
Abstract
The multifaceted roles and mechanisms of necroptosis in cancer cells remain incompletely understood. Here, we demonstrate that FGFR2 inhibition potently inhibits esophageal squamous cell carcinoma (ESCC) by inducing necroptosis in a RIP1/MLKL-dependent manner and show RIP3 is dispensable in this pathway. Notably, MST1 is identified as a necroptotic pathway component that interacts with RIP1 and MLKL to promote necroptosis by phosphorylating MLKL at Thr216. Additionally, FGFR2 inhibition induces Ser518 phosphorylation and triggers ubiquitin-mediated degradation of NF2, culminating in Hippo pathway suppression. Subsequently, YAP activation promotes RIP1 and MLKL transcriptional upregulation, further amplifying necroptosis. Intriguingly, IL-8 derived from necrotic cells stimulates peripheral surviving tumor cells to increase PD-L1 expression. Dual blockade of FGFR2/PD-L1 or FGFR2/IL-8-CXCR1/2 robustly impedes tumor growth in humanized mouse xenografts. Collectively, our findings delineate an alternative FGFR2-NF2-YAP signaling-dependent necroptotic pathway and shed light on the immunoregulatory role of FGFR2, offering promising avenues for combinatorial therapeutic strategies in clinical cancer management.
Similar content being viewed by others


Introduction
Esophageal squamous cell carcinoma (ESCC) is a highly aggressive cancer and the fourth leading cause of cancer-related deaths in China1. Despite decades of effort, the five-year survival rate remains low at 15–25%2. While chemotherapy is the standard treatment, its efficacy is often compromised by acquired resistance to apoptosis, resulting in treatment failure. This underscores the urgent need for strategies to overcome apoptosis resistance or to induce non-apoptotic forms of programmed cell death in ESCC.
Necroptosis is a key player in cancer biology and therapy, specifically in targeting cancer cells resistant to apoptosis. This form of programmed cell death is traditionally regulated by Receptor-Interacting Protein Kinase 1 (RIP1), which teams up with RIP3 to form the necrosome, leading to the phosphorylation of Mixed Lineage Kinase Domain-Like protein (MLKL)3. Activated MLKL then translocates to the plasma membrane, disrupting its integrity and executing necroptosis4,5. However, the pathways governing necroptosis remain under debate. In some scenarios, RIP1 is not essential for necroptosis, such as in responses to viral infections6, TNF/TLR3 induction7,8, or TAK1 deficiency9. Additionally, the interaction between RIP3 and DAI can mediate necroptosis10. Studies also indicate that MLKL-dependent necrosis can occur independently of RIP311, and RIP1/3 does not always impact MLKL-dependent necroptosis12,13. Furthermore, the initiation of inflammation in caspase8-deficient mice has been shown to proceed independently of RIP314, suggesting the presence of alternate regulatory pathways in necroptosis that warrant further exploration. Given the critical role of necroptosis in overcoming apoptosis resistance, exploring its mechanisms and potential targets is crucial for improving survival in ESCC.
Fibroblast growth factor receptors (FGFRs) form a family of four highly conserved transmembrane receptor tyrosine kinases (FGFR1–4). Receptor activation by FGFs initiates a cascade of context-dependent cellular outcomes, including proliferation, survival, migration, and differentiation15. FGFRs are frequently amplified in ESCC16,17, where their overexpression is correlated with poor patient survival18,19, revealing that FGFRs could serve as a potential therapeutic target for ESCC treatment. It is widely accepted that apoptosis induced by downstream pathways blockage is the major death type for FGFR inhibitors to eradicate cancer cells20. However, one study found that the cell death following FGFR blockage is caspase-independent21, and another reported that LY2874455, a pan-FGFR inhibitor, unexpectedly causes necrosis in cholangiocarcinoma cells via downregulating the expression of MCL122. This suggests a potential link between FGFR signaling and necroptosis, though the underlying mechanism remains unclear.
The Hippo signaling pathway, primarily a kinase cascade involving MST1/2 phosphorylation leading to LATS1/2 activation and subsequent YAP/TAZ inactivation23, is increasingly recognized for its role in modulating cell death. Another key player NF2, which facilitates LATS1/2 phosphorylation, has been shown to regulate receptor tyrosine kinase signaling24. While its involvement is well-established in apoptosis25,26 and ferroptosis27,28, its role in necroptosis has not been extensively studied. Recent research, however, indicates that TEAD1 deletion can trigger the necroptotic pathway in cardiomyocytes29, though the specific mechanisms remain to be elucidated.
In this study, we sought to uncover new regulatory mechanisms of necroptosis in ESCC. We also assess the potential anti-tumor effects of necroptosis inducers in cell-derived xenograft (CDX), patient-derived xenograft (PDX), and humanized mouse models. Our research uncovers an interaction between FGFR2 and the Hippo pathway in regulating necroptosis, proposing a targeted therapeutic strategy for ESCC.
Results
FGFR inhibition induces necroptosis
To identify potential therapeutic targets related to necroptosis in ESCC, we analyzed the top 100 genes from the TCGA database that are negatively correlated with the key molecule MLKL in esophageal cancer, focusing particularly on genes with available targeted therapies. Among these, previous research has identified links between necroptosis and the SHH pathway (SMO), calcium influx (TRPV4), autophagy (ULK1 and ULK2), and microtubules (MARK1 and SPAST). FGFR2 emerged as a gene of significant interest from the remaining genes (Fig. 1a). Given the frequent amplification and overexpression of FGFRs in ESCC17, which correlates with poor prognosis18, FGFRs are promising therapeutic targets. Additionally, there are no prior studies exploring FGFR2’s role in regulating necroptosis, making it an area worthy of investigation. We found that mRNA levels (Fig. 1b) and protein levels (Figs. 1c and S1a) of FGFR2 negatively correlated with MLKL in ESCC, leading to the hypothesis that FGFR2 might be linked to a previously unrecognized role in necroptosis. The FGFR1-3 inhibitor, AZD4547, was employed to inhibit FGFR2 activity, showing dose-dependent inhibition of ESCC cell line proliferation (Fig. S1b). Cell lines with high FGFR2 expression, KYSE140 and KYSE180, were selected for subsequent experiments.

a Diagram illustrating candidate genes regulating necroptosis in ESCC. Icons of this figure created in BioRender. Chen, D. (2025) https://BioRender.com/gvkj9ea. b Inverse correlation between FGFR2 and MLKL expression in ESCC (TCGA data). c Protein expression levels of FGFR2 and MLKL in esophageal cell lines. d Transmission electron microscopy of the KYSE140 cells treated for 12 h with DMSO or 10 μM AZD4547. Yellow arrowheads denote loosened nuclei. Red arrowheads indicate swelled cellular organelles. Blue arrowheads indicate plasma membrane rupture. Scale bar: 2 μm. e LDH release as a measure of cellular necroptosis post-AZD4547 treatment for 24 h. n = 3 biological replicates. f FACS analysis of Fluo4 fluorescent cells following 40 μM AZD4547 treatment for 24 h. g Cells were exposured to 40 μM AZD4547 or 0.3 mM EGTA for 24 h to determine the cell survival. n = 4 biological replicates. h Immunoblotting analysis post-24 h AZD4547 treatment. i Immunoblotting of membrane-cytosolic extracts following 24 h AZD4547 treatment. C cytoplasm, M membrane. j AZD4547 (12.5 mg/kg/d, by oral gavage) was given to mice bearing KYSE180 cells xenografts (n = 5 per group) or to PDX_ES0136 models (n = 6 per group) for 14 days. k Mice weights were measured every three days or twice weekly after treatment. All data are mean ± SD. Statistical analyses were performed using one-way ANOVA with multiple comparisons (e, g) or two-way ANOVA with multiple comparisons (j). Source data are provided as a Source Data file.
Post-AZD4547 treatment, transmission electron microscopic and microscopic assessment revealed typical necrotic features, such as disrupted cytoplasmic membranes, swollen mitochondria and cytoplasm, and cytoplasmic vacuolization (Figs. 1d and S1c). The dose-dependent increase in lactate dehydrogenase (LDH) release (Fig. 1e) and the number of AV + PI+ cells (Fig. S1d) further indicated compromised plasma membrane integrity. Calcium influx, a crucial event following membrane rupture4, was significantly enhanced, as evidenced by increased Fluo4 fluorescence (Fig. 1f). Additionally, extracellular calcium removal using the Ca2+ chelator EGTA reduced AZD4547-induced cell death (Fig. 1g). Key necroptotic mediators, RIP1, MLKL, and pMLKL, exhibited dose-dependent and time-dependent protein level increases (Figs. 1h and S1e), with elevated MLKL detected in the membrane fraction (Fig. 1i) and increased MLKL aggregation on the plasma membrane (Fig. S1f). Surprisingly, we found that the basal levels of RIP3 in KYSE140 and KYSE180 cells were nearly undetectable, and its’ protein levels did not upregulate as expected under AZD4547-induced necroptosis (Fig. 1h). These observations suggest that RIP3 may be non-essential in the context of FGFR inhibition-induced necroptosis. Additionally, the antitumor effects of AZD4547 were evaluated in ESCC xenografts. AZD4547 suppressed tumor growth without causing significant weight loss in KYSE180 xenografts and FGFR2-high PDX_ES0136 models (Fig. 1j, k). In contrast, AZD4547 failed to effectively inhibit tumor growth in the FGFR2-low PDX_ES0201 models (Fig. S1g), suggesting that FGFR2 expression is a key biomarker for the efficacy of AZD4547. These results indicate that FGFR inhibitor exert a potent inhibitory effect on ESCC, both in vitro and in vivo, by triggering necroptosis.
The FGFR2-mediated necroptosis is dependent on RIP1 and MLKL
Observing that AZD4547 elevated the expression of RIP1 and MLKL, we aimed to ascertain whether AZD4547-induced cell death was reliant on these proteins. Inhibitors targeting RIP1 kinase (necrostatin-1, Nec-1), MLKL kinase (necrosulfonamide, NSA), and apoptosis (ZVAD-fmk, ZVAD) all restored cell viability post AZD4547 treatment. However, necroptotic inhibitors, particularly Nec-1 and NSA, demonstrated a more pronounced effect in cell viability restoration. In contrast, the ferroptosis inhibitor ferrostatin-1 (Fer-1) did not significantly impact cell viability (Fig. 2a), suggesting that AZD4547’s anticancer activity is primarily dependent on necroptosis induction. Additionally, the application of necroptotic inhibitors led to a decrease in both LDH release (Fig. 2b) and the protein levels of RIP1, MLKL and pMLKL (Fig. S2a) in AZD4547-treated cells. Knockdown of RIP1 and MLKL partially reversed the reduction in cell viability (Figs. 2c and S2b) and significantly curtailed the AZD4547-induced LDH release (Fig. 2d), indicating that AZD4547 triggers RIP1/MLKL-dependent necrotic cell death in ESCC cells.

a KYSE140 and KYSE180 cells were treated with 40 μM AZD4547 in the absence or presence of 30 μM Nec-1, 5 μM NSA, 20 μM ZVAD and 1 μM Fer-1 for 24 h to assay cell viability. b The release of LDH was detected following AZD4547 with Nec-1 and NSA for 24 h. c, d Knockdown of RIP1 and MLKL inhibited AZD4547-induced cell death (c) and LDH release (d). The p value was derived from a comparison with the control group. e–g Cells were transfected with FGFR2 (e), FGFR1 (f) or FGFR3 (g) siRNAs, and whole-cell lysates were subjected to western blot analysis after 48 h. pMLKL was induced by adding 20 μM AZD4547. h Cells were analyzed for proliferation by a real-time cell analyzer (RTCA)-MP system. i Knockdown of FGFR2 inhibited 10 μg/mL cisplatin-induced LDH release. j Transmission electron microscopy of the KYSE140 and KYSE180 cells transfected with FGFR2 siRNAs for 48 h. Yellow arrowheads denote loosened nuclei. Red arrowheads indicate swelled cellular organelles. Blue arrowheads indicate plasma membrane rupture. Green arrowheads indicate vacuolization. Scale bar: 2 μm. k After 48 h transfection with FGFR2 siRNA, membrane-cytosolic extraction proteins were measured using western blot. ns not significant. All data are mean ± SD from n = 3 (b, d), 4 (c, i) or 5 (a) biological replicates. Statistical analyses were performed using one-way ANOVA with multiple comparisons. Source data are provided as a Source Data file.
To further dissect the molecular mechanisms, we examined the effects of eliminating FGFR1, FGFR2, and FGFR3 on the necroptotic pathway. Silencing FGFR2 was found to activate necroptotic signaling, marked by upregulated RIP1, MLKL and pMLKL (under the induction of AZD4547; Fig. 2e), whereas the upregulated RIP1 and MLKL were not observed in cells with knockdown of FGFR1 (Fig. 2f) or FGFR3 (Fig. 2g). Additionally, this activation could be inhibited by NSA (Fig. S2c). Moreover, knocking down FGFR2 was found to significantly suppress the growth of ESCC cells (Fig. 2h) and induce LDH release under cisplatin treatment (Fig. 2i). Electron microscopy highlighted distinct necrotic morphology in FGFR2 knockdown cells, including disrupted cytoplasmic membranes, swollen mitochondria, cytoplasmic vacuolization, and loosened nuclei (Fig. 2j). Further analysis of the cytosolic and membrane fractions revealed an enrichment of MLKL in the membrane fraction of FGFR2 knockdown cells compared to controls (Fig. 2k). These results reinforce the conclusion that FGFR2 inhibition is a crucial driver of necroptosis.
RIP3 is dispensable for FGFR2 inhibition-induced necroptosis
As previous observed, RIP3 protein levels were low in KYSE140 and KYSE180 cells, and AZD4547 did not induce its upregulation (Fig. 1h). To clarify RIP3’s role in FGFR inhibition-induced necroptosis, we expanded our analysis to more esophageal cancer cell lines. Remarkably, unlike RIP1 and MLKL, RIP3 was consistently expressed at low levels across the majority of these cell lines, except for COLO680N (Fig. 3a). Contrary to the canonical necroptosis pathway, which typically requires RIP3, we speculated that RIP3 may be non-essential in FGFR2 inhibitor-induced necroptosis (Fig. 3b). To test this, we selected COLO680N for further study. Post-AZD4547 exposure, COLO680N cells exhibited typical necrotic morphology, including plasma membrane disruption and organelle swelling, with loosened nuclei (Figs. 3c and S2d). Furthermore, both genetic (knockdown of RIP1 or MLKL) and pharmacological (using Nec-1 and NSA) inhibition of necroptosis markedly reversed the AZD4547-induced reduction in cell growth confirmed necroptosis in COLO680N cells (Figs. 3d, e and S2e). However, silencing RIP3 did not significantly affect AZD4547-induced cell death (Fig. 3e) nor prevent MLKL translocation to the membrane (Fig. 3f). Nevertheless, immunoprecipitation analysis showed MLKL interaction with both RIP1 and RIP3 (Fig. 3g), indicating that RIP3 might still participate in necrosome formation in COLO680N cells. Collectively, these results suggest alternative kinases, functioning similarly to RIP3, might play a role in FGFR2 inhibitor-induced necroptosis in ESCC cells with deficient RIP3.

a Western blot analysis of RIP1 and RIP3 expression of esophageal cell lines. b Schematic of the molecules participated in necroptosis under various conditions. Created in BioRender. Chen, D. (2025) https://BioRender.com/ukteuom. c Transmission electron microscopy of the COLO680N cells treated for 12 h with DMSO or 10 μM AZD4547. Yellow arrowheads denote loosened nuclei. Red arrowheads indicate swollen cellular organelles. Blue arrowheads show plasma membrane rupture. Scale bar: 2 μm. d Following transfection with RIP1, RIP3 or MLKL siRNA for 48 h, cell viability was determined after treatment with AZD4547 for another 24 h. The p value was derived from a comparison with the control group. e Cells were treated with 40 μM AZD4547 ± 30 μM Nec-1, 5 μM NSA, 20 μM ZVAD and 1 μM Fer-1 for 24 h to assay cell viability. f Cells were transfected with RIP3 siRNAs for 48 h following AZD4547 treatment for another 24 h and membrane-cytosolic extraction was analyzed by immunoblotting. g Immunoprecipitation assay for MLKL binding to RIP1 and RIP3 post-AZD4547 treatment (40 μM). h Indicated L929s were treated with 10 μM AZD4547 for 24 h, and then cell viability was assayed. The p value was derived from a comparison with the WT group. i Immunoblotting of indicated proteins in L929 RIP3−/− cells following treatment with AZD4547 (0.1–10 μM) for 24 h. j Cells were treated with 1 μM AZD4547 for 24 h and indicated proteins of membrane-cytosolic extraction were analyzed by immunoblotting. k Following transfection with RIP1 and MLKL siRNA for 48 h, 10 μM AZD4547 was added for subsequent 24 h to assay cell viability. The p value was derived from a comparison with the control group. l Cells were treated with AZD4547 with or without Nec-1, NSA, ZVAD, and Fer-1 for 24 h to assay cell viability. ns not significant. All data are mean ± SD from n = 4 (k, l) or 5 (d, e, h) biological replicates. Statistical analyses were performed using one-way ANOVA with multiple comparisons. Source data are provided as a Source Data file.
Moreover, we found that wild type (WT) mouse fibroblast L929 and necroptotic mediator-deficient L929 cells responded differently to AZD4547. RIP1−/− and MLKL−/− cells showed less sensitivity to AZD4547 than WT and RIP3−/− cells (Fig. 3h). Additionally, RIP3−/− L929 cells displayed necrotic features following treatment with AZD4547, characterized by elevated levels of RIP1/MLKL/pMLKL, enhanced translocation of MLKL to the plasma membrane, and conferred resistance to AZD4547 upon knockdown of RIP1 and MLKL, or with Nec-1 and NSA (Fig. 3i–l). This reinforces the notion that RIP3 is unessential for the FGFR2-inhibition necroptosis.
MST1 is involved in the necroptotic pathway
To elucidate the underlying mechanisms of FGFR2 inhibitor-induced necroptosis in cells with low RIP3, we utilized the Integrated Interactions Database (IID) to identify potential protein interactions with both RIP1 and MLKL but not RIP3, which yielded 24 candidates (Fig. 4a). Among these proteins, we reviewed the literature to identify kinases with pro-death functions, and STK4 (also known as MST1) drew our attention. MST1, while traditionally associated with apoptosis30 and inflammation31, has recently been linked to resistance to apoptosis mediated by FGFR4 phosphorylation32 and FGF15 activation through hepatic FGFR4 in response to increased bile acids33. This sheds light on the potential connection between MST1 and FGFR2-mediated necroptosis. We found that MST1 formed a protein complex with RIP1 and MLKL independently of RIP3 expression, with this interaction notably enhanced post-AZD4547 treatment (Fig. 4b, c). Knockdown of MST1 alleviated AZD4547-induced growth inhibition in RIP3-low (KYSE140/KYSE180) and deficient cells (RIP3−/− L929), but not in RIP3-high COLO680N cells (Fig. 4d–g).

a Identification of MST1 through the IID database as one of 24 proteins interacting with RIP1 and MLKL, but not RIP3. b, c Immunoprecipitation and immunoblotting analyses show MST1 complex formation with RIP1 and MLKL following AZD4547 treatment in ESCC (40 μM) and RIP3−/− L929 (10 μM) cells. d–g Cell viability assays post-MST1 siRNA knockdown and subsequent AZD4547 exposure. The p value was derived from a comparison with the control group. h Structure of wild-type MST1 and mutant derivates. i Lysates of HEK293T cells transfected with the Myc-RIP1 alone or together with indicated plasmid, were immunoprecipitated with Flag antibody and analyzed by immunoblotting against Myc antibody. j Lysates of HEK293T cells transfected with the Myc-MLKL alone or together with indicated plasmid, were immunoprecipitated with Myc antibody and analyzed by immunoblotting against Flag antibody. k Lysates of HEK293T cells transfected with the Flag-MST1 alone or together with indicated plasmids were immunoprecipitated and analyzed as in (i). l Lysates of HEK293T cells transfected with the Myc-MLKL alone or together with indicated plasmids were immunoprecipitated and analyzed as in (j). m Mass spectrometry peak photographs showing the phosphorylation at the MLKL T216 site. n Flag-MST1 and MLKL-WT or MLKL-T216A were co-expressed in MLKL−/− L929 cells, followed by treatment with 20 μM AZD4547. The migration shift of MLKL was then analyzed using Phos-tag gel electrophoresis. o Cells were transfected with MLKL-WT or MLKL mutant plasmids and analyzed for proliferation by a real-time cell analyzer (RTCA)-MP system. p Schematic illustrating the molecular participants in FGFR2 inhibition-induced necroptosis under varying conditions. Icons of this figure created with BioRender.com. All data are mean ± SD from n = 4 biological replicates. Statistical analyses were performed using one-way ANOVA with multiple comparisons. Source data are provided as a Source Data file.
In order to delineate the domains of MST1 responsible for binding to RIP1 and MLKL, we generated a series of MST1 truncation mutants as depicted in Fig. 4h. We found that either the kinase domain or the C-terminal fragment containing inhibitory domain and SARAH domain was sufficient for the association with RIP1 and MLKL (Fig. 4i, j). Subsequently, we investigated whether the kinase activity and caspase cleavage of MST1 were required for its interaction with RIP1 and MLKL. As illustrated in Fig. 4h, K59 is the ATP-binding site and D326 is the caspase cleavage site. K59R is the kinase-dead mutant and D326N is the caspase cleavage resistant mutant. We found that the binding of RIP1 and MLKL to MST1 is independent of its kinase activity and caspase cleavage-mediated nuclear translocation. (Fig. 4k, l). These results suggest that MST1 may have multiple binding sites with RIP1 and MLKL independent of the kinase activity or its cleavage by caspase.
Given that MLKL is canonically phosphorylated by RIP3 to execute necroptosis, we hypothesized that MST1 might phosphorylate MLKL upon interaction. An in vitro kinase assay followed by mass spectrometric analysis revealed that MST1 directly phosphorylates MLKL at Thr216 (Fig. 4m), but not at the Ser358 residue. Notably, under AZD4547 induction, Phos-tag immunoblotting revealed that co-transfection of MST1 with MLKL-WT significantly enhanced MLKL phosphorylation, whereas co-transfection with inactive MLKL T216A mutant notably diminished the phosphorylation level of MLKL (Fig. 4n), indicating that MST1 mediates MLKL phosphorylation at T216. To further investigate the functional significance of T216 in necroptosis, we introduced T216 mutations and assessed its impact. The results showed that, in comparison to the WT, transfection with inactive T216A mutant significantly diminished sensitivity to AZD4547 compared to WT, whereas sustained activation at T216 enhanced AZD4547-induced cytotoxicity (Fig. 4o). These findings underscore the critical role of the MLKL T216 site in AZD4547-induced necroptosis and highlight MST1 as a key regulator of necroptosis, particularly as an alternative pathway in the absence of RIP3. However, an upregulation of pMLKLS358 following AZD4547 treatment was observed (Figs. 1h,3i and S1e). Given that the p-S358 is required for MLKL oligomerization, our finding suggests that there may be another kinase may be involved in phosphorylating MLKL at Ser358 in the absence of RIP3, warranting further investigation (Fig. 4p).
MST1 mediates necroptosis in a Hippo pathway-dependent manner
Given the central role of MST1 in the Hippo pathway34, we sought to determine whether this pathway participates in regulating necroptosis. Following AZD4547 exposure, we observed downregulation of MST1, NF2, LATS1/2, and an increase in YAP (Fig. 5a), mirroring the effects of FGFR2 knockdown (Fig. S3a). However, the canonical targets of YAP, CYR61, and CTGF, showed negligible or even reduced expression levels (Fig. S3b), indicating a unique context for FGFR2 inhibition. Additionally, transient knockdown of LATS1/2 also led to an increase in YAP, RIP1, and MLKL (Fig. 5b, c). Conversely, YAP transient knockdown or knockout reduced RIP1 and MLKL expression, while YAP overexpression had the opposite effect (Fig. 5d, e). Furthermore, YAP knockdown decreased cell death induced by AZD4547 (Fig. 5f), suggesting that the Hippo pathway negatively modulates necroptosis.

a Western blot analysis of indicated proteins post-AZD4547 treatment (40 μM) in KYSE140 and KYSE180 cells. b, c. Analysis of LATS1 and LATS2 siRNAs impacts on protein expression. d Indicated proteins expression in YAP overexpressed cells. e Reduced RIP1 and MLKL expression following YAP deletion. f Knockdown of YAP inhibited AZD4547 (40 μM, 24 h)-induced cell death. The p value was derived from a comparison with the control group. g Nuclear-cytosolic protein analysis post-AZD4547 treatment (40 μM). C cytoplasm. N nucleus. h Protein level in cells transfected with wild-type YAP and kinase-inactive YAP mutants (S127A, S397A) for 48 h. i RIP1 and MLKL transcriptional activity in vector and YAP-overexpressed cells were measured by a luciferase assay. j RIP1 and MLKL transcriptional activity in control and siYAP cells were measured by a luciferase assay. k, l ChIP-qPCR assessing TEAD4 (k) or YAP (l) binding to RIP1 and MLKL promoters using control IgG or indicated antibody. m Proposed model for FGFR2-Hippo signaling-mediated necroptosis. Created in BioRender. Chen, D. (2025) https://BioRender.com/n9ng3qk All data are mean ± SD from n = 3 (i, j, k, l) or 4 (f) biological replicates. Statistical analyses were performed using one-way ANOVA with multiple comparisons (f, i, j) or two-tailed unpaired Student’s t-test (k, l). Source data are provided as a Source Data file.
Confirming YAP’s functional activation, we used a nuclear-cytosolic fractionation separation assay, which showed a clear increase in YAP within the nuclear fraction of treated cells (Fig. 5g). YAP-Ser127A and YAP-Ser397A mutants are resistant to LATS1/2 phosphorylation, thereby promoting nuclear retention and transcriptional activity35. In cells transfected with these mutants, RIP1 and MLKL were overexpressed (Fig. 5h). Interestingly, by analyzing the proximal promoter regions of human RIPK1 and MLKL, we identified a conserved TEAD (CATTCC) binding site, respectively. We then constructed dual-luciferase reporter gene plasmids of RIPK1 and MLKL promoters. It showed that overexpressing YAP led to a substantial increase in the luciferase activity with RIPK1 and MLKL promoters (Fig. 5i), while knocking down YAP significantly reduced their luciferase activity (Fig. 5j), indicating that YAP can regulate the transcriptional activities of RIPK1 and MLKL. Further analysis was done through ChIP experiments, and the results showed that the DNA fragments enriched with TEAD4 and YAP antibodies contained sequences from RIPK1 and MLKL (Fig. 5k, l). Corroborating in vitro findings, in vivo results also showed enhanced MLKL and YAP staining post-AZD4547 treatment in CDX and PDX tumor tissue (Fig. S3c) and the finding was corroborated by western blot showing the increased RIP1, MLKL, and YAP (Fig. S3d). These results suggest that YAP transcriptionally regulates the expression of RIP1 and MLKL, thereby modulating the necroptotic pathway. In summary, our findings indicate that FGFR2 governs necroptosis via the Hippo pathway (Fig. 5m). However, the mechanism by which membrane-bound FGFR2 regulates the intracellular Hippo pathway cascade remains unclear.
FGFR2 inhibition modulates the Hippo pathway by promoting NF2 ubiquitination
Probing further into how FGFR2 influences the Hippo pathway cascade, we noted that NF2 has been documented to interact with HER2, affecting Src signaling36. We hypothesized a potential FGFR2-NF2 interaction in mediating necroptosis. Our results indicated that NF2 associates with FGFR2 (Fig. 6a), and NF2 RNA interference or knock out upregulated YAP alongside RIP1 and MLKL (Fig. 6b, c). Moreover, NF2 knockdown increased AZD4547-induced cell death (Fig. 6d). Further analysis revealed that NF2 overexpression mitigates the upregulation of RIP1, MLKL, and pMLKL (under AZD4547 induction) induced by NF2 knockdown or knockout (Fig. 6e). These results supported the notion that NF2 regulates the necroptosis.

a Immunoblot analyses of lysates following binding with antibodies against FGFR2 and NF2 showed association between them. b, c Analysis of the impact of NF2 deletion on protein expression. d Knockdown of YAP increased AZD4547-induced cell death (10 μM in HEK293A cells, 20 μM in ESCC cells). The p value was derived from a comparison with the control or WT group. e Western blotting results exhibiting the protein levels of indicted molecules after re-expressing NF2 in NF2-deleted cells. f Mass spectrometry did not identify a phosphorylation site on NF2. g Increased pNF2 expression following FGFR2 deletion (Induction by adding 20 μM AZD4547). h and i pNF2 levels post-AZD4547 treatment at various concentrations and indicated times. j–l HEK293T cells were co-transfected with indicated plasmids. After 36 h, cells were treated with 10 μM MG132 (j and l) or together with 20 μM AZD4547 (k) for 12 h. Cell lysates were immunoprecipitated with a flag antibody, followed by immunoblotting with an HA antibody to detect ubiquitinated NF2. m HEK293T cells were co-transfected with Myc-FGFR2 and NF2-WT or NF2-S518A and immunoprecipitated with a Flag antibody to detect the binding affinity between FGFR2 and NF2. n Diagram of the relationship between FGFR2 and NF2 in the presence of AZD4547. Created in BioRender. Chen, D. (2025) https://BioRender.com/mpyjqz3. All data are mean ± SD from n = 6 biological replicates. Statistical analyses were performed using two-tailed unpaired Student’s t-test. Source data are provided as a Source Data file.
NF2’s function is mainly regulated through phosphorylation37. We thus speculated that FGFR2 might regulate the Hippo pathway by phosphorylating NF2. However, in vitro kinase assays showed that FGFR2 cannot phosphorylate NF2 (Fig. 6f), while it does affect the protein level of NF2 (Figs. 5a and S3a). We explored whether FGFR2 inhibition affects the ubiquitination of NF2. According to the previous study, the Ser518 phosphorylation was required for the inactivation37 and degradation38 of NF2. Our results showed that FGFR2 knockdown enhanced the Ser518 phosphorylation of NF2 (Fig. 6g), which increased in a AZD4547 dose- and time-dependent manner (Fig. 6h, i). In vitro ubiquitination assay showed that NF2 ubiquitination was reduced upon binding with FGFR2 (Fig. 6j), whereas inhibiting FGFR2 enhanced NF2 ubiquitination (Fig. 6k). The further analysis revealed that increasing FGFR2 concentration led to a gradual decrease in pNF2S518 expression, accompanied by a reduction in NF2 ubiquitination level (Fig. 6l). Notably, mutation at the NF2 Ser518 site did not affect the binding affinity between FGFR2 and NF2 (Fig. 6m). These findings suggest that FGFR2 competitively interacts with NF2, inhibiting Ser518 phosphorylation and subsequently preventing NF2 from ubiquitination and degradation-a process that can be disrupted by AZD4547 treatment. However, this interaction is independent of the Ser518 site (Fig. 6n).
FGFR2 inhibition-induced necroptosis increases PD-L1 expression in surviving tumor cells via paracrine IL-8
Since necroptosis is an inflammatory form of cell death, we next explore whether AZD4547-mediated necroptosis triggers inflammatory activation, which may pave a way for combined immunotherapy. To this end, we conducted RNA-sequencing on AZD4547-treated ESCC cells and their controls. The analysis revealed distinct gene expression profiles in treated cells (Fig. S4a). Enrichment Analysis (GSEA) showed enrichment of TNFα signaling via NF-κB and inflammatory response in the AZD4547-treated phenotype (q < 0.01; Fig. 7a). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis further corroborated this, highlighting several pathways linked to inflammation and immunity, including the NF-κB signaling pathway (Fig. S4b). Meanwhile, we also performed cytokine array to identify differential secreted cytokines upon AZD4547 treatment, which revealed elevated IL-8 secretion (Fig. 7b). The increased release of IL-8 was validated through ELISA. This was observed both under treatment with AZD4547 (Fig. S4c) and upon the knockdown of FGFR2 (Fig. S4d). Consistently, the mRNA level of CXCL8 was also upregulated in AZD4547-treated groups from sequencing data (Fig. 7c). Interestingly, both protein and mRNA levels of CD274 were upregulated post-AZD4547 exposure (Figs. 7d and S4e). And FGFR2 silencing also increased PD-L1 expression (Fig. S4f). Given IL-8 is involved in promoting CD274 expression39, we wonder whether necrotic cells could release IL-8 that cause nearby surviving tumor cells to increase PD-L1 expression and escape immune surveillance. Not surprisingly, we found a correlation between MLKL and PD-L1 levels in ESCC cell lines (Figs. 7e and S4g). Subsequent knockdown of MLKL resulted in decreased PD-L1 expression (Fig. S4h). Immunohistochemistry on a tissue microarray with 104 ESCC cases revealed a positive correlation between MLKL and PD-L1, with 74 (71.2%) specimens showing simultaneous expression (Fig. 7f and Table S1). Subsequently, the intensified PD-L1 expression observed in cells cultured with supernatants from AZD4547-treated cells (Fig. 7g) hinted that the release of IL-8 inducing PD-L1 expression, as IL-8 is the cytokine that shows the most significant change in cell supernatant after AZD4547 treatment. IL-8 was shown to induce PD-L1 expression in a dose- and time-dependent manner (Fig. 7h, i). Neutralizing IL-8 with an anti-IL-8 antibody inhibited the PD-L1 upregulation induced by AZD4547-conditioned medium and IL-8 stimulation (Fig. 7j, k). Additionally, ELISA results revealed that IL-8 knockdown significantly reduced the AZD4547-induced IL-8 release (Fig. S4i, j), and culturing with the AZD4547-conditioned medium from the IL-8 knockdown group had a weaker effect on PD-L1 expression than the control (Fig. 7l). Collectively, these findings suggest that FGFR2 inhibition-induced necroptosis elevates PD-L1 expression in surviving tumor cells via paracrine IL-8.

a GSEA of sequencing data highlighting significant pathways in AZD4547-treated (40 μM) cells. b Cytokine analysis in medium from AZD4547-treated KYSE140 cells, highlighting the significantly increased cytokine (red box). Concentration of cytokines were shown on the bottom. c Differential expression of inflammation-related cytokines between control and AZD4547-treated groups. d Upregulated PD-L1 protein levels post-AZD4547 treatment. e Expression levels of MLKL and PD-L1 in ESCC cell lines. f MLKL and PD-L1 expression in ESCC tissue microarrays. Scale bar, 200 μm. The correlation was shown in table. g PD-L1 expression analysis in cells cultured with supernatant from AZD4547-treated cells (40 μM). Created in BioRender. Chen, D. (2025) https://BioRender.com/qkttth3. h, i PD-L1 levels post-IL-8 treatment at various concentrations and indicated time. j Western blot analysis of PD-L1 in cells treated with AZD-CM and 1 μg/mL IL-8 antibody. k Western blot analysis of PD-L1 in cells treated with 50 ng/mL IL-8 and 1 μg/mL IL-8 antibody. l PD-L1 expression analysis in cells cultured with the AZD4547-conditioned medium from the IL-8 knockdown or control groups. m, n Protein expression analysis following treatment with AZD-CM (m) or 50 ng/mL IL-8 (n). AZD-CM: AZD4547-conditioned medium. All data are mean ± SD from n = 4 biological replicates. Source data are provided as a Source Data file.
Of note, NF-κB pathway was enriched in AZD4547-treated group (Fig. S4b), which was previously implicated in transcriptionally regulated PD-L1 expression40. As Expected, both AZD4547-conditioned medium exposure and IL-8 stimulation consistently upregulated p-p65 (Fig. 7m, n). These results, combined with the sequencing data, indicate that IL-8 may regulate PD-L1 expression through NF-κB p65 activation, potentially serving as an immune evasion strategy by tumor cells under necroptotic inflammation stress.
Synergistic inhibitory effects of FGFR inhibitor combined with PD-L1 inhibition in ESCC Xenografts
Given that FGFR2 inhibition-induced necroptosis can trigger IL-8-dependent PD-L1 expression in surviving cells, we proposed that dual blockade of FGFR2/ PD-L1 or FGFR2/IL-8 could achieve better therapeutic outcomes. Accordingly, we established a humanized mouse model (Fig. S5a, b and Table S2) to assess the therapeutic effectiveness of targeted FGFR2, anti-PD-L1 immunotherapy, targeted IL-8-CXCR1/2 therapy, and their various combinations. Individual treatments with AZD4547, Atezolizumab (a PD-L1 antibody), and Reparixin (an IL-8 receptor CXCR1/2 inhibitor) showed tumor growth inhibition (TGI) rates of 59.9%, 23.8%, and 14.8% in KYSE150 xenografts, respectively. Notably, dual blockade resulted in even greater tumor suppression, with the combination of AZD4547 and Atezolizumab achieving a remarkable TGI of 102.5%, as evidenced by significantly reduced tumor weights and no considerable weight loss in mice (Fig. 8a, b). The immunohistochemistry (IHC) results revealed a significant reduction in Ki67 expression in the combination therapy group compared to the monotherapy groups (Figs. 8c and S5c), further demonstrating that the combination of AZD4547 with a PD-L1 antibody or a CXCR1/2 inhibitor exerts a substantial antitumor effect in ESCC. Corroborating our in vitro findings, in vivo treatment with AZD4547 led to a decrease in FGFR2 expression and a concomitant increase in PD-L1, IL-8, and MLKL levels within the tumor cells (Figs. 8c and S5c). Intriguingly, while YAP expression showed a moderate increase, its localization was primarily nuclear, especially in the AZD4547-Atezolizumab group, suggesting YAP activation. Flow cytometry analysis revealed that infiltrating lymphocytes were predominantly CD3 + T cells (Table S2). IHC findings indicated fewer infiltrating human CD3 + T cells in the AZD4547-treated group compared to controls, while the addition of Atezolizumab or Reparixin increased CD3+ lymphocyte counts. Additionally, granzyme B expression of infiltrating lymphocytes was found to be lower in the AZD4547 group, whereas Atezolizumab or Reparixin addition did not affect granzyme B level. The increased expression of PD-L1 on tumor cells and the reduced expression of CD3 and granzyme B on infiltrating cells further demonstrate that AZD4547 can induce an immunosuppressive microenvironment in tumor tissues. Collectively, these results indicate that combining AZD4547 with PD-L1 antibodies or disrupting PD-L1 expression through IL-8 inhibition could significantly enhance the inhibition of tumor growth.

a Curves showing the xenografts growth of humanized mice bearing KYSE150 cells treated with vehicle control, AZD4547 (12.5 mg/kg/d, by oral gavage), Atezolizumab (5 mg/kg, twice weekly, i.p.), Reparixin (30 mg/kg/d, i.p.), AZD4547 (12.5 mg/kg/d) plus Atezolizumab (5 mg/kg), AZD4547 (12.5 mg/kg/d) plus Reparixin (30 mg/kg/d), or Reparixin (30 mg/kg/d) plus Atezolizumab (5 mg/kg) for 14 days. n = 5 per group. Tumor volumes and mice weights were measured every three days after treatment, and the curves were expressed as mean ± SD. The corresponding TGI were listed. *p < 0.05 by repeated-measures ANOVA. TGI tumor growth inhibition. b Tumors were weighted after removal. c H-scores of Ki67, FGFR2, PD-L1, IL-8, YAP, MLKL, CD3, and granzyme B in xenograft tissues. d Schematic of molecular mechanisms involved in FGFR2 inhibition-induced necroptosis. Created in BioRender. Chen, D. (2025) https://BioRender.com/1wbwjd2. Other data are mean ± SD from n = 3 biological replicates. Statistical analyses were performed using one-way ANOVA with multiple comparisons (b, c) or two-way ANOVA with multiple comparisons (a). Source data are provided as a Source Data file.
Discussion
Oncogenic FGFR2 signaling is a well-established factor in cancer progression. Based on the therapeutic effects observed in our PDX animal model, high FGFR2 expression appears to be an indicator of effective FGFR2-targeted therapy. Thus, FGFR2 may serve as a biomarker for FGFR2-targeted therapy. Therefore, understanding the pathways and cell death modalities resulting from FGFR2 inhibition is crucial for cancer therapy. In this study, we provided compelling evidence demonstrating that inhibition of FGFR2 could induce tumor cell necroptosis, while shaping an immunosuppressive microenvironment. Notably, the concentration of AZD4547 used in our experiments, though higher than in other cancer types41, is clinically achievable42. Traditionally, FGFR inhibition has been associated with apoptosis via blockade of intracellular signaling cascades43,44. Contrary to expectations, our study reveals that FGFR2 inhibition primarily induces necroptosis. This aligns with findings that FGFR inhibitors like PD173074, BGJ398, and AZD4547 induce caspase-independent cell death21, and LY2874455, a pan-FGFR inhibitor, unexpectedly causes necrosis in cholangiocarcinoma by downregulating MCL122. These findings suggest that the mechanisms of FGFR inhibition may vary based on cell type and context.
The role of RIP3-dependent necroptosis is not universal across all cell types. Our results demonstrate that necroptosis following FGFR2 inhibition occurs independently of RIP3. This conclusion is supported by low RIP3 expression in most ESCC cell lines and the lack of significant impact of RIP3 on cell death post-AZD4547 treatment in RIP3 low expressed ESCC cell line and the RIP3−/− L929 cell line. Previous reports indicate that RIP3 levels are low in hepatocytes under basal conditions or during experimentally induced hepatitis45,46, suggesting RIP3 is not necessarily required for necroptosis or inflammation. Najafov A, et al.47 demonstrated that RIP3 expression progressively decreases during tumor growth, with oncogenes BRAF and AXL contributed to this loss. Additionally, MLKL translocation to the membrane still occurred in RIP3-knockdown and RIP3-deficient cells, suggesting the involvement of an alternative kinase for MLKL activation in the absence of RIP3.
We identified MST1 that acted as an alternative pathway transmitting signals from RIP1 to MLKL regardless of whether RIP3 exists or not, highlighting the fine-tuned protein levels of MST1 was crucial for necroptosis. However, the mechanism by which RIP1 recruits and activates MST1 remains unclear, as MST1 lacks a homotypic interaction motif (RHIM) (Fig. 4h), unlike RIP1’s well-established recruitment of RIP3 via RHIM48. MLKL activation appears distinct and may or may not involve RIP1. Additionally, Dovey et al. reported that in inositol phosphate (IP) kinase mutant cells, MLKL failed to oligomerize and localize to membranes despite proper RIP3-dependent phosphorylation, suggesting that necroptosis requires IP-specific kinase activity49. Therefore, in the absence of RIP3, the IP code might contribute to MLKL activation, potentially in collaboration with the unknown kinase proposed in Fig. 4p of our study. These preliminary findings warrant further investigation. Paradoxically, while FGFR2 inhibition led to MST1 downregulation, the association of MST1 with RIP1 and MLKL increased post-AZD4547. A recent study from Xiang Li, et al. found that the quantitative level of RIP1 dictates distinct cell death outcomes (apoptosis or necroptosis), with cells solely undergo apoptosis when RIP1 is below 1000 molecule per cell50. This suggests that MST1 could act as a mediator, switching quantitatively between the Hippo pathway and the necroptotic pathway, particularly when its abundance is low. Further study by SWATH-MS technique, as proposed by Xiang Li et al., may provide insights into this quantitatively regulatory mechanism. Additionally, previous study also showed that MST1 can interact with the glycogen-binding protein laforin and undergo phase separation with glycogen droplets, which suggesting that MST1 may exert different functions via phase separation51.
In this context, YAP, a key component of the Hippo pathway, was found to dictate necrotic cell death by transcriptionally inducing RIP1 and MLKL expression. The relationship between FGFR and YAP was also explored by Yihao Li, et al.52, who found that prolonged FGFR inhibition leads to an epigenetic state that derepresses YAP-associated enhancers. YAP’s role in cancer is multifaceted, with evidence supporting both its oncogenic and tumor suppressor functions. For example, YAP binds p73 to induce the transcription of PUMA in breast cancer cells26, and it activates tumor suppressor PML, contributing to CDDP-induced apoptosis53. Additionally, YAP has been implicated in promoting ferroptosis27, further supporting its tumor suppressor role. However, research on the link between YAP and necroptosis is limit. One notably finding is that RIP3 knockdown reduces nuclear YAP/TAZ levels and activities in breast cancer54. Our study is among the first to explore this relationship, demonstrating that YAP positively regulates necroptosis.
Intriguingly, we observed that FGFR2 inhibition-induced necroptosis, while contributing to the eradication of cancer cells, also posed a risk of promoting an immunosuppressive tumor microenvironment, which characterized by necrotic cells-derived IL-8 inducing increased PD-L1 expression in adjacent surviving tumor cells and decreased CD3 and granzyme B expression in infiltrating lymphocytes. Increased B and T cells and decreased PD-L1 expression also found in macrophages of RIP3 knockout pancreatic models, suggesting an immunoactive microenvironment55. Additionally, RIP1 expression in TAMs facilitates TAM-driven immunosuppression56 and CD8+ TILs are significantly lower in the high-pMLKL group in ESCC57. The release of IL-8 by necrotic tumor cells leads to NF-κB p65 activation in adjacent cancer cells, representing a defense mechanism. To counteract the pro-tumorigenic potential of necroptosis and achieve better antitumor efficacy, combining an immune checkpoint inhibitor with a FGFR2 inhibitor is a rational and synergistic approach, as confirmed in our humanized mouse xenograft models. The combination of targeting FGFR2 with PD1/PD-L1 monoclonal antibodies has already exhibited a synergistic anti-tumor effect in breast cancer58,59.
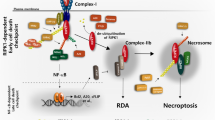
In summary, our study delineates an alternative necroptotic pathway involving the crosstalk between FGFR2-NF2-hippo signaling and the RIP1/MST1/MLKL axis. Under normal conditions, FGFR2 competitively interacts with NF2. However, AZD4547 treatment disrupted the FGFR2-NF2 binding, promoting NF2 phosphorylation at Ser518, which subsequently leads to NF2 ubiquitination and degradation, thereby inhibiting the Hippo signaling cascade. Consequently, YAP translocates to the nucleus, binds to TEAD4, and induces the transcription of RIP1 and MLKL, promoting necroptosis. In this process, MST1 acts as an alternative kinase, interacting with RIP1 and MLKL and subsequently phosphorylating MLKL at Thr216 to mediate necroptosis. Upon cell membrane rupture, IL-8 is released, inducing PD-L1 expression in adjacent surviving tumor cells through NF-κB p65 activation. The increased PD-L1 inhibits the cytotoxic effects of T cells, resulting in immunosuppression. These findings grounded the promising therapeutic strategies of dual blockade of FGFR2/PD-L1 or FGFR2/IL8-CXCR1/2 for clinical treatment (Fig. 8d).
Methods
Ethics
All animal experiments received approval from Peking University Cancer Hospital’s Institutional Animal Care and Use Committee (Approval NO. EAEC 2020–08), with rigorous efforts made to minimize animal suffering.
In vivo animal experiments
Five weeks old female Balb/c nude mice and NOG mice were acquired from Vital River Laboratories (Beijing, China). They were maintained under specific pathogen-free conditions with 22–26 °C, 40–60% humidity and 12 h light/12 h dark cycle and fed regular diet. For CDX tumor generation, 5 × 106 KYSE180 cells in 100 µL PBS were subcutaneously injected into the dorsal right flank of Balb/c nude mice. PDX tumors were initiated by subcutaneous inoculation of tissue samples into the flanks of the mice. When tumors reached approximately 150 mm3, the mice were randomized into two groups for treatment (n = 5 for CDX models, n = 6 for PDX models): (1) control with 100 μL PBS daily, ip; (2) AZD4547 at 12.5 mg/kg/d, by gavage. Human PBMC-engrafted mouse models were created by injecting 7 × 106 PBMCs, sourced from MT-BIO (Shanghai, China), into NOG mice via tail vein. Following cell injection, tumor cells (1 × 106 KYSE150) were implanted subcutaneously. Mice were bled retro-orbitally and the human CD45-positive cell rates in the mouse peripheral blood were analyzed by flow cytometry. Mice with over 25% human CD45-positive cells were deemed successful human PBMC-engrafted models. Three weeks post-injection, with human CD45-positive cell rates between 30–95% (see Fig. S5a, b and Table S2) and tumor volumes around 200 mm3, mice were assigned to the same seven treatment groups (n = 5): (1) control: 100 μL PBS, daily, ip; (2) AZD4547: 12.5 mg/kg/d, by gavage; (3) Atezolizumab: 5 mg/kg, twice weekly, ip; (4) Reparixin: 30 mg/kg/d, i.p.; (5) AZD4547 plus Atezolizumab; (6) AZD4547 plus Reparixin; (7) Reparixin plus Atezolizumab.
We treated animals for 2 weeks and tumor size and body weight were measured every 3 days. Tumor volume was calculated using the formula V = L × W2/2 (V, volume; L, length of tumor; W, width of tumor). In cases where the tumor volume exceeded 2000 mm3, the mice were humanely euthanized for welfare reasons. Post-treatment, mice were euthanized, tumors harvested, and samples processed for FFPE sections or western blot analysis. Tumor growth inhibition (TGI) = (1 − ΔT/ΔC) × 100% (ΔT = tumor volume change of the drug-treated group on the final day of the study, ΔC = tumor volume change of the control group on the final day of the study).
Reagents and antibodies
AZD4547 (AZD), ZVAD-fmk (ZVAD), Necrostatin-1 (Nec-1), Necrosulfonamide (NSA), Ferrostatin-1 (Fer-1), Reparixin (Rep), Atezolizumab (Ate), and MG132 were sourced from Selleck. Cisplatin was purchased from Qilu Pharmaceutical Co. LTD. IL-8 cytokine, antibody and ELISA kit were obtained from Sigma-Aldrich (St. Louis, MO), R&D Systems (Minneapolis, MN, USA), and Boster Biological Technology (California, USA), respectively. Reagents were prepared and stored according to the manufacturers’ instructions. Phos-tag was purchased from FUJIFILM Wako (Japan). The antibodies to FGFR1 (#9740), FGFR2 (#23328), pFRS2 (#3861), RIP1 (#3493), RIP3 (#13526), MLKL (#37705, #26539), Na,K-ATPase (#3010), p-NF-κB (#3039), MST1 (#14946), LATS1 (#3477), LATS2 (#5888), CTGF (#86641), CYR61 (#14479), PD-L1 (#13684), Ki67 (#9027), CD3 (#85061), granzyme B (#46890), secondary horse radish peroxidase (HRP)-conjugated goat anti-rabbit (#7074) and anti-mouse (#7076) antibodies were purchased from Cell Signal Technology (Danvers, MA). pLATS1/2T1079+T1041 (YP1222) was purchased from Immunoway (Plano, TX, USA). Secondary HRP-conjugated affinipure mouse anti-rabbit IgG light chain (AS061), goat anti-mouse IgG light chain (AS062) and pNF2S518 (AP0414) were purchased from ABclonal Technology (Wuhan, China). Primary antibodies against FGFR3 (ab133644), MLKL (ab184718), pMLKLS358 (ab187091), YAP (ab205270), NF2 (ab109244), IL-8 (ab106350), Sodium Potassium ATPase (ab76020) and TEAD4 (ab197589) were purchased from Abcam (Cambridge, UK). Antibodies against β-actin (60008-1-Ig), GAPDH (60004-1-Ig), Lamin B1 (66095-1-Ig), RIP3 (17563-1-AP), Myc (60003-2-Ig), Flag (20543-1-AP), HA (81290-1-RR) and PD-L1 (66248-1-Ig) were purchased from Proteintch Group (Chicago, IL, USA). MLKL antibody (sc-293201) used for immunoprecipitation and NF-κB antibody were purchased from Santa Cruz (Dallas, Texas, USA). The fluorescent antibodies used for flow cytometric analysis, including PE-human CD45 (368509), FITC-human CD3 (300406) and APC-mouse CD45 (10312) were purchased from Biolegend (San Diego, CA, USA).
Cell lines and cell culture
The human ESCC cell lines YES2, KYSE30, KYSE70, KYSE140, KYSE150, KYSE180, KYSE410, KYSE450, KYSE510, and COLO680N were generously provided by Dr. Y. Shimada (Kyoto University). Human normal esophageal epithelial cell lines NE2 and SHEE were kindly provided by Dr. Enmin Li (Shantou University). HET-1A (CRL-2692) and HEK293T (CRL-3216) were purchased from ATCC. L929 wild type (WT), RIP1−/−, RIP3−/−, and MLKL−/− cell lines were gifts from Dr. Jiahuai Han (Xiamen University). HEK293A WT, YAP-KO, and NF2-KO cell lines were gifts from Dr. Faxing Yu (Fudan University). YAP stably overexpressed KYSE140 and KYSE150 cells, along with their control counterparts, were maintained in our laboratory. ESCC cell lines and normal esophageal epithelial cell lines were cultured in RPMI-1640 medium (Hyclone, Logan, UT, USA), whereas L929, HEK293T and HEK293A cell lines were maintained in DMEM (Hyclone). Both media were supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin (Gibco BRL, Gaithersburg, MD, USA). Cell line authentication was conducted via short tandem repeat profiling, and mycoplasma contamination was routinely prevented using a mycoplasma elimination reagent (Yeasen Biotechnology, Shanghai, China).
Cell viability assay
Cells were seeded at a density of 3000–5000 per well in 96-well plates and incubated with specified treatments in complete medium. Cell viability was assessed using MTS assays (Promega Corporation, Madison, WI, USA), following the manufacturer’s protocol. Absorbance was measured at 490 nm using a spectrophotometer (Tecan, Morrisville, NC, USA). The half-maximal inhibitory concentration (IC50) values were calculated using Graphpad Prism Version 9.0 software.
Cell proliferation assay
The proliferation abilities of the ESCC cells were determined using the xCELLigence Real-Time Cell Analyzer (RTCA)-MP system (Acea Biosciences/Roche Applied Science). This platform can measure cellular growth status in real time. Briefly, after transfection for 24 h, 2000 transfected cells were seeded in E-Plate 96 (Roche Applied Science). Cell index was read automatically every 15 min, and the recorded curve was shown as cell index ± s.e.m. For drug sensitivity testing, cell index was normalized at the final detection time point before treatment.
Apoptosis assay
Post-AZD4547 treatment for 24 h, cells were double-stained with Annexin V-Fluorescein isothiocyanate (FITC) and Propidium Iodide (PI) (Beyotime, Shanghai, China), following the manufacturer’s protocol. Analysis was conducted within an hour of staining using flow cytometry (BD Biosciences, NJ, USA), and the proportions of apoptotic cells were determined using FlowJo Version 10.0.7 software (FlowJo, Ashland, OR).
Immunofluorescent staining
Cells at a concentration of 300,000 cells/ml were seeded onto 35 mm glass-bottom dishes (NEST, Jiangsu, China) and incubated overnight. Following 10 μM AZD4547 treatment for 12 h, cells were fixed with 4% paraformaldehyde (Solarbio, Beijing, China) for 15 min, permeabilized with 0.5% Triton X-100 (Amresco, Solon, OH) for 20 min, and blocked with 5% bull serum albumin (BSA, Amresco) for 30 min at room temperature. Cells were then probed with MLKL antibody (1:200) and na,k-ATPase (1:500) overnight at 4 °C, followed by incubation with Alexa Fluor 594-conjugated goat anti-mouse IgG and FITC-conjugated goat anti-rabbit IgG (ZSGB-BIO, Beijing, China, 1:100) for 1 h in the dark at room temperature. Nuclei were counterstained with diamidino-phenyl-indole (DAPI) (Beyotime, 1:10000) for 5 min in the dark. All reagents were diluted in phosphate-buffered saline (PBS), and each step was followed by three 5-min PBS washes. Images were captured using ZEN version 2012 software (Zeiss, Gottingen, Germany) on a laser scanning confocal microscope LSM 780 (Zeiss).
Lactate dehydrogenase (LDH) release assay
Post-treatment, the release of lactate dehydrogenase (LDH) in the culture media was quantified using the LDH Cytotoxicity Assay Kit (Beyotime) as per the manufacturer’s instructions. LDH intensity was measured at 490 nm using a spectrophotometer (Tecan). The percentage cytotoxicity, represented as the percent release of LDH, was calculated relative to control groups as described by the manufacturer.
Analysis of intracellular Ca2+ concentration
For calcium analysis, cells (300,000/well) were plated in 6-well plates and treated with 40 μM AZD4547 for 24 h. The cells were then loaded with 4 µM Fluo4 AM (Beyotime) in PBS for 30 min at 37 °C, followed by triple PBS washes. Intracellular Ca2+ levels were determined by the fluorescence intensity of Fluo-4, using flow cytometry (BD Biosciences) at an excitation wavelength of 488 nm and a 530 nm detection channel.
Plasmids and siRNA transfections
siRNA kits targeting FGFR1, FGFR2, FGFR3, RIP1, RIP3, MLKL, NF2, MST1, YAP, LATS1, LATS2, IL8, and a negative control were sourced from RiboBio (Guangzhou, China). Control vectors, wild type plasmids encoding Ub, FGFR2, RIP1, MLKL, NF2, YAP, MST1, and plasmids encoding various YAP mutants and MST1 mutants were obtained from MailGene (Beijing, China). All plasmids underwent DNA sequencing for verification. Transfections with siRNA or cDNA were performed using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s guidelines. Cells were harvested for immunoblotting or cell viability assays 48 h post-transfection. The siRNA sequences used are listed as follows: FGFR1, #1: CAGATAACACCAAACCAAA #2: GCAAAGAATTCAAACCTGA; FGFR2, #1: GCACCATACTGGACCAACA #2: CTCCGAGTATGAACTTCCA; FGFR3, #1: CGTCGTGGAGAACAAGTTT #2: CCTACGTTACCGTGCTCAA; RIP1, #1: GGAGCAAACTGAATAATGA #2: GGCCAGATGTGGATGACAT; RIP3, #1: GACCGCTCGTTAACATATA #2: GTTTCTGAGTGGCTAAACA; MLKL, #1: CTGGAGATATCCCGTTTCA #2: CGCTGTTACTTCAGGTTGA; NF2, #1: TGGCCAACGAAGCACTGAT #2: GAAGCAGATTTTAGATGAA; MST1, #1: GGCAGCGTATACAAAGCTA #2: CAAGCGGAATACAGTGATA; YAP, #1: GACATCTTCTGGTCAGAGA #2: CCACCAAGCTAGATAAAGA; IL8, #1: CGCCAACACAGAAAUUAUUTT #2: GCCAAGGAGUGCUAAAGAATT; LATS1, GAACCAAACTCTCAAACAA; LATS2, GTTCGGACCTTATCAGAAA.
Western blot
Post-treatment, total cell protein was lysed using RIPA buffer (Beyotime) in the presence of protease and phosphatase inhibitor cocktail tablets (Roche, Basel, Switzerland). Membrane-cytosolic and nuclear–cytosolic fractionation was conducted as per the Membrane and Cytosol Protein Extraction Kit (Beyotime) and the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific), respectively. Protein concentrations were determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Approximately 20 μg of protein per sample were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride (PVDF) membrane (Merck Millipore, Darmstadt, Germany). After blocking with 5% BSA, the membranes were incubated with primary antibodies overnight at 4 °C and with secondary antibodies (1:5000) for 1 h at room temperature. Signal detection was carried out using an Amersham Imager 600 (GE Healthcare, Chicago, IL).
Immunoprecipitation analysis
Cells were lysed in NP-40 buffer (Applygen, Beijing, China) containing protease and phosphatase inhibitors. Equal amounts of protein from these lysates were incubated with protein A/G magnetic beads (MedChemExpress, Monmouth Junction, NJ, USA) at 4 °C for 2 h to facilitate precipitation. The beads were then washed thrice with washing buffer and incubated overnight at 4 °C with gentle rotation using either control IgG or specific antibodies (2 µg). Post incubation, the beads were thoroughly washed, and proteins were eluted by boiling in 5× loading buffer for 10 min before being resolved on SDS-polyacrylamide gels for subsequent western blot analysis.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from cell pellets using Trizol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). The RNA samples with an OD260/OD280 ratio between 1.9 and 2.0 were used for the following cDNA synthesis using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Japan). The qRT–PCR was performed on a 7500 Fast Real-time PCR System (Applied Biosystems) using TB Green Premix Ex TaqTM (Tli RNaseH Plus) (Takara, Japan). The relative expression of PD-L1 was calculated using the comparative Ct method. The primers of PD-L1 and GAPDH were as follows:
PD-L1 forward: CACGGTTCCCAAGGACCTAT,
PD-L1 reverse: CTGATCATGCAGCGGTACAC.
GAPDH forward: GCACCACCAACTGCTTAGC,
GAPDH reverse: GGCATGGACTGTGGTCATA.
Chromatin immunoprecipitation (ChIP) assay
Cells were crosslinked using 1% formaldehyde for 10 min, and glycine was added to a final concentration of 125 mM for 5 min. After washing with cold PBS, cells were lysed, and DNA was sheared to an average fragment size of 200–1000 bp using an ultrasonic homogenizer for 10 min at 20% power on ice. Fifty microliters from each sonicated sample were used to determine DNA concentration and fragment size. The cell lysates were then incubated overnight with 20 μl protein A/G magnetic beads (MedChemExpress) and 10 μg ChIP grade TEAD4 antibody or YAP antibody at 4 °C. Beads were subsequently collected, washed, and treated with proteinase K for 1.5 h at 65 °C and RNase for 1 h at 37 °C. DNA was purified using a PCR purification kit (Qiagen) and assessed by qRT–PCR, and samples were normalized to input DNA. The primers used are as follows:
RIP1 Forward: TGGATGCCCTCAAGTTTTGG,
RIP1 Reverse: GAGGATCTGGAGCACAAGGA,
MLKL Forward: AACACTCACACAGCTTCCCC,
MLKL Reverse: TGGTGGGTATGATAGGGCAGA.
Luciferase assay
Cells were seeded in 12-well plates a day before transfection. A mixture containing the 8xGTIIC promoter-luciferase reporter, pRL-TK-Renilla, and specific plasmids was co-transfected into the cells. After 24 h, cells were lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (#E1960, Promega), as per the manufacturer’s instructions. Luciferase activity was quantified using a Victor Nivo Alpha S (PerkinElmer), with efficiency normalized to Renilla luciferase activity driven by the thymidine kinase promoter (pRL-TK) as an internal control.
In vitro kinase assay
For in vitro kinase assay, NF2 protein was purchased from Elabscience (Wuhan, China), MST1 and MLKL protein were purchased from MCE, and FGFR2 protein was purchased from Abcam. 10 μg substrate protein NF2/MLKL and 0.5 μg kinase FGFR2/MST1 were dissolved with 50 μl 1 × kinase reaction buffer (25 mM Tris pH 7.5, 2 mM DTT, 10 mM MgCl2, 200 μM ATP) containing protease and phosphatase inhibitor (Roche, Germany). The mixture was incubated at 30 °C for 30–60 min. Boiled water bath for 10 min was used to terminate the reaction and then froze with liquid nitrogen. Subsequent mass spectrometric identification and data analysis were completed by SpecAlly Life (Wuhan) Science and Technology Co. Ltd.
Transmission electron microscopy
KYSE140, KYSE180 or COLO680N cells were treated with 10 μM AZD4547 for 12 h or transfected with FGFR2 siRNAs for 48 h, then dissociated from the culture plates, washed once with PBS, and fixed overnight with 2.5% glutaraldehyde (pH 7.2–7.4). Subsequent electron microscopy analysis was completed by HAOMA Biotechnology Co., Ltd (Guangzhou, China).
RNA sequencing
Total RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific) following the manufacturer’s protocol. RNA integrity and concentration were assessed using an Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, CA, USA). Next-generation sequencing was performed on an Illumina HiSeq platform as per Illumina’s guidelines, employing the PE150 sequencing strategy. Data processing and analysis were conducted at Annoroad Gene Technology (Beijing, China).
Flow cytometry
To assess hCD45+, mCD45+, and hCD3+ rates, erythrocytes were lysed immediately post-collection. Then lymphoid cells were stained with corresponding antibodies for 30 min in the dark on ice and washed with PBS. After washing, cells were suspended in 400 μL propidium iodide (BD Biosciences) for flow cytometry analysis.
Human cytokine array
A cytokine array was conducted on medium from KYSE140 cells treated with 40 μM AZD4547 for 24 h. The human cytokine array panel (Wayen Biotech, Shanghai, China) was utilized according to the manufacturer’s guidelines.
ELISA analysis
IL-8 concentrations in cell culture medium post-treatment were measured using an ELISA kit as per the manufacturer’s instructions.
Immunohistochemistry
Tumor sections were deparaffinized, hydrated, and antigen-retrieved, followed by endogenous peroxidase removal. Samples were blocked with 5% BSA and incubated with primary antibodies against Ki-67 (1:400), FGFR2 (1:100), MLKL (1:1000), YAP (1:400), PD-L1 (1:200), CD3 (1:2000), IL-8 (1:400), and Granzyme B (1:1000) at 4 °C overnight. Subsequent detection used IgG polymer and diaminobenzidine (DAB) substrate (ZSGB-BIO). Staining intensity and proportion of positive cells were evaluated and scored by two independent pathologists. The intensity was graded as follows: 0, negative; 1, weak; 2, moderate; 3, strong. The proportion of positive cells was graded: 0, <5%; 1, 5–25%; 2, 26–50%; 3, 51–75%; 4, >75%. Scores were combined, with final scores of 0–3 classified as “negative” and 3–12 as “positive.” H-scores were analyzed using HALO software (Indica Labs, America).
Statistics and reproducibility
The student’s t test was used to compare the differences between two independent groups, one-way analysis of variance (ANOVA) and two-way ANOVA were used for comparisons among three or more groups, and the results are presented as the mean values ± SD. Chi-square test was used to investigate the correlation between two continuous variables. All statistical tests were two-tailed. IB images were performed at least three times as independent experiments with similar results, and representative images are shown. Statistical analyses were performed using GraphPad Prism version 10.0 (La Jolla, CA, USA). All schematic diagrams in the article were generated by PowerPoint or BioRender.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. RNA-seq raw data have been deposited to GSA-Human in Big Data Center (http://bigd.big.ac.cn/), under the accession number: HRA000862. All data are also available from the corresponding author Qimin Zhan zhanqimin@bjmu.edu.cn on request. Source data are provided with this paper.
References
Chen, W. et al. Cancer statistics in China, 2015. CA Cancer J. Clin. 66, 115–132 (2016).
Pennathur, A. et al. Oesophageal carcinoma. Lancet 381, 400–412 (2013).
Chen, D., Yu, J. & Zhang, L. Necroptosis: an alternative cell death program defending against cancer. Biochim. Biophys. Acta 1865, 228–236 (2016).
Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65 (2014).
Chen, X. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121 (2014).
Schmidt, S. V. et al. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 6, 8635–8647 (2015).
Moujalled, D. M. et al. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 4, e465 (2013).
Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Malireddi, R. K. S. et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 217, jem.20191644 (2020).
Ingram, J. P. et al. ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J. Immunol. 203, 1348–1355 (2019).
Gunther, C. et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Invest. 126, 4346–4360 (2016).
Zhang, X. et al. Ceramide nanoliposomes as a MLKL-dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol. Cancer Ther. 17, 50–59 (2018).
Liu, X. et al. Induction of an MLKL mediated non-canonical necroptosis through reactive oxygen species by tanshinol A in lung cancer cells. Biochem. Pharm. 171, 113684 (2020).
Kang, T. B. et al. Caspase-8 deficiency in mouse embryos triggers chronic RIPK1-dependent activation of inflammatory genes, independently of RIPK3. Cell Death Differ. 25, 1107–1117 (2018).
Carter, E. P., Fearon, A. E. & Grose, R. P. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 25, 221–233 (2015).
Lin, D. C. et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 46, 467–473 (2014).
The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175 (2017).
Chen, B. et al. FGFR1 signaling potentiates tumor growth and predicts poor prognosis in esophageal squamous cell carcinoma patients. Cancer Biol. Ther. 19, 76–86 (2018).
Liu, G. et al. Prognostic role of fibroblast growth factor receptor 2 in human solid tumors: a systematic review and meta-analysis. Tumour Biol. 39, 1010428317707424 (2017).
Porta, R. et al. FGFR a promising druggable target in cancer: molecular biology and new drugs. Crit. Rev. Oncol./Hematol. 113, 256–267 (2017).
Packer, L. M. et al. Bcl-2 inhibitors enhance FGFR inhibitor-induced mitochondrial-dependent cell death in FGFR2-mutant endometrial cancer. Mol. Oncol. 13, 738–756 (2019).
Kabashima, A. et al. Fibroblast growth factor receptor inhibition induces loss of matrix MCL1 and necrosis in cholangiocarcinoma. J. Hepatol. 68, 1228–1238 (2018).
Meng, Z., Moroishi, T. & Guan, K. L. Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17 (2016).
Curto, M. et al. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 177, 893–903 (2007).
Udan, R. S. et al. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5, 914–920 (2003).
Matallanas, D. et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975 (2007).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Yang, W.-H. et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 28, 2501–2508.e4 (2019).
Liu, J. et al. TEAD1 protects against necroptosis in postmitotic cardiomyocytes through regulation of nuclear DNA-encoded mitochondrial genes. Cell Death Differ. 28, 2045–2059 (2021).
Wen, W. et al. MST1 promotes apoptosis through phosphorylation of histone H2AX. J. Biol. Chem. 285, 39108–39116 (2010).
Lee, I. Y. et al. MST1 negatively regulates TNFα-induced NF-κB signaling through modulating LUBAC activity. Mol. cell 73, 1138–1149.e6 (2019).
Turunen, S. P. et al. FGFR4 phosphorylates MST1 to confer breast cancer cells resistance to MST1/2-dependent apoptosis. Cell Death Differ. 26, 2577–2593 (2019).
Ji, S. et al. FGF15 activates hippo signaling to suppress bile acid metabolism and liver tumorigenesis. Dev. Cell 48, 460–474.e9 (2019).
Qin, F. et al. Mst1 and Mst2 kinases: regulations and diseases. Cell Biosci. 3, 31 (2013).
Hao, Y. et al. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509 (2008).
Houshmandi, S. S. et al. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Mol. Cell Biol. 29, 1472–1486 (2009).
Sato, T. & Sekido, Y. NF2/Merlin inactivation and potential therapeutic targets in mesothelioma. Int. J. Mol. Sci. 19, 988 (2018).
Laulajainen, M. et al. Multistep phosphorylation by oncogenic kinases enhances the degradation of the NF2 tumor suppressor merlin. Neoplasia 13, 643–652 (2011).
Lin, C. et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut 68, 1764–1773 (2019).
Sun, C., Mezzadra, R. & Schumacher, T. N. Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452 (2018).
Gavine, P. R. et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 72, 2045–2056 (2012).
Saka, H. et al. Safety, tolerability and pharmacokinetics of the fibroblast growth factor receptor inhibitor AZD4547 in Japanese patients with advanced solid tumours: a Phase I study. Invest. N. Drugs 35, 451–462 (2017).
Kwak, Y. et al. Antitumor effects and mechanisms of AZD4547 on FGFR2-deregulated endometrial cancer cells. Mol. Cancer Ther. 14, 2292–2302 (2015).
Pearson, A. et al. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov. 6, 838–851 (2016).
Dara, L. et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 62, 1847–1857 (2015).
Kang, Y. J. et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat. Commun. 6, 8371 (2015).
Najafov, A. et al. BRAF and AXL oncogenes drive RIPK3 expression loss in cancer. PLoS Biol. 16, e2005756 (2018).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Dovey, C. M. et al. MLKL requires the inositol phosphate code to execute necroptosis. Mol. Cell 70, 936–948.e7 (2018).
Li, X. et al. RIP1-dependent linear and nonlinear recruitments of caspase-8 and RIP3 respectively to necrosome specify distinct cell death outcomes. Protein Cell 12, 858–876 (2021).
Liu, Q. et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell 184, 5559–5576.e19 (2021).
Li, Y. et al. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nat. Cell Biol. 23, 1187–1198 (2021).
Lapi, E. et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 32, 803–814 (2008).
Lin, C.-C. et al. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ. 27, 2234–2247 (2020).
Seifert, L. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249 (2016).
Wang, W. et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774.e7 (2018).
Yamauchi, T. et al. Necroptosis in esophageal squamous cell carcinoma: an independent prognostic factor and its correlation with tumor-infiltrating lymphocytes. Cancers (Basel) 13, 4473 (2021).
Lei, J. H. et al. Activation of FGFR2 signaling suppresses BRCA1 and drives triple-negative mammary tumorigenesis that is sensitive to immunotherapy. Adv. Sci. (Weinh.) 8, e2100974 (2021).
Lei, J. H. et al. FGFR2-BRD4 axis regulates transcriptional networks of histone 3 modification and synergy between its inhibitors and PD-1/PD-L1 in a TNBC mouse model. Front. Immunol. 13, 861221 (2022).
Acknowledgements
We express our gratitude to Dr. Jiahuai Han from Xiamen University for generously providing L929 WT, RIP1−/−, RIP3−/−, and MLKL−/− cell lines and to Dr. Faxing Yu from Fudan University for generously providing HEK293A WT, YAP-KO, and NF2-KO cell lines. We appreciate Dr. Chuanhui Han, Shaokun Shu, and Mi Deng from the International Cancer Institute at Peking University Health Science Center for their valuable discussions and insightful suggestions, which have greatly improved the quality of this work. This research was supported by National Key R&D Program of China 2022YFC3401000 (Y.S. & W.Z.), 2023YFC2506404 (R.H.), the National Natural Science Foundation of China 81988101 (Q.Z.), 82303007 (D.C.), 82172930 (W.Z.), 81830086 (Q.Z.), 82173332 (Y.S.), 82372590 (R.H.), the CAMS Innovation Fund for Medical Sciences 2019-I2M-5-081 (Q.Z.), 2023-I2M-2-004 (Y.S.), the Guangdong Basic and Applied Basic Research Foundation 2019B030302012 (Q.Z.), 2022A1515111202 (D.C.), 2023A1515030092 (R.H), the Major Program of Shenzhen Bay Laboratory S201101004 (Q.Z), Noncommunicable Chronic Diseases-National Science and Technology Major Project 2023ZD0501101 (W.Z.), and the “San-ming” Project of Medicine in Shenzhen SZSM202311014 (Q.Z).
Author information
Authors and Affiliations
Contributions
D.C., Z.Z., R.H., Y.S., W.Z. and Q.Z. conceptualized and designed the study. D.C., Z.Z. and R.H. conducted most of the experiments. D.Y., Y.T., H.L., Y.C. and J.L. assisted with experimental work. Y.G. provided plasmids and cells. Y.W., J.C. and Q.W. offered expert consultation. J.F. facilitated IHC experiments and interpretation of results. D.C., Z.Z., R.H., Y.S., W.Z. and Q.Z. drafted and edited the manuscript. W.Z., Y.S. and Q.Z. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tudor Moldoveanu, Zhaocai Zhou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, D., Zhao, Z., Hong, R. et al. Harnessing the FGFR2/NF2/YAP signaling-dependent necroptosis to develop an FGFR2/IL-8 dual blockade therapeutic strategy. Nat Commun 16, 4128 (2025). https://doi.org/10.1038/s41467-025-59318-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59318-9
