
Abstract
Numerous bacteria in the human gut microbiome remain unknown and/or have yet to be cultured. While collections of human gut bacteria have been published, few strains are accessible to the scientific community. We have therefore created a publicly available collection of bacterial strains isolated from the human gut. The Human intestinal Bacteria Collection (HiBC) (https://www.hibc.rwth-aachen.de) contains 340 strains representing 198 species within 29 families and 7 phyla, of which 29 previously unknown species are taxonomically described and named. These included two butyrate-producing species of Faecalibacterium and new dominant species associated with health and inflammatory bowel disease, Ruminococcoides intestinale and Blautia intestinihominis, respectively. Plasmids were prolific within the HiBC isolates, with almost half (46%) of strains containing plasmids, with a maximum of six within a strain. This included a broadly occurring plasmid (pBAC) that exists in three diverse forms across Bacteroidales species. Megaplasmids were identified within two strains, the pMMCAT megaplasmid is globally present within multiple Bacteroidales species. This collection of easily searchable and publicly available gut bacterial isolates will facilitate functional studies of the gut microbiome.
Similar content being viewed by others

Introduction
The cultivation of human gut bacteria has accelerated in the last years1,2,3, providing valuable information on the presence of novel taxa within gut microbiomes. Yet while these published collections of human gut isolates note the novel taxa within their collections1,4, rarely is this novelty made known by describing and validly naming the taxa3. Curating the taxonomic assignment of such collections is essential, as often outdated names are used, or recently described taxa are ignored, causing greater confusion in the current taxonomic sphere5,6,7,8. Such curated taxonomy ensures strains are correctly assigned, allowing strain-level diversity to be studied9. Variation between strains of the same species can lead to functional shifts that, for example, alter the association with host dietary habits10,11. One-way strains can vary due to the presence of mobile genetic elements, which are known to affect the phenotype of the isolates in which they occur12. The study of plasmids within the human gut has been limited until recently13,14.
Accessibility to collections of strains from the human gut is essential for the mechanistic study of microbe-microbe and microbe-host interactions15,16,17. However, accessibility remains problematic, with few strains being deposited in public culture collections and even fewer of the proposed novel taxa being validated. For most of the strains deposited, genomes are available, but their quality is not always high, and metadata such as the source of isolation, cultivation requirements, and full taxonomy are often lacking. The small number of deposited strains limits future confirmatory or comparative studies, while the lack of curated metadata reduces the value of the genomic information. Only the professional acquisition of strains by public collections can ensure that the high-quality standards required for future work are maintained and that valuable strain-associated information is gathered and made freely accessible to researchers and users worldwide18. This is particularly relevant for microbiota research to move beyond associations and towards a more mechanistic understanding, as exemplified by the study of Akkermansia muciniphila19,20.
To provide functional insights into the human gut microbiome based on isolates, we sought to create a publicly accessible collection of bacteria isolated from the human gut. Studying this collection of isolates provided insight into the prevalence and diversity of plasmids within the human gut and the presence of megaplasmids. We also highlight health-associated variation within key genera, including novel species which require further in vivo study.
Results
A diverse range of key commensal species isolated from the human gut
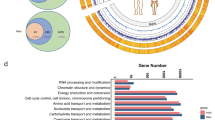
The Human intestinal Bacteria Collection (HiBC) consists of 340 strains, representing 198 species, isolated from human faecal samples (Fig. 1). It contains 29 families from across the seven dominant phyla in the human gut: Bacillota (n = 173 isolates), Bacteroidota (n = 95), Actinomycetota (n = 46), Pseudomonadota (n = 22), Desulfobacterota (n = 2), Fusobacteriota (n = 1), and Verrucomicrobiota (n = 1). Of the 198 species, 29 are novel taxa that we have described and named for validation according to both the SeqCode5 and the ICNP21. High-quality genomes defined as >90% completeness (99.22 ± 1.10%), <5% contamination (0.50 ± 0.77%), >10× genome coverage (380.08 ± 293.97) were generated for all 340 strains. To improve access to the strains, their genomes, information on their cultivation, and isolation source material, we have created the HiBC web interface; https://www.hibc.rwth-aachen.de. The website provides access to the complete collections of 16S rRNA gene sequences, genomes, plasmids, and metadata.

Tree based on all 340 genomes, generated using Phylophlan100. Phyla are indicated with colours. The Bacillota are split due to the placement of Fusobacteriota, which separated strains assigned to ‘Bacillota_A’ by GTDB, however, this is dependent on the method used for tree creation (Supplementary Results). The potential need for splitting the phylum Bacillota is therefore independently supported by the Phylophlan tree and GTDB. Blue circles identify strains belonging to the 29 novel species that are taxonomically described in this paper.
Description of dominant novel taxa within the human gut
There has been a renaissance in isolation of strains from the human gut in the last decade1,2,3,4,22,23,24,25. To place the HiBC isolates in the context of prior studies, we analysed previously published work in which phylogenetically diverse isolates were cultured. Across eight large-scale isolation studies and repositories, a total of 12,565 strains from the human gut were reported (Fig. 2a, Supplementary Data 1). Of these, 9707 (76.1%) isolates were claimed to be requestable in the original publications, but only 1539 (12.2%) have been deposited to a culture collection to ensure long-term availability. However, this includes 1063 isolates that have only been deposited to the China General Microbiological Culture Collection Centre (CGMCC), which prevents the accessibility of risk group 2 organisms, many of which are, to researchers outside China. Many strains within the IHU collection (Institut Hospitalier Universitaire Méditerranée Infection) are also inaccessible under the Nagoya protocol due to concerns surrounding the ethical permission of sampling, which has led to the original paper being retracted26. This means only 476 bacterial isolates (3.8%) from the human gut are currently accessible to the community. While access to many of the isolates is limited, most isolates have been genomes sequenced (11,498, 91.5%), with most being high quality (10,893, 86.7%). To understand the diversity captured by each study, we dereplicated the genomes to estimate the number of species cultured by each study (Fig. 2b). A 33-fold reduction in diversity was found in some studies. Despite the value of capturing variability at the strain level, the limited number of samples used for isolation suggests redundancy within these collections. Therefore, while many studies claim to have a large collection of isolates, this is an overestimate of the true diversity captured.

a The number of strains and genomes produced by eight major isolation projects, along with HiBC, were compared. Strains were deemed requestable if it was claimed in the original publication, although these claims were not substantiated. They were deemed deposited if culture collection identifiers were included in the original paper and were confirmed to exist. Genomes were deemed high quality if they were >90% complete and <5% contaminated. The number of strains within each study is stated, while the percentage meeting each criterion is plotted. Red dots highlight datasets which have barriers to their accessibility, i.e., data available upon request or access limited to specific countries. Strain collections: GMbC, Global Microbiome Conservancy2; BIO-ML, Broad Institute-OpenBiome Microbiome Library24; CGMR, Chinese Gut Microbial Reference25; CAMII, Culturomics by Automated Microbiome Imaging and Isolation26; hGMB, Human Gut Microbial BioBank3; HBC, Human Gastrointestinal Bacterial Collection1; HiBC, Human Intestinal Bacteria Collection (this study); IHU, collection of the Institut Hospitalier Universitaire Méditerranée Infection27,103; HMP, Human Microbiome Project at ATCC. b Number of species per isolate collection, either via manual curation (HiBC) or dereplication of the available genomes (ANI values > 95% indicated identical species). c The cumulative relative abundance of gut metagenomes across 4624 individuals from Leviatan et al.28 covered by all isolated bacteria across studies including HiBC (Global isolates, dark blue), HiBC alone (green), or the subset represented by the 29 novel taxa described in this work (light blue), which had matches within 4583 of the samples. d Relative abundance of dominant (mean relative abundance >0.25%) novel taxa across 4,624 individuals, with the number of positive samples stated. Each strain represents a distinct novel species, described in detail in the protologues at the end of the “Methods” section. e, f Genomic location of proteins significantly differentially prevalent between Crohn’s disease (CD) samples and healthy controls (inner ring), or ulcerative colitis (UC) samples and healthy controls (outer ring). The delta-prevalence (prevalence in healthy donors – prevalence in corresponding patients) is shown in blue (more prevalent in healthy controls), red (UC), or mauve (CD). The species, strain, number of proteins predicted within the genome, and those significantly differentially between health conditions are shown within the circle. Only contigs >10 kp were plotted. In panel c and d, boxplots include a line in the centre indicating the median, the boxes represent the interquartile range, and the whiskers represent the minimum and maximum values, not including outliers.
Next, we assessed the ability of all isolated strains to capture the diversity present in the human gut microbiota (Fig. 2c). This was achieved by mapping the isolates genomes against a MAG collection for which corresponding relative abundance values were available27. The entire landscape of bacteria isolated from the human gut, both from the literature and this study, captured 69.83 ± 18.62% of an individual’s microbiota, while the HiBC alone covered 56.51 ± 20.44%. The 29 novel taxa described within this work enhanced this coverage by 3.75 ± 3.99% on average, and represented >10% of the microbiota from 380 people, which peaked at >40% in two samples. Of the 29 novel taxa, 11 species were identified to have a mean relative abundance >0.15% within positive metagenomic samples (Fig. 2d). The most dominant of these was Ruminococcoides intestinale (mean relative abundance = 2.84%; n = 3023), followed by R. intestinihominis (1.19%; n = 285). Although only recently described28, these results suggest that Ruminococcoides is a dominant but understudied genus within the human gut. A second genus of clear ecological importance to the human gut is Lachnospira, in which we describe two dominant novel species. Interestingly, the two samples in which novel taxa accounted for >40% relative abundance were dominated by either R. intestinale, or the novel species L. rogosae, identified as a medium priority, HMP most wanted taxa (Supplementary Results). Further study of these species is needed to understand their impact on host health and identify the mechanisms, which is now enabled by access to isolated strains.
To uncover associations of the novel taxa with human health conditions, we studied the ecological occurrence of each protein within R. intestinale CLA-AA-H216 (=DSM 117897) (Fig. 2e), as the most abundant novel taxa, and Blautia intestinihominis CLA-AA-H95T (=DSM 111354) (Fig. 2f), as a member of an important genus associated with both health and disease conditions. The association of individual proteins with disease states was determined using InvestiGUT29. This involves studying the prevalence of each individual protein encoded by the strain’s genome, or plasmid, across thousands of metagenomes, then statistically determining if they occur significantly more or less frequently in the gut metagenomes of people with a specific condition or healthy controls. Out of the 2167 proteins encoded by R. intestinale CLA-AA-H216, comparison between Crohn’s disease (CD) patients and healthy controls identified a total of 1902 differentially prevalent proteins, with 1883 being significantly more prevalent in healthy samples, whilst 19 were more prevalent in CD samples. The same pattern was observed with Ulcerative colitis (UC), where a total of 1715 significantly differentially prevalent proteins were identified, with 1684 enriched in healthy samples, and 31 enriched in UC samples. Pathway analysis of the health-associated proteins identified multiple anti-inflammatory pathways, including the ABC transporter for spermidine (PotB/C/D), an anti-inflammatory polyamine30, and biosynthesis of biotin, which has been shown to be immunomodulatory31,32, ameliorating colitis33.
The association of Blautia spp. with inflammatory bowel diseases is more complex, as some species within this genus have been shown to ameliorate colitis34, leading to their inclusion within therapeutic products35. However, recent studies have suggested that some Blautia spp. may exacerbate colitis36. Given the complex interaction of this genus with inflammatory bowel diseases, we studied the association of the novel species, B. intestinihominis CLA-AA-H95T, with both CD and UC. Out of the 3906 proteins, comparison between CD patients and healthy controls identified 523 differentially prevalent proteins, with 488 being significantly more prevalent in healthy samples, whilst 35 were more prevalent in CD samples. In contrast, comparison between UC patients and healthy controls identified 1466 proteins, with 16 being significantly more prevalent in healthy samples, whilst 1450 were more prevalent in UC samples. The functionality of the large number of proteins enriched within UC samples was studied further, identifying ABC transporters for branched-chain amino acids (LivF/G/H/K/M)37, phosphate (PstA/B/C/S)38, and adenosine (NupA/B/C, BmpA)39, each of which has been linked to colitis.
Plasmid landscape in the cultured strains and identification of prevalent megaplasmids
Plasmids are present in the human gut and vary between geographically distinct populations12. However, most analyses have been culture-independent, preventing accurate taxonomic assignment13,14,40. Even studies on horizontal gene transfer using isolates have not reconstructed plasmids and accounted for their impact2. Analysis of bacterial isolates has uncovered many novel plasmids from non-human primates41. As many bacterial genome assembly workflows do not consider plasmids, we developed a genome pipeline that first searches for the presence of plasmids using Recycler42 and plasmidSPAdes43, assembles them, and then removes their reads from consideration during genome assembly (see “Methods” section). This resulted in the reconstruction of plasmids from 46% of the strains (155 out of 340), with a total of 266 plasmids (Fig. 3a). Plasmids were identified in all phyla except Fusobacteriota, for which the HiBC includes only a single isolate. Almost half of the plasmid-positive strains contained more than one plasmid (64 out of 155 strains), up to 6 plasmids in a strain of Phocaeicola vulgatus and two strains of the novel species Enterobacter intestinihominis (Fig. 3b). Across the 266 plasmids identified in HiBC, 3697 proteins were predicted, although only 639 (17.28%) could be functionally annotated. Of these, nine were antibiotic resistance genes, four of which were copies of the APH(2″)-IIIa aminoglycoside resistance gene found on plasmids of identical size (7686 bp) in four strains of Ruthenibacterium lactatiformans (strains CLA-KB-H110, CLA-KB-H15, CLA-KB-H80, CLA-AA-H80).

a Phylum level diversity of plasmid-positive isolates. b Number of plasmids reconstructed within each isolate. The boxplots include a central line indicating the median, boxes represent the interquartile range, and whiskers represent the minimum and maximum values, not including outliers. c Length of reconstructed plasmids in log-10 scale; a bar represents the number; a line represents the distribution. d Network analysis of pBAC plasmid sequence similarity to each other as determined by MobMess. e Sequence alignment of a representative from pBAC cluster 1, 2 and 3. Grey lines show regions of >99% similarity. f The plasmid repertoire purified from 7 strains predicted to contain either a single or multiple pBAC plasmids, as well as additional plasmids. The arrows indicate bands with a size matching the reconstructed pBAC plasmids. g Proteins encoded on pBAC plasmids from different strains with significantly different prevalence between Crohn’s disease (CD) patients and healthy controls. Each point represents a single differentially prevalent protein, coloured based on the pBAC cluster the protein was encoded on (e). The strains are: Bacteroides xylanisolvens (CLA-KB-H139, CLA-SR-H015); Bacteroides caccae (CLA-SR-H005, CLA-KB-H116); Phocaeicola vulgatus (CLA-JM-H27B, CLA-JM-H27,CLA-KB-H143, CLA-AP-H4,4716a, 4896a, HDF, 4889b, CLA-KB-H148); Phocaeicola massiliensis (CLA-AP-H7, CLA-AP-H24); Phocaeicola dorei (CLA-KB-H130, CLA-KB-H95, CLA-KB-H103,CLA-AA-H245); Phocaeicola coprocola (CLA-JM-H3); Bacteroides thetaiotaomicron (CLA-SR-H011); Bacteroides faecis (CLA-KB-H105); Bacteroides cellulosilyticus (CLA-SR-H013); Bacteroides zhangwenhongii (CLA-AA-H144). h Plasmid map of pMMCAT_H253. The innermost rings represent the association of proteins differentially prevalent between CD samples and healthy controls (first ring), or ulcerative colitis (UC) samples and healthy controls (second ring) via InvestiGUT30. Proteins enriched in CD are purple, those enriched in healthy samples are blue, and those enriched in UC are red. The third ring represents the GC content relative to the average of the entire plasmid. Boxes are used to represent genes identified on the plasmid in the forward (outer ring) and reverse (inner ring) strand, with coloured boxes being assigned a COG category, while grey boxes represent COG-unassigned proteins. The fimbriae loci proteins are indicated in dark grey. Enzymes with a single restriction site are indicated on the outer ring in orange. i Quantification of cell adhesion from a pMMCAT-containing strain of P. vulgatus (CLA-AA-H253) and its closest relative strain (H-iso) without the megaplasmid (see “Methods” section). Visualised are the mean values with the 95% confidence intervals. Statistics: paired, one-tailed t-tests. j Representative images of cell adhesion of the two selected strains. Images were taken from the three replicates tested. Green scale bars represent 50 µm.
Megaplasmids (>100 kp) have been described to occur in Lactobacillaceae44 and Bifidobacterium breve45, but their occurrence in a broader range of commensals from the human gut is unknown. We evaluated the length of the reconstructed plasmids and found the average length was 11.74 ± 17.25 kb (Fig. 3c). This included the identification of two megaplasmids, one within a strain of the recently described species Hominifimenecus microfluidus (strain CLA-JM-H9, =DSM 114605; 125.6 kb) and the other in a strain of Phocaeicola vulgatus (strain CLA-AA-H253, =DSM 118718; 103.4 kb). Given the number and size of plasmids a single strain can contain, we assessed their impact on species assignments based on average nucleotide identity, but confirmed that plasmids had only a minor effect on the taxonomic assignment of a genome (Supplementary Results).
Sequence comparison with MobMess14 identified 168 plasmid clusters, the two largest of which were specific to the Bacteroidales (Supplementary Fig. 1a). The largest cluster shared similarities with 1266 plasmids within the comprehensive plasmid database, PLSDB46, but lacked an assigned name, hence, we named it pBAC (plasmid Bacteroidales). The second largest plasmid cluster was identified as the cryptic plasmid, pBI143, which was recently described to be associated with IBD13 and occurred in 14 isolates across eight species. Given that pBAC matches 1266 plasmids within PLSDB, while pBI143 matched only 719, it may be the most prolific plasmid in the human gut. We therefore investigated pBAC further, observing it has been most frequently found in the USA but has also been detected in Denmark, Japan, Ireland, and China (Supplementary Fig. 1b). Investigation of the sequence similarity network identified that pBAC occurred in both a shorter (pBAC-1; ~4220 bp) and longer (pBAC-2; ~5419 bp) form, with the two cluster being connected by two 9494 bp variants (pBAC-3) (Fig. 3d). Comparison of representative sequences from each of these three clusters uncovered that both, pBAC-1 and pBAC-2 shared no similarity, but were identical to regions of pBAC-3 (Fig. 3e). These results suggest that the different forms of pBAC share a common origin, implying they can co-occur within the same strain. We identified B. xylanisolvens CLA-KB-H139 as a strain which was predicted to contain both pBAC-1 and pBAC-2 and confirmed their existence within the isolate by extracting plasmids from a diverse range of species predicted to contain different forms of pBAC (Fig. 3f). This analysis confirmed that pBAC occurs across the Bacteroidales. Given the prevalence of pBAC within common denizens of the human gut, we aimed to study their functional potential. All three pBAC clusters encode mobilisation machinery (MbpA/B/C)47. Interestingly, both pBAC-1 and pBAC-2 encode different toxin-antitoxin pairs, with that in pBAC-1 resembling YoeB and YefM48, while the pBAC-2 encoded system shared similarity with RelE and Phd49. Given their differing protein content, we hypothesised their association with host health may differ, hence studied the prevalence of their encoded proteins across disease cohorts (Fig. 3g). pBAC-1 was observed to encode 2–3 proteins that were significantly more prevalent within CD patients, with the most enriched protein being a replication protein, followed by YefM-like and YoeB-like proteins. Conversely, pBAC-2 plasmids encoded 5–6 proteins within significantly lower prevalence in CD patients, with the most being the RelE-like and PhD-like proteins. Toxin-antitoxin systems within Bacteroidota plasmids have previously been reported, although pBAC shared no similarity with these described plasmids50. These results suggest that the pBAC-encoded toxin-antitoxin pairs are associated with human health, potentially by altering the fitness of their host strain.
In addition to the highly prevalent pBAC plasmid, we studied the megaplasmid present in P. vulgatus (CLA-AA-H253), a prevalent and dominant species in the human gut. The megaplasmid matched 26 plasmids within PLSDB, including multiple designated as pMMCAT (Supplementary Fig. 1a), a recently proposed large plasmid from Bacteroidales, which has yet to be confirmed using sequencing-independent methods51, but has been shown to impact its host's ability to form biofilms52. As such, the P. vulgatus megaplasmid was designated pMMCAT_H253. PacBio sequencing generated a complete genome for strain CLA-AA-H253, which confirmed the reconstruction of pMMCAT_H253. To provide sequencing-independent validation of pMMCAT_H253, we used pulsed field gel electrophoresis which showed a band of ~100 kb after treatment using XbaI, for which the plasmid had a single restriction site allowing for its linearisation (Supplementary Fig. 2a). The bands’ identity was confirmed using southern blot with primers designed based on the predicted pMMCAT_H253 sequence (Supplementary Fig. 2b). Out of the 104 proteins encoded on pMMCAT_H253, only 6 could be assigned to a functional category, highlighting the need for further characterisation of these proteins (Fig. 3h). A locus containing four proteins assigned to ‘fimbrillin family proteins’ was identified within a region of lower GC content compared to the plasmids average, which may suggest this region represents variable cargo and not the backbone. The presence of ‘fimbrillin family proteins’ on pMMCAT_H253 was further investigated as fimbriae can facilitate adhesion of cells53, including to the host epithelium, to enhance colonisation54. We therefore studied the ability of CLA-AA-H253, the pMMCAT-containing strain, and its closest related strain (H-iso, 98.9% ANI), which lacks pMMCAT, to adhere to plastic wells. CLA-AA-H253 adhered to the plastic significantly more than H-iso after only 4 h, with 2-fold more bacteria adherent after 30 h of growth (Fig. 3i). The adherence of strain CLA-AA-H253 was observed in the wells, while H-iso was adhered in patches across the wells (Fig. 3j). These results support the need for greater study of pMMCAT as a potential source for phenotype variation within genera known to contribute to host health55,56.
Enhanced diversity of Faecalibacterium species
The genus Faecalibacterium currently (January 2025) contains seven validly published and correctly named species. HiBC contains representatives for five of these, and isolates representing two novel species. Initial 16S rRNA gene analysis assigned these novel species as Faecalibacterium prausnitzii, a common commensal of the gut microbiota that has been associated with human health conditions ranging from reducing inflammation57 to improving cognitive function in Alzheimer’s disease58. Many of these beneficial observations have been attributed to F. prausnitzii’s ability to produce large amounts of the short-chain fatty acid butyrate59.
Taxonomically, the genomic diversity of F. prausnitzii is proposed to be greater than suggested by 16S rRNA gene sequence analysis60. This has led the GTDB to split this species into 12 proposed species. HiBC contains representatives of the not yet described species defined by GTDB-Tk as F. prausnitzii A (strain CLA-AA-H175) and F. prausnitzii J (strain CLA-AA-H281). This was confirmed using ANI and can be observed in a phylogenomic tree (Fig. 4a). We therefore propose to name these novel species Faecalibacterium tardum (CLA-AA-H175) and Faecalibacterium intestinale (CLA-AA-H281). Protologues for these, and the other 27 novel taxa described in this paper, are provided at the end of the “Methods” section. Both novel species were observed in the majority of microbiota studied at similar relative abundances as F. prausnitzii (F. prausnitzii = 0.24 ± 0.24%; F. tardum = 0.16 ± 0.20%; F. intestinale = 0.14 ± 0.21%) (Fig. 4b).

a The two novel species of Faecalibacterium described within this paper placed within the current landscape of Faecalibacterium spp. with a valid name, along with the type genomes for proposed divisions of F. prausnitzii, as determined by GTDB. The phylogenomic tree was rooted on Ruminococcus bromii ATCC 27255T. Novel species are in bold, and the type strain of F. prausnitzii is underlined. b Relative abundance and prevalence of the genus Faecalibacterium, and each Faecalibacterium species represented within HiBC across 4624 metagenomic samples. The boxplots include a central line indicating the median, boxes represent the interquartile range, and whiskers represent the minimum and maximum values, not including outliers. c Butyrate production pathway in Faecalibacterium with gene names and KEGG ortholog identifiers when possible. d Phylogenomic tree of the Faecalibacterium strains within HiBC, displaying the ability of each strain to produce butyrate over a 48 h-period, along with the OD600 that the strain achieved during the testing period (n = 3 independent batch cultures for each strain; the replicates are shown with individual boxes). The phylogenomic tree was rooted on R. bromii ATCC 27255T. e Sequence comparison of the butyrate production loci across the Faecalibacterium strains. Genes are coloured based on their assignment to each step in the butyrate production pathway in (c). f AlphaFold3 model of the But complex in F. tardum CLA-AA-H175 against the full But protein in F. prausnitzii CLA-AA-H222. The first CLA-AA-H175 But gene is highlighted in yellow in the dashed box, while the second gene is shown in brown. The highlighted protein is indicated in the top right of the dashed box. g Same as in (f), but this time the second CLA-AA-H175 But gene is highlighted in yellow in the dashed box, while the first gene is in brown. The highlighted protein is indicated in the top right of the dashed box. h Correlation of the mean OD600 against the mean butyrate production of each strain with a linear regression and its 95% confidence interval and analysed using a two-sided Pearson correlation coefficient.
Butyrate production by Faecalibacterium spp. is critical for host health, hence, we studied variability between the strains in their ability to produce butyrate (Fig. 4; Supplementary Data 2). Butyrate production by Faecalibacterium is achieved via the acetyl-CoA pathway, which is also the dominant pathway for butyrate production in the human gut (Fig. 4c)61,62. While all Faecalibacterium strains within HiBC produced butyrate, the amounts varied greatly, with F. tardum (strain CLA-AA-H175) producing only 0.8 ± 0.16 mM, whereas strains of F. longum and F. butyricigenerans produced >10 mM (Fig. 4d). Given that butyrate production is a conserved phenotype of this genus, we investigated if variation in the acetyl-CoA pathway loci was responsible for the observed variation between isolates (Fig. 4e). While little variation was observed between most of the strains, F. tardum CLA-AA-H175 encoded two truncated copies of the butryl-CoA:acetate-CoA transferase gene. To understand the cause of these truncated genes, we studied their placement in relation to each other and identified that they occurred on the same strand but in different frames, with an overlap of 55 bp. Interestingly, the second truncated gene is encoded with ‘GTG’ as an alternative start codon, which may lead to lower transcription and hence alter butyrate production further63. Structural modelling of these proteins using AlphaFold3 identified that the truncated proteins form a complex that shares 100% (Fig. 4f) and 90% (Fig. 4g) identity with the full protein from F. prausnitzii CLA-AA-H222, respectively. Acetate utilisation and butyrate production have been linked to the growth of Faecalibacterium spp.64,65, hence we considered whether the ability of the isolate to grow under the testing conditions alters its metabolism, and eventually the ability to produce butyrate. We therefore correlated the average butyrate production of each strain against its average growth, measured by OD600 (Fig. 4h). A strong, significant correlation (r = 0.76; p = 0.01) was identified between the growth of a strain and the amount of butyrate it produced. These results suggest that strains of Faecalibacterium vary greatly in their ability to produce butyrate, either as a result of genetic modification, as in the case of F. tardum, or due to the strain's ability to grow in vitro. This further implies that strains that grow at high abundance in vivo are likely to produce the most butyrate.
Discussion
Over the last decade, the renewed interest in cultivation has expanded the number of genomes available for taxa within the human gut. However, the corresponding strains are rarely accessible to the research community, as highlighted by only 3.8% of human gut isolates being publicly deposited within a culture collection. The process of strain deposition within culture collections has previously been highlighted as a major limiting factor in making strains publicly available66. To overcome this, we initiated a system for bulk deposition of strains at the DSMZ (Supplementary Methods). Initial registration of strains with StrainInfo prior to deposition allows the processing of strains to be monitored. The assignment of DOIs to each strain ensures their traceability, facilitating publication while ensuring strains are consistently referenced. Application of this system allowed for deposition of 340 human gut isolates in the DSMZ, which in turn allowed the naming and description of 29 novel species, including the type species of three novel genera. Based on this, the HiBC is the first collection of human gut isolates that is entirely publicly accessible.
Large-scale cultivation of human gut bacteria in this study led to the isolation of many novel species and genera. Whilst previous cultivation studies report many isolates representing novel diversity, these bacteria are rarely taxonomically described and named. The discovery of prevalent and important novel species in this work will facilitate future experiments on the role of bacteria in health and disease. This included Ruminococcoides intestinale, Blautia intestinihominis, and the isolation of multiple strains of Faecalibacterium, including species with the potential to reduce gastrointestinal inflammation57. We confirmed that F. prausnitzii represents a diverse group of related species that show reduced 16S rRNA gene sequence diversity60. By naming two of these species and describing the butyrate production and growth characteristics of seven different species in this genus, we observed that their ability to produce butyrate was proportional to their ability to grow. Given these results, further quantification of these strains’ ability to grow within the human gut is required to understand their contribution to SCFA levels in vivo, and therefore their potential impact on host health.
The reconstruction of plasmids for almost half of HiBC isolates allows the direct link of plasmids to strains of various species, information previously lacking from studies on plasmids from the human gut12,13,14. Of note, Bacteroidales strains often contain multiple plasmids, particularly copies of pBI143 or pBAC. The pBAC plasmid was the most frequently reconstructed plasmid and was observed to occur in three forms. Investigation of these forms uncovered that both major forms have differing associations with host health. The megaplasmid pMMCAT was also identified within a strain of P. vulgatus, leading to enhanced adhesion to surfaces compared to strains lacking pMMCAT. By making the reconstructed plasmids and strains publicly available, we believe this resource expands the toolbox of methods for genetically modifying non-model commensal gut microbes.
Metagenomic studies have provided important insights into the taxonomic and functional diversity present in the human gut, however, they are unable to provide mechanistic insights. By enabling broad access to human gut isolates in this work, the functionality of taxa can be experimentally validated, facilitating mechanistic studies of microbe-host interactions.
Methods
Ethics
The Ethics Committee of the Medical Faculty of RWTH University Aachen permitted bacterial isolation from human stool under ethical numbers EK 23-055, EK 316-16, and EK 194/19. For strains originating from Vienna, isolation was approved by the University of Vienna ethics committee under ethical number 00161. For strains originating from Braunschweig, isolation was approved by the Ethics Committee of Lower Saxony (MHH permit No. 6794, 8629 and 8750). Written informed consent was signed by all enrolled participants.
Bacterial isolation and cultivation
Human stool was collected in sterile plastic bags and stored in tightly sealed plastic buckets until further processing. An oxygen scavenger sachet (BD Biosciences; ref. 260683) was added to each bucket before sealing to reduce exposure to oxygen. All samples were processed in the lab within 24 h of collection. First, the faecal material was homogenised by manual kneading of the plastic bag. Stool samples were either mixed 1:5 with anaerobic FMT media67, distributed to 1 mL aliquots and stored at −80 °C until further use, or the samples were further processed as described previously68. Hence, approximately 5 g of the samples were dissolved in 50 ml of anoxic PBS supplemented with peptone (0.05% w/v), L-cysteine (0.05% w/v) and dithiothreitol (DTT) (0.02% w/v) by shaking the glass flask intensely. A syringe with needle was used to transfer 5 ml of the slurry through a rubber stopper (previously flamed using ethanol) into a second glass flask containing 45 ml of the same buffer in an anaerobic atmosphere (89.3% N2, 6% CO2, 4.7% H2) to create a 1:100 dilution of the original sample. The flask was moved into an anaerobic workstation (MBraun GmbH, Germany) to prepare 2 ml aliquots under anaerobic conditions, which were mixed with 2 ml of 40% anaerobic glycerol to a final concentration of 20%, then stored at −80 °C until use.
Two different approaches were used for bacterial isolation. For classical isolation, the faecal aliquots were thawed inside the anaerobic workstation and diluted with anoxic PBS (see above) in a tenfold dilution series (10−2–10−6). 50 µl of each dilution step was transferred onto different agar plates and spread using an L-spatula. Single colonies were picked after 1–7 days of incubation at 37 °C under anaerobic conditions. Bacterial cells were re-streaked at least three times to guarantee the purity of the culture. For the second isolation approach, the single-cell dispenser b.sight (Cytena GmbH, Germany) was used as described before69. Details on culture media preparation and ingredients are provided in the Supplementary Methods.
The isolated bacteria were first identified using MALDI-TOF MS (Bruker Daltonics, Bremen, Germany). If a species was not yet present within HiBC, or could not be reliably identified by MALDI-TOF MS, the bacteria were stored as cryo-stocks and DNA extracted for genome sequencing. Isolates from different donors that belonged to the same species were considered to represent individual strains and were kept in the collection. Cryo-stocks were generated by freezing in glycerol media (end concentration 20%) at −80 °C. All HiBC strains have been deposited at the Leibniz Institute-German Collection of Microorganisms and Cell Cultures (DSMZ). In addition, the strains representing novel taxa were deposited at either the Belgian Coordinated Collections of Microorganisms (BCCM) or the Japan Collection of Microorganisms (JCM).
Deposition at the DSMZ was facilitated by developing a bulk-deposition system. In brief, after the DSMZ has assessed a researcher's request to submit a strain collection, it requests the strains in large batches, which are deposited in an iterative process. Once cultures have been received by the DSMZ and passed initial checks, the system allows for the allocation of stable identifiers in the form of Digital Object Identifiers (DOIs) provided by the database StrainInfo (www.straininfo.dsmz.de), where each strain is permanently registered with its metadata. This step ensures that all data (e.g., publications and sequence data) can be linked to the corresponding strain entry when the strains become available. Strain entries are initially displayed with the status ‘Deposition in progress’. When the deposition process is completed (which may take several months) and the strains become available in the DSMZ catalogue, the status of the strains is updated to ‘Published’ in StrainInfo. This process is detailed in greater depth in the Supplementary Methods.
Metabolite production analysis
Concentrations of short-chain fatty acids (SCFAs) (acetate, butyrate, propionate, valerate), branched-chain fatty acids (isobutyrate, isovalerate), intermediate metabolites (ethanol, formate, lactate, 1,2-propandiol, 1-propanol, succinate), as well as mono- and disaccharides (galactose, glucose, lactose) were measured by high-performance liquid chromatography (HPLC). The bacterial strains were grown anaerobically, apart from Robertmurraya yapensis CLA-AA-H227, which was grown under aerobic conditions, in YCFA broth (DSMZ Medium No. 1611) in Hungate tubes for 48 h at 37 °C. Triplicate cultures from each strain were measured. Baseline controls were taken from each sterile Hungate tube before inoculation. Each taken sample was centrifuged (10,000 × g, 10 min, 4 °C), supernatants were collected and stored at −80 °C until HPLC measurement. Samples were prepared and measured (including HPLC settings) as described previously68. External standards were used for concentration determination by comparison of peak retention times (HPLC grade compounds were purchased from Sigma-Aldrich). Peaks were integrated using the Chromaster System Manager Software (Version 2.0, Hitachi High-Tech Science Corporation 2013, 2017). Metabolite concentrations >0.2 mM (limit of detection (LOD) for citrate, 1-propanol), >0.24 mM (LOD for butyrate, formate, galactose, glucose, isobutyrate, isovalerate, lactose, valerate), >0.4 mM (ethanol, 1,2-propandiol) and >0.8 mM (acetate, lactate, propionate, succinate) were considered for statistical analysis if present in all three replicates. Production and consumption of metabolites were calculated by subtracting the baseline values from the sample taken after 48 h of growth.
Cellular fatty acids (CFAs) determination
Cellular fatty acids were measured at the Leibniz Institute DSMZ. The strains representing novel taxa were grown under the conditions indicated in the respective protologues. Approximately 100 mg (wet weight) of cell biomass was extracted according to the standard protocol of the Microbial Identification System (MIDI Inc., version 6.1; technical note #101). CFAs were analysed by converting them into fatty acid methyl esters (FAMEs) through saponification, methylation, and extraction. The resulting FAME mixtures were separated using gas chromatography (GC) and detected with a flame ionisation detector (FID). Subsequent analysis involved the identification of fatty acids using a GC-MS system (Agilent GC-MS 7000D) as described by Vieira et al.70. Further derivatisation methods were used for structural elucidation of unidentified compounds. For branched-chain fatty acids, cyclo-positions, and multiple double bonds, 4,4-dimethyloxazoline (DMOX) derivatives were analysed71.
Isolation of genomic DNA
DNA was isolated using a modified protocol according to Godon et al.72. Frozen cell pellets were mixed with 600 μl stool DNA stabiliser (Stratec biomedical), thawed, and transferred into autoclaved 2-ml screw-cap tubes containing 500 mg 0.1-mm-diameter silica/zirconia beads. Next, 250 µL 4 M guanidine thiocyanate in 0.1 M Tris (pH 7.5) and 500 µL 5% N-lauroyl sarcosine in 0.1 M PBS (pH 8.0) were added. Samples were incubated at 70 °C and 700 rpm for 60 min. A FastPrep® instrument (MP Biomedicals) fitted with a 24 × 2 ml cooling adaptor filled with dry ice was used for cell disruption. The programme was run 3 times for 40 s at 6.5 M/s. After each run, the cooling adaptor was refilled with dry ice. An amount of 15 mg Polyvinylpyrrolidone (PVPP) was added and samples were vortexed, followed by 3 min centrifugation at 15,000 × g and 4 °C. Approximately 650 μl of the supernatant were transferred into a new 2 ml tube, which was centrifuged again for 3 min at 15.000 × g and 4 °C. Subsequently, 500 μl of the supernatant was transferred into a new 2 ml tube and 50 μg of RNase was added. After 20 min at 37 °C and 700 rpm, gDNA was isolated using the NucleoSpin® gDNA Clean-up Kit from Macherey-Nagel. Isolation was performed according to the manufacturer’s protocol. DNA was eluted from columns twice using 40 μl Elution buffer, and concentration was measured with NanoDrop® (Thermo Scientific). Samples were stored at −20 °C.
Genome library preparation and Illumina sequencing
Library preparation and sequencing were conducted using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina with dual index primers and ∼300 ng of DNA (NEB Inc.) on an automation platform (Biomeki5, Beckman Coulter) according to the manufacturer’s instructions. The time used for enzymatic shearing to ca. 500 bp to be used on a MiSeq run was 10 min, and to ca. 200 bp to be used on a NextSeq run was 30 min. PCR enrichment of adaptor-ligated DNA was conducted with five cycles using NEBNext Multiplex Oligos for Illumina (NEB, USA) for paired-end barcoding. AMPure beads (Beckman Coulter, USA) were used for size selection and clean-up of adaptor-ligated DNA. Sequencing was performed either at the IZKF Core Facility Genomics (Uniklinik RWTH Aachen) on a NextSeq platform (Illumina) (PE 150 bp) or on a MiSeq (Illumina) (PE 300 bp) in-house.
Sequencing data processing
Genomes were assembled from paired-end Illumina short reads. Low-quality reads with an expected average quality below 20 over a 5-base window, containing adaptors, and shorter than 50 bases were discarded using Trimmomatic (v0.39)73. Reads with phiX sequences were removed using BBduk from the BBtools suite74. Plasmid sequences were reported only when inferred from plasmidSPades (v3.15.5)75 and then from Recycler (v0.7)42. Reads free from plasmid sequences were then assembled using SPades (v3.15.5)43 with default parameters.
Contigs above 500 bp in assemblies were kept for quality evaluation. Following the MIxS specifications76 as well as requirements from the SeqCode5, genomes with a coverage above 10×, a completion above 90%, and a contamination below 5% based on CheckM (v1.2.2)77 estimation using single-copy marker genes were flagged as high-quality draft genomes. Additional quality flags on the assembly included: (1) <100 contigs, longest contig >100 kp, and N50 > 25 kp using QUAST (v5.0.2)78; (2) detectable 16S and 23S rRNA genes using metaxa2 (v2.2.3)79; (3) >18 unique essential tRNAs genes and a detectable 5S rRNA gene sequence using the annotations of bakta (v1.6.1)80.
The genome assembly pipeline, including the reconstruction of plasmids and quality checks steps, is available as a reproducible workflow using Snakemake (v7.9.0)81: www.github.com/clavellab/genome-assembly.
Genome analysis
Taxonomic, functional, and ecological analysis for all genomes was conducted using Protologger (v1.3)82. For this, 16S rRNA gene sequences were extracted from each strain’s genome using barrnap v0.9 (default settings) (www.github.com/tseemann/barrnap), and the longest 16S rRNA gene sequence from each genome was used as input for Protologger. Phylogenomic trees were generated using Phylophlan v3.0.6083 with proteomes predicted using Prodigal v2.6.3 (default settings)84. Gut metabolic modules presence was based on the identification of all included KOs within a genome, detected by Kofamscan v1.3.085.
Comparison to published isolate collections
Metadata for a previously published collection of microbial isolates from the human gut were obtained from screening literature. To prevent redundancy, we have excluded articles that are a subset of large collections4, and those that lack strain metadata or genome access86. In the case of isolate collections from multiple body sites, the isolation source was checked to be related to the gastrointestinal tract. Of the 12,565 strains identified, 11,498 had genomes and hence could be analysed. Of these, 10,893 had a high-quality genome and were studied. High quality was determined by completion above 90%, and less than 5% contamination based on CheckM (v1.2.2). Strains were determined to be ‘publicly available’ if a genome is available and the strain has been deposited within a culture collection, i.e., DSMZ, CGMCC, etc. The validity of each published culture accession number was checked to ensure strain availability. Genomes within the collections were dereplicated at the species level based on >95% ANI values via FastANI87.
Ecological analysis
The occurrence of each strain across 16S rRNA gene amplicon samples was conducted using Protologger82. In essence, 1000 16S rRNA amplicon samples for each body site (gut, vagina, skin, lung) were processed by IMNGS88. Next, their operational taxonomic unit (OTU) sequences were compared against the 16S rRNA gene sequences of the HiBC strains using BLASTN (>97% identity, >80% coverage). Comparison of HiBC genomes against metagenome-assembled genomes (MAGs) was also conducted using Protologger and its collection of 42,927 MAGs with curated metadata. The 16S rRNA gene sequences of the Human Microbiome Projects “most wanted” taxa were obtained from Fodor et al.89 and their priority taken from the original publication89. The most wanted sequences were compared to the 16S rRNA gene sequences of the HiBC strains using BLASTN (>97% identity, >80% coverage)90. The prevalence and relative abundance of the HiBC strains within shotgun metagenomic samples were determined by comparison of the HiBC strain genomes to the representative genomes from Leviatan et al.27 via FastANI (>95% ANI)87. HiBC strains matching to representative genomes were then connected to the pre-calculated relative abundance of the representative genomes across 4,623 individuals, published in Leviatan et al.27.
Plasmid analysis
Sequence similarity between the isolates plasmids was determined using MobMess14 and clusters were visualised with Cytoscape91. Clustering was conducted first based only on the isolate plasmids, and then including the background previously predicted human gut plasmids. The similarity of each plasmid to known sequences was determined with the ‘mash dist’ option of PLSDB (v2023_11_03_v2)46. The ecology of each plasmid was inferred from the geographical location assigned to each matching plasmid. The sequences of pBAC plasmids were rotated using Rotate, and then aligned with Easyfig92. Only regions of similarity with an identity >90% were studied.
Pulsed field gel electrophoresis
Cultures were grown overnight in 5 mL BHI media, centrifuged at 2400 × g for 5 min (2 mL for P. vulgatus CLA-AA-H253, 5 mL for H. microfluidus CLA-JM-H9), resuspended in 100 µL sterile PBS and incubated at 45 °C for 10 min. Of this, 80 µL was mixed with 320 µL 1% agarose (Bio-Rad Certified Megabase Agarose) at 45 °C, 80 µL transferred to PFGE plug moulds (Bio-Rad CHEF Disposable Plug Molds, 50-Well) and cooled at 4 °C for 30 min. Solidified agarose plugs were transferred to lysis solution inside 2 mL microcentrifuge tubes (20 mM Tris-HCl pH 8.8, 500 mM EDTA pH 8, 1% N-laurylsarcosine, 1 mg/mL Proteinase K) and incubated with shaking at 250 rpm at 45 °C for 4 h. Lysis solution was replaced with fresh lysis solution + RNAse (10 µg/mL) and incubated with shaking at 45 °C overnight. Lysis solution was replaced with wash solution (25 mM Tris-HCl pH 7.5, 100 mM EDTA pH 8) and incubated with shaking at 45 °C. Plugs were then transferred to fresh 2 mL microcentrifuge tubes with wash solution containing phenylmethanesulfonyl fluoride (1 mM) and incubated with shaking at 45 °C for 30 min, twice. Plugs were then transferred to 1 mL wash solution at stored at 4 °C until use in PFGE. For analysis of intact genomic DNA, agarose plugs were subjected to 100 Gy of γ radiation using a 137Cs source (Gammacell 1000), to linearise circular chromosomes93. PFGE was performed using a CHEF Mapper apparatus (Bio-Rad). Intact and XbaI-digested DNA fragments were separated on a 1.2% agarose gel in 0.5× TBE at 14 °C, with a gradient voltage of 6 V/cm, linear ramping, an included angle of 120°, initial and final switch times of 0.64 s and 1 min 13.22 s, respectively, and a run time of 20 h 46 min.
Southern blot
The DNA probe was designed by identifying a 1428 bp DNA sequence unique to the pMMCAT_H258 plasmid sequence. Primers sequences (forward primer: GAATTAACGGCCGAGTTCGC; reverse primer: TACCAGTACCGAGGGGAAGG) were checked against the P. vulgatus genome sequence using SnapGene (www.snapgene.com) to confirm specificity to only plasmid DNA. PCR primers were then designed based on the regions flanking the unique region. P. vulgatus total genomic and plasmid DNA was extracted using the Mericon DNA Bacteria Plus Kit following the manufacturer’s instructions (Qiagen). This DNA was used as a template to amplify the target DNA sequence using Thermo Fisher Scientific Phusion Hot Start 2 DNA polymerase and the above primer set. The amplified DNA was visualised by gel electrophoresis to confirm the expected DNA size, and the DNA was extracted and purified using Thermo Fisher Scientific GeneJET gel extraction kit.
The Southern blot gel was stained with 0.5 µg/ml ethidium bromide and visualised, then acid-nicked in 0.25 M HCl, and subsequently denatured in 1.5 M NaCl, 0.5 M NaOH. The DNA was transferred onto a GE Healthcare Amersham Hybond XL membrane by vacuum transfer using a Vacugene XL gel blotter (Pharmacia Biotech) for 1 h at 40 mBar. The membrane was briefly neutralised in 2× SSPE (20× SSPE: 3 M NaCl, 230 mM NaH2PO4, 32 mM EDTA, pH 7.4) and the DNA then crosslinked with 120 mJ/cm2 UV. The membrane was pre-hybridised for 4 h at 65 °C in 6× SSPE, 1% SDS, 5× Denhardt’s solution (100× Denhardt’s solution: 2% Ficoll 400, 2% polyvinyl pyrrolidone 360, 2% bovine serum albumin), 200 µg/ml salmon sperm DNA (Roche, boiled). The DNA probe used 50 ng of DNA template and was radiolabelled using 0.74 MBq of [α-32P] dCTP (Perkin Elmer) and HiPrime random priming mix (Roche), then purified on a Bio-Rad P-30 column. The membrane was hybridised with the radiolabelled DNA probe overnight at 65 °C in 6× SSPE, 1% SDS, 5% dextran sulphate, 200 µg/ml salmon sperm DNA (Roche, boiled). The membrane was then washed twice for 30 min at 65 °C in 2× SSPE, 0.5% SDS, and twice for 30 min at 65 °C in 0.2× SSPE, 0.5% SDS, before being exposed to a phosphorimager screen (Fujifilm) for 24 h and then scanned on a GE Healthcare Typhoon.
Plasmid extraction and visualisation
Frozen cryo-stocks were plated on mGAM plates and incubated anaerobically at 37 °C for 48 h. A single colony was inoculated into 5 mL of BHI media within a Hungate tube and incubated for 48 h. 2 mL of culture was transferred into 2 mL microcentrifuge tubes and centrifuged at 18,000 × g for 5 min. The medium was removed and the pellet processed using QIAprep Spin Miniprep kit, following the manufacturer’s recommendations (Qiagen, Cat. No. 27106). Depending on DNA concentration, 10–20 µL of extracted DNA was run on a 0.5% agarose gel (Sigma-Aldrich A9539) at 80 V for 120 min (Bio-Rad Biometra) along with GeneRuler 1 kb DNA ladder (Thermo Fisher Scientific SM0312) and stained with a dye (Midori Green Advance, BulldogBio). The gel was imaged with the Bio-Rad GelDoc Go imaging system.
Crystal violet staining to quantify strain adhesion
Strains of interest were tested in triplicate. For each, one colony was picked and grown to saturation overnight in BHI. Cultures were diluted back to an OD600nm of 0.1 in fresh BHI, 3 ml were placed per well in 6-well plates (Nunc cell-culture, treated, flat clear polystyrene plates, Thermo Scientific), and the plates were incubated in static conditions at 37 °C. For each time point, a plate containing the two strains to be compared was removed from the anaerobic cultivation chamber, rinsed with water, and dried at 60 °C for 2 h. Each well was incubated with 1% (w/v) crystal violet solution, then rinsed 3 times with deionised water and air dried. Once all plates were collected, wells were destained by adding 1 ml of a 1:4 acetone:ethanol solution. After gentle mixing, at least three technical replicates of 200 µL per well were placed in wells of a 96-well plate. The optical density of crystal violet staining present in the destaining solution was measured at 590 nm. Wells filled with 200 µL of pure destaining solution were used as blank reference. All wells from the same biological replicate at a given time point were averaged to provide a single point and used for statistical analysis. Cell counting in Neubauer chambers ensured an initial cell concentration varying by less than 5%, and OD600nm measurements in the supernatant at all timepoints of incubation showed similar OD values over all biological replicates, confirming that growth rates were not driving the observed 2-fold difference in attached biomass after 30 h. Representative images of attachment were obtained after 24 h following the above protocol until the first drying stage. Two wells were imaged in their centre using phase contrast (Nikon Ti-E inverted microscope, 40× objective lens).
Protein modelling
Protein sequences were entered into the AlphaFoldServer94. Pairwise structure alignment was conducted using the RCSB PDB web server with the TM-align method95,96.
Website design
Access to taxonomic, cultivation, and genomic information about the HiBC strains is available at https://www.hibc.rwth-aachen.de. This website was created using the Shiny package from R, and the code is available at: https://git.rwth-aachen.de/clavellab/hibc. The 16S rRNA gene sequence and genome for each strain can be downloaded directly from this site, both individually for strains of interest via the research data management platform Coscine, or for the entire collection via the digital repository Zenodo. Further strain-specific metadata provided includes: isolation conditions, source, and risk group.
Description of novel taxa
The description of novel taxa was based on the analysis provided by Protologger v1.382, and manually curated into the protologues below. For each isolate, taxonomy was assigned using the following thresholds: <98.7% 16S rRNA gene sequence identity (as an indication for as-yet undescribed species), <94.5% (undescribed genus), and <86.5% (undescribed family)97. ANI values <95% to separate species91; POCP values <50% for distinct genera98. Phylogenomic trees were also considered to make decisions on genus- and family-level delineation99. All novel taxa have been registered with the SeqCode and will be registered with the ICNP.
Description of Alistipes intestinihominis sp. nov
Alistipes intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, the intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 3.85 Mbp, G+C percentage is 58.29%, with 99.76% completeness and 0.96% contamination. Strain CLA-KB-H122 was determined to be a new species based on 16S rRNA gene analysis, with the closest validly named match being Alistipes timonensis (98.17%). Separation from existing Alistipes species was confirmed by ANI comparison, which gave a value of 91.78% to A. timonensis. GTDB-Tk classified strain CLA-KB-H122 as ‘Alistipes senegalensis’. However, the latter name is as yet not valid. Functional analysis showed the strain has 83 transporters, 17 secretion genes, and predicted utilisation of cellulose and starch along with production of L-glutamate and folate. In total, 395 CAZymes were identified, with 59 different glycoside hydrolase families and 19 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 72 h of growth in DSMZ medium 1611 included 15:0 ISO FAME (19.4%), 15:0 FAME (18.9%), 16:0 ISO FAME (10.6%), 17:0 ISO 3OH FAME (8.8%), 17:0 FAME (7.2%), and 17:0 3OH FAME (6.9%). The type strain, CLA-KB-H122T (phylum Bacteroidota, family Rikenellaceae) (=DSM 118481) (StrainInfo: https://doi.org/10.60712/SI-ID414389.1, genome: GCA_040095975.1), was isolated from human faeces.
Description of Bifidobacterium hominis sp. nov
Bifidobacterium hominis (ho’mi.nis. L. gen. n. hominis, of a human being, pertaining to the human gut habitat, from where the type strain was isolated).
The genome size is 2.03 Mbp, G+C percentage is 55.98%, with 99.77% completeness and 0.45% contamination. The closest relative to strain CLA-AA-H311 was Bifidobacterium pseudocatenulatum (99.07%) based on 16S rRNA gene analysis. However, ANI comparison identified strain CLA-AA-H311 as a novel species within the genus Bifidobacterium, with an ANI value of 93.18% against B. pseudocatenulatum. GTDB-Tk classification as ‘Bifidobacterium sp002742445’ confirmed the proposition of a novel species. Placement within the genus Bifidobacterium was confirmed by the presence of fructose-6-phosphate phosphoketolase (KO1621)100. Functional analysis showed the strain has 90 transporters, 20 secretion genes, and predicted utilisation of starch and production of propionate, acetate, and folate. In total, 137 CAZymes were identified, with 28 different glycoside hydrolase families and 11 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 24 h of growth in DSMZ medium 1203a included 16:0 FAME (28.8%), 18:0 FAME (15.3%), 18:1 CIS 9 DMA (15.1%), 18:1 CIS 9 FAME (14.3%), and 14:0 FAME (7.8%). The type strain, CLA-AA-H311T (phylum Actinomycetota, family Bifidobacteriaceae) (=DSM 118068, =LMG 33596) (StrainInfo: https://doi.org/10.60712/SI-ID414317.1, genome: GCA_040095915.1), was isolated from human faeces.
Description of Blautia aquisgranensis sp. nov
Blautia aquisgranensis (a.quis.gra.nen’sis. L. fem. adj. aquisgranensis, named after the German city of Aachen (Latin name Aquisgranum) where it was isolated).
The genome size is 3.63 Mbp, G+C percentage is 43.76%, with 99.37% completeness and 0.32% contamination. It includes two plasmids (37,495 bp; 1036 bp). The closest relative to strain CLA-JM-H16 was Blautia intestinalis (96.11%) based on 16S rRNA gene analysis. Placement of the strain within Blautia was confirmed based on POCP comparison, as values above 50% to multiple Blautia species were obtained. However, compared to the type species of the genus, Blautia coccoides, gave a value of 42.36%. This inconsistency was also highlighted by GTDB-Tk, which classified strain CLA-JM-H16 as ‘Blautia_A sp900764225’, suggesting Blautia may require splitting into multiple genera in future. As the separation of Blautia would require detailed analysis, which is outside the scope of this manuscript, we propose strain CLA-JM-H16 as a novel species within Blautia. All three novel species of Blautia described within this manuscript were confirmed to represent distinct species based on ANI comparison (B. aquisgranensis Vs. B. caccae, 81.74%; B. aquisgranensis Vs. B. intestinihominis, 76.95%; B. caccae Vs. B. intestinihominis, 78.57%). Functional analysis revealed 157 transporters, 17 secretion genes, and predicted utilisation of arbutin, salicin, sucrose, starch, and production of acetate, propionate, folate, L-glutamate, riboflavin, and cobalamin. In total, 205 CAZymes were identified, with 40 different glycoside hydrolase families and 12 glycoside transferase families represented. The type strain, CLA-JM-H16T (phylum Bacillota, family Lachnospiraceae) (=DSM 114586, =LMG 33033) (StrainInfo: https://doi.org/10.60712/SI-ID414368.1, genome: GCA_040096615.1), was isolated from human faeces.
Description of Blautia caccae sp. nov
Blautia caccae (cac’cae. N.L. fem. n. cacca, human ordure, faeces; from Gr. fem. n. kakkê, human ordure, faeces; N.L. gen. n. caccae, of faeces, referring to the source of isolate).
The genome size is 5.83 Mbp, G+C percentage is 46.73%, with 98.73% completeness and 0.63% contamination. The closest relative to strain CLA-SR-H028 was Blautia hominis (98.66%) based on 16S rRNA gene analysis. ANI comparison identified CLA-SR-H028 as a novel species within the genus Blautia, with all values being below the species threshold. GTDB-Tk classification as ‘Blautia sp001304935’ confirmed the proposition of a novel species within Blautia. Functional analysis showed the strain has 158 transporters, 18 secretion genes, and predicted utilisation of cellobiose, sucrose, starch and production of propionate, acetate, cobalamin, and folate. In total, 353 CAZymes were identified, with 53 different glycoside hydrolase families and 15 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 24 h of growth in DSMZ medium 1203a included 16:0 FAME (20.2%), 14:0 FAME (19.8%), 16:0 DMA (17.5%), 18:1 CIS 11 DMA (6.8%), and 14:0 DMA (5.9%). The type strain, CLA-SR-H028T (phylum Bacillota, family Lachnospiraceae) (=DSM 118556, =LMG 33609) (Straininfo: https://doi.org/10.60712/SI-ID414428.1, genome: GCA_040095955.1), was isolated from human faeces.
Description of Blautia intestinihominis sp. nov
Blautia intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 4.1 Mbp, G+C percentage is 43.49%, with 98.73% completeness and 0.63% contamination. It includes a single plasmid of 22,629 bp. The isolate was assigned to the species Blautia obeum (98.98%) based on 16S rRNA gene analysis. However, ANI comparison to B. obeum clearly identified this isolate as being a separate species (84.16%). This was confirmed by GTDB-Tk classification as ‘Blautia_A sp000436615’, recommending the creation of a novel species. Functional analysis showed the strain has 155 transporters, 18 secretion genes, and predicted utilisation of sucrose and starch, along with production of L-glutamate, folate, propionate, and cobalamin. In total, 177 CAZymes were identified. The type strain, CLA-AA-H95T (phylum Bacillota, family Lachnospiraceae) (=DSM 111354, =LMG 33582) (StrainInfo: https://doi.org/10.60712/SI-ID414326.1, genome: GCA_040096655.1), was isolated from human faeces.
Description of Coprococcus intestinihominis sp. nov
Coprococcus intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 3.6 Mbp, G+C percentage is 43.29%, with 98.43% completeness and 2.52% contamination. A single plasmid of 20,255 bp was detected. The closest relative to strain CLA-AA-H190 was Coprococcus catus (96.76%) based on 16S rRNA gene analysis. ANI comparison identified CLA-AA-H190 as a novel species within the genus Coprococcus, with an ANI value of 90.25% against the closest relative C. catus. GTDB-Tk classification as ‘Coprococcus_A catus_A’ confirmed the proposition of a novel species, but also suggests that the separation of Coprococcus into multiple genera could occur in future. Functional analysis showed the strain has 119 transporters, 18 secretion genes, and predicted utilisation of starch and production of propionate, butyrate, acetate, cobalamin, and folate. In total, 122 CAZymes were identified, with 19 different glycoside hydrolase families and 13 glycoside transferase families represented. The type strain, CLA-AA-H190T (phylum Bacillota, family Lachnospiraceae) (=DSM 114688, =LMG 33015) (StrainInfo: https://doi.org/10.60712/SI-ID414125.1, genome: GCA_040096555.1), was isolated from human faeces.
Description of Enterobacter intestinihominis sp. nov
Enterobacter intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, the intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 4.86 Mbp, G+C percentage is 54.88%, with 99.89% completeness and 0.12% contamination. It contains two plasmids (4416 bp; 2494 bp). Strain CLA-AC-H004 was determined to be a strain of Enterobacter quasihormaechei (99.80%) based on 16S rRNA gene analysis. Separation from existing Enterobacter species was confirmed by ANI comparison, which gave a value of 93.69% to E. quasihormaechei. GTDB-Tk classification of strain CLA-AC-H004 as ‘Enterobacter hormaechei_A’ supports the proposal of a novel species. An ANI value of 99.01% was obtained when compared to Enterobacter hormaechei subsp. hoffmannii. However, given the separation from E. hormaechei we propose these strains represent a separate species and not only a subspecies. ANI comparison also highlighted similarity with Pedobacter himalayensis (95.89%), which has been classified as ‘Enterobacter hormaechei_B’ within GTDB. However, this suggests reclassification of Pedobacter may be required in future. Functional analysis showed the strain has 497 transporters, 98 secretion genes, and predicted utilisation of arbutin, salicin, cellobiose, sucrose, and starch, along with production of L-glutamate, biotin, riboflavin, acetate, propionate, and folate. In total, 316 CAZymes were identified, with 37 different glycoside hydrolase families and 19 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 24 h of growth in DSMZ medium 1203a included 16:0 FAME (38.3%), 17:0 CYCLO CIS 9,10 FAME (18.6%), 18:1 CIS 11 FAME (9.7%), 14:0 FAME (9.0%), 19:0 CYCLO CIS 11,12 FAME (9.0%), and 14:0 3OH (8.8%). The type strain, CLA-AC-H004T (phylum Pseudomonadota, family Enterobacteriaceae) (=DSM 118557, =LMG 33610) (StrainInfo: https://doi.org/10.60712/SI-ID414328.1, genome: GCA_040096145.1), was isolated from human faeces.
Description of Enterocloster hominis sp. nov
Enterocloster hominis (ho’mi.nis. L. gen. n. hominis, of a human being).
The genome size is 6.52 Mbp, G+C percentage is 50.14%, with 99.16% completeness and 2.53% contamination. It includes a single plasmid of 7635 bp. The closest relative to strain CLA-SR-H021 was Enterocloster aldenensis (98.44%) based on 16S rRNA gene analysis. ANI comparison identified CLA-SR-H021 as a novel species within the genus Enterocloster, with all values being below the species threshold. GTDB-Tk classified CLA-SR-H021 as ‘Enterocloster pacaense’, a name derived from the proposed species ‘Lachnoclostridium pacaense’. However, the fact that these two names are not valid supports the proposition of a novel species within Enterocloster. Functional analysis revealed 118 transporters, 14 secretion genes, and predicted utilisation of cellobiose, starch and production of propionate, acetate, and folate. In total, 307 CAZymes were identified, with 39 different glycoside hydrolase families and 14 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 24 h of growth in DSMZ medium 1203a included 16:0 FAME (30.3%), 16:1 CIS 9 DMA (13.7%), 16:1 CIS 9 FAME (10.7%), 14:0 FAME (10.4%), 16:0 DMA (9.6%), 18:1 CIS 11 DMA (5.6%), and 18:1 CIS 11 FAME (5.0%). The type strain, CLA-SR-H021T (phylum Bacillota, family Lachnospiraceae) (=DSM 118482, =LMG 33606) (StrainInfo: https://doi.org/10.60712/SI-ID414422.1, genome: GCA_040096035.1), was isolated from human faeces.
Description of Faecalibacterium intestinale sp. nov
Faecalibacterium intestinale (in.tes.ti.na’le. N.L. neut. adj. intestinale, pertaining to the intestine, from where the type strain was isolated).
The genome size is 2.97 Mbp, G+C percentage is 56.43%, with 100.0% completeness and 0.0% contamination. The isolate was determined to be related to F. prausnitzii (98.08%) based on 16S rRNA gene analysis. ANI comparison to F. prausnitzii was just below species-level assignment (94.46%), and GTDB-Tk classification as ‘Faecalibacterium prausnitzii_J’ recommended the creation of a novel species. Functional analysis showed the strain has 135 transporters, 18 secretion genes, and predicted utilisation of starch and production of L-glutamate, riboflavin, and cobalamin. In total, 159 CAZymes were identified, with 27 different glycoside hydrolase families and 12 glycoside transferase families represented. Production of butyrate (4.74 ± 0.30 mM) was confirmed for strain CLA-AA-H281 when grown in YCFA broth (DSMZ Medium No. 1611) in Hungate tubes for 48 h at 37 °C under anaerobic conditions (6% CO2 and 4.7% H2 in N2). Major (≥5%) cellular fatty acids after 48 h of growth in DSMZ medium 215 included 16:0 FAME (33.2%), 18:1 CIS 11 DMA (11.7%), 18:1 CIS 11 FAME (11.4%), 14:0 FAME (9.3%), 16:0 DMA (8.4%), and 16:1 CIS 9 FAME (6.7%). The type strain, CLA-AA-H281T (phylum Bacillota, family Oscillospiraceae) (=DSM 116193, =LMG 33027) (StrainInfo: https://doi.org/10.60712/SI-ID414306.1, genome: GCA_040096575.1), was isolated from human faeces.
Description of Faecalibacterium tardum sp. nov
Faecalibacterium tardum (tar’dum. L. neut. adj. tardum, pertaining to its slow growth).
The genome size is 3.04 Mbp, G+C percentage is 56.28%, with 99.32% completeness and 0.0% contamination. It contains one plasmid of 14,735 bp in size, which encodes vancomycin resistance via the vanY gene. The isolate was determined to be closely related to Faecalibacterium prausnitzii (98.70%) based on 16S rRNA gene analysis. ANI comparison to F. prausnitzii was on the border of species-level assignment (95.05%), however, GTDB-Tk classification as ‘Faecalibacterium prausnitzii_A’ recommended the creation of a novel species. Strain CLA-AA-H175 was confirmed to represent a distinct species to Faecalibacterium intestinale (CLA-AA-H281, =DSM 116193) (ANI: 94.39%), also described in this paper. Functional analysis showed the strain has 116 transporters, 18 secretion genes, and predicted utilisation of starch and production of L-glutamate. In total, 177 CAZymes were identified, with 28 different glycoside hydrolase families and 12 glycoside transferase families represented. Production of butyrate (0.81 ± 0.16 mM) was confirmed for strain CLA-AA-H175 when grown in YCFA broth (DSMZ Medium No. 1611) in Hungate tubes for 48 h at 37 °C under anaerobic conditions (6% CO2 and 4.7% H2 in N2). The type strain, CLA-AA-H175T (phylum Bacillota, family Oscillospiraceae) (=DSM 116192) (StrainInfo: https://doi.org/10.60712/SI-ID414281.1, genome: GCA_040096515.1), was isolated from human faeces.
Description of Faecousia gen. nov
Faecousia (Faec.ou’si.a. L. fem. n. faex, dregs; N.L. fem. n. ousia, an essence; N.L. fem. n. Faecousia, a microbe associated with faeces).
Based on 16S rRNA gene sequence identity, the closest relatives are members of the genus Vescimonas (Vescimonas fastidiosa, 93.48%). POCP analysis against Vescimonas coprocola (44.75%), the type species of this genus, and V. fastidiosa (41.06%) confirmed that strain CLA-AA-H192 represents a distinct genus from Vescimonas. GTDB-Tk supported the creation of a novel genus, placing strain CLA-AA-H192 within the proposed genus of “Candidatus Faecousia”. The type species of this genus is Faecousia intestinalis.
Description of Faecousia intestinalis sp. nov
Faecousia intestinalis (in.tes.ti.na’lis. N.L. fem. adj. intestinalis, pertaining to the intestines, from where the type strain was isolated).
The genome size is 2.98 Mbp, G+C percentage is 58.44%, with 93.29% completeness and 0.0% contamination. It contains two plasmids (23,094 bp; 3448 bp). Functional analysis showed the strain has 144 transporters, 26 secretion genes, and predicted utilisation of starch, and production of acetate, propionate, L-glutamate, and folate. In total, 116 CAZymes were identified, with 19 different glycoside hydrolase families and 11 glycoside transferase families represented. The type strain, CLA-AA-H192T (phylum Bacillota, family Oscillospiraceae) (StrainInfo: https://doi.org/10.60712/SI-ID414286.1, genome: GCA_040096185.1), was isolated from human faeces.
Description of Flavonifractor hominis sp. nov
Flavonifractor hominis (ho’mi.nis. L. gen. n. hominis, of a human being).
The genome size is 2.94 Mbp, G+C percentage is 58.64%, with 99.33% completeness and 0.0% contamination. Strain CLA-AP-H34 was determined to be a new species based on 16S rRNA gene sequence analysis, with the closest validly named match being Flavonifractor plautii (97.21%). Separation from existing Flavonifractor species was confirmed by ANI comparison, which gave a value of 82.25% to F. plautii. GTDB-Tk classification of strain CLA-AP-H34 as an unknown species within Flavonifractor supports the proposal of a novel species. Functional analysis showed the strain has 112 transporters, 14 secretion genes, and predicted utilisation of starch and production of L-glutamate, riboflavin, butyrate, and folate. In total, 140 CAZymes were identified, with 19 different glycoside hydrolase families and 15 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 72 h of growth in DSMZ medium 1611 included 16:0 DMA (25.2%), 15:0 FAME (15.7%), 15:0 DMA (11.4%), 14:0 FAME (8.9%), 15:0 ISO FAME (8.4%), 17:0 DMA (7.5%), and 16:0 FAME (6.4%). The type strain, CLA-AP-H34T (phylum Bacillota, family Oscillospiraceae) (=DSM 118484, =LMG 33602) (StrainInfo: https://doi.org/10.60712/SI-ID414347.1, genome: GCA_040095835.1), was isolated from human faeces.
Description of Hominiventricola aquisgranensis sp. nov
Hominiventricola aquisgranensis (a.quis.gra.nen’sis. L. masc. adj. aquisgranensis, named after the German city of Aachen, where it was isolated).
The genome size is 3.17 Mbp, G+C percentage is 45.01%, with 99.37% completeness and 0.27% contamination, including a single plasmid of 7983 bp. The closest relative to strain CLA-AA-H78B was Hominiventricola filiformis (96.24%) based on 16S rRNA gene analysis. POCP analysis confirmed genus assignment to Hominiventricola, with a POCP value of 69.06% to the type strain of the only current species within this genus, H. filiformis. GTDB-Tk classified strain CLA-AA-H78B as ‘Choladocola sp003480725’ within “Candidatus Choladocola”. The latter is a heterosynonym of Hominiventricola, a validly published name. ANI comparison confirmed that strain CLA-AA-H78B represents a novel species, as the ANI value to H. filiformis was 82.98%. Functional analysis showed the strain has 134 transporters, 31 secretion genes, and predicted utilisation of cellobiose, starch, arbutin, salicin, and production of L-glutamate, folate, acetate, propionate, and cobalamin. In total, 134 CAZymes were identified. The type strain, CLA-AA-H78BT (phylum Bacillota, family Lachnospiraceae) (=DSM 111355, =LMG 33583) (StrainInfo: https://doi.org/10.60712/SI-ID414323.1, genome: GCA_040096225.1), was isolated from human faeces.
Description of Lachnospira intestinalis sp. nov
Lachnospira intestinalis (in.tes.ti.na’lis. N.L. fem. adj. intestinalis, pertaining to the intestines, from where the type strain was isolated).
The genome size is 3.1 Mbp, G+C percentage is 41.75%, with 99.33% completeness and 0.0% contamination. Strain CLA-AA-H89B was determined to represent a separate species from its closest relative, Lachnospira pectinoschiza (97.48%), based on 16S rRNA gene analysis. This was confirmed based on ANI comparison to all close relatives, which were below the species threshold. GTDB-Tk classification of strain CLA-AA-H89B as ‘Lachnospira sp000437735’ confirmed that this isolate represents a novel species. Functional analysis showed the strain has 112 transporters, 29 secretion genes, and predicted utilisation of starch and cellulose, along with production of L-glutamate, folate, acetate, propionate, and riboflavin. Motility was predicted based on the presence of the following genes: FlhA, FlhB, FlgB, FlgC, FlgD, FlgE, FlgF, FlgJ, FlgK, FlgL, FliC, FliD, FliE, FliF, FliG, FliK, FliM, FliN, MotA, MotB. In total, 162 CAZymes were identified, with 21 different glycoside hydrolase families and 11 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 48 h of growth in DSMZ medium 1611 included 16:0 FAME (32.4%), 18:1 CIS 11 DMA (20.6%), 14:0 FAME (7.8%), 16:0 DMA (6.3%), 18:1 CIS 11 aldehyde (6.2%), and 16:1 CIS 9 DMA (5.5%). The type strain, CLA-AA-H89BT (phylum Bacillota, family Lachnospiraceae) (=DSM 118070) (StrainInfo: https://doi.org/10.60712/SI-ID414325.1, genome: GCA_040095895.1), was isolated from human faeces.
Description of Lachnospira hominis sp. nov
Lachnospira hominis (ho’mi.nis. L. gen. masc. n. hominis, of a human being).
The genome size is 3.15 Mbp, G+C percentage is 37.05%, with 98.66% completeness and 0.67% contamination. Strain CLA-JM-H10 was determined to represent a separate species from its closest relative, Lachnospira rogosae (96.37%), based on 16S rRNA gene analysis. This was confirmed by an ANI of 79.9% between L. rogosa (CLA-AA-H255) and strain CLA-JM-H10. GTDB-Tk classification of CLA-JM-H10 as ‘Lachnospira sp900316325’ confirmed that it represents a novel species. Functional analysis showed the strain has 109 transporters, 27 secretion genes, and predicted utilisation of starch, and production of L-glutamate, folate, and cobalamin. Motility was predicted based on detection of the following genes: FlhA, FlhB, FlgB, FlgC, FlgD, FlgE, FlgF, FlgJ, FlgK, FlgL, FliC, FliD, FliE, FliF, FliG, FliK, FliM, FliN, MotA, MotB. This is consistent with the type species of this genus being motile. In total, 165 CAZymes were identified, with 22 different glycoside hydrolase families and 12 glycoside transferase families represented. The type strain, CLA-JM-H10T (phylum Bacillota, family Lachnospiraceae) (=DSM 114599, =LMG 33585) (StrainInfo: https://doi.org/10.60712/SI-ID414366.1, genome: GCA_040096395.1), was isolated from human faeces.
Description of Lachnospira rogosae sp. nov
Lachnospira rogosae (ro.go’sae. N.L. gen. masc. n. rogosae, of Rogosa).
Strain CLA-AA-H255 was determined to be similar to Lactobacillus rogosae (99.87%) based on 16S rRNA gene analysis. However, the lack of a genome for the type strain of the latter species, along with the lack of the type strain at any established culture collection, prevented further comparison (Tindall, 2014). GTDB-Tk classification of CLA-AA-H255 as ‘Lachnospira rogosae_A’ suggested that it represents a species within a genus distantly related to Lactobacillus. This was reconfirmed by the same placement of a second strain, CLA-AA-H191, which was also assigned to the same placeholder by GTDB-Tk (ANI of 98.86% between our two isolates). Based on these results, we propose L. rogosae was previously misassigned as a member of the genus Lactobacillus. To provide a type strain and correct its placement, we propose the creation of the species Lachnospira rogosae. Functional analysis showed that strain CLA-AA-H255 has 100 transporters, 25 secretion genes, and predicted utilisation of starch, and production of L-glutamate, folate, and riboflavin. The prediction of motility based on genomic analysis is consistent with the observed phenotype of motility in the original type strain of L. ragosae as stated by Holdeman and Moore101. In total, 142 CAZymes were identified, including 20 different glycoside hydrolase families and 12 glycoside transferase families. Ecological analysis of 1000 human gut 16S rRNA gene amplicon samples identified this strain in 1.20% of samples with a relative abundance of 0.43 ± 0.78%. The type strain, CLA-AA-H255T (phylum Bacillota, family Lachnospiraceae) (=DSM 118602, =LMG 33594) (StrainInfo: https://doi.org/10.60712/SI-ID414300.1, genome: GCA_040096455.1), was isolated from human faeces.
Description of Laedolimicola intestinihominis sp. nov
Laedolimicola intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 3.45 Mbp, G+C percentage is 49.65%, with 99.37% completeness and 0.16% contamination. It includes a single plasmid of 5131 bp. Strain CLA-AA-H132 was determined to represent a separate species from its closest relative, Laedolimicola ammoniilytica (98.38%), based on 16S rRNA gene analysis. POCP analysis confirmed that strain CLA-AA-H132 belongs to the recently named genus, Laedolimicola, with a POCP value of 75.19% to the type strain of the only current species within this genus, L. ammoniilytica. GTDB-Tk placement as ‘Merdisoma sp900553635’ suggests that Laedolimicola and Candidatus Merdisoma are homonyms and require future reclassification. ANI comparison confirmed that CLA-AA-H132 represents a novel species, as the ANI value to L. ammoniilytica was only 90.2%. Functional analysis showed the strain has 142 transporters, 21 secretion genes, and predicted utilisation of cellobiose, sucrose, starch, and production of acetate, propionate, L-glutamate, cobalamin, and folate. In total, 153 CAZymes were identified, with 21 different glycoside hydrolase families and 16 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 24 h of growth in DSMZ medium 339 included 16:0 FAME (24.8%), 18:1 CIS 11 DMA (12.8%), 14:0 FAME (10.0%), 16:0 DMA (8.2%), and 16:0 CIS 9 DMA (8.2%). The type strain, CLA-AA-H132T (phylum Bacillota, family Lachnospiraceae) (=DSM 117481, =LMG 33588) (StrainInfo: https://doi.org/10.60712/SI-ID414271.1, genome: GCA_040096155.1), was isolated from human faeces.
Description of Maccoyibacter gen. nov
Maccoyibacter (Mac.co.y.i.bac’ter N.L. fem. gen. n. Maccoyae, referring to the immunologist Kathy McCoy; N.L. masc. n. bacter, a rod; N.L. masc. n. Maccoyibacter, a rod-shaped bacterium named after the immunologist Kathy McCoy, for her many scientific contributions in the field of microbe-host interactions).
Based on 16S rRNA gene analysis, the closest species with a valid name are Roseburia hominis (94.9%) and Eubacterium oxidoreducens (94.23%). POCP values against all close relatives were below the genus delineation threshold of 50%, including many Roseburia spp. (R. hominis, R. faecis, R. porci, R. intestinalis), apart from R. inulinivorans (50.83%). GTDB-Tk placement as “UBA11774 sp003507655” confirmed that strain CLA-AA-H185 represents a novel genus within the family Lachnospiraceae. The type species is Maccoyibacter intestinihominis.
Description of Maccoyibacter intestinihominis sp. nov
Maccoyibacter intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 2.85 Mbp, G+C percentage is 41.17%, with 97.41% completeness and 0.45% contamination. It contains a single plasmid (2341 bp). Functional analysis showed the strain has 99 transporters, 35 secretion genes, and predicted utilisation of starch, and production of acetate, propionate, L-glutamate, cobalamin, folate, and riboflavin. In total, 110 CAZymes were identified, with 17 different glycoside hydrolase families and 13 glycoside transferase families represented. The type strain, CLA-AA-H185T (phylum Bacillota, family Lachnospiraceae) (=DSM 118601) (StrainInfo: https://doi.org/10.60712/SI-ID414284.1, genome: GCA_040096355.1), was isolated from human faeces.
Description of Megasphaera intestinihominis sp. nov
Megasphaera intestinihominis (in.tes.ti.ni.ho’mi.nis. L. gen. neut. n. intestini, of the intestine; L. gen. masc. n. hominis, of a human being; N.L. gen. masc. n. intestinihominis, of the human intestine).
The genome size is 2.38 Mbp, G+C percentage is 53.59%, with 100.00% completeness and 0.00% contamination. The closest relative to strain CLA-AA-H81 was Megasphaera indica (99.09%) based on 16S rRNA gene analysis. ANI comparison to all close relatives was below the species assignment threshold (highest to Megasphaera elsdenii, 90.78%). GTDB-Tk classification as ‘Megasphaera sp000417505’ supports the creation of a novel species. Functional analysis showed the strain has 133 transporters, 16 secretion genes, and predicted utilisation of starch, and production of butyrate, propionate, L-glutamate, folate, riboflavin, and cobalamin. Ecological analysis identified 152 matching (MASH distance <0.05) MAGs, of which 124 originate from the human gut, suggesting this species is most commonly observed within this environment. This is supported by ecological analysis using 16S rRNA gene amplicon datasets, which identified it within 19.0% of 1000 human gut samples, with a relative abundance of 2.14 ± 5.23%. Major (≥5%) cellular fatty acids after 24 h of growth in DSMZ medium 215 included 12:0 FAME (17.1%), 14:0 3OH (14.8%), 18:1 CIS 9 FAME (9.6%), 18:1 CIS 9 DMA (9.3%), 16:0 FAME (8.0%), and 16:1 CIS 7 FAME (5.0%). The type strain, CLA-AA-H81T (phylum Bacillota, family Veillonellaceae) (=DSM 118069, =LMG 33597) (StrainInfo: https://doi.org/10.60712/SI-ID414324.1, genome: GCA_040096415.1), was isolated from human faeces.
Description of Niallia hominis sp. nov
Niallia hominis (ho’mi.nis. L. gen. n. hominis, of a human being).
The genome size is 4.9 Mbp, G+C percentage is 35.33%, with 92.24% completeness and 3.20% contamination. Strain CLA-SR-H024 was assigned to Niallia circulans (100%) based on 16S rRNA gene analysis. However, ANI suggested the isolate represents a novel species compared to all Niallia spp. (values were below 80%). GTDB-Tk classification of strain CLA-SR-H024 as ‘Niallia sp001076885’ confirmed that it represents a novel species. Functional analysis showed the strain has 244 transporters, 43 secretion genes, and predicted utilisation of arbutin, salicin, cellobiose, starch, and dextran, along with production of L-glutamate, folate, and cobalamin. In total, 294 CAZymes were identified, with 34 different glycoside hydrolase families and 14 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 72 h of growth in DSMZ medium 1203a included 15:0 ANTEISO FAME (29.7%), 16:0 FAME (16.8%), 15:0 ISO FAME (14.9%), 16:0 ISO FAME (9.8%), 14:0 FAME (8.4%), 17:0 ANTEISO FAME (6.9%), and 14:0 ISO FAME (5.2%).The type strain, CLA-SR-H024T (phylum Bacillota, family Bacillaceae) (=DSM 118483) (StrainInfo: https://doi.org/10.60712/SI-ID414424.1, genome: GCA_040095995.1), was isolated from human faeces.
Description of Peptoniphilus hominis sp. nov
Peptoniphilus hominis (ho’mi.nis. L. gen. n. hominis, of a human being).
The genome size is 1.99 Mbp, G+C percentage is 33.8%, with 99.3% completeness and 0.0% contamination. Strain CLA-SR-H025 was assigned to Peptoniphilus gorbachii (99.11%) based on 16S rRNA gene analysis. However, ANI suggested the isolate represents a novel species compared to P. gorbachii, giving a value of 88.92%. GTDB-Tk classification of strain CLA-SR-H025 as ‘Peptoniphilus_A grossensis’ confirmed that it represents a novel species, as the name ‘Peptoniphilus grossensis’ was proposed but never validated102. Functional analysis showed the strain has 12 transporters, 1 secretion gene, and no gut metabolic models were identified within the genome. In total, 59 CAZymes were identified, with 10 different glycoside hydrolase families and 8 glycoside transferase families represented. The type strain, CLA-SR-H025T (phylum Bacillota, family Peptoniphilaceae) (=DSM 118555, =LMG 33608) (Straininfo: https://doi.org/10.60712/SI-ID414425.1, genome: GCA_040096015.1), was isolated from human faeces.
Description of Pseudoflavonifractor intestinihominis sp. nov
Pseudoflavonifractor intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, the intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 3.69 Mbp, G+C percentage is 61.54%, with 99.33% completeness and 0.00% contamination. Strain CLA-AP-H29 was assigned to the species Pseudoflavonifractor capillosus (99.53%) based on 16S rRNA gene sequence analysis. However, ANI suggested the isolate represents a novel species compared to P. capillosus, giving a value of 80.96%. GTDB-Tk classified strain CLA-AP-H29 as ‘Pseudoflavonifractor sp944387275’, which confirms that it represents a novel species. Functional analysis predicted that the strain has 121 transporters, 14 secretion genes, and predicted utilisation of starch, and production of L-glutamate, folate, and cobalamin. In total, 180 CAZymes were identified, with 25 different glycoside hydrolase families and 15 glycoside transferase families represented. Major (≥5%) cellular fatty acids after 48 h of growth in DSMZ medium 215 included 14:0 FAME (33.9%), 16:0 DMA (33.8%), 16:0 FAME (9.8%), and 16:0 ALDE (8.0%). The type strain, CLA-AP-H29T (phylum Bacillota, family Oscillospiraceae) (=DSM 118073, =LMG 33601) (Straininfo: https://doi.org/10.60712/SI-ID414342.1, genome: GCA_040096055.1), was isolated from human faeces.
Description of Robertmurraya yapensis sp. nov
Robertmurraya yapensis (yap’ensis. N.L. fem. adj. yapensis, pertaining to Yap trench, which is the geographical position where the first isolate of this species was obtained).
The genome size is 4.74 Mbp, G+C percentage is 37.94%, with 98.85% completeness and 2.13% contamination. It contains a plasmid (2470 bp). The closest relative to strain CLA-AA-H227 was Robertmurraya spiralis (99.23%) based on 16S rRNA gene analysis. The highest POCP scores were for members of the genus Robertmurraya (Robertmurraya kyonggiensis, 84.9%; R. spiralis, 65.33%). The placement of strain CLA-AA-H227 within Robertmurraya was confirmed by GTDB-Tk assignment as ‘Robertmurraya yapensis’, a reclassification of the species ‘Bacillus yapensis’, although neither name has been validated. ANI comparison confirmed that CLA-AA-H227 represents a novel species, as values to close relatives were below the species-level threshold. Functional analysis showed the strain has 246 transporters, 53 secretion genes, and predicted utilisation of arbutin, salicin, cellobiose, starch, and production of butyrate, acetate, propionate, folate, riboflavin, and cobalamin. In total, 238 CAZymes were identified. The type strain, CLA-AA-H227T (phylum Bacillota, family Bacillaceae) (=DSM 113004, =LMG 33018) (Straininfo: https://doi.org/10.60712/SI-ID414150.1, genome: GCA_040096375.1), was isolated from human faeces.
Description of Ruminococcoides intestinale sp. nov
Ruminococcoides intestinale (in.tes.ti.na’le. N.L. neut. adj. intestinalis, pertaining to the intestines, from where the type strain was isolated).
The genome size is 2.32 Mbp, G+C percentage is 40.88%, with 99.33% completeness and 1.01% contamination. The isolate was determined to be similar to Ruminococcus bromii (98.91%) and more distantly related to Ruminococcoides bili (96.76%) based on 16S rRNA gene analysis. While POCP comparison of strain CLA-JM-H38 to R. bromii was 59.79%, and 53.56% to Ruminococcus bovis, suggesting they belong to the same genus, all other comparisons to Ruminococcus species were below 50%, including to the type species, Ruminococcus flavefaciens (27.58%). POCP to R. bili, the type species of the genus Ruminococcoides, was 64.44%. GTDB-Tk classified strain CLA-JM-H38 as “Ruminococcus_E bromii_B”, confirming it is not a member of the genus Ruminococcus. These results support the GTDB assignment that both R. bovis and R. bromii should be reclassified as members of the genus Ruminococcoides. Strain CLA-JM-H38 was confirmed to represent a novel species as all ANI comparisons to close relatives were below 95%, and it represents a distinct novel species from Ruminococcoides intestinihominis described in this work (78.33%). Functional analysis showed the strain has 81 transporters, 15 secretion genes, and predicted utilisation of starch, and production of L-glutamate. In total, 108 CAZymes were identified, with 15 different glycoside hydrolase families and 12 glycoside transferase families represented. Ecological analysis based on 16S rRNA gene amplicons identified this species in 55.20% of 1000 human gut samples with a relative abundance of 1.50 ± 2.49%, suggesting it is a prevalent and dominant bacterial species within the human gut. Major (≥5%) cellular fatty acids after 72 h of growth in DSMZ medium 1611 included 16:0 ISO FAME (49.5%) and 16:0 ISO DMA (23.8%). The type strain, CLA-JM-H38T (phylum Bacillota, family Oscillospiraceae) (=DSM 118486, =LMG 33604) (StrainInfo: https://doi.org/10.60712/SI-ID414376.1, genome: GCA_040096305.1), was isolated from human faeces.
Description of Ruminococcoides intestinihominis sp. nov
Ruminococcoides intestinihominis (in.tes.ti.ni.ho’mi.nis. L. neut. n. intestinum, intestine; L. masc. n. homo, a human being; N.L. gen. n. intestinihominis, of the human intestine).
The genome size is 2.26 Mbp, G+C percentage is 34.26%, with 98.66% completeness and 0.00% contamination. It contains one plasmid (1825 bp). Based on 16S rRNA gene sequence similarity, the isolate was closely related to Ruminococcus bovis (99.3%) and more distant to Ruminococcoides bili (94.8%), the type species of this genus. ANI comparison confirmed the similarity of strain CLA-AA-H171 to R. bovis (95.4%); however, classification by GTDB-Tk as ‘Ruminococcus_E sp934476515’ supported the creation of a novel species. Recently, ‘Ruminococcus_E’ has been validly named as Ruminococcoides, with the type species R. bili28. POCP comparison between the isolate and R. bili provided a value of 51.3%, suggesting that strain CLA-AA-H171 represents a novel species within the genus Ruminococcoides. Functional analysis showed the strain has 75 transporters, 14 secretion genes, and predicted utilisation of starch and production of acetate. In total, 124 CAZymes were identified, with 13 different glycoside hydrolase families and 12 glycoside transferase families represented. Ecological analysis based on 16S rRNA gene amplicons identified this species in 10.40% of 1000 human gut samples with a relative abundance of 0.23 ± 0.68%. The type strain, CLA-AA-H171T (phylum Bacillota, family Oscillospiraceae) (=DSM 114689, =LMG 33587) (StrainInfo: https://doi.org/10.60712/SI-ID414279.1, genome: GCA_040096285.1), was isolated from human faeces.
Description of Ruthenibacterium intestinale sp. nov
Ruthenibacterium intestinale (in.tes.ti.na’le. N.L. neut. adj. intestinale, pertaining to the intestines, from where the type strain was isolated).
The genome size is 3.1 Mbp, G+C percentage is 54.69%, with 98.3% completeness and 0.00% contamination. It contains a plasmid (5063 bp). The closest relative to strain CLA-JM-H11 is Ruthenibacterium lactatiformans (94.93%), the type species of this genus, based on 16S rRNA gene analysis. Placement of the isolate within the genus Ruthenibacterium was confirmed based on POCP comparison, with a value of 56.41% to the type species. Strain CLA-JM-H11 was confirmed to be distinct from R. lactatiformans based on ANI comparison (79.28%). GTDB-Tk classification confirmed this assignment as an unknown species within Ruthenibacterium. Functional analysis showed the strain has 143 transporters, 16 secretion genes, and predicted utilisation of starch, and production of acetate, propionate, folate, and cobalamin. In total, 155 CAZymes were identified, with 27 different glycoside hydrolase families and 13 glycoside transferase families represented. The type strain, CLA-JM-H11T (phylum Bacillota, family Oscillospiraceae) (=DSM 114604, =LMG 33032) (StrainInfo: https://doi.org/10.60712/SI-ID414367.1, genome: GCA_040096265.1), was isolated from human faeces.
Description of Solibaculum intestinale sp. nov
Solibaculum intestinale (in.tes.ti.na’le. N.L. neut. adj. intestinale, pertaining to the intestines, from where the type strain was isolated).
The genome size is 2.81 Mbp, G+C percentage is 54.62%, with 97.99% completeness and 0.67% contamination. The isolate was determined to be a new species based on 16S rRNA gene analysis, with the closest validly named match being Solibaculum mannosilyticum (94.93%). Placement within Solibaculum was confirmed with POCP values above 50% to S. mannosilyticum (55.7%), the type species, and only member of this genus. ANI comparison confirmed the isolate represents a novel species, as no ANI values were above 80%. Functional analysis showed the strain has 82 transporters, 16 secretion genes, and predicted utilisation of starch, and production of acetate, propionate, and L-glutamate. In total, 169 CAZymes were identified, with 20 different glycoside hydrolase families and 15 glycoside transferase families represented. The type strain, CLA-JM-H44T (phylum Bacillota, family Oscillospiraceae) (=DSM 114601, =LMG 33034) (StrainInfo: https://doi.org/10.60712/SI-ID414377.1, genome: GCA_040096205.1), was isolated from human faeces.
Description of Ventrimonas gen. nov
Ventrimonas (Ven.tri.mo’nas. L. masc. n. venter, the belly; L. fem. n. monas, a monad; N.L. fem. n. Ventrimonas, a microbe associated with the belly (intestines/faeces)).
Based on 16S rRNA gene sequence similarity, the closest relatives are members of the genus Hungatella (Hungatella effluvii, 95.15%; Hungatella hathewayi, 94.95%). POCP analysis against H. effluvii (40.38%) and H. hathewayi (39.86%) indicates that strain CLA-AP-H27 represents a distinct genus within Hungatella. GTDB-Tk supported the creation of a novel genus, placing strain CLA-AP-H27 within the proposed genus of “Candidatus Ventrimonas” in the family Lachnospiraceae. The type species of this genus is Ventrimonas faecis.
Description of Ventrimonas faecis sp. nov
Ventrimonas faecis (fae’cis. L. gen. n. faecis, of dregs, pertaining to faeces, from where the type strain was isolated).
The genome size is 3.41 Mbp, G+C percentage is 49.27%, with 98.73% completeness and 0.63% contamination. Functional analysis showed the strain has 156 transporters, 21 secretion genes, and predicted utilisation of starch, and production of acetate, propionate, L-glutamate, cobalamin, and folate. In total, 139 CAZymes were identified, with 21 different glycoside hydrolase families and 12 glycoside transferase families represented. Major (≥10%) cellular fatty acids after 24 h of growth in DSMZ medium 1203a included 16:0 FAME (35.5%), 18:1 CIS 11 DMA (10.0%), 18:1 CIS 11 FAME (9.6%), 18:1 CIS 9 DMA (5.9%), and 16:0 DMA (5.5%). The type strain, CLA-AP-H27T (phylum Bacillota, family Lachnospiraceae) (=DSM 118072, =LMG 33600) (StrainInfo: https://doi.org/10.60712/SI-ID414341.1, genome: GCA_040096075.1), was isolated from human faeces.
Description of Waltera hominis sp. nov
Waltera hominis (ho’mi.nis. L. gen. masc. n. hominis, of a human being, pertaining to the human gut habitat, from where the type strain was isolated).
The genome size is 3.88 Mbp, G+C percentage is 45.72%, with 99.52% completeness and 2.13% contamination. The isolate was assigned to the species Waltera intestinalis (100.0%) based on 16S rRNA gene analysis. However, ANI comparison identified strain CLA-AA-H183 as a novel species within the genus Waltera, with an ANI value of 91.97% against the type species Waltera intestinalis. GTDB-Tk currently lacks the inclusion of Waltera, causing misclassification as ‘Acetatifactor sp003447295’; however, this confirmed the proposition of a novel species. Separation of Waltera from Acetatifactor was revalidated via both phylogenomic analysis (Supplementary Fig. 3), which shows both genera form separate monophyletic groups, and POCP analysis, which shows clear similarity within each genus (Waltera: 68.86%, Acetatifactor: 59.25%), and separation between the genera (39.94 ± 1.63%). Functional analysis showed the strain has 141 transporters, 36 secretion genes, and predicted utilisation of starch, cellulose, and production of butyrate, propionate, acetate, and folate. In total, 228 CAZymes were identified, with 38 different glycoside hydrolase families and 14 glycoside transferase families represented. The type strain, CLA-AA-H183T (phylum Bacillota, family Lachnospiraceae) (=DSM 114684, =LMG 33586) (StrainInfo: https://doi.org/10.60712/SI-ID414283.1, genome: GCA_040096245.1), was isolated from human faeces.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genomes for all strains have been deposited at NCBI under BioProject PRJNA996881. Bulk download of HiBC resources is possible via Zenodo for the genomes (https://doi.org/10.5281/zenodo.12180083), plasmid sequences (https://doi.org/10.5281/zenodo.12187897), 16S rRNA gene sequences (https://doi.org/10.5281/zenodo.12180259), and the isolates metadata (https://doi.org/10.5281/zenodo.12180506). The PacBio genome for P. vulgatus CLA-AA-H253 (=DSM 118718) has been deposited in ENA under accession code PRJEB80480 and Zenodo (https://doi.org/10.5281/zenodo.14674027).
Code availability
The codes for genome processing and creation of the HiBC website are available at https://github.com/ClavelLab/genome-assembly (https://doi.org/10.5281/zenodo.15172576) and https://git.rwth-aachen.de/clavellab/hibc (https://doi.org/10.5281/zenodo.15172609), respectively.
References
Forster, S. C. et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 37, 186–192 (2019).
Groussin, M. et al. Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell 184, 2053–2067.e18 (2021).
Liu, C. et al. Enlightening the taxonomy darkness of human gut microbiomes with a cultured biobank. Microbiome 9, 1–29 (2021).
Browne, H. P. et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546 (2016).
Hedlund, B. P. et al. SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat. Microbiol. 7, 1702–1708 (2022).
Göker, M., Moore, E. R. B., Oren, A. & Trujillo, M. E. Status of the SeqCode in the International Journal of Systematic and Evolutionary Microbiology. Int J. Syst. Evol. Microbiol 72, 10–12 (2022).
Arahal, D. et al. The best of both worlds: a proposal for further integration of Candidatus names into the International Code of Nomenclature of Prokaryotes. Int J. Syst. Evol. Microbiol 74, 1–21 (2024).
Hitch, T. C. A. et al. Harmonious naming across nomenclature codes exemplified by the description of bacterial isolates from the mammalian gut. Syst. Appl. Microbiol. 47, 126543 (2024).
Blanco-Míguez, A. et al. Extension of the Segatella copri complex to 13 species with distinct large extrachromosomal elements and associations with host conditions. Cell Host Microbe 31, 1804–1819.e9 (2023).
De Filippis, F. et al. Distinct genetic and functional traits of human intestinal Prevotella copri strains are associated with different habitual diets. Cell Host Microbe 25, 444–453.e3 (2019).
Fehlner-Peach, H. et al. Distinct polysaccharide utilization profiles of human intestinal Prevotella copri Isolates. Cell Host Microbe 26, 680–690.e5 (2019).
Conteville, L. C. & Vicente, A. C. P. A plasmid network from the gut microbiome of semi-isolated human groups reveals unique and shared metabolic and virulence traits. Sci. Rep. 12, 1–9 (2022).
Fogarty, E. C. et al. Article A cryptic plasmid is among the most numerous genetic elements in the human gut ll ll Article A cryptic plasmid is among the most numerous genetic elements in the human gut. Cell 187, 1206–1222.e16 (2024).
Yu, M. K., Fogarty, E. C. & Eren, A. M. Diverse plasmid systems and their ecology across human gut metagenomes revealed by PlasX and MobMess. Nat. Microbiol 9, 830–847 (2024).
Perez, M. et al. A synthetic consortium of 100 gut commensals modulates the composition and function in a colon model of the microbiome of elderly subjects. Gut Microbes 13, 1–19 (2021).
Clark, R. L. et al. Design of synthetic human gut microbiome assembly and butyrate production. Nat. Commun. 12, 1–16 (2021).
Cheng, A. G. et al. Design, construction, and in vivo augmentation of a complex gut microbiome. Cell 185, 3617–3636.e19 (2022).
Overmann, J. Significance and future role of microbial resource centers. Syst. Appl. Microbiol. 38, 258–265 (2015).
Derrien, M., Vaughan, E. E., Plugge, C. M. & de Vos, W. M. Akkermansia municiphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J. Syst. Evol. Microbiol. 54, 1469–1476 (2004).
Plovier, H. et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 23, 107–113 (2017).
Parker, C. T., Tindall, B. J. & Garrity, G. M. International code of nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 69, S1 (2019).
Hitch, T. C. A. et al. Recent advances in culture-based gut microbiome research. Int. J. Med. Microbiol. 311, 151485 (2021).
Poyet, M. et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med. 25, 1442–1452 (2019).
Huang, P. et al. Gut microbial genomes with paired isolates from China illustrate probiotic and cardiometabolic effects. Cell Genomics 4, 100559 (2024).
Huang, Y. et al. High-throughput microbial culturomics using automation and machine learning. Nat. Biotechnol. 41, 1424–1433 (2023).
Lagier, J. C. et al. Retraction note: Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 10, 601 (2025).
Leviatan, S., Shoer, S., Rothschild, D., Gorodetski, M. & Segal, E. An expanded reference map of the human gut microbiome reveals hundreds of previously unknown species. Nat. Commun. 13, 1–14 (2022).
Molinero, N. et al. Ruminococcoides bili gen. nov., sp. nov., a bile-resistant bacterium from human bile with autolytic behavior. Int J. Syst. Evol. Microbiol 71, 1–11 (2021).
Schmitz, M. A., Dimonaco, N. J., Clavel, T. & Hitch, T. C. A. Lineage-specific microbial protein prediction enables large-scale exploration of protein ecology within the human gut. Nat Commun. 16, 3204 (2025).
Ma, L. et al. Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut Microbes 12, 1–19 (2020).
Wiedmann, S., Eudy, J. D. & Zempleni, J. Biotin supplementation increases expression of genes encoding interferon-γ, interleukin-1β, and 3-methylcrotonyl-CoA carboxylase, and decreases expression of the gene encoding interleukin-4 in human peripheral blood mononuclear cells. J. Nutr. 133, 716–719 (2003).
Agrawal, S., Agrawal, A. & Said, H. M. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am. J. Physiol. Cell Physiol. 311, C386–C391 (2016).
Skupsky, J. et al. Biotin supplementation ameliorates murine colitis by preventing NF-κB activation. Cell Mol. Gastroenterol. Hepatol. 9, 557–567 (2020).
Mao, B. et al. Blautia producta displays potential probiotic properties against dextran sulfate sodium-induced colitis in mice. Food Sci. Hum. Wellness 13, 709–720 (2024).
van der Lelie, D. et al. Rationally designed bacterial consortia to treat chronic immune-mediated colitis and restore intestinal homeostasis. Nat. Commun. 12, 1–17 (2021).
Lee, C. et al. P926 blautia obeum aggravates colitis in a murine model. J. Crohns Colitis 17, i1035–i1035 (2023).
Huang, T. Q. et al. Bergenin alleviates ulcerative colitis by decreasing gut commensal bacteroides vulgatus-mediated elevated branched-chain amino acids. J. Agric. Food Chem. 72, 3606–3621 (2024).
Sugihara, K. et al. Dietary phosphate exacerbates intestinal inflammation in experimental colitis. J. Clin. Biochem. Nutr. 61, 91–99 (2017).
Kolachala, V. L. et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology 135, 861–870 (2008).
Jones, B. V., Sun, F. & Marchesi, J. R. Comparative metagenomic analysis of plasmid encoded functions in the human gut microbiome. BMC Genomics 11, 46 (2010).
Sanders, J. G. et al. A low-cost genomics workflow enables isolate screening and strain-level analyses within microbiomes. Genome Biol. 23, 212 (2022).
Rozov, R. et al. Recycler: an algorithm for detecting plasmids from de novo assembly graphs. Bioinformatics 33, 475–482 (2017).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Computat. Biol. 19, 455–477 (2012).
Li, Y. et al. Distribution of megaplasmids in Lactobacillus salivarius and other Lactobacilli. J. Bacteriol. 189, 6128–6139 (2007).
Bottacini, F. et al. Discovery of a conjugative megaplasmid in Bifidobacterium breve. Appl. Environ. Microbiol. 81, 166–176 (2015).
Schmartz, G. P. et al. PLSDB: advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 50, D273–D278 (2022).
Novicki, T. J. & Hecht, D. W. Characterization and DNA Sequence of the Mobilization Region of PLV22a from Bacteroides Fragilis. J. Bacteriol. 177, 4466–4473 (1995).
Nieto, C. et al. The yefM-yoeB toxin-antitoxin systems of Escherichia coli and Streptococcus pneumoniae: Functional and structural correlation. J. Bacteriol. 189, 1266–1278 (2007).
Smith, J. A. & Magnuson, R. D. Modular organization of the Phd repressor/antitoxin protein. J. Bacteriol. 186, 2692–2698 (2004).
Evans, J. C. et al. A proteolytically activated antimicrobial toxin encoded on a mobile plasmid of Bacteroidales induces a protective response. Nat. Commun. 13, 4258 (2022).
García-Bayona, L., Coyne, M. J. & Comstock, L. E. Mobile type VI secretion system loci of the gut Bacteroidales display extensive intra-ecosystem transfer, multi-species spread and geographical clustering. PLoS Genet. 17, 1–25 (2021).
García-Bayona, L. et al. A pervasive large conjugative plasmid mediates multispecies biofilm formation in the intestinal microbiota increasing resilience to perturbations. Preprint at https://doi.org/10.1101/2024.04.29.590671 (2024).
Sokurenko, E. V., Chesnokova, V., Doyle, R. J. & Hasty, D. L. Diversity of the Escherichia coli type 1 fimbrial lectin. Differential binding to mannosides and uroepithelial cells. J Biol Chem. 272, 17880–17886 (1997).
Jønsson, R. et al. Aggregative adherence fimbriae form compact structures as seen by SAXS. Sci. Rep. 13, 16516 (2023).
Riepe, S. P., Goldstein, J. & Alpers, D. H. Effect of secreted Bacteroides proteases on human intestinal brush border hydrolases. J. Clin. Investig. 66, 314–322 (1980).
Mills, R. H. et al. Multi-omics analyses of the ulcerative colitis gut microbiome link Bacteroides vulgatus proteases with disease severity. Nat. Microbiol. 7, 262–276 (2022).
Sokol, H. et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl Acad. Sci. USA 105, 16731–16736 (2008).
Ueda, A. et al. Identification of Faecalibacterium prausnitzii strains for gut microbiome-based intervention in Alzheimer’s-type dementia. Cell Rep. Med. 2, 100398 (2021).
D’hoe, K. et al. Integrated culturing, modeling and transcriptomics uncovers complex interactions and emergent behavior in a three-species synthetic gut community. eLife 7, 1–30 (2018).
Fitzgerald, C. B. et al. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics 19, 1–20 (2018).
Vital, M., Karch, A. & Pieper, D. H. Colonic butyrate-producing communities in humans: an overview using omics data. mSystems 2, 1–18 (2017).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41 (2017).
Panicker, I. S., Browning, G. F. & Markham, P. F. The effect of an alternate start codon on heterologous expression of a PhoA fusion protein in mycoplasma gallisepticum. PLoS ONE 10, 1–10 (2015).
Duncan, S. H., Barcenilla, A., Stewart, C. S., Pryde, S. E. & Flint, H. J. Acetate utilization and butyryl coenzyme A (CoA): acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol. 68, 5186–5190 (2002).
Afrizal, A. et al. Enhanced cultured diversity of the mouse gut microbiota enables custom-made synthetic communities. Cell Host Microbe 30, 1630–1645 (2022).
Stackebrandt, E. et al. Deposit of microbial strains in public service collections as part of the publication process to underpin good practice in science. Springerplus 3, 1–4 (2014).
Burz, S. D. et al. A guide for ex vivo handling and storage of stool samples intended for fecal microbiota transplantation. Sci. Rep. 9, 1–16 (2019).
Wylensek, D. et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 11, 6389 (2020).
Afrizal, A. et al. Anaerobic single‐cell dispensing facilitates the cultivation of human gut bacteria. Environ. Microbiol. 24, 3861–3881 (2022).
Vieira, S. et al. Usitatibacter rugosus gen. nov., sp. nov. and Usitatibacter palustris sp. nov., novel members of Usitatibacteraceae fam. nov. within the order Nitrosomonadales isolated from soil. Int. J. Syst. Evol. Microbiol. 71, https://doi.org/10.1099/ijsem.0.004631 (2021).
Spitzer, V. Structure analysis of fatty acids by gas chromatography - low resolution electron impact mass spectrometry of their 4,4-dimethyloxazoline derivatives - a review. Prog. Lipid Res. 35, 387–408 (1996).
Godon, J. J., Zumstein, E., Dabert, P., Habouzit, F. & Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 63, 2802–2813 (1997).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bushnell, B. BBMap: a fast, accurate, splice-aware aligner. In 9th Annual Genomics of Energy & Environment Meeting. https://api.semanticscholar.org/CorpusID:114702182 (2014).
Antipov, D. et al. PlasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 32, 3380–3387 (2016).
Yilmaz, P. et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 29, 415–420 (2011).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150 (2018).
Bengtsson-Palme, J. et al. metaxa2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 15, 1403–1414 (2015).
Schwengers, O. et al. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Micro. Genom. 7, 000685 (2021).
Mölder, F. et al. Sustainable data analysis with Snakemake [version 2; peer review: 2 approved]. F1000Res 10, 1–29 (2021).
Hitch, T. C. A. et al. Automated analysis of genomic sequences facilitates high-throughput and comprehensive description of bacteria. ISME Commun. 1, 16 (2021).
Asnicar, F. et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 1–10 (2020).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Aramaki, T. et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Nelson, K. E. et al. A catalog of reference genomes from the human microbiome. Science 328, 994–999 (2010).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90 K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Lagkouvardos, I. et al. IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 6, 1–9 (2016).
Fodor, A. A. et al. The ‘most wanted’ taxa from the human microbiome for whole genome sequencing. PLoS ONE 7, e41294 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011).
Beverley, S. M. Estimation of circular DNA size using γ-irradiation and pulsed-field gel electrophoresis. Anal. Biochem. 177, 110–114 (1989).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Bittrich, S., Segura, J., Duarte, J. M., Burley, S. K. & Rose, Y. RCSB protein data bank: exploring protein 3D similarities via comprehensive structural alignments. Bioinformatics 40, btae370 (2024).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Yarza, P. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645 (2014).
Qin, Q. L. et al. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215 (2014).
Segata, N., Börnigen, D., Morgan, X. C. & Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 1–10 (2013).
Meile, L., Rohr, L. M., Geissmann, T. A., Herensperger, M. & Teuber, M. Characterization of the D-xylulose 5-phosphate/D-fructose 6-phosphate phosphoketolase gene (xfp) from Bifidobacterium lactis. J. Bacteriol. 183, 2929–2936 (2001).
Moore, W. E. & Holdeman, L. V. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 27, 961–979 (1974).
Mishra, A. K., Hugon, P., Robert, C., Raoult, D. & Fournier, P. E. Non contiguous-finished genome sequence and description of Peptoniphilus grossensis sp. nov. Stand Genom. Sci. 7, 320–330 (2012).
Lagier, J. et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol. 1, 16203 (2016).
Acknowledgements
Sequencing was performed with the support of: (i) the DFG-funded NGS Competence Center Tübingen (INST 37/1049-1) and the Institute for Medical Microbiology and Hygiene at the University Hospital (Tübingen, Germany), including help by the Quantitative Biology Center (QBiC) for raw data management and storage; (ii) Nassos Typas and Carlos Geert Pieter Voogdt (EMBL, Heidelberg), for long-read sequencing; (iii) the Genomics Facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University. We are also thankful to: (iv) Patrick Buchta from the Audio-Visual department at the University Hospital of RWTH Aachen for designing the HiBC logo; (v) Marzena Wyschkon and Meina Neumann-Schaal from the Leibniz Institute DSMZ for their help with the deposition of strains and CFA analysis, respectively; (vi) Catherine Gonzalez and Maurice Heizer from the Functional Microbiome Research Group (Institute of Medical Microbiology; University Hospital of RWTH Aachen) for data management with Coscine and for the curation of HiBC, respectively; (vii) Peter Vandamme, Claudine Vereecke, and Anneleen Wieme for handling the deposition of strains at the BCCM/LMG Bacteria Collection. T.C.A.H. received funding from the German Research Foundation (DFG) as part of SFB1382 (Rising Star programme) and from the RWTH Aachen START programme, project “LeakyGut”. T.C. received funding from the DFG, project no. 513892404, no. 445552570, no. 395357507 – SFB1371, no. 403224013 – SFB1382 and no. 460129525 – NFDI4Microbiota, and the German Ministry for Research and Education (BMBF), project Mi-EOCRC (K.Z. 01KD2102D). J.O. received funding from the DFG, project no. 6270054 NFDI 28/1 “NFDI4Microbiota” and 6270048 NFDI 5/1 “NFDI4Biodiversity”, the BMBF, projects no. 8005512901 and 8005512001 of DZIF, and EU/Horizon IRA project MICROBE no. 101094353. M.G. received funding from the DFG, project no. 403224013 – SFB1382. The data used in this publication was managed using the research data management platform Coscine with storage space granted by the Research Data Storage (RDS) of the DFG and Ministry of Culture and Science of the State of North Rhine-Westphalia (DFG: INST222/1261-1 and MKW: 214-4.06.05.08 - 139057). L.M. and T.A. received funding from The Leverhulme Trust: project no. RF-2023-286\2.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
The project was conceptualised and managed by T.C.A.H. and T.C. Isolation and cultivation of bacteria were conducted by J.M.M., J.B., S.N., M.B., S.R., A.A., A.M.A., D.W., A.C., S.A.V.J., A.P., A.V., M.S., E.C.D., K.B., M.W., K.A.W., L.A., and A.R. M.B. performed analysis of Bacteroidales plasmids. Confirmation of plasmid pMMCAT presence was conducted by L.M. and T.A., with M.D.C. and M.G. evaluating its impact on adhesion. Cultivation of strains was supervised by T.C.A.H., D.B., T.S., and T.C., with samples for isolation from S.T., Th.Cr., and J.S. N.K. and N.T. performed genome sequencing. T.C.A.H., J.M.M., N.T., A.A., and N.K. conducted stool sample acquisition. Deposition of strains at the DSMZ was handled by S.N., A.L., I.S., J.W., T.R., M.P., B.A., L.C.R., and J.O. J.M.M., C.P., S.N., M.B., D.W., A.C., T.R., B.A., and T.C. curated isolates. The website and metadata curation were conducted by T.C.A.H., J.M.M., C.P., J.H., and L.C.B. Bioinformatic analysis was conducted by T.C.A.H., C.P., and M.A.S. C.P. deposited the sequencing data. T.C. coordinated the project. J.O. and T.C. provided essential infrastructure and secured funding. The manuscript was written by T.C.A.H. and T.C., and reviewed by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hitch, T.C.A., Masson, J.M., Pauvert, C. et al. HiBC: a publicly available collection of bacterial strains isolated from the human gut. Nat Commun 16, 4203 (2025). https://doi.org/10.1038/s41467-025-59229-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59229-9
