
Abstract
The quality of Li–solid electrolyte interface is crucial for the performance of solid-state lithium metal batteries, particularly at low stack pressure, but its dynamics during cell operation remain poorly understood due to a lack of reliable operando characterization techniques. Here, we report the evolution of Li–electrolyte interface with high spatial resolution using operando scanning electron microscopy under realistic operating conditions. By tracking the stripping process of both Li and Li-rich Li-Mg alloy anodes, we show that multiple voids coalesce into a single gap and eventually delaminate the interface in Li, whereas the voids split and collapse to partially recover interfacial contact in Li-Mg. Density functional theory calculations show that the stronger Mg-S interaction at the metal–electrolyte interface attracts Mg toward the interface and repels Li-vacancies into the bulk, resulting in a reduced number of voids. The pressure-dependent voltage profiles of Li and Li-Mg stripping suggest that loss of contact due to void formation, rather than Mg accumulation at the interface, is the origin of high overpotential that limits the utilization of metal anodes. Improved interfacial contact enables stable cycling of all-solid-state lithium full cell at low stack pressure (1 MPa) and moderate rate (2 mA cm−2) simultaneously. The real-time visualization of Li–electrolyte interface dynamics provides critical insights into the rational design of solid-state battery interfaces.
Similar content being viewed by others


Introduction
All-solid-state lithium batteries stand out as next-generation battery technology due to their potential for high energy density1,2. However, the unstable Li–electrolyte interface has been one of the major obstacles limiting cell performance3,4,5,6,7. The root cause of this challenge is the large partial volume of lithium (12.97 cm3 mol−1)8. During stripping, voids form at the interface when the rate of Li stripping exceeds the rate at which Li is supplied from the bulk metal due to Li vacancy hopping, resulting in contact loss and subsequent non-uniform plating9,10,11,12,13.
Li transport at the interface can be modified by chemical interactions between the metal anode and the solid electrolyte interphase (SEI) that governs interface adhesion, wetting, and void generation. Modeling has revealed that a lithiophilic interphase attracts Li atoms toward the Li/SEI interface, thus effectively repelling vacancies away from the interface and mitigating void generation14. A lithiophobic interphase does the opposite by trapping and accumulating vacancies into voids14. Naturally formed SEI, such as Li2CO315 on oxide electrolyte and Li2S16 on sulfide electrolyte, are often lithiophobic, facilitating void formation during stripping16,17. As a result, the metal–electrolyte interface is susceptible to contact loss that induces current focusing and uneven Li-plating morphology in the successive plating process, further exacerbated by the rough electrolyte surface and native passivation layer on lithium17,18. Eventually, interfacial instability may lead to Li metal penetrating the solid electrolyte and short-circuiting the cell12,19.
Li solid solutions, such as Li-Mg alloy, have been investigated as alternative materials to Li metal due to their similar redox potential to pure Li and improved interfacial mechanical stability20,21,22,23. Unlike many other metals with limited solubility24,25,26, Mg exhibits a solubility of up to 70 atom% in the Li body-centered cubic (BCC) structure27, enabling Li-Mg alloy to remain in a single phase and maintain a low potential over a wide range of stripping capacities. Moreover, Li-Mg alloy shows improved interfacial contact compared to Li after stripping, as supported by ex situ morphological characterization and indirect impedance-based techniques20,21. However, the underlying mechanisms that alter the post-stripping interface morphology remain unclear, particularly given the comparable volume change of Li and Li-Mg during stripping.
The improved interfacial contact was initially attributed to higher effective Li diffusivity in the Li-Mg alloy20, based on a study in liquid electrolyte using neutron tomography28. However, secondary ion mass spectrometry (SIMS) showed that Li has lower tracer diffusivity in Li-Mg alloy21. This discrepancy suggests that the simple Fick’s Law28 treatment of Li diffusion may not be sufficient to model the interfacial contact evolution at the Li-Mg alloy–electrolyte interface. Instead, Darken’s equations should be used to describe interdiffusion in binary alloys29,30. Given the inconsistency in the reported Li diffusivity in Li-Mg alloy, it has been speculated that the contact is maintained because of the non-strippable Mg21.
To date, the interface dynamics are poorly understood due to the lack of reliable operando characterization techniques with high spatial and temporal resolution. Current in situ methods, such as X-ray computed tomography (X-ray CT), face significant challenges in sample preparation in order to achieve sufficient resolution to accurately capture void dynamics9,10. Meanwhile, most characterization techniques with high spatial resolution, such as scanning electron microscopy (SEM), are used ex situ and cannot track the evolution of individual voids31. Both SEM and X-ray CT are important techniques that complement each other to provide a comprehensive picture of the interface dynamics.
Here, we investigate the dynamics at the metal–electrolyte interface with high spatial resolution using operando SEM under low stack pressure. We reveal the mechanisms behind the improved interfacial contact enabled by a Li-rich Li-Mg alloy. Density functional theory (DFT) calculations indicate that Mg atoms preferentially bond with sulfur at the interface, move toward it, repel Li vacancies, and consequently reduce void formation. As a result, we demonstrate stable cycling of all-solid-state cells at a high rate (1 C, 2 mA cm−2) under the lowest reported stack pressure (1 MPa). Postmortem analysis confirms the absence of interfacial voids and a uniform Mg distribution in the Li-Mg anode after 260 cycles.
Results
Void dynamics in Li and Li-Mg under 0.2 MPa
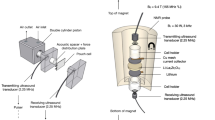
We characterize the void dynamics at the metal–electrolyte interface with operando SEM. A miniature cell is loaded in an air-free transfer vessel custom designed by our group, as shown in Supplementary Fig. 1. The cell mounting stage is designed to apply low stack pressure (0.2 MPa) to the cell to prevent metal extrusion that could disrupt the operando observation (Supplementary Fig. 2). The cell was polished by Ar+ beam to allow for a large cross-sectional area (>1000 µm in width), as compared to focused ion beam (FIB) methods21,31, adding statistical significance to the observed void geometry.
Voltage profiles significantly differ between Li and Li-Mg electrodes during stripping. At the current of 0.5 mA cm−2, the voltage profile in Fig. 1a shows the Li electrode experiences a rapid overpotential increase at a small areal capacity (1 mAh cm−2), followed by voltage fluctuation, whereas the Li-Mg electrode shows a stable potential up to 3 mAh cm−2. This difference in voltage profiles can be attributed to the contact difference at the interface. Figure 1b shows the contact loss at the metal–electrolyte interface with respect to stripped areal capacity. The overpotential drastically increases when contact loss reaches 90%.

a Voltage profile and interface contact loss during stripping. The Li electrode experiences a severe overpotential increase after stripping 1 mAh cm−2. Li-Mg electrode remains at a relatively stable potential up to 3.5 mAh cm−2. b Contact loss calculated based on cross-section SEM images over a 160-µm-wide interface (Supplementary Video 1 zoom out location 1 and Video 2 zoom out location 1). c Number of voids in the imaged area. The decrease in the number indicates void coalescence, while the increase suggests void splitting. d, e Box chart of void width (e) and height (f) evolution. Horizontal lines represent maximum, median, and minimum, while box boundaries represent quartiles. When small voids coalesce into larger ones, the void width and height increase. When large voids split or collapse, the void width and height decrease. f Snapshots of the operando SEM images at the Li–electrolyte interface (Supplementary Video 1). g Snapshots of the operando SEM images at the Li-Mg–electrolyte interface (Supplementary Video 2). The scale bar is 10 µm for all images.
As soon as stripping begins, voids immediately appear at the interface for both metals (Supplementary Videos 1, 2) due to their similar partial volume at approximately 13 cm3 mol−1 (Supplementary Note 1). In Li metal, a large number of small voids (2.2 ± 1.3 µm, 16–18 voids per 100 µm width) initially form at the interface (Fig. 1f and Supplementary Fig. 3a). These small voids then grow and coalesce into larger voids (Supplementary Fig. 3b) as the stripped capacity increases (0.25 mAh cm−2). Void coalescence results in the current focusing at the remaining contact area, which exacerbates non-uniform Li removal and eventually creates a gap between metal and electrolyte (1.25 mAh cm−2, Supplementary Fig. 4) that leads to a severe overpotential increase32. The stack pressure of 0.2 MPa was insufficient to close the gap, likely due to the low deviatoric stress33, as well as the tensile stress state at the edge as illustrated in Supplementary Fig. 5, making the contact loss observed in the operando SEM appears more pronounced.
In contrast, the Li-Mg alloy shows a markedly different behavior (Fig. 1g). Initially, voids also form at the interface, but they are fewer in number and smaller in size compared to the pure Li case (3.8 ± 1.0 µm, 2–3 voids per 100 µm width). The presence of Mg alters the void dynamics, as voids in the Li-Mg alloy tend to grow to ~20 µm in width, then split into smaller voids (0.5 mAh cm−2) or collapse (Supplementary Fig. 6) rather than coalesce, maintaining better interfacial contact. The void dynamics are illustrated in Supplementary Fig. 7a, b. The correlation between the overpotential and contact loss is summarized in Supplementary Fig. 7c.
The number, width, and height of voids are statistically analyzed across a larger interface width (160 µm) in Supplementary Videos 1–2 and are plotted in Fig. 1c–e. In Li metal (purple curve), a large number of small voids are generated immediately after stripping begins (Fig. 1c). The mean, quartile, and maximum values of both width (Fig. 1d) and height (Fig. 1e) increased with areal capacity, followed by a decrease in the number of voids. At 1.25 mAh cm−2, all voids coalesced into a single gap across the entire field of view. In contrast, the number of voids in Li-Mg gradually increases with the stripped capacity. However, the maximum void width value decreases three times at the capacities of 0.625, 1, and 2.375 mAh cm−2, respectively, suggesting the splitting of large voids. Similarly, the maximum void height value also drops at 0.5, 1, and 2.625 mAh cm−2, corresponding to void collapse and restored interfacial contact. The width and height of voids in Li-Mg are observed to be limited within a certain range throughout the entire stripping process. Postmortem analysis also shows the shape of the void inside the sample, which is similar to the shape of the voids on the surface (Supplementary Fig. 8) and in ex situ samples (Supplementary Fig. 9). Operando stripping tests conducted after prior Li plating on Li-Mg also demonstrated an interface without delamination. However, voids were not visible due to the roughened surfaces (Supplementary Fig. 10).
Deshpande et al.32 recently theoretically predicted void coalescence as the origin of interface delamination and suggested that voids could collapse when they grow beyond a critical size. Here, we provide direct experimental evidence in support of this prediction. In Li-Mg, the small number of voids are less likely to encounter each other during stripping, allowing for growth in width. The velocity field created by stripping tends to collapse large voids, as predicted by the void growth model32, and consequently, interfacial contact can be partially recovered. Such void collapse is also recently studied in Li-Mg anode by Zhu et al. with a phase field model34. In Li metal, however, the large number of small voids are more likely to coalesce before reaching the critical size required for void collapse to occur. Thus, the number of voids formed during stripping is a critical factor governing interfacial morphology. The reason for the smaller number of voids in Li-Mg can be elucidated through the subsequent DFT calculations.
Mg suppresses the accumulation of vacancies at interface
To understand the difference in void formation and dissipation in the Li and Li-Mg alloy, the preferential distribution of Li, Mg, and vacancies at the metal–electrolyte interface were simulated via DFT calculations, assuming Li2S stoichiometrically dominates the electrolyte surface after SEI formation on argyrodite (~80 vol%)35,36 at near-zero electrochemical potential (vs. Li+/Li) in Fig. 2a, b. Vacancy accumulation and void formation are thermodynamically favored at the lithiophobic Li–Li2S interface16. The DFT calculated work of adhesion is 0.39 J m−2, corresponding to a non-wetting interface with a wetting angle (θ) of 93.6°. The lower Li vacancy formation energy (0.28 eV in Fig. 2c) than that of the bulk Li metal (0.50 eV, Supplementary Table 1)14 suggests the vacancies prefer to accumulate at the Li-Li2S interface instead of migrating into the Li-bulk. Moreover, when the vacancy at the interface exchanges its location with the Li in the second layer, the vacancy formation energy increases by 0.15 eV (Fig. 2d). Based on our previous DFT-informed kinetic Monte Carlo (KMC) simulations, this energetics should lead to vacancy accumulation and large number of void formation during stripping. These voids will ultimately coalesce and result in complete delamination32, as observed in the operando stripping experiment. Li creep under compressive stress will help Li vacancies migrate into the bulk37,38.

a, b (top panel, simulation cells), a vacancy was generated on the first layer (L1) during stripping (starting structures). c The enlarged view of the region in the blue box in the simulation cell (d), the vacancy in (c) was filled by a Li atom from the second layer (L2). e The enlarged view of the region in the orange box in the simulation cell. f The vacancy in (e) was filled by a Li atom from L2. g The vacancy in (e) was filled by a Mg atom from L2. The total energy differences were calculated compared to the starting structures. The distances between the vacancy and the Mg atom are labeled in (e–g). Li: purple, Mg: orange, S: yellow, Li vacancy: unfilled black circles.
In contrast, the addition of Mg atoms to Li leads to a lithiophilic Li-Mg/Li2S interface (θ = 87.1°, Supplementary Table 2), which prevents vacancy accumulation at the interface. Figure 2b illustrates the energetics of the Li vacancy generated in the first layer adjacent to Mg (3.3 Å) (Fig. 2e) being filled by either Li (Fig. 2f) or Mg (Fig. 2g) (also listed in Supplementary Table 3). A more negative energy value compared to the initial stripped structure (Fig. 2e) indicates a more stable structure. Thus, the Li vacancy generated at the interface during stripping is thermodynamically favored to be filled by Mg (Fig. 2g, −0.67 eV) rather than Li (Fig. 2f, −0.16 eV). This preferential Mg filling can lead to Mg accumulation at the interface. This thermodynamic driving force is consistent with the more stable Mg-S bonds (−1.677 eV/bond in MgS) compared to Li-S bonds (−1.504 eV/bond in Li2S), considering their most stable sulfide species. Additional sampling of Li vacancy structures is shown in Supplementary Table 3 and Supplementary Fig. 11. When a Li vacancy is generated far away (4.9 Å) from the Mg atom, it tends to stay at the interface due to an increase in energy by 0.13 eV when filled by an adjacent Li atom. This behavior reflects the lithiophobic nature observed at the pure Li/Li2S interface. Hence, the thermal stability of the Li-Mg/Li2S interface during stripping depends on the distance between Mg and vacancies.
It turns out that Mg tends to repel nearby Li vacancies. The Li-vacancy formation energy (\({E}_{{vac}}^{{form}}\)) is 0.53 eV when a vacancy is adjacent to Mg (3.0 Å, first nearest neighbor) in the BCC-Li lattice, while it decreases to 0.50 eV when the vacancy is located 4.9 Å (third nearest neighbor) away from the Mg. The Li-Mg alloy structure and the vacancy positions are shown in Supplementary Fig. 12. The \({E}_{{vac}}^{{form}}\) decreases with Mg–vacancy distance in Li-Mg alloy and eventually converges to the value in pure Li (0.50 eV), suggesting the Mg–vacancy interactions become negligible when separated by more than 4.9 Å. Similar Mg–vacancy repulsion is also observed at the interface. In the most stable configuration (Fig. 2g), the Mg–vacancy distance is 7.2 Å, whereas the distances in less stable configurations (Fig. 2e, f) are 3.3 and 4.0 Å, respectively. Therefore, Mg atoms in Li-Mg alloy repel vacancies away from the interface during stripping and suppress local void formation24. At the stripped sites, increased local Mg concentration reduces Li diffusivity21, as the region with more non-strippable Mg atoms cannot provide sufficient vacancies to assist the Li diffusion to the surface39. As a result, vacancies accumulate into voids that grow in width. Void geometry statistics in Fig. 1 also show only a small number of voids, but with larger widths emerge in Li-Mg as stripping starts. Moreover, the non-strippable nature of Mg and strong Mg–S interaction further prevent total contact loss at large areal capacities24.
Accumulation of Mg at interface
Considering the crucial role of Mg distribution on vacancy formation in Li-Mg alloy, we investigated the circumstances under which Mg accumulation occurs during stripping. Energy-dispersive spectroscopy (EDS) was used to characterize the Mg distribution in the Li-Mg anode after stripping at different current densities, as shown in Fig. 3. Increasing the current density from 0.05 to 0.5 mA cm−2 leads to an overpotential rise at around 3 mAh cm−2 (Fig. 3b), consistent with the voltage profile in operando tests. Three-electrode analysis further verifies that the overpotential originates from the stripped Li-Mg anode (Supplementary Fig. 13). Pristine Li-Mg shows uniform Mg distribution (Fig. 3f), verified by the flat Mg intensity profile across the metal anode (Fig. 3i). Stripping for 4 mAh cm−2 resulted in a decrease in metal thickness and slightly higher Mg intensity at the interface at both current densities (Fig. 3g, h, j, k).

a Schematical illustration of the preparation process of imaged samples in (c–e). b Voltage profile of stripping test conducted at room temperature and 1 MPa stack pressure for 4 mAh cm−2. c–e Zoom-in SEM images of Li-Mg negative electrode at pristine state (c), after 0.05 mA cm−2 stripping (d), and after 0.5 mA cm−2 stripping (e) corresponding to black, red, and blue circles, respectively, illustrated in (b). f–h Zoom-out SEM image and Mg mapping of Li-Mg negative electrode at pristine state (f), after 0.05 mA cm–2 stripping (g), and after 0.5 mA cm−2 stripping (h). i–k Normalized EDS intensity profile of (f–h). The maximum values of Cu (current collector) and S (electrolyte) intensities are normalized to unity. The intensity profiles are aligned based on metal–electrolyte interface. The Mg intensity at 20 µm from the current collector is set to unity to represent Mg concentration in the bulk electrode. The average composition of the stripped Li-Mg (4 mAh cm−2 from 90 µm Li-Mg) is Li0.96Mg0.04. The error bar represents the standard deviation of EDS intensity across the imaged region. l Li stripping at 0.2, 1, 1.5, and 7 MPa. m Li-Mg stripping at 0.2, 1, 14, 50 MPa.
The accumulation of Mg at the interface is not caused by bulk diffusion or creep deformation. Our diffusion barrier calculations showed that the Li atom hopping barrier (\({E}_{{hop}}\)) to fill a Li vacancy is only 0.055 eV in bulk Li. The Mg atom hopping barrier is twice as high (0.102 eV) in Li-Mg alloy in dilute concentration. The hopping barrier of Li next to an Mg atom is 0.061 eV (Supplementary Table 1), suggesting slower Li diffusion in Li-Mg alloys but still faster than Mg diffusion, agreeing with the tracer diffusion measurements21. Therefore, vacancies should be filled more by Li if we only consider interdiffusion. Because the Mg concentration profile did not show strong dependence on striping rates, the observed Mg accumulation is more likely driven by Mg–S chemical interactions at the interface rather than diffusion kinetics, which is typically rate-dependent. This cannot be caused by creep either, as the creep deformation in Li-Mg alloy drives both Li and Mg atoms indiscriminately toward the interface. Further, the Li-Mg phase diagram (Supplementary Fig. 14) shows that the melting point of BCC-Li alloy increases with Mg fraction, suggesting a decreased homologous temperature (Tambient/Tmelting) and a slower thermally activated creep deformation with Mg alloying40.
Origin of pressure-dependent overpotential
Previously, the overpotential in Li-Mg anode has been attributed primarily to compositional changes in Li-Mg that result in the formation of hexagonal close-packed (HCP) Mg phase with low Li diffusivity20. It is worth noting that Mg accumulation at the interface does not necessarily lead to an overpotential increase. Instead, the overpotential increase is accompanied by loss of contact at metal–electrolyte interface (Fig. 3e and Supplementary Fig. 8). Overpotential arising from loss of contact area can be eliminated by increasing stack pressure, which supplies additional lithium flux via creep deformation of metal anode10,41,42,43. We compared the voltage profiles of metal anode stripping at different stack pressures at 0.5 mA cm−2. Li metal shows severe overpotential increase at low pressures (0.2 and 1 MPa) during stripping, while a flat voltage profile is observed up to 4 mAh cm−2 at 1.5 MPa or higher (Fig. 3l). In contrast, Li-Mg merely shows overpotential increase within 3 mAh cm−2 at any pressures due to the suppressed void formation by Mg accumulation. The overpotential at large areal capacities can still be eliminated with higher stack pressure (Fig. 3m)20,44.
The pressure-dependent overpotential behavior confirms that void formation-induced contact loss is the primary origin of overpotential increase in solid-state batteries instead of phase transition induced by composition change20. Interestingly, no HCP-Mg phase was detected in stripped Li-Mg alloy20, even at a very high capacity (19 mAh cm−2 from a 100 ± 10 µm Li-Mg, Supplementary Fig. 15). In solid-state batteries, Li stripping no longer occurs at points of lost contact, naturally limiting the composition change in Li-Mg alloy. This differs from Li-Mg alloy stripped in liquid electrolyte, where BCC-to-HCP phase transition was experimentally verified with X-ray diffraction, and the overpotential increase is mainly driven by the low Li diffusivity in HCP-Mg phase28.
Reversibility in morphology and composition
To understand the effect of interfacial contact on the subsequent plating process, we conducted operando SEM studies for both Li and Li-Mg samples. Figure 4a shows the voltage profile during 0.5 mA cm−2 plating for the same samples that underwent stripping in Fig. 1. The Li electrode experienced a short-circuit at around 1.0 mAh cm−2, while the Li-Mg survived more than 3.0 mAh cm−2. Operando SEM video (Supplementary Video 3) and snapshots of SEM images (Fig. 4b) reveal Li filament growth at multiple sites. Such filaments, with a diameter of 1.1 ± 0.4 µm, triggered local current focusing that leads to cell shorting (Supplementary Fig. 16)19,45,46. In contrast, Fig. 4c shows bulk Li plating in a pillar-like fashion at the Li-Mg–electrolyte interface.

a Voltage-capacity profile of the plating test after the stripping test in Fig. 1. Li electrode experiences short-circuiting after plating about 1 mAh cm−2. Li-Mg electrode remains at a relatively stable voltage up to 3 mAh cm−2. The decrease in voltage beyond 3.2 mAh cm−2 originates from the Li depletion in the counter electrode. b Li–electrolyte interface during plating. Li filaments are highlighted with arrows. The scale bar is 50 µm. c Li-Mg–electrolyte interface during plating. The scale bar is 50 µm. d, e SEM image, Mg and S mapping of an ex situ plated Li-Mg (2.6 mAh cm−2 at 0.28 mA cm−2, corresponding to 13 µm of Li) at 25 °C (d) and at 60 °C (e). The scale bar is 10 µm. f Normalized EDS intensity profile of Mg and S at 25 and 60 °C. Vertical dash lines indicate the boundary of newly plated Li. The error bar represents the standard deviation of EDS intensity across the imaged region.
To ensure reversible cycling, Mg needs to diffuse into the plated Li to achieve uniform composition distribution. DFT calculation shows Mg diffusivity in Li (DMg in Li) is 2.06 × 10−13 and 2.42 × 10−12 cm2 s−1 at 25 and 60 °C, respectively (Supplementary Table 1). The strong temperature dependence is attributed to the high Mg hopping barrier as previously discussed. Estimating the average diffusion length using \(\sqrt{4{Dt}}\) results in an apparent difference (1.7 vs 5.9 µm) at 25 and 60 °C for a 10-h interdiffusion. We conduct postmortem analysis of Li-Mg anode after plating 2.6 mAh cm−2 of Li, corresponding to a 13-µm thick region shown between dash lines in the cross-sectional SEM images, EDS mapping, and line analysis (Fig. 4d–f). We observed a clear temperature effect on the distribution of Mg. At 25 °C, the Mg intensity averaged between the dashed lines is 50% of the bulk intensity. In contrast, the percentage increases to 80% at 60 °C.
During plating, Mg diffusion appears slower compared to stripping, as only a slight increase in Mg intensity is observed at the interface in Fig. 3j, k. This is because Li and Mg diffusion needs to overcome the combined energy barrier of \({E}_{{vac}}^{{form}}\) + \({E}_{{hop}}\) during plating, with the high \({E}_{{vac}}^{{form}}\) dominating the process. In contrast, sufficient vacancies are already present during stripping, and diffusion is governed solely by \({E}_{{hop}}\), enabling much faster Li and Mg diffusion. Taken together, the behavior in Li and Li-Mg stripping and plating is summarized in Supplementary Fig. 17; these observations underscore the importance of alloying elements in stabilizing the metal–electrolyte interface in solid-state batteries.
High-rate electrochemical cycling at 1 MPa
High stack pressure up to several tens of megapascals is often required to ensure high-rate operation of all-solid-state batteries. This high-pressure requirement poses significant challenges in practical applications because these redundant pressurizing devices lower the energy density of batteries and increase the cost. We summarize the stack pressure and cumulative areal capacity of reported all-solid-state cells using Li and alloy anodes in Fig. 5a and Supplementary Table 4. To focus on the metal anode–solid electrolyte interface, we exclude the reports using the interlayer or liquid catholyte approach. Alloy anodes that form intermetallic phases during lithiation, such as Li-Si47, Li-In48, Li-Al49, often require a high stack pressure (≥50 MPa) to enable high rate because intermetallic phases are typically mechanically rigid40. In contrast, solid solution alloys, e.g., Li0.97Mg0.03, remain as Li BCC phase during lithiation and are mechanically ductile, which is beneficial for high-rate cycling at low stack pressure. To demonstrate this point, we fabricated all-solid-state cells comprised of LiNi0.83Mn0.06Co0.11O2 composite cathode (NMC), Li6PS5Cl (LPSCl) electrolyte, and a Li-Mg anode.

a Stack pressure and cumulative capacity of full all-solid-state batteries with Li or alloy anodes. Cycling temperature is represented in colors. The circle radius in the inset corresponds to the current density. Further details are listed in Supplementary Table 4. b Voltage profile at 0.1, 0.2, 0.33, 0.5, and 1 C (2 mA cm−2) with LiNi0.83Mn0.06Co0.11O2 cathode. The cathode active material loading is 10 mg cm−2 or 2 mAh cm−2. c Capacity retention and Coulombic efficiency of full cell. d NMC composite cathode after 260 cycles at 1 MPa, and e after 770 cycles at 14 MPa. Bright particles are NMC, dark particles are Li6PS5Cl, and black particles are cracks. f Rate performance of full cells at 1 MPa. g–i Postmortem analysis of Li-Mg anode after 260 cycles at 1 MPa. SEM images (g), Mg mapping (h), and S mapping (i).
Figure 5b shows voltage profiles with specific capacity of 208 mAh g−1 at 0.1 C and 163 mAh g−1 at 1 C at a stack pressure of 1 MPa. Li-Mg anode enables stable cycling at 2 mA cm–2 (1 C) and 1.6 mAh cm–2, with capacity retention of 76% for 260 cycles (Fig. 5c). Postmortem analysis at the discharged state shows the decay is mainly attributed to failure of NMC composite cathode (Fig. 5d, e) rather than the anode–electrolyte interface (Fig. 5g–i). Large transverse cracks are observed in the composite cathode due to chemo-mechanical mismatch between the cathode active material and the solid electrolyte. Higher stack pressure of 14 MPa can effectively mitigate crack formation, as shown in Fig. 5e, enabling 80% capacity retention over 750 cycles at 1 C rate (Supplementary Fig. 18). The rate performance at 1 MPa shown in Fig.5f, however, is nearly identical to that at 14 MPa (Supplementary Fig. 22), suggesting that cycling at low pressure with Li-Mg anode does not lead to significant overpotential increase. Figure 5g–i shows the cross-sectional SEM and EDS images of the anode–electrolyte interface. No voids were found, and uniform Mg distribution was observed in the anode, confirming both morphological and compositional reversibility of Li-Mg. These findings suggest that cell failure at low stack pressure is predominantly cathode-driven. Improving cycle life under such conditions would require zero-strain cathode materials or solid electrolytes capable of reversible deformation to accommodate chemo-mechanical stresses in the composite cathode.
In this work, we investigate the morphological evolution of pure Li and Li-Mg alloy anodes during stripping and plating using operando SEM and high-rate cycling in all-solid-state cells under a stack pressure of 1 MPa. We provide direct evidence that void coalescence is the origin of interfacial delamination in Li metal anodes. In contrast, voids in Li-Mg alloys split into smaller ones that subsequently collapse, thereby preserving interfacial contact. The difference is attributed to the suppressed void formation in Li-Mg alloys. DFT calculations show that vacancies tend to accumulate into voids at Li–argyrodite interface with lithiophobic Li2S as SEI. In Li-Mg, Mg prefers to accumulate at the interface and repels vacancies, suppressing the growth of vacancies into voids and ensuring a uniform plating after stripping. Pressure-dependent voltage profiles of both anodes during stripping further support the link between void formation and the high overpotential that hinders anode utilization. Elevated temperatures promote Mg diffusion into freshly plated Li, restoring compositional homogeneity and mitigating void formation in subsequent cycles. The ability of the Li-Mg anode to recover both morphological and compositional reversibility enables stable full cell cycling, even at a low stack pressure of 1 MPa. Under these conditions, our cells achieve 250 cycles at 2 mA cm−2 and 1.6 mAh cm−2. The real-time visualization of Li–electrolyte interfacial dynamics reported in this work offers a powerful tool for guiding the rational design of solid-state battery interfaces.
Methods
Materials
Li metal foil (99.9%) and Indium ingots (99.99%) were purchased from Alfa Aesar. Li-Mg foil (100 ± 10 µm thick, 10 wt% Mg, Li0.97Mg0.03), Li6PS5Cl solid electrolyte (d50 < 1 µm), and LiNi0.83Mn0.06Co0.11O2 (NMC, d50 = 3−4 µm) was purchased from MSE Supplies. The grain sizes of Li and Li-Mg are both around 100 µm, as shown in Supplementary Fig. 19. Vapor-grown carbon fiber (VGCF, PR-19-XT-HHT) was purchased from Applied Sciences. All materials are processed under an Ar-filled glovebox with O2 and H2O <1 ppm.
Solid-state battery fabrication
About 127 mg of solid electrolyte was first pressed in a Φ12.7 mm (1.267 cm2) polyether ether ketone (PEEK) die at 375 MPa for two minutes. Electrolyte conductivity under such densification condition is 3.5 ± 0.1 mS/cm at room temperature, and 12.4 ± 0.3 mS/cm at 60 °C, with measurement details in Supplementary Fig. 20. The titanium plunger used for pressing was polished with 3000 grit sandpaper until a mirror surface finishing (RMS roughness <0.1 μm). The as-pressed pellet is 500 µm in thickness. For Li–In or Li-Mg–In cells, In ingots are flattened at 200 MPa into a sheet (40 ± 5 mg cm−2, 9.4 mAh cm−2 Li is required to reach LiIn) to avoid phase transition50. An Indium sheet is cut to a Φ12.7 mm disk and pressed onto the solid electrolyte at 150 MPa for 1 min. Li metal foil was rolled to 5.3 mg cm–2 using a stainless steel rod. Li and Li-Mg electrodes of the same thickness (100 ± 10 µm, 6 mg cm–2) were then cut to Φ12.7 mm disks and pressed to the counter side of the pellet at 50 MPa for 30 s. The cell structure is (Li or Li-Mg)|Li6PS5Cl|In. The open-circuit potential of the lithiated indium counter electrode is 0.623 V vs. Li+/Li. For three-electrode testing, both In and Li/Li-Mg disks with a smaller diameter (Φ3.96 mm) is used. A piece of Li metal was placed at the edge of the solid electrolyte pellet as the reference electrode. For full cell fabrication, NMC, ball-milled LPSCl, and VGCF (70:27:3 wt%) are mixed in an agate mortar, then densified with electrolyte at 375 MPa for 5 min. The Li-Mg disk was pressed onto the counter side at 50 MPa. The full cell structure is Li-Mg|Li6PS5Cl|LiNi0.83Mn0.06Co0.11O2.
Operando SEM characterization
As-prepared Li–In or Li-Mg–In cells were removed from the PEEK die and tailored to a 3 mm × 3 mm × 4 mm miniature cell with a razor blade. One side was polished with an Ar+ beam at −30 °C (JEOL, IB-10520CCP), resulting in a 1100 µm × 700 µm flat surface. The miniature cell was loaded into an air-free vessel (Solid Design Instruments, Model FV-003A) shown in Supplementary Fig. 1. A Small stack pressure (0.2 MPa) was applied with a plastic bolt. The air-free vessel was loaded into an SEM (Axia ChemiSEM) and connected to a potentiostat (Biologic SP300) via feedthrough. SEM images were taken every 15 min at the same location. Postmortem analysis shown in Supplementary Fig. 8 was carried out by cutting and polishing the cell after the operando test.
Electrochemical characterization
Solid-state die cells were tested in ambient conditions (25 ± 1 °C) or in an environment chamber (60 °C). The stack pressure was monitored with a force sensor (Autoda) as shown in Supplementary Fig. 21. Long-term cycling was performed with a Landt battery cycler with a stack pressure of 1 and 14 MPa. For the 1 MPa test, silicone rubber disks wrapped in copper foil were used as the current collector to assure uniform pressure distribution. The cycling rate was gradually increased from 0.1 to 1 C during formation cycles. Full cell with NMC was cycled between 2.6 and 4.25 V vs. Li+/Li. Reproducing the cell test is shown in Supplementary Fig. 22. Specific capacity is calculated based on the mass of NMC.
EDS analysis
EDS analysis is performed on an Ar+-polished cross-section to minimize the impact of surface morphology on signal yield. The same working distance, electron gun voltage, and current are used for all EDS samples to enable the semi-quantitative analysis of Mg signal intensity. All samples in Figs. 3, 4 are immediately cooled down to −30 °C to stop Mg diffusion. Samples are imaged within 2 h after −30 °C-polishing to minimize the impact of Mg diffusion during sample preparation.
DFT calculations
The VASP (Vienna Ab initio Simulation Package) was employed to perform the plane-wave DFT calculations51. The generalized gradient approximation (GGA)52 in the Perdew–Burke–Ernzerhof (PBE) flavor53 was used as the exchange-correlation functional. The projector-augmented-wave (PAW) potentials54 were chosen for all elements. A cutoff energy of 500 eV was sufficient to reach the convergence criteria of 10−6 eV for self-consistent electronic minimization and 0.01 eV/Å for force relaxation. A single Gamma point and the Gaussian smearing with a width of 0.1 eV were applied in the calculations. More details are included in Supplementary Information Note 2. The construction of interface structures is included in Supplementary Information Note 3. Atomic coordinates for the optimized structure are available in Supplementary Data 1.
Data availability
All the data generated or analyzed during this study are included in this published article and its Supplementary Information files. Source data are provided with this paper.
References
Janek, J. & Zeier, W. G. Challenges in speeding up solid-state battery development. Nat. Energy 8, 230–240 (2023).
Albertus, P., Babinec, S., Litzelman, S. & Newman, A. Status and challenges in enabling the lithium metal electrode for high-energy and low-cost rechargeable batteries. Nat. Energy 3, 16–21 (2018).
Krauskopf, T., Richter, F. H., Zeier, W. G. & Janek, J. Physicochemical concepts of the lithium metal anode in solid-state batteries. Chem. Rev. 120, 7745–7794 (2020).
Doux, J. et al. Stack pressure considerations for room‐temperature all‐solid‐state lithium metal batteries. Adv. Energy Mater. 10, 1903253 (2020).
Lu, Y. et al. Critical current density in solid‐state lithium metal batteries: mechanism, influences, and strategies. Adv. Funct. Mater. 31, 2009925 (2021).
Ye, L. & Li, X. A dynamic stability design strategy for lithium metal solid state batteries. Nature 593, 218–222 (2021).
Ye, L., Lu, Y., Wang, Y., Li, J. & Li, X. Fast cycling of lithium metal in solid-state batteries by constriction-susceptible anode materials. Nat. Mater. 23, 244–251 (2024).
Han, S. Y. et al. Stress evolution during cycling of alloy-anode solid-state batteries. Joule 5, 2450–2465 (2021).
Kasemchainan, J. et al. Critical stripping current leads to dendrite formation on plating in lithium anode solid electrolyte cells. Nat. Mater. 18, 1105–1111 (2019).
Jolly, D. S. et al. Temperature dependence of lithium anode voiding in argyrodite solid-state batteries. ACS Appl. Mater. Interfaces 13, 22708–22716 (2021).
Wang, M. J., Choudhury, R. & Sakamoto, J. Characterizing the Li-solid-electrolyte interface dynamics as a function of stack pressure and current density. Joule 3, 2165–2178 (2019).
Zhang, X., Wang, Q. J., Harrison, K. L., Roberts, S. A. & Harris, S. J. Pressure-driven interface evolution in solid-state lithium metal batteries. Cell Rep. Phys. Sci. 1, 100012 (2020).
Krauskopf, T., Hartmann, H., Zeier, W. G. & Janek, J. Toward a fundamental understanding of the lithium metal anode in solid-state batteries-an electrochemo-mechanical study on the garnet-type solid electrolyte Li6.25Al0.25La3Zr2O12. ACS Appl. Mater. Interfaces 11, 14463–14477 (2019).
Yang, C.-T. & Qi, Y. Maintaining a flat Li surface during the Li stripping process via interface design. Chem. Mater. 33, 2814–2823 (2021).
Sharafi, A. et al. Surface chemistry mechanism of ultra-low interfacial resistance in the solid-state electrolyte Li7La3Zr2O12. Chem. Mater. 29, 7961–7968 (2017).
Yang, X. et al. Phase evolution of a prenucleator for fast Li nucleation in all‐solid‐state lithium batteries. Adv. Energy Mater. 10, 2001191 (2020).
Otto, S.-K. et al. Storage of lithium metal: the role of the native passivation layer for the anode interface resistance in solid state batteries. ACS Appl. Energy Mater. 4, 12798–12807 (2021).
Singh, D. K. et al. Li6PS5Cl microstructure and influence on dendrite growth in solid-state batteries with lithium metal anode. Cell Rep. Phys. Sci. 3, 101043 (2022).
Ning, Z. et al. Visualizing plating-induced cracking in lithium-anode solid-electrolyte cells. Nat. Mater. 20, 1121–1129 (2021).
Krauskopf, T., Mogwitz, B., Rosenbach, C., Zeier, W. G. & Janek, J. Diffusion limitation of lithium metal and Li–Mg alloy anodes on LLZO type solid electrolytes as a function of temperature and pressure. Adv. Energy Mater. 9, 1902568 (2019).
Siniscalchi, M. et al. On the relative importance of Li bulk diffusivity and interface morphology in determining the stripped capacity of metallic anodes in solid-state batteries. ACS Energy Lett. 7, 3593–3599 (2022).
Yang, C. et al. An electron/ion dual‐conductive alloy framework for high‐rate and high‐capacity solid‐state lithium‐metal batteries. Adv. Mater. 31, 1804815 (2019).
Aspinall, J. et al. The impact of magnesium content on lithium-magnesium alloy electrode performance with argyrodite solid electrolyte. Nat. Commun. 15, 4511 (2024).
Yan, K. et al. Selective deposition and stable encapsulation of lithium through heterogeneous seeded growth. Nat. Energy 1, 16010 (2016).
Huang, Y., Shao, B. & Han, F. Li alloy anodes for high-rate and high-areal-capacity solid-state batteries. J. Mater. Chem. A 10, 12350–12358 (2022).
Pan, H. et al. Carbon-free and binder-free Li-Al alloy anode enabling an all-solid-state Li-S battery with high energy and stability. Sci. Adv. 8, eabn4372 (2022).
Nayeb-Hashemi, A. A., Clark, J. B. & Pelton, A. D. The Li-Mg (lithium-magnesium) system. Bull. Alloy Phase Diagr. 5, 365–374 (1984).
Zhang, Y., Chandran, K. S. R., Jagannathan, M., Bilheux, H. Z. & Bilheux, J. C. The nature of electrochemical delithiation of Li-Mg alloy electrodes: neutron computed tomography and analytical modeling of Li diffusion and delithiation phenomenon. J. Electrochem. Soc. 164, A28–A38 (2016).
Darken, L. S. Diffusion, mobility and their interrelation through free energy in binary metallic system. Trans. AIME 184, 201 (1948).
Zhong, W., Zhang, Q. & Zhao, J.-C. A simple yet general model of binary diffusion coefficients emerged from a comprehensive assessment of 18 binary systems. Acta Mater. 215, 117077 (2021).
Lu, Y. et al. The void formation behaviors in working solid-state Li metal batteries. Sci. Adv. 8, eadd0510 (2022).
Agier, J. A. B., Shishvan, S. S., Fleck, N. A. & Deshpande, V. S. Void growth within Li electrodes in solid electrolyte cells. Acta Mater. 240, 118303 (2022).
Haslam, C. G., Wolfenstine, J. B. & Sakamoto, J. The effect of aspect ratio on the mechanical behavior of Li metal in solid-state cells. J. Power Sources 520, 230831 (2022).
Fang, R. et al. Modeling the electro-chemo-mechanical failure at the lithium-solid electrolyte interface: Void evolution and lithium penetration. J. Mech. Phys. Solids 192, 105799 (2024).
Zhu, Y., He, X. & Mo, Y. Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfaces 7, 23685–23693 (2015).
Schwietert, T. K. et al. Clarifying the relationship between redox activity and electrochemical stability in solid electrolytes. Nat. Mater. 19, 428–435 (2020).
Feng, M., Yang, C.-T. & Qi, Y. The critical stack pressure to alter void generation at Li/solid-electrolyte interfaces during stripping. J. Electrochem. Soc. 169, 090526 (2022).
Feng, M., Liu, X., Harris, S. J., Sheldon, B. W. & Qi, Y. A multiscale model to understand the interface chemistry, contacts, and dynamics during lithium stripping. J. Mech. Phys. Solids 193, 105878 (2024).
Shi, S. et al. Direct calculation of Li-ion transport in the solid electrolyte Interphase. J. Am. Chem. Soc. 134, 15476–15487 (2012).
Deysher, G. et al. Transport and mechanical aspects of all-solid-state lithium batteries. Mater. Today Phys. 24, 100679 (2022).
Sakamoto, J. More pressure needed. Nat. Energy 4, 827–828 (2019).
Wang, M. J., Kazyak, E., Dasgupta, N. P. & Sakamoto, J. Transitioning solid-state batteries from lab to market: linking electro-chemo-mechanics with practical considerations. Joule 5, 1371–1390 (2021).
Masias, A., Felten, N., Garcia-Mendez, R., Wolfenstine, J. & Sakamoto, J. Elastic, plastic, and creep mechanical properties of lithium metal. J. Mater. Sci. 54, 2585–2600 (2019).
Zhao, L. et al. Taming metal–solid electrolyte interface instability via metal strain hardening. Adv. Energy Mater. 13, 202300679 (2023).
Kim, S. H. et al. In situ observation of lithium metal plating in a sulfur-based solid electrolyte for all-solid-state batteries. J. Mater. Chem. A 7, 13650–13657 (2019).
McConohy, G. et al. Mechanical regulation of lithium intrusion probability in garnet solid electrolytes. Nat. Energy 8, 241–250 (2023).
Tan, D. H. S. et al. Carbon-free high-loading silicon anodes enabled by sulfide solid electrolytes. Science 373, 1494–1499 (2021).
Luo, S. et al. Growth of lithium-indium dendrites in all-solid-state lithium-based batteries with sulfide electrolytes. Nat. Commun. 12, 6968 (2021).
Liu, Y. et al. Aluminum foil negative electrodes with multiphase microstructure for all-solid-state Li-ion batteries. Nat. Commun. 14, 3975 (2023).
Santhosha, A. L., Medenbach, L., Buchheim, J. R. & Adelhelm, P. The indium−lithium electrode in solid‐state lithium‐ion batteries: phase formation, redox potentials, and interface stability. Batter. Supercaps 2, 524–529 (2019).
Kohn, W., Becke, A. D. & Parr, R. G. Density functional theory of electronic structure. J. Phys. Chem. 100, 12974–12980 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Ernzerhof, M. & Scuseria, G. E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 110, 5029–5036 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B Condens. Matter 50, 17953–17979 (1994).
Acknowledgements
Y.Y. and Y.Q. acknowledges the US Department of Energy’s Battery 500 Consortium under the Vehicle Technologies Program (No. DE-AC05-76RL01830) as the subcontractor to PNNL (subcontract number 679154). YQ also acknowledges the support from the Assistant Secretary for Energy Efficiency and Renewable Energy, Vehicle Technologies Office of the US Department of Energy, under Contract DE-EE0008863 under the Battery Material Research (BMR) Program.
Author information
Authors and Affiliations
Contributions
Y.Y. directed the project. L.Z. designed the experiment, performed operando SEM characterization, solid-state cell fabrication, and electrochemical tests. M.F. and Y.Q. performed DFT calculations and theoretical analysis. C.W., S.R., and Z.F. assisted with operando SEM characterization. Q.A. and J. L. conducted some postmortem SEM imaging. C.W. and L.G. repeated stripping and cycling tests. Z.C. optimized positive electrode fabrication. L.Z. and M.F. wrote the first draft of the manuscript. L.Z., Y.Q., and Y.Y. revised the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y.Y. has equity interest in LiBeyond, LLC. F.Z. and Y.Y. have equity interests in Solid Design Instruments, LLC. The University of Houston reviewed and approved their relationship in compliance with its conflict-of-interest policy. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Cheol-Min Park and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, L., Feng, M., Wu, C. et al. Imaging the evolution of lithium-solid electrolyte interface using operando scanning electron microscopy. Nat Commun 16, 4283 (2025). https://doi.org/10.1038/s41467-025-59567-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59567-8
This article is cited by
-
Solid-State Lithium Batteries with Liquid Additives: A Critical Review of Progress and Challenges
Electrochemical Energy Reviews (2026)
-
Recent advances in corrosion behaviour and interfacial failure of rubber–metal composites
npj Materials Degradation (2025)
