
Abstract
The essential role of RIPK3 in necroptosis makes its inhibition a promising therapeutic strategy. However, the development of RIPK3 inhibitors has been hampered by on-target apoptosis and limited kinase selectivity. Inspired by the R69H mutation, which prevents on-target apoptosis by disrupting RIPK3 dimerization, we design LK-series inhibitors that effectively inhibit RIPK3 in biochemical assays and block TNF-α-induced necroptosis in both mouse L929 and human HT29 cells without inducing apoptosis. The representative compound, LK01003, shows high selectivity across a panel of 379 kinases. Our structural studies reveal that LK compounds act as Type I1/2 inhibitors, engaging a unique hydrophobic site and stabilizing an inactive conformation of RIPK3. Moreover, several type II inhibitors are also revealed to maintain RIPK3 in the inactive conformation and do not induce on-target apoptosis. These findings suggest a promising strategy for rational design of safe and selective inhibitors by locking the inactive conformation of RIPK3.
Similar content being viewed by others

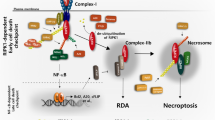

Introduction
Necroptosis is a crucial form of programmed cell death involved in various diseases, including neurodegenerative disorders1, ischemia-reperfusion injuries2, inflammatory diseases3,4,5, and cancer6. Necroptosis is typically triggered when apoptosis is inhibited7, and begins with TNF-α and other ligands binding to receptors to activate receptor-interacting serine/threonine-protein kinase 1 (RIPK1). RIPK1 then forms a complex with RIPK3, termed the necrosome, where RIPK3 phosphorylates mixed lineage kinase domain-like protein (MLKL), permeabilizing the plasma membrane and ultimately leading to rupture of the cell8,9,10,11,12,13,14. Moreover, RIPK3 can drive this pathway independently of RIPK1. For example, Z-DNA-binding protein 1 (ZBP1) activates RIPK3 when infected with the influenza A virus, which in turn activates MLKL, causing cell death13,14. Toll-like receptor 3 (TLR3) and TLR4 engagement by viral RNA or bacterial LPS recruits the TLR adaptor molecule, Toll/IL-1R domain-containing adaptor inducing interferon-β (TRIF), which also interacts with RIPK3 to induce necroptosis in sepsis15. Accordingly, the indispensable role of RIPK3 in the necroptosis pathway demonstrates its essential importance for managing this form of cell death, leading to the involvement of RIPK3 in pathogenesis of a diverse range of diseases16, such as Alzheimer’s and Parkinson’s disease17,18,19, myocardial infarction20, stroke21, inflammatory bowel disease3,22, rheumatoid arthritis23,24, and multiple sclerosis25. Together, these findings highlight the potential of RIPK3 as a crucial therapeutic target for indications associated with necroptosis-mediated cell death and inflammation26.
Nevertheless, numerous efforts to develop drug-like RIPK3 inhibitors have proven challenging. More than ten years ago, a high-throughput screening followed by structural optimization carried out by GSK researchers led to the discovery of GSK'87227, a pioneer RIPK3 inhibitor with nanomolar potency that has since been extensively investigated for its efficacy in multiple necroptosis-related disease models3,28,29. Unexpectedly, it has been observed that GSK'872, as well as other RIPK3 inhibitors, such as GSK'843 and GSK'840 with chemical scaffolds different from GSK'872, can trigger RIPK3-mediated apoptosis at concentrations approximately twice their EC50 values30. This kind of apoptosis occurs mainly through the recruitment of RIPK1 and the assembly of a death-inducing signaling complex, including FADD and caspase 8, resulting in caspase 8 activation and apoptotic cell death30. Such an on-target toxicity has a significant impact on the safety and viability of RIPK3 inhibitors for clinical use, preventing them from further development. Recently, TAK-63231 and its derivatives32 have been identified as RIPK3 inhibitors without causing distinct apoptotic effects, but their selectivity towards RIPK3 over other kinases remains insufficiently high for further development. It is worth noting that the mechanism by which the inhibitors induce or do not induce cell apoptosis has not yet been elucidated. Accordingly, general principles are not currently available for the design and development of new RIPK3 inhibitors that consistently evade apoptotic effects.
By contrast, the relationship between RIPK3 mutants and apoptosis provides alternative insights into on-target toxicity caused by apoptosis, i.e., on-target apoptosis. It has been reported that mutations in certain residues, such as D161N at the ATP-binding site, result in ATPase-inactive RIPK3 but paradoxically induce severe apoptosis, similar to the effects observed with RIPK3 inhibitors like GSK'87230,33. Such a mutation makes RIPK3 adopt conformations that facilitate the recruitment of RIPK1 and subsequent activation of caspase 8, ultimately resulting in the induction of apoptosis. Other kinase-inactive residue mutations at the ATP-binding site, like K51A, D161G, and D143N, do not trigger apoptosis unless RIPK3 inhibitors are present30. Notably, the R69H mutation, situated near the ATP-binding site, does not affect the function of RIPK3 but uniquely prevents on-target apoptosis triggered by RIPK3 inhibitors, which is presumably attributed to the disruption of RIPK3 kinase domain (RIPK3KD) homodimerization by the mutation34. These findings suggest that on-target apoptosis might be associated with the conformations of the inhibitor binding pocket, of which specific conformations can trigger apoptosis while others do not.
Kinase inhibitors are typically categorized into types I-IV based on their distinct binding modes with the kinase35. Among these, Type I, Type I1/2, and Type II inhibitors primarily target the ATP-binding pocket but differ in their interactions with two key regulatory motifs: the DFG motif (Asp-Phe-Gly) in the activation loop and the αC-helix. Type I inhibitors bind to the kinase and stabilize its active conformation (“DFG-in, αC-helix-in”), essentially mimicking ATP binding. Type II inhibitors bind to a hydrophobic pocket adjacent to the ATP-binding site in the inactive conformation (“DFG-out, αC-helix-in”) of the kinase, providing a different mechanism of inhibition. Type I1/2 inhibitors emerge as a novel subclass that simultaneously engages the ATP-binding site and an adjacent allosteric site, resulting in a unique “αC-helix-out” conformation with partial DFG-in features. This innovative mechanism bypasses ATP competition (a limitation inherent to Type I inhibitors) while avoiding the reliance on full DFG-out accessibility (a prerequisite for Type II inhibitors). On the other hand, Type III and IV inhibitors bind to sites distal to the ATP-binding pocket, allosterically modulating kinase activity. Given the diversity of inhibitor binding modes, it is plausible that binding of different types of RIPK3 inhibitors may induce conformations in the inhibitor binding pocket similar to those observed with various mutations, some of which trigger on-target apoptosis and others do not. In this context, a systematic exploration and classification of binding conformations of RIPK3 inhibitors, along with an investigation of their potential to induce on-target apoptosis, are expected to reveal the conformational changes responsible for apoptosis and provide an effective solution for on-target apoptosis induced by the inhibitors.
In this work, we first investigate the molecular mechanism by which the R69H mutation prevents on-target apoptosis induced by GSK'872. Subsequently, we carry out structure-based design of a series of inhibitors derived from GSK'872 to achieve the conformational change in the inhibitor binding pocket similar to that induced by the R69H mutation. Among them, LK01003 stands out for its lack of on-target apoptosis, excellent selectivity, and potent inhibition against RIPK3. Crystallographic studies reveal that the absence of on-target apoptosis and the improved selectivity of LK01003 compared to GSK'872 are mainly ascribed to its unique binding mode and ability to occupy a hydrophobic site that has not been targeted by existing inhibitors. Binding to this newly identified site is critical for the inhibitor to abolish on-target apoptosis, which was further validated by designing additional inhibitors that possess scaffolds different from GSK'872 and are derived from known inhibitors with severe apoptotic effects. Our study thus provides a new and broadly applicable paradigm for structure-based rational design and development of RIPK3 inhibitors with excellent selectivity and a lack of on-target apoptosis.
Results
Co-crystal structures of mRIPK3KD and mRIPK3KD R69H in complex with GSK'872
A previous study by Raju et al. suggestes that the R69H mutation eliminates GSK'872-induced apoptosis by dissociating RIPK3KD dimers into monomers34. In this context, we first examine whether the R69H mutation could prevent on-target apoptosis in L929 cells, a widely used cell line for studying RIPK3 inhibitors. Our results show that GSK'872, at low micromolar concentrations (e.g., 1.6 μM), significantly reduces the viability of cells expressing the ATPase-inactive RIPK3 K51A mutant (Fig. 1a). Importantly, the loss of cell viability is reversed by the addition of zVAD, a pan-caspase inhibitor that prevents apoptosis, suggesting that the observed cell death is a result of apoptosis induced by GSK'872. However, GSK'872 does not cause such apoptosis in cells expressing RIPK3 with the double mutation of K51A and R69H (Fig. 1b). This confirms that the R69H mutation can rescue cells from apoptosis triggered by GSK'872.

a, b Viability of L929 cells with RIPK3 knockout (RIPK3-/-) and overexpressed RIPK3 K51A (a) or RIPK3 K51A/R69H (b), treated with GSK'872 with (open triangle) and without (open circle) zVAD. c Superimposition of two co-crystal structures of mRIPK3KD (magenta cartoons) and mRIPK3KD R69H (green cartoons) in complex with GSK'872 with enlarged views of bound GSK'872 and R69 or H69 with its surroundings. Hydrogen bonds are represented by black dashed lines. d Overlaid size-exclusion chromatograms (Superdex 200) of mRIPK3KD with DMSO (black lines), mRIPK3KD with GSK'872 (magenta lines), and mRIPK3KD R69H with GSK'872 (green lines). The protein is concentrated to 10 mg/mL and GSK'872 is added to a final compound concentration of 1 mM. Three independent experiments are performed for (a, b), each with three technical replicates, and data are shown as mean ± SD. The data shown in d represent one of three independent experiments, all of which yield similar results. Statistical differences are determined by multiple t-tests. **q < 0.01, ***q < 0.001, q < 0.0001. Source data are provided as a Source Data file.
Subsequently, to investigate whether the R69H mutation affects the binding mode of the inhibitor, we determine co-crystal structures of mouse RIPK3 kinase domain (mRIPK3KD) and mRIPK3KD R69H in complex with GSK'872. Superimposition of the two crystal structures shows that the bound conformations of GSK'872 and overall structures (backbone) of the protein are almost identical in both complexes (Fig. 1c). This suggests that the protective effect of the R69H mutation against inhibitor-induced on-target toxicity is not resulting from altering the inhibitor’s binding mode or the overall structure of the kinase domain.
We further examine interactions of R69 with its surroundings in the complex structure of mRIPK3KD bound with GSK'872. It is found that R69 is located at the dimer interface of the kinase domain, where it forms hydrogen bonds with the main chain of V66 and L76 from another protomer (Fig. 1c). Mutation of R69 to H69 disrupts these hydrogen-bonding interactions (Fig. 1c), which provides a structural basis for understanding how the R69H mutation impairs dimerization of mRIPK3KD. In addition, we conduct size-exclusion chromatography experiments. The results show that both apo mRIPK3KD and its complex with GSK'872 elute at a position corresponding to the molecular weight of a dimer (Fig. 1d). In contrast, the peak position for eluted mRIPK3KD R69H in complex with GSK'872 aligns with a monomer (Fig. 1d). Taken together, these results demonstrate that the R69H mutation promotes the disassembly of the mRIPK3KD dimers into monomers by disrupting the dimer interface of the kinase domain.
Structure-based design of inhibitors to mimic the R69H mutation
The co-crystal structure of mRIPK3KD in complex with GSK'872 shows that R69 is located behind an αC-helix, while the thiazole group of GSK'872 is well positioned on the other side of the αC-helix (Fig. 2a). In order to mimic the R69H mutation, we introduce hydrophobic groups on the thiazole group of GSK'872 to interfere with the αC-helix by forming direct interactions with multiple hydrophobic amino acids of the helix (Fig. 2a). As a result, compounds LK01001, LK01002, and LK01003 are designed, synthesized, and evaluated by the ADP-Glo assay, demonstrating significant inhibition against the kinase activity of RIPK3 with IC50 values of 11, 3, and 2 nM, respectively (Fig. 2b, c). The potency of these three compounds is comparable or superior to that of GSK'872, which is also tested as a reference compound and has an IC50 value of 6 nM.

a Strategy for the design of R69H-mimicking inhibitors based on GSK'872. b Chemical structures of inhibitors with their IC50 values determined by the ADP-Glo assay and EC50 values against TNF-α-induced necroptosis in L929 and HT29 cell lines determined by the CCK-8 assay. c–e Representative inhibitory profiles of GSK'872 (magenta solid circle), LK01001 (green solid triangle), LK01002 (blue solid inverted triangle), LK01003 (purple solid diamond), and LK01045 (cyan open inverted triangle) against mRIPK3KD in the ADP-Glo assay (c), and against TNF-α-induced necroptosis in L929 (d) and HT29 (e) cells. In L929 cells, necroptosis is induced by treatment with mouse TNF-α (20 ng/mL) and zVAD (10 μM). Necroptosis in HT29 cells is induced by treatment with zVAD (20 μM), human TNF-α (40 ng/mL), and Smac mimetic SM-164 (100 nM). f, g Western blot analysis of RIPK3 and MLKL phosphorylation in L929 (f) and HT29 (g) cells with or without RIPK3 inhibitors. Cells are treated with inhibitors at concentrations of 3 and 10 µM, followed by induction of necroptosis for 3 hours. h Overlaid size-exclusion chromatograms (Superdex 200) of mRIPK3KD with GSK'872 (magenta lines), LK01002 (blue lines), LK01003 (purple lines), and LK01045 (cyan lines), alongside mRIPK3KD R69H mutant with GSK'872 (green lines). The protein is concentrated to 10 mg/mL and the inhibitors are added to a final compound concentration of 1 mM. The data in panel c are plotted as the mean ± SD of triplicate wells. Three independent experiments are performed for (d, e), each with three technical replicates, and data are shown as mean ± SD. Data shown in panels (f–h) represent one of the three independent experiments, all of which showed similar results. Source data are provided as a Source Data file.
Necroptosis inhibition tests on these compounds are subsequently conducted on L929 and HT29 cell lines, which are widely used for the assessment of cellular necroptosis and potency of RIPK3 inhibitors. Necroptosis in L929 cells is induced using mouse TNF-α and zVAD. The anti-necroptotic activities of the compounds in L929 cells are determined with an EC50L929 value of 7.0 μM for LK01001, 4.8 μM for LK01002, and 3.7 μM for LK01003 (Fig. 2b, d), similar to the EC50L929 value of GSK'872 (3.6 μM). In HT29 cells, necroptosis is induced using zVAD, human TNF-α, and Smac mimetic SM-164. The EC50HT29 value of LK01001, LK01002, LK01003, and GSK'872 is 4.1, 2.0, 0.85, and 7.9 μM, respectively (Fig. 2b, e). This indicates that LK01002 and LK01003 are much more potent than GSK'872 to block necroptosis in HT29 cells. Given the higher potency of LK01002 over its enantiomer LK01001, shown in both the ADP-Glo assay and necroptosis inhibition tests in HT29 cells, we focused on LK01002 and LK01003 for the subsequent experiments. Western blot analysis confirms that LK01002 and LK01003 effectively inhibit RIPK3 and MLKL phosphorylation induced by TNF-α in both L929 and HT29 cells (Fig. 2f, g).
In addition, we introduce an oxyethanol group onto the quinoline ring of LK01003 that is facing the solvent to improve the solubility of the compound. This modification results in the development of LK01045 (Fig. 2b), which also demonstrates strong inhibition against RIPK3 kinase activity with an IC50 value of 16 nM (Fig. 2b, c). More importantly, LK01045 shows improved inhibition against TNF-α-induced necroptosis in both L929 and HT29 cells, with EC50 values of 0.61 and 0.16 µM, respectively (Fig. 2b, d, e). These EC50 values represent approximately 6-fold and 5-fold stronger potency compared to LK01003, and approximately 6-fold and 49-fold stronger potency compared to GSK'872. Western blot analysis further confirms that LK01045 effectively inhibits RIPK3 and MLKL phosphorylation induced by TNF-α in both L929 and HT29 cells (Fig. 2f, g). Together, these results show the potent activity of LK01002, LK01003, and LK01045 in preventing cell necroptosis by inhibiting the kinase activity of RIPK3.
Further investigation is carried out to evaluate the potential of LK01002, LK01003, and LK01045 to mimic the effects of the R69H mutation by examining the impact of these three compounds on the dimerization of the RIPK3 kinase domain. As expected, the binding of LK01002, LK01003, or LK01045 disrupts the dimerization of mRIPK3KD, as evidenced by the delayed peak positions in the size-exclusion chromatography, which overlap well with that of the mRIPK3KD R69H bound with GSK'872 (Fig. 2h). These results suggest that these three compounds effectively reproduce the R69H mutation’s ability to impede the dimer formation of the kinase domain.
Moreover, in the cell viability assay, toxicity due to on-target apoptosis is observed with GSK'872 at a concentration of 3.1 μM (Fig. 3a). However, none of LK01002, LK01003, and LK01045 exhibits on-target toxicity at concentrations up to 50 μM (Fig. 3b-d). Such a significant increase in the tolerated concentration level represents a breakthrough in our attempts to prevent on-target toxicity induced by the inhibitors. We further tested these compounds in L929 cells with RIPK3 knockout (L929RIPK3−/−). The CC50 value of GSK'872 significantly increases from 6.3 μM in L929 to 22 μM in L929RIPK3−/− cells (Fig. 3a, e), indicating reduced toxicity in the absence of RIPK3. In contrast, the CC50 values of LK01002, LK01003, and LK01045 in both L929 and L929RIPK3−/− cells remain largely unchanged (Fig. 3b-d, f-h), further demonstrating that these compounds do not cause on-target toxicity. We also tested LK01002, LK01003, and LK01045 in L929 cells expressing the K51A/R69H mutants and found that their CC50 values are almost identical to those in the K51A mutant cell line, further supporting the conclusion that they do not induce on-target toxicity (Supplementary Fig. 1). In addition, GSK'872 activates caspase 3 at a concentration of 5 μM (Fig. 3i). In stark contrast, none of LK01002, LK01003, and LK01045 triggers caspase 3 activation even at 50 μM (Fig. 3i), which is approximately 10, 14, and 82 times their respective EC50 values in L929 cells. Moreover, immunoprecipitation analysis reveals that GSK'872 enhances the interaction between RIPK3 and RIPK1 (Fig. 3j), thereby activating the apoptotic pathway and leading to the cleavage of caspase 8 and caspase 3 (Fig. 3k). In contrast, these three compounds neither increase the interaction of RIPK3 with RIPK1 (Fig. 3j) nor activate the apoptotic pathway (Fig. 3k). Accordingly, the absence of increased RIPK3-RIPK1 interaction and the lack of activation of apoptosis-related proteins when treated with LK01002, LK01003, and LK01045 lent strong support to the observation that the three compounds can potently inhibit RIPK3 without triggering the undesired apoptotic signaling. Also, the successful discovery of this new series of inhibitors suggests that mimicking the effect of R69H mutation offers a promising strategy for the identification of new RIPK3 inhibitors without on-target toxicity. Although LK01045 shows a more potent EC50 value compared to LK01003, subsequent experiments are primarily carried out with LK01003 that differs from GSK'872 only in the addition of a cyclopentane ring, in order to explore the significant effect caused by the introduction of this group relative to GSK'872.

a–d Cell viability of L929 cells exposed to various concentrations of GSK'872 (a), LK01002 (b), LK01003 (c), and LK01045 (d), with (open triangle) and without (open circle) the addition of zVAD. e–h Cell viability of L929 cells with RIPK3 knockout (L929RIPK3-/-) exposed to various concentrations of GSK'872 (e), LK01002 (f), LK01003 (g), and LK01045 (h), with (open triangle) and without (open circle) the addition of zVAD. i The activity of caspase 3 in L929 cell lysates treated with 10 µM GSK'872 (magenta solid circle), 5 µM GSK'872 (pink open circle), 50 µM LK01002 (blue open inverted triangle), 50 µM LK01003 (purple open diamond), 50 µM LK01045 (blue open square), or DMSO (black open triangle). j Western blot analysis of RIPK3 co-immunoprecipitation with RIPK1 in L929 cells treated with GSK'872, LK01002, LK01003, or LK01045. Cells are treated with inhibitors at 10 µM for 5 hours. Normalized quantification plots represent the mean ± SD of three independent experiments. k Western blot analysis of caspase 3 and caspase 8 cleavage in L929 cells treated with GSK'872, LK01002, LK01003, or LK01045 for 3 hours. Data in panels (a–i) are presented as mean ± SD from three independent experiments, each with three technical replicates. Data shown in (j, k) represent one of the three independent experiments, all of which show similar results. Statistical differences in (a–h) are assessed using multiple t-tests, and those in (j) using an unpaired t-test. **q < 0.01, ***q < 0.001, ****q < 0.0001. Source data are provided as a Source Data file.
Selectivity is a key consideration in the development of small-molecule kinase inhibitors. The selectivity profile of LK01003 against a panel of 379 kinases demonstrates significantly enhanced on-target selectivity of LK01003 compared to GSK'872. Specifically, LK01003 exhibits an inhibition rate greater than 65% only against RIPK2 and breast tumor kinase (BRK) at a concentration of 1 μM, yielding an S (35) score of approximately 0.005 (Fig. 4a), whereas at the same concentration, GSK'872 inhibits 13 kinases from the overlapping panel of kinases30 (Supplementary Fig. 2). This highlights the superior selectivity of LK01003 for RIPK3. Additionally, using the ADP-Glo assay, the IC50 values of LK01003 against RIPK2 and BRK are determined to be 48 and 248 nM, leading to the selectivity index (SI) of 24 and 124, respectively. We further test LK01003 at a concentration of 10 μM against the panel of 379 kinases (Supplementary Fig. 3). The results show that LK01003 exhibits over 65% inhibition rate towards 12 kinases, yielding an S(35) score of approximately 0.03. Together, these data demonstrate that LK01003 is a highly specific inhibitor of RIPK3 with potent activity.

a Selectivity profile of LK01003 against 379 kinases assayed at 1 μM. The color gradient represents the inhibitory ratio of LK01003 against kinases: magenta represents 100% inhibition, white represents 50% inhibition, green represents no inhibition (0%), and the gradient colors in between represent varying levels of inhibition. The experiment is carried out in duplicate. b Comparison of interaction patterns of GSK'872 and LK01003 with mRIPK3KD by superimposition of their co-crystal structures. c Enlarged view of the DFG motif and αC-helix in the co-crystal structures of mRIPK3KD bound with GSK'872 and LK01003. GSK'872 and LK01003 are shown as magenta and green sticks. d Specific interactions formed by the cyclopentyl group of LK01003 with mRIPK3KD revealed by the co-crystal structure. e Molecular surface representation of the binding pockets for GSK'872 and LK01003 in the co-crystal structures. The overlapped binding site of the two inhibitors is colored in yellow. The unique binding site for the cyclopentyl group of LK01003 is highlighted in red. f The superimposed conformations of the αC-helix and R69 in mRIPK3KD bound with GSK'872 and LK01003. g Interaction patterns of R69 in the structure of mRIPK3KD in complex with LK01003. Source data are provided as a Source Data file.
Mechanism of action of LK01003 with mRIPK3KD and mRIPK3KD R69H
The co-crystal structure of mRIPK3KD in complex with LK01003 provides valuable insights into the molecular mechanism by which LK01003 mimics the R69H mutation and achieves remarkably high selectivity (Supplementary Table 1). In the complex structure, LK01003 mainly occupies the ATP-binding pocket located between the N-lobe and C-lobe of RIPK3KD (Fig. 4b). Superimposition of the co-crystal structures of mRIPK3KD bound with LK01003 and GSK'872 reveals almost identical binding modes in the binding pocket where LK01003 overlaps well with GSK'872 (Fig. 4b, c). However, the introduction of an additional cyclopentyl group in LK01003 induces a significant conformational change in the αC-helix as well as the DFG motif, pushing them away from the ATP-binding site (Fig. 4b). This conformational shift leads to the disorder of the αC-helix and the formation of a hydrophobic binding site between the αC-helix and the DFG motif. The cyclopentyl group establishes hydrophobic interactions with residues M65, L74, and F162 at this new site (Fig. 4b-d), which not only strengthens the binding of LK01003 with the pocket but also renders the kinase domain retain inactive conformations associated with the disordered αC-helix. This in turn changes the conformation of R69, thereby preventing it from forming stable hydrogen bonds with the main chains of V66 and L76, as seen in the complex structure of mRIPK3KD bound with GSK'872 (Figs. 1c, 4f, g). Therefore, like the R69H mutation, LK01003 effectively impedes the dimerization of mRIPK3KD by destabilizing the ordered structure of the αC-helix and reducing the key hydrogen bonds necessary for the stable dimer of the kinase domain. The mechanism of action of LK01003 with RIPK3KD reinforces the notion that the significant conformational changes of the αC-helix and the DFG motif at the dimer interface induced by residue mutation or ligand binding is able to prevent on-target apoptosis. Moreover, based on the binding mode of LK01003 with mRIPK3KD revealed by the co-crystal structure, it can be categorized as a relatively rare Type I1/2 inhibitor.
Although LK01003 disrupts mRIPK3KD dimerization in solution, the complex of mRIPK3KD with LK01003 seems dimeric in the co-crystal structure. It is similar to the binding of GSK'872 to mRIPK3KD R69H, which also appears predominantly as a monomer in solution but as a dimer in the co-crystal structure. This suggests that the assembly of the kinase domain in solution may differ from those in the crystal, with the latter possibly being influenced by crystallization conditions or crystal packing that helps stabilize the dimeric form. To this end, we determine the co-crystal structure of mRIPK3KD R69H in complex with LK01003 (Supplementary Table 1). As in solution, the complex of mRIPK3KD R69H bound with LK01003 also adopts a monomer in the co-crystal structure. Moreover, the binding mode of LK01003 with mRIPK3KD R69H, as shown in the co-crystal structure, closely resembles its binding mode with mRIPK3KD (Supplementary Fig. 4a). Both co-crystal structures of LK01003 in complex with mRIPK3KD and mRIPK3KD R69H show similar conformational changes in the αC-helix and DFG motif of the kinase domain, when compared to the co-crystal structure of mRIPK3KD in complex with GSK'872 (Fig. 3c and Supplementary Fig. 4b). These findings indicate the dual effects of the R69H mutation and the binding of LK01003 render mRIPK3KD R69H sufficiently stable in a monomeric state, not only in solution but also in crystals.
Link between inhibitor-induced conformational changes and on-target toxicity, and unique features of LK series inhibitors
The comparative structural study of mRIPK3KD in complex with GSK'872 and LK01003 reveals distinct conformational changes in the ATP-binding pocket, suggesting that inhibitor-induced conformational changes may be linked to the ability to induce on-target apoptosis. This observation prompts us to comprehensively investigate the relationship between the conformational changes upon binding of RIPK3 inhibitors and their ability to induce apoptosis. We compile a panel of existing RIPK3 inhibitors, including GSK'87230, GSK'84330, Dabrafenib36, GW'39B37, PP238, TAK-63231, and compound 1839, for the measurement of inhibitory activity against RIPK3 as well as on-target apoptosis in cells and the investigation of the ligand binding modes (Fig. 5a). These compounds inhibit RIPK3 ATPase activity with IC50 values ranging from 2 to 56 nM (Fig. 5b) in the ADP-Glo assay and block TNF-α-induced necroptosis in L929 cells with EC50 values ranging from 1.1 to 31 μM (Fig. 5c). We then evaluate these compounds for on-target apoptosis in L929 cells with or without zVAD. The results show that five of these seven compounds, namely GSK'872, GSK'843, Dabrafenib, GW'39B, and PP2, induce on-target apoptosis at concentrations of 3.1, 12.5, 25, 3.1, and 12.5 µM, respectively, which are about 0.4-4 times their respective EC50 values (Fig. 5a, d and Supplementary Fig. 5). This on-target apoptosis is reversed by the addition of zVAD (Fig. 5d and Supplementary Fig. 5), suggesting that these five RIPK3 inhibitors trigger apoptosis alongside their inhibition of necroptosis. Western blot analysis also confirms the activation of the apoptotic pathway, showing the cleavage of caspases 8 and 3 after treatment with these compounds (Fig. 5e). Conversely, TAK-632 and compound 18 do not exhibit on-target apoptosis or induce caspase cleavage, even at high concentrations of 100 and 50 μM, respectively (Fig. 5d, e and Supplementary Fig. 5), which are 26 and 45 times their respective EC50 values (Fig. 5a, c). Therefore, these two compounds are similar to LK01003 in terms of cellular anti-necroptotic activity but do not induce on-target apoptosis, although their IC50 values are weaker than that of LK01003.

a Chemical structures and inhibitory activities of compounds against RIPK3 ATPase activity (IC50) and necroptosis of L929 cells (EC50L929). The crystal structure of mRIPK3KD in complex with Dabrafenib is not resolved, and therefore, the specific type of inhibition by Dabrafenib is not defined. b, c Representative inhibitory profiles of GSK'872 (red solid circle), GSK'843 (blue solid triangle), Dabrafenib (purple solid diamond), GW'39B (orange open circle), PP2 (olive open square), TAK-632 (brown open triangle), and compound 18 (pink open inverted triangle) against RIPK3, measured by the ADP-Glo assay (b) and the necroptosis assay in L929 cell line (c). Necroptosis in L929 cells is induced by treatment with mouse TNF-α (20 ng/mL) and zVAD (10 μM). d Cell viability of L929 cells exposed to RIPK3 inhibitors in the presence (open triangle) and absence (open circle) of zVAD. The concentrations of the inhibitors used are 3.1 µM for GSK'872, 12.5 µM for GSK'843, 25 µM for Dabrafenib, 3.1 µM for GW'39B, 12.5 µM for PP2, 100 µM for TAK-632, and 50 µM for compound 18. e Western blot analysis of caspases 3 and 8 cleavage by RIPK3 inhibitors in L929 cells. The ADP-Glo assay in panel b is performed in triplicate. Panels c and d show data from three independent experiments, each performed with three technical replicates. Panel e presents representative results from one of three independent experiments, all of which yield similar outcomes. Statistical differences are determined by multiple t-tests. **q < 0.01, ***q < 0.001, q < 0.0001. Source data are provided as a Source Data file.
Next, we determine co-crystal structures of mRIPK3KD in complex with GSK'843, GW'39B, PP2, TAK-632, and compound 18 in addition to GSK'872 (Supplementary Table 1). Superimposition of the six complex structures reveals that all compounds bind into the ATP-binding pocket between the N-lobe and C-lobe (Fig. 6), but only the binding of LK01003 disturbs the ordered structure of the αC-helix and extends into a previously unoccupied hydrophobic region (Figs. 4e, 6a, b). However, besides LK01003, the treatment with TAK-632 and compound 18 also does not induce on-target apoptosis of cells, prompting us to dig deeper into the conformational changes of the kinase domain associated with on-target apoptosis. GSK'872, GSK'843, GW'39B, and PP2, which induce on-target apoptosis, exclusively occupy the front region of the ATP-binding site with similar ligand binding modes, leading mRIPK3KD to adopt an active conformation with the DFG motif locked in an “in” state by a salt bridge formed between residues K51 and D161 (Fig. 6a). Given the active conformation of mRIPK3KD upon inhibitor binding, these four compounds are classified as Type I inhibitors. In contrast, the binding of TAK-632 and compound 18 extends into the backside of the ATP-binding pocket, flipping the DFG motif outward and maintaining mRIPK3KD in an inactive conformation (Fig. 6b). These two compounds are thus categorized as Type II inhibitors. Notably, the binding of LK01003, a Type I1/2 inhibitor, also locks mRIPK3KD in an inactive conformation (Fig. 4b-e). Accordingly, the inactive conformations of mRIPK3KD upon binding of inhibitors appear to be key to eliminating on-target apoptosis in cells. Otherwise, apoptosis occurs alongside the anti-necroptotic activity. This finding provides valuable insights into the structural basis of on-target toxicity associated with certain RIPK3 inhibitors.

a Binding modes of Type I inhibitors (GSK'872, GSK'843, GW'39B, and PP2) to mRIPK3KD. b Binding modes of Type II inhibitors (TAK-632 and compound 18) to mRIPK3KD. c Binding modes of Type I1/2 inhibitors (LK01003 and LK01004) to mRIPK3KD. d Binding mode of a Type I1/2 inhibitor (LK01003) to hRIPK3KD. The N-lobe and C-lobe of the RIPK3KD are depicted as yellow and light green cartoons, respectively. The activation loop (AC loop) and αC-helix are displayed as light navy, and orange cartoons, respectively. The residues K51, E61, D161, and F162 in mRIPK3KD and K50, E60, D160, and F161 in hRIPK3KD are shown as sticks. GSK'872, GSK'843, GW'39B, PP2, TAK-632, compound 18, LK01003, and LK01004 are represented by magenta, olive, orange, teal, white, slate, green, and brown sticks, respectively. Hydrogen bonds are marked by dashed lines.
As Type I1/2 inhibitors like LK01003 and LK01045 can disrupt the dimerization of mRIPK3KD, we performed gel-filtration analysis to investigate whether Type II inhibitors have the same effect. The results show that, upon the addition of compound 18, mRIPK3KD is eluted at a position corresponding to the molecular weight of a dimer, similar to the DMSO control, indicating no effect of dimerization disruption found with compound 18 (Supplementary Fig. 6). Similarly, mRIPK3KD is also eluted as a dimer upon the addition of TAK-632 (Supplementary Fig. 6). These results indicate that disruption of dimerization caused by the inhibitors is not a prerequisite for their ability to prevent on-target apoptosis.
In addition, to investigate whether the unique binding pose of LK01003 with mRIPK3KD is also applicable to hRIPK3KD, we solve the co-crystal structure of hRIPK3KD in complex with LK01003 (Supplementary Table 1). The binding modes of LK01003 in two co-crystal structures of mRIPK3KD and hRIPK3KD are almost identical (Fig. 6c, d). The cyclopentyl group of LK01003 also causes a significant shift in the αC-helix and the DFG motif, pushing them away from the ATP-binding site (Fig. 6a, d and Supplementary Fig. 7a) and stabilizing an inactive conformation of hRIPK3KD. This shift also triggers substantial conformational changes in the activation loop (Fig. 6d) and exposes a previously unrecognized hydrophobic binding site between the αC-helix and the DFG motif. The cyclopentyl group establishes hydrophobic interactions with residues M64, L73, and F161 at this new site (Supplementary Fig. 7b), similar to the interactions formed with mRIPK3KD (Fig. 4d). Therefore, the co-crystal structures show that the unique binding mode of LK01003 in mRIPK3KD is also present in hRIPK3KD.
A potential strategy to eliminate on-target apoptosis
To verify the effectiveness of targeting the newly discovered hydrophobic site to avoid on-target toxicity, we conduct the structure-based design of new scaffold RIPK3 inhibitors based on PP2 and Zharp-99 which show significant on-target apoptosis in cells. By superimposing the co-crystal structures of RIPK3KD in complex with multiple inhibitors, we observe that the phenyl ring of PP2 aligns well with the phenyl ring of GSK'872 (Supplementary Fig. 8a). On this basis, we design and synthesize two additional compounds, LK01004 and LK01005, in order to occupy the newly identified site as LK01003 does (Supplementary Fig. 8b). These new compounds demonstrate potent inhibition against RIPK3 in the ADP-Glo assay, with IC50 values of 41 and 81 nM, respectively. Moreover, they exhibit EC50 values of 3.0 and 2.1 μM, respectively, in L929 cells, where necroptosis is induced by treatment with mouse TNF-α and zVAD, confirming their cellular potency against necroptosis by inhibiting RIPK3. To investigate whether these compounds induce on-target toxicity, we perform the cell viability assay. Remarkably, unlike PP2, the CC50 values of LK01004 and LK01005 do not change significantly in the presence or absence of zVAD, indicating that these new compounds do not induce on-target apoptosis in cells (Supplementary Fig. 8c, d). Furthermore, Western blot analysis provides the compelling evidence that at a concentration of 100 μM, LK01004 and LK01005 do not activate the apoptotic pathway, whereas PP2 causes the cleavage of caspases 8 and 3 at a concentration of 25 μM (Supplementary Fig. 8e).
The co-crystal structure of mRIPK3KD in complex with LK01004 is successfully determined at a resolution of 2.78 Å (Supplementary Table 1). A comparable structural analysis shows that the regions shared between LK01004 and PP2 adopt a similar binding conformation (Fig. 6a, c). The newly introduced isobutyl group in LK01004 with flexibility occupies the same binding site as the cyclopentyl group in LK01003, resulting in the outward displacement of the αC-helix and significant conformational changes in the DFG motif (Fig. 6c). In this co-crystal structure, we successfully resolve the structure of the activation loop following the DFG motif. When compared to the co-crystal structure of mRIPK3KD bound with PP2, the activation loop also undergoes substantial conformational changes (Fig. 6a, c).
The selectivity profile of LK01004 at 10 µM is assessed against a panel of 379 kinases (Supplementary Fig. 3). The results demonstrate that LK01004 displays an S (35) selectivity score of approximately 0.18, which is significantly lower than the reported S(35) score of 0.41 for PP240, indicating a significant improvement in kinase selectivity for LK01004 compared to PP2. It should be noted that both LK01005 and LK01004 still exhibit a substantial portion of RIPK3-independent cell killing (Supplementary Fig. 8a, d), likely due to relatively inferior kinase selectivity compared to LK01003, which leads to off-target toxicity.
Analogously, we introduced a cyclopentyl group into Zharp-99 to occupy this miraculous binding site, yielding the new compound LK01033 (Supplementary Fig. 9a). Zharp-9941, a compound derived from GSK'872, is reported to exhibit enhanced anti-necroptotic activity compared to GSK'872, but it triggered stronger on-target apoptosis. As expected, LK01033 shows strong activity in inhibiting necroptosis in both L929 and HT29 cells induced by TNF-α and zVAD, with EC50 values of 0.39 µM and 0.11 µM, respectively, and for reference, the EC50 values of Zharp-99 are 0.07 µM and 0.22 µM, respectively (Supplementary Fig. 9a). Importantly, LK01033 does not induce on-target apoptosis, even at a concentration of 10 µM, whereas 0.1 µM Zharp-99 activates the apoptotic pathway as evidenced by cleavage of caspase 8 and caspase 3 (Supplementary Fig. 9b). The CC50 value of LK01033 in L929 cells does not change significantly in the presence or absence of zVAD, while for Zharp-99 it differs by ~36-fold (Supplementary Fig. 9c, d).
The success of these two additional cases highlights the robustness of the strategy that occupies the newly identified site to eliminate on-target apoptosis while reducing off-target effects on other kinases, paving the way for the development of highly safe and selective RIPK3 inhibitors.
Discussion
The central role of RIPK3 in cell death pathways, particularly necroptosis, makes it a promising target for the treatment of various necroptosis-mediated diseases. However, the development of RIPK3 inhibitors is hindered by on-target toxicity, which primarily results from activation of apoptosis pathways upon inhibitor binding. Previous studies show that the R69H mutation can mitigate on-target apoptosis induced by GSK'872 by disrupting the dimerization of RIPK3KD. In the present study, X-ray protein crystallography together with gel-filtration analysis explicitly reveals that the R69H mutation significantly weakens the hydrogen-bonding interactions between R69 and neighboring residues at the dimer interface of mRIPK3KD bound with GSK'872, which promotes the dissociation of dimers into monomers. Guided by this structural insight, we design and synthesize a series of new RIPK3 inhibitors derived from GSK'872 to replicate the conformational changes induced by the R69H mutation. As expected, these new inhibitors, represented by compounds LK01003 and LK01045, exhibit potent inhibition of RIPK3 ATPase activity, effective anti-necroptosis activity, but are absent of on-target apoptosis. Notably, LK01045 shows stronger cellular activity than LK01003, possibly due to improved physicochemical properties. Moreover, the co-crystal structure of mRIPK3KD in complex with LK01003 reveals that the compound occupies a hydrophobic site and induces conformational changes of key motifs in the kinase domain, including the αC-helix and DFG motif. In addition to the absence of on-target toxicity, occupation of the new site also leads to enhanced selectivity compared to GSK'872, as revealed by kinase profiling assays. In particular, LK01003 exhibits excellent kinase selectivity.
The broad applicability of this newly identified site is further demonstrated by the design of a second class of inhibitors (LK01004 and LK01005) that target this site but with scaffolds distinct from that of LK01003. In LK01004, the introduction of an isobutyl group to the PP2 scaffold effectively occupies the newly identified site and significantly prevents RIPK3-mediated apoptosis. Like LK01003, LK01004 also shows a notable improvement in the kinase selectivity profile compared to PP2, with an S (35) value improving from 0.41 to 0.18. Nevertheless, it is important to note that LK01004 is not so selective as LK01003, possibly due to the poor selectivity of the PP2-scaffold. The generality of this strategy is further confirmed through modification of Zharp-99. Introducing a cyclopentyl group to generate LK01033 abolishes the severe apoptosis induced by Zharp-99. Therefore, our study identifies a previously uncharacterized site in RIPK3KD and, more importantly, demonstrated through multiple examples that inhibitors designed to target this site not only eliminate on-target apoptosis but also enhance kinase selectivity, providing a targetable site and strategy for the rational design of safer and more selective RIPK3 inhibitors. While these findings are currently derived from the molecular and cellular experiments, future studies are needed to evaluate the pharmacokinetic properties and therapeutic potential of these type I1/2 inhibitors in animal models relevant to RIPK3-mediated diseases.
Conformational changes of kinases upon inhibitor binding or residue mutagenesis and their impact on kinase-independent pathways have emerged as a fascinating research area in the field of kinase biology and drug discovery. Traditionally, kinase inhibitors have been designed to target the ATP-binding site, aiming to directly inhibit ATPase activity of the kinase. However, studies reveal that the binding of different types of inhibitors can trigger distinct conformational changes, leading to the modulation of pathways unrelated to the ATPase activity. A notable example is that some BRAF inhibitors are shown to induce a specific conformational change in the kinase domain, promoting the formation of BRAF-CRAF heterodimers and subsequently leading to the paradoxical activation of the MAPK pathway in BRAF wild-type cells42. Here, our X-ray protein crystallography studies reveal the distinct conformational changes in mRIPK3KD upon binding of GSK'872 and LK01003, which inspired us to systematically explore the relationship between the binding modes of various inhibitors and their associated toxicity. We found that all type I inhibitors (including GSK'872, GSK'843, GW'39B, and PP2) stabilize mRIPK3 in an active conformation, which is tightly associated with the induction of apoptosis. In contrast, type II inhibitors and our LK series inhibitors (e.g., Type I1/2 inhibitors) do not induce apoptosis. They both stabilize mRIPK3KD in an inactive conformation but through distinct structural changes: type II inhibitors primarily affect the activation loop, while LK series inhibitors impact both the αC-helix and the activation loop. It thus clearly shows how different RIPK3 inhibitors, with distinct binding modes, can induce various conformational changes that influence ATPase-independent pathways, such as on-target apoptosis. Notably, LK series inhibitors can disrupt dimerization of mRIPK3KD, while Type II inhibitors do not cause such dimerization disruption, suggesting that the dimerization disruption is not a prerequisite for preventing apoptosis. This indicates that there may be alternative mechanisms, such as those employed by Type II inhibitors, that could also avoid apoptosis and deserve further investigation.
Currently, the mechanisms by which RIPK3 inhibitors or mutations induce apoptosis are primarily studied in mouse species30,33,34,43, while those mechanisms remain unconfirmed in human species, largely due to no human cell lines available for the induction of apoptosis by the inhibitors. To explore potential species-specific differences, we determine the co-crystal structures of both mRIPK3KD and hRIPK3KD in complex with LK01003 in order to explore the difference that might be caused by different species. Notably, although the R69 residue that plays key roles at the dimer interface of mRIPK3KD is replaced by D68 in hRIPK3KD, the binding mode of LK01003 and overall kinase domain structure remain very similar in two co-crystal structures. LK01003 binding induces conformational changes in the αC-helix and DFG motif in both mRIPK3KD and hRIPK3KD, keeping them in an inactive conformation. While our study confirms that Type I1/2 inhibitors prevent on-target apoptosis in murine cells by disrupting kinase dimerization, further studies are needed to determine if this mechanism applies to human cells.
Methods
Chemistry
GSK'872, GSK'843, Dabrafenib, PP2, TAK-632, and compound 18 were purchased from MedChemExpress. Procedures and synthetic schemes for compounds GW'39B, Zharp-99, LK01001–LK01005, LK01033, and LK01045 are provided in the Supplementary Methods section of the Supplementary Information. Spectral data for compounds GW'39B, Zharp-99, LK01001–LK01005, LK01033, and LK01045 are included in the Supplementary Note 1. All commercially available chemicals and solvents were used directly without further purification. All reactions were monitored by thin-layer chromatography (TLC) on silica gel plates (GF-254). All compounds were characterized by their NMR and MS spectra. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 400, a Bruker Avance 500 or a Bruker Avance 600 using TMS as the internal standard. NMR data were recorded using TopSpin software (v3.0 or v3.2) and analyzed using MestReNova (v12.0.0-20080). HPLC traces were collected and analyzed on an Elite 3150 system using Chromsoft software (v8.2.0.82). High-resolution mass spectrometry (HRMS) spectra were acquired with an Agilent G6520 Q-TOF mass spectrometer using MassHunter Workstation software (vB.05.01) and analyzed with Xcalibur software (v2.0.5). Low-resolution mass spectrometry (LRMS) spectra were acquired with a Thermo Fisher FINNIGAN LTQ spectrometer and analyzed using Xcalibur software (v2.0.5). The purities of compounds used in the biochemical tests were over 95%. Purity was determined by the following method: column, WondaSil C18 Superb (5 μm, 4.6 mm × 250 mm); mobile phase A: water containing 0.1% trifluoroacetic acid (TFA); mobile phase B: acetonitrile containing 0.1% trifluoroacetic acid (TFA); gradient, 10% B to 100% B over 15 minutes, 100% B for 5 minutes, 100% B to 10% B over 5 minutes; flow rate, 1.0 mL/min; detection wavelength, 254 nm; column temperature, 25 °C.
Cell lines and cell culture
HT29 (HTB 38) and mouse fibrosarcoma L929 (CCL-1) cell lines were purchased from the American Type Culture Collection (ATCC). RIPK3-knockout L929 cell lines were generated using the CRISPR/Cas9 system with an sgRNA (5′-GAGAACGTTCTGCTCCTGCT-3′) targeting the mouse RIPK3 gene. Disruption of the target gene was confirmed by sequencing the gene loci and by immunoblotting cell lysates with antibodies. HT29 cells were cultured in RPMI 1640 medium supplied by Gibco, while L929 cells were maintained in Minimum Essential Medium (MEM) from Gibco. All media were supplemented with 10% (v/v) fetal bovine serum (FBS). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. For all experiments, cells were harvested during the exponential growth phase.
Antibodies
Antibodies used in this study included: anti-human RIPK3 (Cell Signaling, #13526, 1:1000 for WB), anti-mouse RIPK3 (Abcam, ab62344, 1:1000 for WB), anti-human phospho-RIPK3 (Ser227) (Cell Signaling, #93654, 1:1000 for WB), anti-mouse phospho-RIPK3 (Thr231/Ser232) (Abcam, ab222320, 1:1000 for WB), anti-RIPK1 (Cell Signaling, #3493, 1:1000 for WB, 1:100 for IP), anti-human MLKL (Abcam, ab184718, 1:1000 for WB), anti-mouse MLKL (Cell Signaling, #37705, 1:1000 for WB), anti-human phospho-MLKL (Ser358) (Abcam, ab187091, 1:1000 for WB), anti-mouse phospho-MLKL (Ser345) (Abcam, ab196436, 1:1000 for WB), anti-caspase-3 (Cell Signaling, #9662, 1:1000 for WB), anti-cleaved caspase-3 (Asp175) (Cell Signaling, #9661, 1:1000 for WB), anti-caspase-8 (Cell Signaling, #4790, 1:1000 for WB), anti-cleaved caspase-8 (Asp387) (Cell Signaling, #8592, 1:1000 for WB), anti-GAPDH (Arigo, ARG10112, 1:1000 for WB), anti-β-actin (Arigo, ARG65683, 1:1000 for WB), HRP-conjugated secondary antibodies (Beyotime, A0208 and A0216, 1:10000 for WB), and mouse anti-rabbit IgG-HRP (Abmart, M21006, 1:1000 for WB).
Recombinant protein expression and purification
The mouse RIPK3 kinase domain (1-313) with C111A mutation or C111A/R69H double mutations was subcloned into the pFastBacHT B Vector (Invitrogen) with a C-terminal 10×His-tag. Bacmids were generated in DH10Bac cells using the Bac-to-Bac system, and the resultant baculoviruses were produced and amplified in Sf-9 insect cells. Cells were transfected with baculoviruses for 72 hours, harvested, and then homogenized in lysis buffer containing 25 mM Tris (pH 7.8), 500 mM NaCl, 5% glycerol, 5 mM β-mercaptoethanol, and protease inhibitors. The lysate was incubated with Ni-NTA beads (GE Healthcare) for 30 minutes on ice, then the protein-bound resins were pelleted by centrifugation at 120 g for 5-10 minutes. The resins were packed into a gravity column and washed twice with lysis buffer supplemented first with 20 mM and then with 50 mM imidazole. The bound proteins were eluted using lysis buffer plus 300 mM imidazole. The concentrated eluate was then subjected to size exclusion chromatography using a pre-equilibrated HiLoad™ 16/600 Superdex™ 75 pg column (GE Healthcare) with 25 mM Tris (pH 8.0), 150 mM NaCl, and 5 mM DTT. The purified proteins were finally concentrated to 10 mg/mL for crystallization.
The recombinant human RIPK3 kinase domain (2-328) with an N-terminal 6×His and TEV tag along with a C-terminal 10×His tag for the ADP-Glo assay, and RIPK2 (1-310) with a C-terminal 10×His tag for the ADP-Glo assay, were also subcloned into the pFastBacHT B Vector. Both proteins were expressed and purified using the same protocol established for preparing mouse RIPK3. Briefly, baculoviruses were produced in Sf-9 cells via the Bac-to-Bac system, followed by cell lysis and protein purification using Ni-NTA beads. The eluted proteins were then subjected to size exclusion chromatography on a HiLoad™ 16/600 Superdex™ 75 pg column for further purification. The purified proteins were used for the ADP-Glo assay.
The recombinant human RIPK3 kinase domain (2-315) with C110A and C3S mutations was subcloned into the pFastBacHT B Vector (Invitrogen) with N-terminal 10×His and TEV tag. Bacmids were generated in DH10Bac cells using the Bac-to-Bac system, and the resultant baculoviruses were produced and amplified in Sf-9 insect cells. Cells were transfected with baculoviruses for 72 hours, harvested, and then homogenized in lysis buffer containing 50 mM Tris (pH 8.0), 1 M NaCl, 10% glycerol, 5 mM β-mercaptoethanol, and protease inhibitors. The lysate was incubated with Ni-NTA beads (GE Healthcare) for 30 minutes on ice, then the protein-bound resins were pelleted by centrifugation at 120 g for 5-10 minutes. The resins were packed into a gravity column and washed twice with lysis buffer supplemented with 20 mM first and then with 50 mM imidazole. The bound proteins were eluted using lysis buffer plus 300 mM imidazole. The concentrated elution was then subjected to size exclusion chromatography using a pre-equilibrated HiLoad™ 16/600 Superdex™ 200 pg column (GE Healthcare) with 50 mM HEPES (pH 7.5), 200 mM NaCl, 5% glycerol, and 1 mM TCEP. The purified proteins were finally concentrated to 10 mg/mL for crystallization.
The human BRK kinase domain (185-446) with C433T mutation was subcloned into the pFastBacHT B Vector (Invitrogen) with a C-terminal 6×His-tag. Bacmids were generated in DH10Bac cells using the Bac-to-Bac system, and the resultant baculoviruses were produced and amplified in Sf-9 insect cells. Cells were transfected with baculoviruses for 72 hours, harvested, and then homogenized in lysis buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, 5% glycerol, 5 mM β-mercaptoethanol, and protease inhibitors. The lysate was incubated with Ni-NTA beads (GE Healthcare) for 30 minutes on ice, then the protein-bound resins were pelleted by centrifugation at 120 g for 5-10 minutes. The resins were packed into a gravity column and washed with lysis buffer supplemented with 20 mM imidazole. The bound proteins were eluted using lysis buffer plus 300 mM imidazole. The concentrated elution was then subjected to size exclusion chromatography using a pre-equilibrated HiLoad™ 16/600 Superdex™ 75 pg column (GE Healthcare) with 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM TCEP, and 5% glycerol. The purified proteins were finally concentrated to 8 mg/mL and then cryopreserved with 20% glycerol for the ADP-Glo assay.
Protein crystallization and structure determination
Co-crystals of the mouse and human RIPK3 kinase domains in complex with various inhibitors were grown using the hanging-drop or sitting-drop vapor diffusion methods. The protein was incubated with the inhibitors at concentrations ranging from 1 to 5 mM for 30 to 60 minutes. Co-crystals of mRIPK3(1-313)-C111A in complex with GSK'872, GSK'843, LK01003, GW'39B, PP2, and LK01004 were obtained under conditions of 10-20% PEG 3350, 50 mM magnesium formate, and pH 5.0-7.0. Additionally, co-crystals of compound 18 and TAK-632 in complex with mRIPK3(1-313)-C111A were grown under conditions of 10% PEG 1500, 17.1% PEG 300, 50 mM BICINE (pH 8.4), and 18-22% w/v PEG 3350, 10-15 mM L-proline, respectively. Co-crystals of mRIPK3(1-313)-C111A/R69H in complex with GSK'872 and LK01003 were obtained under conditions of 25% PEG 1500, 100 mM MIB buffer (pH 7.0), and 0.2 M ammonium acetate, 0.1 M MES, 30% v/v glycerol ethoxylate (pH 6.5), respectively. Co-crystals of hRIPK3(2-315)-C110A/C3S in complex with LK01003 were grown in 25% PEG 3350, 0.2 M MgCl2, and 0.1 M Tris buffer (pH 7.5). X-ray diffraction data were collected at beamlines BL02U1, BL10U2, and BL18U1 at the Shanghai Synchrotron Radiation Facility using Blu-Ice software. X-ray diffraction data were processed with Finback software (v1.0). The diffraction data were auto-processed by the Aquarium pipeline44. The structures were solved by molecular replacement using the program CCP4 suite (v7.0.078)45, with the coordinates of PDB entry 4M66 as the search model. The final structures were manually refined with COOT (v0.8.9.2)46 and PHENIX (v1.17.1-3660)47, and further analyzed with PyMOL (v2.4.0). The refined structures were subsequently deposited in the Protein Data Bank with accession codes listed in Supplementary Table 1. Diffraction data, refinement statistics, and the quality of the structures are detailed in Supplementary Table 1.
ADP-Glo kinase assay
The recombinant human RIPK3, RIPK2, or BRK kinase domain was incubated with either DMSO or a compound in kinase reaction buffer that contains 40 mM Tris (pH 7.5), 20 mM MgCl2, and 0.1 mg/mL BSA. Subsequently, ATP was added to reach a final concentration of 20 µM, and the reaction mixture was incubated at room temperature for 1 hour. To measure ATPase activity, luminescence was recorded following the addition of the ADP-Glo kinase assay kit. This assay includes two steps: (i) stop the kinase reaction and deplete the remaining ATP; (ii) add a luminescent reagent to detect the ADP produced during the reaction. The process, following the manufacturer’s instructions (Promega), ensures accurate measurement of ATPase activity based on ADP production. Each compound was tested in triplicate across at least eight different concentrations. The IC50 values were determined using nonlinear regression analysis with GraphPad Prism software (v8.0).
Chromatographic analysis
During the protein purification process, 10 µM of the compound or DMSO was added to the cell lysate. The proteins eluted from the Ni-NTA column were concentrated to 10 mg/mL, incubated with 1 mM of each compound, and centrifuged at 17,000 g for 15 minutes. Following centrifugation, 100 μL of the sample was injected into a pre-equilibrated HiLoad™ 10/300 Superdex™ 200 pg Increase column (GE Healthcare), which had been equilibrated with buffer (25 mM Tris (pH 8.0), 150 mM NaCl, and 5 mM DTT) supplemented with 10 µM of the compound. To ensure the consistency of experiments, the injection volume for all samples was maintained at 100 μL. Three independent experiments were performed for each compound.
Cell viability assay
L929, L929 RIP3-/-, L929 RIP3−/− + RIPK3 K51A, or L929 RIP3-/- + RIPK3 K51A/R69H cells were seeded at a density of 10,000 to 20,000 cells per well in a 96-well plate with a volume of 100 µL per well. The cells were allowed to adhere for 24 hours to ensure proper attachment and recovery. After that, cells were treated with either the compound alone or in combination with zVAD (a pan-caspase inhibitor) for 18-20 hours. Following treatment, 10 µL of CCK-8 (Cell Counting Kit-8) solution was added to each well, and the cells were incubated at 37 °C for an additional 1-2 hours. The absorbance was then measured at a wavelength of 450 nm using a microplate reader. The absorbance measurement is indicative of cell viability, with higher absorbance indicating more viable cells. For each compound, three independent experiments and each independent experiment in triplicate were performed for the determination of CC50 values. The CC50 values were expressed as the mean ± SD and calculated via nonlinear regression analysis using GraphPad Prism software (v8.0).
Necroptosis induction and inhibition assay in L929 and HT29
Necroptosis in HT29 cells was induced by pretreating the cells with zVAD (20 μM) for 1 hour, followed by treatment with human TNF-α (40 ng/mL) and Smac mimetic SM-164 (100 nM) for 18-20 hours. In L929 cells, necroptosis was induced by pretreatment with zVAD (10 μM) for 1 hour, followed by treatment with mouse TNF-α (20 ng/mL) for 18-20 hours. L929 and HT29 cells were seeded at a density of 10,000 to 20,000 cells per well in 96-well plates, with 100 μL of culture medium per well. The cells were allowed to adhere and grow for 24 hours in a humidified incubator set at 37 °C with 5% CO2. Then, the cells were exposed to the compound at various concentrations along with zVAD for 1 hour. Necroptosis was then induced for an additional 18-20 hours. Cell viability was assessed using the CCK-8 assay according to the manufacturer’s protocols. Briefly, 10 μL of CCK-8 reagent was added to each well, and the cells were cultured for 1-2 hours. The absorbance was measured at 450 nm using a microplate reader. For each compound, three independent experiments and each independent experiment in triplicate were performed for the determination of EC50 values. The EC50 values were expressed as the mean ± SD and calculated via nonlinear regression analysis using GraphPad Prism software (v8.0). The analysis was performed with 100% as the top value and the survival ratio of cells treated under necroptosis induction conditions as the bottom value.
Assessment of necroptosis inhibition by Western blot analysis
L929 and HT29 cells were seeded in 6- and 12-well plates, respectively, and allowed to reach approximately 80% confluency. Subsequently, cells were treated with either the compound, DMSO, or blank culture medium along with zVAD. After 1-2 hours of treatment, necroptosis in L929 cells was induced by adding mouse TNF-α (20 ng/mL) for 3 hours. Necroptosis in HT29 cells was induced by adding human TNF-α (40 ng/mL) and Smac mimetic SM-164 (100 nM) for 3 hours, respectively. Following induction, the culture medium was aspirated, and cells were washed three times with cold PBS. Cell lysates of HT29 cells were prepared by adding 100 μL of RIPA lysis buffer containing protease inhibitors, phosphatase inhibitors, and SDS loading buffer to each well, followed by incubation at room temperature for 10 minutes. L929 cells were lysed in a buffer containing 10 mM Tris-HCl (pH 8.0), 1% SDS, 1 mM sodium orthovanadate, protease inhibitors, and phosphatase inhibitors. The cell lysates were then boiled for 10 minutes, followed by freezing at -80 °C. Protein samples were separated by 10% SDS-PAGE gel electrophoresis, and the proteins were transferred onto PVDF membranes using a rapid wet transfer system (eBlot L1, GenScript). After overnight incubation with primary antibodies, species-specific antibodies conjugated with horseradish peroxidase (HRP) were used for detection. Three independent experiments were conducted for each compound. Immunoblot images were obtained using the Image Analysis System 5200CE (Tanon) with AllDoc_x software. Uncropped and unprocessed blot scans are available in the Source Data.
Immunoprecipitation
For RIPK1 immunoprecipitation, L929 cells were treated with 10 μM of the compound for 5 hours and washed with cold PBS, then lysed in the following buffer: 20 mM Tris-HCl (pH 7.5), 137 mM NaCl, 1 mM EDTA, 1.5 mM MgCl2, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM NaF, 1 mM Na3VO4, and protease inhibitors, followed by centrifugation at 17,000 g for 15 minutes at 4 °C. The soluble fraction was collected, and a portion was taken to prepare the input sample. Anti-RIPK1 antibody was incubated with Protein A/G Magnetic Beads (MedChemExpress, HY-K0202) for 2 hours at 4 °C. Then, the beads were washed three times in wash buffer (1 × PBS with 0.5% Tween-20, pH 7.4) and the soluble fraction was incubated with RIP1 antibody-bound magnetic beads at 4 °C for another 2 hours. Subsequently, the beads were washed three times with wash buffer, then directly boiled in 1× SDS loading buffer and prepared as immunoprecipitation (IP) samples, followed by immunoblot analysis. Immunoblot images were obtained using the Image Analysis System 5200CE (Tanon) with AllDoc_x software. Uncropped and unprocessed blot scans are available in the Source Data.
Assessment of caspase 3 and caspase 8 cleavage by Western blot analysis
L929 cells were seeded in 12-well plates, with 1 mL of medium per well. After growing to 80% confluency, cells were treated with either the compound, DMSO, or blank culture medium along with 10 μM zVAD. After 3 hours, the supernatant was discarded, and cells were washed once with pre-chilled PBS. The cell lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM Na2EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4) was used to prepare a 1× SDS sample buffer, with 100 µL added per well. After lysing at room temperature for 10 minutes, the samples were collected and boiled for 10 minutes. Proteins were separated using 15% SDS-PAGE and transferred to a PVDF membrane with a rapid wet transfer system (GenScript, eBlot L1). The membrane was blocked with 5% skim milk for 1 hour at room temperature to prevent non-specific binding, and then incubated with primary antibodies overnight at 4 °C. Detection was carried out using species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies. Three independent experiments were carried out for each compound. Immunoblot images were obtained using the Image Analysis System 5200CE (v12.0.0-20080) with AllDoc_x software. Uncropped and unprocessed blot scans are available in the Source Data.
Analysis of caspase 3 activity in L929 cell lysates
L929 cells were seeded and cultured in 12-well microplates for 12 hours, then treated with DMSO or the compound for 0, 1, 2, 3 or 4 hours. After treatment, the cells were collected and lysed in the following buffer: 50 mM HEPES, 5 mM DTT, 1 mM EDTA, 0.1% Triton X-100, 0.1% CHAPS, pH 7.4, followed by centrifugation at 17,000 g for 15 minutes at 4 °C. The supernatants were collected and protein concentrations were measured using the BCA Protein Quantification Kit (Yeasen). Finally, cell lysates were used to detect caspase 3 activity according to instructions of the caspase 3 activity assay kit (Beyotime). To assess caspase 3 activity, 50 µL of cell lysate supernatant was taken from each well, and 10 µL of the caspase 3 chromogenic substrate Ac-DEVD-pNA (Beyotime, P9710) was added to each well of a 96-well plate. The plate was gently shaken to mix the contents and then incubated overnight at 37 °C. The next day, absorbance was measured at 405 nm, and caspase 3 activity was determined by subtracting the blank control reading from that of the test group. Three independent experiments and each experiment in triplicate were conducted for each compound.
Selectivity profiling
The selectivity evaluations of LK01003 and LK01004 against 379 human kinases were conducted following Eurofins’ standard KinaseProfiler assays (Eurofins Cerep, 50-005KP10). For protein kinases, the assay employed a radiometric format, whereas lipid kinases, along with ATM (h), ATR/ATRIP (h), and DNA-PK (h), were analyzed using a time-resolved fluorescence (HTRF) format. LK01003 at final concentrations of 1 μM and 10 μM was incubated with the kinase of interest and its respective substrate at 37 °C. LK01004 at a final concentration of 10 μM was incubated with the kinase of interest and its respective substrate at 37 °C. The reaction was initiated by adding ATP to each well, reaching a final concentration equivalent to the Km values of the kinase hydrolysis ATP. Following the reaction, the signal from each well was measured using a microplate reader. This signal was then compared to that of DMSO (the reference) to determine the extent of kinase inhibition by LK01003 and LK01004.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic coordinates and structure factors generated in this study have been deposited into the Protein Data Bank under accession codes 9IWW (mRIPK3KD in complex with GSK'872), 9IWX (mRIPK3KD R69H in complex with GSK'872), 9IWY (mRIPK3KD in complex with LK01003), 9IWZ (mRIPK3KD in complex with GSK'843), 9IX0 (mRIPK3KD in complex with GW'39B), 9IX1 (mRIPK3KD in complex with PP2), 9IX2 (mRIPK3KD in complex with TAK-632), 9IX3 (mRIPK3KD in complex with compound 18), 9LFW (mRIPK3KD R69H in complex with LK01003), 9LFV (mRIPK3KD in complex with LK01004) and 9LFU (hRIPK3KD in complex with LK01003). The structural model used for molecular replacement in this study is available in the Protein Data Bank under accession code 4M66. The mRNA sequence of mouse RIPK3 used in this study is available in the GenBank database under accession code AAF03133.1 [https://www.ncbi.nlm.nih.gov/protein/AAF03133.1]. The mRNA sequences of human RIPK2, RIPK3, and BRK used in this study are available in the NCBI Gene database under accession codes 8767, 11035, and 5753, respectively. All data supporting the findings of this study are available within the paper, its Supplementary Information, and Source Data files. Source data are provided with this paper.
References
Rajesh, Y. & Kanneganti, T. D. Innate immune cell death in neuroinflammation and Alzheimer’s Disease. Cells 11, 1885 (2022).
Linkermann, A. et al. Necroptosis in immunity and ischemia-reperfusion injury. Am. J. Transplant. 13, 2797–2804 (2013).
Wang, R. et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 580, 386–390 (2020).
Weinlich, R., Oberst, A., Beere, H. M. & Green, D. R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Gautam, A. et al. Necroptosis blockade prevents lung injury in severe influenza. Nature 628, 835–843 (2024).
Gong, Y. et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 18, 100 (2019).
Kaiser, W. J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Zhang, D. W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014).
Zhang, T. et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180, 1115–1129 (2020).
Jiao, H. et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395 (2020).
He, S. et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 108, 20054–20059 (2011).
Morgan, M. J. & Kim, Y. S. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 54, 1695–1704 (2022).
Zhang, S. et al. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis 8, e2905 (2017).
Caccamo, A. et al. Necroptosis activation in Alzheimer’s Disease. Nat. Neurosci. 20, 1236–1246 (2017).
Iannielli, A. et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s Disease models. Cell reports 22, 2066–2079 (2018).
Piamsiri, C. et al. Targeting necroptosis as therapeutic potential in chronic myocardial infarction. J. Biomed. Sci. 28, 25 (2021).
Leng, Y. et al. Receptor interacting protein kinases 1/3: the potential therapeutic target for cardiovascular inflammatory diseases. Front. Pharmacol. 12, 762334 (2021).
Xu, J. et al. Epithelial Gab1 calibrates RIPK3-dependent necroptosis to prevent intestinal inflammation. JCI insight 8, e162701 (2023).
Lee, S. H. et al. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Scientific reports 7, 10133 (2017).
Jhun, J. et al. RIPK1 inhibition attenuates experimental autoimmune arthritis via suppression of osteoclastogenesis. J. Transl. Med. 17, 84 (2019).
Ofengeim, D. et al. Activation of necroptosis in multiple sclerosis. Cell reports 10, 1836–1849 (2015).
He, S. & Wang, X. RIP kinases as modulators of inflammation and immunity. Nature immunology 19, 912–922 (2018).
Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Chen, J. X. et al. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J. Transl. Med. 16, 233 (2018).
Huang, Z. J. et al. RIP3-mediated microglial necroptosis promotes neuroinflammation and neurodegeneration in the early stages of diabetic retinopathy. Cell Death Dis. 14, 227 (2023).
Mandal, P. et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell. 56, 481–495 (2014).
Chen, X. et al. Identification of the Raf kinase inhibitor TAK-632 and its analogues as potent inhibitors of necroptosis by targeting RIPK1 and RIPK3. Br. J. Pharmacol. 176, 2095–2108 (2019).
Zhang, H. et al. N-(7-Cyano-6-(4-fluoro-3-(2-(3-(trifluoromethyl)phenyl)acetamido)phenoxy)benzo[d]thiazol-2-yl)cyclopropanecarboxamide (TAK-632) analogues as novel necroptosis inhibitors by targeting receptor-interacting protein kinase 3 (RIPK3): synthesis, structure-activity relationships, and in vivo efficacy. J. Med. Chem. 62, 6665–6681 (2019).
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Raju, S. et al. Kinase domain dimerization drives RIPK3-dependent necroptosis. Sci. Signal. 11, eaar2188 (2018).
Roskoski, R. Jr Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 103, 26–48 (2016).
Li, J. X. et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5, e1278 (2014).
Rodriguez, D. A. et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 23, 76–88 (2016).
Li, W. et al. Targeting RIPK3 oligomerization blocks necroptosis without inducing apoptosis. FEBS Lett 594, 2294–2302 (2020).
Hart, A. C. et al. Identification of RIPK3 type II inhibitors using high-throughput mechanistic studies in hit triage. ACS Med. Chem. Lett. 11, 266–271 (2020).
Brandvold, K. R. et al. Development of a highly selective c-Src kinase inhibitor. ACS Chem. Biol. 7, 1393–1398 (2012).
Xia, K. et al. Discovery of a potent RIPK3 inhibitor for the amelioration of necroptosis-associated inflammatory injury. Front. Cell Dev. Biol. 8, 606119 (2020).
Yao, Z. et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 25, 284–291 (2019).
Chen, X. et al. Mosaic composition of RIP1-RIP3 signalling hub and its role in regulating cell death. Nat. Cell Biol. 24, 471–482 (2022).
Yu, F. et al. Aquarium: an automatic data-processing and experiment information management system for biological macromolecular crystallography beamlines. J. Appl. Crystallogr. 52, 472–477 (2019).
Collaborative, C. P. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 50, 760–763 (1994).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D. 58, 1948–1954 (2002).
Acknowledgements
We thank the staff from beamlines BL02U1, BL10U2, and BL18U1 at Shanghai Synchrotron Radiation Facility for assistance in data collection. This work was supported by the National Key R&D Program of China (No. 2023YFF1205104) to Y.X., Research Program of Shanghai Institute of Materia Medica, Chinese Academy of Sciences (No. SKLDR-2024-TT-03) to Y.X., the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0830000) to Y.X., Research Program of Shanghai Institute of Materia Medica, Chinese Academy of Sciences (No. SIMM0320231006) to Y.X., and the National Natural Science Foundation of China (No.21807104) to Y.X.
Author information
Authors and Affiliations
Contributions
Y. X., Q. Z., and H. S. conceived and design the project. Y. X. and H. S. designed the experiments; Y. X., H. S., and G. C. performed the drug design; G. C. and J. H. performed the chemical experiments and collected the data; H. S., H. X., W. L., H. H., W. Z. and M. L. carried out protein expression and purification, crystallization, X-ray diffraction data collection, solved and analyzed the crystal structures; H. X., H. S., and W. L. determined the IC50 values of the compounds and performed cellular experiments; M. X. and Q. S. assisted with data analysis; Y. X. and H. S. wrote and revised the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, H., Chen, G., Xie, H. et al. Structure-based design of potent and selective inhibitors targeting RIPK3 for eliminating on-target toxicity in vitro. Nat Commun 16, 4288 (2025). https://doi.org/10.1038/s41467-025-59432-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59432-8
