
Abstract
Fragment screening by crystallography has recently skyrocketed. Multiple synchrotrons have built specialized screening platforms, established workflows, and assembled compound libraries. Crystallographic fragment screening is now widely accessible to groups that had previously not considered the approach. While hundreds of crystallographic fragment-screening campaigns have been conducted in the last few years, most of the underlying data have neither been published nor made publicly accessible. This perspective highlights the importance of establishing effective mechanisms for preserving large and often heterogeneous groups of datasets intrinsic to crystallographic fragment-screening campaigns, thereby ensuring their accessibility for advancing research and enabling applications such as training AI-based models.
Similar content being viewed by others


Introduction
The discovery of small organic molecules that bind to a macromolecular target is one of the first steps in drug discovery. For decades, the dominant approach has been high-throughput screening (HTS) using hundreds of thousands of drug-sized molecules1. Nearly 30 years ago, Shuker and colleagues2 demonstrated that NMR could be used to identify smaller molecules called fragments (molecules with a molecular weight of typically less than 300 Da or consisting of less than 23 non-hydrogen atoms) that bind weakly but efficiently to a target. Because the number of possible small molecules grows exponentially with the number of heavy atoms, screening small libraries of fragments is a much more efficient means of exploring chemical space than screening large libraries of millions of compounds3,4,5. A few years later, X-ray crystallography was used for the first time as the primary screening method to identify fragment binders6. Following that, several papers heralded high-throughput X-ray crystallography as a starting point for drug discovery7,8,9,10,11,12. At that time, these developments were primarily driven by industry, with new companies such as Astex Therapeutics and SGX Pharmaceuticals, Inc. leading the way. There were two limitations, however. The first was that X-ray crystallography was nowhere near as rapid as it is today. To efficiently screen hundreds, let alone thousands of fragments by X-ray crystallography required cocktails (i.e., mixtures of up to ten structurally dissimilar fragments). Compound cocktails, however, have the disadvantages that the concentration of the individual components in the mixture cannot be as high as one might like, and that individual components need to be unambiguously identified in electron density maps. Moreover, if one cocktail component destroys the crystal packing, possible binding information for the remaining components would be lost. Because of these throughput limitations, X-ray crystallography was often relegated to a confirmatory step after higher-throughput biophysical screening methods, such as Nuclear Magnetic Resonance (NMR), Surface Plasmon Resonance (SPR), Thermal Shift Assay (TSA), or Microscale Thermophoresis (MST). The second limitation was that academic researchers did not initially embrace these new developments, perhaps because of the resources required. Consequently, it fell to the private sector to develop the method further and prove its value for discovering drug leads and chemical probes13. In retrospect, this state of affairs is surprising. Due to their lower complexity, fragments often engage in higher-quality interactions with their target than do larger molecules (see Figure 2 in de Souza Neto et al., 2020)14. At the same time, negative or suboptimal interactions are more easily avoided. This phenomenon is described by the term ligand efficiency, which is defined as the binding free energy normalized for the number of non-hydrogen atoms15. Even though fragments often bind weakly due to their small size, they often exhibit greater ligand efficiency than larger molecules. Also, optimization strategies such as growing fragments, merging fragments, or linking two or more fragments (see Figure 3 in de Souza Neto et al., 2020)14 can improve the binding affinity substantially, as was proposed more than 40 years ago16. By the mid-2010s, the situation had changed. Developments at synchrotron radiation sources, including beamline instrumentation, pixel array detectors, sample-handling robotics, and software for data processing and structure determination, made it possible to collect an entire dataset from a single crystal in seconds or minutes, thereby increasing daily throughput from a few dozen to several hundred datasets17,18,19.
At about the same time as crystallographic data collection throughput was increasing, there was growing recognition that biophysical methods used for pre-screening often proved to be suboptimal. An important paper by Schiebel and colleagues20 compared seven methods, including NMR and X-ray crystallography, to assess the binding of 361 fragments to endothiapepsin. Surprisingly, the overlap among all seven methods was zero, meaning that not a single fragment could be detected by all seven methods. Thus, Schiebel and colleagues20 concluded that any biophysical method used to pre-screen before crystallography would lead to a loss of potential hits, bolstering the case for “crystallography first.” A second and equally important argument for “crystallography first” is the wealth of structural information one can obtain about the fragment binding site and its binding pose, directly informing and enabling downstream medicinal chemistry optimization strategies. Indeed, a 2019 paper noted that more than a third of researchers would not even begin optimizing fragments without crystallographic information21. If anything, this number has only grown22. Technology development groups at synchrotrons seized this opportunity and established workflows and procedures for screening libraries of up to 1000 fragments and offered access to academic and industrial user communities23,24,25,26,27,28,29,30.
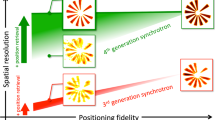
At present, more than ten major synchrotrons around the globe have installed or are about to install a fragment-screening facility with a dedicated workflow (Fig. 1). While Europe is clearly ahead with six facilities, other parts of the world are ramping up similar developments. In terms of crystallographic throughput, the UK-based XChem facility at the Diamond Light Source25,29 is currently the leader, having been responsible for more than 50% of all publicly disclosed crystallographic fragment-screening campaigns worldwide to date. Somewhat surprising is the low level of activity at synchrotron radiation sources in North America. While one of us (JSF) has developed a facility in his UCSF lab, synchrotron-based facilities, which would offer access to and could support a large user community, are mostly lacking, except for the facility currently being developed at NSLS-II (https://wiki-nsls2.bnl.gov/MX/index.php?title=Fragment_Screening). Already today, at least 150 crystallographic fragment-screening campaigns are conducted annually by the academic and industrial users of various facilities. Once all the facilities listed in Fig. 1 operate at full capacity, the annual number could easily increase to 1000 campaigns or more. Assuming 1000 compounds per campaign, this level of utilization would translate into 106 individual diffraction datasets, and assuming a not-unreasonable 10% fragment hit rate, fragment screening alone would yield 105 individual protein-ligand structures every year. Additionally, even the unliganded structures may be valuable and worth depositing, for example, in establishing background electron densities for automatic data processing and hit-finding tools such as the Pan-Dataset Density Analysis approach PanDDA31, as discussed below.

#Number of diffraction datasets, which can be collected in a 24-h period. $Number of fragment-screening campaigns currently being executed by the respective facility per year.
Discussion on sharing and archival of data arising from fragment-screening campaigns
Data sharing and archival in structural biology
Most reputable scientific journals have established data-sharing requirements for the publication of new macromolecular X-ray crystal structures. Atomic coordinates must be deposited in the Protein Data Bank or PDB32, together with experimentally determined structure factor amplitudes. Upon submission of the manuscript to a journal, the official Worldwide Protein Data Bank (wwPDB) Validation Report must frequently be submitted along with the manuscript, together with proof of submitting the relevant data to the PDB. The purpose of this requirement is to enable anyone to examine the evidence that the deposited atomic coordinates are supported by the underlying experimental data. These procedures were originally implemented in response to community requests and are now well established and work to the benefit of data producers, journals, manuscript referees, and PDB data consumers. Indeed, PDB data are so robust that they have been used to train artificial intelligence/machine-learning methods for predicting protein structures based on amino acid sequences with an accuracy comparable to that of experimental methods33. No one could have foreseen this opportunity when the PDB was established in 1971, demonstrating that vast quantities of high-quality scientific data can underpin important advances and enable unexpected breakthroughs.
The success of rigorous validation and expert biocuration of 3D biostructure data is also evidenced by PDB growth statistics. Of the nearly 230,000 macromolecular structures archived in the PDB (as of December 2024), more than 190,000 have been determined by crystallography, with ~10,000 new crystal structures being released to the public annually.
Fragment-screening data challenge the current procedures and databases!
Considering the sheer numbers of datasets and structures mentioned above, fragment screening precipitates two key, interrelated issues in terms of distinct data-associated challenges: (1) data growth at an ever-accelerating pace and (2) concomitantly increased structure refinement effort.
Fragment screening could potentially increase the influx of X-ray structures into the PDB by nearly an order of magnitude. Even with the use of RCSB PDB GroupDep, which expedites deposition, validation and biocuration of tens to hundreds of similar structures in parallel, it could be challenging for the wwPDB to manage tens of thousands of additional fragment co-crystal structures using current protocols. A typical wwPDB biocurator needs about 3 h to validate and biocurate each protein-ligand structure deposited via OneDep and about half an hour for a protein-ligand structure entered via GroupDep. This productivity level translates into ~700 OneDep structures or ~4000 GroupDep structures per biocurator per year. These numbers are impressive, but they are nowhere near what would be needed if 105 additional protein-ligand structures were added annually.
Furthermore, many structures from high-throughput fragment screening may not be directly comparable to traditionally determined, fully refined PDB structures. Protein-ligand structures from fragment screens are often only partially refined, and typically only in the vicinity of the ligand binding site to determine if, where, and how a fragment is bound. Extra effort devoted to refining such a structure to convergence, which could easily add an extra one to two days per structure, may not immediately return sufficiently useful information to be warranted. This reality means that such structures may be flagged as being of “lower quality” in wwPDB Validation Reports.
Another difficulty for deposition is that most fragments bind at sub-stoichiometric occupancy. This partial occupancy results in compositional and often accompanying conformational heterogeneity of the crystal34,35. These two related types of heterogeneity are difficult to simultaneously encode using current refinement procedures. The use of the “altloc” field to describe both types leads to ambiguities during validation by PDB curators, which slows down the exchange for structures that explicitly represent all heterogeneity that can be modeled. With new refinement procedures and deposition standards that directly confront these problems, tradeoffs between completeness and quality that currently confront crystallographers could be reduced.
An additional complication is that the experimental evidence for the presence of a fragment in a fragment-screening campaign may not solely be contained in an individual diffraction dataset measured from a single fragment-soaked crystal. This is when the Pan-Dataset Density Analysis PanDDA approach31 comes in. The PanDDA approach relies upon a specialized type of difference electron density map, known as the “event map”, wherein the unbound or ground-state background density, which is constructed from dozens or more “unbound” datasets, is subtracted from fragment-bound data to reveal evidence for the presence of the fragment, which may have sub-stoichiometric occupancy. This added complication necessitates the deposition of “unbound” datasets alongside fragment-bound datasets to enable data consumers to reproduce fragment-screening results from archived data. Additionally, there is potential value in the curation and archival of “unbound” datasets, which may contain interesting structural features far from the target site of interest and beyond what could be expected. The importance of depositing “unbound” datasets is further underscored by their potential use for future discovery. In addition to enabling background map calculations, there are cases where the presence of ligands only becomes evident when the entire fragment-screening campaign is pre-clustered into subgroups of datasets36. For each subgroup, a ground-state model needs to be constructed, and an independent PanDDA analysis needs to be performed. Such cases may occur, for example, when slight variations in cell dimensions lead to non-isomorphism, or they could simply be the consequence of a statistical distribution of slightly different crystal forms arising from non-uniform crystal soaking procedures or cryocooling conditions. A remarkable example, where the number of positive hits was increased by about 50% as a result of data clustering, was recently published37. This was only made possible because all datasets had been archived on Zenodo.
What do people do to satisfy current publication and deposition requirements?
The following examples taken from the literature or from anecdotal reports describe how some fragment-screening teams currently operate. Simply put, there are currently no commonly established procedures.
Approach 1
Refine the protein structures automatically without the ligand as much as possible, identify the ligand by difference electron density analysis, place the ligand, and refine the liganded structure to convergence. This is the traditional approach, resulting in fully refined protein-ligand structures. However, as the number of liganded structures exceeds one or two dozen, this approach becomes impractical and too time-consuming. As mentioned above, refining to convergence can easily add a day or two of work per structure. This approach was reported by Schiebel and colleagues38 and was routinely followed at SGX Pharmaceuticals, Inc.
Approach 2
Determine the protein structure by automated Molecular Replacement using DIMPLE39. When features appear in the difference electron density map indicating the presence of a ligand, refine the structure further until the ligand density becomes clearer. Place the ligand and refine the liganded structure further, but not necessarily to convergence. This approach is favored at the SPring-8 (Japan) fragment-screening facility.
Approach 3
Refine the protein structures automatically without the ligand as much as possible and identify the ligand in a PanDDA event map31. Use the PanDDA event map to place the ligand and make minor modifications to the structure only in the ligand binding site. From here onwards, there are two possible routes. Approach 3A: Adjust the ground-state model to the average PanDDA map. Combine the ground state with the ligand-bound model and refine using giant.refine (PanDDA). Check the resulting model for peaks >5 sigma, adjust and re-refine using giant.refine. This approach was described by Wollenhaupt and colleagues40. Approach 3B: Deposit just the ligand-bound structure along with the final refined mtz file from the auto-refinement pipeline used. 3B is merely a simplified version of 3A. Barthel and colleagues41 used this approach to deposit about 270 protein-ligand structures from a 1000-compound screening campaign.
Approach 4
Determine the protein structure by automated Molecular Replacement using DIMPLE39. Use the average PanDDA map to adjust the ground-state model, and re-run DIMPLE with the updated ground-state model. Then, use the PanDDA event map to place the ligand and to make some minor modifications to the structure in the ligand binding site only. Combine the ground state and bound-state models and refine using giant.refine (PanDDA). Check the resulting model, adjust, and re-refine using giant.refine. Deposit the ligand-bound structure together with the experimental structure factors, the final refined mtz file, and the PanDDA event map (stored as distinct data blocks in one cif file). This approach is currently used by the XChem facility at Diamond Light Source29.
Clearly, this list is far from being complete. Researchers may follow modified flavors of the mentioned approaches, or they could proceed along different routes. The important message is that there are currently no commonly established, standardized procedures.
What are the best options for FAIR archiving of crystallographic fragment-screening data?
Principally, the two key questions in this context are (1) What is the minimum data assemblage that must be captured to enable results to be replicated and to have value for future method development and data analyses, and (2) How to make the output from high-throughput fragment screens accessible to the wider scientific community?
The current situation is that different researchers, sometimes even from the same lab, have different answers to the questions above and follow different procedures. The lack of agreed-upon standards is far from satisfactory. Given the vast amount of data, the big question is: are there possibilities for management of fragment-screening data that are both practical and FAIR (Findable, Accessible, Interoperable, and Reusable)42? Four options for addressing current challenges are outlined below. In some, the PDB would play the central role, while in others, deposition and archiving would happen either partially or completely independent of the PDB.
Option 1
Archiving fragment screening hits as fully refined protein-ligand co-crystal structures. For lower-occupancy ligands, doing so would involve refining the structure at least as a two-state (bound and unbound) model. Currently, available software is limited in this respect, but developments are ongoing to remedy this. Then, such structures could be handled by the PDB the same as any other structure. For protein-ligand structures for which PanDDA was used to identify the presence of the ligand, the PanDDA evidence also needs to be deposited, to enable the data consumer to evaluate the evidence. Two possibilities for doing so are briefly outlined immediately after this section; see below. This option is the most conservative and would place the greatest burden on the fragment-screening team, adding an estimated one to two days per structure for refinement to convergence and the time necessary to prepare parallel deposition of the structures.
Option 2
Archiving fragment-screening hits as partially refined protein-ligand structures. Most of the atomic coordinates would be the result of some auto-refinement procedure, then the ligand would be placed based on a traditional difference Fourier map or based on the PanDDA event map, and small modifications to the protein structure in the vicinity of the ligand binding site(s) would be carried out by hand. Then, the structure would be refined for one more cycle and deposited as is. Adherence to current wwPDB validation practices would result in “inferior” wwPDB Validation Reports for these data. As for Option 1, where PanDDA was used for ligand identification, deposition of the PanDDA event map would be necessary. Because such structures would not be comparable in quality to most of the fully refined structures archived in the PDB, it may be worth considering whether they should be segregated into a separate branch of the archive or flagged as originating from a fragment-screening experiment. This option would entail less work for depositors than Option 1, but more work for the wwPDB consortium.
Option 3
Archiving fragment-screening campaign information in its entirety (or as pre-clustered subsets of the data) in a single data repository. Doing so would entail management of all processed diffraction data and all fully and/or partially refined co-crystal structures. Scientifically, this approach would appear to be ideal. It would make both positive and negative results available to the community and enable data consumers to reproduce the evidence for each fragment hit. Moreover, method developers would have access to all the fragment-screening data to improve fragment hit detection (possibly by AI-based approaches). An important requirement here is that the entire screen be understood as a single investigation, not as an amalgamation of individual experiments. Given the number of fragment-screening campaigns on the horizon, it is an open question whether these data should be housed in the PDB or elsewhere. If they were to be housed in the PDB, two questions arise. First, would partially refined structures with “inferior” wwPDB Validation Reports have to be segregated from fully refined structures? Second, how would the “unbound” datasets be managed?
Option 4
Archiving fully refined protein-ligand follow-up structures based on fragment hits in the PDB and preservation of remaining fragment-screening campaign information in a separate data resource(s). This “hybrid” approach would avoid overloading wwPDB biocurators and flooding the PDB with partially refined structures with “inferior” wwPDB Validation Reports, etc. But it would create challenges for the fragment-screening research community. Immediately, there would be the challenge of making data freely available to satisfy current publication requirements. Other databases such as CHEMBL43, BindingDB44, Github, Zenodo, or XRDa could potentially play a role here, or such data could be added as supplementary information to a publication (e.g., Füsser and colleagues45). More than likely, this approach would require adaptations to those databases and development of clear guidelines for data depositors. An even greater concern would be how the fragment-screening research community will ensure adherence to the FAIR data principles42 with such a “hybrid” approach.
Importantly, the four options described are neither exhaustive nor mutually exclusive. For example, adoption of Option 1 does not preclude making all remaining data available under Option 4.
Current possibilities for preserving the PanDDA evidence for the presence of a ligand in a deposition
As mentioned above, an important consideration in the context of data preservation and deposition is that the experimental evidence for the presence of the ligand must be extractable from the deposited data. For protein-ligand structures in which the ligand was identified in a difference electron density map, this is easily ensured, because the file with the experimental structure factor amplitudes already contains all necessary information. In cases where PanDDA was used for ligand identification, it is not so obvious because the information on the presence of the ligand is distributed over many datasets, including “unbound” datasets. Without them being deposited as well, there are two principal possibilities:
(i) The structure factor amplitude file of a protein-ligand structure needs to be supplemented with the corresponding amplitudes and phases of the Fourier-transformed PanDDA event map. These could be simply added as two additional columns (PanDDA_eventmap_F and PanDDA_eventmap_PHI) to the file containing the experimental data, so the meaning of these columns is obvious for any downstream program that is used to visualize this map.
(ii) The coefficients of the Fourier-transformed PanDDA event map need to be added as separate data blocks to the cif file for deposition. In this case, data consumers have to first extract the event map from the cif file before the map can be visualized.
The second option is most common today, because the PanDDA event map is typically calculated in symmetry P1 and therefore the amplitudes and phases of the Fourier-transformed map are not directly compatible with the experimental structure factor amplitudes file. However, an elegant solution could be to transform the event map to the symmetry of the crystal and then proceed with the first option.
New possibilities for preserving the PanDDA evidence for the presence of a ligand in a deposition
It may turn out that the currently provided facilities and mechanisms are not sufficient to address the two key questions in the previous section in a scientifically and administratively satisfactory manner. In this case, one needs to think beyond what is currently possible and feasible and explore new territory as well.
Outlook and perspectives
Because there is no established procedure for how to treat structures produced by crystallographic fragment-screening campaigns nor for preservation of results from these campaigns, we call for a community-wide discussion to agree on best practices and accepted procedures. As representative of the central Structural Biology archive, the wwPDB is in the pole position for initiating this. Such a discussion should involve researchers involved in producing fragment-screening data, facility operators, representatives of relevant data resources, such as PDB, BindingDB, etc., scientific publishers, and data consumers. A “white-paper” describing the outcome of such a discussion could recommend community consensus guidelines, which can be implemented at the PDB (or elsewhere) and to which reputable publishers would subscribe. No matter the outcome, it is imperative that the FAIR principles are fully embraced by fragment-screening researchers.
These guidelines are urgently needed! Crystallographic fragment-screening campaigns have become ever more popular in recent years, and we anticipate a looming tsunami of fragment-screening campaigns with potentially millions of datasets and hundreds of thousands of protein-ligand co-crystal structures on the horizon. If these data are to be captured and made available to the community, we need to ensure that relevant, easy-to-use, and robust tools and standards are in place. A further and more involved question is how crystallographic fragment-screening data can be made interoperable with outcomes of complementary biophysical fragment screens (e.g., from NMR and cryo-electron microscopy).
Finally, one should always bear in mind that a fragment-screening campaign is just the beginning of a lead discovery project. While specific protein-fragment complexes are the near-term goal for fragment screens, there is no telling what valuable insights might be drawn from such a massive collection of data over the coming years and decades. For this reason, we advocate making as much data as widely available as possible.
References
Entzeroth, M., Flotow, H. & Condron, P. Overview of high-throughput screening. Curr. Protoc. Pharmacol. 44, 9.4.1–9.4.27 (2009).
Shuker, S. B., Hajduk, P. J., Meadows, R. P. & Fesik, S. W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534 (1996).
Hall, R. J., Mortenson, P. N. & Murray, C. W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 116, 82–91 (2014).
Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W. & Jhoti, H. Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15, 605–619 (2016).
Leach, A. R. & Hann, M. M. Molecular complexity and fragment-based drug discovery: ten years on. Curr. Opin. Chem. Biol. 15, 489–496 (2011).
Nienaber, V. L. et al. Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nat. Biotechnol. 18, 1105–1108 (2000).
Blundell, T., Jhoti, H. & Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 1, 45–54 (2002).
Blundell, T. L. & Patel, S. High-throughput X-ray crystallography for drug discovery. Curr. Opin. Pharmacol. 4, 490–496 (2004).
Hartshorn, M. J. et al. Fragment-based lead discovery using X-ray crystallography. J. Med. Chem. 48, 403–413 (2005).
Mooij, W. T. et al. Automated protein-ligand crystallography for structure-based drug design. ChemMedChem 1, 827–838 (2006).
Burley, S. K., Hirst, G., Sprengeler, P. & Reich, S. Fragment-based structure-guided drug discovery: strategy, process, and lessons from human protein kinases in Drug Design: Structure- and Ligand-Based Approaches (eds Merz, K. M. Jr et al.) (Cambridge University Press, 2010).
Spurlino, J. C. Fragment screening purely with protein crystallography. Methods Enzymol. 493, 321–356 (2011).
Brown, P. J. & Müller, S. Open access chemical probes for epigenetic targets. Future Med. Chem. 7, 1901–1917 (2015).
de Souza Neto, L. R. et al. In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 8, 93 (2020).
Hopkins, A. L., Groom, C. R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 9, 430–431 (2004).
Jencks, W. P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. USA 78, 4046–4050 (1981).
Helliwell, J. R. & Mitchell, E. P. Synchrotron radiation macromolecular crystallography: science and spin‐offs. IUCr J. 2, 283–291 (2015).
Grabowski, M. et al. Synchrotron radiation as a tool for macromolecular X‐ray crystallography: a XXI century perspective. Nucl. Instrum. Methods Phys. Res. B 489, 30–40 (2021).
Grimes, J. M. et al. Where is crystallography going? Acta Crystallogr. D74, 152–166 (2018).
Schiebel, J. et al. Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: a case study. ACS Chem. Biol. 11, 1693–1703 (2016).
Erlanson, D. A., Davis, B. J. & Jahnke, W. Fragment-based drug discovery: advancing fragments in the absence of crystal structures. Cell Chem. Biol. 26, 9–15 (2019).
Erlanson, D. A. Poll results: fragment finding methods and structural information needed for fragment-to-lead efforts. Practical Fragments. https://practicalfragments.blogspot.com/2024/11/poll-results-fragment-finding-methods.html (2024).
Lima, G. M. A. et al. FragMAX: the fragment-screening platform at the MAX IV Laboratory. Acta Crystallogr. D76, 771–777 (2020).
Cornaciu, I. et al. The automated crystallography pipelines at the EMBL HTX facility in Grenoble. J. Vis. Exp. 172, e62491 (2021).
Douangamath, A. et al. Achieving efficient fragment screening at XChem Facility at Diamond Light Source. J. Vis. Exp. 171, e62414 (2021).
Wollenhaupt, J. et al. Workflow and tools for crystallographic fragment screening at the Helmholtz-Zentrum Berlin. J. Vis. Exp. 169, e62208 (2021).
Kaminski, J. W. et al. Fast fragment- and compound-screening pipeline at the Swiss Light Source. Acta Crystallogr. D78, 328–336 (2022).
Barthel, T. et al. The HZB F2X-facility—an efficient crystallographic fragment screening platform. Appl. Res. 3, e202400110 (2024).
Fearon, D. et al. Accelerating drug discovery with high-throughput crystallographic fragment screening and structural enablement. Appl. Res. 4, e202400192 (2025).
Huang, L. et al. Novel starting points for fragment-based drug design against human heat-shock protein 90 identified using crystallographic fragment screening. IUCrJ 12, 177–187 (2025).
Pearce, N. M. et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 8, 15123 (2017).
wwPDB Consortium. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 47, D520–D528 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pearce, N. M., Krojer, T. & von Delft, F. Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallogr. D73, 256–266 (2017).
Wankowicz, S. A. & Fraser, J. S. Comprehensive encoding of conformational and compositional protein structural ensembles through the mmCIF data structure. IUCrJ 11, 494–501 (2024).
Ginn, H. M. Pre-clustering data sets using cluster4x improves the signal-to-noise ratio of high-throughput crystallography drug-screening analysis. Acta Crystallogr. D76, 1134–1144 (2020).
Mehlman, T., Ginn, H. M. & Keedy, D. A. An expanded trove of fragment-bound structures for the allosteric enzyme PTP1B from computational reanalysis of large-scale crystallographic data. Structure 32, 1231–1238 (2024).
Schiebel, J. et al. High-throughput crystallography: reliable and efficient identification of fragment hits. Structure 24, 1398–1409 (2016).
Wojdyr, M., Keegan, R., Winter, G. & Ashton, A. DIMPLE—a pipeline for the rapid generation of difference maps from protein crystals with putatively bound ligands. Acta Crystallogr. A69, s299 (2013).
Wollenhaupt, J. et al. F2X-Universal and F2X-Entry: structurally diverse compound libraries for crystallographic fragment screening. Structure 28, 694–706 (2020).
Barthel, T. et al. Large-scale crystallographic fragment screening expedites compound optimization and identifies putative protein-protein interaction sites. J. Med. Chem. 65, 14630–14641 (2022).
Wilkinson, M. et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2023).
Liu, T. et al. BindingDB in 2024: a FAIR knowledgebase of protein-small molecule binding data. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae1075 (2025).
Füsser, F. T., Wollenhaupt, J., Weiss, M. S., Kümmel, D. & Koch, O. Novel starting points for fragment-based drug design against mycobacterial thioredoxin reductase identified using crystallographic fragment screening. Acta Crystallogr. D79, 857–865 (2023).
Acknowledgements
The authors would like to thank—in alphabetical order—Drs. Johanna Hakanpää (DESY, Germany), Jae-Hee Jeong and Suk-Youl Park (Pohang Accelerator Laboratory, Korea), Tobias Krojer (MAX IV, Sweden), Josan Marquez (EMBL-GR, France), Andrey Nascimento (SIRIUS, Brazil), Wenming Qin (SSRF, Shanghai, China), Alan Riboldi-Tunnicliffe and Rachel Williamson (Australian Synchrotron, Australia) and May Elizabeth Sharpe (PSI, Switzerland) for providing information about their respective facilities. Many thanks also to Dr. Tatjana Barthel (HZB, Germany) for her insights and discussion on current refinement practices. The authors are also particularly grateful to the following members of the wwPDB leadership team, Drs. Jeffrey C. Hoch (BMRB, USA), Genji Kurisu (PDBj, JP), Kyle Morris (EMDB, EBI, UK), and Sameer Velankar (PDBe, EBI, UK) for critical and helpful comments and feedback on the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: D.A.E. and M.S.W. Investigation: M.S.W., D.F., D.K., M.C.N., N.S., J.S.F., J.W. and S.K.B. Writing and editing: all.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mathew Martin, Cameron Mura and Marc O’Reilly for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Erlanson, D.A., Burley, S.K., Fearon, D. et al. Where and how to house big data on small fragments. Nat Commun 16, 4179 (2025). https://doi.org/10.1038/s41467-025-59233-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59233-z
