions, the path of allosteric communication between the two halves is prominently observable and routes through the di-Cu site in the NB-protein. B-factors for individual amino acids are plotted for the BchN/BchN′ (top panel) or the BchB/BchB′ (bottom panel) subunits from each half in the (C) DPOR turnover and (D) DPOR transition state structures. The BchN subunits show more differences compared to BchB.
We observe intrinsic asymmetry in the NB-protein that is enhanced upon Pchlide binding, and further propagated by asymmetric binding of L-protein. Thus, we sought to understand how asymmetry is used for ET and substrate reduction in DPOR. Aromatic residues are commonly used as relays for long-range ET36. Thus, we analyzed the conformational changes in key aromatic residues in the NB-protein situated between the [4Fe-4S] clusters and the di-Cu site (Fig. 7A and Supplementary Fig. 11). An overlay of the structural changes in NB-protein between the apo, Pchlide-bound, and turnover cryo-EM structures reveals a snapshot of how allosteric communication is likely propagated in DPOR. A direct comparison of these changes between the two halves reveals asymmetric movements in key residues (Supplementary Movie 1). Trp-6, Phe-28, Trp-36, Phe-67, Tyr-277, Trp-390, Phe-396, and Phe-418 all show major asymmetric sidechain conformations. In addition, the imidazolium ring of His residues also participates in ET37. His-13, His-378, His-413, and His-56 residues all display asymmetric side-chain conformations. Finally, His-404 and Met-408 that coordinate the di-Cu site also display asymmetric conformational changes, suggesting that the Cu ions and the associated helix are integral to allosteric control in DPOR (Supplemental Movie 1).

A A superposition of the NB-protein from the three cryo-EM structures (apo, Pchlide-bound, and under single-turnover) is shown, and the key residues, the [4Fe-4S]NB cluster, and the di-Cu site are depicted. Overall changes in the environment surrounding the substrate and clusters are visible, and asymmetric motions are observed between the two halves of the NB-protein. The residues are colored to match the individual NB-protein subunits and shaded to correspond to the NB-protein apo (light), Pchlide-bound NB-protein (medium), and DPOR turnover (dark) structures. Residues colored pink, blue, purple, and green represent BchN, BchB, BchN′, and BchB′, respectively. The * denotes residues that interact across the two halves in trans. We propose that the transfer of an electron in the left half of the NB-protein (bound to L-protein) is promoted due to the proper alignment of residues that favor ET. These residues are not aligned in the other half leading to the allosteric suppression of L-protein binding and conditions that do not favor ET. B Superposition of two NB-protein halves in the DPOR-turnover structure. Select amino acids that show conformational changes between the two halves are shown. Asp-374 (red) and the C17 = C18 double bond in Pchlide (green) are highlighted. Distances between the di-Cu cluster and the C17 = C18 bond (and His-378) and the [4Fe-4S]NB cluster are shown. The blue and pink arrows denote speculative paths of electron transfer from the [4Fe-4S]NB to Pchlide.
We propose that structural asymmetry is used to facilitate ET in one half, while suppressing it in the other. Residues situated between the [4Fe-4S]NB and Pchlide (and likely the di-Cu site) would serve as conduits for ET. Thus, we compared the two halves of the NB-protein in the DPOR turnover structure to identify potential residues in this path (Fig. 7B and Supplementary Fig. 12). The electron must be transferred from the [4Fe-4S]NB to the C17 = C18 double bond, which is situated adjacent to Asp-274, which was shown to donate a proton to Pchlide (Fig. 1A)11,12. An Asp-274 to Ala substitution stalls DPOR activity after the first ET in one half of the complex17. His-38, Trp-36, Phe-396, & Trp-390 from BchN and Phe-418, Tyr-277 & Asp-274 from BchB are aligned to form one potential path of ET. In this path, Tyr-277 and Asp-274 act in trans. These residues accommodate different conformations in one-half versus the other during turnover (Fig. 7B). Tyr-38, Trp-6, Leu-42, and His-378 from BchB are positioned in line for ET along another potential path. His-404 (in trans) is positioned to coordinate the di-Cu site. These residues also accommodate different conformations in the two halves during turnover. So, movement of one or more of these residues in either path within one-half would block ET. These alternating movements are likely coupled to L-protein binding and ATP turnover, thus forming the basis for asymmetric ET in DPOR. We emphasize again that this structure of DPOR was solved during turnover, and thus, a few technical caveats need to be accounted for. Unlike in the case of nitrogenase18, the substrate for DPOR is a large Pchlide molecule. Therefore, the movements of residues in the active site are extensive during ET and Pchlide reduction. Our interpretations of the side chain conformations around the active site are based on lower resolution structural data (Supplementary Fig. 12). In addition, we have also not captured the L-protein in a state when ET occurs between the [4Fe-4S]L and [4Fe-4S]NB clusters (discussed below). Thus, one should also consider that additional conformational changes likely transpire in the path of allostery.
Binding of L-protein is promoted through asymmetric interactions with NB-protein and regulated by the disordered N-terminus
In both our turnover and transition-state DPOR structures, the position of L-protein bound on NB-protein is rotated by ~ 45–50° in comparison to the crystal structures (Fig. 8A and Supplementary Fig. 13). As a consequence, the distance between the [4Fe-4S]L and [4Fe-4S]NB clusters is ~ 39 Å compared to ~ 14 Å in the crystal structure (PDB: 2YNM; Supplementary Fig. 14). Closer positioning of the two FeS clusters likely promotes ET. Thus, a rolling motion of L-protein between the two states might be required for ET, as established for the nitrogenase complex14,18,38,39,40,41. However, while the different nucleotide-dependent engagement states of the two proteins and the rolling-motion phenomenon have been established for the nitrogenase system39, such movements have not been shown for DPOR. In our structures, the two BchL subunits are positioned such that they make contact with only the BchB subunit (Fig. 8B and C, and Supplementary Fig. 15). In the turnover complex, the binding is promoted through several salt-bridges between L-protein and the BchB subunit (Fig. 8B). In the transition-state complex, additional contacts further stabilize binding, suggesting a slightly different binding conformation (Fig. 8C). Interestingly, these contacts vary between the two docked L-proteins in contrast to symmetrical contacts observed in the X-ray structure (PDB:2YNM)7.

A Superposition of the P. marinus transition-state DPOR crystal structure (PDB: 2YNM in gray) with the cryo-EM transition-state structure (both solved in the presence of ADP-AlF3) shows a large ~ 45° difference in the docking positions of the respective L-proteins on the NB-protein. Salt-bridge interactions between the L-protein and the NB-protein in the (B) turnover complex and (C) transition-state complex are shown. D In the cryo-EM structure of DPOR under turnover conditions, the disordered N-terminal regions of BchL (colored green; residues 7 to 30) are ordered and situated at the binding interface between the two component proteins. E A minor fraction of the 2D classes in the transition-state cryo-EM dataset show L-protein bound in the two conformations shown in panels (C, E). The bottom panel is a high contrast image shown for better visualization. The white arrows denote the L-proteins.
Why are the L-proteins positioned differently in our cryo-EM structures of R. sphaeroides DPOR compared to the crystal structure of P. marinus DPOR? We had previously uncovered that a disordered region in the N-terminus of L-protein is autoinhibitory to function and binds across the [4Fe-4S]L cluster42. We showed that ATP binding to L-protein remodels this interaction, relieves auto-inhibition, and promotes L-protein binding and subsequent ET to the NB-protein. In our cryo-EM structures, the [4Fe-4S]L cluster is buried by the disordered region (Fig. 8D). Thus, the disordered N-terminal region of L-protein must be released in order to transition to the ET-permissive state. So, our interpretation is that during turnover, the L-protein accommodates a minimum of two states. In state-1, the L-protein docks on top of the NB-protein, transfers the electron, and rapidly slots into state-2, an orientation where it is on the side of the NB-protein as seen in our structures (Fig. 8). Such structural transitions are also coupled to ATP binding and hydrolysis by the L-protein as shown by our in-solution biochemical analysis42.
A potential explanation for not observing state-1 in our cryo-EM studies could be its transient nature. We reanalyzed the 2D classes in the transition-state cryo-EM data and found a very minor subset (~ 3.8 %) to showcase additional hazy density that corresponds to L-protein sampling two positions on the NB-protein (Fig. 8E). Thus, the state primed for ET that is driven upon release of the N-terminal disordered region of the L-protein is transient and, therefore, not populated in our cryo-EM dataset. We speculate that this feature might be distinct to Rhodobacter DPOR and evolved to protect the integrity of the cluster42. Since the L-protein is dynamic in both our cryo-EM structures, precise determination of the nucleotide occupancy was difficult. Using a linked L-protein where the ATPase site was mutated in only one subunit, we showed that substrate reduction activity is lost42. Thus, L-protein binding and sampling these two states (possibly rolling) on the NB-protein is likely coupled to asymmetric ATP binding and hydrolysis within the L-protein and further regulated by the disordered N-terminal region.
Discussion
DPOR and other nitrogenase-like enzymes share a common overall structural resemblance to nitrogenase. Recent cryo-EM data on nitrogenase along with data for DPOR, presented here, show that these protein complexes function asymmetrically18,43. For DPOR, asymmetry appears to be intrinsic to the NB-protein and enhanced upon Pchlide binding. This asymmetry from within the NB-protein then dictates which half interacts with the L-protein. Based on our structural work, we propose that allosteric communication can transpire through dual paths. The first arises from changes within the active site upon Pchlide binding, that is then propagated to the L-protein binding interface on the NB-protein. The second level of regulation is initiated upon L-protein binding, which likely suppresses L-protein binding on the opposite BchNB′ interface, which is situated ~ 100 Å away. The discovery of the di-Cu site at the heart of the NB-protein tetramer and the associated helices that interact with Pchlide and the active sites in trans adds another layer of complexity to the ET and substrate reduction mechanisms.
The relationships between electron flow from the [4Fe-4S]NB cluster of the NB-protein to Pchlide and/or from the di-Cu site remain to be resolved. The Pchlide is situated ~ 18.7 Å from the di-Cu site compared to ~ 9–12 Å from the [4Fe-4S]NB cluster of NB-protein (Supplementary Fig. 16). Therefore, it is not unreasonable to assume that the di-Cu site might also play a direct role in the ET mechanism from the FeS cluster to Pchlide. The [4Fe-4S]NB cluster of NB-protein is pre-reduced, and we have shown that this electron is transferred to Pchlide in the absence of L-protein using a deficit spending-like mechanism as shown for nitrogenase17,44. This is also evident in our single-turnover Pchlide reduction experiments, where half the Pchlide is reduced within 10 min whereas the reduction of the second Pchlide takes more than an hour (Fig. 1H). This would suggest that the enzyme is able to track the number of electrons transferred at any given point in the reaction.
The number of electrons transferred to the substrate could be sensed through differential conformational changes and/or bending/puckering of the Pchlide and Chlide molecules. While speculative, we envision that transfer of the first electron upon Pchlide binding enhances the asymmetry and differential bending of the Pchlide molecules (Fig. 4). We also propose that such conformational sensing of substrate versus product within the active site is relayed through the helices that interact in trans and coordinated by the di-Cu site. The bent versus straight conformations of this helix, captured in both our study and previous X-ray structures of DPOR11 (Fig. 5) lend support this is mechanistic feature.
The second electron required to complete Pchlide reduction is provided upon L-protein binding. This second ET step transpires rapidly in one half. The long-range allosteric changes and negative cooperativity occur on a much slower time scale, resulting in slower binding of L-protein on BchNB′. Another puzzle is the coupling of ATP utilization to the remodeling of the inhibitory N-terminal region of the L-protein. We established that ATP binding to the two BchL subunits releases the inhibition in both subunits, resulting in the rapid docking and immediate ET (Fig. 9)42. The subsequent asymmetric ATP hydrolysis in one BchL subunit triggers the movement to a post-ET state where the N-terminal disordered region has collapsed to the inhibitory state captured in our cryo-EM structures. When and how this movement triggers L-protein binding in the opposite half of BchNB′, along with a host of other mechanistic queries, remain a fascinating mystery to be interrogated. How does the di-Cu site and its redox state control asymmetry? How and when is ET activated in the suppressed half? Does this occur upon ATP hydrolysis and L-protein rolling/dissociation in the first BchNB half upon Pchlide to Chlide reduction?

A model for asymmetric events during the electron transfer (ET) cycle in DPOR. Release of the disordered N-terminal regions in the L-protein upon ATP binding is a prerequisite for binding to the NB-protein to initiate ET. Since the [4Fe-4S]NB cluster is pre-reduced, the first electron is transferred to Pchlide through a deficit-spending-like mechanism. Whether this happens on both sides or allosterically suppressed in one half remains to be elucidated. The Binding of the reduced L-protein drives the second ET event leading to Pchlide reduction in one active site. The docking position of the L-protein on the NB-protein that is permissive for ET and how these movements are coupled to ATP hydrolysis remains to be established. The timing of L-protein binding to the other half, the associated regulatory events, and when the L-protein dissociates are speculative.
The structural work presented here opens up a fascinating series of questions regarding the mechanisms of allosteric control in long-range ET in nitrogenase-like enzymes. The asymmetry shown here for the nitrogenase-like DPOR and in nitrogenase14,15,18,43 have also been shown in other enzymes that catalyze long-range ET reactions. Ribonuclease reductase (RNR)45,46 and Nitric oxide synthase (NOS)47 are two examples where structural and biochemical studies have unraveled similar asymmetric mechanisms of action. Thus, such structural and functional principles of asymmetric ET are likely a feature of other oxidoreductase enzyme complexes in nature.
Methods
Plasmids
Plasmids for recombinant overproduction of BchL and BchNB were generated as previously described17,48,49,50. Briefly, the open reading frames for BchB and BchN were PCR amplified from Rhodobacter sphaeroides genomic DNA and cloned into the multiple cloning sites 1 and 2 of a pRSF-Duet1 vector, respectively. BchB was designed to carry an N-terminal 6X poly-histidine tag followed by a 3 C protease cleavage site. Please note that the addition of an affinity tag to either the N- or C-terminus of BchN leads to loss in activity17. The BchL open reading frame was similarly PCR amplified and cloned into pRSF-Duet1 with an N-terminal 6X poly-histidine tag followed by a 3 C protease cleavage site. The H404A-M408A NB-protein double point mutant was generated using Q5 site-directed mutagenesis (New England Biolabs).
Protein expression and purification
Wild-type and mutant DPOR component proteins (L-protein and NB-protein) were purified separately using SufFeScient E. coli BL21(DE3) cells (PK11466) as previously described17,48,49,50 with minor modifications. Transformants were grown in 4 to 6 L Luria-Bertani (LB) medium in acid-washed (1 % HCl in water) baffled flasks, supplemented with 1 mM L-cysteine and 1 mM iron (III) citrate. Cultures were grown at 37 °C with shaking at 200 rpm until the optical density of the cultures at 600 nm reached 0.6-0.8. Surfactants should be avoided during cell growth as they lead to degradation/oxidation of the FeS clusters. Protein expression was induced by the addition of Isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. Cultures were grown for an additional 12 h with shaking at 100 rpm at 17 °C. The cells were transferred to centrifuge bottles with rubber gaskets and incubated with 1.7 mM sodium dithionite for 3 h at 17 °C. Care should be taken after this step, and the bottles should not be opened to avoid exposure to air. Cells were harvested by centrifugation, and the bottles were moved into a Coy Lab anaerobic chamber (glove box) under a 95 % N2/5 % H2 mixture before decatenation. Further lysis and purification steps were all performed under anoxygenic conditions and inside the glove box as previously described17,42,48,49. All buffers were purged of air under ultrahigh-purity N2 on a home-built Schlenk line and stored under N2. Cell pellets were resuspended in ~ 100 ml of Buffer A (100 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, 10% glycerol, and 1.7 mM of sodium dithionite) supplemented with 3X protease inhibitor cocktail (Sigma Inc.) and 0.1 mg/ml Lysozyme. After incubation at room temperature for 30 min, the cells were lysed by sonication (pulse of 1 sec ON and 3 sec OFF for a total of 3 min inside the glove box), and the lysate was centrifuged at 37,157xg for 60 min at 4 °C. The clarified supernatant was loaded onto a 5 ml Ni2+-NTA column (Gold Biotechnology Inc.) equilibrated with Buffer A. Both NB-protein and L-protein complexes were eluted with a step-gradient of imidazole. Fractions containing protein were brown in color and were analyzed on SDS-PAGE for purity (~ 95% pure). These fractions were pooled and concentrated using an Amicon spin concentrator (30 kDa cutoff) and further fractionated on a S200 HiLoad 26/600 size exclusion chromatography column (Cytiva Healthcare) pre-equilibrated with Buffer A containing no glycerol. Protein-containing fractions were pooled, concentrated, and aliquoted into 1.2 ml cryo-tubes with a gasket-sealed cap (Corning Scientific). Closed tubes with protein were flash frozen in the glove box gas exchanger chamber using liquid nitrogen and stored under liquid nitrogen. Protein concentrations were determined using Bradford reagent with bovine serum albumin (BSA) as the reference.
Generation of Pchlide
Pchlide was generated from a Rhodobacter capsulatus ZY-5 strain harboring a deletion of the BchL gene4,51 (a kind gift from Dr. Carl Bauer, Indiana University) and purified as described17,48,49,50. Briefly, ZY-5 cells from a frozen glycerol stock were streaked onto an RCV-2/3 agar plate with 0.5 mg/L kanamycin and incubated overnight at 37 °C. The plate was then moved to room temperature, covered with aluminum foil, and allowed to incubate for 2-3 more days. Dark brown colored colonies usually appear in ~ 2-3 days. A single brown-colored transformant was first grown overnight in 10 ml RCV-2/3 media with 0.5 mg/L kanamycin at 37 °C. This starter culture was then transferred to an aluminum foil-wrapped flask containing 250 mL RCV 2/3 media with 0.5 mg/L kanamycin and grown overnight at 34 °C at 130 RPM. After overnight incubation, the media turned green/dark brown in color, indicating the accumulation of Pchlide. Supernatant containing Pchlide was collected after centrifuging the culture at 4200 RPM for 30 min. At this stage, if more Pchlide is required, the pelleted cells can be resuspended with fresh RCV-2/3 media containing 0.05 mg/L of kanamycin, and the growth can be extended overnight. The supernatant from 5 such collections were pooled, covered in aluminum foil, and stored at 4 °C till further processing. The pooled supernatant solution was spun at 5000 RPM for one hour and filtered through a 0.44 μm Whatman #2 paper filter. The supernatant containing Pchlide was extracted with 1/3 volume of di-ethyl ether in a separating funnel inside a fume hood. The mixture was shaken vigorously and allowed to separate. The aqueous and organic phase layers separate over time. The organic phase contains Pchlide and is subsequently dried using a flow of nitrogen. The dried Pchlide is a dark green/black flaky substance, which is then dissolved in 100 % DMSO. The concentration of Pchlide is determined at 626 nm using ε=30,400 M-1 cm-1. Pchlide was stored at 4 °C in amber colored tubes.
Preparation of Pchlide-bound NB-protein for cryo-EM and substrate reduction analysis
The poly-histidine affinity tag was first removed from NB-protein by treatment with 3 C protease (2 µM protease for 20 µM NB-protein) and incubated overnight at RT in the glove box, followed by separation on a SEC column using buffer A with no glycerol. Fractions containing cleaved NB-protein were pooled and concentrated for cryo-EM sample preparation. NB-protein (67 μM tetramer; 134 μM total Pchlide binding sites) was mixed with an excess of Pchlide (520 μM) and passed over a pre-equilibrated SEC column (Superose 6 Increase 5/150 GL; Cytiva Inc.) with Buffer A with no glycerol at room temperature. Peak fractions containing Pchlide-bound NB-protein were pooled and concentrated to ~ 20 μM using an Amicon spin concentrator (30 kDa cutoff). Protein concentration was estimated using a Bradford assay with BSA as reference.
Pchlide reduction assay
20 μM NB-protein (tetramer concentration; 40 μM total L-protein binding sites) pre-bound to Pchlide (as described above) was mixed with 80 μM L-protein (dimer concentration) in buffer A (100 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2, 10% glycerol, and 1.7 mM of sodium dithionite). 1 ml of total reaction was prepared, and the reactions were initiated by the addition of 3 mM ATP (final concentration). At the defined time points, 200 μl of the reaction was extracted and quenched with 800 μl of 100% acetone. The acetone/reaction mixture vortexed and spun down in a table-top centrifuge at 13,226 × g for 4 min. The supernatant was transferred to a quartz cuvette and absorbance scans (600–700 nm) were recorded on a Cary 100 UV-Vis spectrophotometer (Agilent Technologies). Chlide formation was quantified using the molar extinction coefficient ε666nm = 74,900 M−1cm−1. The Pchlide reduction traces were normalized using background spectra recorded in the absence of ATP.
ICP-MS
The metal analysis was performed on a 7800 ICP-MS coupled to an SPS4 autosampler (Agilent Technologies) as previously described52. Briefly, the wild-type and mutant DPOR complexes were diluted to the same concentration, and 1435 µg of each sample was digested in 5% optima grade HNO3 (Fisher Chemical) at 99 °C for 30 min. After incubation, samples were cooled to room temperature, briefly spun down, and 100 µL of the clear supernatant (~ 1400 µg of protein) was diluted to 4 ml (2% HNO3, 0.5% HCl in water) for metal analysis. The signal for 63Cu ions were monitored for 30 sec with an integration time/mass of 1.5 sec/ion in helium mode. The mobile phase was 2% HNO3, 0.5% HCl in water, with a flow rate of 1 ml/min. The 4 ml volume allowed for collection of two measurements per complex (n = 2). This experiment was repeated twice for reproducibility and error analysis.
EPR spectroscopy
EPR spectra were obtained at 10 K on an updated Bruker EMX-AA-TDU/L spectrometer equipped with an ER4112-SHQ resonator (9.48 GHz). Temperature was maintained with a ColdEdge/Bruker Stinger S5-L recirculating helium refrigerator and an Oxford MercuryITC temperature controller. Spectra were recorded with 1.2 G (0.12 mT) digital field resolution, 5.2 mW incident microwave power, and 11 G (1.1 mT) magnetic field modulation at 100 kHz. The sample for EPR spectroscopy contained NB-protein at a concentration of 278 μM, that contained 1.7 mM sodium dithionite. Protein samples were prepared and transferred to the EPR tubes in the glove box and capped with a butyl rubber stopper. Samples were removed from the glove box and immediately flash frozen in liquid nitrogen and analyzed by EPR.
Cryo-EM
The apo-NB-protein and Pchlide-bound NB-protein samples were prepared as described above and used for cryo-EM studies. For the turnover complex, Pchlide-bound NB-protein and L-protein were mixed as described above for the Pchlide reduction assay reactions, but in a smaller total volume (10 µl). Glycerol was omitted from the buffer for cryo-EM experiments. The sample was prepared in a sealed glass vial inside the anaerobic chamber. Similarly, a stock of 100 mM ATP in Buffer A was prepared in a separate sealed glass vial. ATP was mixed with the DPOR protein using a Hamilton gas-tight syringe and incubated for 5 min, and immediately applied onto the grid. Samples for the transition-state DPOR structure determination were prepared similarly, but ADP-AlF3 was used instead of ATP. A stock of ADP-AlF3 was first prepared under anaerobic conditions as described7,8 by mixing 8 mM ADP, 1.6 mM AlCl3, and 40 mM NaF3 in water at room temperature. Cryo-EM samples were prepared on Quantifoil R2/2 (Q350CR2, Electron Microscopy Sciences) holey carbon grids that were plasma cleaned for 60 s using a Solarus 950 (Gatan) with an H2/O2 mixture. Samples were applied onto the grids using a Vitrobot Mark IV (Thermo Fisher Scientific) with the chamber at 4 °C and 100% humidity. To reduce the exposure to the air, a Hamilton gas-tight syringe was used to rapidly apply 3 μl of the sample on the grid from the sealed glass vial. After a 20 s incubation, each grid was blotted for 2 s with a blot force of 0 and immediately plunged into liquid ethane cooled by liquid nitrogen. The total time that each sample spent outside of sealed glass vial during grid preparation and freezing was less than 50 sec. Grids were stored under liquid N2 until data collection. Grids were clipped and loaded into an image-corrected 300 kV Titan Krios G3i cryo-TEM (Thermo Fisher Scientific). Movies were recorded with a Gatan K3 Bioquantum direct electron detector (Gatan Ametek) in an automated fashion using EPU software 2.12.1 (Thermo Fisher Scientific) with a pixel size of 0.825 Å and a nominal defocus range of − 1.0 to − 2.4 µm. Data were acquired with a dose rate of 22 e-/Å2 over a 2.3 s exposure time, yielding a total dose of 50 e-/Å2 over 40 fractions. This sample preparation strategy was optimal in achieving consistent uniform thin ice, and no air-water interface issues were encountered. Excellent distribution of diverse and alternate views of the protein in 2D classification were observed for all the samples.
Image processing of NB-protein apo structure
The single-particle cryo-EM data was processed using cryoSPARC v4.4.1. For the apo NB-protein dataset, 4376 raw movies were motion corrected using patch motion correction, and CTF estimation was performed using patch CTF estimation. Particles were picked initially using blob particle picking and extracted using a box size of 400 pixels. The resulting particle stack (2,532,257 particles) was subjected to several rounds of 2D classification to generate an initial clean particle stack (551,140 particles) for 3D construction using ab initio. Initial 3D volumes were further cleaned using heterogeneous refinement. Volumes were examined in ChimeraX (4), and a 3D volume-particle stack combination (357,713 particles) was selected for further processing. Low-quality particles in the stack were further removed using several rounds of 2D classification. This was followed by ab initio and hetero-refinement by separating the volume into one clean volume. This clean volume was then subjected to homo-refinement and non-uniform refinement using 160,048 particles (final particle stack) with well-defined NB-protein density. The resulting cryo-EM density yielded a map with an overall resolution of 2.7 Å at a Fourier shell correlation cut-off of 0.143.
Image processing of NB-protein bound to Pchlide
For the Pchlide-bound NB-protein dataset, 5394 raw movies were motion corrected using patch motion correction, and CTF estimation was performed using patch CTF estimation. Particles were picked initially using blob particle picking and extracted using a box size of 400 pixels. The resulting particles stack (3,107,286 particles) was subjected to several rounds of 2D classification to generate an initial clean particle stack (342,672 particles) for initial 3D construction using ab initio. Initial 3D volumes were further cleaned using heterogeneous refinement. Volumes were examined in ChimeraX (4), and a 3D volume-particle stack combination (160,124 particles) was selected for further processing. Low-quality particles in the stack were further removed using several rounds of 2D classification. This was followed by non-uniform refinement of the consensus volume using 72,187 particles (final particle stack) with well-defined NB-protein and bound-Pchlide density. The resulting cryo-EM density yielded a map with an overall resolution of 3.02 Å at a Fourier shell correlation cut-off of 0.143. C2 symmetry was enforced on this volume by using non-uniform refinement with no symmetry relaxation, with an overall resolution of 2.92 Å at a Fourier shell correlation cut-off of 0.143. This volume was then further refined using DeepEMhancer.
Image processing of DPOR structure under turnover
For the DPOR-turnover (ATP-Mg2+) dataset, 3725 raw movies were motion corrected using patch motion correction, and CTF estimation was performed using patch CTF estimation. Particles were picked initially using the template picker with the NB-protein structure as an input model and extracted using a box size of 480 pixels. The resulting particle stack (856,671 particles) was subjected to several rounds of 2D classification to generate an initial clean particle stack (190,994 particles) for 3D construction using ab initio. Note that the initial 2D classes were a combination of unbound L-protein and NB-protein, along with a complex of NB-protein and L-protein at a 1:1 ratio (a NB-protein tetramer bound to one L-protein homodimer). Surprisingly, no 2D classes showing two L-protein homodimers bound to the NB-protein tetramer (as seen in former X-ray crystallography studies) were observed. 2D classes corresponding to the complex were selected and processed further. Initial 3D volumes were further cleaned using heterogeneous refinement. Volumes were examined in ChimeraX (4), and a 3D volume particle stack combination (84,350 particles) was selected for further processing. This was followed by homo-refinement with a particle stack of 83,885 particles. The particle stack was further 3D classified into four classes, followed by hetero refinement on a particle stack of 21,290 particles. This was followed by final homo refinement of the consensus volume using 18,965 particles (final particle stack) with well-defined NB-protein with Pchlide and bound to only one L-protein homodimer. The resulting cryo-EM density yielded a map with an overall resolution of 3.82 Å at a Fourier shell correlation cut-off of 0.143. As mentioned in the main text, we also observed free NB from the dataset. This 2D classes were separated, and the resulting particle stack (263,237 particles) was subjected to several rounds of 2D classification to generate an initial clean particle stack (158,811 particles) for 3D construction using ab initio. This was followed by hetero-refinement, which gave us two volumes, with the cleanest volume having 96,908 particles. This volume was further cleaned and separated into two volume and the final particle stack here had 77,395 particles. This volume was extracted to 1X, followed by home-refinement and NU-refinement. The resulting cryo-EM density yielded a map (71,585 particles) with an overall resolution of 3.22 Å at a Fourier shell correlation cut-off of 0.143.
Image processing of DPOR structure under transition state
For the DPOR dataset collected under turnover conditions (ADP-AlF3), 10,488 raw movies were motion corrected using patch motion correction, and CTF estimation was performed using patch CTF estimation. Particles were picked initially using the template picker with a model of DPOR as input and extracted using a box size of 480 pixels. The resulting particle stack (1,071,772 particles) was subjected to several rounds of 2D classification to generate an initial clean particle stack (73,354 particles) for initial 3D construction using ab initio. Note that the initial 2D classes were a combination of unbound L-protein and NB-protein, along with a complex of NB-protein and L-protein at a 1:2 ratio (a NB-protein tetramer bound to two L-protein homodimers). In contrast to the turnover dataset, no 1:1 complexes were observed. 2D classes of the 1:2 complex were selected for further processing. Initial 3D volumes were further cleaned using heterogeneous refinement. This was followed up by homo- and non-uniform refinement with a particle stack of 44,953 particles. The resulting particle stack was further 3D classified into two classes followed by homo refinement on a particle stack. This was followed by final non-uniform refinement of the consensus volume using 20,571 particles (final particle stack) with well-defined NB-protein bound to Pchlide and two BchL dimers. The resulting cryo-EM density yielded a map with an overall resolution of 3.68 Å at a Fourier shell correlation cut-off of 0.143.
Model building and refinement
ChimeraX was used to dock the NB-protein (PDB: 3AER) model into the electron microscopy density maps for NB-protein apo and NB-protein Pchlide-bound complexes. Similarly, L-protein (PDB:6UYK) along with NB-protein (PDB: 3AER) models were combined and docked in the electron density for the full DPOR complex under turnover and transition-state conditions. Subsequent models were iteratively refined and rebuilt in COOT-0.9.8.5 EL53 and further refined and validated using Phenix. Structural figures were generated using Chimera54 or ChimeraX55.
Density functional theory calculations
Density functional theory (DFT) calculations were performed using ORCA 6.0.056. The composite electronic structure method B97-3c was chosen, which combines the B97 GGA functional, a triple-ζ mTZVP basis set, and both DFT-D3 and short-range basis corrections57. To simulate the Pchlide binding pocket environment, the Conductor-like Polarizable Continuum Model (C-PCM) was applied with a dielectric constant of 658. The cluster model included amino acid residues within 4 Å of Pchlide in chain A of the PDB file 8VQJ (turnover structure of DPOR). These include Phe-28 (chain A), Thr-32 (A), Ser-60 (A), Ala-61 (A), Leu-375 (A), Trp-390 (A), Ile-392 (A), Glu-393 (A), Phe-396 (A), Tyr-38 (chain B), Leu-41 (B), Met-45 (B), Ile-46 (B), Val-379 (B), Val-273 (chain D), and Asp-274 (D). H atoms were added to all heavier atoms as necessary, and the α-carbons of boundary amino acid residues were truncated by removing their carboxyl and amine groups. In addition, a water molecule was positioned near the Mg2+, reflecting the typical coordination of Mg2+ by water ligands in Pchlide analogs29,30,31,32,33, leading to a five-coordinate Mg2+ center.
Geometry optimization was carried out in four steps, guided by the fitting uncertainty of each amino acid residue (indicated by colors) shown in the PDB validation report:
-
1.
Non-hydrogen atoms in Pchlide, yellow residues, green residues, and the α-carbons of orange residues were fixed, then the orange residues were optimized.
-
2.
Non-hydrogen atoms in Pchlide, green residues, and the α-carbons of yellow and orange residues were fixed, then yellow and orange residues were optimized.
-
3.
Non-hydrogen atoms in Pchlide and the α-carbons of green, yellow, and orange residues were fixed, then green, yellow, and orange residues were optimized.
-
4.
α-carbons of green, yellow, and orange residues were fixed, then the entire model was optimized (normal optimization convergence criteria were used).
The energy minimization profile and the final Pchlide conformation from step 4 above are shown in Supplementary Fig. 6. The ring unfolds in the first 20 steps, resulting in an energy stabilization of ~ 230 kcal/mol. Further relaxation of amino acid residues and the ring leads to an additional ~ 20 kcal/mol stabilization. The optimized conformation is ~ 248 kcal/mol lower in energy than the puckered turnover structure of DPOR. The Mg2+ is pulled away from the Pchlide ring plane by ~ 0.43 Å by the axial water molecule, consistent with Pchlide analogs in the CCDC database29,30,31,32,33. Geometry optimizations were also performed in the absence of the water molecule, in which case, the carbonyl oxygen of Ala61 coordinates to the Mg2+. The Ala-61 is in the binding pocket in the cryo-EM structure, and coordinates on the other side of the ring as the water molecule, pulling the Mg2+ out by ~ 0.37 Å. Furthermore, the carboxylate group of the Pchlide ring is hydrogen-bonded with the amide group of Ile-46 (chain B), distorting the Pchlide slightly from planarity (structure not included).
Hybrid quantum mechanics/molecular mechanics calculations
An all-atom simulation box (Supplementary Table 3) was constructed using the MCPB.py script in AmberTools 202459, where the molecular mechanics (MM) parameters of Pchlide were derived. The simulation box contains the NB-protein extracted from PDB file 8VQJ, retaining only Pchlide from chain A while removing Pchlide from chain C as well as the Cu ions and the Fe-S clusters. The protein protonation state at pH 7.5 was used. The protein was then solvated, with sodium ions added to ensure charge neutrality. The system underwent energy minimization (using a nonbonded cutoff of 8.0 Å), followed by gradual heating to 300 K in the canonical ensemble (using Langevin thermostat), and 3 ns equilibration under 1 atm conditions in the isothermal-isobaric ensemble (using Berendsen barostat). The temperature, pressure, and box density were stable at the end of equilibration. Pchlide structure was fixed during simulation. The equilibrated structure was then used for hybrid quantum mechanics/molecular mechanics (QM/MM) calculations performed in ORCA 6.0.056.
The QM region in the calculations includes Pchlide and a water molecule, which diffuses into Pchlide’s binding pocket and coordinates with the Mg2+ center during MM equilibration. The active MM region contains amino acid residues, solvents, and ions within ~ 10 Å of the Mg2+ in the MM equilibrated structure. The rest of the simulation box is inactive (structurally fixed) in the calculation. The QM region is described using the B97-3c composite method57, and the MM region is treated with the AMBER force field60, which is widely used for protein systems. No covalent bonds crossed the QM/MM boundary, and electrostatic embedding was applied. Geometry optimization was carried out in two steps:
-
1.
Pchlide was fixed, and the water molecule and the active MM region were optimized.
-
2.
Both QM and MM regions were optimized.
The energy minimization profile and the final Pchlide conformation from step 2 above are shown in Supplementary Fig. 6. Similar to the DFT calculations, the ring unfolds in the first 20 steps, lowering the system energy by ~ 220 kcal/mol. Further relaxation of amino acid residues leads to an additional ~ 20 kcal/mol energy stabilization. The optimized conformation is ~ 243 kcal/mol lower in energy than the starting puckered structure. The Mg2+ is pulled away from the center of the Pchlide plane by ~ 0.49 Å by the water molecule.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The single particle cryo-EM maps and models have been deposited into the PDB and EMDB with the following PDB and EMDB codes: apo NB-protein PDB: 8VQH & EMD-43443; Pchlide-bound NB-protein (C1 symmetry) PDB: 8VQI & EMD-43444; Pchlide-bound NB-protein (C2 symmetry) PDB: 9EFU & EMD-47980; DPOR under turnover PDB: 8VQJ & EMD-43446; DPOR under transition state PDB: 9BUO & EMD-44913. An additional structure of the free NB-protein from the turnover dataset is also deposited into the PDB: 9E7H & EMD-47669. Results from 3D variability analysis (3DVA) are included in the Supplementary Data 1 and Supplementary Data 2 files provided with the manuscript. AMBER input files for energy minimization, heating under NVT ensemble, equilibration under NPT ensemble, and coordinate and parameter files are available on Zenodo (https://doi.org/10.5281/zenodo.14816647). PDB codes of previously published structures used in this study are 2YNM, 3AER, 3AEQ, and 3AEK. Source data are provided as a Source Data File. Source data are provided in this paper.
References
Reinbothe, C. et al. Chlorophyll biosynthesis: spotlight on protochlorophyllide reduction. Trends Plant Sci. 15, 614–624 (2010).
Masuda, T. & Takamiya, K. Novel insights into the enzymology, regulation and physiological functions of light-dependent protochlorophyllide oxidoreductase in angiosperms. Photosynth. Res. 81, 1–29 (2004).
Fujita, Y. Protochlorophyllide reduction: a key step in the greening of plants. Plant Cell Physiol. 37, 411–421 (1996).
Fujita, Y. & Bauer, C. E. Reconstitution of light-independent protochlorophyllide reductase from purified bchl and BchN-BchB subunits. In vitro confirmation of nitrogenase-like features of a bacteriochlorophyll biosynthesis enzyme. J. Biol. Chem. 275, 23583–23588 (2000).
Seefeldt, L. C. et al. Reduction of substrates by nitrogenases. Chem. Rev. 120, 5082–5106 (2020).
Einsle, O. & Rees, D. C. Structural enzymology of nitrogenase enzymes. Chem. Rev. 120, 4969–5004 (2020).
Moser, J. et al. Structure of ADP-aluminium fluoride-stabilized protochlorophyllide oxidoreductase complex. Proc. Natl. Acad. Sci. USA 110, 2094–2098 (2013).
Moser, J. & Layer, G. Enzymatic systems with homology to nitrogenase: Biosynthesis of bacteriochlorophyll and coenzyme F(430). Methods Mol. Biol. 1876, 25–35 (2019).
Ghebreamlak, S. M. & Mansoorabadi, S. O. Divergent members of the nitrogenase superfamily: Tetrapyrrole biosynthesis and beyond. Chembiochem 21, 1723–1728 (2020).
Sarma, R. et al. Crystal structure of the L protein of Rhodobacter sphaeroides light-independent protochlorophyllide reductase with MgADP bound: a homologue of the nitrogenase Fe protein. Biochemistry 47, 13004–13015 (2008).
Muraki, N. et al. X-ray crystal structure of the light-independent protochlorophyllide reductase. Nature 465, 110–114 (2010).
Nomata, J. et al. Dark-operative protochlorophyllide oxidoreductase generates substrate radicals by an iron-sulphur cluster in bacteriochlorophyll biosynthesis. Sci. Rep. 4, 5455 (2014).
Brocker, M. J. et al. Crystal structure of the nitrogenase-like dark operative protochlorophyllide oxidoreductase catalytic complex (ChlN/ChlB)2. J. Biol. Chem. 285, 27336–27345 (2010).
Seefeldt, L. C. et al. Energy transduction in nitrogenase. Acc. Chem. Res. 51, 2179–2186 (2018).
Danyal, K. et al. Negative cooperativity in the nitrogenase Fe protein electron delivery cycle. Proc. Natl. Acad. Sci. USA 113, E5783–E5791 (2016).
Duval, S. et al. Electron transfer precedes ATP hydrolysis during nitrogenase catalysis. Proc. Natl. Acad. Sci. USA 110, 16414–16419 (2013).
Corless, E. I., Bennett, B. & Antony, E. Substrate recognition induces sequential electron transfer across subunits in the nitrogenase-like DPOR complex. J. Biol. Chem. 295, 13630–13639 (2020).
Rutledge, H. L., Cook, B. D., Nguyen, H. P. M., Herzik, M. A. Jr. & Tezcan, F. A. Structures of the nitrogenase complex prepared under catalytic turnover conditions. Science 377, 865–869 (2022).
Hordt, A. et al. Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of alphaproteobacteria. Front. Microbiol. 11, 468 (2020).
Xu, W. et al. A closer look at the spectroscopic properties of possible reaction intermediates in wild-type and mutant (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase. Biochemistry 51, 4835–4849 (2012).
Xu, W. et al. Paramagnetic intermediates of (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase (GcpE/IspG) under steady-state and pre-steady-state conditions. J. Am. Chem. Soc. 132, 14509–14520 (2010).
Adedeji, D. et al. Possible direct involvement of the active-site [4Fe-4S] cluster of the GcpE enzyme from Thermus thermophilus in the conversion of MEcPP.FEBS Lett. 581, 279–283 (2007).
Li, J. et al. Isoprenoid biosynthesis: ferraoxetane or allyl anion mechanism for IspH catalysis? Angew. Chem. Int. Ed. Engl. 52, 6522–6525 (2013).
Nomata, J., Terauchi, K. & Fujita, Y. Stoichiometry of ATP hydrolysis and chlorophyllide formation of dark-operative protochlorophyllide oxidoreductase from Rhodobacter capsulatus. Biochem. Biophys. Res. Commun. 470, 704–709 (2016).
MacGowan, S. A. & Senge, M. O. Contribution of bacteriochlorophyll conformation to the distribution of site-energies in the FMO protein. Biochim. Biophys. Acta 1857, 427–442 (2016).
Haddad, R. E. et al. Origin of the red shifts in the optical absorption bands of nonplanar tetraalkylporphyrins. J. Am. Chem. Soc. 125, 1253–1268 (2003).
Ishizuka, T. et al. Nonplanar porphyrins: synthesis, properties, and unique functionalities. Chem. Soc. Rev. 51, 7560–7630 (2022).
Kingsbury, C. J. & Senge, M. O. The shape of porphyrins. Coord. Chem. Rev. 431, 213760 (2021).
Kratky, C. & Dunitz, J. D. Comparison of the results of two independent analyses of the ethylchlorophyllide a dihydrate crystal structure. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 31, 1586–1589 (1975).
Chow, H. C., Serlin, R. & Strouse, C. E. Crystal and molecular structure and absolute configuration of ethyl chlorophyllide a-dihydrate. Model for the different spectral forms of chlorophyll a. J. Am. Chem. Soc. 97, 7230–7237 (1975).
Serlin, R., Chow, H. C. & Strouse, C. E. Crystal and molecular structure of ethyl chlorophyllide b-dihydrate at-153.deg. J. Am. Chem. Soc. 97, 7237–7242 (1975).
Kratky, C. & Dunitz, J. D. Methylchlorophyllide a dihydrate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 33, 545–547 (1977).
Kratky, C., Isenring, H. P. & Dunitz, J. D. Methylpyrochlorophyllide a monohydrate monoetherate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 33, 547–549 (1977).
Sun, J. et al. Evidence that histidine forms a coordination bond to the A(0A) and A(0B) chlorophylls and a second H-bond to the A(1A) and A(1B) phylloquinones in M688H(PsaA) and M668H(PsaB) variants of Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 1837, 1362–1375 (2014).
Borisov, V. A., D’yachenko, A. N. & Kraidenko, R. I. Reaction of ammonium chloride with the copper(II) sulfide and oxide, and identification of the reaction products. Russ. J. Gen. Chem. 81, 1430–1433 (2011).
Shipps, C. et al. Intrinsic electronic conductivity of individual atomically resolved amyloid crystals reveals micrometer-long hole hopping via tyrosines. Proc. Natl. Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2014139118 (2021).
Onidas, D., Stachnik, J. M., Brucker, S., Kratzig, S. & Gerwert, K. Histidine is involved in coupling proton uptake to electron transfer in photosynthetic proteins. Eur. J. Cell Biol. 89, 983–989 (2010).
Rutledge, H. L. & Tezcan, F. A. Electron transfer in nitrogenase. Chem. Rev. 120, 5158–5193 (2020).
Tezcan, F. A. et al. Nitrogenase complexes: multiple docking sites for a nucleotide switch protein. Science 309, 1377–1380 (2005).
Rees, D. C. et al. Structural basis of biological nitrogen fixation. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 363, 971–984 (2005). discussion 1035-1040.
Tokmina-Lukaszewska, M. et al. Fe protein docking transduces conformational changes to MoFe nitrogenase active site in a nucleotide-dependent manner. Commun. Chem. 6, 254 (2023).
Corless, E. I. et al. The flexible N-terminus of BchL autoinhibits activity through interaction with its [4Fe-4S] cluster and released upon ATP binding. J. Biol. Chem. 296, https://doi.org/10.1074/jbc.ra120.016278 (2021).
Warmack, R. A. et al. Structural consequences of turnover-induced homocitrate loss in nitrogenase. Nat. Commun. 14, 1091 (2023).
Danyal, K., Dean, D. R., Hoffman, B. M. & Seefeldt, L. C. Electron transfer within nitrogenase: evidence for a deficit-spending mechanism. Biochemistry 50, 9255–9263 (2011).
Funk, M. A., Zimanyi, C. M., Andree, G. A., Hamilos, A. E. & Drennan, C. L. How ATP and dATP act as molecular switches to regulate enzymatic activity in the prototypical bacterial class Ia ribonucleotide reductase. Biochemistry 63, 2517–2531 (2024).
Brignole, E. J. et al. 3.3-A resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. Elife 7, https://doi.org/10.7554/elife.31502 (2018).
Smith, B. C., Underbakke, E. S., Kulp, D. W., Schief, W. R. & Marletta, M. A. Nitric oxide synthase domain interfaces regulate electron transfer and calmodulin activation. Proc. Natl. Acad. Sci. USA 110, E3577–E3586 (2013).
Corless, E. I., Mettert, E. L., Kiley, P. J. & Antony, E. Elevated expression of a functional suf pathway in Escherichia coli BL21(DE3) enhances recombinant production of an iron-sulfur cluster-containing protein. J. Bacteriol. 202, https://doi.org/10.1128/jb.00496-19 (2020).
Corless, E. I. et al. The flexible N-terminus of BchL autoinhibits activity through interaction with its [4Fe-4S] cluster and relieved upon ATP binding. J. Biol. Chem. 296, https://doi.org/10.1074/jbc.ra120.016278 (2020).
Corless, E. I. & Antony, E. Methods for heterologous overproduction of Fe-S proteins. Methods Mol. Biol. 2353, 69–78 (2021).
Nomata, J., Swem, L. R., Bauer, C. E. & Fujita, Y. Overexpression and characterization of dark-operative protochlorophyllide reductase from Rhodobacter capsulatus. Biochim. Biophys. Acta 1708, 229–237 (2005).
Larson, J. et al. Arsenic exposure causes global changes in the metalloproteome of Escherichia coli. Microorganisms 11, 382 (2023).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 73–78 (2011).
Brandenburg, J. G., Bannwarth, C., Hansen, A. & Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 148, 064104 (2018).
Barone, V. & Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 (1998).
Li, P. & Merz, K. M. Jr. MCPB.py: A Python based metal center parameter builder. J. Chem. Inf. Model 56, 599–604 (2016).
Ponder, J. W. & Case, D. A. Force fields for protein simulations. Adv. Protein Chem. 66, 27–85 (2003).
Acknowledgements
This study was support by funding from the Department of Energy, Office of Basic Energy Sciences DE-SC0020965 (E.A.), FWP-100593 (R.S.), and DE-SC0019342 (B.M.H.). Department of Energy, Office of Biological and Environmental Research, KP1607011 (LBMS). The National Science Foundation, CHE-2204023 (B.B), CHE-1532168 (B.P.B.), and CHE-2333907 (B.M.H.). The National Institutes of Health, Office of the Director, S10 OD030343 (E.A.) and The National Institute of General Medical Sciences, P30 GM133894 (SSRL). Funding for the Montana State University Mass Spectrometry Facility (RRID:SCR_012482) used in this publication was made possible in part by the MJ Murdock Charitable Trust and the National Institute of General Medical Sciences (P20GM103474 and S10OD28650), and the MSU Office of Research and Economic Development. We thank Dr. Summer Brock and Dr. Katherine Basore at the Washington University Center for Cellular Imaging and Dr. Liguo Wang at the Laboratory for BioMolecular Structure (LBMS) at Brookhaven National Labs for cryo-EM data collection. We also thank Dr. Bassem Mohammed for advice on data processing. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The Laboratory for BioMolecular Structure (LBMS) is supported by the DOE Office of Biological and Environmental Research (KP1607011). We also acknowledge generous financial support from the Doisy Research Fund of the Edward A. Doisy Department of Biochemistry and Molecular Biology at Saint Louis University School of Medicine.
Author information
Authors and Affiliations
Contributions
Conceptualization: E.A. and RK. Methodology: R.K., J.D., M.T.L., B.B., B.B., B.M.H., K.Z., and E.A. Investigation: R.K., N.W., Y.J.G., N.W., B.B., B.B., J.Z., K.Z., and E.A. Visualization: R.K., J.D., and E.A. Funding acquisition: E.A., B.B., B.B., B.M.H., and R.S. Project administration: E.A. Supervision: E.A., R.K., B.B., B.M.H., and R.S. Writing – original draft: E.A. and R.K. Writing – review & editing: All authors.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jorge López-Alonso, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kashyap, R., Walsh, N., Deveryshetty, J. et al. Cryo-EM captures the coordination of asymmetric electron transfer through a di-copper site in DPOR. Nat Commun 16, 3866 (2025). https://doi.org/10.1038/s41467-025-59158-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59158-7
