
Abstract
Highly regio-, diastereo- and enantioselective iron-catalyzed sequential hydrosilylation of o-alk-n-enyl-phenyl silanes with alkynes is reported for various 5-, 6-, and 7-membered benzosilacycles in 60-94% yields with up to 95:5 rr, 95:5 dr, and 99% ee. Chiral fully carbon-substituted silicon-stereogenic benzosilacycles could also be obtained via triple hydrosilylation reactions. The unique electronic effect of ligands is observed while adjusting the regioselectivity and enantioselectivity in hydrosilylation reactions. A possible mechanism has been proposed by variable time normalization analysis (VTNA) and H/D exchange experiment.
Similar content being viewed by others
Introduction
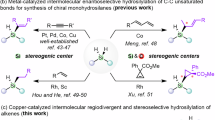
Silasubstitution strategy was regarded as an innovative strategy to develop new materials and pesticides, and has found interesting applications in many fields (Fig. 1A)1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16. A series of 5-, 6-, and 7-membered benzosilacycles had been constructed using transition metal catalysis, such as Rh17,18,19,20,21,22,23,24,25,26,27,28,29, Pd30,31,32,33,34,35,36,37, Ru38, Ir39, Sc40, and Ni41 etc. (Fig. 1B). It should be noted that neither the catalytic cyclization for the synthesis of benzosilacycles containing two Si-H bonds which could be further derivatized, nor the construction of vicinal chiral carbon and silicon-centers in benzosilacycles has been reported. Although intramolecular hydrosilylation of alkenes containing mono-substituted silanes could be proposed to deliver the benzosilacycles containing two Si–H bonds which could undergo further sequential reactions42, there were several challenges: (1) The side reactions, such as hydrogenation, intermolecular hydrosilylation, and over functionalization of Si–H bonds, could significantly affect the production of benzosilacycles containing two Si–H bonds. (2) Compared to the C–C bond, the transition state was more flexible due to the longer C–Si bond which prompted more hardship in differentiating two enantiotopic faces. (3) For the sequential reactions, the difficulty to achieve the compatibility and control the sequence was increased in the presence of the multiple active Si–H bond and similar unsaturated C–C bonds in one pot. Additionally, although iron was the most earth-abundant transition metal43, neither the iron-catalyzed hydrosilylation for the construction of benzosilacycles nor sequential hydrofunctionalization have been reported until now. Here, we reported an iron-catalyzed sequential hydrosilylation of n-alkenyl-phenyl silanes with alkynes to afford 5-, 6-, and 7- membered various benzosilacycles in good yields with excellent diastereo- and enantioselectivitiy, and sequential reactions in one pot for the construction of fully carbon-substituted silicon center, as shown in Fig. 1C. In this process, while changing the electronic effects on ligands, the regioselectivity and enantioselectivity of hydrosilylation reactions could be controlled.

A Silasubstitution strategy, B Metal-catalyzed construction of benzosilacycles, C This work: Construction of 5-, 6-, 7-membered benzosilacycles by iron catalysts. The green colour is about the construction of 5-membered benzosilacycles, blue colour is aboult the construction of 6-membered benzosilacycles and purple colour is about the construction of 7-membered benzosilacycles. The pink colour is about “Si”.
Results: the iron-catalyzed sequential hydrosilylation to construct benzosilacycles
Results 1: the construction of 5- and 6-membered various benzosilacycles benzosilacycles
At the beginning of the screening, simple (2-vinylphenyl)silane (1a) and 1,2-diphenylethyne (2a) were chosen as model substrates. Lithium tert-butoxide and toluene were used as a mild activator and solvent, respectively. The reaction using bidentate bipyridine iron dichloride complex did not provide hydrosilylation products (entry 1, Table 1). The PDI ligand promoted the intramolecular hydrosilylation to afford 3a′ in 35% yield without sequential hydrosilylation product 3a (entry 2). Both Pybox and OIP ligands could promote the sequential reaction to deliver 3a in 44% and 54% yield, respectively. Increasing the steric hindrance on oxazoline and imine could improve the reactivity (entries 5 and 6). The reaction using 1.2 equiv of 1a afforded 3a in 96% yield (entry 7). Finally, the standard conditions were identified as using silane 1a (1.2 equiv), 1,2-diphenylethyne 2a (0.5 mmol), Fe-3 (5 mol%), and LiOtBu (15 mol%) in a solution of toluene (0.5 M) stirring at room temperature for 24 h. It should be noted that the catalyst Fe-3 could distinguish the intermolecular hydrosilylation and intramolecular hydrosilylation, and also raise the activity of the reactions. More details are shown in supplementary information (Table S2)
Under the optimized conditions, the substrate scope was explored in Fig. 2A. Reactions of various diaryl alkynes containing ethers, halogens, heterocycles, diheteroaryl and dialkyl alkynes efficiently proceeded to afford desired anti-Markovnikov products 3a–3o in 68-93% yields. The 5-methyl and 4-chloro group on aryl silane could be tolerated to access 3q and 3r in 85 and 81% yield, respectively. Time course is shown in supplementary information (SI Figs. 1 and 2)

aStandard conditions: 1a (0.60 mmol), 2a (0.50 mmol), cat. Fe-3 (0.025 mol), LiOtBu (0.075 mmol), toluene (1 mL). bStandard conditions: 4a (0.3 mmol), 2a (0.3 mmol), cat. Fe-4 (0.015 mmol), LiOtBu (0.045 mmol), toluene (0.6 mL). cStandard conditions: 4a (0.30 mmol), 2a (0.30 mmol), cat. Fe-5 (0.015 mmol), LiOtBu (0.045 mmol), toluene (0.6 mL). dIsolated yield in the parentheses. eYields were determined by 1H NMR using TMSPh as an internal standard. A The scope of 2-vinylphenyl silanes; B The scope of 2-allylphenyl silanes.
As shown in supporting information Table S4, the reaction using Fe-7 and LiOtBu in a solution of toluene at room temperature for 24 h afforded the mixed product in 78% yield with 50:50 rr. While using Fe-16 with minor hindrance of oxazoline, only 22% yield and 50:50 rr are provided. While adjusting the steric hindrance of imine could realize the control of Markovnikov selectivity to deliver the 5-membered benzosilacycle 5a in 91% yield with >95:5 rr and 92:8 dr. Then the use of Fe-17 which was modified with the electronic donating group could increase the anti-Markovnikov regioselectivity to 15:85. The use of more sterically hindered on oxazoline Fe-5 could improve the regioselectivity to afford 6-membered benzosilacycle 6a in 75% yield with 95:5 rr (Fig. 2B).
Results 2: The construction of 6- and 7-membered various benzosilacycles benzosilacycles
When (2-(but-3-en-1-yl)phenyl)silane (7a) was used as a substrate, the asymmetric iron-catalyzed intramolecular hydrosilylation could occur smoothly to deliver 6-membered dihydro-benzosilacycle 8a. As shown in Table 2, the use of Fe-8 afford the product with 79% ee. While electronic donating group was added to the ligand, 90% ee was provided. Electronic effects on the ligand might have a positive impact on enantioselectivity of intramolecular hydrosilylation reactions. The reactions of substrates with 4-methory, 3-fluoro, and 4-TBSO- groups were pushed on smoothly to afford products 8b–8d in 87–90% yields with 93:7 to >95:5 rr, and 90–97% ee (Fig. 3A). The absolute configuration was consistent with that previously reported by our group43.

aStandard conditions: 7a (0.50 mmol), cat. Fe-5 (0.010 mmol), NaBHEt3 (0.030 mmol). bStandard conditions: 7a (0.30 mmol), cat. Fe-5 (0.006 mmol), NaBHEt3 (0.018 mmol), cat. Fe-3 (0.015 mmol), 2a (0.30 mmol), LiOtBu (0.045 mmol), toluene (0.6 mL). cStandard conditions: 11a (0.30 mmol), L (0.006 mmol), NaBHEt3 (0.018 mmol), THF (0.6 mL). dStandard conditions: 7a (0.36 mmol). eYields were determined by 1H NMR using TMSPh as an internal standard. fIsolated yield in the parentheses. A The scope of benzosilacycles with two Si–H bonds; B The scope of benzosilacycles with vicinal chiral carbon and silicon-centers; C The scope of 7-membered benzosilacycles.
After the first intramolecular hydrosilylation completed, the complex Fe-3 (5 mol%), LiOtBu (15 mol%), alkynes (1 eq.) and toluene (0.5 M) were added in the system. The sequential reactions could be carried out to provide the 6-membered monohydro-benzosilacycle 10a with vicinal chiral carbon and silicon centers in 65% yield with >95:5 rr, >95:5 dr, and 93% ee (Fig. 3B)44,45. The sequential reactions of substrates with 4-methory, 3-fluoro, and 4-TBSO- groups were carried out smoothly to afford 10b–e in 60–70% yields with excellent regio-, diastereoselectivity (>95:5 rr, >95:5 dr), and 86-93% ee. The substrates with heterocycle and alkyl alkyne could also be pushed on efficiently. For the unsymmetrical alkyne but-1-yn-1-ylbenzene, sequential reaction afforded the α-syn-selective product 10j in 75% yield. The complex molecular empagliflozin ntermediate was also easily transformed into the corresponding chiral silanes 10k with uncompromised 80% yield with 93% ee. The absolute configuration of the resulting carbon and silicon-stereogenic center in 10l was unambiguously determined by an X-ray diffraction. Excitedly, 7-membered carbon-stereogenic benzosilacycle 12a could also be constructed in 73% yield with 99% ee while using (2-(pent-4-en-1-yl)phenyl)silane (11a) in the presence of iron catalyst (Fig. 3C).
The reaction of 1a with 2a could be carried out in a gram scale to afford 3a with 1.28 g in 82% yield (Fig. 4A, eq.1). The reaction of 7a could be performed on gram scale, providing 6-membered benzosilacycles 10a with 1.06 g in 62% yield (Fig. 4A, eq.2). Treatment of 3a with 1-ethynyl-4-methoxybenzene in the presence of a dicobalt carbonyl N-heterocyclic carbene [(IPr) Co2(CO)7] (IPr = 1,3-di(2,6-diisopropylphenyl)imidazol-2-ylidene) complex provided 5-membered heterocycles derivatives 14a in 74% yield (Fig. 4Beq.3)46,47,48,49. Terminal alkene, terminal alkyne and α, β unsaturated ketone could be used as partners to convert to fully carbon-substituted silicon-stereogenic heterocycles derivatives (15a, 16a and 17a) in the presence of Karstedt’s catalyst (Fig. 4B, eq.4–6)50,51,52. It is interesting that three stereocenters benzosilacycles 17a could be obtained, which was quite difficult to be afforded by using other methods.

aeq.1 Gram scale reaction of 1a, beq.2 Gram scale reaction of 7a, ceq.3 further derivatization of 3a, deq.4-6 further derivatization of 10a. A. Gram scale reaction of 1a and 7a. B further derivatization of 3a and 10a.
Deuterium-labeling experiments were conducted to explore the mechanism in those asymmetric hydrosilylation processes as shown in Fig. 5. The silicon-deuterated product 19a-d3 or 21a-d3 was afforded by the asymmetric sequence hydrosilylation of unsaturated C-C bonds with silane-d3 (18a or 20a) catalyzed by iron catalyst. The result suggested that insertion was regioselective and irreversible (eq.7 and eq.8)53. The fact that the product 21a′ with deuterium incorporation into the α-carbon was not generated indicated the impossibility of a Fe-H or Fe-D intermediate in the catalytic cycle, because facile H/D exchange between Cα–H and Fe–D would otherwise occur through reversible olefin insertion into the Fe-D bond54,55. When intramolecular hydrosilylation with a 1:1 mixture of silane-d3 (20a) and silane 7b was carried out and followed by the intermolecular hydrosilylation, silicon-deuterated products 21a and 21b were obtained (in Supporting Information). The construction of silicon-deuterated product 21b and the next deuterium-labeling experiment in eq.9, giving 24a-d2 and 25a-d2, indicated that Si-Fe species might participate in the catalytic cycle.

eq.7-8 Deuterium-labeling experiments.
Mechanistic interrogation was realized through kinetic studies using variable time normalization analysis (VTNA)56,57,58,59,60,61,62. VTNA was used to determined the reagent orders of the reaction components, from which we hypothesized the turnover-limiting step. This method was used to determined the kinetic order of the reaction components in the second step. Two different slopes were observed in Fig. 6a, where the kinetic profile of the reaction with two different iron catalyst concentrations was shown. When a first-order factor of catalyst concentration (t [Fe]1) was used (Fig. 6c), two reaction progress curves failed to overlap. While using a 0.5-order correlation (t [Fe]0.5), two reaction progress curves overlapped (Fig. 6b), indicating that the kinetic order for iron is a 0.5-order rather than the first order. It was speculated that the active intermediates of iron catalyst might exist in the form of dimers63. It also meant that the rate of reaction was relatively insensitive to changes in the concentration of the reactants, but not completely unaffected. Next, a superior reaction was observed when increasing substrate 8a loading (Fig. 6d–f), corresponding to a first order. Same with substrate 8a, the result that a first order for substrate 2a was obtained from VTNA (Fig. 6g–i). The observation of [8a]1 and [2a]1 suggested that one molecular 8a and one molecular 2a were involved in the turnover limiting step. More information were shown in (SI Figs. 3–5)

a–c VTNA analysis of catalyst, d-f VTNA analysis of silane, g–i VTNA analysis of alkyne.
Based on the above mechanisms, the proposed mechanism was shown in Fig. 7. The Fe–Si species (A) was proposed through reducing Fe-5 by NaBHEt3 and silane64,65,66,67. Then alkene inserted in Fe–Si bond to generate iron alkyl species (B), which wonder go δ-bond metathesis with silane to regenerate species (A) and afford the hydrosilylation product (8a). The Fe–Si (C) obtained from (8a). Because the kinetic order for iron is a 0.5-order rather than the first order, the active intermediates Fe–Si (C) might exist in the form of dimers, which was proposed and this proposal would be tested in our future work. Then alkyne inserted into Fe–Si bond to generate iron alkenyl species (D), which reacted with silane to regenerate Fe–Si species (C) and afforded the final product (10a). More studies were continuously under-going in our laboratory to gain an accurate understanding of the mechanism. The mechanism of Fe(0) species cannot be completely ruled out.

The proposed mechanism starts from intramolecular hydrosilylation, followed by intermolecular hydrosilylation.
The primary models to predict the regiochemical outcome about the construction of 5- and 6-membered benzosilacycle were shown in Fig. 8. The primary models PM(A) and (B) provided similar possibility of Markovnikov and anti-Markovnikov hydrosilylation products 5a and 6a. While increasing the steric hindrance of imine, the primary models PM(C) was disfavored. While electronic donating groups added to ligand, electronegativity on Fe turned over. Then the primary models PM(E) become the dominant conformation and anti-Markovnikov product was provided. In the primary models, electronic donating groups on the ligand may exist in a resonant form, resulting in the electronegativity of Fe turning over. This resonant form might alter the size of ligand cavity, thus making the transition state more compact and improving the enantioselectivity of the reaction Fig. 8. More information were shown in (SI Figs. 6 and 7).

A–H The primary models to predict the regiochemical outcome to explain the selectivity in the construction of 5- and 6-membered benzosilacycles.
Discussion
In this work, we developed a mild iron-catalyzed system for the construction of 5-, 6-, and 7-membered benzosilacycles with highly regio-, diastereo- and seteroselectivity. Eight kinds of benzosilacycles including silacycles with vicinal chiral carbon and silicon-centers were successfully synthesized. The regioselectivity of hydrosilylation reactions could be controlled by iron catalyst step by step, and the kinetic order of the reaction components in the second step was determined by using variable time normalization analysis (VTNA). The development of more asymmetric iron-catalyzed reactions is ongoing in our laboratory.
Methods
A 25 mL Schlenk flask equipped with a magnetic stirrer and a flanging rubber plug is dried with flame. When cooled to ambient temperature, it is transferred into glove box. Then Fe-3 complex (0.025 mmol, 5 mol%), LiOtBu (0.075 mmol, 15 mol%), 1a (0.6 mmol, 1.2 equiv), alkynes (0.5 mmol), toluene (1 mL, 0.5 M) are added sequentially. The mixture is stirred at room temperature for 24 h. Then the resulting solution is transferred into room and quenched with 10 mL of PE and filtered through a pad of silica gel, washed with PE/AcOEt (10/1) (or other suitable solvent) (5 × 10 mL). The combined filtrate is concentrated and the yield is monitored by 1H NMR analysis using TMSPh as an internal standard. The resulting solution is purified by flash column chromatography to give the corresponding product.
Data availability
The authors declare that the data Supplementary the findings of this study are available within the paper and its Supplementary Information file. The X-ray crystallographic coordinates for the structure of compound 10I reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2383336. These data can be obtained free of charge from https://www.ccdc.cam.ac.uk. The Cambridge Crystallographic Data Centre via The experimental procedures and characterization of all new compounds are provided in the Supplementary Information. Data supporting the findings of this manuscript are also available from the authors upon request.
References
Wang, D. & Chan, T.-H. Chiral organosilicon compounds in asymmetric synthesis. Chem. Rev. 92, 995–1006 (1992).
Oestreich, M. Silicon-stereogenic silanes in asymmetric catalysis. Synlett 11, 1629–1643 (2017).
Li, L., Lai, G.-Q., Jiang, J.-X. & Xu, L.-W. The recent synthesis and application of silicon-stereogenic silanes: A renewed and significant challenge in asymmetric synthesis. Chem. Soc. Rev. 40, 1777–1790 (2011).
Zhao, W.-T., Gao, F. & Zhao, D.-B. Recent progress in σ-bond cross-exchange reactions to access diverse silacycles. Synlett 29, 2595–2600 (2018).
Ye, F. & Xu, L.-W. A glimpse and perspective of current organosilicon chemistry from the view of hydrosilylation and synthesis of silicon-stereogenic silanes. Synlett 32, 1281–1288 (2021).
Wu, Y.-C. & Wang, P. Silicon-stereogenic monohydrosilane: synthesis and applications. Angew. Chem. Int. Ed. 61, e202205382 (2022).
Yuan, W. & He, C. Enantioselective C–H functionalization toward silicon-stereogenic silanes. Synthesis 54, 1939–1950 (2022).
Ge, Y.-C., Huang, J. K. & He, C. Transition-metal-catalyzed enantioselective C−H silylation. Chem. Catal. 2, 2898–2928 (2022).
Huang, W.-S., Wang, Q., Yang, H. & Xu, L.-W. State-of-the-art advances in enantioselective transition-metal-mediated reactions of silacyclobutanes. Synthesis 54, 5400–5408 (2022).
Gao, J.-H. & He, C. Chem. Chiral silanols: strategies and tactics for their synthesis. Eur. J. 29, e202203475 (2023).
Showell, G. A. & Mills, J. S. Chemistry challenges in lead optimization: silicon isosteres in drug discovery. Drug Discov. Today 8, 551–556 (2003).
Gately, S. & West, R. Novel therapeutics with enhanced biological activity generated by the strategic introduction of silicon isosteres into known drug scaffolds. Drug Dev. Res. 68, 156–163 (2007).
Franz, A. K. & Wilson, S. O. Organosilicon molecules with medicinal applications. J. Med. Chem. 56, 388–405 (2013).
Rémond, E., Martin, C., Martinez, J. & Cavelier, F. Silicon-containing amino acids: synthetic aspects, conformational studies, and applications to bioactive peptides. Chem. Rev. 116, 11654–11684 (2016).
Fujii, S. & Hashimoto, Y. Progress in the medicinal chemistry of silicon: C/Si exchange and beyond. Future Med. Chem. 9, 485–505 (2017).
Ramesh, R. & Reddy, D. S. Quest for novel chemical entities through Incorporation of silicon in drug scaffolds. J. Med. Chem. 61, 3779–3798 (2018).
Onoe, M. et al. Rhodium-catalyzed carbon–silicon bond activation for synthesis of benzosilole derivatives. J. Am. Chem. Soc. 134, 19477–19488 (2012).
Kuninobu, Y., Nakahara, T., Takeshima, H. & Takai, K. Rhodium-catalyzed Intramolecular silylation of unactivated C(sp3)–H Bonds. Org. Lett. 15, 426–428 (2013).
Zhao, W.-T., Lu, Z.-Q., Zheng, H., Xue, X.-S. & Zhao, D.-B. Rhodium-catalyzed 2-arylphenol-derived six-membered silacyclization: straightforward access toward dibenzooxasilines and silicon-containing planar chiral metallocenes. ACS Catal. 8, 7997 (2018).
Chang, X., Ma, P. L., Chen, H. C., Li, C. Y. & Wang, P. Asymmetric synthesis and application of chiral spirosilabiindanes. Angew. Chem. Int. Ed. 59, 8937–8940 (2020).
Mu, D. et al. Streamlined construction of silicon-stereogenic silanes by tandem enantioselective C–H silylation/alkene hydrosilylation. J. Am. Chem. Soc. 142, 13459–13468 (2020).
Tang, R.-H. et al. Catalytic asymmetric trans-selective hydrosilylation of bisalkynes to access AIE and CPL-active silicon-stereogenic benzosiloles. iScience 23, 101268 (2020).
Yang, B., Yang, W., Guo, Y.-H., You, L.-J. & He, C. Enantioselective silylation of aliphatic C−H bonds for the synthesis of silicon-stereogenic dihydrobenzosiloles. Angew. Chem. Int. Ed. 59, 22217–22222 (2020).
Chen, S. Y. et al. Enantioselective construction of six- and seven-membered triorgano-substituted silicon-stereogenic heterocycles. Nat. Commun. 12, 1249–1258 (2021).
Guo, Y. H., Liu, M. M., Zhu, X. J., Zhu, L. R. & He, C. Catalytic asymmetric synthesis of silicon-stereogenic dihydrodibenzosilines: silicon central-to-axial chirality relay. Angew. Chem. Int. Ed. 60, 13887–13891 (2021).
Yuan, W. et al. Asymmetric synthesis of silicon-stereogenic monohydrosilanes by dehydrogenative C–H silylation. Org. Lett. 23, 1367–1372 (2021).
Huang, Y. H. et al. Enantioselective synthesis of silicon-stereogenic monohydrosilanes by rhodium-catalyzed intramolecular hydrosilylation. Angew. Chem. Int. Ed. 61, e202113052 (2022).
Gou, F.-H., R, F., W, Y. C. & Wang, P. Catalytic kinetic resolution of monohydrosilanes via rhodium catalyzed enantioselective intramolecular hydrosilylation. Angew. Chem. Int. Ed. 63, e202404732 (2024).
Zeng, Y. et al. Rhodium-catalyzed dynamic kinetic asymmetric hydrosilylation to access silicon-stereogenic center. Angew. Chem. Int. Ed. 61, e202214147 (2022).
Meng, T. H., Ouyang, K. B. & Xi, Z. F. Palladium-catalyzed cleavage of the Me–Si bond in ortho-trimethylsilyl aryltriflates: synthesis of benzosilole derivatives from ortho-trimethylsilyl aryltriflates and alkynes. RSC Adv. 3, 14273 (2013).
Zhao, W. T., Gao, F. & Zhao, D. B. Intermolecular σ-bond cross-exchange reaction between cyclopropenones and (benzo)silacyclobutanes: straightforward access towards sila(benzo)cycloheptenones. Angew. Chem. Int. Ed. 57, 6329 (2018).
Qin, Y., Han, J. L., Ju, W. C. & Zhao, D. B. Ring Expansion to 6-, 7-, and 8-membered benzosilacycles through strain-release silicon based cross-coupling. Angew. Chem. Int. Ed. 59, 8481–8485 (2020).
Qin, Y., Li, L. H., Liang, J.-Y., Li, K. L. & Zhao, D. B. Silacyclization through palladium-catalyzed intermolecular silicon-based C(sp2)–C(sp3) cross coupling. Chem. Sci. 12, 14224–14229 (2021).
Wang, X. C., Wang, H. R., Xu, X. F. & Zhao, D. B. Ring expansion to 8-membered silacycles through formal cross-dimerization of 5-membered palladacycles with silacyclobutanes. Eur. J. Org. Chem. 2021, 3039–3042.
Shi, Y. F. et al. Sila-spirocyclization involving unstrained C(sp3)−Si bond cleavage. Nat. Commun. 13, 6697 (2022).
Yin, K.-L. et al. Enantioselective construction of sila-bicyclo[3.2.1] scaffolds bearing both carbon- and silicon-stereocenters. ACS Catal. 12, 13999–14005 (2022).
Zhang, J. Y. et al. Reversing site-selectivity in formal cross-dimerization of benzocyclobutenones and silacyclobutanes. CCS Chem. 5, 1753–1762 (2023).
Fang, H. Q., Hou, W. J., Liu, G. X. & Huang, Z. Ruthenium-catalyzed site-selective intramolecular silylation of primary C–H bonds for synthesis of sila-heterocycles. J. Am. Chem. Soc. 139, 11601–11609 (2017).
Su, B. & Hartwig, J. F. Ir-catalyzed enantioselective, intramolecular silylation of methyl C–H bonds. J. Am. Chem. Soc. 139, 12137–12140 (2017).
Zhan, G. et al. Enantioselective construction of silicon-stereogenic silanes by scandium-catalyzed intermolecular alkene hydrosilylation. Angew. Chem. Int. Ed. 57, 12342–12346 (2018).
Zhang, J. Y., Yan, N., Ju, C. W. & Zhao, D. B. Nickel(0)-catalyzed asymmetric ring expansion toward enantioenriched silicon-stereogenic benzosiloles. Angew. Chem. Int. Ed. 60, 25723–25728 (2021).
Lu, W. X., Zhao, Y. M. & Meng, F. K. Cobalt-catalyzed sequential site- and stereoselective hydrosilylation of 1,3- and 1,4-enynes. J. Am. Chem. Soc. 144, 5233–5240 (2022).
Cheng, B., Liu, W. B. & Lu, Z. Iron-catalyzed highly enantioselective hydrosilylation of unactivated terminal alkenes. J. Am. Chem. Soc. 140, 5014–5017 (2018).
Lgawa, K., Yoshihiro, D., Ichikawa, N., Kokan, K. & Tmooka, K. Catalytic enantioselective synthesis of alkenylhydrosilanes. Angew. Chem. Int. Ed. 51, 12745 (2012).
Wen, H. N., Wan, X. L. & Huang, Z. Asymmetric synthesis of silicon-stereogenic vinylhydrosilanes by cobalt-catalyzed regio- and enantioselective alkyne hydrosilylation with dihydrosilanes. Angew. Chem. Int. Ed. 57, 6319 (2018).
Wang, D. Y. et al. Markovnikov hydrosilylation of alkynes with tertiary silanes catalyzed by dinuclear cobalt carbonyl complexes with NHC ligation. J. Am. Chem. Soc. 143, 12847 (2021).
Gao, Y. F., Wang, L. J. & Deng, L. Distinct catalytic performance of cobalt(I)–N heterocyclic carbene complexes in promoting the reaction of alkene with diphenylsilane: selective 2,1-hydrosilylation, 1,2-hydrosilylation, and hydrogenation of alkene. ACS Catal. 8, 9637–9646 (2018).
Liu, Y. & Deng, L. Mode of activation of cobalt(II) amides for catalytic hydrosilylation of alkenes with tertiary silanes. J. Am. Chem. Soc. 139, 1798–1801 (2017).
Sun, J. & Deng, L. Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 6, 290–300 (2016).
Downing, C. M. & Kung, H. H. Diethyl sulfide stabilization of platinum-complex catalysts for hydrosilylation of olefins. Catal. Commun. 12, 1166–1169 (2011).
Sommer, L. H., Lyons, J. E. & Fujimoto, H. Stereochemistry of asymmetric silicon. XV. stereospecific hydrosilation and exchange reactions of R3Si*H(D) Catalyzed by Group VIII Metal Centers. J. Am. Chem. Soc. 91, 7051 (1969).
Book, A. G., Pannell, K. H. & Anderson, D. G. α-Silyl-cis-stilbenes from silylcarbonium ions and from the platinum-catalyzed addition of silanes to diphenylacetylene. J. Am. Chem. Soc. 90, 4374 (1968).
Lu, P., Wang, H. L., Mao, Y. H., Hong, X. & Lu, Z. Cobalt-catalyzed enantioconvergent hydrogenation of minimally functionalized isomeric olefins. J. Am. Chem. Soc. 144, 17359–17364 (2022).
Du, X. Y., Zhang, Y. L., Peng, D. J. & Huang, Z. Base–metal-catalyzed regiodivergent alkene hydrosilylations. Angew. Chem. Int. Ed. 55, 6671–6675 (2016).
Hu, M. Y. et al. Iron-catalyzed regiodivergent alkyne hydrosilylation. J. Am. Chem. Soc. 142, 16894–16902 (2020).
Choudhary, S., Cannas, D. M., Wheatley, M. & Larrosa, I. A manganese(I) tricarbonyl-catalyst for near room temperature alkene and alkyne hydroarylation. Chem. Sci. 13, 13225–13230 (2022).
Burés, J. A simple graphical method to determine the order in catalyst. Angew. Chem. Int. Ed. 55, 2028–2031 (2016).
Burés, J. Variable time normalization analysis: general graphical elucidation of reaction orders from concentration profiles. Angew. Chem. Int. Ed. 55, 16084–16087 (2016).
Blackmond, D. G. Kinetic profiling of catalytic organic reactions as a mechanistic tool. J. Am. Chem. Soc. 137, 10852–10866 (2015).
Baxter, R. D., Sale, D., Engle, K. M., Yu, J. Q. & Blackmond, D. G. Mechanistic rationalization of unusual kinetics in Pd-catalyzed C−H olefination. J. Am. Chem. Soc. 134, 4600–4606 (2012).
Lin, Z. P., Oliveira, J. C. A., Scheremetjew, A. & Ackermann, L. Palladium-catalyzed electrooxidative double C−H arylation. J. Am. Chem. Soc. 146, 228–239 (2024).
Pan, Z. H., Liu, M. L., Zheng, C. Y., Gao, D. Q. & Huang, W. Study of Karstedt’s catalyst for hydrosilylation of a wide variety of functionalized alkenes with triethoxysilane and trimethoxysilane. Chin. J. Chem. 35, 1227–1230 (2017).
Doll, J. S., Heldner, M. L., Scherr, M., Ballmann, J. & Rosca, D.-A. The atmosphere matters: the effect of “Inert” gas on the catalytic outcomes in cobalt-mediated alkyne and olefin hydroboration. ACS Catal. 13, 8770–8782 (2023).
Guo, J., Cheng, Z. Z., Chen, J. H., Chen, X. & Lu, Z. Iron- and cobalt-catalyzed asymmetric hydrofunctionalization of alkenes and alkynes. Acc. Chem. Res. 54, 2701–2716 (2021).
Chen, C. H. et al. Iron-catalyzed asymmetric hydrosilylation of vinylcyclopropanes via stereospecific C-C Bond cleavage. iScience 23, 100985 (2020).
Cheng, B., Lu, P., Zhang, H. Y., Cheng, X. P. & Lu, Z. Highly enantioselective cobalt-catalyzed hydrosilylation of alkenes. J. Am. Chem. Soc. 139, 9439–9442 (2017).
Chen, J. H., Cheng, B., Cao, M. Y. & Lu, Z. Iron-catalyzed asymmetric hydrosilylation of 1,1-disubstituted alkenes. Angew. Chem. Int. Ed. 54, 4661–4664 (2015).
Acknowledgements
The authors thank National Key Research and Development Program of China (2021YFA1500200), National Natural Science Foundation of China (NSFC) (22271249), Zhejiang Provincial Natural Science Foundation of China (LDQ24B020001), the Fundamental Research Funds for the Central Universities (226-2022-00224 and 226-2024-00003), the Center of Chemistry for Frontier Technologies, and Xinjiang Key Laboratory of Organosilicon Functional Molecules and Materials. We thank Tongtong Li for repeating the construction of 3a, Zhanchen Ye for repeating the construction of 10a, Lixuan Zheng for the synthesis of 2s, Zikai Qian for the synthesis of 2r. We also thank Jiyong Liu for X-ray crystallographic analysis; Luling Wu, Yaqin Liu for NMR analysis; Qiaohong He for ESI-HRMS analysis from Analysis and Test Platform of Department of Chemistry, Zhejiang University.
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.L.; Methodology, X.W, J.J.Z., D.Y.W. L.D.; Investigation, X.W., and J.J.Z.; Writing-original draft, Z.L. and X.W.; Writing-review & editing, Z.L., and X.W.; Funding Acquisition, Z.L. and L.D.; Supervision, Z.L. and L.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Liangbin Huang, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Zhao, J., Wang, D. et al. Iron-catalyzed sequential hydrosilylation. Nat Commun 16, 4338 (2025). https://doi.org/10.1038/s41467-025-59364-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59364-3
