
Abstract
Advancements in chimeric antigen receptor (CAR) T-cell therapy for treating diffuse large B-cell lymphoma (DLBCL) have been limited by an incomplete understanding of CAR T-cell differentiation in patients. Here, we show via single-cell, multi-modal, and longitudinal analyses, that CD8+ CAR T cells from DLBCL patients successfully treated with axicabtagene ciloleucel undergo two distinct waves of clonal expansion in vivo. The first wave is dominated by an exhausted-like effector memory phenotype during peak expansion (day 8–14). The second wave is dominated by a terminal effector phenotype during the post-peak persistence period (day 21–28). Importantly, the two waves have distinct ontogeny from the infusion product and are biologically uncoupled. Precursors of the first wave exhibit more effector-like signatures, whereas precursors of the second wave exhibit more stem-like signatures. We demonstrate that CAR T-cell expansion and persistence are mediated by clonally, phenotypically, and ontogenically distinct CAR T-cell populations that serve complementary clinical purposes.
Similar content being viewed by others


Introduction
Diffuse large B-cell lymphoma (DLBCL), the most common non-Hodgkin’s lymphoma in the United States, is characterized by diffusely proliferating and malignant B cells at nodal or extranodal sites1. Although up-front chemoimmunotherapy is often curative, patients with DLBCL that is refractory to up-front treatment or relapse following remission (r/r DLBCL) have limited treatment options and poor outcomes2,3. Effective treatment options were limited until the United States Food and Drug Administration approved autologous CD19-directed chimeric antigen receptor (CAR) T-cell therapies for r/r DLBCL in 2017. Autologous CD19-directed CAR T-cell therapy involves virally transducing a patient’s T cells ex vivo with a CD19-directed CAR—an engineered receptor consisting of an extracellular anti-CD19 single-chain variable fragment, a hinge/transmembrane region, and intracellular costimulatory (CD28 or 4-1BB) and activation (CD3ζ) domains. CAR-transduced T cells (i.e., CAR T cells) are cultured and infused into the patient, where they lyse CD19+ lymphoma cells4. CD19-directed CAR T-cell therapy has achieved complete response rates of 40–54%5,6,7,8 for r/r DLBCL, but non-response rates6,9 and treatment toxicities10 remain as challenges11.
Development of CAR T-cell formulations with higher response rates and fewer toxicities requires a thorough understanding of how CAR T cells differentiate in patients with r/r DLBCL. The pioneering ZUMA-1 trial (NCT02348216) for axicabtagene ciloleucel (autologous CD28-costimulated CAR T cells) demonstrated that peripheral blood CAR T cells expand, contract, and sometimes persist.5 Greater expansion predicted higher response rates, but also higher likelihood of developing treatment toxicities5,12. Longer persistence predicted durable remission and long-term immunosurveillance with leukemias, but its relevance for preventing DLBCL relapse remains obscure9,13. Although many factors are associated with expansion and persistence (including CAR design14,15 and CAR T-cell phenotypes16,17,18,19,20, among others21,22,23), how and why CAR T cells differentiate into expansive or persistent phenotypes in vivo is still an open question. Addressing this question requires a single-cell approach that integrates CAR T-cell phenotypes and clonal kinetics over longitudinal timepoints. However, existing studies have either focused on CAR T-cell phenotypes19,24,25 or clonal kinetics26 without integrating both data modalities, or lacked the temporal resolution required to decipher cell fates over longer periods12,27. Consequently, a complete and longitudinal understanding of CAR T-cell differentiation in vivo has remained elusive.
To comprehensively elucidate CAR T-cell differentiation in vivo, we perform single-cell, multi-modal (paired RNA-seq/CITE-seq/TCR-seq), and longitudinal analyses of CD28-costimulated CAR T cells from infusion product and peripheral blood of seven patients with r/r DLBCL who were complete responders under treatment with axicabtagene ciloleucel. Peripheral blood CAR T cells are sorted using CD19 antigen-tetramers28. Importantly, we report that the CD8+ CAR T cells observed during the peak expansion period (day 8–14) have distinct clonotypic repertoires compared to the later CD8+ CAR T cells observed during the post-peak persistence period (day 21–28). We further analyze how these two CAR T-cell populations differ with respect to phenotypes, transcriptional profiles, regulatory networks, and infusion product precursors. Our findings not only offer a finer understanding of CAR T-cell biology in vivo, but also inform efforts to develop CAR T cells with improved expansion and persistence.
Results
Study design and clinical findings
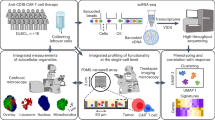
To interrogate CD28-costimulated CAR T-cell differentiation in vivo, we longitudinally interrogated the phenotypes and clonal dynamics of CAR T cells from seven patients (P1-7) who achieved complete responses under treatment with axicabtagene ciloleucel (Fig. 1a). Patients were diagnosed with r/r DLBCL and treated at the University of Chicago Medicine between 2019 and 2021 (Table 1). Clinical response was determined by positron emission tomography/computed tomography imaging 30 days after infusion product administration. Complete response was defined as no detectable lymphoma (Fig. S1). To capture phenotypic heterogeneity and clonal dynamics longitudinally, we performed single-cell multi-modal analyses (paired RNA-seq/CITE-seq/TCR-seq via the 10x Genomics platform) on infusion product and peripheral blood biospecimens at three timepoints: peak expansion (Texp, day 8–14), early post-peak persistence (Tper1, day 21), and late post-peak persistence (Tper2, day 28) (Table 2). The three peripheral blood timepoints were available for all patients, except P3 (only Texp and Tper1). CAR T cells were sorted from peripheral blood using CD19 antigen-tetramers (representative staining in Fig. 1b, Fig. S2)28. Compared to CD19 antigen-tetramer-negative T cells, CD19 antigen-tetramer-positive T cells specifically expressed the CAR transgene, which validates our sorting strategy (Fig. 1c).

a Schematic depicting sorting strategy, data generation, and single-cell multi-modal analysis of CAR T cells in peripheral blood from seven CAR T-cell therapy patients (P1-7) who exhibited complete responses with axicabtagene ciloleucel. Created in BioRender. Hu, Y. (2025) https://BioRender.com/hqo12oz. b Representative flow plots depicting anti-CD3 and CD19 antigen-tetramer staining of peripheral blood mononuclear cells from P3 versus a healthy donor. CD3+Tet+ (“CAR”) and CD3+Tet− (“ENDO”) patient cells were sorted from the indicated gates. c Violin plots depicting normalized CAR transgene mRNA expression of sorted CAR and ENDO T cells, split by CD4+ (left) and CD8+ (right) subsets. Expression levels were compared by two-sided Wilcoxon Rank-Sum test with Bonferroni correction, whereby **** indicates p < 0.0001. d Line plots depicting expansion and contraction of peripheral blood CAR abundance over the course of therapy. e, f Bar graphs and heatmaps depicting overlap coefficients for TCR clonotypes comparing CAR (left) and ENDO (right) repertoires between Texp, Tper1, and Tper2. Overlap coefficients were compared by two-sided t test, whereby * indicates p < 0.05 and ns indicates not significant. Each dot represents a measurement from a single patient (n = 7). Data are presented as mean values ± SEM. Exp expansion stage, Per persistence stage. Source data are provided as a Source Data file.
CD8+ CAR T cells undergo a clonotypic shift between Texp and Tper
To analyze CAR T-cell population dynamics, we tracked CAR abundance (% CAR+ of CD3+ T cells) in peripheral blood throughout the course of therapy. CAR abundance at peak expansion ranged from 11% to 73% (Fig. 1d). Peak expansion occurred at day 8–14, which is consistent with prior clinical findings5. For each timepoint, we quantified proportions of CD8+ and CD4+ T cells within the total CAR T-cell population by single-cell RNA-seq and CITE-seq. CAR T cells were predominantly CD8+ across most patients and timepoints (Fig. S3).
We next analyzed the dynamics of the CAR T-cell clonal repertoire across longitudinal timepoints (Texp at day 8–14, Tper1 at day 21, Tper2 at day 28) using single-cell TCR-seq. Clone sizes were similar across timepoints (Fig. S4a). Clonotypes did not overlap between patients (Fig. S4b). Repertoire overlap analysis indicated that Texp clonotypes were significantly distinct from Tper1 or Tper2 clonotypes (Fig. 1e, f, left). In sharp contrast, Tper1 and Tper2 clonotypes overlapped substantially more. As a control, we also analyzed endogenous (non-CAR) T cells from matched timepoints. Unlike with CAR T cells, endogenous T cells did not show distinctive clonotypic patterns (Fig. 1e, f, right), indicating that the shift in clonotypes between Texp and Tper is CAR-specific. This CAR-specific clonotypic shift was consistent across patients (Fig. S4b). Moreover, the distinction between Texp and Tper clonotypes is driven by CD8+ T cells, and not by CD4+ T cells (Fig. S4c). Collectively, these findings indicate that CD8+ CAR T cells undergo a clonotypic shift between Texp and Tper.
CD8+ CAR T cells undergo a phenotypic shift from exhausted-like effector memory to terminal effector
Having shown a shift in CD8+ CAR T-cell clonotypes between Texp and Tper, we hypothesized that unique CD8+ CAR T-cell phenotypes dominate Texp and Tper. To test this hypothesis, we filtered CD8+ CAR T cells for Uniform Manifold Approximation and Projection (UMAP) and identified six T-cell clusters (Fig. 2a) based on gene and protein markers (Fig. 2b, expanded marker set in Fig. S5a). No cluster was patient-specific (Fig. S5b). All clusters expressed CAR transgene and CD8α, validating our sorting and filtering processes, respectively. All clusters expressed CXCR3, a chemokine receptor that demarks activated T cells.

UMAPs depicting single-cell transcriptomes of CD8+ (a) and CD4+ (e) CAR T cells colored by cell cluster. Inset depicts distribution of transcriptomes across timepoints. Violin plots depicting normalized expression levels of key genes and proteins for annotating and phenotyping CD8+ (b) and CD4+ (f) CAR T cells. For extended versions, see Fig. S5a and S6a. Stacked bar graphs depicting proportions of each CD8+ (c) and CD4+ (g) CAR T-cell phenotype at different timepoints. Boxplots depicting proportion of CD8+ (d) and CD4+ (h) CAR T cells of a given phenotype at different timepoints. Each dot represents a measurement from a single patient (n = 7). The central line indicates the median. The bounds of the box indicate the 25th–75th percentile. The length of the whiskers indicates 1.5 times the inter-quartile range from the first and third quartiles. Proportions are compared between timepoints by two-sided t test, whereby *** indicates p < 0.001, ** indicates p < 0.01, * indicates p < 0.05, and ns indicates not significant. CM central memory, EM effector memory, TE terminal effector, Mem memory, ISG interferon stimulated genes, Th1 type 1 helper-like. Source data are provided as a Source Data file.
Examination of gene and protein markers revealed that one of the six clusters represented proliferating T cells (MKI67+TOP2A+). The remaining five non-proliferating clusters were annotated as central memory (CM, TCF7+TBX21–), effector memory (EM, TCF7+TBX21+), or terminal effector (TE, TCF7-TBX21+) T cells. The TCF7+TBX21– CM cluster exhibited markers of stemness (IL7R, high CD127) and minimal markers of effectorness (GZMB, CX3CR1). The two TCF7+TBX21+ EM clusters occupied the lower half of the UMAP and uniquely expressed CXCR6, a chemokine receptor that facilitates trafficking into solid tumors29. One of these EM clusters upregulated markers consistent with early exhaustion (NR4A2, TOX, GZMK, low TIM-3), hence it was designated “exhausted-like EM”. Lastly, the two TCF7-TBX21+ TE clusters occupied the upper half of the UMAP and uniquely downregulated GZMK. One of these TE clusters upregulated markers consistent with late exhaustion (TOX, PDCD1, high TIM-3), hence it was designated “exhausted-like TE”. The other TE cluster was highly clonal (some clone sizes >100), suggesting expansion through proliferation (Fig. S7a). Overall, most CD8+ CAR T cells were TBX21+ EM or TE, which is consistent with the established link between CD28 costimulation and effector memory (rather than central memory) differentiation14,15.
Phenotypic compositions of CD8+ CAR T cells at Texp and Tper were compared. CAR T cells at Texp were predominantly exhausted-like EM (64%) whereas CAR T cells at Tper were predominantly TE (63% for Tper1, 77% for Tper2) (Fig. 2c). These findings were statistically significant and consistent across all seven patients (Fig. 2d, Fig. S5c). Moreover, the large clone sizes within the Tper-specific TE cluster (Fig. S7a) suggest active TE proliferation at Tper. CAR T cells at Tper1 were enriched for EM. From Tper1 to Tper2, the EM proportion decreased while the TE proportion increased, suggesting progressive differentiation from EM to TE. However, the overall phenotypic compositions at Tper1 and Tper2 were more similar than different, which is concordant with findings from repertoire overlap analysis (Fig. 1e, f). Moreover, we observed decreasing proliferating proportions and increasing CM proportions over time (Fig. 2d), though this was not always statistically significant. These changing proportions may suggest some CAR T cells were returning from an activated to a resting phenotype. In conclusion, CD8+ CAR T cells phenotypically shifted from exhausted-like EM to TE between Texp and Tper.
CD4+ CAR T cells maintain a memory phenotype with CAR Treg persistence
We next performed clustering, annotation, and longitudinal analyses of CD4+ CAR T cells to investigate phenotypes at Texp and Tper. We identified five T-cell clusters, all expressing CAR transgene and CD4, based on gene and protein markers (Fig. 2e, f; expanded marker set in Fig. S6a). No cluster was patient-specific (Fig. S6b). All clusters expressed CXCR3, indicating T-cell activation and type 1 helper polarization.
One of the five clusters represented proliferating T cells (MKI67+TOP2A+). The remaining four non-proliferative clusters were annotated as memory (Mem, TBX21–FOXP3–), type 1 helper (Th1, TBX21+FOXP3–), or regulatory (Treg, TBX21–FOXP3+) T cells. The two TBX21–FOXP3– Mem clusters at the center of the UMAP comprised most of the cells. One of the Mem clusters upregulated IRF7 and interferon-stimulated genes (MX1, OAS1, ISG15), indicating response to type I interferon signaling. This signature suggests dynamic interferon secretion in vivo and is in concordance with type I interferon’s role in memory CD4+ T-cell differentiation30. The TBX21+FOXP3– Th1 cluster along the lower half of the UMAP upregulated cytolytic genes (PRF1, GNLY, GZMK, NKG7) and tissue-homing chemokine receptors (CX3CR1, CXCR6), resembling the cytotoxic CD4+ CAR T cells described by Melenhorst et al.19. Lastly, the TBX21–FOXP3+ Treg cluster expressed classic Treg markers (IL2RA, CD25, low IL7R, low CD127) and upregulated IKZF2, indicating a suppressive phenotype31. CAR Tregs inhibit conventional CAR T cells27,32 and portend progressive disease12,27.
Phenotypic compositions of CD4+ CAR T cells at Texp and Tper were compared. CD4+ CAR T cells predominantly exhibited a Mem phenotype (49–82%) across all timepoints (Fig. 2g). This observation was consistent across all patients (Fig. S6c). Between Texp and Tper1, the Mem proportion significantly decreased while the Th1 proportion significantly increased, indicating that CD4+ CAR T cells may be increasingly polarized during the early contraction period (Fig. 2h). Moreover, across all patients, the Treg proportion steadily increased from Texp (5%) to Tper1 (11%) to Tper2 (21%), indicating that CAR Tregs were maintained in peripheral blood after peak expansion. The CAR Treg cluster was predominantly non-clonal (Fig. S7b), suggesting CAR Tregs were maintained through persistence rather than proliferation. While previous studies have only investigated CAR Tregs at day 712,27, this current study affirms and extends the presence of CAR Tregs until at least day 28. Importantly, persistence of CAR Tregs points towards their possible involvement in decreasing acute inflammation and restoring immune homeostasis after peak expansion. In conclusion, we discovered that CD4+ CAR T cells did not exhibit an abrupt Texp-to-Tper phenotypic shift. Rather, they consistently exhibited a memory phenotype with CAR Tregs persisting over the course of therapy.
Integration of clonotypic and phenotypic shifts in CD8+ CAR T cells supports a two-stage differentiation model
Having observed shifts in both CD8+ CAR T-cell clonotypes and phenotypes between Texp and Tper, we hypothesized the existence of two distinct waves of in vivo clonal expansion. To test this hypothesis, we linked the clonotype and phenotype of single CD8+ CAR T cells in our dataset using their cell barcodes as unique indices. After this linking process, we confirmed that the proportions of T cells with exhausted-like EM (EM-exh) and terminal effector (TE) phenotypes (Fig. 3a) are consistent with prior results (Fig. 2c). A phenotype label was subsequently assigned to each CD8+ CAR T-cell clone based on its predominant phenotype at each timepoint. For each patient, we tracked the total abundances of clones sharing a common phenotype label across timepoints. Notably, the abundance of EM-exh clones at Texp was significantly reduced at Tper1 and Tper2 (Fig. 3b, left), while the abundance of TE clones at Tper1 and Tper2 was significantly reduced at Texp (Fig. 3b, middle and right). In addition to redemonstrating the shifts in CD8+ CAR T-cell clonotypes and phenotypes, these findings support the existence of two distinct waves of in vivo clonal expansion. (Fig. 3c).

All figure panels are based on the clonotype-phenotype linked dataset (n = 32432 cells). a Proportion of CAR T cells with an exhausted-like effector memory (EM-exh), terminal effector (TE), or other phenotypes at each timepoint. b Total abundance of all clones that were predominantly EM-exh at Texp (left), TE at Tper1 (middle), or TE at Tper2 (right), measured across timepoints. A clone’s predominant phenotype was defined as the phenotype with the greatest representation. Each point represents clones from a patient (n = 6). Abundance across timepoints were compared by repeated measures ANOVA with paired two-sided post hoc t tests, whereby **** indicates p < 0.0001, *** indicates p < 0.001, ** indicates p < 0.01, * indicates p < 0.05, and ns indicates not significant. c Overlay of clonal abundance dynamics with 95% confidence intervals for clones annotated as EM-exh from Texp (red) or TE from Tper1/Tper2 (blue). d Pie chart depicting proportion of the top 500 clones classified as Wave 1, Wave 2, or Other. e Heatmap depicting normalized CAR abundance across timepoints for the top 500 largest clones. f Clonal dynamics and phenotypic distribution for the top 500 clones, grouped into Wave 1 and Wave 2. Left panels depict the change in clonal abundance across timepoints, while right panels show the predominant phenotype distribution at each timepoint. g Cartoon summarizing the two-stage model for CAR T-cell differentiation. Bulk CAR T-cell expansion and contraction (black line) masks the dynamics of Wave 1 (EM-exh, expansion phase timeframe, red) and Wave 2 (TE, persistence phase timeframe, blue) clones. Created in BioRender. Hu, Y. (2025) https://BioRender.com/rwp4v6g. Source data are provided as a Source Data file.
Next, we evaluated the explanatory power of our hypothesis for two distinct clonal waves. To minimize noise, we filtered for the top 500 largest clones (mean size 10.8, minimum size 4). Resulting clones were annotated with the following three definitions, based on our hypothesis for two waves: “Wave 1” (phenotypically EM-exh with highest abundance at Texp), “Wave 2” (phenotypically TE with highest abundance at Tper1/Tper2), or “Other” (not Wave 1 or Wave 2). Wave 1 and Wave 2 clones together accounted for a significant majority (76%) of the top 500 clones (Fig. 3d). In accordance with our definitions, Wave 1 clones were maximally abundant at Texp, while Wave 2 clones were maximally abundant at Tper (Fig. 3e). Wave 1 clones predominantly exhibited an EM-exh phenotype at Texp and sharply decreased in abundance at Tper (Fig. 3f, left). In contrast, Wave 2 clones predominantly exhibited a TE phenotype at Tper and sharply decreased in abundance at Texp (Fig. 3f, right). To investigate whether these findings are dependent on clone size, we expanded our analysis to the top 3000 largest clones (mean size 3.7, minimum size 2). Under this filter, Wave 1 and Wave 2 clones together continued to account for a significant majority (79%) of the top 3000 clones (Fig. S8a) and reaffirm the expected clonal and phenotypic dynamics (Fig. S8b, c). Therefore, our hypothesis for two distinct waves of in vivo clonal expansion has high explanatory power and is robust to clone size.
In conclusion, the two distinct waves of in vivo clonal expansion strongly substantiate a two-stage CD8+ CAR T-cell differentiation model (Fig. 3g). Under this model, some CAR T cells (Wave 1) with an exhausted-like EM phenotype expand earlier to dominate the peak expansion timeframe (Texp), while other CAR T cells (Wave 2) with a TE phenotype expand later to dominate the post-peak persistence timeframe (Tper). Of note, the two-stage differentiation model uncouples CD8+ CAR T cells from peak expansion and post-peak persistence by designating these two waves as separate lineages. Moreover, these findings provide evidence against the intuitive idea that the post-peak contraction in CAR abundance is solely apoptosis or extravasation of short-lived CAR T cells from peak expansion. Rather, even as total CAR abundance contracts after peak expansion, a distinct subset of CAR T-cell clones simultaneously expands to eventually dominate the peripheral blood CAR T-cell repertoire.
Texp- and Tper-specific transcriptional signatures and regulatory networks
We set out to identify the molecular determinants underlying CD8+ CAR T cells at Texp and Tper using gene set enrichment analysis (GSEA), differential gene expression analysis (DGEA), and regulatory network analysis. For internal validation, the average expression of timepoint-specific genes (see Methods) for each sample (patient by timepoint) were hierarchically clustered and compared via correlation (Fig. 4a). Samples largely clustered by timepoint, validating the existence of patient-independent molecular signatures. We then validated our dataset against external data from Maus et al., which consisted of CD8+ CAR T cells from patients with large B-cell lymphoma (Fig. 4a, b, colored in tan and brown, six in total after filtering)27. Based on their sample collection timing (day 7), external data from Maus et al. should resemble our samples at Texp, rather than at Tper. Consistent with expectations, external data resembled Texp transcriptomes on both pseudo-bulk (Fig. 4a, via correlation) and single-cell (Fig. 4b, via label transfer) levels, which increases the external validity of our findings.

a Heatmap depicting correlation between pseudo-bulk transcriptome of each sample (patient by timepoint). Samples were ordered along columns and rows by hierarchical clustering. Transcriptomes of day 7 samples from Maus et al. were added for external validation. b Stacked bar graph depicting label transfer of timepoint (Texp or Tper) from this study’s dataset onto single-cell transcriptomes of samples from Maus et al. c Gene set enrichment analysis comparing CAR T cells between Texp and Tper. Gene sets were ordered by direction of upregulation and magnitude of enrichment. d Tile map depicting normalized expression of genes (columns) among different cell clusters at Texp and Tper (rows). Genes were manually grouped into modules according to known functions. e Schematic for regulon construction and Texp versus Tper classification. After transcriptomes were transformed into regulomes, regulon scores were calculated to train a machine-learning model to classify CAR T cells from Texp and Tper. Key regulons were identified based on importance for the model’s predictions. Created in BioRender. Hu, Y. (2025) https://BioRender.com/3ltrr3g. f Bar graph depicting the SHapley Additive exPlanation (SHAP) values for the top eight regulons underlying Texp and Tper predictions. g Tile map depicting normalized signature scores of regulons (columns) among different cell clusters at Texp and Tper (rows). Regulons were grouped as Texp-determining (left) and Tper-determining (right). Target networks for the top eight Texp-determining (h) and Tper-determining regulons (i). In each regulatory network, only the top differentially expressed genes are depicted. Each gene is colored according to log2 fold-change between expression in Texp (red) and Tper (blue). Source data are provided as a Source Data file.
To elucidate the immunological processes at each timepoint, CD8+ CAR T-cell transcriptomes at Texp and Tper from our seven patients were compared via GSEA (Fig. 4c and data file S1 for single-cell method, Fig. S9a and data file S2 for pseudo-bulk method) and DGEA (Fig. 4d, Fig. S9b, data file S3-4). CAR T cells at Texp upregulated gene sets for cell cycling and apoptosis, consistent with a short-lived phenotype. Upregulation of the cell cycling gene set remained true even when only comparing the proliferating cell cluster at Texp and Tper (Fig. S9d). This indicates that, while proliferating cells are present at both Texp and Tper, those at Texp are more proliferative than those at Tper. We also observed Texp-specific and cluster-independent upregulation of genes related to T-cell activation (including CD69, CD44, CD74) and exhaustion (including PRDM1, CTLA4, NR4A2), implicating antigen-engagement and CAR-mediated signaling during Texp. On the other hand, CAR T cells at Tper upregulated gene sets for cytotoxicity and immune regulation. Expression of cytotoxicity-related genes (including KLRB1, FCGR3A, PRF1, GZMB, NKG7) was restricted to TE and exhausted-like TE clusters, indicating that this gene signature arises from Tper-specific preponderance of the terminal effector phenotype. We also observed Tper-specific upregulation of regulatory genes, including GTPase immune-associated proteins (GIMAP5, GIMAP7) which predict long-term persistence33, as well as sphingosine-1-phosphate receptors (S1PR1, S1PR5) which indicate blood localization34. These signatures suggest that CAR T cells at Tper are functional and capable of long-term persistence in peripheral blood. Lastly, downregulation of genes related to T-cell activation and exhaustion implicate decreased in vivo antigen load and/or CAR-mediated signaling during Tper.
Interestingly, some differentially expressed gene signatures between CD8+ CAR T cells at Texp and Tper pertained to cytokine signaling (Fig. 4c, d, see Fig. S9c). CAR T cells at Texp upregulated TNF response genes (including NFKBIA, NFKBIE, DUSP4, RELB). TNF can be secreted by CAR T cells for autocrine signaling20, predicts efficacious CAR T cells20, and contributes to CRS35. On the other hand, CAR T cells at Tper upregulated type I interferon (IFN-I) response genes (including STAT1, IFITM1, IFITM2, IRF7). Many IFN-I response genes have known antiviral functions. The temporally specific and cluster-independent upregulation of TNF and IFN-I response genes indicates a dynamic in vivo cytokine environment during the CAR T-cell immune response.
Next, we investigated Texp- and Tper-specific regulatory networks using a machine-learning model (Fig. 4e, see Methods). Single-cell transcriptomes were transformed into regulomes to calculate regulon scores. A machine-learning model was trained using the regulon scores to classify CD8+ CAR T cells at Texp and Tper. This strategy resulted in high (~90%) classification accuracy across patients (Fig. S10a). Top Texp- and Tper-determining regulons were identified based on each regulon’s importance (SHAP value) for the model’s prediction (Fig. 4f, data file S5). Sizes of timepoint-determining regulons span approximately 10–1000 genes (Fig. S10b). Regulon expression was compared between cell clusters and timepoints (Fig. 4g). Texp- and Tper-determining regulons were broadly upregulated at Texp and Tper, respectively. However, expression of some regulons was cluster-specific (e.g., FLI1 regulon among EM clusters).
The top three Texp-determining regulons (for JUND, RELB, BHLHE40) exhibited higher expression among non-proliferating clusters and included co-regulated inflammation-associated genes (including NFKBIA, TNFAIP3, DUSP5) (Fig. 4h, see Fig. S11a)36. The RELB and BHLHE40 genes themselves are included in the JUND regulon, indicating they may be directly upregulated by JUND. These three regulons were upregulated at Texp and suggest response to pro-inflammatory cytokines (such as TNF, discussed above) or CAR signaling. BHLHE40 rewires mitochondrial metabolism for tissue residency37, which may contribute to CAR T-cell fitness after entering the lymphoma. Shared genes between the JUND and BHLHE40 regulons included AP-1 transcription factors (JUN, JUNB, FOSL2), which have been implicated in T-cell and CAR T-cell exhaustion38,39, and may contribute to the exhausted-like EM phenotype that predominates the first clonal wave at Texp. Among remaining Texp-determining regulons, FLI1 is notable for antagonizing effector T-cell differentiation40. Consistent with known biology, expression of the FLI1 regulon was upregulated among EM clusters, downregulated among TE clusters, and did not significantly change between Texp and Tper within TE clusters.
On the other hand, Tper-determining regulons were more obscure (Fig. 4i, see Fig. S11b). Among top Tper-determining regulons, KLF13 regulates T-cell apoptosis41, BPTF activates a stemness gene expression program42 and maintains peripheral T-cell homeostasis43, and ELF4 promotes memory CD8+ T cells by regulating quiescence44. Altogether, the putative functions of the KLF13, BPTF, and ELF4 regulons may underlie the delayed expansion and persistence of the second clonal wave at Tper. Moreover, these functions indicate restoration of immune homeostasis during Tper. The TBX21 regulon was notable for effector-associated genes (including NKG7, ZEB2) and was upregulated within non-CM clusters, consistent with preponderance of the TE phenotype at Tper. The TBX21 regulon also includes S1PR5, which is a known T-BET target45 and retains T cells in peripheral blood34. Lastly, the STAT1 regulon was notable for interferon-associated antiviral genes (including OAS2, ISG20), in concordance with upregulation of IFN-I signaling gene sets at Tper.
CD8+ CAR T cells at Texp and Tper originate from distinct precursors in the infusion product
The distinct clonotypes, phenotypes, transcriptional signatures, and regulatory networks underlying the two waves of CD8+ CAR T cells led us to hypothesize that they originate from distinct precursors in the infusion product. To test this hypothesis, we first investigated infusion product phenotypes. After filtering for infusion product CD8+ T cells, we identified four clusters based on gene expression density (Fig. 5a, b, Fig. S12b) and levels (Fig. 5c, Fig. S12a): proliferating (high MKI67), activated naïve-like (naïve-act, high TCF7), type 1 effector (EFF-Tc1, high TBX21), and type 2 effector (EFF-Tc2, high GATA3) T cells. The TCF7hi naïve-act cluster upregulated markers of naiveness (IL7R, LEF1, CCR7, CD45RA) and T-cell activation (CD38, CD95), consistent with an early activated or stem cell-like memory phenotype. The two EFF clusters upregulated cytolytic molecules (GZMB, PRF1, GNLY) and downregulated markers of naiveness (IL7R, LEF1). EFF-Tc1 and EFF-Tc2 were distinguished by expression density of lineage-specific transcription factors (TBX21 for Tc1, GATA3 for Tc2) and receptors (KLRG1 for Tc1, CCR4 for Tc2). The EFF-Tc1 cluster exhibited lower CAR transgene expression than the EFF-Tc2 cluster (Fig. S12b, c), which may impact their functional phenotypes in vivo46,47. The three non-proliferating clusters upregulated IRF7 and anti-viral genes (ISG20, IFITM1, IFITM2), indicating type I interferon signaling during ex vivo transduction and/or expansion. Cluster proportions varied between patients (Fig. S12d). In general, proliferating T cells (54%) were the most prevalent, while proportions of non-proliferating phenotypes (naïve-act, 14%; EFF-Tc1, 11%; EFF-Tc2, 21%) were mutually comparable (Fig. 5e, top row).

a UMAP depicting single-cell transcriptomes of infusion product CD8+ T cells colored by cell cluster. Density maps (b) and violin plots (c) depicting expression levels of key genes and proteins for annotation and phenotyping. For extended version, see Fig. S11a, b. d Cartoon depicting identification of Pre-Texp and Pre-Tper using endogenous TCR clonotypes as unique indices. Created in BioRender. Hu, Y. (2025) https://BioRender.com/49klewt. e Stacked bar graph depicting proportion distribution of all infusion product (top row) or precursors of peripheral blood CD8+ CAR T cells (bottom two rows) among the four cell clusters. f Colored UMAPs depicting distribution of Pre-Texp, Pre-Tper, and non-linked infusion product cells (“IP only”) on the overall UMAP. g Tile map depicting normalized expression of genes (columns) among Pre-Texp, Pre-Tper, and IP only infusion product cells (rows). Genes were manually grouped into modules according to known functions. h Violin plots depicting expression levels of select differentially expressed gene sets between Pre-Texp (n = 1813 cells), Pre-Tper (n = 2502 cells), and IP only (n = 8464 cells) infusion product cells. Expression levels were compared by two-sided Wilcoxon Rank-Sum test with p values adjusted for multiple hypotheses testing using the Benjamini-Hochberg method, whereby **** indicates p < 0.0001 and ** indicates p < 0.01. The central line indicates the median. The bounds of the box indicate the 25th–75th percentile. The length of the whiskers indicates 1.5 times the inter-quartile range from the first and third quartiles. i Gene set enrichment analysis comparing Pre-Texp and Pre-Tper. Gene sets were ordered according to direction of upregulation and magnitude of enrichment. j Enrichment plots for select gene sets differentially expressed between Pre-Texp and Pre-Tper infusion product cells. Enrichment scores are computed by the Kolmogorov-Smirnov test with false discovery rate (FDR) adjusted q-values. k Cartoon depicting fates of CD8+ CAR T cells over the entire course of therapy, from infusion product precursors to peripheral blood CAR T cells at Texp and Tper. Created in BioRender. Hu, Y. (2025) https://BioRender.com/3dtq7c4. NES normalized enrichment score. Source data are provided as a Source Data file.
Having established infusion product phenotypes, we next performed TCR lineage tracing analysis to developmentally link infusion product CAR T cells with peripheral blood CAR T cells using their endogenous TCR clonotypes as unique indices. This method delineates infusion product precursors of CD8+ CAR T cells at Texp (“Pre-Texp”) and at Tper (“Pre-Tper”) (Fig. 5d). Both Pre-Texp and Pre-Tper comprised all four infusion product clusters (Fig. 5e, bottom rows) and largely overlapped on the UMAP (Fig. 5f). Hence, we conclude that coarse cluster phenotype alone cannot accurately predict in vivo differentiation. Compared to T cells not linked to peripheral blood (“IP only”), Pre-Texp and Pre-Tper cells upregulated expression of effectorness (including GZMB, TNF, GNLY) and type 1 polarization (including IFNG, KLRG1) genes, as well as lower expression of stemness (including SELL, IL7R) and type 2 polarization (including GATA3, IL4R) genes (Fig. 5g). The significance of type 1 versus type 2 polarized CD8+ CAR T cells is not well-understood in the literature and warrants future studies. Pathway analysis indicated that Pre-Texp and Pre-Tper upregulated gene sets for T-cell effector function (“PD-1 signaling”, “T-cell-mediated cytotoxicity”), response to interferon γ (“interferon γ signaling”, “antigen processing and presentation”), and DNA replication, whereas non-linked CAR T cells upregulated gene sets for stemness (Fig. 5h, data file S6). Our discovery of CD8+ CAR T-cell precursors with an effector-like phenotype is corroborated by findings from Thomas et al., who also identified effector-like precursors from CAR T-cell patients treated for B-cell acute lymphoblastic leukemia48. Possible fates for the non-linked, naïve-like, IP-only CAR T cells include extravasation into the lymphoma, expansion in peripheral blood outside the Texp-Tper window (day 8–28), or failure to persist in vivo.
Lastly, we directly compared transcriptomic signatures of Pre-Texp and Pre-Tper via GSEA. Although both Pre-Texp and Pre-Tper broadly exhibited an effector phenotype (Fig. 5g, h), Pre-Texp upregulated effectorness-associated gene sets (including TCR, PD-1, interferon γ, and IL-1 signaling), whereas Pre-Tper upregulated stemness-associated gene sets (Fig. 5i). In concordance, DGEA showed that Pre-Texp upregulated effector molecules (GZMB, GZMK, FCGR3A, NKG7, KLRG1), AP-1 transcription factors (JUN, JUND, FOS), and MHC class II expression (HLA-DRB1, HLA-DRB5, HLA-DRA), whereas Pre-Tper upregulated naïve-like markers (SELL, IL7R, S1PR1) (Fig. S12e, data file S7). Upregulation of JUN in Pre-Texp confers exhaustion resistance38, potentially underlying their eventual efficacy at Texp. Interestingly, Pre-Tper exhibited greater CAR transgene expression (Fig. S12e), which may reflect transduction differences among apheresis precursors or a link between CAR expression and in vivo differentiation47. Next, we compared Pre-Texp and Pre-Tper using transcriptomic signatures from a human CD8+ differentiation atlas from Giles et al.49. Pre-Texp more closely resembled an effector memory state, whereas Pre-Tper more closely resembled either a stem-cell memory or central memory state (Fig. 5j). Altogether, these patterns indicate that, while both Pre-Texp and Pre-Tper exhibited an effector phenotype in the infusion product, Pre-Texp were more differentiated with greater effectorness, whereas Pre-Tper were less differentiated with greater stemness.
In conclusion, TCR lineage tracing analysis supports our hypothesis that CD8+ CAR T cells at Texp and Tper originate from different infusion product precursors. Integrating these findings into the two-stage differentiation model paints a more complete picture of CD8+ CAR T cells over the month following CAR T-cell administration (Fig. 5k). Effector CD8+ CAR T cells exist along a gradient of effectorness and stemness in the infusion product. Following infusion into the patient, effector CAR T cells with greater effectorness rapidly expand until peak expansion (Texp, days 8–12), adopting a functional and cytotoxic EM phenotype with exhaustion-like characteristics upon antigen stimulation. Lymphoma infiltration and in vivo killing50, CRS5, and ICANS5 coincide with the Texp timeframe, suggesting that these CAR T cells mediate tumor clearance and side effects. Subsequently, this first wave of expanded CAR T cells diminishes through apoptosis or extravasation. Simultaneously, the remaining effector CAR T cells with greater stemness from the infusion product expand during the post-peak persistence timeframe (Tper, day 21–28). These newer and longer-lived CAR T cells adopt effector characteristics and persist in vivo, where they may ensure a durable response through long-term immunosurveillance.
CD8+ Exhausted-like EM CAR T cells at Texp exhibit characteristics of early exhaustion
Discovery of the predominant exhausted-like EM phenotype in peripheral blood at Texp may be surprising since exhaustion was conventionally defined in lymphoid organs or tumor-infiltrating lymphocytes51. To verify this annotation, we further characterized the molecular signatures of the exhausted-like EM cluster. The exhausted-like EM cluster was densely situated at the lower half of the UMAP (Fig. 6a, b). We used RNA-seq/CITE-seq to examine gene and protein expression density, respectively (Fig. 6c, Fig. S13a for additional markers). The exhausted-like EM cluster highly expressed exhaustion-associated transcription factors (NR4A2, TOX, IRF4) and inhibitory receptors (ENTPD1, PDCD1, TIGIT, LAG3, CTLA4). Intermediate expression of memory (TCF7, LEF1, CD27, IL7R) and effector (TBX21, GZMB, PRF1, GNLY, IFNG, NKG7) genes was also observed. Additionally, the cluster exhibited low expression of CX3CR1 (effector lineage marker) and B3GAT1 (senescence marker). CITE-seq measurements of protein expression for cell surface receptors (including CD39, PD-1, TIGIT, LAG3, CD27, CD57, CD127, TIM-3, CXCR3) were largely consistent with corresponding gene expression. Sequencing data indicated that high expression of CD39 (protein for ENTPD1) and low expression of CD57 (protein for B3GAT1) most clearly differentiated the exhausted-like EM cluster from other clusters. To validate these characteristics, flow cytometry was employed to analyze a set of longitudinal patient PBMCs (Fig. S13b for gating). Consistent with sequencing data, flow cytometry data demonstrated that the CD39+CD57– phenotype was highest among CD8+ CAR T cells at Texp (75%) compared to at Tper1 (33%) and Tper2 (18%) (Fig. 6d, top row). Furthermore, flow cytometry showed decreased CX3CR1 expression at Texp, and increased expression at Tper1 and Tper2, in agreement with sequencing data (Fig. 6d, bottom row). This mixed expression pattern of exhaustion, memory, and effector markers resembles the circulating PD1+CD39+ Tex cells described by the Wherry group’s human T-cell differentiation atlas49.

UMAP depicting single-cell transcriptomes of peripheral blood CD8+ T cells colored by cell cluster (a) or density contours of each cluster (b). The exhausted-like EM cluster is located on the lower half of the UMAP. c Density maps depicting expression levels of major T-cell genes and proteins, divided into categories. In the “receptor” category, proteins are placed directly beneath the corresponding gene. d Flow plots for validating transcriptomic data. Plots depict expression of CD39 and CD57 (top row) or CX3CR1 (bottom row) in CD8+ CAR T cells at each timepoint from P2. Exhausted-like EM CAR T cells were expected to be Texp-specific with high CD39 and low CD57/CX3CR1 expression. e, f Heat map depicting normalized expression of major gene sets from Wherry et al. (e). Expression of two of the gene sets were depicted as violin plots (f), ordered by decreasing expression level per cluster (EM, exh-like: n = 7163 cells; CM: n = 1883 cells, EM: n = 4937 cells; TE, exh-like: n = 552 cells; TE: n = 21019 cells). The central line indicates the median. The bounds of the box indicate the 25th–75th percentile. The length of the whiskers indicates 1.5 times the inter-quartile range from the first and third quartiles. Expression levels were compared to that of the cluster with highest expression via two-sided Wilcoxon Rank-Sum test, with p values adjusted for multiple hypotheses testing using the Benjamini-Hochberg method, whereby **** indicates p < 0.0001, *** indicates p < 0.001. g Volcano plot depicting differentially expressed genes found via two-sided Wilcoxon Rank-Sum test with Bonferroni adjustment between EM and exhausted-like EM CD8+ CAR T cells. Genes were colored according to direction of upregulation. h Enrichment plots for select gene sets differentially expressed between EM and exhausted-like EM CD8+ CAR T cells. Enrichment scores are computed by the Kolmogorov-Smirnov test with false discovery rate (FDR) adjusted q-values. Source data are provided as a Source Data file.
Next, we conducted GSEA using gene sets from Wherry et al. that identified T-cell differentiation states in the Armstrong/clone 13 LCMV model (Fig. 6e).52 These gene sets constitute comprehensive references for defining T-cell exhaustion because they (1) incorporate standing knowledge from the entire field, (2) integrate single-cell RNA-seq and ATAC-seq data from serial timepoints, and (3) originate from the Wherry group where comprehensive transcriptional signatures of T-cell exhaustion and most exhausted subsets were first defined. The two EM clusters (EM and exhausted-like EM) in our dataset correlated with different states from the Wherry group’s model. Our EM cluster resembled the “transitional I” T cells from the Armstrong model, indicating that these cells are not exhausted. Conversely, our exhausted-like EM cluster resembled the “precursor exhausted” T cells from the clone 13 model (Fig. 6f, left). Simultaneously, the exhausted-like EM cluster did not resemble the “terminally exhausted” T cells from the same model (Fig. 6f, right), which instead correlated with our exhausted-like TE cluster. Given clear differences between the EM and exhausted-like EM clusters, we directly compared their transcriptomic signatures through DGEA and GSEA. The exhausted-like EM cluster upregulated genes associated with T-cell activation (CD69, CD38, DUSP4, NFKBIA) and exhaustion (TOX, NR4A2, PRDM1, CTLA4) (Fig. 6g, data file S8). In contrast, the EM cluster upregulated genes associated with effector function (TBX21, CX3CR1, GZMB, PRF1, FCGR3A), memory (IL7R, S1PR1), and type I interferon signaling (STAT1, IFITM1, IFITM3). GSEA revealed that the exhausted-like EM cluster exhibited more exhaustion signatures and fewer effector signatures compared to the EM cluster (Fig. 6h, left column, Fig. S13c). GSEA also confirmed that the exhausted-like EM cluster resembled the “exhausted progenitor” and “exhausted precursor” subsets from Wherry et al. (whereas the EM cluster resembled the “transitional I” subset) (Fig. 6h, middle and right columns). Our findings suggest that the exhausted-like EM cluster exhibits robust gene and protein signatures of early exhaustion (justifying the “exhausted-like” annotation), yet is also phenotypically distinct from effector memory T cells.
Discussion
In this study, we use a combination of single-cell RNA-seq/CITE-seq/TCR-seq and longitudinal analyses to investigate CD28-costimulated CAR T-cell differentiation in seven patients with r/r DLBCL. Our findings are summarized in our two-stage model for CAR T-cell differentiation (Fig. 3g). Specifically, CD8+ CAR T cells undergo two distinct clonal expansion waves (at peak expansion and post-peak persistence timeframes), as revealed by clonotypic, phenotypic, and linked clonotypic-phenotypic analyses. The two waves are dominated by exhausted-like effector memory and terminal effector phenotypes, respectively. The exhausted-like effector memory annotation is supported by a CD39+CD57- flow phenotype, low CX3CR1 expression, and an early exhaustion signature. We also identify transcription factors and regulatory networks associated with the first wave (including JUND, RELB, BHLHE40, FLI1 regulons) and second wave (including KLF13, BPTF, ELF4, TBX1, STAT1 regulons). Lastly, lineage tracing analysis determined that CD8+ CAR T cells from both waves derive from effector precursors in the infusion product. However, effector precursors of the first wave exhibit more effector-like signatures, whereas effector precursors of the second wave exhibit more stem-like signatures (Fig. 5k), suggesting that pre-infusion heterogeneity mediates two-stage differentiation. Our two-stage model implies that manipulating the phenotypic composition of the infusion product may allow more precise control over in vivo CAR T-cell differentiation for modulating therapeutic efficacy.
Although two-stage differentiation is a phenomenon that has not yet been reported in the literature, the individual elements of our model are consistent with current understanding in the field. Previous studies on 4-1BB-costimulated CAR T-cell differentiation described bursts of CAR T-cell clonal expansion that lead to significant changes in the CAR T-cell clonal repertoire over time19,53. Our study not only generalizes this observation to CD28-costimulated CARs, but also directly pairs changes in the clonal repertoire with changes in T-cell phenotypes. Furthermore, previous reports have described CAR T-cell clones with memory-like phenotypes that exhibit delayed expansion after infusion23, which corroborates with the second clonal expansion wave at Tper. Finally, the Texp- and Tper-specific phenotypes described in our model are consistent with prior phenotyping studies25,27,48,54,55. We also report agreement between our transcriptomes at Texp and external data from Maus et al.27. By integrating our findings from multiple data modalities with established studies in the CAR T-cell field, we present a more complete and coherent two-stage model for in vivo CD28-costimulated CAR T-cell differentiation.
The two-stage differentiation model we present in this study has an important implication. CD28-costimulated CD8+ CAR T cells from the peak expansion and post-peak persistence timeframes are biologically uncoupled. This uncoupling significantly informs how we understand CAR T-cell expansion and persistence. Expansion facilitates rapid tumor clearance but can also cause CRS and ICANS5,17, while persistence facilitates long-term immunosurveillance but can also cause B-cell aplasia and hypogammaglobulinemia9,13,56. Our findings suggest that expansion and persistence, both of which serve complementary clinical purposes, are mediated by distinct CD8+ CAR T-cell populations. Effective and personalized CAR T-cell therapies balance expansion and persistence, taking into consideration each patient’s tumor burden, tolerance for side effects, and risk of relapse. Our findings suggest that expansion and persistence may be independently tuned to meet a patient’s needs. On the other hand, our findings also suggest that engineering CD8+ CAR T cells that simultaneously expand and persist competently may be challenging, given that these characteristics originate from uncoupled CAR T-cell populations in vivo.
In addition to clonal kinetics and phenotypic heterogeneity, we characterized Texp- and Tper-specific upregulation of TNF and IFN-I response gene sets and regulons, respectively. Notably, TNF-secreting CD8+ CAR T cells have been associated with complete responders20, suggesting that TNF may be signaling in an autocrine or paracrine manner during Texp. Upregulation of IFN-I response genes during Tper was intriguing. IFN-I response signatures in the apheresis of patients with B-cell acute lymphoblastic leukemia predict poor CAR T-cell persistence57, but IFN-I signaling may also enhance CAR T-cell efficacy in vivo58. Complicating this story further, we also identified IFN-I response signatures in the infusion product, which implies that interferons are secreted by an unknown source during ex vivo transduction and/or expansion. Hence, the possible roles of IFN-I in CAR T-cell differentiation are likely complex, context-dependent, or time-dependent, and warrant further mechanistic investigation.
We concluded our study by using single-cell TCR-seq to link CD8+ CAR T cells at Texp and Tper with effector precursors in the infusion product that exhibit more effector-like or more stem-like signatures, respectively. Discovery of effector precursors is externally corroborated by findings from Thomas et al.48. Importantly, this linkage suggests that the magnitude or duration of peak expansion and post-peak persistence can be modulated by manipulating the relative quantities of precursors in the infusion product. However, we cannot rule out the possibility that the in vivo peripheral blood environment in a patient with DLBCL also plays an instrumental role in determining whether a CAR T cell clonally expands at Texp or Tper. Future studies can investigate this possibility by analyzing interactions between CAR T cells and other blood cells (such as myeloid cells, B cells, or NK cells) at Texp and Tper.
The present study focuses on CD28-costimulated CAR T cells from complete responders, but two-stage differentiation may be more widely applicable. Others forms of adoptive cell therapy, such as 4-1BB-costimulated CAR T cells for B-cell leukemias59 or systemic lupus erythematosus60, may also exhibit a two-stage differentiation pattern in vivo. Indeed, our transcriptomic signatures at Texp were highly consistent with external data from day 7 tisagenlecleucel CD8+ CAR T cells from Maus et al27, hinting that two-stage differentiation may be more general. Different forms of adoptive cell therapy and disease contexts may exhibit different temporal dynamics or favor one wave over the other. Two-stage differentiation may also be aberrant in non-responders. Future studies are needed to explore potential differences in temporal dynamics, cell phenotypes, or major regulatory pathways at longitudinal timepoints in alternative therapeutic contexts.
Although this current study represents an advance for the CAR T-cell therapy field, we acknowledge four limitations. Firstly, we analyzed CAR T cells in two locations: peripheral blood and infusion product. However, CAR T cells are also found elsewhere, including lymph nodes and lymphoma foci. Importantly, CAR T-cell differentiation may be influenced by interactions with antigen-presenting cells in lymph nodes or with tumor cells in the lymphoma. Our study was ultimately limited by sample availability. Future studies can focus on less accessible locations and determine CAR T cell fates beyond peripheral blood. Secondly, we studied CAR T-cell intrinsic factors (RNAs, proteins, clonotypes) that can influence differentiation. However, differentiation may also depend on interactions between CAR T cells and a complex in vivo environment that includes other leukocytes (myeloid cells, B cells, NK cells), lymphoma cells, and cytokines. Thirdly, we focused on CAR T cells longitudinally between peak expansion and post-peak persistence timeframes. Hence, we cannot rule out additional waves of clonal expansion in the near (<1 week after infusion) or long (>1 month after infusion) terms. Lastly, we lack knowledge on whether two-stage differentiation applies to other contexts, such as non-responders, 4-1BB-costimulated CAR T cells, or TCR-transduced T cells. Future studies can explore alternative clinical contexts and test the generality of the two-stage differentiation model.
Methods
All methods complied with ethical guidelines established by the institutional review board at the University of Chicago Medicine.
Patient biospecimen collection
Deidentified biospecimens were obtained from a cellular therapy biobank in accordance with the institutional review board at the University of Chicago Medicine. Consent was obtained for biospecimen collection. Ethical guidelines were followed.
Longitudinal peripheral blood mononuclear cells (PBMCs) were collected from peripheral blood biospecimens by Ficoll-Paque PLUS (Cytiva, 95021-205) and stored in freezing media (RPMI, 10% FBS, 10% DMSO). Residual infusion product cells were collected from the patient’s spent infusion product bag and stored in CELLBANKER 1 (Amsbio, 11888). Cells were cryopreserved in liquid-phase nitrogen.
Generation of CD19-tetramers
Tetramers28 were constructed from AviTag-biotinylated human His-tagged CD19 (Acro Biosystems, CD9-H82E9) and Alexa Fluor 647-labeled streptavidin (BioLegend, 405237). Biotinylated CD19 was added to the tetrameric streptavidin at a 4:1 molar ratio for 30 min at 4 °C in the dark. This mixture was diluted with PBS to convenient concentrations for staining. For each batch of biotinylated CD19, quality controls were performed by generating CD19-tetramers and quantifying their ability to stain anti-CD19 CAR-transduced Jurkat T cells.
Single-cell RNA-seq/CITE-seq/TCR-seq
Cryopreserved biospecimens were thawed (RPMI, 10% FBS) and washed with cold FACS buffer (PBS, 2% BSA, 0.05% sodium azide). Fc receptors were blocked with Human TruStain FcX (BioLegend, 422301) at 1:50 dilution for 5 min at 4 °C. Then, cells were incubated for 30 min at 4 °C in the dark with a staining solution containing CITE-seq antibodies (described below), BV421-labeled anti-CD3ε (clone SK7, BioLegend, 344833), and AF647-labeled CD19-tetramers (3 nM final concentration) for CAR T-cell phenotyping and detection. Subsequently, stained cells were conjugated with LIVE/DEAD Fixable Near-IR viability dye (Invitrogen, L34975) at 1:1000 dilution in PBS for 5 min at room temperature. Finally, cells were washed three times in cold cell media (RPMI, 10% FBS) before fluorescence-activated cell sorting (BD Biosciences, FACSAria Fusion). CAR+ sorting gates were drawn based on fluorescence of PBMCs from a similarly stained healthy donor.
Sorted endogenous T cells (CD3+CAR-) and CAR T cells (CD3+CAR+) were separately partitioned into droplets for single-cell RNA-seq/CITE-seq/TCR-seq via Chromium Next GEM Single-Cell 5’Kit v2 (10x Genomics, 1000263). RNA-seq libraries were prepared according to manufacturer protocols. CITE-seq libraries were prepared via the 5’ Feature Barcode Kit (10x Genomics, 1000256). TCR-seq libraries were prepared via the Chromium Single-Cell Human TCR Amplification Kit (10x Genomics, 1000252). All libraries (RNA-seq, CITE-seq, TCR-seq) were quantified via the Qubit dsDNA HS Assay Kit (Invitrogen, Q32851), quality-checked for fragment sizes via high-sensitivity D5000 screentapes (Agilent, 5067-5592), pooled, and sequenced (Illumina, Novaseq-6000 and Nextseq-550).
CITE-seq antibody preparation
Thirty-one human “Cellular Indexing of Transcriptomes and Epitopes by Sequencing” (CITE-seq61) antibodies were obtained from BioLegend (TotalSeq-C reagents): anti-CD3ε (clone UCHT1, 300479), anti-CD5 (clone UCHT2, 300637), anti-TCRα/β (clone IP26, 306743), anti-TCRγ/δ (clone B1, 331231), anti-CD4 (clone SK3, 344651), anti-CD8α (clone SK1, 344753), anti-CD45RA (clone HI100, 304163), anti-CD45RO (clone UCHL1, 304259), anti-CCR7 (clone G043H7, 353251), anti-CD95 (clone DX2, 305651), anti-CD57 (clone QA17A04, 393321), anti-CD25 (clone BC96, 302649), anti-CD127 (clone A019D5, 351356), anti-CD103 (clone Ber-ACT8, 350233), anti-CXCR3 (clone G025H7, 353747), anti-CCR4 (clone L291H4, 359425), anti-CCR6 (clone G034E3, 353440), anti-PD-1 (clone EH12.2H7, 329963), anti-TIM-3 (clone F38-2E2, 345049), anti-LAG-3 (clone 11C3C65, 369335), anti-CD39 (clone A1, 328237), anti-TIGIT (clone A15153G, 372729), anti-CD27 (clone O323, 302853), anti-CD40L (clone 24-31, 310849), anti-GITR (clone 108-17, 371227), anti-OX40 (clone Ber-ACT35, 350035), anti-4-1BB (clone 4B4-1, 309839), anti-CD28 (clone CD28.2, 302963), mouse IgG1-κ isotype control (clone MOPC-21, 400187), mouse IgG2a-κ isotype control (clone MOPC-173, 400293), and mouse IgG2b-κ isotype control (clone MPC-11, 400381). All antibodies were diluted at a 1:100 ratio. Residual infusion product cells were stained with all 31 CITE-seq antibodies. Patient peripheral blood mononuclear cells were stained with all CITE-seq antibodies minus anti-CD3ε (30 in total) because T cells sorted from these samples were stained with BV421-labeled anti-CD3ε. To prepare the CITE-seq staining solution, the antibody pool was constructed and centrifuged at 14,000 × g for 10 min at room temperature in FACS buffer (PBS, 2% BSA, 0.05% sodium azide) to remove aggregates. The antibody supernatant was extracted and diluted with fresh FACS buffer to appropriate staining concentrations.
Analysis of CAR T-cell phenotype by flow cytometry
Cryopreserved biospecimens were thawed (RPMI, 10% FBS) and washed with cold FACS buffer (PBS, 2% BSA, 0.05% sodium azide). Fc receptors were blocked with Human TruStain FcX (BioLegend, 422301) at 1:50 dilution for 5 min at 4 °C. Then, cells were incubated for 30 min at 4 °C in the dark with a staining solution containing AF647-labeled CD19-tetramers (3 nM final concentration), BV421-labeled anti-CD3ε (clone SK7, BioLegend, 344833), AF488-labeled anti-CD8α (clone SK1, BioLegend, 344716), PE-labeled anti-CX3CR1 (clone 2A9-1, BioLegend, 341603), PE/Cy7-labeled anti-CD39 (clone A1, BioLegend, 328211), and BV605-labeled anti-CD57 (clone QA17A04, BioLegend, 393303). Subsequently, stained cells were conjugated with LIVE/DEAD Fixable Near-IR viability dye (Invitrogen, L34975) at 1:1000 dilution in PBS for 5 min at room temperature. Finally, cells were washed three times in cold FACS buffer (RPMI, 10% FBS) before flow cytometer analysis (BD Biosciences, FACSAria Fusion).
Single-cell RNA-seq data processing
RNA-seq and paired CITE-seq reads were aligned to the GRCh38 reference genome, which was modified with the CAR transgene sequence used in axicabtagene ciloleucel, and quantified using the cellranger count (10x Genomics, version 7.0.0)62. Only filtered gene/feature-barcode matrices that contained barcodes with unique molecular identifier (UMI) counts that passed the quality control were used for downstream analyses.
UMAP analysis and clustering on single-cell RNA-seq data
UMAP analysis and clustering were performed using the Seurat package (Version 4.3.0)63. Raw count matrices were first converted to Seurat objects before being further merged into one Seurat object, with protein expression added as the antibody-derived tag (ADT) assay. Cells with <300 genes detected or >10% mitochondrial RNA content were excluded from further analysis.
The raw count was log-normalized using the NormalizeData function with default options. The top 5000 variable features were then identified using the FindVariableFeatures function with the default “vst” method. The data were centered and scaled using the ScaleData function, with additional regression against 1) the percent of mitochondrial RNA content and 2) difference in cell cycle S-phase score and G2M-phase score. Scaled data were then used as input for principal component analysis (PCA) based on variable genes using the RunPCA function. Data harmonization to remove patient-specific effects was performed on the principal components using the Harmony package (Version 1.0) through the RunHarmony function64. Next, UMAP was constructed based on the first 30 harmony components. The same harmony components were used to construct the shared nearest neighbor (SNN) graph using the FindNeighbors function, which was then partitioned to identify clusters using the FindClusters function with default Louvain algorithm. These clusters were manually aggregated and classified as T-cell subsets based on known markers. CD8 and CD4 T-cell classifications were based on both gene and protein expression of CD4, CD8α, and CD8β.
For subset analysis, the corresponding subsets were extracted from the master Seurat object using the subset function. The above detailed preprocessing steps were repeated to generate the corresponding UMAP and subset annotations.
Gene expression visualization
Gene expression was visualized in three ways: violin plots, heatmaps, and density plots. Violin plots were made based on normalized expression. Heatmaps were made based on average scaled expression, both using Seurat internal functions. Density plots were constructed using the Nebulosa package (version 1.6.0) to enhance visibility and mitigate sparsity of gene expression on UMAP. This is necessary for datasets with large number of cells (e.g. our CD8+ CAR-T dataset has ~38k cells).
Differential gene expression (DEG) analysis on single-cell RNA-seq data
DEG analyses were by default performed using the FindMarkers function in Seurat package, with default parameters and the appropriate ‘ident.1” and “ident.2” set as contrast. Unless otherwise stated, the results were filtered with p_val_adj <0.05 and abs(avg_log2FC) > 0.25. Moreover, we also performed additional DEG analyses through a pseudo-bulk approach to better control for patient-specific effects, using the LibraDEG package (version 1.0.0)65 with default parameters (edgeR with LRT method). This was mainly performed when extracting timepoint-specific genes. Areas where this pseudo-bulk method is used are clearly stated in the figure legend.
Timepoint-specific gene signature validation
To validate the timepoint-specific molecular signatures shared across patients and against external data, we adopted two independent and complementary approaches: (1) correlation of timepoint-specific gene expression, (2) label transfer from our data to external data.
For the correlation approach, we first conducted pseudo-bulk DEG analyses of Texp vs Tper across all patients and kept all the statistically significant genes (padj <0.05) as timepoint-specific genes. Note that we did not filter by log fold-change to include even weak timepoint-specific signatures. Essentially, the goal here is to remove all non-timepoint-specific genes that may confound correlation analysis. The normalized gene expressions of these timepoint-specific genes were extracted from both our data and the external data. Following that, average expression was calculated for each sample (patient by timepoint). Correlations were then calculated pairwise for all samples.
For the label transfer approach, we adapted the Seurat standard data integration and label transfer workflow with default parameters. This is a complementary approach where, rather than at patient-level expression, individual cells in the external data and our data could be compared and similar timepoint-labels could be assigned.
GSEA and pathway enrichment analysis
Gene Set Enrichment Analysis (GSEA) and pathway enrichment analysis were carried out using the clusterProfiler (version 4.4.4)66 package based on the msigdb database built in msigdbr package (version 7.5.1)67.
Exhaustion signatures from reference datasets
Besides standard msigdb exhaustion-related pathways, we obtained T-cell subset-specific signatures from Wherry et al. (see Supplementary Table 1 in their study)52. The reference study provides the most updated and comprehensive CD8+ (exhausted) T cell subsets, in the standard LCMV/Cl13 mouse models where Tex was first defined. To refine the signature, we converted the mouse genes to their human orthologous genes and filtered the list to retain only the top 500 (by descending avg_log2_FC) statistically significant (p_val_adj <0.05) genes for each subset.
Regulon analysis
To construct regulons in a specific cell subset, we first extracted the raw count matrix of that subset. Then, we supplied it as input for the pySCENIC package (version 0.10.4) and ran through the workflow as detailed in its documentation and publication68,69. More specifically, the base script was adapted from the “PBMC10K” example script on the pyScenic website70. In addition, the arboreto component was used to speed up analysis. Each resultant regulon is a gene list with a central transcription factor and all its putative target genes determined through the SCENIC algorithm, and each set of regulons is sample/subset-specific and represents a putative regulatory unit in that specific sample/subset.
Regulon or general gene set signature scoring
Individual cells were scored using the AUCell package (version 1.10.0)68 for a particular gene set from the msigdb database or from a single-cell-derived regulon as follows. The normalized gene expression was first used as input into the AUCell_buildRankings function to score each cell for gene set enrichment and to build a ranking matrix. The signature score was then calculated as an AUC score using the AUCell_calcAUC function with all default parameters. In later revisions, Ucell package (version 2.0.1)71 was used in some re-analyses, as it is computationally more efficient and generates similar results as the AUCell package.
Machine learning classification on regulon scores
To classify CD8+ CAR T cells at Texp and Tper, we first built a regulon set for each subset independently, assuming that regulatory networks for each timepoint may be different. Then, we merged all regulon sets into a master regulon set. We calculated the regulon scores using the master regulon set for all cells from the two subsets, resulting in a regulon score matrix. Each cell then served as an observation and each regulon as a feature for machine learning.
The machine learning classification models were built in the caret framework using the caret package (version 6.0–90)72. Briefly, we divided the input data into training (75%), validation (15%), and test sets (10%). We trained and optimized xgboost models73 through grid hyperparameter search and with a 5-fold cross-validation until reaching a best accuracy of >85% (refer to figures for specific accuracies).
We interrogated the final models for feature importance evaluation using the Shapley Additive Explanation (SHAP) method74 implemented in the SHAPforxgboost package (version 0.1.1)75. Average SHAPley values were used to rank the regulons. Top timepoint-determining regulons were visualized and further analyzed.
Single-cell TCR-seq data processing
The TCR-seq reads from each sample were aligned to the 10x curated GRCh38 vdj reference genome and quantified using the cellranger vdj (10x Genomics, version 6.0.0). The results were then aggregated using the cellranger aggr function, with the source patient (donor) and timepoint (origin) of each sample supplied in the metadata to guide the clonotype assignment. The resultant clonotype and filtered contig annotation data were used for downstream analyses.
Single-cell TCR-seq tracing and phenotype linking
Each unique clonotype is defined by the amino acid and nucleotide sequence of CDR3 regions for paired productive TCRα and TCRβ chains. TCR clonality overlap and tracing analyses were carried out using the immunarch package (version 0.9.0)76. Clones with the same TCR CDR3 sequences from the same patient at different timepoints were re-grouped and traced from infusion product to the post-infusion samples.
Phenotype-to-clonotype linking: To identify Texp and Tper precursors, clonotypes among post-infusion Texp and Tper cell populations were identified respectively and traced back to the infusion product cells. When linking the TCR clonotype to the corresponding cell phenotype, we keep only data with matching cell barcodes from both the TCR-seq and RNA-seq data.
Abundance tracing: Clonal abundance for each CAR T clone is calculated as the percentage of all CAR T cells occupied by a clone at a given timepoint. Each clone was assigned a phenotype at each timepoint based on the most frequently occurring phenotype. Texp clones that were predominantly EM-exh had their abundances tracked at Tper1/Tper2. Tper1/Tper2 clones that were predominantly TE had their abundances tracked at Texp.
CAR transgene mapping
The axicabtagene ciloleucel CAR design is documented77. Its sequence was confirmed by Sanger sequencing of genomic DNA extracted from axicabtagene ciloleucel infusion products. The CAR sequence was then added to the GRCh38 FASTA and GTF files accordingly. A custom reference for cellranger was built from these annotation files using cellranger mkref (10x Genomics, version 7.0.0). The resultant custom reference was used for CAR transgene mapping through cellranger count.
Inclusion and ethics statement
Roles and responsibilities were agreed amongst collaborators in advance. Our findings are not expected to result in stigmatization, incrimination, or discrimination.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All single-cell RNA-seq, CITE-seq, and TCR-seq data have been deposited in the Gene Expression Omnibus (GEO) and are available with the accession code GSE207192. Source data are provided with this paper.
Code availability
For reproducibility, code used for our analysis and figure-making is available on Zenodo: https://doi.org/10.5281/zenodo.15116870.
References
Diffuse Large B-Cell Lymphoma - Cancer Stat Facts. https://seer.cancer.gov/statfacts/html/dlbcl.html
Liu, Y. & Barta, S. K. Diffuse large B‐cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 94, 604–616 (2019).
Longo, D. L., Sehn, L. H. & Salles, G. Diffuse large B-cell lymphoma. https://doi-org.proxy.uchicago.edu/10.1056/NEJMra2027612, 384, 842–858 (2021).
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Schuster, S. J. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380, 45–56 (2019).
Abramson, J. S. et al. Pivotal safety and efficacy results from transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood 134, 241–241 (2019).
Abramson, J. S. et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396, 839–852 (2020).
Locke, F. L. et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 20, 31–42 (2019).
Schubert, M. L. et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 32, 34–48 (2021).
Milone, M. C. et al. Engineering-enhanced CAR T cells for improved cancer therapy. Nat. Cancer 2021 28 2, 780–793 (2021).
Good, Z. et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat. Med. 28, 1860–1871 (2022).
Gupta, A. & Gill, S. CAR-T cell persistence in the treatment of leukemia and lymphoma. https://doi-org.proxy.uchicago.edu/10.1080/10428194.2021.1913146, 62, 2587–2599 (2021).
Kawalekar, O. U. et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016).
Salter, A. I. et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 11, 1–18 (2018).
Fraietta, J. A. et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571 (2018).
Rossi, J. et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132, 804–814 (2018).
Deng, Q. et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 26, 1878–1887 (2020).
Melenhorst, J. J. et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 602, 503–509 (2022).
Finney, O. C. et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Invest 129, 2123–2132 (2019).
Neelapu, S. S. CAR-T efficacy: is conditioning the key? Blood 133, 1799–1800 (2019).
Turtle, C. J. et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 126, 2123–2138 (2016).
Fraietta, J. A. et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 1 https://doi.org/10.1038/s41586-018-0178-z (2018).
Michelozzi, I. M. et al. The enhanced functionality of low-affinity CD19 CAR T cells is associated with activation priming and polyfunctional cytokine phenotype. Blood 136, 52–53 (2020).
Jackson, Z. et al. Sequential single cell transcriptional and protein marker profiling reveals TIGIT as a marker of CD19 CAR-T cell dysfunction in patients with non-Hodgkin’s lymphoma. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-21-1586/696778/SEQUENTIAL-SINGLE-CELL-TRANSCRIPTIONAL-AND-PROTEIN (2022).
Sheih, A. et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 11, 1–13 (2020).
Haradhvala, N. J. et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 28, 1848–1859 (2022).
Hu, Y. et al. Antigen multimers: Specific, sensitive, precise, and multifunctional high-avidity CAR-staining reagents. Matter 4, 1–24 (2021).
Lesch, S. et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021 511 5, 1246–1260 (2021).
Davis, A. M., Ramos, H. J., Davis, L. S. & Farrar, J. D. Cutting edge: A T-bet-independent role for IFN-α/β in regulating IL-2 secretion in human CD4 + central memory T cells. J. Immunol. 181, 8204–8208 (2008).
Kim, H. J. et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science (80-) 350, 334–339 (2015).
Lee, J. C. et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 71, 2871–2881 (2011).
Ciucci, T. & Bosselut, R. Gimap and T cells: A matter of life or death. Eur. J. Immunol. 44, 348–351 (2014).
Evrard, M. et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J. Exp. Med. 219, (2021).
Zhang, L. et al. Etanercept as a new therapeutic option for cytokine release syndrome following chimeric antigen receptor T cell therapy. Exp. Hematol. Oncol. 10, 4–7 (2021).
Daniels, M. A., Luera, D. & Teixeiro, E. NFκB signaling in T cell memory. Front. Immunol. 14, 1–12 (2023).
Park, S. L. & Mackay, L. K. Bhlhe40 keeps resident T cells too fit to quit. Immunity 51, 418–420 (2019).
Lynn, R. C. et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019).
Stelekati, E. et al. Long-term persistence of exhausted CD8 T cells in chronic infection is regulated by MicroRNA-155. Cell Rep. 23, 2142–2156 (2018).
Chen, Z. et al. In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280.e22 (2021).
Zhou, M., Clayberger, C. & Krensky, A. M. Krüppel-like transcription factor 13 regulates T lymphocyte survival in vivo. J. Immunol. 178, 5496–5504 (2007).
Xu, B. et al. The chromatin remodeler BPTF activates a stemness gene-expression program essential for the maintenance of adult hematopoietic stem cells. Stem Cell Rep. 10, 675–683 (2018).
Wu, B. et al. BPTF is essential for T cell homeostasis and function. J. Immunol. 197, 4325–4333 (2016).
Mamonkin, M., Puppi, M. & Lacorazza, H. D. Transcription factor ELF4 promotes development and function of memory CD8+ T cells in Listeria monocytogenes infection. Eur. J. Immunol. 44, 715–727 (2014).
Jenne, C. N. et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J. Exp. Med. 206, 2469–2481 (2009).
Walker, A. J. et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther. 25, 2189–2201 (2017).
Rodriguez-Marquez, P. et al. CAR density influences antitumoral efficacy of BCMA CAR T cells and correlates with clinical outcome. Sci. Adv. 8 https://www.science.org (2022).
Wilson, T. L. et al. Common trajectories of highly effective CD19-specific CAR T cells identified by endogenous T cell receptor lineages. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-21-1508/3166017/cd-21-1508.pdf (2022).
Giles, J. R. et al. Human epigenetic and transcriptional T cell differentiation atlas for identifying functional T cell-specific enhancers. Immunity 55, 557–574.e7 (2022).
Chen, P. H. et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight 5, (2020).
Blank, C. U. et al. Defining ‘T cell exhaustion. Nat. Rev. Immunol. 19, 665–674 (2019).
Giles, J. R. et al. Shared and distinct biological circuits in effector, memory and exhausted CD8+ T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat. Immunol. 23, 1600–1613 (2022).
Shah, N. N. et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 3, 2317–2322 (2019).
Goldberg, L. et al. Single-cell analysis by mass cytometry reveals CD19 CAR T cell spatiotemporal plasticity in patients. Oncoimmunology 11, (2022).
Li, X. et al. Single-cell transcriptomic analysis reveals BCMA CAR-T cell dynamics in a patient with refractory primary plasma cell leukemia. Mol. Ther. 29, 645–657 (2021).
Chavez, J. C., Bachmeier, C. & Kharfan-Dabaja, M. A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 10, 2040620719841581 (2019).
Chen, G. M. et al. Integrative bulk and single-cell profiling of pre-manufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov. 1677.2020 https://doi.org/10.1158/2159-8290.cd-20-1677 (2021).
Zhao, Z. et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28, 415–428 (2015).
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Mackensen, A. et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 28, 2124–2132 (2022).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017 149 14, 865–868 (2017).
What is Cell Ranger? -Software -Single Cell Gene Expression -Official 10x Genomics Support. https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger.
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, (2021).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. 2, (2021).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Aibar, S. et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020 157 15, 2247–2276 (2020).
PBMC10k_downstream-analysis. http://htmlpreview.github.io/?. https://github.com/aertslab/SCENICprotocol/blob/master/notebooks/PBMC10k_downstream-analysis.html.
Andreatta, M. & Carmona, S. J. UCell: Robust and scalable single-cell gene signature scoring. Comput. Struct. Biotechnol. J. 19, 3796–3798 (2021).
Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 28, 1–26 (2008).
Tianqi, C. & Guestrin, C. XGBoost: A scalable tree boosting system. KDD (2016).
Lundberg, S. M. & Lee, S. I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst. 2017, 4766–4775 (2017).
Liu, Y. & Just, A. SHAPforxgboost: SHAP Plots for ‘XGBoost’. (2020).
ImmunoMind Team. immunarch: An R package for painless bioinformatics analysis of T-cell and B-cell immune repertoires. https://doi.org/10.5281/zenodo.3367200 (2019).
Kochenderfer, J. N. et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 32, 689–702 (2009).
Acknowledgements
We thank Phi Beta Psi, the Ullman Fund in Cancer Immunology, the Hoogland Lymphoma Research Pilot Projects, Chicago Immunoengineering Innovation Center, American Cancer Society Scholar Award (SG-22-136-01-1BCD) and NIH New Innovator award (1DP2AI144245) for financial support (to J.H.). Y.H. was supported by the University of Chicago MSTP Training Grant (T32GM007281). T.P. was supported by the University of Chicago CSTR training grant (T32HL007381). N.A. was supported by the University of Chicago MTCR training grant (T32CA009594). This project was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health through Grant Number 5UL1TR002389-02 that funds the Institute for Translational Medicine. We thank the UChicago Human Immunologic Monitoring Facility for patient biospecimen cryopreservation, as well as the Pritzker School of Molecular Engineering Single-Cell Immunophenotyping Core and UChicago Genomics Facility for instrumentation to generate single-cell sequencing data. This work was completed in part with resources provided by the University of Chicago’s Research Computing Center. The authors thank Dr. Karen M. Watters for scientific editing of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.H., G.C., J.P.K., and J.H. conceived and designed all experiments. J.H. supervised the project. T.A., P.A.R., and M.R.B. coordinated patient biospecimens for these studies. G.C. and T.P. performed all single-cell and machine-learning analyses. Y.H. organized execution of all experiments. Y.H. and E.T. sorted CAR T cells and generated single-cell data. G.C. and Y.H. jointly interpreted data. N.A. and Y.H. designed cartoons. E.T. performed flow cytometry validation. Y.H. and G.C. prepared the manuscript. J.H. and J.P.K. edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
P.A.R. reports Research Support/Funding: BMS, Kite Pharma, Inc./Gilead, MorphoSys, Calibr, Tessa Therapeutics, Fate Therapeutics, Xencor, and Novartis Pharmaceuticals Corporation. Speaker’s Bureau: Kite Pharma, Inc./Gilead; Consultancy on advisory boards: AbbVie, Novartis Pharmaceuticals Corporation, BMS, Janssen, BeiGene, Karyopharm Therapeutics Inc., Takeda Pharmaceutical Company, Kite Pharma, Inc./Gilead, Sana Biotechnology, Nektar Therapeutics, Nurix Therapeutics, Intellia Therapeutics, and Bayer. Honoraria: Novartis Pharmaceuticals Corporation. M.R.B. reports Membership on an Advisory Board or Consultancy for Kite/Gilead, Novartis, CRISPR Therapeutics, Autolus Therapeutics, BMS, Incyte, Sana Biotechnology, Iovance Biotherapeutics. He has served on a Speakers Bureau for BMS, Kite/Gilead, Agios, and Incyte. J.P.K. receives research support from Merck, Verastem, and iTeos; has served on a speaker’s bureau for Kite/Gilead; and has served on advisory boards for Verastem, Seattle Genetics, MorphoSys, and Karyopharm. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, G., Hu, Y., Pan, T. et al. Two-stage CD8+ CAR T-cell differentiation in patients with large B-cell lymphoma. Nat Commun 16, 4205 (2025). https://doi.org/10.1038/s41467-025-59298-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59298-w
