
Abstract
DNA damage repair is a critical physiological process closely linked to aging. The accumulation of DNA damage in renal proximal tubular epithelial cells (PTEC) is related to a decline in kidney function. Here, we report that DNA double-strand breaks in PTECs lead to systemic metabolic dysfunction, including weight loss, reduced fat mass, impaired glucose tolerance with mitochondrial dysfunction, and increased inflammation in adipose tissues and the liver. Single-cell RNA sequencing analysis reveals expansion of CD11c+ Ccr2+ macrophages in the kidney cortex, liver, and adipose tissues and Ly6Chi monocytes in peripheral blood. DNA damage in PTECs is associated with hypomethylation of macrophage activation genes, including Gasdermin D, in peripheral blood cells, which is linked to reduced DNA methylation at KLF9-binding motifs. Macrophage depletion ameliorates metabolic abnormalities. These findings highlight the impact of kidney DNA damage on systemic metabolic homeostasis, revealing a kidney-blood-metabolism axis mediated by epigenetic changes in macrophages.
Similar content being viewed by others

Introduction
DNA damage repair is an essential physiological process for the maintenance of an individual’s life. DNA damage can be induced by various internal and external stresses, and an increase in DNA damage sites due to an increase in DNA damage or a decrease in DNA repair has been observed in aging and certain pathological conditions1. Recent reports showed that an increase in DNA damage can lead to a loss of chromatin structural balance, namely, epigenomic changes, which can trigger aging phenotypes2.
The incidence of chronic kidney disease (CKD) is increasing worldwide to varying degrees depending on regions as the population ages. CKD increases the risk for end-stage renal failure as well as cardiovascular complications, strongly affecting patient prognosis. The increase in the prevalence of hypertension and diabetes with aging and the consequent increase in conditions such as nephrosclerosis and diabetic nephropathy contribute to this trend3. With aging, the glomerular filtration rate (GFR) decreases, and nephrosclerosis increases4. In contrast, recent reports have suggested that CKD may accelerate systemic aging. Although previous reports have identified prominent immune aging in patients on dialysis5,6, recent large-scale data analysis using artificial intelligence (AI) has revealed that CKD is an important factor in the progression of systemic aging7,8. However, the underlying mechanisms remain unclear.
We have previously investigated the important role of DNA damage and subsequent epigenetic changes in kidney diseases, especially focusing on glomerular podocytes9,10,11,12,13. Podocytes form a slit diaphragm and play a crucial role in the urinary protein barrier. These terminally differentiated cells fundamentally do not divide or replenish, making them a focal point in the study of renal aging14. DNA double-strand breaks in podocytes not only lead to changes in the renal immune microenvironment centered on the activation of CD8+ T cells but also induce the proliferation of CD44+ memory cells in peripheral blood, together with blood DNA methylation changes15. Furthermore, changes in DNA methylation in peripheral blood may be involved in CD8+ T-cell activation and differentiation. Another model of increased DNA damage in podocytes also suggests an increase in CD44+ CD8+ T cells16, similar to what has been previously described as immune aging17. This finding is consistent with the results of previously reported epigenome-wide association studies, which indicated that blood DNA methylation changes are associated with a decrease in kidney function18,19,20. Given the high cell type specificity of DNA damage repair pathways, investigating the role of DNA damage repair in proximal tubular epithelial cells (PTEC) compared to that in podocytes is important. PTECs are metabolically active cells with abundant mitochondria and retain some degree of proliferative capacity, unlike podocytes. Damage to PTECs is a key mechanism that triggers tubulointerstitial fibrosis, the final common pathway of renal failure. Therefore, we focused on DNA damage in PTECs to elucidate its unique roles in renal function and systemic homeostasis.
In this study, we find that repeated DNA damage repair in PTECs not only leads to changes in the renal immune and metabolic environment that are different from those associated with podocyte DNA damage but also increases the number of activated monocytes associated with DNA methylation changes in peripheral blood cells, ultimately resulting in systemic metabolic disorders, similar to a phenotype of metabolic aging.
Results
Continuous nonmutagenic DNA damage in proximal tubular epithelial cells (PTECs) causes systemic metabolic changes
I-PpoI is a homing endonuclease that cuts the target 15-bp DNA motif, and the mouse genome contains 19 canonical I-PpoI sites and 112 degenerate sites21. To investigate the effect of continuous DNA DSBs in proximal tubular epithelial cells (PTECs) on renal and systemic homeostasis, we generated mice with PTEC-specific I-PpoI expression (γGT-Cre, ROSA26-STOP-IPpoI, referred to as I-PpoI mice). DNA double-strand breaks (DSBs) were confirmed to occur at known I-PpoI cleavage sites, including ribosomal DNA (rDNA) in the PTECs isolated from I-PpoI mice compared to their peripheral blood cells (Fig. 1A). The levels of γH2AX, a DNA DSB marker, were greater in PTECs from the I-PpoI mice than in those from the wild-type (WT) mice (Fig. 1B). Additionally, DNA DSBs at I-PpoI cleavage sites did not increase in the liver, peripheral blood, macrophages or adipose tissues of the I-PpoI mice (Supplementary Figs. 1A–E), and the levels of γH2AX were not increased in the visceral or subcutaneous adipose tissue from the I-PpoI mice (Supplementary Fig. 1F). It was also confirmed that the cre expression was observed in PTECs, not in adipose tissues in the I-PpoI mice, using gGT-Cre+Td-tomato mice (Supplementary Fig. 2).

A I‒PpoI cutting efficacy of rDNA in isolated PTECs from the I-PpoI and WT mice. B Representative photomicrographs of immunofluorescence (IF) staining of γH2AX (green)/AQP1 (red) in the kidney cortex. The experiment was repeated with similar results three times. C Real-time RT‒PCR analysis of biomarkers of renal proximal tubule injury in the renal cortex. D Serum BUN and Cr levels at 16 weeks of age. E The amount of albuminuria at 16 weeks of age. F Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (upper) and Gene Ontology enrichment analysis (lower) for upregulated differentially expressed genes. The graph shows the log p-value calculated using the Benjamini‒Hochberg-corrected two-tailed t test for the enrichment of a specific pathway. G Real-time RT‒PCR analysis of genes related to fatty acid metabolism in the renal cortex. H Time course of body weight. I The weights of VAT (left), SAT (middle) and BAT (right) at 16 weeks of age. J Serum ALT, ALP, TG and FFA levels at 16 weeks of age. K (left) Ip-GTT and area under the curve (AUC) for glucose values 0–120 min after glucose load at 16 weeks of age. (right) Fasting serum insulin levels at 16 weeks of age. L Hepatic FC and the ratio of FC to TC at 16 weeks of age. M, N Real-time RT‒PCR analysis of genes related to fatty acid metabolism in the liver (M) and genes involved in lipolysis and adipose differentiation in VAT (N). O (a) Western blot analysis of PPARγ protein. b Band intensity of PPARγ relative to GAPDH. P The RQ (VCO2/VO2) values determined by indirect metabolic calorimetry (CLAMS). Q, R Real-time RT‒PCR analysis of genes associated with cellular senescence in the kidney cortex (Q) and SASP-associated genes and senescence markers in the kidney (Ra), the liver (Rb) and VAT (Rc). Tg (the I-PpoI), 16-week-old γGT-Cre+ ROSA26-STOP-I-PpoI; WT, 16-week-old wild type. In A, C–E, G–I, L–N, (Q), and R, n = 6 samples in each group. In K and P, n = 4 in each group. In (J), n = 8 in each group. The data are presented as the means ± SEMs. *p < 0.05, **p < 0.01 vs. the control; N.S., not significant. Scale bars, 20 µm in (B). Source data are provided as a Source Data file.
Even at 1 year of age, there were no obvious differences in renal tissue between the I-PpoI mice and the WT mice according to periodic acid-Schiff (PAS) and Masson trichrome (MT) staining (Supplementary Fig. 1G); however, KIM-1 and NGAL expression and serum urea nitrogen and creatinine levels were elevated, and mild albuminuria was detected in the I-PpoI mice at 16 weeks of age (Fig. 1C–E), suggesting some phenotypic changes in the kidney of the I-PpoI mice as supported by the results of the immunohistochemical staining of vimentin, which is a marker for epithelial-mesenchymal transition22 (Supplementary Fig. 1H). Urine glucose and amino acid levels were not increased in the I-PpoI mice compared to the WT mice (Supplementary Fig. 1I). RNA sequence (RNA-seq) analysis of the renal cortex indicated that changes in pathways related to fatty acid metabolism were prominent in the I-PpoI mice, and these findings were confirmed by RT‒PCR analysis, suggesting decreased expression of carnitine palmitoyl-transferase 1 A (CPT1a), the rate-limiting enzyme in fatty acid β-oxidation, CPT2, CCAAT/ enhancer-binding protein-alpha (CEBPa), CCAAT/enhancer-binding protein-alpha (CEBPb), peroxisome proliferator-activated receptor (PPAR)α and PPARγ (Fig. 1F, G).
Notably, the I-PpoI mice exhibited weight loss, which was evident at ~16 weeks of age (Fig. 1H), despite no changes in food intake (Supplementary Fig. 1J). Impedance methods indicated that weight loss was attributable to a decrease in fat mass (Supplementary Fig. 1K), and indeed, visceral and subcutaneous fat weight was reduced in the I-PpoI mice (Fig. 1I), whereas the amount of muscle was not significantly altered in these mice (Supplementary Fig. 1L). Biochemical tests revealed that the serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), fasting triglyceride (TG) and free fatty acid (FFA) levels were elevated, indicating enhanced lipolysis (Fig. 1J), and impaired glucose tolerance was suggested in the I-PpoI mice due to insulin resistance, as evaluated by an intraperitoneal glucose tolerance test (ip-GTT) and fasting serum insulin levels (Fig. 1K). The free cholesterol (FC) as well as FC/total cholesterol ratio, a marker of fatty liver diseases23, was significantly increased in the liver of the I-PpoI mice, and the expression of CPT1a, CPT2, PPARα, and PPARγ was reduced in the livers of these mice, indicating impaired fatty acid metabolism (Fig. 1L, M). Although no histological changes were observed at 16 weeks of age, the number of hepatocytes with a single large fat droplet increased in the livers of 2-year-old I-PpoI mice compared to those of WT mice (Supplementary Fig. 1M). Free cholesterol in the kidney tended to be increased in the I-PpoI mice (Fig. 1L), and partial ectopic myocardium fat was observed in 6-month-old I-PpoI mice (Supplementary Fig. 1N).
Adipose tissues revealed no significant changes in the I-PpoI mice at 16 weeks of age, but the structures showed mild fibrosis in 1-year-old I-PpoI mice (Supplementary Fig. 1O, P). The expression of genes involved in lipolysis and adipose differentiation, including CEBPa and CEBPb, was significantly decreased in visceral adipose tissue of the I-PpoI mice, suggesting impaired adipose differentiation (Fig. 1N). The I-PpoI mice exhibited decreased gene expression of PPARγ, along with its target genes and key regulators involved in de novo lipogenesis and lipid droplet formation in visceral adipose tissue (VAT) (Fig. 1N, O). Energy expenditure was evaluated using indirect calorimetry, which revealed a reduced diurnal variation in the respiratory exchange ratio (RER) in the I-PpoI mice. This finding suggests a loss of metabolic flexibility, characterized by impaired switching from carbohydrates to lipids as an energy source during the night, which resembles patterns observed in metabolically unhealthy lean individuals and aging24,25 (Fig. 1P). These results suggest that impaired adipocyte differentiation and function may lead to an overflow of free fatty acids, which could result in the seemingly contradictory phenotype of decreased fat mass and ectopic fat deposition in other organs.
The accumulation of DNA double-strand breaks is known to induce cell senescence and contribute to the aging process. Then, the expression of genes associated with cellular senescence, including cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING), was examined, and the results revealed that the expression of these genes was elevated in the renal cortex of the I-PpoI mice (Fig. 1Q). The expression of senescence-associated secretory phenotype (SASP)-associated genes, such as tumor necrosis factor (TNF)α and interleukin (IL)-1β, and senescence markers, including p16 and p21, was also increased not only in the renal cortex but also in the liver and visceral fat of the I-PpoI mice (Fig. 1R). These findings suggested that DNA damage in PTECs caused systemic metabolic alterations with elevated inflammatory and senescence markers in the kidney, the liver and adipose tissues.
DNA damage in PTECs causes altered glucose and lipid metabolism in the kidney and liver
Next, to gain insight into the metabolic alterations occurring in the kidney and liver, we conducted a metabolomic analysis using samples from the renal cortex and liver of 16-week-old I-PpoI mice and WT controls. High anaerobic glycolysis and low tricarboxylic acid (TCA) cycle activity are characteristic features of the I-PpoI mice (Fig. 2A). In both the renal cortex and the liver, we observed a significant increase in the levels of lactate and pyruvate, which is consistent with risk factors for glucose intolerance26. In the kidney of the I-PpoI mice, an increase in the expression of key rate-limiting enzymes in the glycolytic pathway, such as hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK) were observed (Fig. 2B), suggesting a compensatory upregulation of glycolysis in response to reduced aerobic respiration due to mitochondrial dysfunction. In contrast, in the liver of the I-PpoI mice, there was a decrease in the expression of glycolytic enzymes alongside an increase in the expression of gluconeogenic enzymes including glucose-6-phosphatase (G6Pase), phosphoenolpyruvate carboxykinase (PEPCK) and pyruvate carboxylase (PCK) (Fig. 2C). This shift indicates enhanced gluconeogenesis, a process that typically relies on substrates such as lactic acid, amino acids or fat in conditions of impaired glucose tolerance. Furthermore, the levels of many intermediates and products associated with the TCA cycle, such as citrate and α-ketoglutaric acid, decreased in the I-PpoI mice27. Branched-chain amino acids (BCAAs) were also found to be increased in both the renal cortex and the liver (Fig. 2D). According to a recent report, BCAAs are negatively correlated with insulin sensitivity, independent of BMI, and BCAA levels are elevated in the serum of individuals with type 2 diabetes28,29. Moreover, increased levels of ornithine and citrulline (Fig. 2A) and elevated expression of branched-chain amino acid transaminases (BCAT1 and BCAT2) (Fig. 2B, C) indicate an upregulated urea cycle. These results suggest that amino acids were utilized as substrates for the TCA cycle and gluconeogenesis, compensating for impaired fatty acid uptake in the kidney and liver. Importantly, the NADH/NAD ratio increased, while the energy charge, calculated as ATP + ½ ADP/ATP + ADP + AMP, decreased in the I-PpoI mice (Fig. 2E). These findings suggested that the I-PpoI mice exhibited impaired fatty acid metabolism and were unable to meet their energy requirements, leading to metabolic reprogramming and subsequent changes in key metabolites in the kidney cortex and liver.

A The metabolic steps of glycolysis, the TCA cycle (upper left) and the urea cycle (upper right). Red and light red squares show that the levels of each metabolite increased in the I-PpoI mice (Tg) (p < 0.01 or < 0.05, respectively); blue and light blue squares show that the levels of each metabolite decreased in the I-PpoI mice (p < 0.01 or < 0.05, respectively). (lower) The apparent contents per tissue weight of representative metabolites involved in glycolysis and the TCA cycle [nmol/g tissue weight]. B, C Real-time RT‒PCR analysis of key rate-limiting enzymes in the glycolytic pathway, such as HK, PFK, PK, BCAT1 and BCAT2 and gluconeogenic enzymes including G6Pase, PEPCK and PCK in the kidney (B) and the liver (C). D The apparent contents per tissue weight of essential amino acids in the kidney and the liver [nmol/g tissue weight]. E The apparent ratio of NADH/NAD in the kidney and liver (left). The apparent energy charge was calculated by the following equation: apparent energy charge = (ATP + [0.5 × ADP])/(ATP + ADP + AMP) (right). All values are presented as the means ± SEMs. *p < 0.05, **p < 0.01 vs. the control; N.S., not significant. n = 6 samples in each group. Source data are provided as a Source Data file.
DNA damage in PTECs causes mitochondrial dysfunction in cultured tubular cells
To quantitatively analyze the metabolic changes caused by DNA damage in PTECs in vitro, we used a flux analyzer to evaluate the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), which are indicators of glycolysis and mitochondrial respiration, respectively, in cultured PTECs, immortalized cell-line, overexpressing I-PpoI with increased DNA damage as confirmed by the long-distance PCR method (Fig. 3A). The I-PpoI-expressing PTECs exhibited increased NGAL and KIM-1 expression as well as decreased CPT1a expression, as observed in the I-PpoI mice (Fig. 3B). Cultured PTECs transfected with I-PpoI had a reduced OCR at baseline and decreased ATP production, as measured by a Seahorse Extracellular Flux Analyzer (Fig. 3C). The fluoro carbonyl cyanide phenylhydrazone (FCCP)-induced maximal respiration and spare respiration capacity were markedly reduced in the cultured PTECs overexpressing I-PpoI (Fig. 3D). It was also revealed that the capacity of fatty acid oxidation (FAO) was reduced in cultured PTECs with increased DNA damage (Figs. 3E, F, and Supplementary Fig. 3A), although fatty acid was the main source of energy production in healthy PTECs. Then, the mitochondrial DNA copy number was evaluated, indicating that significant reduction in the kidney and the liver of the I-PpoI mice compared to controls (Fig. 3G). Furthermore, the activities of electron transport chain complexes were decreased, accompanied by reduced ATP production and impaired expression of PGC-1α, a key regulator of mitochondrial biogenesis (Fig. 3H). These results indicate that I-PpoI-induced DNA damage disrupts mitochondrial respiratory capacity in the hepatocytes as well as PTECs, leading to metabolic reprogramming, consistent with the previously reported age-related mitochondrial dysfunction associated with insulin resistance.

A Schematic diagram outlining the experimental design for cultured PTECs. Created in BioRender. https://BioRender.com/egn26ya (B) Real-time RT‒PCR analysis of biomarkers of renal proximal tubule injury and the rate-limiting enzyme involved in fatty acid β-oxidation in cultured PTECs overexpressing I-PpoI and controls. C OCR of cultured PTECs overexpressing I-PpoI and controls measured in a Seahorse XF24 system. Oligomycin, an inhibitor of ATP synthesis (1.5 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), a mitochondrial oxidative phosphorylation uncoupler (1.0 µM) and rotenone/antimycin A (0.5 μM), a complex I/III inhibitor, were injected sequentially at the indicated time points. D Basal and maximal OCRs, ATP production and storage respiration capacity in cultured PTECs overexpressing I-PpoI and controls. E OCR of cultured PTECs overexpressing I-PpoI and controls measured in a Seahorse XF24 system. Etomoxir, an irreversible inhibitor of CPT1 (4 µM), Oligomycin (1.5 μM), FCCP (1.0 µM) and rotenone/antimycin A (0.5 μM) were injected sequentially at the indicated time points. F Fatty acid oxidation (FAO) dependency in cultured PTECs overexpressing I-PpoI and controls. G Mitochondrial DNA copy number in the kidney and the liver. H Real-time RT‒PCR analysis of PGC-1α, a key regulator of mitochondrial biogenesis in the kidney and the liver. In A, G and H, n = 6 samples in each group. In B, E and F, n = 4 samples in each group. In C and D, n = 8 samples in each group. Each data point represents the mean ± SEMs. *p < 0.05, **p < 0.01 vs. the control. Source data are provided as a Source Data file.
DNA damage in PTECs activates and promotes the proliferation of CD11c+ and Ccr2+ macrophages in the kidney cortex
As DNA DSBs in PTECs cause elevated expression of inflammatory and aging factors in the kidney, single-cell RNA sequencing (scRNA-seq) was performed on the renal cortex of 16-week-old I-PpoI mice to investigate alterations in cell populations in the kidney after DNA DSBs in PTECs. We visualized the single-cell datasets using UMAP and identified 10 cell-type clusters, which were classified using established cell-specific markers of murine kidneys as previously described (Fig. 4A)30,31. The total numbers of cells were 7803 and 4719 in the WT mice and the I-PpoI mice, respectively. In the I-PpoI mice, the proportion of PTECs was reduced, and the expression of Egf, a biomarker of tubular cell health, was reduced. By repeating the UMAP clustering analysis of macrophages, dendritic cells (DC), monocytes and neutrophils, we found marked expansions of Ccr2hi CD11c+ macrophages and inflammatory Ly6Chi Ccr2+ monocytes in the renal cortex of the I-PpoI mice. Neutrophils were also increased in the renal cortex of the I-PpoI mice (Fig. 4B, C). Ccr2hi CD11c+ macrophages were reported to originate from recruited Ly6Chi Ccr2+ monocytes30, which are associated with increased levels of circulating proinflammatory cytokines such as TNF, IL-1β and Ccr2. CD11c+ macrophages were shown to play an important role in metabolic disturbances and insulin resistance in adipocytes32. CD11b expression was also increased in DCs, monocyte and neutrophil clusters (Fig. 4C). Immunohistochemical staining also revealed that CD11b+ cells and CD11c+ cells were increased in the kidney, the liver, and visceral adipose tissue and that the expressions of Ccr2 and Ly6C were elevated in the kidney and visceral adipose tissue of the I-PpoI mice (Figs. 4D, E, and Supplementary Fig. 4A, B). Fluorescence-activated cell sorting (FACS) analysis revealed increased numbers of CD11b+ and CD11c+ macrophages in the kidney and visceral adipose tissue, as well as an increased population of Ly6Chi CD11b+ monocytes in the peripheral blood cells of the I-PpoI mice compared to those in the peripheral blood cells of the WT mice (Fig. 4F). In order to further elucidate the mechanism responsible for metabolic derangement in the I-PpoI mice, the characteristics of adipose tissue macrophages (ATM) defined by F4/80+ and CD11b+ isolated from the I-PpoI mice, ob/ob mice and wild-type controls at 16 weeks of age were analyzed. Interestingly, even though the increased proportion of CD11c+ cells was observed in the ATMs of both the I-PpoI mice and ob/ob mice (Supplementary Fig. 3B), ATMs in the I-PpoI mice exhibited different gene expression profiles compared to those seen in ob/ob mice. In ATMs isolated from obese adipose tissue, trends toward the up-regulation of most of M1-associated genes and the down-regulation of M2-associated genes were observed. On the other hand, in those isolated from the I-PpoI mice, M1- associated genes were mildly upregulated and M2- associated genes were randomly regulated in a different way from that in ob/ob mice (Supplementary Fig. 3C). Moreover, similarly to lipodystrophic mice, which exhibit insulin resistance and glucose intolerance in lean phenotypes, the proportion of CD40+ cells in F4/80+ CD11b+ ATMs was not increased in the I-PpoI mice, in contrast to the marked increase observed in ob/ob mice (Supplementary Fig. 3D). These findings suggest that PTEC DNA damage causes the activation of macrophages and monocytes not only in the kidney but also in the peripheral circulation, leading to the change in systemic immune environment in a different way from that of obesity. Furthermore, F4/80+ CD11b+ ATMs of 16-week-old I-PpoI mice, the increased expression of Trem2 was observed (Supplementary Fig. 3E), which are reported to be observed in aged humans and are hallmarks of lipid-associated macrophages involved in inflammation and fibrotic remodeling33. The results are consistent with the promoted fibrosis observed in visceral adipose tissue of 1-year-old I-PpoI mice compared to control mice of the same age (Supplementary Fig. 1H).

A UMAP plots of the single-cell data classified by cell clusters (left), relative proportions of cells in the I-PpoI (Tg) and WT mice (middle left) and sample information (middle right). Expression of selected marker genes for each cell classification (right). The color of the dot is proportional to the average expression value, and the size is expressed as a percentage. B UMAP plots of subclusters of macrophages (left) and proportions of the subgroups in the I-PpoI and WT mice (middle left). C UMAP plots of subclusters of DCs, monocytes and neutrophils (left) and proportions of the subgroups in the I-PpoI and WT mice (middle left). Expression of selected marker genes for each classification (middle right). The color of the dot is proportional to the average expression value, and the size is expressed as a percentage. Violin plots showing the expression levels of selected genes characterized in the clusters (right). The y-axis shows the log-scale normalized read count. D, E Representative immunohistochemical staining images with an anti-CD11b antibody (D) an anti-CD11c antibody (E) and in the kidney cortex (a), the liver b and VAT c at 16 weeks of age. F Representative FACS plots of the kidney cortex a, VAT b, and peripheral blood mononuclear cells (PBMCs) (c) of I-PpoI and WT mice at 16 weeks of age (left). Proportions of CD11b+ or CD11c+ macro`phages in the kidney and VAT and those of CD11b+ Ly6chi monocytes in PBMCs, as assessed by flow cytometry (right). In a–c n = 4 samples in each group. The data are presented as the means ± SEMs. *p < 0.05, **p < 0.01 vs. the control. Scale bars, 20 µm (left) and 100 µm (middle and right) in D and E. The same results were obtained by four different samples in each group. Source data are provided as a Source Data file.
DNA methylation of the KLF9-binding motif is decreased in the I-PpoI mice
We next examined the effects of I-PpoI-induced DSBs on changes in DNA methylation in peripheral blood cells of the I-PpoI mice. According to reduced representation of bisulfite sequencing (RRBS) analysis, a total of 5,488 significant differentially methylated regions (DMR) were identified in the peripheral blood of the I-PpoI mice. DMRs were detected in pathways related to amino acid metabolism, insulin resistance and secretion (Fig. 5A, B). Notably, using motif analysis, we identified a Krüppel-like factor 9 (KLF9)-binding motif in hypomethylated DMRs in the peripheral blood of the I-PpoI mice (Fig. 5C). Gasdermin D (GSDMD), a target gene of the transcription factor KLF9, which is reportedly involved in macrophage activation, was confirmed to be hypomethylated (Fig. 5D). These results suggest that DNA damage in PTECs causes altered DNA methylation in peripheral blood cells related to macrophage activation.

A Significant signaling pathways of hypermethylated DMRs specifically in blood cells of the I-PpoI mice (Tg) compared to those in blood cells of WT mice. B Significant signaling pathways of hypomethylated DMRs specifically in blood cells of the I-PpoI mice compared to those in blood cells of WT mice. C The top 5 enriched motifs in hypomethylated DMRs, including promoter regions and CpG islands, of blood cells in the I-PpoI mice according to regulatory sequence analysis tools. For each motif, the corresponding Bonferroni-adjusted p value (E value) and the number of input sequences containing the specific motif (n in parenthesis) are listed on top of the motif logo. The known JASPAR transcription factors are listed below the corresponding logo. D Relative methylation rate of Gasdermin D (GSDMD) in blood cells of the I-PpoI mice compared to those in blood cells of WT mice. **p < 0.01 vs. the control. Source data are provided as a Source Data file.
DNA damage in cultured PTECs changes adipocyte phenotypes through activated macrophages
To confirm the potential role of activated macrophages in the altered phenotypes of adipocytes, we conducted an in vitro analysis using cultured murine RAW 264.7 macrophages that had been transferred into the medium of murine PTECs following transfection with I-PpoI and murine adipocytes cultured in conditioned medium from the activated macrophages (Fig. 6A). RAW 264.7 macrophages cultured in the medium of PTECs overexpressing I-PpoI for 48 h exhibited increased expression of inflammatory cytokines such as IL-1β, TNFα and Ccr2. As shown in the I-PpoI mice, GSDMD and KLF9 expression was increased in activated macrophages (Fig. 6B), which was associated with decreased DNA methylation of the GSDMD promoter region (Fig. 6C). To further elucidate the precise mechanism underlying DNA damage-induced macrophage activation, we investigated the release of damage-associated molecular patterns (DAMPs) from DNA-damaged PTECs, as these molecules have strong potential to activate macrophages. Our results showed that the concentration of high mobility group box protein-1 (HMGB1), a well-known DAMP34, was significantly elevated in the culture supernatant of I-PpoI-transfected PTECs compared to that of controls (Fig. 6D). Next, the promoter activity of the GSDMD gene with or without DNA methylation was examined using a CpG-free plasmid vector system, which suggested that promoter methylation of the investigated region plays a functional role in the control of GSDMD promoter activity (Fig. 6E). Regarding the factors mediating changes in DNA methylation, we focused on exosomes released by injured PTECs. Interestingly, we observed decreased mRNA expression of DNA methyltransferase (DNMT)1 and DNMT3A not only in exosomes released by cultured DNA-damaged PTECs but also in RAW 264.7 macrophages cultured with the exosomes (Fig. 6F).

A Schematic diagram outlining the experimental design for cultured adipocytes in conditioned medium from activated murine macrophage RAW 264.7 cells that had been transferred into the medium of murine PTECs following transfection with I-PpoI. Created in BioRender. https://BioRender.com/7yyff15 (B) Real-time RT‒PCR analysis of inflammatory cytokines such as IL-1β, TNFα, Ccr2, GSDMD and KLF9 in the RAW 264.7 cells. C Real-time RT‒PCR analysis of DNA methylation levels of the GSDMD promoter region in the RAW 264.7 cells. D The concentration of high mobility group box protein-1 (HMGB1) in the culture supernatant of cultured murine PTECs. E Relative promoter activity of pCpG-free promoter plasmid containing GSDMD promoter without or with methylase SssI (−/+). (F) a Real-time RT‒PCR analysis of DNA methyltransferases (DNMTs) in exosomes released by cultured murine PTECs following transfection with I-PpoI. b Real-time RT‒PCR analysis of DNMTs in the murine RAW 264.7 macrophages cultured with the exosomes. G Representative photomicrographs of IF staining of NT-GSDMD (green) in the RAW 264.7 cells. Scale bar, 10 µm. The experiment was repeated with similar results three times. H IL-1β levels in the supernatant of the RAW 264.7 cells to which the supernatant of cultured murine PTECs with or without DNA damage was added. I Real-time RT‒PCR analysis of inflammatory cytokines such as IL-1β and TNFα and genes involved in lipolysis and adipose differentiation in the cultured murine adipocytes in conditioned medium with the supernatant of macrophages activated by PTECs transfected with I-PpoI. The data are presented as the means ± SEMs. In B, (D–H) and I, n = 6 samples in each group. In C, n = 4 samples in each group. *p < 0.05, **p < 0.01 vs. the control; N.S., not significant. Source data are provided as a Source Data file.
Moreover, GSDMD is one of the important factors in pyroptosis by facilitating its cleavage and pore formation in the cell membrane of macrophages, which enables the release of IL-1β35. In vitro analysis showed that the N-terminal cleavage product (GSDMD-NT) was localized to the plasma membrane of murine macrophages that had been transferred into the culture medium of murine PTECs with DNA damage (Fig. 6G). Also, IL-1β levels were elevated in the supernatant of macrophages to which the supernatant of PTECs with DNA damage was added (Fig. 6H). Subsequently, we cultured mouse adipocytes derived from OP9 cells, a line of bone marrow-derived mouse stromal cells, in conditioned medium from the activated macrophages. This procedure led to increased expression of inflammatory cytokines such as IL-1β and TNFα, while the expression of PPARγ and CEBPa decreased, indicating inflammation and impaired differentiation of adipocytes (Fig. 6I). The alterations in the phenotype of cultured mouse adipocytes were not observed when adipocytes were directly cultured in the medium of murine PTECs with increased DNA damage (Supplementary Fig. 3F). Collectively, these findings suggest that DAMPs such as HMGB1 released from DNA-damaged PTECs activates macrophages and induces pyroptosis through DNA methylation changes and that inflammatory cytokines, such as TNFα and IL-1β, secreted by activated macrophages may impair adipocyte differentiation and function.
Macrophage depletion restored metabolic alterations in the I-PpoI mice
To investigate the pathogenic effects of inflammatory macrophages on metabolic alterations in the I-PpoI mice, we performed macrophage depletion using clodronate liposomes in 12-week-old I-PpoI mice (Fig. 7A). Administration of clodronate reduced the number of inflammatory CD11c+ macrophages and Ly6Chi Ccr2+ monocytes in both the I-PpoI mice and the WT mice, as confirmed by FACS analysis (Fig. 7B). Body weight decreased in both the I-PpoI mice and the WT controls (Fig. 7C). Macrophage deletion caused no significant differences in the serum BUN or Cr levels between the I-PpoI mice and the controls, while the serum triglyceride levels were markedly decreased in the I-PpoI mice (Fig. 7D). Furthermore, impaired glucose tolerance was restored one week after macrophage depletion (Fig. 7E). Depletion of macrophages also markedly reduced TNF-α and IL-1β expression in the liver and visceral adipose tissue of the I-PpoI mice (Fig. 7F, G). These findings suggested an important role for activated macrophages in metabolic alterations in the I-PpoI mice.

A Schematic diagram outlining the experimental design for macrophage depletion. Created in BioRender. https://BioRender.com/442s6k4 (B) Representative FACS plots of the kidney cortex a, VAT b, and PBMCs c of the I-PpoI (Tg) and WT mice at 16 weeks of age with or without clodronate (left). Proportions of CD11b+ and CD11c+ macrophages in kidney and visceral adipose tissue and of CD11b+ Ly6chi monocytes in PBMCs, as assessed by flow cytometry (n = 3, 3). C Time course of body weight. D Serum BUN, Cr and TG levels of the I-PpoI and WT mice at 16 weeks of age with or without clodronate. E (left) ip-GTT and AUC for glucose values 0–120 min post glucose load of the I-PpoI and WT mice at 16 weeks of age with or without clodronate. (right) Fasting serum insulin levels of the I-PpoI and WT mice at 16 weeks of age with or without clodronate. F Real-time RT‒PCR analysis of proinflammatory markers in the liver. G Real-time RT‒PCR analysis of proinflammatory markers in visceral adipose tissue. In C–G, n = 4 samples in each group. The data are presented as the means ± SEMs. *p < 0.05, **p < 0.01 vs. the control; N.S., not significant. Source data are provided as a Source Data file.
Promotion of DNA DSBs in PTECs was associated with increased numbers of CD11b+ and CD11c+ cells, accompanied by reduced DNA methylation of GSDMD in peripheral blood in humans
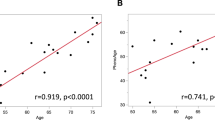
Next, the association between DNA DSBs in PTECs and macrophage activation was investigated using human samples. DNA DSBs in PTECs were detected by double immunostaining with anti-gH2AX, and immunohistochemical staining with anti-CD11b or anti-CD11c antibodies was performed on kidney biopsy samples from patients diagnosed with diabetic nephropathy or minor glomerular abnormalities (Fig. 8A, B). The patient profiles included in the study are listed in Supplementary Table 1. PTEC DNA DSB levels were positively correlated with CD11b+ and CD11c+ areas (Fig. 8B). In addition, the association between PTEC DNA DSBs and peripheral blood DNA methylation was examined. The profiles of the CKD patients included in this study are listed in Supplementary Table 2. DNA methylation of peripheral blood cells was assessed using the Infinium Methylation EPIC array, and DNA DSBs in the SGLT2 gene, which were used to indicate PTEC DNA damage, were evaluated using long-distance PCR of shed urinary cells. When evaluating DNA methylation in the promoter region of GSDMD, we observed that DNA methylation at the CpG site cg22012530 in the TSS1500 region was inversely associated with the extent of PTEC DNA damage (Fig. 8C), which is consistent with the results of the in vitro study. Furthermore, in the analysis of nine patients except ESKD listed in Supplementary Table 2, DNA methylation at the GSDMD promoter region tended to be negatively correlated with the hepatic steatosis index (HSI) (Fig. 8C).

A (Left) Representative photomicrographs of IF double staining of γH2AX (green)/AQP1 (red) in the kidney cortex of patients with DN (a) or MGA (b). Scale bar, 50 µm. (right) Quantification of the immunolabeled double-positive area. B (left) Representative photomicrographs of immunohistochemical staining with anti-CD11b or anti-CD11c antibodies in the kidney cortex of patients diagnosed with DN (a) or MGA (b). Scale bar, 50 µm. (right) Correlation between the γH2AX-positive area and the CD11b- or CD11c-positive area. γH2AX-, CD11b- and CD11c-positive areas were counted per field of view. Scale bar, 50 µm. C (a) Correlation between the extent of DNA damage in PTECs using the DNA damage in the SGLT2 gene and DNA methylation at the CpG site cg22012530 in the TSS1500 region. b Correlation between DNA methylation at the CpG site cg22012530 in the TSS1500 region and the hepatic steatosis index (HSI). D Graphical abstract of the study. DNA DSBs in PTECs cause not only metabolic alterations in the kidney but also DNA methylation in blood cells, including reduced DNA methylation at KLF9-binding motifs, such as GSDMD, which triggers the expansion of activated Ly6Chi Ccr2+ monocytes in blood. The activated monocytes subsequently trigger systemic metabolic disturbances associated with macrophage activation. Created in BioRender. https://BioRender.com/rrfwed6. In A and B, n = 5 samples in each group. In C, n = 12 samples in each group. The clinical data are presented in Supplementary Table 1 and 2, respectively. The data are presented as the means ± SEMs. **p < 0.01 vs. the control. Source data are provided as a Source Data file.
Discussion
The present study elucidates the systemic metabolic consequences of continuous nonmutagenic DNA damage in PTECs, underscoring a aspect of kidney-cell DNA damage affecting overall body metabolism. Our findings revealed that PTEC-specific expression of I-PpoI, which induces DNA DSBs, not only disrupts local renal metabolic homeostasis but also precipitates broader metabolic disturbances through macrophage activation associated with altered DNA methylation (Fig. 8D). This systemic impact highlights the integral role of the kidney in maintaining metabolic equilibrium.
The importance of PTEC DNA damage in various kidney diseases, including cisplatin nephropathy, ischemia‒reperfusion injury (IRI), and diabetic nephropathy, has been reported, and the repair pathways of PTECs have been suggested to be possible therapeutic targets36,37,38,39. PTEC DNA damage is significantly associated with a decrease in the eGFR39. However, using these pathological models, it was difficult to clarify the systemic effects of PTEC DNA damage itself.
In our study, along with DNA DSBs, PTECs exhibited downregulated expression of key enzymes related to fatty acid metabolism. Previous studies have indicated that PTEC injury induces impaired fatty acid metabolism in PTECs. CPT1, the key rate-limiting enzyme in fatty acid oxidation, is reportedly decreased in a murine IRI model, and the increase in CPT1 alleviated renal damage40. In humans, decreased CPT1a expression has been reported via transcriptome analysis of tubulointerstitial microdissection samples from diabetic and hypertensive CKD patients41 and is associated with the progression of decreased eGFR and renal fibrosis42. Additionally, inhibition of CPT2 has been reported to induce lipid toxicity and sustained cellular stress43, and lipid toxicity-induced PTEC injury could be a major event in diabetic nephropathy44. Thus, metabolically active proximal tubules, which are dependent on fatty acid oxidation, are interconnected with cell damage and lipid metabolic abnormalities. Our metabolomic analysis further revealed mitochondrial dysfunction resulting from DNA damage in PTECs. DNA damage activates numerous proteins, leading to extensive downstream changes in various cellular pathways. Among these pathways, nuclear-to-mitochondria signaling (NM signaling) involves factors such as PARP1, ataxia telangiectasia mutated (ATM), and the transcription factor p53, which contribute to the development of age-related diseases such as aging, neurodegeneration, and cancer45. When the electron transport chain (ETC) activity of damaged mitochondria decreases, the amount of nicotinamide adenine dinucleotide (NAD) necessary for glycolysis decreases, impairing the continuous production of ATP from the oxidation of fatty acids or glucose. Nicotinamide mononucleotide (NMN), a precursor of NAD, has shown therapeutic potential for AKI in mouse models46,47. Therefore, DNA damage in PTECs could trigger lipid metabolic abnormalities, consistent with previous findings.
In the I-PpoI mouse model, PTEC DNA damage changes not only renal metabolism but also systemic metabolism, including weight loss, reduced fat mass, increased liver fat, and impaired glucose tolerance, resembling changes associated with aging48. These changes are reminiscent of those observed in aging cells, where mitochondrial dysfunction and altered energy metabolism are prevalent. In vitro experiments with cultured PTECs overexpressing I-PpoI showed that the impairment in mitochondrial function, as evidenced by reduced OCR and ECAR, aligns with the metabolic shifts observed in vivo. This mitochondrial dysfunction, potentially linked to aging-associated insulin resistance, reinforces the idea that PTEC DNA damage may strongly contribute to systemic metabolic aging. In general, aging is associated with increased fat mass or obesity until advanced old age49, whereas extremely aged individuals have decreased fat mass and may display a metabolically-unhealthy phenotype50, suggesting a complex relationship between adiposity and metabolic health during aging. Recent reports indicate that deactivation of PPARγ function could drive a metabolically-unhealthy lean phenotype51,52,53. Therefore, the metabolic phenotype of continuous PTEC DNA damage may be caused through a decrease in fat PPARγ expression due to adipose inflammation, leading to impaired adipocyte differentiation and ectopic fat accumulation. The accumulation of ectopic fat in the liver is thought to be a cause of metabolic disorders, including hepatic insulin resistance54,55,56.
Single-cell analysis of the renal cortex in the I-PpoI mice revealed the infiltration of Ccr2hi CD11c+ macrophages. Macrophages are traditionally classified into two functionally opposing phenotypes: the classically activated proinflammatory (M1) phenotype and the alternatively activated anti-inflammatory (M2) phenotype. Although there is no established classification for kidney macrophages, recent single-cell analysis results suggest that in kidney injury and disease, such as acute kidney injury and diabetic nephropathy, M1 macrophages are deleterious and that M2 macrophages facilitate recovery57,58,59. In contrast, CD11c+ macrophages have been identified in the renal cortex after ischemia‒reperfusion, and their contribution to tissue repair has been reported60. Interestingly, exposure to tumor cell-derived DNA through CD11c-mediated endocytosis has been shown in mature human THP-1 cells61. Repetitive DNA damage in PTECs suggests the potential contribution of endocytosis of PTEC-derived DNA fragments via CD11c to the activation of monocytes and macrophages. Furthermore, our findings indicate that DAMPs released from DNA-damaged PTECs contribute to macrophage activation. In contrast, DNA-damaged podocytes exhibit increased accumulation of cytoplasmic dsDNA and preferentially activate T cells15, highlighting cell type-specific differences in immune responses. Ccr2+ macrophages are infiltrating macrophages derived from monocytes mobilized from the peripheral circulation in response to C-C motif chemokine ligand 2 (CCL2)62. An increase in inflammatory monocytes in the peripheral circulation was also observed in the I-PpoI mice, suggesting that the Ccr2+ macrophages in the kidneys are derived from monocytes. Interestingly, in the heart and liver, during aging, embryonic-derived resident macrophages are replaced by monocyte-derived macrophages63. The increase in Ccr2+ macrophages in response to DNA damage in PTECs may parallel the macrophage transition observed in aging tissues. This observation is particularly intriguing because it offers a cellular mechanism for inflammageing—a chronic low-grade inflammation state associated with aging—in the kidney and systemic organs.
The program of differentiation and activation from monocytes to macrophages involves crucial epigenetic remodeling associated with DNA methylation64. In blood cells from the I-PpoI mice, a decrease in methylation at the binding site of KLF9, including the GSDMD gene, was observed, which is implicated in macrophage activation34,65. Interestingly, a GWAS identified a correlation between GSDMD gene mutation and diabetes, indicating a potentially important role for GSDMD in impaired glucose intolerance66,67. Our results indicate that the macrophages activated by DNA-damaged PTECs, may upregulate GSDMD expression through alterations in DNA methylation, and promote pyroptosis and inflammation.
In the I-PpoI mice, an increase in CD11c-positive macrophages was observed not only in the kidneys but also in metabolic tissues such as adipose tissue and the liver. This finding implies that changes in systemic inflammatory macrophages, mediated by the methylation of blood cells originating from DNA-damaged PTECs, may be involved in metabolic alterations. Insulin resistance is associated with adipose tissue macrophages (ATMs), and inflammatory CD11c-positive macrophages are considered a marker of insulin resistance68. TNFα secreted by ATM induces PPARγ downregulation, leading to adipogenic dysfunction, promoting lipolysis, and increasing circulating TG and FFA levels, thereby contributing to insulin resistance. Notably, the surface markers and gene expression profiles of ATMs in the I-PpoI mice differed from those in obese mice but were similar to those observed in lipodystrophic mice, showing decreased fat mass, insulin resistance, dyslipidemia, hepatic steatosis, and an increased risk of diabetes and atherosclerosis69. There had been few reports on the detailed profile of macrophages infiltrating adipocytes in lean elderly individuals, but a very recent paper has shown the expression of ATMs of non-obese aged humans33. Interestingly, in the ATMs of 16-week-old I-PpoI mice, the increased expression of Trem2 was observed, which are reported to be observed in aged humans and are hallmarks of lipid-associated macrophages involved in inflammation and fibrotic remodeling. The pivotal role of activated macrophages in mediating these systemic changes is underscored by the reversal of metabolic alterations upon macrophage depletion in the I-PpoI mice. This intervention suggests that targeting macrophage activation and the subsequent inflammatory cascade could be promising therapeutic strategies for managing systemic metabolic disturbances caused by DNA damage in PTECs.
Moreover, this study has implications for human pathology. To date, it has been reported that in CKD mouse models and patients, chronic inflammation is triggered in visceral fat, which may lead to a reduction in the visceral fat mass and potentially increase the risk of cardiovascular disease70,71,72. In this study, we elucidated that increased insulin resistance may be significantly influenced by macrophage activation subsequent to tubular DNA damage, with DNA methylation changes playing a role. The correlation between DNA DSBs in PTECs and increased CD11b+ and CD11c+ cells in human kidney biopsy samples, along with changes in DNA methylation patterns in peripheral blood cells, mirrors our findings in the mouse model. Moreover, DNA methylation of the GSDMD gene was associated with hepatic steatosis index (HSI), suggesting that CKD with PTEC DNA damage may predispose individuals to insulin resistance and hepatic steatosis through activated macrophages with altered DNA methylation, providing valuable insights into the prediction of future metabolic disorders and cardiovascular complications. The association between DNA methylation changes in blood cells and renal function has already been reported in many human EWASs73. In particular, DNA methylation changes in blood cells associated with renal prognosis in diabetic nephropathy patients and diabetic complications have been reported74,75. In addition, a recent study suggested that PTEC DNA damage was associated with age acceleration, as evaluated by epigenetic clocks using blood cell DNA methylation profiles76. Although DNA methylation changes in peripheral blood cannot be conclusively established as either the primary cause or a consequence of macrophage activation, the DNA methylation changes observed following PTEC DNA damage may be implicated in enduring metabolic alterations, peripheral blood DNA methylation changes are considered valuable predictive markers for assessing the extent of DNA damage in PTECs and the systemic metabolic disorders they may induce. Further studies are necessary to evaluate its potential as a therapeutic target for other organ complications of CKD with PTEC DNA damage.
In conclusion, our study provides compelling evidence that DNA damage in PTECs can initiate a cascade of events leading to systemic metabolic alterations. These findings underscore the potential impact of DNA damage in PTECs on metabolic aging. Future studies should explore the therapeutic potential of modulating macrophage activation and inflammation in managing metabolic disorders linked to kidney disease with PTEC DNA damage.
Methods
Generation of PTEC-specific I-PpoI-expressing mice
R26R-I-PpoI knock-in mice (accession no. CDB0059E; https://large.riken.jp/distribution/mutant-list.html) were generated via CRISPR/Cas9-mediated knock-in in zygotes as previously described77. First, we generated loxP-STOP-loxP I-PpoI mice. In order to insert loxP-STOP-loxP I-PpoI into the Rosa26 locus of C57BL/6 zygotes, the CRISPR‒Cas9-assisted knock-in system was used (Supplementary Fig. 5). For homologous recombination-mediated knock-in, a donor vector consisting of homology arms and the SA/loxP/PGK-Neo-tpA (STOP)/loxP/I-PpoI cDNA-bpA was generated to insert the cassette 6 bases upstream of the PAM sequence. STOP represents a transcription terminator sequence that is removed by Cre/loxP recombination. For microinjection, a mixture of CRISPR RNA (crRNA) (50 ng/µl), trans-activating crRNA (tracrRNA) (100 ng/µl), donor vector (10 ng/µl) and Cas9 protein (100 ng/µl) was injected into the pronucleus of a zygote at the one-cell stage. From 228 zygotes, 65 F0 mice were obtained, 15 of which were knock-in mice, as confirmed by PCR. PCR was performed by using the following primers: 5’-FW: 5’- TCA GAG AGC CTC GGC TAG-3’ and 5’-RV: 5’-CTA CTG CGC CCT ACA GAT C-3’ (1.2 kbp), and 3’-FW: 5’-GGG GGA TTG GGA AGA CAA TAG C-3’ and 3’-RV: 5’- AGA ACT GCA GTG TTG AGG-3’ (714 bp) (Supplementary Fig. 5). crRNA (5’- CGC CCA UCU UCU AGA AAG ACg uuu uag agc uau gcu guu uug -3’) and tracrRNA (5’-AAA CAG CAU AGC AAG UUA AAA UAA GGC UAG UCC GUU AUC AAC UUG AAA AAG UGG CAC CGA GUC GGU GCU-3’) were purchased from FASMAC. The germline transmission of 3 knock-in F0 mice was confirmed by genotyping F1 mice. For expression of I-PpoI in specific tissues via the Cre/loxP system, R26R-I-PpoI mice were bred with C57BL/6-background γGT-Cre mice expressing Cre recombinase under the control of the γGT promoter (Jackson Laboratory, #: 012841) (γGT-Cre mice), which were obtained from the Jackson Laboratory. Mice were group-housed under standard housing conditions (12 h light/dark cycle, temperature 20–22 °C, humidity 40–60%, and free access to water and food (CLEA, Rodent Diet #:CE-2)). Mice were euthanized using non-narcotic anesthetic combination (Me/Mi/Bu) of medetomidine (Me), midazolam (Mi), and butorphanol (Bu).
In order to confirm the expression of I-PpoI is PTEC-specific, γGT-Cre mice were bred with Rosa-CAG-LSL-tdTomato mice (Jackson Laboratory, #: 007914). Rosa-CAG-LSL-tdTomato is a Cre reporter tool strain designed to have a loxP-flanked STOP cassette preventing transcription of a CAG promoter-driven red fluorescent protein variant (tdTomato) - all inserted into the Gt (ROSA) 26Sor locus. Rosa-CAG-LSL-tdTomato mice express robust tdTomato fluorescence following Cre-mediated recombination. This strain is congenic on the C57BL/6 J genetic background. All γGT-Cre (+) Rosa-CAG-LSL-tdTomato mice were heterozygous. Kidneys, livers, spleens, gastrocnemius muscles and adipose tissues were removed from the littermates and frozen sections were prepared and stained with DAPI. Then they were photographed using a laser confocal microscope (LSM980; Zeiss), and ×20 field images were acquired (Supplementary Fig. 4). 16-week-old male obese (ob/ob) C57BL/6 J mice were purchased from Jackson Laboratory. (B6.Cg-Lepob/J, #:000632)
PTEC isolation
PTECs were isolated by means of a previously described protocol with some modifications78. Briefly, following endothelial cell depletion using anti-CD31 antibodies (BL102503; BioLegend), PTECs were isolated from minced murine kidney tissue with magnet-activated cell sorting using an anti-prominin-1 antibody (130-123-793; Miltenyi Biotec).
Histological studies
The kidneys were removed, fixed in 4% paraformaldehyde, and then embedded in paraffin blocks. The paraffin sections were stained with periodic acid-Schiff (PAS), Masson trichrome (MT) or the indicated antibodies listed in Supplementary Data, followed by incubation with a secondary antibody for immunostaining. Stained paraffin sections were photographed using a laser confocal microscope (LSM980; Zeiss), and five ×20 field images were acquired and, for measuring positive areas, quantified using color channel analysis and pixel counting using Photoshop CC 2024 software (Adobe)79.
Biochemical studies
Mice were housed in a pathogen-free environment under controlled conditions (temperature, 20–26 °C; humidity, 40–70%; light/dark cycle, 12 h/12 h). For urine collection, the mice were individually housed in metabolic cages for 24 h. Urinary albumin was measured with a murine albumin ELIZA kit (Albuwell; Exocell). Urinary Cr levels and serum urea nitrogen (BUN), creatinine, alanine aminotransferase (ALT), alkaline phosphatase (ALP), triglyceride and free fatty acid levels were measured enzymatically.
Impedance methods
The in vivo body composition, including inguinal fat mass, epididymal fat mass, total fat mass and free fat mass, of the I-PpoI and WT mice was determined by bioelectrical impedance analysis (BIA) (ImpediVET, ImprdiMed, Ltd., Brisbane, Australia).
An intraperitoneal glucose tolerance test (ip-GTT) and assessment of insulin resistance
An ip-GTT was performed in the I-PpoI and WT mice at 16 weeks of age. Mice were fasted for 16 h to achieve baseline blood glucose levels. After fasting, the blood glucose levels were measured in blood collected from the tail vein by a glucometer; subsequently, the mice were intraperitoneally injected with 1 g/kg body weight glucose (Sobel et al. 2006). Blood glucose levels were determined after 30, 60, 90, and 120 min.
Real-time quantitative PCR, RNA-seq analysis, and long-distance PCR
RNA was isolated using ISOGEN (Nippon Gene, Japan) according to the manufacturer’s instructions. RNA was reverse-transcribed using TaqMan Reverse Transcription Reagents (Invitrogen). Real-time RT‒PCR was performed using StepOne instruments from Applied Biosystems (Thermo Fisher Scientific). The PCR data were normalized to GAPDH gene expression, and the comparative Ct quantitation method was used. The primers and probes used are listed in Supplementary Data.
For determination of the changes in PTEC gene expression caused by DNA DSBs, RNA-seq analysis was performed. Total RNA isolated from the kidney cortex was used to prepare an RNA-seq library using the TruSeq Stranded mRNA LT Sample Prep Kit according to the TruSeq Stranded mRNA Sample Preparation Guide, part #15031047 Rev. E (Illumina, San Diego, CA, USA). The libraries were sequenced for 100 cycles (paired ends) using a NovaSeq 6000 instrument (Illumina). The sequence reads from each RNA-seq library were mapped onto the mouse genome (mm9) using HISAT2 version 2.2.11, and the number of sequence reads for each gene was calculated using featureCounts version 1.5.2 of the Subread package2. DEGs, heatmaps, and PCAs were analyzed using the DESeq2 package version 1.28.13. The enrichment of GO biological processes and the correlation network of enriched GO terms were analyzed with the clusterProfiler package, version 3.16.14, with p values of 0.01 and 0.05 and q-values of 0.05 (BH methods).
DNA was extracted with a NucleoSpin Tissue Kit (TaKaRa, Japan). The quantitative long-distance PCR method used to detect DNA damage is based on the assumption that DNA with fewer lesions will amplify to a greater extent than more damaged DNA if equal amounts of DNA from different samples are amplified under identical conditions, as described previously80. For evaluation of the cutting efficiency of I-PpoI, DNA from PTECs isolated from I-PpoI mice was analyzed by qPCR using primers spanning the I-PpoI site on the rDNA (Supplementary Data). The data were normalized to the relative DNA level based on the average Ct value of three non-I-PpoI-associated genomic DNA segments (Chr2-Ppp1r1c, Chr6-Hoxa9, and Chr7-H19-ICR; Supplementary Data), and subsequently normalized to the relative DNA level of peripheral blood cells of I-PpoI mice, as described previously21.
Capillary electrophoresis-based western blotting using the WES system
Capillary Western analyzes were performed using the Protein Simple WES System. Samples were diluted with 0.1× Sample Buffer and mixed with 5× Fluorescent Master Mix (containing 5× sample buffer, 5× fluorescent standard and 200 mM DTT) at a ratio of 4:1. Mixed samples were heated at 95 °C for 5 min. Primary antibodies were diluted at a 1:25 dilution for PPARγ (#2435; mouse monoclonal; Cell Signaling Technology) and a 1:50 GAPDH (#2118; mouse monoclonal; Cell Signaling Technology). The size-based assay was then performed in a WES instrument as described in the manufacturer’s instruction. The capillaries were filled with separation matrix for 200 s, stacking matrix for 15 s and sample for 9 s. Separation was then performed at 375 V for 25 min in each capillary. After separation, the capillaries were exposed to ultraviolet light for 200 s. Subsequently, the matrix was removed and washed with washing buffer three times. The capillaries were then blocked with antibody dilution buffer for 5 min. Finally, the capillaries were incubated with primary and secondary antibodies for 30 min, respectively. The signal was captured at 15 s and calculated by Compass software.
Indirect calorimetry (comprehensive lab animal monitoring system)
The rates of oxygen consumption (VO2) and carbon dioxide production (VCO2) and food intake were determined by using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments). Mice were individually placed into the sealed metabolic chambers with free access to food and water. The study was carried out in an experimental room with 12-hour-dark (18:00–6:00) and 12-hour-light (6:00-18:00) cycles. Respiratory quotient (RQ) was calculated as the ratio of VCO2 to VO2 (VCO2/VO2).
Representation of bisulfite sequencing (RRBS) analysis
RRBS analysis was performed according to the manufacturer’s instructions (Rhelixa). In brief, DNA was extracted and adjusted to a concentration of ≥ 30 ng/µL, OD 260/280 ≥ 1.8 and OD260/230 ≥ 1.0. Then, for methylation analysis of CpG regions, library preparation was performed according to the Zymo Research Zymo-Seq RRBS Library Kit (Cat No. D5460/5461) manual (Ver. 1.0.0), i.e., enrichment of CpG-rich regions by fragmentation of genomic DNA using the restriction enzyme Mspl, addition of the TruSeq adapter, bisulfite treatment and addition of index sequences. The enriched DNA samples were amplified, and next-generation sequencing was performed. The methylated regions were identified using the MACS peak finding algorithm with a cutoff of p = 1e-7. Regions of DNA methylation that were different from one another were identified using a peak signal fold change. The cutoff criteria for was fold change ≥ 2 for hypermethylation and ≤ 0.5 for hypomethylation, as described previously81.
De novo motif analysis was performed using 15 hypermethylated DMRs, including CpG islands and promoter regions. The sequences of the resulting genomic regions were used as input in Motif Analysis of Large Nucleotide Datasets (MEME, https://meme-suite.org/meme/tools/meme-chip) to determine enriched motifs using default settings. The statistical significance of a motif identified by MEME was indicated by an E value < 0.05.
Single-cell RNA sequencing analysis
The digestion buffer was a mixture of Dulbecco’s PBS with collagenase type II (3 mg/ml, Worthington), ProNase E (1.5 mg/ml, Millipore), DNase (62.5 U/mL, TaKaRa), and 5 mM calcium chloride. Kidneys were harvested, minced into 1 mm3 cubes, and digested using digestion buffer with shaking at 37 °C for 20 min. Then, the digestion mixture was incubated in a thermomixer for 5 min at 37 °C with shaking at 1200 RPM. The solution was successively passed through 70- and 30-μm cell strainers. The solution was then centrifuged at 300 × g for 5 min, and the resulting cell pellet was diluted with DMEM–high-glucose media (Gibco) supplemented with 10% FBS (Sigma‒Aldrich). The solution was passed through 30-μm cell strainers three times, and a single-cell suspension was obtained. The single-cell suspension was loaded onto a chip on a 10× Chromium Single Cell instrument (10× Genomics). Bar coding and cDNA amplification were performed using Chromium Single Cell 3’Reagent Kits v3.1 (10× Genomics) according to the manufacturer’s instructions. Sequencing was performed on an MGI tech DNBSEQ-G400 at Azenta Japan Corp. Cell Ranger v3.1 (10× Genomics) software with a mouse mm10 reference sequence in prebuilt Cell Ranger reference packages was used for initial data processing. After initial data processing, all the processing steps were performed using Seurat v3. We analyzed each sample separately and retained only cells that had more than 200 genes and fewer than 2500 genes. We next normalized the data and identified 2000 highly variable features per dataset for principal component analysis (PCA). We assigned an identifier for the I-PpoI or WT mice to distinguish them. We then merged the samples with the standard workflow using “Merge”. Subsequently, we performed PCA on the integrated data and identified the true dimensionality of the data on the basis of jackstraw. We clustered the cells using “FindNeighbors” and “FindClusters” with a resolution of 0.5. We next visualized cell clustering with uniform manifold approximation and projection (UMAP). To assign cluster identities, we assessed the expression of known canonical markers using “FindAllMarkers”. Macrophage, dendritic cell (DC), monocyte and neutrophil clusters were extracted, and subclustering was performed in the same way.
Macrophage depletion
We performed macrophage depletion in the I-PpoI or WT mice by intraperitoneal injection of clodronate liposomes (Hygieia Bioscience, Osaka, Japan), which are bisphosphonates encapsulated by liposomes that are known to induce macrophage depletion in vivo, or control liposomes once weekly from 12 to 15 weeks of age82,83.
Sample preparation and CE-MS analysis
Snap-frozen tissue was immediately homogenized with methanol (1 ml) containing internal standards (300 µM each of methionine sulfone and MES) to inactivate enzymes. After addition of deionized water (500 µl) and chloroform (1 ml), the suspensions were centrifuged at 12,000 × g for 15 min at 4 °C. The upper aqueous layer was centrifugally filtered through a 5-kDa cutoff filter (Human Metabolome Technologies, Tsuruoka, Japan) to remove proteins. The filtrate was lyophilized and dissolved in 50 µl of Milli-Q water containing reference compounds (200 µM each of 3-aminopyrrolidine and trimesate) before CE-MS analysis.
Metabolites were analyzed by using an Agilent CE time-of-flight MS (CE-TOFMS) system equipped with an Agilent G7100A CE instrument and an Agilent 6530 Q-TOF LC/MS system (Agilent Technologies, Waldbronn, Germany) described in a previous report84.
Cell culture
Cultured murine proximal tubular endothelial cells (mPTEC), immortalized cell-line, were kindly provided by Dr. Nobuya Sasaki from Kitasato University85. mPTECs were cultured at 37 °C in renal epithelial cell growth medium (REGM supplemented with 0.5% fetal bovine serum, recombinant human epidermal growth factor, insulin, hydrocortisone, epinephrine, triiodothyronine, transferrin, 30 µg/mL gentamicin and 15 ng/mL amphotericin) purchased from Lonza (Basel, Switzerland). pCNPpo6 was purchased from the RIKEN Center for Life Science Technologies (Hyogo, Japan), and transient transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. I-PpoI-transfected or control PTECs were cultured in 6-well plates, and gene expression was evaluated via real-time RT‒PCR after 24 h, as described above.
Mouse macrophage RAW 264.7 cell lines were purchased from American Type Culture Collection (ATCC, #TIB-71) and cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) at 37 °C. Then, culture medium from I-PpoI-transfected or control PTECs was added to the RAW 264.7 cells. After incubation for 48 h, the expression of proinflammatory cytokines was evaluated via real-time RT‒PCR. Culture medium from RAW 264.7 cells was added to mouse adipocytes differentiated from OP9 cell line, a line of mouse bone marrow-derived stromal cells, which was obtained from ATCC(#CRL-2749). After incubation for 48 h, the expressions of proinflammatory cytokines and lipogenesis and fatty acid oxidation markers were evaluated.
Cell lines purchased from ATCC were authenticated by Short Tandem Repeat (STR) prior to purchase, and mPTECs were authenticated prior to receiving them as a gift.
Measurement of the oxygen consumption rate
mPTECs were seeded at a confluency of 70–90%; 5 × 104/well onto an XF24 cell culture microplate (Seahorse Bioscience) and incubated in REBM™ Renal Epithelial Cell Growth Basal Medium at 37 °C overnight. The cultured medium was changed to Seahorse XF DMEM medium supplemented with glucose, pyruvate and glutamine. The cells were analyzed using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience), and the OCR and ECAR were measured using a Mito Stress Kit (Seahorse Bioscience) according to the manufacturer’s protocol. In the Palmitate Oxidation Stress Test (Seahorse Bioscience), mPTECs cultured in Seahorse XF DMEM medium are added with L-carnitine and palmitate conjugated BSA or BSA control of a final concentration of 0.5 mM and 0.2 mM prior to assay, respectively, according to the manufacturer’s instructions. The difference between the basal OCR and the OCR after added with 4 µM etomoxir, an irreversible inhibitor of carnitine palmitoyltransferase-1 (CPT1) (shown by the arrows in the Fig. 3E), means the dependency of the mPTECs on fatty acid oxidation. The OCR and ECAR were normalized by counted cell numbers.
Mitochondrial copy number determination
Mitochondrial copy number was determined using Mouse Mitochondrial DNA Copy Number Kit (Detroit R&D, Cat No. MCN3) following the manufacturer’s instructions using genomic DNA extracted from the kidney and the liver of the I-PpoI mice or controls.
Fluorescence detection of the FAO activity in PTECs
The FAO activity in murine PTECs with DNA damage was examined using the fatty acid oxidation detection reagent, FAO blue™ (Funakoshi Cat No. FDV-0033) according to the manufacturer’s instructions86. Briefly, the PTECs following transfection with or without I-PpoI were incubated in palmitate -conjugated-BSA-supplemented REBM™ Renal Epithelial Cell Growth Basal Medium at 37 °C for 8 h. The culture medium was removed and the PTECs were washed with the HBS buffer, added with 20 µM FAO blue™ -containing HBS and incubated at 37 °C for 30 min. Then, the PTECs were washed with the HBS buffer and observed with blue fluorescence using a laser confocal microscope (LSM980; Zeiss), and ×40 field images were acquired. All confocal images are representative of three independent experiments.
Infinium methylation EPIC array
Genomic DNA was extracted from whole blood cells using the QIAamp DNA Blood Mini 139 Kit (Qiagen, USA). DNA methylation profiling was performed using the Infinium Methylation EPIC BeadChip array (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. The EPIC array is a comprehensive methylation platform that interrogates over 850,000 CpG sites across the human genome, covering 99% of RefSeq genes and 95% of CpG islands.
Genomic DNA (500 ng) was bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA). The bisulfite-converted DNA was then amplified, fragmented, and hybridized to the EPIC BeadChip. The arrays were scanned using the Illumina iScan system, and the raw intensity data (IDAT files) were imported into R for further processing and analysis.
The raw data were processed and normalized using the minfi package in R. Probes with p values > 0.01 in more than 5% of the samples, cross-reactive probes, and probes targeting SNPs were excluded from the analysis. Normalization was performed using the functional normalization method, which adjusts for technical variation while preserving biological variation. Beta values, representing the ratio of the methylated probe intensity to the overall intensity (sum of the methylated and unmethylated probe intensities), were calculated for each CpG site.
Differential methylation analysis was conducted using the limma package in R, with adjustments for multiple testing using the Benjamini‒Hochberg method to control the false discovery rate (FDR). CpG sites with an adjusted p value < 0.05 and an absolute beta value difference > 0.1 between groups were considered differentially methylated.
HMGB1 immunoassay
High mobility group box protein-1 (HMGB1) levels were analyzed in the culture supernatant of mPTEC were collected 48 h after transfection with or without I-PpoI using the mouse HMGB1 enzyme-linked immunosorbent assay (ELIZA) kit (Invitrogen, #EEL102) according to the manufacturer’s instructions.
Exosome purification and characterization
Urinary specimens were collected from 20-week-old WT mice and I-PpoI mice for exosome extraction. Murine podocytes were cultured in serum-free RPMI after the medium was collected to purify the exosomes. Exosomes were extracted from supernatant fractions from cell cultures 24–48 h after transient transfection. For exosome purification and mRNA extraction, we used a SeraMir Exosome RNA Amplification Kit (System Biosciences) according to the manufacturer’s protocol.
IL-1β immunoassay
IL-1β levels were analyzed in the culture supernatant of murine RAW 264.7 macrophages cultured with the supernatant of the murine PTECs following transfection with or without I-PpoI for 48 h using the mouse IL-1β enzyme-linked immunosorbent assay (ELIZA) kit (Proteintech Group, inc. #KE10003) according to the manufacturer’s instructions.
Immunostaining of macrophages
The murine RAW 264.7 macrophages seeded on chambered coverslips and supplemented with the supernatant of the murine PTECs following transfection with or without I-PpoI were fixed for 40 min with 4% paraformaldehyde in PBS, permeabilized for 5 min in 0.1% Triton X-100 in PBS and blocked using 5% BSA for 1 h. Then, the cells were stained with Cleaved Gasdermin D (Asp 276) primary antibody diluted at a 1:2500 (#10137 Rabbit monoclonal; Cell Signaling Technology), followed by incubation with fluorescent-conjugated goat anti-mouse IgG (Invitrogen). Then, the macrophages were washed with the HBS buffer and observed with blue fluorescence using a laser confocal microscope (LSM980; Zeiss), and ×40 field images were acquired. All confocal images are representative of three independent experiments.
Flow cytometry
Cell suspensions were prepared using phosphate-buffered saline (PBS) containing 1% bovine serum albumin and 0.05% sodium azide and stained for fluorescence-activated cell sorting (FACS) analysis of cell surface markers with the following fluorochrome-conjugated Abs (purchased from BioLegend): anti-F4/80, anti-CD11b, anti-CD11c, anti-Ly6C and anti-CD40. Flow cytometry samples were analyzed using CytoFLEX S.
Human studies
Human diabetic nephropathy samples were obtained from patients who were definitively diagnosed with diabetic nephropathy via kidney biopsy. As controls, kidney samples were obtained from patients without diabetes mellitus who were diagnosed with minimal change disease with normal light microscopy findings on renal biopsy. The clinical data are presented in Supplementary Table 1. Paraffin sections were stained with γH2AX and AQP1 and photographed using a laser confocal microscope (LSM980; Zeiss), and five ×20 field images were acquired and quantified via color channel analysis and pixel counting using Photoshop 2024 software (Adobe) as previously described79.
Urine sample collection and processing
Urine samples were prospectively collected from the outpatients who signed a consent form at the clinic visit, with the clinical data presented in Supplementary Table 2. All samples were assigned a study number that connected them to clinical information from the chart to deidentify them. The samples are processed immediately without freezing to maintain the integrity of the cellular components and prevent crystal formation and left over from routine urine collections, which were midstream samples. The samples are processed as described before76.
Hepatic steatosis index (HSI)
HSI was calculated by the following formula: 8×(ALT/AST) + BMI + 2 (if diabetes) + 2 (if female) using the clinical data of the outpatients listed in Supplementary Table 2. The high HSI indicate that a positive diagnosis of hepatic steatosis is highly likely87.
Study approval
All animal studies were approved by the Animal Research Committee, School of Medicine, Keio University (Approval No. A2022-198), and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). Human studies were approved by the Ethics Committee of Keio University School of Medicine (approval number:2020-0147). Written informed consent was obtained from participants prior to inclusion in the study. Participants were identified using a number rather than their name. This study was conducted in adherence with the Declaration of Helsinki. All methods were carried out in accordance with the institutional guidelines of the ethics committee at Keio University (Tokyo, Japan).
Quantification and statistical analysis
The results are expressed as the mean ± SEM. Statistical comparisons were made by ANOVA followed by Scheffe’s post hoc test to compare more than 2 groups and by a 2-tailed Student’s t test to compare 2 groups. Statistical significance was defined as p < 0.05.
All the statistical details of the experiments can be found in the Figure legends. Statview 5.0 software (SAS Institute) was used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The research data supporting this study can be accessed within the manuscript and associated Supplementary Materials. Source data are provided as a Source Data file. All datasets generated and analyzed in this investigation are available from the corresponding author on reasonable request. The authors declare that all the data supporting the findings of this study are available within the paper and its supplementary information files. All raw sequencing dataset in this study have been deposited in the Gene Expression Omnibus (GEO). Single-cell RNA sequencing (scRNA-seq) data have been deposited in GEO under accession code [GSE275550], Reduced representation of bisulfite sequencing (RRBS) analysis have been deposited in GEO under accession code [GSE284752], RNA-seq analysis data have been deposited in GEO under accession code [GSE285052] and Infinium DNA methylation analysis data using EPIC array have been deposited in GEO under accession code [GSE285416]. Source data are provided with this paper.
References
Sedelnikova, O. A. et al. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170 (2004).
Yang, J. H. et al. Loss of epigenetic information as a cause of mammalian aging. Cell 186, 305–326.e327 (2023).
Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020).
Asghar, M. S. et al. Age-Based Versus Young-Adult Thresholds for Nephrosclerosis on Kidney Biopsy and Prognostic Implications for CKD. J. Am. Soc. Nephrol. 34, 1421–1432 (2023).
Xiang, F. et al. Premature aging of circulating T cells predicts all-cause mortality in hemodialysis patients. BMC Nephrol. 21, 271 (2020).
Crépin, T. et al. Uraemia-induced immune senescence and clinical outcomes in chronic kidney disease patients. Nephrol. Dial. Transpl. 35, 624–632 (2020).
Oh, H. S. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
Tian, Y. E. et al. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat. Med. 29, 1221–1231 (2023).
Hishikawa, A. et al. Decreased KAT5 expression impairs DNA repair and induces altered DNA methylation in kidney podocytes. Cell Rep. 26, 1318–1332 (2019).
Hishikawa, A. et al. DNA damage and expression of DNA methylation modulators in urine-derived cells of patients with hypertension and diabetes. Sci. Rep. 10, 3377 (2020).
Hayashi, K. et al. Association of glomerular DNA damage and DNA methylation with one-year eGFR decline in IgA nephropathy. Sci. Rep. 10, 237 (2020).
Hishikawa, A. et al. DNA repair factor KAT5 prevents ischemic acute kidney injury through glomerular filtration regulation. iScience 24, 103436 (2021).
Yoshimoto, N. et al. Significance of podocyte DNA damage and glomerular DNA methylation in CKD patients with proteinuria. Hypertens. Res. 46, 1000–1008 (2023).
Shankland, S. J. et al. Podocyte aging: why and how getting old matters. J. Am. Soc. Nephrol. 32, 2697–2713 (2021).
Nakamichi, R. et al. DNA-damaged podocyte-CD8 T cell crosstalk exacerbates kidney injury by altering DNA methylation. Cell Rep. 42, 112302 (2023).
Hama, E. Y. et al. Podocyte Ercc1 is indispensable for glomerular integrity. Biochem Biophys. Res Commun. 704, 149713 (2024).
Mellouk, A. & Bobé, P. CD8(+), but not CD4(+) effector/memory T cells, express the CD44(high)CD45RB(high) phenotype with aging, which displays reduced expression levels of P2X(7) receptor and ATP-induced cellular responses. Faseb j. 33, 3225–3236 (2019).
Gluck, C. et al. Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat. Commun. 10, 2461 (2019).
Heylen, L. et al. Age-related changes in DNA methylation affect renal histology and post-transplant fibrosis. Kidney Int 96, 1195–1204 (2019).
Park, J. et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight 4, e128886 (2019).
Kim, J., Sturgill, D., Tran, A. D., Sinclair, D. A. & Oberdoerffer, P. Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability. Nucleic Acids Res. 44, e64 (2016).
de Matos, A. C. et al. Vimentin expression and myofibroblast infiltration are early markers of renal dysfunction in kidney transplantation: an early stage of chronic allograft dysfunction? Transpl. Proc. 42, 3482–3488 (2010).
Ho, C. M. et al. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J. Inflamm. (Lond.) 16, 7 (2019).
Franczyk, M. P. et al. Importance of adipose tissue NAD+ biology in regulating metabolic flexibility. Endocrinology 162, bqab006 (2021).
Yan, L., Rust, B. M. & Palmer, D. G. Time-restricted feeding restores metabolic flexibility in adult mice with excess adiposity. Front Nutr. 11, 1340735 (2024).
Hinder, L. M., Vivekanandan-Giri, A., McLean, L. L., Pennathur, S. & Feldman, E. L. Decreased glycolytic and tricarboxylic acid cycle intermediates coincide with peripheral nervous system oxidative stress in a murine model of type 2 diabetes. J. Endocrinol. 216, 1–11 (2013).
Zhao, L. et al. Metabolomic analysis identifies lactate as an important pathogenic factor in diabetes-associated cognitive decline rats. Mol. Cell Proteom. 17, 2335–2346 (2018).
Lotta, L. A. et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a Mendelian randomisation analysis. PLoS Med 13, e1002179 (2016).
Wang, X., Hu, Z., Hu, J., Du, J. & Mitch, W. E. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 147, 4160–4168 (2006).
Conway, B. R. et al. Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. J. Am. Soc. Nephrol. 31, 2833–2854 (2020).
Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018).
Bu, Y. et al. Insulin regulates lipolysis and fat mass by upregulating growth/differentiation factor 3 in adipose tissue macrophages. Diabetes 67, 1761–1772 (2018).
Whytock, K. L. et al. Aging human abdominal subcutaneous white adipose tissue at single cell resolution. Aging Cell 23, e14287 (2024).
Yang, H., Wang, H., Czura, C. J. & Tracey, K. J. The cytokine activity of HMGB1. J. Leukoc. Biol. 78, 1–8 (2005).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Wang, P., Ouyang, J., Jia, Z., Zhang, A. & Yang, Y. Roles of DNA damage in renal tubular epithelial cells injury. Front Physiol. 14, 1162546 (2023).
Yin, Q. et al. MiR-155 deficiency protects renal tubular epithelial cells from telomeric and genomic DNA damage in cisplatin-induced acute kidney injury. Theranostics 12, 4753–4766 (2022).
Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V. & Bonventre, J. V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543 (2010).
Kishi, S. et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Invest 129, 4797–4816 (2019).
Idrovo, J. P., Yang, W. L., Nicastro, J., Coppa, G. F. & Wang, P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J. Surg. Res. 177, 157–164 (2012).
Kang, H. M. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46 (2015).
Miguel, V. et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Invest 131, e140695 (2021).
Rinaldi, A. et al. Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight 7, e161783 (2022).
Wang, H., Zhang, S. & Guo, J. Lipotoxic proximal tubular injury: a primary event in diabetic kidney disease. Front Med (Lausanne) 8, 751529 (2021).
Fang, E. F. et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 17, 308–321 (2016).
Guan, Y. et al. Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a Sirtuin 1-dependent manner. J. Am. Soc. Nephrol. 28, 2337–2352 (2017).
Jia, Y. et al. Nicotinamide mononucleotide attenuates renal interstitial fibrosis after AKI by suppressing tubular DNA damage and senescence. Front Physiol. 12, 649547 (2021).
Yamamuro, T. et al. Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat. Commun. 11, 4150 (2020).
Flegal, K. M., Kit, B. K., Orpana, H. & Graubard, B. I. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. Jama 309, 71–82 (2013).
Tchkonia, T. et al. Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684 (2010).
Qi, N. et al. Adipocyte-specific inactivation of NAMPT, a key NAD(+) biosynthetic enzyme, causes a metabolically-unhealthy lean phenotype in female mice during aging. Am. J. Physiol. Endocrinol. Metab. 327, E81–E88(2024).
Gilardi, F. et al. Systemic PPARγ deletion in mice provokes lipoatrophy, organomegaly, severe type 2 diabetes and metabolic inflexibility. Metabolism 95, 8–20 (2019).
Wang, F., Mullican, S. E., DiSpirito, J. R., Peed, L. C. & Lazar, M. A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc. Natl Acad. Sci. USA 110, 18656–18661 (2013).
Janssen, J. The causal role of ectopic fat deposition in the pathogenesis of metabolic syndrome. Int J. Mol. Sci. 25, 13238 (2024).
Mathew, H., Farr, O. M. & Mantzoros, C. S. Metabolic health and weight: understanding metabolically unhealthy normal weight or metabolically healthy obese patients. Metabolism 65, 73–80 (2016).
Ding, C., Chan, Z. & Magkos, F. Lean, but not healthy: the ‘metabolically obese, normal-weight’ phenotype. Curr. Opin. Clin. Nutr. Metab. Care 19, 408–417 (2016).
Wang, Y. & Harris, D. C. Macrophages in renal disease. J. Am. Soc. Nephrol. 22, 21–27 (2011).
Lee, S. et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 22, 317–326 (2011).
Lech, M. et al. Macrophage phenotype controls long-term AKI outcomes-kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 25, 292–304 (2014).
Kim, M. G. et al. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transpl. 25, 2908–2921 (2010).
Laursen, M. F. et al. CD11c-targeted delivery of DNA to dendritic cells leads to cGAS- and STING-dependent maturation. J. Immunother. 41, 9–18 (2018).
Serbina, N. V. & Pamer, E. G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317 (2006).
Poller, W. C., Nahrendorf, M. & Swirski, F. K. Hematopoiesis and cardiovascular disease. Circ. Res. 126, 1061–1085 (2020).
Bakshi, C., Vijayvergiya, R. & Dhawan, V. Aberrant DNA methylation of M1-macrophage genes in coronary artery disease. Sci. Rep. 9, 1429 (2019).
Qu, R. et al. Down-regulation of KLF9 ameliorates LPS-caused acute lung injury and inflammation in mice via reducing GSDMD expression. Autoimmunity 55, 587–596 (2022).
Yan, M. et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 8, 258 (2022).
Liu, Z., Chen, Y., Mei, Y., Yan, M. & Liang, H. Gasdermin D-mediated pyroptosis in diabetic cardiomyopathy: molecular mechanisms and pharmacological implications. Molecules 28, 7813 (2023).
Guilherme, A., Virbasius, J. V., Puri, V. & Czech, M. P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9, 367–377 (2008).
Herrero, L., Shapiro, H., Nayer, A., Lee, J. & Shoelson, S. E. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc. Natl Acad. Sci. USA 107, 240–245 (2010).
Xiang, D. M. et al. Chronic kidney disease promotes chronic inflammation in visceral white adipose tissue. Am. J. Physiol. Ren. Physiol. 312, F689–f701 (2017).
Witasp, A. et al. Increased expression of pro-inflammatory genes in abdominal subcutaneous fat in advanced chronic kidney disease patients. J. Intern Med. 269, 410–419 (2011).
Gertow, J. et al. Altered protein composition of subcutaneous adipose tissue in chronic kidney disease. Kidney Int Rep. 2, 1208–1218 (2017).
Chu, A. Y. et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 8, 1286 (2017).
Li, K. Y. et al. DNA methylation markers for kidney function and progression of diabetic kidney disease. Nat. Commun. 14, 2543 (2023).
Chen, Z. et al. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat. Metab. 2, 744–762 (2020).
Hishikawa, A. et al. Predicting exacerbation of renal function by DNA methylation clock and DNA damage of urinary shedding cells: a pilot study. Sci. Rep. 14, 11530 (2024).
Abe, T., Inoue, K. I., Furuta, Y. & Kiyonari, H. Pronuclear microinjection during S-phase increases the efficiency of CRISPR-Cas9-assisted knockin of large DNA donors in mouse zygotes. Cell Rep. 31, 107653 (2020).
Legouis, D. et al. Ex vivo analysis of renal proximal tubular cells. BMC Cell Biol. 16, 12 (2015).
Hayashi, K. et al. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J. Clin. Invest 124, 2523–2537 (2014).
Furda, A. M., Bess, A. S., Meyer, J. N. & Van Houten, B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 920, 111–132 (2012).
Kim, C. H. et al. Short-term calorie restriction ameliorates genomewide, age-related alterations in DNA methylation. Aging Cell 15, 1074–1081 (2016).
Hu, Z., Zhan, J., Pei, G. & Zeng, R. Depletion of macrophages with clodronate liposomes partially attenuates renal fibrosis on AKI-CKD transition. Ren. Fail 45, 2149412 (2023).
Sumida, T. et al. Complement C1q-induced activation of β-catenin signalling causes hypertensive arterial remodelling. Nat. Commun. 6, 6241 (2015).
Fujii, K. et al. Xanthine oxidase inhibitor ameliorates postischemic renal injury in mice by promoting resynthesis of adenine nucleotides. JCI Insight 4, e124816 (2019).
Sasaki, H., Sugiyama, M. & Sasaki, N. Establishment of renal proximal tubule cell lines derived from the kidney of p53 knockout mice. Cytotechnology 71, 45–56 (2019).
Uchinomiya, S. et al. Fluorescence detection of metabolic activity of the fatty acid beta oxidation pathway in living cells. Chem. Commun. (Camb.) 56, 3023–3026 (2020).
Lee, J. H. et al. Hepatic steatosis index: a simple screening tool reflecting nonalcoholic fatty liver disease. Dig. Liver Dis. 42, 503–508 (2010).
Acknowledgements
We acknowledge Dr. Mari Fujiwara (Core Facility, Collaborative Research Resources, Keio University School of Medicine) for technical assistance with the flow cytometry experiments, Dr. Sasaki Nobuya (Laboratory of Laboratory Animal Science and Medicine, School of Veterinary Medicine, Kitasato University) for kindly providing the mouse PTECs, and Dr. Takako Hishiki (Department of Biochemistry, Keio University School of Medicine) for technical assistance with the metabolome analysis. This study was supported by the JST FOREST Program (Grant Number JPMJFR210V, Japan), Grants for Scientific Research (23K24351, 22H03091, 20H00535, 19K08688, and 20K17291) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, AMED (Grant Number JP24gm6710029h0001), Keio University Research Grants for Life Science and Medicine, the Takeda Science Foundation, Terumo Life Science Foundation, Uehara Memorial Foundation, Astellas Foundation for Research on Metabolic Disorders, and Japan Kidney Association, and 2024 Young Researcher’s Award of Japanese Society of Women Nephrologist (JSWN).
Author information
Authors and Affiliations
Contributions
E.S.N. performed most of the experiments, analyzed the data and wrote the manuscript. A.H.I. generated the I-PpoI mice, performed the DNA methylation array using human samples and interpretation of the metabolome data, and wrote the manuscript. R.N. performed the analysis of the RNA-seq and scRNA-seq data and wrote the manuscript. R.A. performed the in vitro analysis. S.C. contributed to the immunological analysis. A.H.A. contributed to the pathological analysis of human biopsies. N.Y., E.Y.H., T.M., W.I., S.Y., J.Y., and H.I. contributed to the drafting and revision of the manuscript. K.H. conceived and designed the research studies, performed the experiments, analyzed and interpreted the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liang Ma, Gopala Rangan and Chengguo Wei for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nishimura, E.S., Hishikawa, A., Nakamichi, R. et al. DNA damage in proximal tubules triggers systemic metabolic dysfunction through epigenetically altered macrophages. Nat Commun 16, 3958 (2025). https://doi.org/10.1038/s41467-025-59297-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59297-x
