
Abstract
While hierarchical ordering is a distinctive feature of natural tissues and is directly responsible for their diverse and unique properties, efforts to synthesize biomaterials have primarily focused on using molecular-based approaches with little emphasis on multiscale structure. Here, we report a bottom-up self-assembly process to produce highly porous hydrogel fibers that resemble extracellular matrices both structurally and mechanically. Physically crosslinked nanostructured micelles form the walls of micrometer-sized water-rich pores with preferred orientation along the fiber direction. Low elastic moduli (<1 kPa), high elasticity (extending by more than 12 times the initial length), non-linear elasticity (e.g., hyperelasticity), and completely reversible extension are derived from unevenly distributed strain between the micrometer-sized pores and the polymer chains, which is reminiscent of cellular solids. Control of the material microstructure and orientation over many orders of magnitude (e.g., nm–μm), while holding the nanostructure constant, reveals how the multiscale structure directly impacts mechanical properties.
Similar content being viewed by others

Introduction
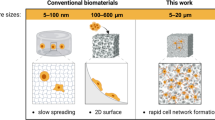
Biological tissues exhibit complex and hierarchically ordered structures while producing a diverse range of properties from a limited number of components1,2,3. Several key properties are critical to properly replicate soft tissues for biomedical applications: biomimetic strain-hardening4, highly reversible deformation5,6, and a microstructure conducive to cell proliferation7. Hydrogels are desirable materials for mimicking natural tissues due to their high water content3,8,9—enabling transport of nutrients and waste products to and from cells—but crosslinked hydrogels composed of a single polymer network exhibit a limited range of mechanical properties10,11. Advances in synthetic chemistry have led to materials that closely mimic the properties of natural tissues12,13,14,15, but these hydrogels frequently lack any structure or ordering beyond their molecular networks. The creation of hierarchical, anisotropic structures within hydrogels has been demonstrated, but is typically achieved via external forces such as shear or directional ice growth, which are difficult to scale up and limited in application16. Here, we reveal that pore size and microstructure in a physically cross-linked, hierarchically ordered hydrogel prepared via rapid self-assembly result in reversible deformation and dramatically lead to non-linear elasticity when uniaxially deformed (i.e., hyperelasticity)17 in what would otherwise be a stiff and brittle hydrogel18.
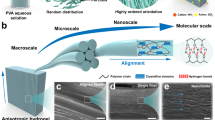
When an amphiphilic ABA triblock copolymer containing hydrophobic A and hydrophilic B domains is dissolved in a water-miscible organic solvent and injected into water (Fig. 1a), the block copolymer will self-assemble at both the nano and microscales19,20,21. The hydrophobic end-blocks self-assemble into micelle cores at the nanoscale, which function as physical cross-links bridged by the hydrophilic mid-block to form a network18,19,20,21. At the microscale, pores are formed within the micelle network due to macrophase separation induced by solvent exchange with the water bath (Fig. 1b), resulting in a porous network reminiscent of the endomysium, the extracellular matrix of skeletal muscles (Fig. 1c)22. The TEM image, cryo-SEM image, and photograph shown in Fig. 1ai–iii are results from the work described here.

a Hydrogels exhibiting hierarchical order are prepared using a rapid injection processing method in which an amphiphilic triblock copolymer, poly(styrene)-poly(ethylene)-poly(styrene) (PS-PEO-PS) is initially dissolved in a water miscible organic solvent and injected into water using a syringe pump. Hydrogel fibers exhibit a multiscale structure spanning many orders of magnitude where (i) randomly oriented micelles organize into a network at the nanoscale that comprises (ii) the walls of the water-rich cavities that form a preferentially aligned porous microstructure in the direction of (iii) the prepared hydrogel fiber after rapid injection processing. The spherical micellar domains in (1a–i) were characterized via TEM and stained with uranyl acetate. The cryo-SEM image of the porous microstructure shown in (1a–ii) was prepared using a 10.5 wt% polymer solution in DMF. b Scheme representing the different levels of ordering during self-assembly, starting from a single triblock copolymer chain and ending with micrometer-sized pores. c Decellularized muscle tissue (i.e., endomysium) with natural hierarchical structure mirroring the synthetic hydrogel structure using rapid injection processing. Reproduced with permission from ref. 22. Copyright 2005 John Wiley and Sons.
The reported hierarchical, porous hydrogels are prepared in a single step without the need for external forces or templating, which allows tailoring of material properties and scalability20. Due to the water-activated self-assembly, hierarchical hydrogels are easily prepared using molds or dip coatings20, but hydrogel fibers produced via injection were selected for this work due to their uniformity and for consistency with prior research. Previous work from our group has described the necessary experimental procedures to produce highly elastic porous hydrogels using rapid injection processing19,20,21, but the effect of microstructure/nanostructure on the mechanical response is unknown. Here, we reveal how initial polymer solution conditions for rapid injection processing impact the hierarchical structure, and thus dictate the mechanical properties, using a single triblock copolymer at constant molecular weight and block volume fraction. The reported hydrogels are distinctly different from all previous works in that the reports either synthesize a library of polymers or use complex processing procedures to achieve the range of mechanical properties23. Excitingly, the porous hydrogels exhibit structurally dependent mechanical properties using a bottom-up approach as opposed to top-down additive manufacturing techniques24. A fundamental understanding of multiscale structure formation and the impact of morphology on mechanical properties is currently unknown but is critical in realizing the potential of these hydrogels as transformative biomaterials.
Here, we demonstrate control over the microstructure and resulting mechanical properties by using either N,N-dimethylformamide (DMF) or tetrahydrofuran (THF) as the initial solvent and varying the concentration of the poly(styrene)-poly(ethylene oxide)-poly(styrene) (SOS) triblock copolymer in the initial solution. These parameters produce significant differences in the microstructure, but interestingly, there is minimal variation in the nanoscale ordering of the micelle network with respect to processing conditions, which is attributed to the disordered sphere morphology resulting from the high-volume fraction of the hydrophilic midblock in the triblock copolymer. Thus, the changes in mechanical properties are directly linked to the changes within the microstructure (i.e., pore size and morphology). Hydrogels produced with DMF and THF exhibit distinct pore morphologies, with only the THF morphology exhibiting changes in pore size with respect to initial polymer solution concentration. The pores within the hydrogels produce an additional reversible deformation mechanism that increases the extensibility of the hydrogel fibers, where the pores deform initially under applied stress, then, after the pores are collapsed, stress is transferred to the micelle network. Simultaneously controlling multiscale structure and orientation over drastically different length scales using a single, bottom-up processing step is critically important in creating next-generation biomaterials.
Results
Preparing hierarchical hydrogels
Hierarchical hydrogels were produced using an SOS polymer with a number-average molecular weight (Mn), O-block volume fraction (fO), and a dispersity (Đ) of 192 kg/mol, 0.9, and 1.05, respectively (Fig. S1 and Table S1). A large PEO mid-block volume fraction was targeted to promote the formation of a disordered spherical micelle morphology at the nanometer scale20. Additionally, the S-end blocks were selected as the hydrophobic moiety in the triblock copolymer due to the high glass transition temperature (Tg), preventing chain pull-out and improving mechanical stability20.
Two different water-miscible organic solvents, THF and DMF, were used to form the hydrogel to explore the impact of processing conditions on hydrogel structure25,26. The physically crosslinked spherical micelle network that forms at the nanoscale is primarily dictated by the molecular weight and fO of the block copolymer. Previous studies indicate the initial polymer solution must be at or above the entanglement concentration for the hydrogel to form19,20. Thus, all hydrogels were prepared using initial solution concentrations between 8% and 15% by weight. The needle diameter and polymer solution injection rate were constant for all prepared hydrogel fibers. This range allowed both the polymer solution concentration and viscosity to be held constant to isolate the effects of varying the organic solvent (Fig. S2). The fibers remained in water for at least 24 h prior to any experiments being performed to ensure diffusion was complete. The water content of the prepared hydrogel fibers was unusually high at ≈98% water by weight and consistent across sample conditions (Fig. S3). In contrast, physically crosslinked triblock copolymer hydrogels prepared using traditional preparation methods (e.g., thermally annealing the neat SOS polymer and then hydrating) are typically 4–40% water by weight18.
Characterizing the hierarchical structure
The nanoscale structure of the hydrogels was visualized using transmission electron microscopy (TEM) and assessed by small-angle X-ray scattering (SAXS). Micelles were imaged via TEM by drop-casting micellar aggregate precursors formed at low initial polymer concentrations onto TEM grids and drying, after which they were stained using a 2 wt% solution of uranyl acetate in water. The micelle diameter in both solvents was measured to be 20 ± 1 nm (Fig. 2a). While the diameter of spherical micelles is known to depend on polymer concentration and solvent27, the relatively narrow 8–15 wt% concentration range and large O-block volume fraction resulted in a consistent micelle size27.

a TEM of micelles formed from a low concentration initial solution of polymer in DMF. b 1D synchrotron SAXS data for all six hydrogel samples, indicating that the domain spacing, as indicated by the primary peak position (q*), is similar regardless of initial solution parameters (solvent, concentration). c SAXS patterns for an annealed SOS material in the dried and the hydrated state. A SAXS pattern of a DMF-11.5 wt% hierarchical hydrogel sample, shown in red, is added for comparison. d Representative 1D and 2D SALS data, using a 625 nm laser, from hydrogels made using 12.5 wt% and 14.5 wt% initial solutions of polymer in THF, showing strong signal and alignment in the fiber direction. Scale: 50 μm–1. e Cross-sectional imaging of the porous microstructure of a THF-11.0 wt% sample in the hydrated state obtained via fluorescence confocal microscopy at 63× magnification. f 3D rendering of the THF-11.0 wt% sample from Z-stack showing alignment of hydrogel pores largely parallel to the fiber axis z, in agreement with the SALS data.
Synchrotron SAXS measurements confirmed the disordered organization of the micelle network in the hydrogels. The domain spacing d of the micelle network was determined from the one-dimensional (1D) scattering intensity versus scattering vector (q) shown in Fig. 2b using the relationship d = 2π/q*, where q* is the primary scattering peak associated with the center-to-center micelle distance. The average center-to-center micelle distance was calculated to be 80 nm. In-depth SAXS analysis was conducted using a spherical form factor and Percus–Yevick hard sphere approximation structure factor28, but fitting the full q-range was challenging due to the significantly low-q scattering and smeared spherical form factor oscillations at higher-q. The low-q upturn is indicative of hydrogel heterogeneity and larger-scale features, which are consistent with the hierarchical structure. Therefore, the SAXS patterns were interpreted by tracking the change in q*. Variations in domain spacing with solvent and concentration are evident in the change of q* but are within a 5 nm range. The consistency in the domain spacing, despite variations in the viscosity and solvent, confirms that the nanostructure is insensitive to the sample conditions used in the study. In contrast, there is a distinct difference when hydrogels are produced using conventional methods from the same polymer (Fig. 2c). When a dry, annealed polymer sample showing disordered sphere morphology is hydrated, the morphology remains, but d increases due to swelling (identified by a shift in the primary scattering peak q* to lower q) from 43 nm to 104 nm. This value is larger than the hierarchical gels despite having a much lower swelling ratio. The difference in domain spacing is rationalized by the existence of pores in the network, creating polymer-rich regions in the hydrogels.
While hydrogel nanostructures were characterized using SAXS, micron-sized features were probed using small-angle light scattering (SALS)29. 2D and 1D data obtained from hydrogels prepared using THF at concentrations of 12.5 wt% and 14.5 wt% polymer in THF is shown in Fig. 2d. The high degree of anisotropy in the 2D plots indicates the pore walls are preferentially oriented along the long axis of the hydrogels (i.e., “fiber axis” of the hydrogels). While the domain spacing and average pore diameter are calculated from the primary scattering peaks for the THF samples within the quantifiable q-range, DMF samples exhibited multiple peaks, preventing quantification of the domain spacing. To directly confirm the presence of the pores in the hydrated state, a 1 mm3 sample of THF-12.5 wt% hydrogel fiber was stained in 0.1 wt% aqueous solution of Nile Red. The fiber sample was arranged with the fiber axis parallel to the glass slide and coverslip, allowing the pores to be imaged along the long axis. Pores were clearly visible in this sample, with the results showing the pores to be highly interconnected (Fig. 2e). The z-stack composite image further shows a high degree of orientation in the pores, roughly parallel with the fiber axis (Fig. 2f).
Cryo-SEM revealed more detailed real-space images of the porous microstructure perpendicular to the fiber axis. Samples (~5 mm-thick) were first vitrified in liquid nitrogen, then cryo-milled to obtain a smooth surface (see “Methods”). While fluorescence confocal microscopy confirmed the presence of the pores in the hydrated state, cryo-SEM provided a more resolved microstructure that was influenced by the initial solvent and concentration. Samples produced using DMF show a coaxial, or “core-sheath,” morphology (Fig. 3a–c), where the outer edge of each fiber is comprised of large pores, while the inner core contains exclusively small pores. The bimodal pore size distribution explains the difficulty in the SALS analysis. The demarcation between regions is sharp and distinct, and the outer region width averages 200–240 µm. The outer and inner pore sizes are consistent across all DMF concentrations, approximately 30 μm and 5 µm, respectively (Fig. 3h). In contrast, hydrogels prepared using THF exhibit a monomodal pore distribution (Fig. 3e–g), where the averaged pore size decreases from 30 ± 13 μm to 9 ± 4 μm as concentration increases from 11 wt% to 14.5 wt% (Fig. 3h). Furthermore, the pores size distribution in hydrogels prepared using THF narrows with increasing concentration, which is supported by SALS. At the highest initial solution concentration for the THF sample, the pores resemble those found in the inner, small-pore region of the samples prepared using DMF (Fig. 3d), though slightly larger (Fig. 3h).

a, b Cryo-SEM images of hydrogels prepared using DMF. Hydrogel fibers prepared using DMF display a coaxial morphology where larger and smaller pores reside in the outer and inner regions. Scale bar: 100 µm. c Schematic representation of the hydrogel microstructure for DMF. d Higher magnification cryo-SEM image highlighting the more uniform pore size in the core region of the hydrogel fiber prepared using the 11.5 wt% polymer solution in DMF. e, f Cryo-SEM images of hydrogels prepared using THF exhibit a highly porous microstructure where the pore sizes are monomodal and the size decreases with increasing concentration. Scale bar: 100 µm. g Schematic representation of the hydrogel microstructures of THF samples. h Change in average pore diameter with respect to initial polymer concentration when using either THF or DMF as the solvent, measured via cryo-SEM and SALS. Pore diameter values shown in (h) determined from SEM measurements, are represented as mean values with error bars representing standard deviation. Standard deviation values in (h) for DMF outer pores are ±13 μm (n = 165), ±17 μm (n = 287), and ±9 μm (n = 125) for 9.5 wt%, 10.5 wt%, and 11.5 wt%, respectively. Inner pore diameters for DMF are ±2 μm (n = 121), ±3 μm (n = 1020), and ±3 μm (n = 463) for 9.5 wt%, 10.5 wt%, and 11.5 wt%, respectively. Pore diameters for THF samples are ±13 μm (n = 72), ±10 μm (n = 374), and ±4 μm (n = 174) for 11.0 wt%, 12.5 wt%, and 14.5 wt%, respectively.
The exact reason for the microstructural differences between hydrogel fibers prepared with THF and DMF is still under debate. We hypothesize that at the initial stages of solvent exchange, PS micelles form due to water infiltration into the fiber. Figure 2a shows the formation of micelles at lower initial polymer solution concentrations. The mechanism of micrometer-sized pore formation in the reported hydrogels is posited to be analogous to macropore formation in separation membranes prepared using nonsolvent-induced phase separation (NIPS)30. The reason for the stark differences in microstructural morphology between fibers prepared using DMF and THF is not immediately apparent and is being actively explored. There is a general rule of thumb in the NIPS community that polymer solution viscosity and solvent exchange rates control structure, but both parameters are nearly identical under these experimental conditions (Figs. S2 and S17), and neither parameter holds explanatory power30. Thus, microstructural variations are expected to be due to changes in the ternary phase diagrams (i.e., polymer, solvent, and water) and solvent exchange trajectories where the compositional pathways during solvent exchange traverse phase boundaries at different times.
Relating mechanical properties to microstructure
The porous microstructure plays a critical role in determining the mechanical properties of the hydrogels (Fig. 4a, b). Despite their low elastic modulus (E < 1 kPa), all samples reversibly stretch to many times their initial length (see Supplementary Movie 1), which, to our knowledge, has never been reported in highly porous hydrogels. Hydrogel samples produced using DMF display both non-linear elasticity at large strains and a significantly higher elongation at break, which is attributed to their coaxial morphologies31. Samples produced with DMF possess near-identical microstructures and have highly consistent deformation responses, while the samples produced using THF show a steady increase in the ultimate tensile strength of the hydrogels with concentration as the average pore size decreases. The Young’s modulus, E, and toughness of the hydrogels follow a similar trend (Fig. 4b), which is inversely correlated with average pore size (Fig. 3h). Though the micrometer pore dimensions result in stiffness changes, the fraction of mid-blocks bridging adjacent micelles may potentially vary with concentration and polymer molecular parameters, which are currently under investigation.

a Representative stress-strain curves of hydrogel fibers prepared at different initial polymer concentrations and in different solvents, all tested at a strain rate of 3/min. DMF samples exhibit high extensibility and non-linear elasticity at large strains. b Plots showing the change in Young’s Modulus (E) and toughness with varying solvent and concentration. THF samples show an increase in E with increasing concentration, while DMF samples are constant and within error. Young’s Modulus and toughness values shown in (b) were determined from uniaxial extension measurements and are represented as mean values with error bars representing standard deviation. The number of replicate uniaxial extension measurements (n) for DMF is n = 8, n = 12, and n = 7 for 9.5 wt%, 10.5 wt%, and 11.5 wt%, respectively. THF samples with mean values and error bars are n = 10, n = 5, and n = 8 for 11.0 wt%, 12.5 wt%, and 14.5 wt%, respectively. c Cyclic loading and unloading uniaxial extension curves for a hydrogel fiber prepared using DMF at 11 wt%. The hydrogel sample was elongated into the strain-hardening regime, yet shows no hysteresis after 5 cycles. d In situ straining of a hydrogel sample prepared using a 12.5 wt% solution of polymer in THF, imaged via transmission light microscopy. The images, as well as a representative diagram, show an increase in pore alignment with respect to elongation, which is reversed when the gel fiber is relaxed. e In situ SAXS of a hydrogel sample prepared from a 12.5 wt% THF solution. No measurable change can be seen in the micelle center-to-center distance at elongation ratios of up to λ = 5. f Ashby plot of ultimate tensile strength versus elongation at break for the reported hierarchical hydrogels and different hydrogel systems. Hierarchical hydrogels are soft and highly extendable when compared to a large set of hydrogel literature.
All hydrogel fibers reversibly extend for at least five loading and unloading cycles when deformed up to six times the original length with no hysteresis (Fig. 4c). Drying in thinner samples prevented additional cycles in some instances. Reversible deformation and non-linear elasticity at large strains, two hallmark traits of natural tissues, are highly difficult to mimic simultaneously in hydrogel materials, and here result from the hierarchical ordering present in the fibers. The hydrogel deformation response is analogous to an elastomeric foam, where first cells buckle under tension, followed by a rapid rise in stress as the spaces within the pores disappear and the walls begin to deform32. The deformation of pores under strain is observed directly via in situ transmission light microscopy (Fig. 4d), where the fibers were stretched to five times their original length in water and imaged in situ. Though the contrast between the water and the pores themselves is somewhat weak, the edges of the pore walls (Fig. 4d, i) can be observed to deform under stress when the fiber is extended (Fig. 4d, ii) and return when the stress is removed (Fig. 4d, iii). Thus, we conclude that the total strain is unevenly portioned between the pores and the bridging PEO chains. The initial pore deformation requires very little force, which is insufficient to cause chain extension. Chain extension, therefore, occurs at larger strains, which is expected to result in non-linear elasticity behavior at large strains. The proposed mechanism is supported by in situ SAXS measurements, where there is no measurable change in the micelle center-to-center distance at up to λ = 5 when uniaxially extended (Fig. 4e). Interestingly, the non-linearity stress-strain response in the high-strain region for the samples prepared using THF (Fig. 4a) is steeper for samples exhibiting smaller pores (i.e., higher initial polymer/THF concentrations) and that the non-linearity stress-strain response occurs at larger strains for samples prepared using DMF as compared to THF (Fig. 4a). The difference in the onset of strain-stiffening between THF and DMF samples is attributed to the pore size and pore microstructure, highlighting alternative avenues for controlling mechanical responses in hydrogels through microstructure as opposed to molecular modifications.
Discussion
The complex structuring produced from self-assembly in these materials produces low moduli and tensile strength, high extensibility, minimal hysteresis, and non-linear elasticity at large strains. While one or more of these properties may be produced in synthetic hydrogels, the combination of these properties in a porous hydrogel is challenging. Physical hydrogels without the hierarchical ordering demonstrated here exhibit a narrow range of properties18,33,34,35, and adding pores to hydrogels typically negatively impacts the mechanical properties36, although recent results indicate that open pores can enhance the properties of hydrogels (Fig. 4f)16,37,38,39,40,41,42,43. The flexibility of the hierarchical hydrogels is rivaled only by supramolecular44,45,46 and interpenetrating network hydrogels47,48,49,50, but these materials frequently suffer from limited reversibility due to viscous dissipation. Self-assembling bottlebrush elastomer hydrogels demonstrate strain-stiffening responses similar to natural tissue4,51, but these materials lack the porous structure or hierarchical ordering that is critical for synthetic biomaterials. A wide range of properties is accessible by changing the conditions of the polymer, and initial solution, while additional techniques exist to strengthen hydrogels and increase their rigidity, including tailoring the relative composition of the hydrophobic and hydrophilic groups in the block copolymer. While future investigations are needed to determine the precise impact of molecular architecture and expand the accessible mechanical property range, these hydrogels represent an exciting class of biomaterials where structure dictates properties, not molecular modifications.
Hierarchical porous hydrogels, prepared using rapid injection processing, exhibit tunable micrometer pores that dictate the mechanical properties. The structural dependence of the mechanical properties demonstrates an alternative avenue to tailoring the properties of synthetic biomaterials beyond molecular modification. The preferentially aligned porous structure of these hydrogels enables completely reversible elastic deformation at strains easily up to six times the initial length. While the reported hydrogels are significantly more elastic than many natural tissues, the system possesses many avenues for modification of the microstructure and thus mechanical properties. Despite significant variations in microstructure and mechanical properties, these hydrogels exhibit no change in the micelle morphology and water swelling ratio, allowing the microstructure to be tuned independently of the nanostructure. The reported hydrogels and rapid injection processing will potentially lead to customizable cell scaffolds and biomedical materials, with a diverse range of mechanical properties tuned through the selection of the initial amphiphilic block copolymer architecture and subsequent processing parameters.
Methods
Amphiphilic triblock copolymer synthesis
SOS triblock copolymers were chosen as a representative amphiphilic triblock copolymer for this system due to the enhanced mechanical properties derived from the high glass transition of poly(styrene), which aids in stabilizing the micelle structure20. The triblock copolymer used in this research was synthesized via sequential living anionic polymerization, which has been previously described20. After each step in the synthesis process, the molecular weight and dispersity were determined using size-exclusion chromatography, while the volume fraction of the polymers in the material was characterized via proton nuclear magnetic resonance spectroscopy. Supplementary Information contains more detailed synthetic information and characterization results.
Characterization of the block copolymer solutions
Two solvents were used as water-miscible organic solvents for the precursor solutions: THF (Sigma Aldrich, ≥99.9%) and DMF (Sigma Aldrich, anhydrous 99%). The critical water concentrations for micellization for the polymer in different solvents were determined by light scattering experiments of dilute amphiphilic block copolymer solutions in organic solvents with increasing addition of water, similar to previous reported studies27. The viscosity of the polymer solutions at high concentrations and solvents was determined using a Discovery Hybrid Rheometer at room temperature with parallel plate geometry. A solvent trap was used to prevent the evaporation of the more volatile THF solvent. The trends calculated from this data were used to match the viscosities of the solutions between the solvents.
Hydrogel production
Polymer solutions were injected into a coagulation bath containing RO water using a syringe pump set to an injection speed of 1 mL/min and a needle gauge of 19. Initial solutions were used with concentrations ranging from 8 wt% to 15 wt%. Though the gel fibers formed almost instantly on contact with water, they were left in the water bath overnight to ensure complete solvent diffusion, then transferred to vials of RO water for storage at 4 °C.
Conventional physically crosslinked triblock copolymer hydrogels were produced by thermally annealing the neat SOS polymer above the O-block and S-block crystallization and glass transition temperature, respectively, and then swelling with water. Specifically, hydrogels were prepared by first annealing the triblock copolymer in a thermal press at 100 psi and 150 °C for 30 min, followed by annealing under vacuum for 24 h at 150 °C. After cooling, the annealed sample was hydrated in pure RO water for 3 days to ensure total hydration.
Nanoscale characterization
The micellar nanostructure was characterized using a synchrotron X-ray source at the National Source for Light Scattering II at Brookhaven National Laboratory, both under static conditions, and in situ under extension using a Linkam tensile stage. Microgels formed from a semi-dilute solution of the triblock copolymer injected into water, were drop-cast onto a TEM grid could be used to approximate the micelle diameter. The microgel samples were stained with a 2 wt% solution of uranyl acetate in water and imaged using an FEI Tecnai BioTwin TEM.
SALS experiments
Figure S5 shows the SALS setup, which has been published previously in ref. 29 The ray diagram shows the initial laser of 625 nm scattered from the sample at an angle θ through a convex lens L1 (Edmund Optics-NT67-245) with a numerical aperture, N.A. = 0.85 that captures up to 58° scattered light. The converging scattered light then passes through the beam stop and is refocused through another convex lens L2 (Thorlabs-LA 1951A). The beam stop is at a focal length away from lens L1 and blocks all direct light from the laser. The l1 and l2 can be changed to optimize the q-range. The scattered light is then projected onto a CMOS detector, D (Basler acA800–510 um), which is a focal length away from L2.
A control image is taken using a diffraction grating with 100 lines per mm. This was later used to determine the pixel-to-q conversion for the scattering patterns. The hydrogel was then loaded into the sample setup. The l1 was set to ~25 mm and l2 to ~75 mm. The room lights were then turned off to avoid any background scattering. A scattering image was then taken for each sample. The hydrogel is held in the setup using plate grips. These plate grips are 0.5 in wide, offering enough space to load the hydrogel. The nuts can be moved, allowing the top mount (movable) to move in the z-direction. The hydrogel can then be stretched to different lengths.
Microstructure imaging
The hierarchical hydrogel microstructure was characterized using fluorescence confocal microscopy, SALS, and cryogenic scanning electron microscopy (Cryo-SEM).
Fluorescence confocal measurements were performed on 5 mm sections of a fiber that had never been frozen, prepared using an initial solution concentration of 11.0 wt% polymer in THF and stained overnight in a 0.01 mg/mL solution of Nile Red in water. The samples were laid lengthwise along the glass slide with a coverslip placed on top. The samples were imaged approximately parallel to the fiber axis at 10× and 63× magnification, producing cross-sectional images showing the porous structure. The composite z-stack images at both magnifications show a high degree of alignment roughly parallel to the fiber axis, indicating an additional level of hierarchy potentially caused by the shear flow during injection.
Vitrified hydrogel cross-sections were prepared by mounting the sample on aluminum pins, submerging it in liquid nitrogen, and cryo-milling the surface perpendicular to the fiber axis to reveal the hydrogel cross-section. Hydrogels were stored under liquid nitrogen, then transferred to a Zeiss Cryo-SEM for imaging at −125 °C. Water was partially sublimated at −95 °C, then the sample was sputter-coated with a gold-palladium alloy. Pore sizes were measured using Image J. An example of pore size measurement is shown in the Supplementary Information.
In situ TLM measurements (Fig. 4d) were performed by combining a customized tensile tester with a confocal Raman microscope (Alpha 300R, WITec, Germany). The tensile tester was made using two linear piezo actuators (Physical Instrument, Germany) combined with L-shape clamps, allowing stretching of the samples while they were immersed underwater. The TLM images were captured using a 63× water immersion objective lens (Zeiss, Germany) while the sample was exposed to transmission white light. A displacement rate of 0.5 mm/s was applied for stretching of the fibers.
Uniaxial extension tests
The mechanical properties of the hydrogels were characterized via uniaxial extension under ambient conditions using an MTS Criterion load frame equipped with a 10 N force transducer. The hydrogels were secured with spring-action tensile grips to minimize hydrogel slip during extension and strained at a rate of 3/min. To determine the cross-sectional area, hydrogel cross-sections were placed on a glass slide and imaged using an Olympus 100 optical microscope. The areas of the cross-sections were calculated using ImageJ. Hysteresis was determined by repeatedly straining the hydrogel to five times its original length for five cycles.
Rheological characterization
The viscosity of polymer solutions at various polymer concentrations was measured using a Discovery HR-3 Hybrid Rheometer at room temperature, equipped with a solvent trap to prevent evaporation, which is critical for THF samples at high polymer concentrations. Solutions in the entangled regime were measured with a 2 cm parallel plate geometry, while semi-dilute solutions used a 6 cm parallel plate geometry. The solutions were tested in a flow sweep experiment with a shear rate range of 0.05 ≤ \(\dot{\gamma }\) ≤ 500 Hz. The values were averaged to determine the viscosity of the solution.
To measure the storage and loss moduli of the equilibrium samples, the shear modulus of these hydrogels was measured via oscillatory shear rheology using a 3 mm plate on a TA Ares-G2 rheometer, lowered just until contact with the sample resulted in a slight increase in force. Approximately 4 mm2 pieces of both the equilibrium and hierarchical gel samples were tested, first with a dynamic strain sweep to determine the strain region appropriate for testing, and next with a dynamic frequency sweep from 0.1 rad/s to 10 rad/s (see Fig. S22 in Supplementary Information).
Data availability
The polymer characterization, solution viscosity, X-ray scattering, and mechanical measurement data generated in this study have been deposited in the Data Commons at Penn State database under accession code, https://doi.org/10.26208/5JEG–X325. The data is also available from the authors on request.
References
Eder, M., Amini, S. & Fratzl, P. Biological composites—complex structures for functional diversity. Science 362, 543–547 (2018).
Meyers, M. A., McKittrick, J. & Chen, P.-Y. Structural biological materials: critical mechanics-materials connections. Science 339, 773–779 (2013).
Shin, H., Jo, S. & Mikos, A. G. Biomimetic materials for tissue engineering. Biomaterials 24, 4353–4364 (2003).
Sheiko, S. S. et al. Mechanically diverse gels with equal solvent content. ACS Cent. Sci. 8, 845–852 (2022).
Lee, K. Y., Peters, M. C., Anderson, K. W. & Mooney, D. J. Controlled growth factor release from synthetic extracellular matrices. Nature 408, 998–1000 (2000).
Guimarães, C. F., Gasperini, L., Marques, A. P. & Reis, R. L. The stiffness of living tissues and its implications for tissue engineering. Nat. Rev. Mater. 5, 351–370 (2020).
Lu, P., Weaver, V. M. & Werb, Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012).
Armiento, A. R., Stoddart, M. J., Alini, M. & Eglin, D. Biomaterials for articular cartilage tissue engineering: learning from biology. Acta Biomater. 65, 1–20 (2018).
Slaughter, B. V., Khurshid, S. S., Fisher, O. Z., Khademhosseini, A. & Peppas, N. A. Hydrogels in regenerative medicine. Adv. Mater. 21, 3307–3329 (2009).
Sakai, T. et al. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules 41, 5379–5384 (2008).
Cai, S., Niu, B., Ma, X., Wan, S. & He, X. High strength, recyclable, anti-swelling and shape-memory hydrogels based on crystal microphase crosslinking and their application as flexible sensor. Chem. Eng. J. 430, 132957 (2022).
Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 374, 212–216 (2021).
Lin, S. et al. Anti-fatigue-fracture hydrogels. Sci. Adv. 5, eaau8528 (2019).
Appel, E. A., del Barrio, J., Loh, X. J. & Scherman, O. A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 41, 6195–6214 (2012).
Gong, J. P. Why are double network hydrogels so tough? Soft Matter 6, 2583–2590 (2010).
Sano, K., Ishida, Y. & Aida, T. Synthesis of anisotropic hydrogels and their applications. Angew. Chem. Int. Ed. 57, 2532–2543 (2018).
Melly, S. K., Liu, L., Liu, Y. & Leng, J. A review on material models for isotropic hyperelasticity. Int. J. Mech. Syst. Dyn. 1, 71–88 (2021).
Guo, C. & Bailey, T. S. Highly distensible nanostructured elastic hydrogels from AB diblock and ABA triblock copolymer melt blends. Soft Matter 6, 4807–4818 (2010).
Lang, C., Kumar, M. & Hickey, R. J. Influence of block sequence on the colloidal self-assembly of poly(norbornene)-block-poly(ethylene oxide) amphiphilic block polymers using rapid injection processing. Polym. Chem. 11, 375–384 (2020).
Lang, C. et al. Solvent-non-solvent rapid-injection for preparing nanostructured materials from micelles to hydrogels. Nat. Commun. 10, 3855 (2019).
Lang, C. et al. Nanostructured block copolymer muscles. Nat. Nanotechnol. 17, 752–758 (2022).
Trotter, J. A. & Purslow, P. P. Functional morphology of the endomysium in series fibered muscles. J. Morphol. 212, 109–122 (1992).
Foudazi, R., Zowada, R., Manas-Zloczower, I. & Feke, D. L. Porous hydrogels: present challenges and future opportunities. Langmuir 39, 2092–2111 (2023).
Jiao, P., Mueller, J., Raney, J. R., Zheng, X. & Alavi, A. H. Mechanical metamaterials and beyond. Nat. Commun. 14, 6004 (2023).
Yu, Y., Zhang, L. & Eisenberg, A. Morphogenic effect of solvent on crew-cut aggregates of apmphiphilic diblock copolymers. Macromolecules 31, 1144–1154 (1998).
Hickey, R. J., Haynes, A. S., Kikkawa, J. M. & Park, S.-J. Controlling the self-assembly structure of magnetic nanoparticles and amphiphilic block-copolymers: from micelles to vesicles. J. Am. Chem. Soc. 133, 1517–1525 (2011).
Mai, Y. & Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 41, 5969–5985 (2012).
Ilavskya, J. & Jemiana, P. R. Irena: tool suite for modeling and analysis of small-angle scattering. J. Appl. Crystallogr. 42, 347–353 (2009).
Dhakal, S., Chen, Z., Estrin, D. & Morozova, S. Spatially-resolved network dynamics of poly(vinyl alcohol) gels measured with dynamic small angle light scattering. Gels 8, 394 (2022).
Müller, M. & Abetz, V. Nonequilibrium processes in polymer membrane formation: theory and experiment. Chem. Rev. 121, 14189–14231 (2021).
Naeimirad, M. et al. Recent advances in core/shell bicomponent fibers and nanofibers: a review. J. Appl. Polym. Sci. 135, 46265 (2018).
Gent, A. N. & Thomas, A. G. The deformation of foamed elastic materials. J. Appl. Polym. Sci. 1, 107–113 (1959).
Luo, J. et al. Biomimetic strain-stiffening hydrogel with crimped structure. Adv. Funct. Mater. 31, 2104139 (2021).
Kamata, H., Akagi, Y., Kayasuga-Kariya, Y., Chung, U.-I. & Sakai, T. Nonswellable” hydrogel without mechanical hysteresis. Science 343, 873–875 (2014).
Pan, J. et al. Hierarchical multiscale hydrogels with identical compositions yet disparate properties via tunable phase separation. Adv. Funct. Mater. 32, 2110277 (2022).
LaNasa, S. M., Hoffecker, I. T. & Bryant, S. J. Presence of pores and hydrogel composition influence tensile properties of scaffolds fabricated from well-defined sphere templates. J. Biomed. Mater. Res. Part B 96B, 294–302 (2011).
Han, L. et al. Tough, self-healable and tissue-adhesive hydrogel with tunable multifunctionality. NPG Asia Mater. 9, e372–e372 (2017).
You, F., Wu, X. & Chen, X. 3D printing of porous alginate/gelatin hydrogel scaffolds and their mechanical property characterization. Int. J. Polym. Mater. Polym. 66, 299–306 (2017).
Song, X. et al. A novel human-like collagen hydrogel scaffold with porous structure and sponge-like properties. Polymers. https://doi.org/10.3390/polym9120638 (2017).
Luo, R., Wu, J., Dinh, N.-D. & Chen, C.-H. Gradient porous elastic hydrogels with shape-memory property and anisotropic responses for programmable locomotion. Adv. Funct. Mater. 25, 7272–7279 (2015).
Lu, M. et al. Phase-separation-induced porous hydrogels from amphiphilic triblock copolymer with high permeability and mechanical strength. Chem. Mater. 34, 10995–11006 (2022).
Baur, E., Hirsch, M. & Amstad, E. Porous 3D printable hydrogels. Adv. Mater. Technol. 8, 2201763 (2023).
Alsaid, Y. et al. Tunable sponge-like hierarchically porous hydrogels with simultaneously enhanced diffusivity and mechanical properties. Adv. Mater. 33, 2008235 (2021).
Ying, G. et al. Bioprinted injectable hierarchically porous gelatin methacryloyl hydrogel constructs with shape-memory properties. Adv. Funct. Mater. 30, 2003740 (2020).
Zhang, W., Zhang, K., Yan, S., Wu, J. & Yin, J. A tough and self-healing poly(l-glutamic acid)-based composite hydrogel for tissue engineering. J. Mater. Chem. B 6, 6865–6876 (2018).
Liu, C. et al. Tough hydrogels with rapid self-reinforcement. Science 372, 1078–1081 (2021).
Cui, Y., Tan, M., Zhu, A. & Guo, M. Strain hardening and highly resilient hydrogels crosslinked by chain-extended reactive pseudo-polyrotaxane. RSC Adv. 4, 56791–56797 (2014).
Feig, V. R., Tran, H., Lee, M. & Bao, Z. Mechanically tunable conductive interpenetrating network hydrogels that mimic the elastic moduli of biological tissue. Nat. Commun. 9, 2740 (2018).
Park, N. & Kim, J. Anisotropic hydrogels with a multiscale hierarchical structure exhibiting high strength and toughness for mimicking tendons. ACS Appl. Mater. Interfaces 14, 4479–4489 (2022).
Gong, J. P., Katsuyama, Y., Kurokawa, T. & Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155–1158 (2003).
Vashahi, F. et al. Injectable bottlebrush hydrogels with tissue-mimetic mechanical properties. Sci. Adv. 8, eabm2469 (2022).
Acknowledgements
The research was supported by the National Science Foundation through the DMREF program (CMMI 2119717). R.J.H. would especially like to thank the Max Plank Institute for Colloids and Interfaces and the Alexander von Humboldt Foundation for supporting his research experience in Potsdam, Germany, to conduct hydrogel characterization. X-ray scattering experiments were performed at the National Synchrotron Light Source II (NSLS II) facility located at sector 11-BM of Brookhaven National Lab. NSLS II is a US Department of Energy (DOE) Office of Science user facility operated for the DOE Office of Science under contract number DE-SC0012704. The authors would like to acknowledge Dr. Masafumi Fukuto and Dr. Ruipeng Li for helping to run X-ray scattering experiments at BM-11. The authors would also like Dr. Missy Hazen and Dr. John Cantolina at the Pennsylvania State University Huck Institute of Materials for assistance in cryo-milling and cryo-SEM characterization of hydrogel cross-sections.
Author information
Authors and Affiliations
Contributions
E.C.L. and R.J.H. conceived the research. E.C.L. synthesized, prepared, and characterized the hydrogels. S.D. and S.M. conducted SALS and fluorescence confocal experiments and provided analysis of the data. S.A. conducted the in situ transmission light microscopy measurements. R.A. and D.R.T. theoretically helped to understand the self-assembly process during solvent exchange. P.F. provided critical insight into understanding and explaining the impact of pore deformation on hydrogel mechanical response. R.J.H. supervised the research. E.C.L. and R.J.H. wrote the manuscript. All authors contributed to the analysis and discussion of the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lloyd, E.C., Dhakal, S., Amini, S. et al. Porous hierarchically ordered hydrogels demonstrating structurally dependent mechanical properties. Nat Commun 16, 3792 (2025). https://doi.org/10.1038/s41467-025-59171-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59171-w
This article is cited by
-
Cellulose hydrogel with in-situ confined nanopores for boosting moist-electric conversion
Nature Communications (2025)
