
Abstract
Record power conversion efficiencies (PCEs) of perovskite solar cells (PSCs) are usually achieved using organic spiro-OMeTAD. However, conventional doping with hygroscopic dopants (LiTFSI and tBP) leads to compromised device stability. We introduce a synergistic mixed doping strategy that utilizes a combination of metal-TFSI dopants—LiTFSI, KTFSI, NaTFSI, Ca(TFSI)2, and Mg(TFSI)2—to enhance doping efficiency while effectively removing hygroscopic contaminants from the Spiro-OMeTAD solution. This approach achieves PCEs exceeding 25% and significantly improves stability under harsh environmental conditions. Notably, Ca(TFSI)2 and Mg(TFSI)2 facilitate enhanced oxidative doping, while NaTFSI promotes interstitial doping in the bulk perovskite. Additionally, KTFSI serves as a catalytic agent, lowering the reaction energy barrier for the other dopants, thereby accelerating spiro-OMeTAD ion radical production. These findings underscore the potential of synergistic doping in optimizing the performance and longevity of photovoltaic devices.
Similar content being viewed by others
Introduction
Metal halide perovskite solar cells (PSCs) have achieved remarkable power conversion efficiencies (PCEs) exceeding 26% in both n-i-p and p-i-n architectures1,2,3,4,5. However, achieving long-term stability alongside high efficiency remains a significant barrier6,7,8, particularly in n-i-p PSCs9,10,11,12,13,14,15,16,17. Spiro-OMeTAD is the most commonly used hole transport layer (HTL) in these high-performance devices, but traditional doping methods—such as lithium bis(trifluoromethane)sulfonimide (LiTFSI) and 4-tert-butylpyridine (tBP)—have notable limitations18,19,20,21,22. While these approaches improve conductivity, they also introduce hygroscopic by-products, leading to moisture sensitivity and thermal degradation, ultimately compromising the stability of PSCs.
Recent efforts to improve doping strategies were developed23,24, such as CO2 doping25, which removes reactive lithium species to reduce hygroscopicity, ion-modulated radical doping26, which uses external ions to control radical formation in Spiro-OMeTAD, and photo-redox doping27, which employs photoactive complexes to catalyze the symmetry-breaking charge separation, enhancing charge transport. Although these methods show promise in improving stability, enhancing efficiency remains an ongoing challenge.
To address these issues, we developed a doping method termed the synergistic mixed doping strategy. This approach leverages two functionally complementary dopants. The first type comprises divalent metal TFSI salts, which exhibit significantly higher doping efficiency than conventional LiTFSI. These salts effectively oxidize Spiro-OMeTAD, enhancing its conductivity and work function. The second type, potassium TFSI (KTFSI), is non-doping-active but plays a catalytic role, boosting the doping efficiency of the other metal TFSI salts. Additionally, KTFSI forms aggregates that absorb residual metal TFSI and by-products, effectively expelling them from the HTL, resulting in a cleaner and more stable environment.
Using this doping strategy, we achieved PSCs with high efficiency (PCE > 25%) and remarkable stability. Our devices demonstrated a T100 lifetime of approximately 1200 hours under 25 ± 5% relative humidity without encapsulation, a T90 lifetime exceeding 2184 hours at 65° ± 5 °C with simple packaging, and a T90 lifetime over 2184 hours under continuous illumination in an N2 glovebox. This approach significantly reduces the trade-off between oxidizing Spiro-OMeTAD efficiently while maintaining stability in PSCs. This marks a breakthrough in developing more durable, high-performing organic conductive materials through innovative doping strategies.
Results and discussion
PSC performance based on 5D spiro-OMeTAD
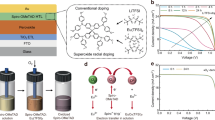
Figure 1a illustrates the chemical structures of the various dopants used in the spiro-OMeTAD solution, along with the size of the central metal ions, highlighting the distinctions between synergistic mixed doping and conventional doping (For details regarding the rationale behind selecting these dopant combinations, please refer to Supplementary Note 1). The preparation process for the doped spiro-OMeTAD solution is schematically depicted in Supplementary Fig. 1, outlining the key steps involved in implementing synergistic mixed doping strategy. Device performance parameters for standard LiTFSI (54 mol%) and low-concentration (36.7 mol%) LiTFSI, alongside single-doped spiro-OMeTAD with Ca(TFSI)₂, Mg(TFSI)₂, NaTFSI, or KTFSI, as well as LiTFSI-based binary, ternary, quaternary, and quinary-doped solutions, are provided in Supplementary Figs. 2-6. Details of the preparation of the various spiro-OMeTAD solutions are given in Methods. Among the five single dopants tested, KTFSI exhibited the lowest efficiency, while Ca(TFSI)₂ and Mg(TFSI)₂ showed better doping efficiencies than LiTFSI; however, the low-concentration (36.7 mol%) device efficiencies remained inferior to the reference device using standard LiTFSI. Notably, multi-dopant devices (ternary, quaternary, quinary) at low concentrations outperformed the reference, suggesting a synergistic effect among mixed dopants in enhancing performance.

a Chemical structures of various dopants. The total TFSI concentration of the standard reference spiro-OMeTAD solution is set at a ratio of 1:0.68 relative to the total TFSI concentration of the 5D dopants. b Current density-voltage (J-V) curves of PSCs utilizing a commercial SnO₂ electron transport layer and Cs₀.₀₅FA₀.₉₅PbI₃ prepared via sequential deposition, comparing standard LiTFSI doping and 5D doping of spiro-OMeTAD. c J-V curves of PSCs with a chemical bath-deposited (CBD) SnO₂ electron transport layer and Cs₀.₀₅FA₀.₉₅PbI₃ prepared via sequential deposition, evaluating standard LiTFSI doping against 5D doping of spiro-OMeTAD. d J-V curves of PSCs using FAPbI₃ prepared via antisolvent deposition in air, illustrating the differences between standard LiTFSI doping and 5D doping of spiro-OMeTAD. e J-V curves of PSCs incorporating Cs₀.₀₅MA₀.₀₅FA₀.₉₀PbI₃ prepared via antisolvent deposition in nitrogen, comparing the performance of standard LiTFSI doping with 5D doping of spiro-OMeTAD. f Power conversion efficiency (PCE) tracking of unencapsulated PSCs based on standard LiTFSI doping and 5D doping of spiro-OMeTAD, assessed under dark conditions (20–30% RH in air), continuous illumination (in nitrogen, 25~30 °C), and 65 ± 5 °C heat (sealed in a vial in a heating oven with no light source).
We focused on the quinary-doped (5D) spiro-OMeTAD devices, which demonstrated the best performance and highest consistency in PCE statistics. Figure 1b compares the reference and 5D devices using Cs₀.₀₅FA₀.₉₅PbI₃ PSCs with a planar commercial SnO₂ electron transport layer (ETL). The reference device required approximately 24 hours of oxidation to achieve optimized PCE (reward scan: 24.59%, forward scan: 23.96%). In contrast, the 5D device reached optimal PCE after only 12 hours, with reward and forward scans of 25.65% and 25.44%, respectively. The reduced hysteresis in the 5D device may result from the interstitial doping of Na⁺ in the bulk perovskite, a point we will discuss later. In addition to SnO₂/Cs₀.₀₅FA₀.₉₅PbI₃ devices, we applied the 5D spiro-OMeTAD to PSCs with various perovskite compositions, electron transport layers (ETLs), and fabrication methods (one-step, two-step, N₂ or air environments). These included SnO₂(CBD)/Cs₀.₀₅FAPbI₃ (two-step, in N₂), SnO₂(CBD)/FAPbI₃ (one-step, in air), and SnO₂(CBD)/Cs₀.₀₅FA₀.₉₀MA₀.₀₅PbI₃ (one-step, in N₂). In all cases, the PCEs matched or slightly exceeded those of standard LiTFSI-doped reference devices, demonstrating the versatility of the 5D doping strategy across various systems (Fig. 1c–e, detailed photovoltaic parameters were provided in Supplementary Figs. 7 and 8, and Supplementary Table 1).
Due to the reduced dopant concentration in the 5D spiro-OMeTAD solution (5D: ref = 0.68:1 (TFSI-), 5D: ref = 0.48:1 (metal ionic)) and the precipitation of aggregations formed during doping, hygroscopic by-products were largely filtered out of the HTL system, contributing to improved device stability (this will be explained in detail later). A systematic comparison of device stability between the 5D and standard LiTFSI devices under storage, heat, and illumination is presented in Fig. 1f. The reference device exhibited PCE degradation after 600 hours of dark storage at approximately 25 ± 5% RH, while the 5D device maintained stable performance for 1200 hours (ISOS-D-1). Under continuous illumination (ISOS-L-1I) or at 65 ± 5 °C (ISOS-T-1), the 5D device extended lifetime (T90) from approximately 500 hours to over 2184 hours.
Scanning electron microscopy (SEM) further illustrated the protective effect of the 5D-doped spiro-OMeTAD HTL (Supplementary Fig. 9). The reference device, after exposure to ~45 ± 5% RH and 60 °C thermal stress, exhibited significant morphological changes, including corroded metal electrodes and voids in the perovskite layer. This suggests that residual LiTFSI and by-products absorbed moisture, while the volatilization of tBP accelerated perovskite degradation. In contrast, the 5D devices maintained a compact, uniform morphology, indicating that the 5D-doped HTL effectively protected the perovskite layer.
Electrical properties of 5D spiro-OMeTAD
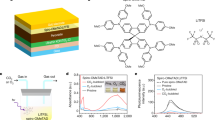
We compared the electrical properties of standard LiTFSI-doped spiro-OMeTAD films with those of the 5D spiro-OMeTAD. Various techniques were employed, including UV-vis absorption spectroscopy, Kelvin probe force microscopy (KPFM), conductive atomic force microscopy (C-AFM), space-charge-limited current (SCLC) analysis for hole mobility, and two-probe measurements for bulk conductivity. The characteristic peak at 520 nm28 in the 5D spiro-OMeTAD solution confirmed the formation of radical cations (spiro-OMeTAD•+) (Fig. 2a). The bulk conductivity (σ) of the spiro-OMeTAD films was monitored over aging time (Fig. 2b). As the aging period increased, the conductivity of the 5D sample consistently remained higher than that of the standard LiTFSI-based film. After more than 300 hours, the σ of the LiTFSI-doped sample spiked, likely due to leakage resulting from void formation in the film. SCLC analysis indicated that the hole mobility of the 5D spiro-OMeTAD film (7.28 × 10⁻³ cm² V⁻¹ s⁻¹) was slightly higher than that of the standard LiTFSI-doped film (4.42 × 10⁻³ cm² V⁻¹ s⁻¹) (Fig. 2c).

a Ultraviolet-visible absorption spectra of spiro-OMeTAD solutions employing both conventional and 5D doping strategies, with an insect displaying photograph of the solutions. b The evolution of electrical conductivity (σ) over aging time for spiro-OMeTAD films prepared using conventional versus 5D doping. c Space charge limited current (SCLC) hole mobilities measured in hole-only devices (ITO/PEDOT:PSS/spiro-OMeTAD/Au) for both doping strategies. KPFM surface topography images of spiro-OMeTAD films/ITO samples based on conventional (d) and 5D doping (e). Conductive atomic force microscopy (CAFM) current maps of spiro-OMeTAD films/ITO samples based on conventional (f) and 5D doping (g). h Photoluminescence (PL) intensity of perovskite/glass samples with spiro-OMeTAD for both doping methods. i Time-resolved photoluminescence (TRPL) measurements of perovskite/glass samples with spiro-OMeTAD utilizing conventional and 5D doping strategies.
Using conductive atomic force microscopy (C-AFM) and Kelvin probe force microscopy (KPFM), we observed that both films displayed comparable conductivity (Fig. 2d, e); however, the 5D spiro-OMeTAD exhibited a higher work function, suggesting a weakened p-type character (Fig. 2f, g). In situ KPFM and C-AFM measurements under 40% relative humidity (RH) demonstrated that, within 30 minutes, the standard-doped spiro-OMeTAD film experienced significant changes in work function and conductivity, while the 5D film remained stable (Supplementary Figs. 10 and 11).
We further evaluated charge extraction from the perovskite to the HTL using steady-state photoluminescence (PL) and time-resolved PL (TRPL). Similar PL intensities indicated comparable charge extraction efficiencies, likely due to negligible differences in the concentration of spiro-OMeTAD radical cations (Fig. 2h). The carrier lifetime (τ) of the perovskite was measured at 687 ns. The 5D spiro-OMeTAD-based perovskite film exhibited a two-exponential decay with a fast lifetime (τ₁ = 69 ns) and a slow lifetime (τ₂ = 2280 ns). The shorter τ₁ confirmed efficient charge extraction, while the longer τ₂ suggested improved suppression of non-radiative recombination at the interface. In contrast, the standard LiTFSI-doped spiro-OMeTAD-based film displayed only a fast decay, likely due to residual LiTFSI causing interface degradation and non-radiative recombination29,30 (Fig. 2i). The differential lifetime τPL derived from the TRPL decay also clearly demonstrates the difference in carrier dynamics between the ref and 5D (Supplementary Fig. 12). The charge extraction efficiency and suppressed recombination in the 5D device were further corroborated by transient photovoltage (TPV) and transient photocurrent (TPC) measurements (Supplementary Fig. 13), demonstrating that the 5D device maintains the same charge extraction efficiency as the reference while contributing to a higher open-circuit voltage (VOC) in PSCs using the 5D spiro-OMeTAD (Supplementary Table 2).
Ultraviolet photoelectron spectroscopy (UPS) measurements revealed that the valence band energy (EvB) of the 5D spiro-OMeTAD (−5.15 eV) was lower than that of the reference spiro-OMeTAD (−5.00 eV) (Supplementary Fig. 14). This reduction decreased the energy-level gap (ΔEvB) between the perovskite (−5.97 eV) and spiro-OMeTAD from 0.97 eV (LiTFSI) to 0.82 eV, contributing to reduced VOC loss in PSCs (Supplementary Fig. 15). Consequently, PSCs incorporating 5D spiro-OMeTAD demonstrated enhanced PCEs and improvements in VOC (Supplementary Table 2).
The mechanism of doping spiro-OMeTAD
UV-visible spectra and bulk conductivity measurements ranked the doping efficiency as Mg²⁺ > Ca²⁺ > Li⁺ > Na⁺ > K⁺ (Supplementary Figs. 16 and 17), reflecting their varying abilities to generate spiro-OMeTAD radicals. We quantitatively compared the formation kinetics of spiro-OMeTAD•+TFSI⁻ based on Mg²⁺, Li⁺, Na⁺, and 5D dopants (Supplementary Figs. 18–21). Except for the NaTFSI doped sample, which showed undetectable EPR signals, a singlet EPR signal with giso = 2.0033 ± 0.0002, typical for the organic cation radical of spiro-OMeTAD31, was observed in all other samples, confirming that the oxidation of spiro-OMeTAD occurs under illumination. The maximal (equilibrium) EPR values were 5.71 for Mg(TFSI)2, 0.98 for LiTFSI, and approximately 0 for NaTFSI (Supplementary Fig. 22). Given these EPR values and the efficiency rankings, we predicted a theoretical EPR value for the 5D dopant system to be below 2.48, calculated as follows: [(0.2 × 5.71 (Mg)) + (<0.2 × 5.71 (Ca)) + (0.2 × 0.98 (Li)) + (0 (Na)) + (0 (K))]. However, our experimental value of 4.5 suggests a synergistic effect among the mixed dopants, enhancing doping efficiency beyond their individual contributions.
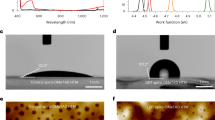
We then employed high-resolution liquid nuclear magnetic resonance (NMR) to determine whether the reaction mechanisms differed between individual and mixed dopants. Six types of hydrogen atoms in spiro-OMeTAD (Fig. 3a) were analyzed across undoped, Li-doped, Ca-doped, and 5D-doped samples, revealing a consistent mechanism: regardless of dopant type, the nitrogen atom serves as the reaction site. Chemical shifts of the hydrogen atoms supported this conclusion, ordered as 6 > 5 > 3 > 1 > 2 > 4. Building on this, we derived the doping mechanisms of other dopants from the known LiTFSI oxidation pathway according to Equation 1 and 2 (Supplementary Fig. 23)32. Differences primarily arise in the second step, with reaction energies per TFSI calculated as follows: -3.23 eV (Mg(TFSI)2), -2.27 eV (Ca(TFSI)2), −1.36 eV (LiTFSI), −0.34 eV (NaTFSI), and 0.43 eV (KTFSI) (Fig. 3b), in agreement with our conductivity and UV-visible data. The positive energy for KTFSI means its inability to oxidize spiro-OMeTAD directly.

a 1H NMR spectra of undoped spiro-OMeTAD, LiTFSI-doped spiro-OMeTAD, Ca(TFSI)₂-doped spiro-OMeTAD, and 5D doped spiro-OMeTAD solutions. The molecular structure of spiro-OMeTAD is illustrated on the right, with H types attributed and labeled in the spectra. b Density functional theory (DFT) calculated reaction energies for various dopants based on Equation (2) from the classical oxidation reaction of spiro-OMeTAD, which is provided in supplementary information Fig. 23. c Comparison of the reaction energy diagram for LiTFSI doping of spiro-OMeTAD with and without the presence of non-doping active KTFSI. d Comparison of the reaction energy diagram for Ca(TFSI)₂ doping of spiro-OMeTAD with and without non-doping active KTFSI. e Electron paramagnetic resonance (EPR) signals of doped spiro-OMeTAD solutions (72 mg/mL) in the presence and absence of KTFSI. f Evolution of EPR signals from doped spiro-OMeTAD solutions (72 mg/mL) with and without KTFSI under continuous illumination. g Schematic representation illustrates the mechanism by which KTFSI lowers the reaction energy barrier: a Additional energy required to break the ionic bond between Li⁺ and TFSI⁻. b KTFSI attracts the unsaturated Li⁺ ions from LiTFSI, facilitating the formation of a TFSI dimer that creates a transition state, thereby promoting the dissociation of one TFSI to bind with spiro-OMeTAD.
Interestingly, our experiments showed that devices containing KTFSI, despite its lack of doping activity, demonstrated superior performance compared to those without it. In binary dopant systems, Li+K outperformed both Li+Ca and Li+Mg, while in ternary systems, Li+K+Mg was more effective than Li+Ca+Mg. To understand KTFSI’s role, we examined the change in reaction barriers for LiTFSI with and without KTFSI. Although the reaction energy for LiTFSI is negative (−1.36 eV), overcoming the barrier to form the intermediate transition state [spiro⁺-O₂⁻… TFSI… Li] is necessary. DFT calculations indicate that KTFSI reduces this barrier from 2.45 eV to −0.54 eV (Fig. 3c, with optimized structures illustrated in Supplementary Figs. 24 and 25). A similar effect was observed with the more active Ca(TFSI)₂, where the reaction barrier was reduced from 0.54 eV to −1.57 eV (Fig. 3d, with optimized structures illustrated in Supplementary Figs. 26 and 27).
Based on the findings from LiTFSI and Ca(TFSI)₂, we propose that KTFSI may be applicable across a broader range of doping-active dopants. Moreover, a comparative analysis of EPR intensity over time in the Mg/Ca/Li/Na quaternary system, both with and without the inclusion of KTFSI, further substantiates its promoting effect. The presence of KTFSI notably enhances both the yield (Fig. 3e) and the generation rate (Fig. 3f) of spiro-OMeTAD radical ions. This enhancement can be attributed to two mechanisms (Fig. 3g): first, as Li⁺ and TFSI⁻ ions partially dissociate, the unsaturated Li⁺ ion is drawn toward KTFSI, forming a transition state characterized by one Li⁺ and two TFSI⁻ ligands. This arrangement facilitates the dissociation of a single TFSI⁻ ion, allowing it to bind effectively with spiro-OMeTAD. Second, due to KTFSI’s lack of oxidizing properties towards spiro-OMeTAD, its concentration remains stable, thereby functioning as a catalyst within the doping system.
Removal of metal ions improve device reliability
Figure 4a presents spiro-OMeTAD solutions doped with various dopants, all of which appear clear, except for those containing KTFSI, which exhibit turbidity. Dynamic light scattering (DLS) analyses reveal the presence of micron-sized aggregates in the turbid solutions (Supplementary Fig. 28). These aggregates likely form due to the larger ionic radius and reduced coordination ability of K⁺ compared to Li⁺. Additionally, the higher zeta potentials associated with these aggregates suggest their inherent instability (Supplementary Fig. 29). Importantly, the aggregates induced by KTFSI are expected to effectively adsorb impurities, such as unreacted dopants and reaction by-products, from the solution. These impurities can subsequently be removed from the HTL system through filtration.

a Photographs depicting spiro-OMeTAD solutions with various dopants alongside filtered 5D spiro-OMeTAD solutions. b X-ray photoelectron spectroscopy (XPS) atomic ratios of spiro-OMeTAD films prepared from precursor solutions, illustrating changes before and after filtration. c Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis detailing the concentrations of Li, Na, K, Mg, and Ca atoms in both reference and 5D spiro-OMeTAD solutions before and after filtration. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profile analysis showcasing the distribution of Li (d) and Na (e) within the hole transport layer (HTL)/perovskite/electron transport layer (ETL)/ITO samples based on the reference and 5D spiro-OMeTAD HTL. Further analysis of other elements is provided in Supplementary information Fig. 33. f Maximum power point (MPP) tracking stability of PSCs utilizing the reference and 5D spiro-OMeTAD.
X-ray photoelectron spectroscopy (XPS) analysis of films prepared from filtered and unfiltered 5D spiro-OMeTAD solutions reveals a significant reduction in Na, K, Ca, and Mg signals following filtration, alongside a notable decrease in oxygen content (Fig. 4b and Supplementary Fig. 30). This suggests that these dopants primarily exist in the HTL as oxide products. In contrast, films doped with LiTFSI exhibit only a slight reduction in Li and oxygen signals after filtration, indicating that minimal amounts of LiTFSI or its oxidation by-products are removed. Given the interactions between tBP and the dopants (Supplementary Fig. 31)20, we speculate that these dopants or their by-products may associate with tBP molecules, adsorbing onto the KTFSI-induced aggregates and facilitating their removal during filtration.
Recognizing the low sensitivity of XPS for trace elements, particularly Li, we further employed inductively coupled plasma-atomic emission spectrometry (ICP-AES) to quantitatively compare the concentrations of various dopants in solutions before and after filtration, as well as to calculate the filtration efficiency of each dopant (Fig. 4c). The concentration of Li atoms in the 5D spiro-OMeTAD solution is markedly lower than that in the reference solution, accounting for merely one-seventh of the latter’s concentration, consistent with the actual feed ratio (1:(0.68/5)). This disparity becomes even more pronounced post-filtration. Interestingly, Na⁺ emerges as the predominant ion in the filtered 5D solution. The calculated reduction ratios for different dopants are detailed in Supplementary Table 3. For reference LiTFSI-doped spiro-OMeTAD, Li shows minimal changes in concentration, with a reduction ratio of only 5%, indicating limited removal by the filtration process. In contrast, the 5D-doped spiro-OMeTAD solution exhibits reduction ratios of 94% for K, 72% for Mg, 35% for Ca, 29% for Na, and 20% for Li following filtration. The high reduction ratio for K suggests that KTFSI-induced aggregates can be effectively filtered. The greater reduction of Li in the 5D solution compared to the reference indicates that KTFSI-induced aggregation enhances the filtration efficiency of other dopants, likely due to the adsorptive purification of KTFSI-based aggregates (Supplementary Fig. 32). Additionally, in the Spiro-OMeTAD solution with 5D dopants, the filtration efficiencies of the other dopants exceed that of LiTFSI. These observations suggest that the implementation of the 5D doping strategy may contribute to the formation of a more “purified” HTL.
We also conducted time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profiling to assess the distribution of residual metal ions in PSCs. For PSCs with reference spiro-OMeTAD, a stronger signal corresponding to Li ions was detected throughout the active layer and both charge-extraction layers, consistent with literature results33. In contrast, the 5D samples exhibited a drastic reduction in Li ion signals across the active layer and both charge-extraction layers (Fig. 4d), attributable to the significantly reduced LiTFSI concentration in the spiro-OMeTAD solution and enhanced filtration efficiency due to KTFSI aggregation. Although Mg(TFSI)₂, Ca(TFSI)₂, KTFSI, and NaTFSI were introduced into the 5D solution, the first three did not significantly affect the system (see Supplementary Fig. 33 for detailed analysis), with only the distribution signal of Na being enhanced throughout the active layer and both charge-extraction layers (Fig. 4e). Considering the role of Na+ in interstitial doping34,35, we posit that the increased Na+ doping contributes to the observed reduction in hysteresis effects in the 5D devices.
Scanning electron microscopy (SEM) mapping reveals the formation of numerous holes in the reference spiro-OMeTAD film after 72 hours of storage, attributed to the hygroscopic nature of residual dopants and by-products. Fourier-transform infrared (FTIR) analysis further confirms that the LiTFSI-doped spiro-OMeTAD film absorbs moisture during storage, whereas the 5D-doped film does not (Supplementary Fig. 34). In addition to the improvements in storage stability, heat stability, and light stability illustrated in Fig. 1c, d, the 5D dopant-based spiro-OMeTAD HTL demonstrated long-lasting maximum power point (MPP) tracking under continuous illumination at 60 ± 5 °C. The MPP for a reference device with a LiTFSI-doped spiro-OMeTAD HTL decreased to below 80% of its initial value within 28 hours. In contrast, the perovskite solar cell employing a 5D-doped spiro-OMeTAD HTL exhibited a significantly slower decline in MPP, retaining approximately 80% of its initial maximum power even after 1000 hours of MPP tracking. We attribute the enhanced reliability of the device to the reduced presence of metal ions in solar cells utilizing synergistic mixed dopants (Fig. 4f).
We further expand our discussion to encompass the universality of the 5D strategy. Although our initial selection of mixed dopant components was somewhat arbitrary, detailed characterization and mechanistic studies of the LiTFSI, NaTFSI, KTFSI, Mg(TFSI)₂, and Ca(TFSI)₂ combination now indicate that multiple alternative formulations are viable. In particular, to achieve the self‐cleaning function, potential alternative 5D should incorporate an metal-TFSI featuring a large metal ion and minimal doping activity (e.g., KTFSI, RbTFSI, or CsTFSI). For highly efficient doping and oxidation of spiro‐OMeTAD, all of the remaining dopants are viable, provided that their concentrations are eventually optimized to yield an appropriate spiro‐OMeTAD⁺ concentration. In addition, considering that the metal ions from the dopants can migrate into the perovskite layer, one can select the Metal-TFSI species based on the desired level of ionic doping into the perovskite.
The presence of non-doping-active KTFSI enhances the doping efficiency of other dopants by lowering their reaction energy barrier, facilitating the generation of sufficient spiro-OMeTAD⁺TFSI⁻ radical cations even at reduced concentrations. Notably, the incorporation of KTFSI leads to the formation of micro-sized aggregates within the spiro-OMeTAD solution. These aggregates can effectively sequester unreacted metal TFSI salts and metal oxide by-products, which can compromise device stability, allowing for their removal through simple filtration prior to application. Consequently, the synergistic mixed doping strategy effectively reconciles the balance between efficiency and stability, significantly broadening the range of options available for doping organic semiconductors.
Methods
Materials
The transparent indium tin oxides glass (ITO, 15 Ω per square) and glass/fluorine-doped tin oxide (FTO, 10–15 Ω per square) were purchased from Advanced Election Technology Co., Ltd. The perovskite raw materials including lead iodide (PbI2, purity 99.999%), formamidinium iodide (FAI, purity 99.99%), methylammonium iodide (MAI, purity 99.9%), methylammonium chloride (MACl, purity 99.9%) were all purchased from Advanced Election Technology Co., Ltd. Cesium iodide (CsI, purity 99.999%), 2-Phenethylammonium iodide (PEAI, purity 99.5%), Octylammonium p-toluenesulfonate (OATsO, purity 99.5%) and Spiro-OMeTAD (99.8%) were purchased from Xi’an Polymer Light Technology Corp. The required solvent including N, N-Dimethylformamide (DMF, extra dry, purity 99.8%) and isopropanol (IPA, extra dry, purity 99.8%) were purchased from J&K Scientific. Dimethyl sulfoxide (DMSO, anhydrous, ≥99.9%), anisole (anhydrous, ≥99.9%), chlorobenzene (CB) (anhydrous, ≥99.9%), FK209, water (ACS reagent), t-BP (99%), Li-TFSI, NaTFSI, KTFSI, Ca(TFSI)2, Mg(TFSI)2, silver (Ag, purity 99.99%) and gold (Au, purity 99.999%) were purchased from Sigma-Aldrich Inc. Tin Oxide (SnO2) nanoparticles (15 wt% in water) were obtained from Alfa-Aesar Inc. All chemical reagents were used as received without further purification unless otherwise specified.
Preparation of electron transport layer
Glass/ITO substrates (15 Ω sq-1) were sequentially cleaned by sonication with detergent, deionized water, acetone, and isopropyl alcohol for 20 min each and treated in ultraviolet-ozone for 30 min. A thin layer (ca. 30 nm) of SnO2 was spin-coated onto the ITO glass and baked at 160 °C for 35 min. After cooling to room temperature, the glass/ITO/SnO2 substrates were treated with ultraviolet-ozone for 15 min and finally transferred to a N2-filled glovebox before use. Chemical bath deposition (CBD) of SnO2 layers were carried out as follows: FTO substrates were cleaned by sonicating in detergent solution, deionized water, acetone, and IPA for 20 min each. CBD solution was prepared by mixing 625 mg of urea, 625 μL of HCl, 12.5 μL of TGA, and 137.5 mg of SnCl2·2H2O per 50 mL of DI water. The FTO substrates and the CBD solution were loaded onto a glass reaction vessel and reacted at 90 °C for 5 h. After the reaction, the substrate was removed from the solution and washed by deionized water and isopropanol sequentially in the ultrasonic bath for 5 min each. Before use, CBD-SnO2 layers were annealed in the air (RH = 25–35%) at 170 °C for 60 min, then UV-O3 treated for 15 min. In this work, FTO/SnO2-based devices were only used for efficiency; device characterization and stability tests were carried out using ITO/SnO2-based devices.
Preparation of perovskite films
Cs0.05FA0.95PbI3 perovskite (sequential deposition): PbI2 solution was made by dissolving 1.4 M PbI2 and 0.07 M CsI in 1 mL DMF/DMSO mixed solvent (v/v 94/6). The FAI/MACl solution was prepared by dissolving 80 mg FAI and 13 mg MACl in 1 mL IPA. The solutions were stirred overnight before use. The PbI2 solution was spin-coated onto the electron transport layer substrate at 1500 rpm for 40 s and the FAI/MACl solution was spin-coated onto the substrate at 1800 rpm for 40 s. No annealing was performed for PbI2. The film was annealed in a glove box at 90 °C for 60 s, and then annealed in the open at 150 °C for 10 min with 30-40% humidity.
Cs0.05MA0.05FA0.9PbI3 perovskite (antisolvent deposition): 1.65 M perovskite precursor solution was prepared by dissolving CsI, FAI, MAI, and PbI2 in a mixed solvent of 800 µL DMF and 200 µL DMSO. An additional 5 mol% PbI2, 15 mol% MACl, and 1 mg MASCN were added to the precursor solution and the solution was stirred for 1 hour. The filtered perovskite precursor was then spin-coated on the substrate at 1000 rpm for 10 s and 5000 rpm for 40 s in nitrogen glovebox. 300 µL of anisole anti-solvent was dropped quickly at 25 s during the second spinning step. Afterwards, the film was annealed at 110 °C for 30 min in N2 glovebox.
FAPbI3 perovskite (antisolvent deposition in air): the precursor solution (1.8 M) was prepared by mixing 911 mg PbI2, 309.6 mg FAI, 4 mg PACl, 30 mg MACl in mixed solvents of 840 µl DMF and 160 µl NMP. The as-prepared solution was vigorously shaken for 1 hour before use. A two-step rotation procedure of 1000 rpm at 10 s followed by 4000 rpm at 40 s was employed to spin-coating the precursor solution. During spinning, 200 µl of ether was dropped onto the film 24 s prior to the end of the second step. The as-cast pristine films were annealed at 150 °C for 10 min. Spin-coating and annealing were both conducted in the open with 20-30% humidity.
Fabrication of PSCs devices
After depositing perovskite films on this ETL substrate following the abovementioned method, the passivator solutions were deposited on the perovskite surface by spin-coating. For the surface passivation layer, 20 μL of passivator solution (0.5 mg/mLPEAI or 1 mg/mL OATsO) was spin-coated on top of the as-prepared perovskite at 4000 rpm for 40 s. No annealing was performed for the passivation layer. Finally, the spiro-OMeTAD solution was spun onto the passivated perovskite film at 3000 rpm for 30 s. Typically, the reference spiro-OMeTAD solution was made of 72 mg spiro-OMeTAD in 1 mL CB with 28.8 µL t-BP and 32 mM Li-TFSI (17.5 µL of 520 mg/mL in ACN). The 5D spiro-OMeTAD solution was made of 72 mg spiro-OMeTAD in 1 mL CB with 28.8 µL t-BP and 21.76 mM M-TFSI (4.35 mM LiTFSI, 4.35 mM NaTFSI, 4.35 mM KTFSI, 2.17 mM Ca(TFSI)2, and 2.17 mM Mg(TFSI)2). For single-doped spiro-OMeTAD solution, the dopants consist of 21.76 mM LiTFSI, or 21.76 mM NaTFSI, or 21.76 mM KTFSI, or 10.88 mM Ca(TFSI)2 or 10.88 mM Mg(TFSI)2. For LiTFSI-based binary doped spiro-OMeTAD solution, the dopants consist of 10.88 mM LiTFSI and 10.88 mM NaTFSI, or 10.88 mM KTFSI, or 5.44 mM Ca(TFSI)2, or 5.44 mM Mg(TFSI)2. For LiTFSI-based ternary doped spiro-OMeTAD solution, the dopants consist of 7.25 mM LiTFSI+7.25 mM NaTFSI+7.25 mM KTFSI (Li+Na+K), or 7.25 mM LiTFSI+7.25 mM NaTFSI+3.62 mM Ca(TFSI)2 (Li+Na+Ca), or 7.25 mM LiTFSI+7.25 mM NaTFSI+3.62 mM Mg(TFSI)2 (Li+Na+Mg), or 7.25 mM LiTFSI+7.25 mM KTFSI + 3.62 mM Ca(TFSI)2 (Li+K+Ca), or 7.25 mM LiTFSI+7.25 mM KTFSI + 3.62 mM Mg(TFSI)2 (Li+K+Mg), or 7.25 mM LiTFSI+3.62 mM Ca(TFSI)2 + 3.62 mM Mg(TFSI)2 (Li+Ca+Mg). For LiTFSI-based quaternary doped spiro-OMeTAD solution, the dopants consist of 5.44 mM LiTFSI+5.44 mM KTFSI + 2.72 mM Ca(TFSI)2 + 2.72 mM Mg(TFSI)2 (Li+K+Ca+Mg). Regardless of how many dopants are mixed, the total amount of TFSI contained in the solution is constant. After oxidizing in a dryer with humidity below 20% for 12~24 h, the devices were completed by evaporating 10 nm MoO3 and then 100 nm silver or gold in a vacuum chamber (base pressure, 2 × 10−4 Pa).
Device characterization
J-V characteristics. J-V characteristics of photovoltaic cells were taken using a Keithley 2400 source measure unit under a simulated AM 1.5 G spectrum, with an Oriel 9600 solar simulator. The illumination light was calibrated to 100 mW cm−2 AM 1.5 G using a silicon reference cell. The devices based on commercial SnO2 were measured both in reverse scan (1.20 V → 0 V, step 0.02 V) and in forward scan (0 V → 1.2 V, step 0.02 V), while the devices based on CBD SnO2 were measured only in reverse scan (1.20 V → 0 V, step 0.02 V) due to the large hysteresis induced by such ETL. All the devices were measured without pre-conditioning, such as light-soaking, and applied a bias voltage. The aperture area of the device was determined by the shadow mask to be 0.072 cm2 for solar cells.
Stability test. The devices for stability test were fabricated with a structure of ITO/SnO2/Cs0.05FA0.95PbI3/spiro-OMeTAD/Au. The MPP tracking of the devices was conducted under 1 sun in N2 glovebox at 60 ± 5 °C. The voltage at the MPP was automatically adjusted and applied, and the power output of the devices was tracked over time.
Materials characterization
Steady-state PL. Steady-state PL measurements were conducted using a FLS1000 fluorescence spectrometer (Edinburgh Instruments). A 450 nm monochromatic light was used as the excitation source by filtering the white light from a 450 W xenon arc lamp. The intensity of the excitation light can be adjusted by changing the slit width. PL emission spectra with wavelengths ranging from 680 nm to 890 nm were collected. The slit width for the excitation source was fixed at 2 nm to ensure the same irradiation intensity for different samples.
Time resolved photoluminescence (TRPL). Time resolved photoluminescence (TRPL) spectra measurements were taken on a 1 μm2 laser spot size at room temperature using commercially spectrometer (FLS 1000) with 405 nm laser source (less than 100 ps, 0.1 MHz).
Bulk conductivity (two-probe methods). The substrate of the sample films for conductivity measurement was the etched ITO glass with a channel 300 μm wide. Considering the low conductivity values of the doped spiro-OMeTAD films relative to the ITO electrodes, contact resistance of the probes is negligible, so a two-probe method was adopted with ITO as both electrodes, i.e., ITO/ doped spiro-OMeTAD/ITO. The current-voltage curves from ‒2 V to 2 V were collected by the source meter (2400 Series Source Meter, Keithley Instruments). Conductivity was calculated according to Ohm’s law (eqn (1)):
where I, W, V, d and L represent the current injected, width of the channel (300 µm), voltage read out, thickness of the film in the channel, and length of the channel (2 cm), respectively.
The photoelectron spectroscopic experiments. UPS (HeI hv = 21.22 eV) and XPS (monochromatized Al Kα hν = 1486.6 eV), were carried out using a ThermoFisher ESCALAB Xi+ system and calibrated by referencing to Fermi level and Au 4f7/2 peak position of the Ar+ion sputter-clean gold foil. The XPS experimental condition was set so that the full-width at half-maximum of the clean Au 4f7/2 line 15(at the binding energy of 84.00 eV) was 0.65 eV. UPS was performed to study the interfacial alignment and the frontier electronic structure features with an error margin of ±0.05 eV. The work function and the vertical IP are derived from the secondary electron cut-off and the frontier edge of the occupied density of states. All spectra were measured at a photoelectron take-off angle of 0° (normal emission).
Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) measurements were performed on a TOF-SIMS instrument from PHI nano TOF3, operated in the spectral mode using a 30 keV Bi3+ primary ion beam with an ion current of 7.6 nA. A mass-resolving power of approximately 6,000 m/∆m was reached. For depth profiling a 2 keV GICB sputter beam with a current of 20 nA was used to remove the material layer-by-layer in interlaced mode from a raster area of 400 × 400 µm. This raster area was chosen to ensure a flat crater bottom over an area of 100 × 100 µm used for the mass spectrometry. The position of the ITO substrate interface in the sputter depth profile was defined by the half-maximum of the In2+secondary ion count rate.
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements were performed on an ICP-AES instrutment from ThermoFisher ICAP PRO XP. The sample was prepared as shown below: 3 mL of deionized water was added to 1 mL of Spiro-OMeTAD solution, stirred overnight, and left to stand for 1 h before the upper clear solution (aqueous phase) was aspirated with a syringe for testing.
Computational methods. We use density function theory (DFT) to calculate oxidation thermodynamics of Spiro-OMeTAD with metal-TFSI. The exchange-correlation interactions are considered by a semi-local gradient approximation (Perdew-Burke-Ernzohof 1996)36. we use only Gamma point for the integration of energy in geometry optimization. We use the Gaussian type double-zeta valence polarized function as basis set (DZVP), and the cutoff energy is set to be the 600 eV. To accelerate calculations, we used a pseudopotential to replace the interactions involving cores and inner electrons. All calculations are performed with CP2K37.
Other characterizations. SEM images were obtained using a scanning electron microscope (Zeiss Gemini 450) with an accelerating voltage of 5 kV and a beam current of 200 pA. The FTIR spectra were acquired using a Fourier transform infrared spectrometer (Nicolet iS50, Thermo Scientific). NMR spectra were measured by a 500 MHz solution NMR spectrometer with a room temperature probe (Bruker Biospin). Ultraviolet-visible (UV-Vis) absorption spectra were measured with a PerkinElmer model Lambda 900 fitted with a reflection/transmission accessory. Conductived atomic force microscopy (C-AFM) and kelvin probe force microscopy (KPFM) measurements were carried out using an AFM (Bruker Dimension Icon) in air with controlled humidity at around 30 ~ 40 RH%. The Spiro-OMeTAD sample contains hygroscopic dopants, and during C-AFM testing in air (35-45% RH), a wet electrolyte layer may form on the surface of the sample or in localized regions enriched with dopants. This electrolyte layer is typically conductive, which could lead to a stronger current signal than normal. Due to the influence of moisture on the electrolyte layer, the current may become more unstable and difficult to control. In such cases, the current response in C-AFM may show higher background currents, causing abnormal high-current regions in the image, thus reducing the contrast and image quality. The appearance of localized high currents in the Spiro-OMeTAD sample’s C-AFM images indicates that the sample is likely highly hygroscopic. The EPR spectra were measured using a CW electron paramagnetic resonance spectrometer ESP-100 spectrometer and the solutions for EPR testing do not require additional N2 gas protection, and illumination is added to the test to reduce total test time. Mobility measurements were conducted based on the ITO/PEDOT:PSS/doped spiro-OMeTAD/Au. The current-voltage curves from 0 V to 8 V were collected by the source meter (2400 Series Source Meter, Keithley Instruments).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All (other) data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Source data are provided with this paper.
References
Park, J. et al. Controlled growth of perovskite layers with volatile alkylammonium chlorides. Nature 616, 724–730 (2023).
Shen, Y. et al. Strain regulation retards natural operation decay of perovskite solar cells. Nature, https://doi.org/10.1038/s41586-024-08161-x (2024).
Chen, H. et al. Improved charge extraction in inverted perovskite solar cells with dual-site-binding ligands. Science 384, 189–193 (2024).
Zheng, Y. et al. Towards 26% efficiency in inverted perovskite solar cells via interfacial flipped band bending and suppressed deep-level traps. Energy Environ. Sci. 17, 1153–1162 (2024).
Green, M. A. et al. Solar cell efficiency tables (Version 64). Prog. Photovoltaics 32, 425–441 (2024).
Gao, D. et al. Long-term stability in perovskite solar cells through atomic layer deposition of tin oxide. Science 386, 187–192 (2024).
Wang, M., Fei, C., Uddin, M. A. & Huang, J. Influence of voids on the thermal and light stability of perovskite solar cells. Sci. Adv. 8, 5977 (2022).
Ding, Y. et al. Cation reactivity inhibits perovskite degradation in efficient and stable solar modules. Science 386, 531–538 (2024).
Song, W. et al. Improving the morphology stability of Spiro-OMeTAD films for enhanced thermal stability of perovskite solar cells. ACS Appl. Mater. 13, 44294–44301 (2021).
Ye, L. et al. Superoxide radical derived metal-free spiro-OMeTAD for highly stable perovskite solar cells. Nat. Commun. 15, 7889 (2024).
Jena, A. K., Numata, Y., Ikegami, M. & Miyasaka, T. Role of spiro-OMeTAD in performance deterioration of perovskite solar cells at high temperature and reuse of the perovskite films to avoid Pb-waste. J. Mater. Chem. A 6, 2219–2230 (2018).
Liu, X. et al. Perovskite solar cells based on spiro-OMeTAD stabilized with an alkylthiol additive. Nat. Photonics 17, 96–105 (2023).
Jeong, M. et al. Stable perovskite solar cells with efficiency exceeding 24.8% and 0.3-V voltage loss. Science 369, 1615–1620 (2020).
Arora, N. et al. Perovskite solar cells with CuSCN hole extraction layers yield stabilized efficiencies greater than 20%. Science 358, 768–771 (2017).
Saliba, M. et al. A molecularly engineered hole-transporting material for efficient perovskite solar cells. Nat. Energy 1, 15017 (2016).
Jeong, M. et al. Large-area perovskite solar cells employing spiro-Naph hole transport material. Nat. Photonics 16, 119–125 (2022).
Jeon, N. J. et al. A fluorene-terminated hole-transporting material for highly efficient and stable perovskite solar cells. Nat. Energy 3, 682–689 (2018).
Kim, H. S. et al. Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9%. Sci. Rep. 2, 591 (2012).
Sakai, N. et al. Adduct-based p-doping of organic semiconductors. Nat. Mater. 20, 1248–1254 (2021).
Wang, S. et al. Unveiling the role of tBP–LiTFSI complexes in perovskite solar cells. J. Am. Chem. Soc. 140, 16720–16730 (2018).
Nakka, L., Cheng, Y., Aberle, A. G. & Lin, F. Analytical review of Spiro-OMeTAD hole transport materials: paths toward stable and efficient perovskite solar cells. Adv. Energ. Sust. Res. 3, 2200045 (2022).
Seo, J.-Y. et al. Dopant engineering for Spiro-OMeTAD hole-transporting materials towards efficient perovskite solar cells. Adv. Funct. Mater. 31, 2102124 (2021).
Chen, X. et al. Binary hole transport layer enables stable perovskite solar cells with PCE exceeding 24%. DeCarbon 1, 100004 (2023).
Guo, B. et al. Green solvent accelerates Spiro-OMeTAD oxidation for efficient perovskite solar cells. Solar RRL 8, 2300934 (2024).
Kong, J. et al. CO2 doping of organic interlayers for perovskite solar cells. Nature 594, 51–56 (2021).
Zhang, T. et al. Ion-modulated radical doping of spiro-OMeTAD for more efficient and stable perovskite solar cells. Science 377, 495–501 (2022).
Kim, S. G. et al. Photo-doping of spiro-OMeTAD for highly stable and efficient perovskite solar cells. Joule 8, 1707–1722 (2024).
Howie, W. H., Harris, J. E., Jennings, J. R. & Peter, L. M. Solid-state dye-sensitized solar cells based on spiro-MeOTAD. Sol. Energy Mater Sol. Cells 91, 424–426 (2007).
Rombach, F. M., Haque, S. A. & Macdonald, T. J. Lessons learned from spiro-OMeTAD and PTAA in perovskite solar cells. Energy Environ. Sci. 14, 5161–5190 (2021).
Liu, M. et al. Beyond hydrophobicity: how F4-TCNQ doping of the hole transport material improves stability of mesoporous triple-cation perovskite solar cells. J. Mater. Chem. A 10, 11721–11731 (2022).
Namatame, M. et al. Direct observation of dramatically enhanced hole formation in a perovskite-solar-cell material spiro-OMeTAD by Li-TFSI doping. Appl. Phys. Lett. 110, 123904 (2017).
Ouedraogo, N. A. N. et al. Oxidation of Spiro-OMeTAD in high-efficiency perovskite solar cells. ACS Appl. Mater. 14, 34303–34327 (2022).
Ahläng, C., Nyman, M. & Österbacka, R. Influence of the electric potential on charge extraction and interface recombination in perovskite solar cells. Phys. Rev. Appl. 16, 014041 (2021).
Zhao, Y. et al. Suppressing ion migration in metal halide perovskite via interstitial doping with a trace amount of multivalent cations. Nat. Mater. 21, 1396–1402 (2022).
Li, C. et al. Emerging alkali metal ion (Li+, Na+, K+ and Rb+) doped perovskite films for efficient solar cells: recent advances and prospects. J. Mater. Chem. A 7, 24150–24163 (2019).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kühne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 0007045 (2020).
Acknowledgements
All the authors thank Dr. Changle Mu and Dr. Taofei Zhou from Instrumentation and Service Center for Physical Sciences (ISCPS), Dr. Xiaohuo Shi, Dr. Danyu Gu, Dr. Yinjuan Chen, Dr. Xin Li, Dr. Min Zhou and Dr. Xingyu Lu from Instrumentation and Service Center for Molecular Sciences at Westlake University for the assistance in the characterizations. Funding: R.W. acknowledges the grant by National Natural Science Foundation of China (Grant No. 62474143). J.X. and R.W. acknowledge the grant (LD24E020001) by Natural Science Foundation of Zhejiang Province of China. J.X. acknowledges the grant by the Natural Science Foundation of Zhejiang Province of China (LR24F040001), National Natural Science Foundation of China (Grant No. 62274146). R.W. acknowledges the support of Key R&D Program of Zhejiang (2024SSYS0061), the Zhejiang Key Laboratory of Low-Carbon Intelligent Synthetic Biology (2024ZY01025) and Muyuan Laboratory (programme ID 14136022401).
Author information
Authors and Affiliations
Contributions
S.W. conceived the idea, did the fabrication and characterization of perovskite films and devices, did the data analysis, and wrote the manuscript under the supervision of R.W. and J.X. S.Z. contributed to the fabrication of devices based on single-doped, LiTFSI-based binary, ternary, and quaternary-doped spiro-OMeTAD and partial characterization (C-AFM and KPFM). X.S. performed the NMR measurements and data analysis. C.Y. contributed to the simulated calculations. D. G. performed the EPR measurements. D. J. conducted the XPS and UPS tests. Y.C. performed the TOF-SIMS measurements. J.Z.X. provided assistance on DLS measurements. R.W. and J.X. reviewed and edited the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kuan Sun, Jihuai Wu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Zhang, S., Shi, X. et al. Self-cleaning Spiro-OMeTAD via multimetal doping for perovskite photovoltaics. Nat Commun 16, 4167 (2025). https://doi.org/10.1038/s41467-025-59350-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59350-9
