
Abstract
Rheumatoid arthritis (RA) is characterized by massive intra-articular infiltration of pro-inflammatory macrophages, leading to articular immune dysfunction, severe synovitis, and ultimately joint erosion. Comprehensive remodeling of articular immune homeostasis and bone homeostasis is essential for alleviating RA. Here we report on Spirulina platensis (SP), a natural microorganism commercially farmed worldwide as a food, as an efficient regulator of both synovial inflammation and osteoclast differentiation in male RA mouse models. SP reduces excessive reactive oxygen species and downregulates pro-inflammatory cytokines in synovial macrophages. Moreover, SP reprograms pro-inflammatory M1-like macrophages to anti-inflammatory M2-like phenotype, suppressing synovitis and remodeling redox balance. Notably, SP inhibits osteoclast activation effectively and blocks the progression of bone erosion. SP is then engineered with macrophage membranes (SP@M) to enable immune evasion and RA-targeting in vivo. SP@M increases LC3-mediated autophagy as well as strengthens ubiquitin-mediated proteasomal degradation toward KEAP1, which promotes the expression and nuclear translocation of NRF2. The NRF2 further activates antioxidant enzymes to terminate macrophages-initiated pathological cascades and reestablish intra-articular immune homeostasis, conferring a bone recovery and chondroprotective effect in collagen-induced arthritis mouse models. This work shows the therapeutic activity of FDA-approved functional food of SP in suppressing synovial inflammation and osteoclast differentiation, offering an off-the-shelf strategy for RA treatment.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a devastating systemic autoimmune disease characterized by serious synovitis and bone loss, leading to joint destruction and deformity1,2. It causes substantial health and economic burden to patients and modern society. The therapeutic market worldwide is projected to reach US$33.96 billion by 20253. The primary pathophysiological process of RA involves the abnormal activation of the immune system, leading to synovial inflammation and subsequent bone destruction. Numerous pro-inflammatory cytokines and immune cells are involved in this process4. Among them, synovial macrophages (SMs) are known as the most prominent cells responsible for lesion formation and aggravation. SMs secrete various types of proinflammatory cytokines, overproduce reactive oxygen species (ROS), and then harass the arthritic synovial microenvironment via complicated inflammation networks5,6. The excessive infiltration of SMs into synovia causes articular immune dysfunction, accelerates the progression of synovitis, and leads to osteoclast activation and ultimate bone erosion and joint destruction7,8. Osteoclasts are multinucleated giant cells differentiated from the monocyte-macrophage lineage, responsible for bone resorption and playing a central role in bone homeostasis9. In the pathological environment of RA, due to synovial inflammation, the osteoclast-osteoblast axis is severely disrupted, leading to enhanced osteoclast function and RA-related bone destruction10.
Nowadays, clinical treatment for symptomatic RA faces numerous challenges. The most frequently used treatment relies on the combination of several immunosuppressants such as corticosteroids, leflunomide, and standard disease-modifying antirheumatic drugs (DMARDs) like methotrexate (MTX)11 to address joint inflammation. However, only two-thirds of patients with RA reach satisfactory clinical remission12. Moreover, these immunosuppressive treatments often increase the risk of infections due to the inhibition of the immune system13,14. Managing bone remodeling in RA progression suffers the same difficulty. The indirect control of bone remodeling via inflammation inhibition is ambiguous. For example, although the tight control of synovial inflammation has proven to arrest the progression of bone erosions in some cases (for instance TNF-α inhibitor has a promising effect on mitigating bone erosion in RA)15, the use of corticosteroids and MTX causes even more severe bone destruction in RA patients16,17. Denosumab (anti-receptor activator of nuclear factor-κB ligand antibody) is a RANKL monoclonal antibody that directly inhibits osteoclast-medicated bone resorption by binding to osteoblast-produced RANKL. It is used in another osteoclast-related disease, osteoporosis, but shows a limited effect in clinical use for RA18. This might be attributed to the uncontrolled inflammation persistently activating inflammatory osteoclast formation. To this end, the development of new therapies, ideally targeting synovitis and inflammatory-related bone erosion in joints, as well as regulating those inflammation-independent factors that can also interfere with bone homeostasis in RA, such as the general RANKL signaling pathway, would be promising for remodeling bone homeostasis in RA.
Natural spirulina platensis (SP) is the only microorganism commercially farmed worldwide as a food19. It has been widely used in preclinical therapy for tumors, intestinal diseases, and anemia via oral administration20,21,22. Recent studies have expanded the application of SP via intravenous injection for magnetic actuation and cargo delivery20,23,24, which demonstrated its biocompatibility and biosafety as an intravenous medication. SP has been found to contain multiple functional compounds including polysaccharides, proteins and lipids with potential anti-oxidant, anti-inflammatory and immunomodulatory effects25, making it a great candidate for handling the complicated pathology of RA.
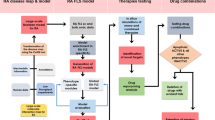
The present study unveils that SP alleviates intracellular oxidative stress and promotes the pro-inflammatory M1-like to anti-inflammatory M2-like repolarization of SMs, remodeling the redox balance and suppressing synovial inflammation (Fig. 1A). It is noteworthy that SP is also efficient in directly inhibiting RANKL-induced osteoclast differentiation, which plays a synergistic role in remodeling bone homeostasis in RA (Fig. 1A). SP can target the macrophages to accelerate the protein degradation of Kelch-like ECH-associated protein 1 (KEAP1) via microtubule-associated protein 1 light chain 3 (LC3)-mediated autophagy and ubiquitin-mediated proteasome degradation. Following the KEAP1 inactivation, the expression and the nuclear translocation of nuclear factor-erythroid 2 p45-related factor 2 (NRF2) are increased, which subsequently promotes the transcription of antioxidant proteins like superoxide dismutase 2 (SOD2) and heme oxygenase 1 (HO-1), and leads to the mitigation of synovial inflammation. The resolution of joint inflammation could inhibit inflammatory cytokines-induced osteoclast activation and facilitate the remission of bone erosion. Importantly, SP is engineered with macrophage cell membranes (SP@M) to enable immune evasion and RA targeting in vivo. The SP@M recovers the balance of synovial microenvironment, and finally achieves bone remodeling and cartilage restoration in collagen-induced arthritis (CIA) mouse models (Fig. 1A). Compared with current therapies for RA, which show success in targeting various aspects of inflammatory pathways, the SP-based therapy in this work targeting both the synovial inflammation and RANKL-induced osteoclast differentiation is more promising as an off-the-shelf therapeutic for the alleviation of RA.

A Schematic illustration of the targeting delivery of macrophage cell membranes-engineered SP (SP@M) toward inflammatory arthritis, which acts as a targeted therapeutic to remodel bone homeostasis in RA by relieving synovial inflammation and independently suppressing osteoclast activation. The SP accelerates the protein degradation of Kelch-like ECH-associated protein 1 (KEAP1) via microtubule-associated protein 1 light chain 3 (LC3)-mediated autophagy and ubiquitin-mediated proteasome degradation. Following the KEAP1 inactivation, the nucleus translocation of transcription factor nuclear factor-erythroid 2 p45-related factor 2 (NRF2) is increased, leading to the mitigation of synovial inflammation and amelioration of RA in collagen-induced arthritis (CIA) mouse models. B The bright-field microscopic image of SP cells. C The scanning electron microscopy image of the single SP cell. D The fluorescence image of SP cells. E UV–vis absorption spectrum of SP cells. For B–E, three independent experiments were repeated.
Results
SP restores synovial homeostasis by inflammatory modulation of macrophages
The prokaryotic and photosynthetic microorganism of spirulina platensis (SP) has a naturally helical morphology and a length of 100–500 μm, as shown by the bright-field microscopy and scanning electron microscopy images (Fig. 1B, C, and Supplementary Fig. 1). Importantly, the autofluorescence of chlorophyll in SP endows it with strong red fluorescence (Fig. 1D), which suggests its potential application in biological imaging. The UV–vis absorption spectrum of SP indicates obvious absorption around 440 and 680 nm, which is attributed to the abundant chlorophyll in SP cells (Fig. 1E). The broad absorption band around 620 nm can be ascribed to the specific phycocyanin present in SP.
The main trigger of articular bone erosion is synovitis, including the overproduction of ROS and proinflammatory cytokines, as well as RANKL, thereby creating a highly inflammatory and tissue-damaging microenvironment of RA. Comprehensive regulation of oxidative stress and production of proinflammatory cytokines is essential for restoring synovial homeostasis. Raw264.7 cells were selected to simulate SMs in vitro, and the inflammatory modulation ability of SP was explored in Raw264.7 cells. Inflammation was induced by the stimulation of lipopolysaccharides (LPS, 100 ng/mL), which led to the generation of inflammatory mediators and oxidative stress, including ROS. The intracellular ROS level was initially investigated by the 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe. It was observed that LPS stimulation brought about the overproduction of ROS, as evidenced by obvious green fluorescence in Raw264.7 cells. However, the fluorescence intensity in cells was significantly decreased following the SP treatment, showing a dose-dependent manner (Fig. 2A, B). Moreover, Real-time reverse transcription polymerase chain reaction (RT-qPCR) analysis revealed a significant up-regulation in the mRNA expression of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), interleukin-1 (IL-1), interleukin-6 (IL-6), and interleukin-12 (IL-12) in LPS-stimulated Raw264.7 cells (Fig. 2C). In contrast, SP treatment resulted in a dose-dependent down-regulation of these pro-inflammatory cytokines, demonstrating the anti-inflammatory effect of SP (Fig. 2C). Macrophages are phenotypically plastic. In the case of RA, they differentiated into a pro-inflammatory M1-like phenotype because of the over-produced ROS and chemotaxis factors. Next, we evaluated the modulatory effect of SP on the phenotypic conversion of Raw264.7 cells using flow cytometry. Most Raw264.7 cells were polarized into the M1-like phenotype upon LPS stimulation, evidenced by the high expression of CD86 (phenotype marker of M1-like macrophages). As shown in Fig. 2D–F, dose-dependent treatment with SP reduced the proportions of M1-like macrophages as well as increased the proportions of anti-inflammatory M2-like macrophages (mannose receptor CD206 as the phenotypic marker of M2-like macrophages), suggesting that SP inhibited the polarization of proinflammatory M1-like macrophages and promoted the differentiation of anti-inflammatory M2-like macrophages. Additionally, RT-qPCR analysis indicated that the mRNA expressions of CD206, interleukin-10 (IL-10), and arginase-1 (Arg-1), commonly recognized markers of M2-like macrophages, were significantly upregulated among all SP treatment groups (Fig. 2G). Overall, the findings supported our hypothesis that SP could regulate oxidative stress and promote reprogramming of macrophages to an anti-inflammatory phenotype, which restricted inflammation in an RA-specific context and restored synovial homeostasis.

A Fluorescence analysis of 2′,7′-dichlorofluorescein diacetate (DCFH-DA) in macrophages stimulated with lipopolysaccharides (LPS, 100 ng/mL) and different doses of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5.0 × 106, 1.0 × 107) for 24 h. Scale bar = 75 μm. B Quantification of relative fluorescence intensity of DCFH-DA shown in panel A. C Real-time fluorescence quantitative PCR (RT-qPCR) analysis of TNF-α, iNOS, IL-1, IL-6, and IL-12 in macrophages stimulated with LPS (100 ng/mL) and different doses of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5.0 × 106, 1.0 × 107) for 24 h. D Flow cytometry of CD86 marker in macrophages stimulated with LPS (100 ng/mL) and different doses of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5.0 × 106, 1.0 × 107) for 24 h. E Flow cytometry of CD206 and CD11b markers in macrophages stimulated with interleukin-4 (IL-4, 20 ng/mL) and different doses of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5.0 × 106, 1.0 × 107) for 24 h. F Quantification of CD86- or CD206-labeled positive cells shown in panels D and E. G RT-qPCR analysis of CD206, IL-10, and Arg-1 in macrophages stimulated with IL-4 (20 ng/mL) and different doses of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5.0 × 106, 1.0 × 107) for 24 h. All data presented as mean (SD) are from three replicates. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc test. The exact p-values were labeled.
SP blocks the progression of bone erosion by suppressing osteoclast activation
Articular bone erosion is a central clinical feature of RA and can occur soon after disease onset. Herein, bone marrow-derived macrophages (BMMs) were employed to investigate the effects of SP on suppressing osteoclast activation in vitro. As shown in Fig. 3A, B, RANKL-induced BMMs showed apparent osteoclastic characteristics, including more tartrate-resistant acid phosphatase (TRAP)-positive cells by TRAP staining and mature osteoclast formation by F-actin staining. In contrast, the number and area of TRAP-positive cells, and the average osteoclast area indicated by F-actin staining were significantly reduced after the SP treatment, showing a dose-dependent manner (Fig. 3A, B). This observation indicated the inhibitory effects of SP on the differentiation of osteoclasts. To further explore the effect of SP on the osteoclastogenesis in BMMs, the expressions of critical osteoclastogenesis-related genes like nuclear factor of activated T cells 1 (NFATc1), c-Fos proto-oncogene (c-Fos), and cathepsin K (CTSK) were analyzed by RT-qPCR. It was found that SP treatment downregulated the expression of these genes in RANKL-induced BMMs (Fig. 3C). Besides, compared with the control group, the protein expression levels of NFATc1, c-Fos, and CTSK in cells after the SP treatment were also suppressed at 0, 1, 3, and 5 days (Fig. 3D, E), suggesting an obvious inhibition of osteoclast formation. Mitogen-activated protein kinase (MAPK) and nuclear factor κB (NF-κB) signaling pathways are the major signal transduction steps in osteoclast differentiation. To clarify these signaling pathways involved in osteoclastogenesis, we pretreated RANKL-induced BMMs with various concentrations of SP for 24 h. Then, Western blotting (WB) analysis was employed to quantify the phosphorylation levels of P65 within the NF-κB signaling pathway, along with those of ERK and P38 in the MAPK signaling pathway. It was observed that SP blocked the phosphorylation of all these proteins (Fig. 3F, G), suggesting suppressed osteoclastogenesis. This result was further verified by the fluorescence staining of phosphorylation of P65 and P38, which showed that SP could inhibit the phosphorylation and translocation of P38 and P65 proteins in RANKL-induced BMMs (Fig. 3H, I). Collectively, these results indicated that SP effectively blocked the differentiation of osteoclasts, thereby restoring bone balance and alleviating the progression of bone erosion in RA. Since MTX, one of the DMARDs used in clinical practice, showed no obvious influences on the differentiation of osteoclasts (Supplementary Fig. 2A–D), the inhibition capacity of SP on both the synovial inflammation and the osteoclast differentiation makes it an ideal candidate for RA treatment.

A Tartrate-resistant acid phosphatase (TRAP) staining and fluorescent F-actin staining of bone marrow-derived macrophages (BMMs) stimulated with macrophage colony-stimulating factor (M-CSF, 30 ng/mL) and receptor activator of nuclear factor-κ B ligand (RANKL, 50 ng/mL), followed by treatment with different doses of SP for 5 days. B Quantification of number and area of osteoclasts in TRAP stain and area of F-actin belt shown in panel A. C RT-qPCR analysis of Nfatc1, C-fos, and Ctsk in BMMs stimulated with M-CSF and RANKL, followed by treatment with SP for 5 days. D Western-blot (WB) analysis of NFATc1, c-Fos, and Ctsk in BMMs stimulated with M-CSF and RANKL, followed by treatment with SP. (For c-Fos, the samples derive from the same experiment and that gels/blots were processed in parallel, and image was cropped at the dotted line only for the purpose of this figure.) E Semi-quantification of greyscale value in NFATc1, c-Fos, and Ctsk in panel D. F WB analysis of phospho-P38, P38, p-ERK, ERK, IκBα, p-P65, and P65 in BMMs stimulated with M-CSF and RANKL, followed by treatment with SP. G Semi-quantification of greyscale value in p-P38/P38, p-ERK/ERK, IκBα, and p-P65/P65 in panel F. H Immunofluorescence analysis of p-P38 and p-P65 in BMMs stimulated with M-CSF and RANKL, followed by treatment with SP. I Quantification of the integrated optical density (IOD)/DAPI of p-P38 and p-P65 as shown in panel H. All data presented as mean (SD) are from three replicates. Statistical analysis was performed using the Kruskal–Wallis test with Dunn’s post hoc test for ordinal data such as osteoclasts number and the one-way ANOVA followed by Tukey’s post-hoc test for continuous data. The exact p-values were labeled.
SP remodels bone homeostasis in vitro via NRF2/KEAP1 signaling pathways
NRF2 is a redox-sensitive transcription factor that codes for versatile cytoprotective proteins, including antioxidant, detoxification, and anti-inflammatory proteins26. Besides, NRF2 acts as a vital transcription factor that regulates bone homeostasis27. The deficiency of NRF2 promotes osteoclast differentiation and osteoclast activity, leading to an increase in bone resorption. In this work, SP was found to remodel bone homeostasis in vitro by alleviating oxidative stress levels, suppressing synovial inflammation, and inhibiting osteoclast differentiation. Given the comprehensive role of NRF2 in stress response, inflammation, and bone metabolism, we thus hypothesized that NRF2 might be involved in the biological activity of SP. To confirm this, the expression of important proteins involved in redox homeostasis, including the NRF2/KEAP1 complex, SODs, and glutathione peroxidase (GPX) enzymes families in both macrophages and RANKL-stimulated BMMs was analyzed by WB. As shown in Fig. 4A, B, SP upregulated the expression of SOD2, HO-1, NRF2, GPX1, and GPX4, while it downregulated the KEAP1 dose-dependently in macrophages. Similarly, SP suppressed the KEAP1, whereas it increased the expression of SOD2, HO-1, NRF2, GPX1, and GPX4 in RANKL-stimulated osteoclasts (Fig. 4C, D). These alterations in protein expression could potentially be ascribed to the intricate regulatory network of the NRF2/KEAP1 complex: Under homeostatic conditions, NRF2 is continuously ubiquitinated and targeted for 26S proteasomal degradation by its negative regulator KEAP1. SP might inactivate KEAP1, which allowed NRF2 to escape ubiquitination, accumulate within the cell, and translocate to the nucleus28. It was found that SP had no impact on the mRNA transcription of Keap1, whereas the transcription levels of Nrf2 and Homx1 were significantly elevated (Supplementary Fig. 3A–C). Thus, SP may function by interfering with the protein degradation process of KEAP1. To verify this hypothesis, the interactions between KEAP1 and NRF2, and the effect of SP on KEAP1 production and NRF2 stabilization were then studied. Given that the degradation of KEAP1 encompasses two distinct pathways, namely autophagy29 and proteasomal degradation30, we employed the protein synthesis inhibitor cycloheximide (CHX), the proteasome inhibitor MG132, and the autophagy inhibitor chloroquine (Chlq) to further explore the underlying mechanism in macrophages (Fig. 4E). As shown in Fig. 4F, G, SP effectively facilitated the degradation of the KEAP1 protein, yet this process could be impeded by MG132, demonstrating that the degradation of KEAP1 is dependent on the proteasome-mediated pathway. Moreover, we transfected 293T cells with Myc-labeled ubiquitin and Flag-labeled KEAP1 plasmids for further ubiquitination assay. It was found that KEAP1 was poly-ubiquitinated, and the ubiquitinated modification level of KEAP1 increased with SP in a dose-dependent manner. This result confirmed that SP could enhance the ubiquitinated modification of KEAP1 (Fig. 4H). Interestingly, we transfected 293T cells with Myc-labeled ubiquitin, HA-labeled KEAP1, and Flag-labeled NRF2 plasmids and observed that the upregulation of KEAP1 promoted the poly-ubiquitination of NRF2, and SP could down-regulate the KEAP1 expression and block the NRF2 ubiquitination dose-dependently (Supplementary Fig. 4). These findings suggested that SP regulated the NRF2 expression mainly via the modulation of KEAP1. Moreover, as shown in Fig. 4I, J, Chlq could also block the SP-mediated degradation of KEAP1, indicating that the autophagy-mediated pathway was also involved. To further verify the autophagy pathway, macrophages were labeled with an RFP-GFP-LC3 probe (LC3 thereafter). It was observed that SP significantly upregulated the number of LC3-labeled autophagosomes (Supplementary Fig. 5). Besides, the protein expression of LC3 increased with SP in a dose-dependent manner, regardless of the existence of Chlq (Supplementary Figs. 6, S7). Moreover, immunofluorescence assays showed that SP decreased the mean fluorescence intensity of KEAP1, whereas it increased the co-localization of KEAP1 and ubiquitin dose-dependently (Fig. 4K, L). Meanwhile, SP enhanced the mean fluorescence intensity of LC3. Moreover, it promoted the co-localization of LC3 with KEAP1 (Fig. 4M, N). These results collectively suggested that SP could promote the engulfment of KEAP1 by autophagosome.

A WB analysis of SOD2, HO-1, NRF2, GPX1, GPX4, and KEAP1 in macrophages stimulated with varying doses of SP. B Semi-quantification of grayscale value in SOD2, HO-1, NRF2, and KEAP1 in panel A. C WB analysis of SOD2, HO-1, NRF2, GPX1, GPX4, and KEAP1 in BMMs stimulated with M-CSF, RANKL, and SP for 24 h. D Semi-quantification of grayscale value in SOD2, HO-1, NRF2, and KEAP1 in panel C. E Illustration of the protein degradation assay of KEAP1 (Created in BioRender. Yang, X. (2025) https://BioRender.com/nriylby). F WB analysis of HO-1, NRF2, KEAP1, and GPX4 in macrophages stimulated with varying doses of SP, cycloheximide (CHX, 50 nM), and MG132 (10 μM) for 8 h. G Semi-quantification of grayscale value in NRF2 and KEAP1 in panel F. H Ubiquitylation analysis of KEAP1 in 293T cells treated with MG132 (10 μM) and varying doses of SP using HA-KEAP1 and Myc-ubiquitin plasmids with HA-tagged beads. Three independent experiments were repeated. I WB analysis of HO-1, NRF2, KEAP1, and GPX4 in macrophages stimulated with varying doses of SP, CHX (50 nM), and chloroquine (Chlq, 25 nM) for 8 h. J Semi-quantification of greyscale value in NRF2 and KEAP1 in panel I. K Immunofluorescence analysis of KEAP1 and ubiquitin in macrophages stimulated with varying doses of SP for 24 h. Scale bar = 20 μm. Zoomed-in scale bar = 5 μm. L Quantification of the mean fluorescence intensity of KEAP1 and the merged color, as shown in panel K. M Immunofluorescence analysis of KEAP1 and LC3 in macrophages stimulated with varying doses of SP for 24 h. Scale bar = 20 μm. Zoomed-in scale bar = 5 μm. N Quantification of the mean fluorescence intensity of KEAP1, LC3, and the merged color, as shown in panel M. All data presented as mean (SD) are from three replicates. Statistical analysis was performed using the one-way ANOVA followed by Tukey’s post-hoc test. The exact p-values were labeled.
Preparation of SP@M and target delivery of SP@M to inflammatory ankle joints in CIA mice
The modulation of immune homeostasis and the blockade of osteoclast differentiation of SP inspired us to evaluate its in vivo performance in RA. The lack of specificity of SP in vivo, however, may lead to adverse effects that must be carefully managed for clinical application. Encouragingly, cell membrane coating provides an effective top-down platform functionalization strategy, producing biomimetic formulations and enabling them to navigate complex biological environments effectively31. In the progression of RA, a large proportion of infiltrating cells are macrophages, which play a pivotal role in RA. Therefore, live macrophages might be an ideal choice for the biomimetic modification of SP due to their immune response and proactive migration to inflammatory sites. Herein, the SP was engineered with macrophage cell membranes (MCMs) to avoid immune clearance and increase the targeted accumulation of the SP at disease sites. To realize the modification of MCMs on SP, MCMs were initially isolated and purified. MCMs were derived from murine macrophage cell line (RAW264.7 cells) by sequential hypotonic lysis, homogenizer extrusion, gradient centrifugation, and low-temperature sonication processes, following established literature protocols31,32 (Fig. 5A). Simultaneously, SP was processed under continuous stirring, resulting in the generation of fragmented SP with a length of 5–100 μm (Fig. 5B, C). After this treatment, the autofluorescence and the viability of SP were still well-maintained (Fig. 5C, Supplementary Fig. 8). The fragmented SP was then filtrated through a 5 μm cell strainer. Afterward, the concentrated SP cells and the isolated MCMs were repeatedly coextruded through a 5 μm porous polycarbonate membrane (as depicted in Fig. 5A), resulting in the generation of MCMs-engineered SP (denoted as SP@M). This fragmentation and membrane modification process did not impair the biological effects of SP (Supplementary Figs. 9, 10). The microstructure of native SP and SP@M was visualized by transmission electron microscopy. Compared with native SP, it is clear that macrophage membranes were partially modified on the SP surface in the SP@M (Fig. 5D, E, and Supplementary Fig. 11). The study conducted by Liu et al. revealed that up to 90% of the biomimetic nanoparticles are only partially coated by cell membranes33, which is in contradiction to the common assumption of perfect coating. The partial coating of SP with macrophage membranes in our study was consistent with the findings reported in previous literature33. The zeta potential of SP@M was more negative than that of native SP, while consistent with the zeta potential of MCMs (Supplementary Fig. 12). Maintaining the proteins of MCMs on the SP surface enables them to effectively navigate complex biological environments. In this regard, the protein composition of SP@M was determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). As shown in Fig. 5F, protein banding patterns of MCMs and SP@M were nearly identical according to the Coomassie blue staining assay, demonstrating the effective translocation of MCMs proteins onto the SP substrates. Meanwhile, the SDS-PAGE assay also indicated that important protein bands, such as TNFR2 (50 kDa), CCR2α (35 kDa), and CD36 (36 kDa) in MCMs, were well maintained in the as-prepared SP@M (Fig. 5G).

A Schematic illustration of the preparation process of SP@M (Created in BioRender. Yang, X. (2025) https://BioRender.com/ndq30fv). B, C The bright-field and fluorescence microscopy images of SP@M. D, E Transmission electron microscopy images of SP (D) and SP@M (E). F Protein composition of SP, MCMs, and SP@M was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining. G WB analysis of TNFR2, CCR2α, and CD36 in SP, MCMs, and SP@M groups. H In vivo fluorescence imaging of CIA mice after intravenous administration of Cy5-labeled SP or Cy5-labeled SP@M (100 μL, at an SP concentration of 5.0 × 107 cells/mL). Low = 5*107. High = 5*108. I The time-dependent quantitative data of in vivo fluorescence signals in paws, and the quantitative data of fluorescence signals in limbs and other tissues at 24 h. All data presented as mean (SD) are from three biological replicates. Statistical analysis was performed using the one-way ANOVA followed by Tukey’s post-hoc test. The exact p-values were labeled. For B–G, three independent experiments were repeated.
The collagen-induced arthritis (CIA) mouse model (at both paws of the mouse) was established in vivo. The targeting ability of SP@M toward the inflammatory ankle joints of CIA mice was then investigated. Mice were exposed to intravenous injection of Cy5-labeled SP and Cy5-labeled SP@M, respectively, and then subjected to in vivo imaging. As shown in Fig. 5H, I, within the initial 0.5 h, fluorescence signals were detected in the paws of the SP@M group. This clearly indicated that SP@M rapidly accumulated at the inflammatory sites of RA. Additionally, even after 24 h, the fluorescence signals in the paws of the SP@M group remained strongly maintained. In contrast, the fluorescence signals in the paws of SP group were weak during the 24 h observation. The enhanced targeting efficiency of SP@M was likely ascribed to the functional proteins on the MCMs, which showed an inherent immune affinity for the inflammation in RA. The study conducted by Liu et al. indicated that partially coated NPs could still be internalized by the target cells33. The in vivo imaging of SP@M further validated the successful modification of MCMs and the targeting ability of SP@M toward the inflammatory sites.
SP@M enables synovial homeostasis, bone recovery, and chondroprotection in vivo
Next, we investigated the in vivo therapeutic efficiency of SP and biomimetic SP@M using the abovementioned CIA mouse model (Fig. 6A). The CIA mice showed typical RA symptoms and developed severe swelling and erythema in paws (Fig. 6B). Following this, saline, free MCMs, SP, and SP@M were then intravenously injected into CIA mice every three days. MTX, one of the standard DMARDs for RA in clinics, was used as the gold-standard treatment group (Fig. 6A, B). A sham intervention was also carried out as a sham control. The degree of paw swelling was observed over time as a direct indicator of RA. As shown in Fig. 6B, C, the paw swelling thickness was increased over time in the saline group. Besides, MCMs and SP showed no obvious reduction in paw inflammation and edema. In contrast, SP@M markedly ameliorated paw swelling and suppressed the severity of RA by the end of the treatment period, achieving better results than MTX (Fig. 6B, C). The therapeutic efficacy of SP@M was further quantitatively evaluated using a clinically tested scoring system. The SP@M group obtained a significantly lower score than the MTX group, indicating its superior therapeutic efficacy (Fig. 6C). Bone erosion is a central feature of RA and is associated with disease severity. Accordingly, the rate of bone erosion in each group was determined by micro-computed tomography analysis (Fig. 6D). For bone surface erosion, we could see that SP@M could considerably inhibit bone erosion in the ankle and toe joints of CIA mice, which is even better than that in the MTX group. In contrast, SP could only partially recover the bone erosion while MCMs showed no effect. Quantitative analyses of the erosion index demonstrated that the SP@M treatment showed the best bone erosion recovery (Fig. 6E). Moreover, the significantly higher bone volume/total volume (BV/TV) of talus bone levels after the SP@M treatment further suggested its superior performance to ameliorate the RA-induced bone degradation (Fig. 6D, E). Furthermore, the functional assessments of lower limbs of mice, including the reflexive mechanical pain-related responses experiment34 and thermal pain-related responses experiment35, were conducted using the von Frey test and hot plate test, respectively. The standard response value for the von Frey test is 5.0–10.0 g, and the normal response value for the thermal test is 5–9 s. As shown in Fig. 6F, the response value of CIA mice in both the saline and the MCMs groups decreased to approximately 2.0 g in the von Frey test and to 4 s in the hot plate test, indicating a pain-sensitive phenotype of chronic inflammation. Encouragingly, the SP@M and MTX treatments could mitigate these pains and increase the threshold and latency of pain reflection, respectively (Fig. 6F). Additionally, gait analysis was carried out using a CatWalk system to assess the gait performance in mice. The gait parameters, including maximum contact area and print area, were collected to analyze the walking stability. A smaller maximum contact and print area of the four limbs, manifested as a shorter stance phase and a longer swing phase when the mice make contact with the glass plate, generally indicates that the mice avoid putting weight on their affected limbs due to pain36. Compared to the saline, MCMs, and SP groups, the SP@M treatment could satisfactorily increase the maximum contact and print area, even higher than that of the MTX treatment (Supplementary Fig. 13A–C).

A Scheme of the collagen-induced arthritis (CIA) mouse model and relative therapy strategy (Created in BioRender. Yang, X. (2025) https://BioRender.com/bab9l0q). n = 6, biological repeats. B Gross photo of right lower limb in mice shown in panel A. Scale bar = 2 cm. C Quantification of the mean paw thickness and clinical score of mice shown in panel A. D Micro-computed tomography analysis of right ankle joint and focused talus bone in mice shown in panel A. Scale bar = 1 mm. Zoomed-in scale bar = 500 μm. E Quantification of erosion index and bone volume/total volume (BV/TV) of talus bone in mice shown in panel C. F Quantification of threshold of mice in mechanical and thermal pain-related responses experiments shown in panel A. G Hematoxylin and eosin (HE) stain (scale bar = 50 μm), tartrate-resistant acid phosphatase (TRAP) stain (scale bar = 50 or 25 μm), and Safranin O/Fast green (SO/FG) stain (scale bar = 200 μm) of focused area in ankle joints of mice shown in panel A. *Tibia bone. H Quantification of ‘SMASH’ score (synovitis, cartilage destruction, and bone erosion) of mice paraffin section shown in panel G. I Quantification of number of TRAP positive osteoclasts in mice paraffin section shown in panel G. J Immunohistochemistry stain of TNF-α in the focused area in ankle joints of mice shown in panel A. Scale bar = 25 μm. K Quantification of relative integrated optical density (IOD) value of TNF-α in ankle joints of mice shown in panel J. All data presented as mean (SD) are from six biological replicates. Statistical analysis was performed using the Kruskal–Wallis test with Dunn’s post hoc test for ordinal data such as osteoclasts number, erosion index, SMASH scoring, and the one-way ANOVA followed by Tukey’s post-hoc test for continuous data. The exact p-values were labeled.
Then, the histopathological analysis was performed and scored according to the Standardized Microscopic Arthritis Scoring of Histological sections (‘SMASH’). According to the hematoxylin and eosin (HE) staining, tartrate-resistant acid phosphatase (TRAP) staining, and Safranin O/Fast green (SO/FG) staining, the ankle joint tissues in the saline group exhibited extensive inflammatory cell infiltration, synovial hyperplasia, severe bone erosion, and cartilage destruction (Fig. 6G, H). The SP@M treatment markedly reduced synovitis, mitigating SMs infiltration in the synovium. Besides, bone surface erosion, talus bone destruction, and articular cartilage degeneration were significantly recovered in the ankle joint, suggesting the bone recovery and chondroprotection roles of SP@M (Fig. 6G, H). Moreover, the number of osteoclasts in the inflammatory area was significantly reduced in the SP@M group compared to the saline group, demonstrating a promising osteoclast inhibition effect of SP@M in vivo (Fig. 6G, I). Since proinflammatory cytokines play an essential role in re-balancing immune cells and osteoclasts, we subsequently evaluated the proinflammatory cytokine expression of TNF-α by immunohistochemistry. As shown in Fig. 6J, K, the expression of TNF-α was significantly elevated in the saline group, whereas the positive cells labeled by TNF-α were decreased considerably after the SP@M treatment. This result was consistent with the alleviated synovial inflammation and attenuated cartilage erosion in the SP@M group (Fig. 6G, H). Moreover, Cathepsin K (CTSK), a well-known key enzyme highly expressed in the bone resorption process in RA, was analyzed to mark the osteoclast differentiation. The expression of CTSK was upregulated in the saline group, while the SP@M treatment effectively suppressed this upregulation (Supplementary Fig. 14). Overall, these findings suggested that SP@M showed a promising rescuing effect on RA in vivo by enabling synovial homeostasis, bone recovery, and chondroprotection. More importantly, the alleviation effect of SP@M in the CIA mouse model was comparable to, or even better than that of MTX used in clinics. The hemolytic assay revealed that SP and SP@M did not show hemolytic activity (Supplementary Fig. 15). The histopathological analysis indicated that major organs after the SP or SP@M treatment remained normal tissue structures without obvious inflammatory lesions or damage (Supplementary Fig. 16). Moreover, the SP@M exerted no significant influence on mice serum based on the biochemical analysis (Supplementary Fig. 17). It is reasonable to infer that SP@M is biosafe in vivo as an intravenous medication and is promising for future application in clinics.
To explore the underlying mechanism of SP@M in alleviating RA, the synovium and proliferated inflammation tissue around the joints in upper limbs in the saline group and SP@M group were isolated, respectively, for RNA sequencing analysis (Supplementary Fig. 18A). An initial investigation was conducted on the whole-transcriptome changes in these two groups. The differentially expressed genes (DEGs) were identified and shown in the volcano plot (Supplementary Fig. 18B). Compared to the saline group, 1907 genes were upregulated and 1765 genes were downregulated in the SP@M group. The Gene Ontology (GO) analysis showed that these DEGs were involved in multiple mitochondrial respiration-related processes, including respiratory chain complex I and electron transport (Fig. 7A). Moreover, GO analysis of the cellular component also showed that mitochondrion, mitochondrial inner membrane, mitochondrial matrix, and mitochondrial respiratory complex I were enriched in the top 20 cellular components, which suggested the regulation of cellular homeostasis (Supplementary Fig. 18C). In accordance with this, oxidative phosphorylation was enriched according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Fig. 7B) and the gene set enrichment analysis (GSEA) (Fig. 7C), confirming the oxidative redox regulation after the SP@M treatment. Importantly, the osteoclast differentiation signaling pathway was also involved according to the KEGG analysis (Fig. 7B), which was consistent with the inhibitory effect of SP@M on osteoclast differentiation. Moreover, “the immune system process”, “inflammatory response” and “negative regulation of MAP kinase activity” were also involved in the biological processes via GO analysis (Fig. 7A). Collectively, following the SP@M treatment, oxidative stress, inflammatory response, and osteoclast differentiation were regulated, which restored the osteoimmunology homeostasis for RA alleviation.

A Gene Ontology (GO) analysis of biological processes of the saline and SP@M groups (n = 3, biological repeats). B Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis between the saline and SP@M groups. C Gene set enrichment analysis (GSEA) of oxidative phosphorylation signaling pathway between the saline and SP@M groups. D Immunofluorescence analysis of KEAP1 and NRF2 in the focused area of ankle joints in RA mice paraffin section. Ta: Talus bone, Tb: Tibia bone, *synovium. Scale bar = 500 μm. E Integrated optical density (IOD)/DAPI quantification of KEAP1 and NRF2 as shown in panel D. F Immunofluorescence analysis of KEAP1, ubiquitin, and LC3 in the focused area of ankle joints in RA mice paraffin section. Ta Talus bone, Tb Tibia bone, *synovium. Scale bar = 500 μm. Zoomed-in scale bar = 25 μm. Six independent biological experiments were repeated. G SP@M regulates the macrophages and osteoclasts to suppress synovial inflammation and osteoclast differentiation, leading to inflammation antagonism, bone protection, and cartilage recovery in joints of RA mice in vivo. Mechanistically, SP@M suppresses KEAP1 via LC3-mediated autophagy and ubiquitin-mediated proteasomal degradation, promoting the nuclear translocation of NRF2 for antioxidant system activation (Created in BioRender. Yang, X. (2025) https://BioRender.com/vdfpv0u). All data presented as mean (SD) are from six biological replicates. Statistical analysis was performed using the one-way ANOVA followed by Tukey’s post-hoc test. The exact p-values were labeled.
Immunofluorescent staining of tissues was used to further clarify the specific remodeling of inflammatory response. The SP@M treatment led to a significant decrease in the fluorescence intensity of CD86 (the phenotypic marker of M1-like macrophages) and an increase in the intensity of CD206 (the phenotypic marker of M2-like macrophages), indicating the remission of pro-inflammatory microenvironment (Supplementary Fig. 19A, B). Following the re-balance of the macrophage phenotype, we found that SP@M could inhibit the upregulated KEAP1 signal intensity and recover the decreased NRF2 signal intensity, compared to the saline group (Fig. 7D, E). Further immunofluorescence assay showed that SP@M upregulated the co-localization of ubiquitin to KEAP1 (Fig. 7F). Meanwhile, the interaction of LC3 to KEAP1 was increased after the SP@M treatment (Fig. 7F), which was in good agreement with the in vitro results.
Discussion
Significant progress in targeted therapy of RA has led to improved quality of life and outcomes for RA patients. Despite this, a notable percentage of affected individuals still experience persistent inflammation and progressive disability, clinically manifested as synovial inflammation and joint damage primarily. Unmet needs include partial response and non-response to treatment in many patients, failure to achieve immune homeostasis or drug-free remission, and inability to repair damaged tissues. Nowadays, the development of drug delivery systems has brought new insights into RA treatment. A variety of carriers based on bioengineered probiotics, dendrimers, polymersomes, and nanocarriers (such as nanoparticles, nanocapsule, nanoliposome, and nanopolymer) have been explored to deliver cargos (such as small molecule drugs, peptides, miRNA, siRNA, and plasmid DNA) for the treatment of RA and autoimmune diseases37,38,39,40,41,42,43,44,45. These anti-RA drug delivery systems demonstrated the critical role of anti-inflammation and immunomodulation for effective RA treatment and underlined the importance of SMs-targeting therapy. For instance, Zhang et al. constructed an innovative macrophage-hitchhiking IL-10 pDNA delivery system, which could effectively accumulate at inflammatory sites of RA and alleviate symptoms of inflammation in vivo by inducing the re-polarization of macrophages45. Wang et al. found a targetable method for M1-like to M2-like repolarization via clearance of cell-free DNA in RA treatment46.
Apart from SMs, osteoclast represents another joint-specific pathogenic cell, which is responsible for bone resorption in RA. However, current treatments targeting osteoclasts still face a lot of challenges for clinical usage and this therapy still needs long-term observation. For example, denosumab, a RANKL mono-antibody, can hardly control the inflammation of RA in clinical practices18. Interfering with the metabolism in osteoclasts showed promising curing effects for RA but only on animal models for pre-clinical usage47. Emerging therapies like certolizumab pegol (Cimzia) focus on the inflammatory cytokines to co-regulate the bone and immune environment and achieve dramatic effects in clinical trials48. Recent research has indicated that treatment focused on only the bone or the inflammation is not enough for the treatment of RA49. Systematic management of synovial inflammation and concurrent and independent regulation of osteoclast activation are highly desirable for remodeling bone homeostasis in RA. However, no therapy has yet been reported.
In our study, we explored the innate function of SP toward SMs and osteoclasts, and offered a targeted therapy of RA by a macrophage-cell-membrane-cloaked Spirulina platensis (denoted as SP@M) (Fig. 1A). SP is a natural microorganism and is widely cultured around the world. It is a rich source of protein and several fatty acids, vitamins, and minerals, making it a valuable superfood resource and dietary supplement. SP has been reported to function in reducing cholesterol and regulating blood sugar and is widely used in preclinical therapy for tumors. We unveiled that SP could remodel bone homeostasis in RA by simultaneously targeting SMs and osteoclasts in vitro. On one hand, it could regulate oxidative stress and promote the reprogramming of SMs to an anti-inflammatory phenotype, which restricted inflammation in an RA-specific context and restored synovial homeostasis (Figs. 1 and 2). On the other hand, it effectively blocked the differentiation of osteoclasts, thereby restoring bone balance and alleviating the progression of bone erosion in RA (Figs. 1 and 3).
How to efficiently deliver SP to inflammatory lesions in RA in vivo is another problem that urgently needs to be solved. Biomimetic engineering techniques with immune cell membranes are now receiving more and more attention for the advantages of drug delivery50. Camouflaging with cell membranes could fully utilize the ligands, such as antibodies and peptides, to enhance specific binding to receptors overexpressed in the tumor and inflammation51. These cell membranes exert multiple functions, such as immune evasion, inflammation/tumor targeting, and antigen presentation52. For example, Gao et al. applied macrophage membrane-coated nanoparticles for tumor immunotherapy with great targetable effect53, Su et al. developed stem cell membrane-coated micro-ribbon scaffolds for bone defect regeneration due to the abilities of cell chemotaxis, adhesion, and collection54.
In the progression of RA, SMs are dramatically increased in the synovium, and they invade joints and proliferate in cartilage and bones, causing synovium swelling, cartilage destruction, and bone erosion55. Macrophages are dynamic cells and are frequently used as a “ship” for chemotaxis toward inflammation and tumors56,57. In this work, SP was engineered with macrophage cell membranes (MCMs) to enhance the inflammation-targeting ability of SP in the synovial microenvironment (Fig. 5). This MCMs engineering was expected to help camouflage the antigenicity of SP to improve the pharmacokinetics of SP@M and strengthen its bioavailability in in vivo RA treatment. Consequently, SP@M could domesticate SMs into an anti-inflammatory phenotype of M2-like, and suppress synovial inflammation in vivo (Fig. 6). Remarkably, this inflammation-targeting SP@M restored osteoimmunology homeostasis, inhibited the activation of osteoclasts, protected bone formation, and promoted cartilage recovery in CIA mouse models in vivo (Fig. 6). Notably, the alleviation effect of RA in the mouse model by SP@M was comparable to, or even better than that by MTX used in clinical practice (Fig. 6). Since MTX showed no obvious influence on osteoclasts’ differentiation (Supplementary Fig. 2), the inhibition capacity of SP on both synovial inflammation and osteoclast differentiation made it an ideal candidate for RA treatment.
SP possesses high biocompatibility and is widely used as a carrier to deliver materials or drugs into the focus of disease58. Its effects relied on the “cargo on the ship”. Differently, the SP alone in our work acted as a missile attacking the inflammatory lesion of RA and led to bone homeostasis remodeling. We wonder how SP exerts its biological functions. We initially compared the therapeutic effects of live SP and inactivated SP (including SP powder and boiled SP), trying to clarify whether SP needs to be biologically active to exert its therapeutic effects. Unlike the live SP, which displayed remarkable antioxidant effects and osteoclastogenesis-inhibitory properties, both SP powder and boiled SP lost these biological functions (Supplementary Figs. 9, 10). It is likely that SP functions by secreting substances or producing metabolic byproducts. Exosomes are extracellular vesicles (EVs) that are mainly composed of nucleic acids, proteins, lipids, amino acids, and metabolites. They are secreted by all cells59. Exosomes could mediate the substance exchange between cells, thereby affecting the biological properties and activities of recipient cells59. We therefore hypothesized that SP exerted its biological effects through the secretion of exosomes. To verify this hypothesis, we proceeded to extract SP-derived exosomes (as shown in Supplementary Fig. 20) and then evaluate their biological properties. Remarkably, these SP-derived exosomes (denoted as SP EVs) showed similar anti-inflammatory and anti-osteoclastogenic properties when compared with the live SP (Supplementary Figs. 9, 10). Consequently, it can be inferred that SP might exert its therapeutic effects in part through the secretion of exosomes. The more exact mechanisms will be further explored in our future studies.
Mammalian cells have evolved elaborate mechanisms for restoring redox homeostasis and resolving acute inflammatory responses. Among them, NRF2/KEAP1 is a classical signaling pathway to regulate the redox balance in cells26. KEAP1 modifies NRF2 via specific cysteinyl residues, leading to the ubiquitination of NRF2 for proteasomal degradation. Under oxidative stress, NRF2 was released from KEAP1, translocated into and accumulated in the nucleus to induce expression of its target genes, and increased the expression of antioxidant enzymes like SODs and HOs families60. This redox stress-sensing adaptive response system has been extensively studied in terms of molecular mechanism and biological significance61. It is a ubiquitous, evolutionarily conserved intracellular defense mechanism, a master regulator of cytoprotective enzymes that regulates redox metabolism, inflammation, bone remodeling, and detoxification in multiple auto-immune diseases like inflammatory bowel disease and asthma62. Indeed, we observed increased KEAP1 and decreased NRF2 in both macrophages and RANKL-induced osteoclasts, as well as in ankle joints of CIA mouse models, indicating an unbalanced redox environment (Fig. 6). It has been demonstrated that NRF2 over-activation may be therapeutically beneficial for ameliorating RA owing to its anti-inflammatory and anti-oxidative activity associated with limiting cytokine production, cartilage-matrix degradation, and bone resorption63. In this work, we found that SP and SP@M could down-regulate the expression level of KEAP1 in vitro (Fig. 4) and in vivo (Fig. 7), which played a key role in SP@M-mediated RA alleviation. Specifically, SP and SP@M increased the LC3-mediated autophagy as well as strengthened the ubiquitin-mediated proteasomal degradation toward KEAP1 (Figs. 4, 7). Protein degradation is a complicated post-translational process in cells. Directly regulating the protein expression level could be beneficial in treating diseases64. For example, a lysosome-targeting chimera was created to engulf membrane protein65. Our previous work also revealed that proteasomal modification of GPX4 and autophagy to AGEs is helpful in managing intervertebral disc degeneration66. In this work, the post-translational modification toward KEAP1 was observed, which subsequently promoted the expression and nuclear translocation of NRF2. The NRF2 further activated the antioxidant enzymes like SODs and GPXs to recover bone homeostasis and suppress the progression of RA both in vitro and in vivo (Fig. 7G).
Natural Spirulina platensis (SP) was found to act as an off-the-shelf therapeutic to remodel bone homeostasis in RA by simultaneously suppressing synovial inflammation and osteoclast activation. SP regulated oxidative stress and reprogrammed SMs to an anti-inflammatory M2-like phenotype, which restricted inflammation in an RA-specific context and restored synovial homeostasis. Notably, it effectively blocked the differentiation of osteoclasts, thereby restoring bone balance and alleviating the progression of bone erosion in vitro. We engineered SP with macrophage cell membranes (MCMs) to construct an inflammation-targeting “missile” (SP@M) to attack the inflammatory lesion in vivo. It was unveiled that SP@M downregulated the expression of KEAP1 via LC3-mediated autophagy and ubiquitin-mediated proteasomal degradation, which subsequently promoted the accumulation and nuclear translocation of NRF2 to activate the antioxidant systems. Consequently, SP@M restored the osteoimmunology homeostasis, suppressed the activation of osteoclasts, protected bone formation, and promoted cartilage recovery in CIA mouse models in vivo. This work offers insights for RA treatment by targeting synovial inflammation and osteoclast differentiation via FDA-approved and natural microalgae, showing promise for clinical application in RA treatment.
Methods
Cell cultures
The Raw264.7 cell line is immortalized cells bought from Shanghai Fuheng Biological Company (cat. No. FH0328). Dulbecco’s modified Eagle’s medium (DMEM) with 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) was used for the culturing of the Raw264.7 cells.
Primary bone marrow‑derived macrophages (BMMs) cells were extracted from the whole bone marrow of the tibia and femur bone in male 6-week-old C57BL/6 mice (GemPharmatech, Nanjing, China). Then, cells were cultured in minimal essential medium alpha (α-MEM) supplied with 30 ng/mL macrophage colony-stimulating factor (M-CSF; cat. no. 416-ML; R&D Systems, Minneapolis, MN, USA), 10% FBS, and 1% penicillin–streptomycin. Both cells were cultured in an incubator at 37 °C and 5% CO2.
Intracellular ROS detection
Raw264.7 cells (30-40% confluence) seeded onto a confocal dish were stimulated with lipopolysaccharide (LPS; 100 ng/mL; cat. no. S1732; Beyotime Institute of Biotechnology, China) and treated via different concentrations of spirulina (SP, cells/mL, 1.2 × 106, 2.5 × 106, 5.0 × 106, 1 × 107) for 24 h. Then, these cells were incubated with 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; 10 μM; 30 min; Beyotime Institute of Biotechnology, China), and the ROS level was analyzed per the manufacturer s protocol. Finally, the pictures were captured using a Leica DM4000 B epifluorescence microscope (Leica Microsystems, Wetzlar, Germany), and the relative fluorescence intensity was calculated using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Quantitative real-time PCR analysis
Raw264.7 were cultured in six-well plates at the density of 3 × 105 cells/well. Then these cells were stimulated to M1-like (LPS; 100 ng/mL) or M2-like (Interleukin-4; IL-4; 20 ng/mL; Beyotime Institute of Biotechnology, China) polarization and treated via different concentrations of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5 × 106, 1 × 107) for 24 h.
For Keap1, Nrf2, and Homx1 detection, Raw264.7 were cultured in six-well plates at a density of 3 × 105 cells/well. Then, these cells were treated with different concentrations of SP (cells/mL, 6.2 × 105, 1.2 × 106, 2.5 × 106, 5 × 106, 1 × 107) for 24 h.
Primary BMMs were seeded onto six-well plates at a density of 3 × 105 cells/well. Then, these cells were stimulated to the osteoclast phenotype with 30 ng/mL M-CSF and 50 ng/mL receptor activator of nuclear factor-κB ligand (RANKL; R&D Systems, Minneapolis, MN, USA) and treated by SP (2.5 × 106 cells/mL) or MTX (4 μM) for 0, 1, 3, and 5 days.
For RNA extraction, TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) was used to isolate total RNA from both cells. The complementary DNA (cDNA) was reverse transcribed from the obtained total RNA using the cDNA Synthesis Kit. RT-qPCR was performed using the TB Green Premix Ex Taq Kit on an Applied Biosystems QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) per the following conditions: Denaturation at 95 °C for 30 s; 40 cycles of 95 °C for 3 s and 60 °C for 34 s; and then 95 °C for 15 s, 60 °C for 60 s and finally, 95 °C for 15 s. Specific primer pairs were designed using NCBI blast, and the sequences are provided in Supplementary Table 1. β actin gene expression was used as the internal control. Target gene expression levels were determined using the 2−ΔΔCq method.
Flow cytometry analysis
Raw264.7 were cultured in six-well plates at the density of 3 × 105 cells/well. Then these cells were stimulated to M1-like or M2-like polarization and treated via different concentrations of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5 × 106, 1 × 107) for 24 h. Next, cell suspensions were prepared and blocked in a 0.3% bovine serum albumin (BSA) solution, incubated with primary antibodies (including PE-CD86, APC-CD206, and FITC-CD11b; BD Biosciences, USA) for 30 min, washed with fresh 0.3% BSA, and applied to flow cytometry analysis on an LSRFortessa flow cytometer (BD Biosciences, USA) to count at least 10 000 events. Finally, the FlowJo software (Version 10.4, BD Biosciences, USA) was used to analyze the result with a gating strategy shown in Supplementary Fig. 21.
TRAP staining assay
Primary BMMs were seeded onto 96-well plates at the density of 8 × 103 cells/well (or onto a 24-well plate at the density of 2 × 104 cells/well). Then these cells were stimulated to osteoclast phenotype with 30 ng/mL M-CSF and 50 ng/mL RANKL and treated via different concentrations of SP (cells/mL, 6.2 × 105, 1.2 × 106, 2.5 × 106, 5 × 106, 1 × 107) or different concentrations of MTX (μM; 0.25, 0.5, 1, 2, 4) for 5–7 days until the matured osteoclasts could be observed in the plates. Then, plates were washed with phosphate-buffered saline (PBS) 3 times and fixed with 4% paraformaldehyde for 15 min. Then, tartrate-resistant acid phosphatase (TRAP) staining was applied immediately at 37 °C for 30 min at dark. Images were captured using an optical microscope (Olympus, Tokyo, Japan). The osteoclasts were TRAP-positive cells with more than three nuclei, and the area and number of these osteoclasts were quantified using the ImageJ software (NIH, Bethesda, MD, USA).
Immunofluorescence of podosome actin belt
Primary BMMs were seeded onto 24-well plates at the density of 2 × 104 cells/well. Then these cells were stimulated to osteoclast phenotype with 30 ng/mL M-CSF and 50 ng/mL RANKL and treated via different concentrations of SP (cells/mL, 6.2 × 105, 1.2 × 106, 2.5 × 106, 5 × 106, 1 × 107) or different concentrations of MTX (μM; 0.25, 0.5, 1, 2, 4) for 5–7 days until the matured osteoclasts could be observed in the plates. Then, plates were washed with PBS, fixed with 4% paraformaldehyde, permeabilized by 0.2% Triton X-PBS solution, repeatedly washed by PBS, and stained with FITC-labeled phalloidin to bounds with the F-actin ring in cells for 1 h in darkness. The nuclei were stained at RT for 10 min without light by 4′, 6-diamidino-2-phenylindole (DAPI). Images were captured using a Leica DM4000 B epifluorescence microscope.
Protein extraction and western blot (WB) analyses
Primary BMMs were seeded onto six-well plates at a density of 3 × 105 cells/well. Then, these cells were stimulated to the osteoclast phenotype with 30 ng/mL M-CSF and 50 ng/mL RANKL and treated by SP (2.5 × 106 cells/mL) or MTX (4 μM) for 0, 1, 3, 5 days; or 0, 10, 20, 40 min, or 24 h.
Raw264.7 were cultured in six-well plates at a density of 3 × 105 cells/well. Then these cells were treated via different concentrations of SP (cells/mL, 6.2 × 105, 1.2 × 106, 2.5 × 106, 5 × 106, 1 × 107) for 24 h.
Total cellular proteins were extracted from cultured cells using a RIPA lysis buffer supplemented with phosphatase and protease inhibitors (Roche, Basel, Switzerland). and the content of proteins was quantified with BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). The exact amount of proteins was resolved on 4–20% SDS–PAGE gel for separation and was electroblotted onto 0.22-μm PVDF membranes (Merck-Millipore, CA, USA). Membranes were blocked with 5% skimmed milk-PBS at room temperature for 1 h and then incubated overnight at 4 °C with primary antibodies (diluted 1:1000 in 5% BSA-TBST). Primary antibodies included NFATc1 (cat. no. ab2796; mouse mAb), c-Fos (cat. no. 9F6; rabbit mAb), CTSK (cat. no. E7U5N; rabbit mAb), phospho-P38 (cat. no. Thr180/Tyr182, D13.14.4E; rabbit mAb), P38 (cat. no. D13E1; rabbit mAb), phospho-ERK (cat. no. Thr202/Tyr204, and D3F9; rabbit mAb), ERK (cat. no. 137F5; rabbit mAb), IκBα (cat. no. L35A5; mouse mAb), phospho-P65 (cat. no. Ser536, 93H1; rabbit mAb), P65 (cat. No. D14E12; rabbit mAb), SOD2 (cat. no. 10701-1-AP; rabbit mAb), HO-1 (cat. no. 10701-1-AP; rabbit mAb), NRF2 (cat. no. 80593-1-RR; rabbit mAb), KEAP1(cat. no. 80744-1-RR; rabbit mAb), GPX1 (cat. no. ab22604; rabbit mAb), GPX4 (cat. no. ab125066; rabbit mAb), HA-tag (cat. no. C29F4; rabbit mAb), Ubiquitin (cat. no. 10201-2-AP; rabbit mAb), LC3 (cat. no. #4108; rabbit mAb), and β-actin (cat. no. D6A8; rabbit mAb). Membranes were washed three times using Tris-buffered saline-Tween20 (TBST) and subsequently incubated with anti-rabbit or anti-mouse IgG (H + L; DyLight™ 800 4 × PEG conjugate; Cell Signaling Technology, Danvers, MA, United States) secondary antibody (1:5000 dilution) for 1 h at RT in the dark. Membranes were washed extensively in TBST, and protein immunoreactivity was detected on a LI-COR Odyssey fluorescence imaging system (LI-COR Biosciences, Lincoln, NE, USA). The gray values of each protein were measured using Image J software (National Institutes of Health, United States). The original scans of blots and gels were displayed in the Source data file.
Confocal fluorescence microscopy
Primary BMMs (30–40% confluence) seeded onto a confocal dish were stimulated to osteoclast phenotype with 30 ng/mL M-CSF and 50 ng/mL RANKL and treated with 2.5 × 106 cells/mL SP for 30 min.
Raw264.7 cells (30–40% confluence) seeded onto a confocal dish were treated via different concentrations of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5 × 106) for 24 h.
Then, the dishes were washed by PBS, fixed with 4% paraformaldehyde, permeabilized by 0.2% Triton X-PBS solution, and then incubated with primary antibodies (p-P38, p-P65, KEAP1, Ubiquitin, and LC3) at 4 °C overnight. The next day the dishes were repeatedly washed by PBS and stained with Alexa Fluor 488 and 555 Conjugate secondary antibodies (anti‐rabbit, anti-mouse, 1:500; Cell Signaling Technology) for 1 h in darkness. The nuclei were stained at RT for 10 min without light by DAPI. Images were captured using a Leica DM4000 B epifluorescence microscope. The IOD/DAPI and the relative fluorescence intensity were calculated using Image-Pro Plus 6.0 software.
Protein degradation assay
Raw264.7 cells were cultured in six-well plates at the density of 3 × 105 cells/well. Then these cells were pretreated with cycloheximide (CHX; 50 nM; cat. no. S7418; Selleck Chemicals, China) for 2 h to inhibit the synthesis of proteins, and the next step the lower three well cells were treated with MG132 (10 μM; cat. no. S2619; Selleck Chemicals, China) or chloroquine (Chlq; 25 nM; cat. no. S6999; Selleck Chemicals, China) for 10 h. Concurrently, cells were treated via different concentrations of SP (cells/mL, 2.5 × 106 and 5 × 106) for 12 h. Finally, the proteins were extracted and applied to WB analysis.
Ubiquitylation modification assay
293 T cells were transfected with HA-PLVC, HA-KEAP1, and Myc-Ubiquitin or transfected with Flag-PLVC, Flag-NRF2, HA-KEAP1, and Myc-Ubiquitin (synthesized, purchased from Shanghai Ai Bosi Biological Technology Co., Ltd.) using lipofectamine 3000 (cat. no. L3000015; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The next day, cells were treated via different concentrations of SP (cells/mL, 2.5 × 106 and 5 × 106). After 24 h, cells were stimulated with MG132 (10 μM) for 8 h at 37 °C and then suspended and washed three times with PBS. The proteins were extracted with 1.2 ml NETT solution (1% Triton X-100, 2.5 M NaCl, 0.9 M Tris–HCl [pH 7.5], 0.25 M EDTA, 100 mM PMSF), 200 μL lysate was used as input whilst the remaining 1000 μL samples were incubated with 30 μL HA-tagged or Flag-tagged magnetic beads at 4 °C overnight. The samples were then boiled at 99 °C for 10 min and finally subjected to WB analysis.
RFP-GFP-LC3B assay
RFP-GFP-LC3B (Component A, Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B Kit, Invitrogen Waltham, MA, USA; #P36239) were added to Raw264.7 cells (30–40% confluence) seeded onto a confocal dish for 16 h, then these cells were treated via different concentrations of SP (cells/mL, 1.2 × 106, 2.5 × 106, 5 × 106) for 24 h. Finally, cells were processed using confocal laser scanning microscopy with Hoechst stain to nuclei.
Isolation of cell membrane from macrophage
RAW 264.7 cells were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin (Beyotime). The macrophage cell membranes (MCMs) were prepared using previously reported methods with some modifications. Briefly, RAW 264.7 cells were washed three times with phosphate buffer saline (PBS, Corning) and then incubated in ice-cold hypotonic lysing buffer (50 mM Tris–HCl, 10 mM magnesium sulfate at pH 7.5) for 4 h at 4 °C. Then, sucrose was added to the cell suspension and eventually reached a sucrose concentration of 0.25 M. RAW264.7 cells were repeatedly disrupted using a dounce homogenizer with a tight-fitting pestle. After that, the cell suspension was centrifugated at 300 × g for 20 min (4 °C). The supernatant was collected while the precipitate was resuspended in the hypotonic lysing buffer and subjected to the dounce homogenizer treatment again. The supernatants were saved and centrifuged at 18,000×g for 40 min (4 °C) to collect the cell membrane. The supernatant was washed twice with cold PBS and stored at −80 °C for further use. The protein quantity of the purified MCMs was determined by a bicinchoninic acid protein (BCA, Beyotime) assay.
Synthesis of macrophage cell membranes (MCMs)-engineered SP
Spirulina platensis (SP) and Zarrouk’s media were obtained from Guangyu Biological Technology (Shanghai, China). SP was maintained in Zarrouk’s medium at 25 °C under light irradiation (3000–6000 lx). SP powder was purchased from the Macklin Biochemical Technology (Shanghai, China). The macrophage cell membranes (MCMs)-cloaked SP were synthesized by a reported extrusion method28,29,30. Briefly, SP was cracked into small fragments by continuous magnetic stirring at room temperature for 12 h. Afterward, the fractured SP was filtrated through a 5 μm cell strainer and concentrated to a concentration of 5 × 108 cells/mL. To prepare MCMs-cloaked SP, MCMs were physically extruded through a 5 μm porous polycarbonate membrane (Membrane Space). Then, MCMs were cloaked onto the SP by co-extruding the two components through a 5 μm porous polycarbonate membrane. The extrusion process was repeated for at least 20 times. The SP@M was then collected and redispersed in DI water at 4 °C for further use. Fresh SP@M was recommended to be prepared before use.
Dead/alive assay of SP
Fragmented SP was obtained by continuously stirring SP for 12 h. Boiled SP was obtained by boiling the SP for 10 min. Then, SP, fragmented SP, and boiled SP were gathered and stained with fluorescein diacetate (FDA) for 30 min at 37 °C. Then, the CLSM images were captured using a Leica DM4000 B epifluorescence microscope.
Protein identification of MCMs and SP@M
Then, SP, MCMs, and SP@M protein were subjected to SDS–PAGE gel electrophoresis for separation and following the electroblotting process for further WB assay. The protein pattern was identified with Coomassie blue staining. The primary antibodies used were TNFR2 (cat. no. 19272-1-AP), CCR2α (cat. no. 16153-1-AP), and CD36 (cat. no. 18836-1-AP) purchased from Proteintech Inc, Wuhan, China.
Isolation and characterization of SP-derived exosomes
100 mL of SP cell culture media (1 × 107 cells/mL, free of FBS, cultured for 24 h) was harvested and centrifuged at 2000×g for 30 min to remove cells and debris. Then, the supernatant containing the cell-free culture media was transferred to a new tube, and 50 mL of the Total Exosome Isolation reagent (Cat. no. 478359; Thermo Fisher Scientific, Waltham, MA, United States) was added, mixed, vortexed, and incubated at 4 °C overnight. We centrifuged this mixture at 10,000×g for 1 h at 4 °C, and gathered and resuspended the extracted exosomes in 400 μL saline. Then, the TEM test was carried out and images were captured using a Hitachi TEM system (HC/HR select = HC-1, accelerating voltage = 80,000, emission = 10.2, vacuum = 6.9E−05 Pa). The nanoparticle size distribution and concentration were measured using a ZetaView instrument (Particle Metrix).
Establishment of CIA model
All animal experiments were authorized by the Institutional Animal Ethics Review Board of the Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (Approve No. SH9H-2024-T246-1). Bovine type-II collagen solution (2 mg/mL) and complete Freund’s adjuvant (CFA) (4 mg/mL) were shaken to emulsion. For the first immunization, male mice (n = 36; DBA/1 J; divided into six groups: Sham group without immunization; saline group; Macrophage membrane [MCMs] group; macrophage membrane-cloaked spirulina [SP@M] group; Spirulina [SP] group; Methotrexate [MTX] group, GemPharmatech, Nanjing, China) were treated by intradermal injection of the complete emulsion at the end of the tail. For the boost immunization, the mice were injected with the emulsion of bovine type-II collagen solution (2 mg/mL) with incomplete Freund’s adjuvant (IFA) on day 21.
Biodistribution in the CIA mouse model
SP was initially labeled with Cy5-PEI to obtain Cy5-labeled SP. Then, Cy5-labeled SP@M was prepared using the previously described macrophage cell membrane modification process. CIA mice were intravenously injected with Cy5-labeled SP or Cy5-labeled SP@M (100 μL, at an SP concentration of 5 × 107 cells/mL). The biodistribution of Cy5-labeled SP and Cy5-labeled SP@M was then tested by in vivo imaging measurement of fluorescence intensity using a Caliper IVIS Lumina III In Vivo Imaging System (Perkin Elmer, USA) at specific time points (0.5, 6, and 24 h post-injection) (n = 3 in each group). Then, the major organs and the four limbs were dissected to evaluate the fluorescence intensity.
Assessment of paw swelling and clinical score
The CIA mice were administered 100 µL of PBS, MCMs, SP@M (4 × 107 cells/mL), SP (4 × 107 cells/mL), and MTX (5 mg/kg) intravenously every three days for a total of 10 times. Non-immunized mice in the sham group were administered PBS intravenously. And mice were evaluated every 3 days for paw swelling scores and clinical scores, and the hind paw diameters were measured by a digital caliper. A blinded researcher gave paw scorings (score ~ 0-4) based on the following criteria: 0, normal; 1, mild redness of ankle or tarsal joints; 2, mild redness and swelling extending from the ankle to the tarsals; 3, moderate redness and swelling from ankle to metatarsal joints; 4, severe redness and swelling encompassing the ankle, foot, and digits67. Clinical severity was graded as 0 (normal), 0.5 (erythema and edema in only one digit), 1 (erythema and mild edema of the footpad, or ankle or two to five digits), 2 (erythema and moderate edema of two joints [footpad or ankle, two to five digits]), 3 (erythema and severe edema of the entire paw), or 4 (reduced swelling and deformation leading to incapacitated limb)68. At the end, images of the left hind paws of mice were captured.
Behavioral assessment
On day 60, the behaviors of the mice were analyzed via the Von Frey test, hot plate test, and gait analysis. In this study, the von Frey test was performed on each animal to measure mechanical allodynia in mice. We used flexible nylon von Frey filaments in logarithmic increments of force (0.04–2 g), and the “up-down method” was used. Every result was recorded to assess the 50% mechanical paw withdrawal threshold response (g).
To determine behavioral response to noxious heat, the surface temperature of the hot plate was maintained at 55 °C. We placed the mice on the hot plate and started timing until a quick withdrawal or flick of the paw was observed, which is defined as latency (s). Twenty seconds was set as a cutoff time to prevent mouse paw injury. For the accuracy of the results, we measured three times at 10-min intervals, and the results were averaged.
All our gait analyses were based on a CatWalk runway; animals were trained daily on a CatWalk runway until they were able to make consecutive uninterrupted runs for at least one week. Valid data were recorded at least four times for each animal, with an average of six animals per group; all trials marked by the software as compliant were reviewed manually; if the animal stopped or turned back during the experiment, the recording was discarded. Print area and max contact area parameters were collected for each paw, respectively, and blindly evaluated.
μCT
In the end, the right ankles of mice were collected and applied to μCT analysis. A high-resolution Skyscan 1275 micro-CT scanner (Bruker micro-CT, Kontich, Belgium) was used to perform the micro-computed tomography (CT) scanning with the following parameter settings: source voltage, 46 kV; source current, 75 μA; AI, no filter A; A pixel A size A, 9.000546 μm; rotation step, 0.4°. The bone volume (BV)/total volume (TV) of talus bone was calculated with software (Version: 6.5-3, SCANCO Medical AG, Switzerland) by evaluating and analyzing the three-dimensional region of interest (ROI). For the erosion index, six sites in the ankle joint were scored: the talus, navicular bone, medial cuneiform bone, and the bases of the first, second, and third metatarsals. Each site was scored on a scale of 0–3 (0 = normal; 1 = pitting; 2 = full thickness holes in small-medium areas; and 3 = full thickness holes in medium-large areas) with a maximum score of 18. The scores of the arthritic hind paw determined by the two observers were averaged to obtain the final erosion score69.
Histology and immunofluorescence staining
Ankles from mice were fixed in 4% paraformaldehyde for 48 h and embedded into paraffin. Then, they were cut into 8 µm sections and stained with Safranin O/Fast Green (SO/FG), TRAP, and Hematoxylin and eosin (H&E). The SMASH score, including synovitis, cartilage destruction, and bone erosion, was analyzed according to a previous article48. The number of osteoclasts and TRAP-positive multinucleated osteoclasts per field (Oc.S/BS) was calculated.
These tissue sections were deparaffinized in graded xylene, rehydrated in graded alcohol solutions, and then incubated in antigen retrieval buffer (Roche, Basel, Switzerland) at 37 °C for 30 min. After natural cooling, the slides were placed in PBS (pH 7.4) and washed thrice for 5 min each. An autofluorescence quencher was added to the tissue sections for 5 min, rinsed with running water for 10 min, and then blocked with BSA for 30 min. The sections were subsequently incubated with primary antibodies in a wet box at 4 °C overnight. Primary antibodies were used at 1:100 dilution and included Anti-NRF2, Anti-KEAP1, anti-ubiquitin, anti‐LC3, anti‐CD86, and anti-CD206. The next day, the slides were immersed in PBS and washed three times, then incubated with Alexa Fluor 488 and 555 Conjugate secondary antibody (anti‐rabbit, anti‐mouse, 1:500; Cell Signaling Technology) for 50 min at room temperature in the dark. The slides were again immersed in PBS, washed three times in PBS, and then counterstained in the dark with DAPI solution for 10 min at room temperature. Finally, sections were observed using a Leica DM4000 B epifluorescence microscope. The IOD/DAPI was calculated using Image-Pro Plus 6.0 software.
Immunohistochemistry staining
The paraffin sections were processed using an immunohistochemistry kit (cat. no. G1215-200T; Servicebio Technology) according to the manufacturer’s instructions. The primary antibodies used were anti-TNF-α and anti-CTSK. Images were captured using a Leica DM4000 B microscope. The IOD value and positive cells (%) were calculated using Image-Pro Plus 6.0 software.
RNA seq analysis
Total RNA of ankles from the saline group (n = 3) and SP@M group (n = 3) were extracted as per the manufacturer’s protocol. Then, they were analyzed via RNA (transcriptome) sequencing as Wuhan Huada Gene Technology Co., Ltd. (China): Volcano Plot (|log2FC| ≥ 1, FDR ≤ 0.001), Gene Oncology (GO) of biological pathway and cellular component analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and gene set enrichment analysis (GSEA) was used by mRNA relative expression as Transcripts PerKilobase Million (TPM) to review further pathways involved on the Mybgi platform (WuhanHuada Gene Technology, https://mybgi.bgi.com/tech/login).
Hemolysis assay
The hemolytic toxicity of SP and SP@M on mice red blood cells (RBCs) was preliminarily assessed using a hemolysis assay. Briefly, 500 μL of mice with 5% erythrocytes (Shanghai Lab, Animal Research Center Co., Ltd) was centrifuged at 200×g for 15 min, after which the supernatant was removed. The erythrocytes were resuspended to 10 mL with saline following three washes of the precipitate. The SP and SP@M were added to the erythrocyte suspension at a final concentrations range of 1.25 × 106, 2.5 × 106, 5.0 × 106, 1.0 × 107 cells/mL, whereas the RBCs treated with saline as negative control and Triton X-100, 2% (v/v) as positive control. The RBCs of each group were incubated at 37 °C for 1 h and then centrifuged at 200×g for 15 min. Subsequently, 100 µL of the supernatant was taken, and the absorbance at 540 nm was measured via Infinite M200 Pro microplate reader (Nanoquant, Tecan, Swiss). The percentage of hemolysis was calculated using the following formula:
Biocompatibility assay
The major organs, including the heart, liver, spleen, lungs, and kidneys of the previous CIA mouse models, were harvested from the euthanized mice for further H&E staining and histology assay to evaluate the toxicity of MCMs, SP, and SP@M.
The supplementary biochemical analysis was applied to the DBA/1J mice (n = 12; divided into 2 groups: saline group and macrophage cell membranes-cloaked spirulina [SP@M] group). 100 μL saline and SP@M (4 × 107 cells/mL) were administrated intravenously in the tail once every three days (10 times). When injections finished, all mice were euthanized, and the serum was harvested. Then the levels of albumin (ALB), total bilirubin (TBIL), alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyltransferase (γ-GT), triglyceride (TG), cholesterol (CHOL), glucose (GLU), urea nitrogen (BUN), and creatinine (CRE) ion Calcium (Ca), and ion Phosphote (P) were measured using the Pointcare V3 automatic biochemical analyzer (Tianjin MNCHIP Technologies Ltd., Tianjin, China) with commercial diagnostic kits.
Statistics and reproducibility
Data in this study were obtained from three to six independent experiments or repeated measurements. Significant differences among groups were analyzed using one-way analysis of variance (ANOVA) with Tukey’s post hoc test or the Kruskal–Wallis test with Dunn’s post hoc test. All analyses were calculated with GraphPad software (version 8.0; NY, USA). Differences were defined as significant with a p-value of <0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study are available on the National Center for Biotechnology Information (NCBI) database with SRA accession code: PRJNA1212571. Source data are provided with this paper.
References
Uhlig, T. & Kvien, T. K. Is rheumatoid arthritis really getting less severe? Nat. Rev. Rheumatol. 5, 461–464 (2009).
Hsieh, P.-H. et al. Economic burden of rheumatoid arthritis: a systematic review of literature in biologic era. Ann. Rheum. Dis. 79, 771–777 (2020).
Safiri, S. et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 78, 1463–1471 (2019).
Willemze, A., Trouw, L. A., Toes, R. E. M. & Huizinga, T. W. J. The influence of ACPA status and characteristics on the course of RA. Nat. Rev. Rheumatol. 8, 144–152 (2012).
Zhou, F. et al. Redox homeostasis strategy for inflammatory macrophage reprogramming in rheumatoid arthritis based on ceria oxide nanozyme-complexed biopolymeric micelles. ACS Nano 17, 4358–4372 (2023).
Zhang, S. et al. Remodeling articular immune homeostasis with an efferocytosis-informed nanoimitator mitigates rheumatoid arthritis in mice. Nat. Commun. 14, 817 (2023).
Alivernini, S., Firestein, G. S. & McInnes, I. B. The pathogenesis of rheumatoid arthritis. Immunity 55, 2255–2270 (2022).
Catrina, A. I., Svensson, C. I., Malmström, V., Schett, G. & Klareskog, L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 13, 79–86 (2017).
Salhotra, A., Shah, H. N., Levi, B. & Longaker, M. T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 21, 696–711 (2020).
Komatsu, N. & Takayanagi, H. Mechanisms of joint destruction in rheumatoid arthritis-immune cell–fibroblast–bone interactions. Nat. Rev. Rheumatol. 18, 415–429 (2022).
van Vollenhoven, R. F. Treatment of rheumatoid arthritis: state of the art 2009. Nat. Rev. Rheumatol. 5, 531–541 (2009).
Hudson, M., Tascilar, K. & Suissa, S. Comparative effectiveness research with administrative health data in rheumatoid arthritis. Nat. Rev. Rheumatol. 12, 358–366 (2016).
Atzeni, F. et al. Different effects of biological drugs in rheumatoid arthritis. Autoimmun. Rev. 12, 575–579 (2013).
Breedveld, F. C. & Dayer, J. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 59, 841–849 (2000).
Smolen, J. S. Insights into the treatment of rheumatoid arthritis: a paradigm in medicine. J. Autoimmun. 110, 102425 (2020).
Wysham, K. D., Baker, J. F. & Shoback, D. M. Osteoporosis and fractures in rheumatoid arthritis. Curr. Opin. Rheumatol. 33, 270–276 (2021).
Su, Y.-J., Lin, C.-Y. & Hsu, C.-Y. Analyzing the risk of osteoporosis and fracture in rheumatoid arthritis patients who have been treated with various biologics. Int. J. Rheum. Dis. 27, e15055 (2024).
Takeuchi, T. et al. Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): a randomised, double-blind, placebo-controlled phase 3 trial. Ann. Rheum. Dis. 78, 899–907 (2019).
Jester, B. W. et al. Development of spirulina for the manufacture and oral delivery of protein therapeutics. Nat. Biotechnol. 40, 956–964 (2022).
Zhong, D., Li, W., Qi, Y., He, J. & Zhou, M. Photosynthetic biohybrid nanoswimmers system to alleviate tumor hypoxia for FL/PA/MR imaging-guided enhanced radio-photodynamic synergetic therapy. Adv. Funct. Mater. 30, 1910395 (2020).
Selmi, C. et al. The effects of Spirulina on anemia and immune function in senior citizens. Cell. Mol. Immunol. 8, 248–254 (2011).
Chen, Q.-W., Cao, M.-W., Qiao, J.-Y., Li, Q.-R. & Zhang, X.-Z. Integrated cascade catalysis of microalgal bioenzyme and inorganic nanozyme for anti-inflammation therapy. Nanoscale Horiz. 8, 489–498 (2023).
Zhong, D., Zhang, D., Xie, T. & Zhou, M. Biodegradable microalgae-based carriers for targeted delivery and imaging-guided therapy toward lung metastasis of breast cancer. Small 16, e2000819 (2020).
Zhang, X. et al. Engineered oxygen factories synergize with STING agonist to remodel tumor microenvironment for cancer immunotherapy. Adv. Funct. Mater. 33, 2300833 (2023).
Finamore, A., Palmery, M., Bensehaila, S. & Peluso, I. Antioxidant, immunomodulating, and microbial-modulating activities of the sustainable and ecofriendly Spirulina. Oxid. Med. Cell. Longev. 2017, 3247528 (2017).
Motohashi, H. & Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549–557 (2004).
Dong, Y. et al. A clinical-stage Nrf2 activator suppresses osteoclast differentiation via the iron–ornithine axis. Cell Metab. 36, 1–17 (2024).
Baird, L. & Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 40, e00099–20 (2020).
Bae, S. H. et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 17, 73–84 (2013).
Cai, C. et al. USP25 regulates KEAP1-NRF2 anti-oxidation axis and its inactivation protects acetaminophen-induced liver injury in male mice. Nat. Commun. 14, 3648 (2023).
Liu, Y., Luo, J., Chen, X., Liu, W. & Chen, T. Cell membrane coating technology: a promising strategy for biomedical applications. Nano-Micro Lett. 11, 100 (2019).
Cao, Z., Cheng, S., Wang, X., Pang, Y. & Liu, J. Camouflaging bacteria by wrapping with cell membranes. Nat. Commun. 10, 3452 (2019).
Liu, L. et al. Cell membrane coating integrity affects the internalization mechanism of biomimetic nanoparticles. Nat. Commun. 12, 5726 (2021).
Zhu, W. et al. Rapid continuous 3D printing of customizable peripheral nerve guidance conduits. Mater. Today 21, 951–959 (2018).
Shin, M. J. et al. Inflammation-targeting mesenchymal stem cells combined with photothermal treatment attenuate severe joint inflammation. Adv. Mater. 36, e2304333 (2024).
Wu, J. et al. miR-100-5p-abundant exosomes derived from infrapatellar fat pad MSCs protect articular cartilage and ameliorate gait abnormalities via inhibition of mTOR in osteoarthritis. Biomaterials 206, 87–100 (2019).
Hou, M. et al. Immuno-engineered nanodecoys for the multi-target anti-inflammatory treatment of autoimmune diseases. Adv. Mater. 34, e2108817 (2022).
Koo, S. et al. Ceria-vesicle nanohybrid therapeutic for modulation of innate and adaptive immunity in a collagen-induced arthritis model. Nat. Nanotechnol. 18, 1502–1514 (2023).
Yang, L. et al. Mannose-mediated nanodelivery of methotrexate to macrophages augments rheumatoid arthritis therapy. Biomater. Sci. 11, 2211–2220 (2023).
Han, H., Xing, J., Chen, W., Jia, J. & Li, Q. Fluorinated polyamidoamine dendrimer-mediated miR-23b delivery for the treatment of experimental rheumatoid arthritis in rats. Nat. Commun. 14, 944 (2023).
Wang, Y. et al. A bioengineered probiotic for the oral delivery of a peptide Kv1.3 channel blocker to treat rheumatoid arthritis. Proc. Natl Acad. Sci. USA 120, e2211977120 (2023).
Wei, Y. et al. Oral delivery of siRNA using fluorinated, small-sized nanocapsules toward anti-inflammation treatment. Adv. Mater. 35, e2206821 (2023).
Meng, S. et al. Surface-decorated nanoliposomal leonurine targets activated fibroblast-like synoviocytes for efficient rheumatoid arthritis therapy. Biomater. Sci. 11, 7099–7113 (2023).
Yang, J., Yang, B. & Shi, J. A nanomedicine-enabled ion-exchange strategy for enhancing curcumin-based rheumatoid arthritis therapy. Angew. Chem. Int. Ed. 62, e202310061 (2023).
Zhang, X. et al. Macrophage-hitchhiking interleukin-10 plasmid DNA delivery system modulates rheumatoid arthritis microenvironment via the re-polarization of macrophages. Nano Today 54, 102068 (2024).
Wang, Z. et al. A Targeted exosome therapeutic confers both CfDNA scavenging and macrophage polarization for ameliorating rheumatoid arthritis. Adv. Mater. 35, e2302503 (2023).
Cao, S. et al. l-arginine metabolism inhibits arthritis and inflammatory bone loss. Ann. Rheum. Dis. 83, 72–87 (2024).
Østergaard, M. et al. Certolizumab pegol, abatacept, tocilizumab or active conventional treatment in early rheumatoid arthritis: 48-week clinical and radiographic results of the investigator-initiated randomised controlled NORD-STAR trial. Ann. Rheum. Dis. 82, 1286–1295 (2023).
Brown, P., Pratt, A. G. & Hyrich, K. L. Therapeutic advances in rheumatoid arthritis. Br. Med. J. 384, e070856 (2024).
Zhang, F. et al. Biomembrane-functionalized micromotors: biocompatible active devices for diverse biomedical applications. Adv. Mater. 34, e2107177 (2022).
Oroojalian, F., Beygi, M., Baradaran, B., Mokhtarzadeh, A. & Shahbazi, M.-A. Immune cell membrane-coated biomimetic nanoparticles for targeted cancer therapy. Small 17, e2006484 (2021).
Fang, R. H., Gao, W. & Zhang, L. Targeting drugs to tumours using cell membrane-coated nanoparticles. Nat. Rev. Clin. Oncol. 20, 33–48 (2023).
Gao, C. et al. Conjugation of macrophage-mimetic microalgae and liposome for antitumor sonodynamic immunotherapy via hypoxia alleviation and autophagy inhibition. ACS Nano 17, 4034–4049 (2023).
Su, N. et al. Stem cell membrane-coated microribbon scaffolds induce regenerative innate and adaptive immune responses in a critical-size cranial bone defect model. Adv. Mater. 35, e2208781 (2023).
Alivernini, S. et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 26, 1295–1306 (2020).
Mei, J. et al. Biofilm microenvironment-responsive self-assembly nanoreactors for all-stage biofilm associated infection through bacterial cuproptosis-like death and macrophage re-rousing. Adv. Mater. 35, e2303432 (2023).
Li, S. et al. A novel cargo delivery system-AnCar-ExoLaIMTS ameliorates arthritis via specifically targeting pro-inflammatory macrophages. Adv. Sci. 11, e2306143 (2024).
Zhang, D. et al. Oral microalgae-nano integrated system against radiation-induced injury. ACS Nano 17, 10560–10576 (2023).
Kalluri, R. & LeBleu, V. S. The biology, function, and biomedical applications of exosomes. Science 367, eaau6977 (2020).
Geertsema, S. et al. The NRF2/Keap1 pathway as a therapeutic target in inflammatory bowel disease. Trends Mol. Med. 29, 830–842 (2023).
Sies, H., Berndt, C. & Jones, D. P. Oxidative stress. Annu. Rev. Biochem. 86, 715–748 (2017).
Athari, S. S. Targeting cell signaling in allergic asthma. Signal Transduct. Target. Ther. 4, 45 (2019).
Saha, S. An overview of therapeutic targeting of Nrf2 signaling pathway in rheumatoid arthritis. ACS Omega 9, 10049–10057 (2024).
Wells, J. A. & Kumru, K. Extracellular targeted protein degradation: an emerging modality for drug discovery. Nat. Rev. Drug Discov. 23, 126–140 (2024).
Ahn, G. et al. Elucidating the cellular determinants of targeted membrane protein degradation by lysosome-targeting chimeras. Science 382, eadf6249 (2023).
Yang, X. et al. Polydopamine nanoparticles targeting ferroptosis mitigate intervertebral disc degeneration via reactive oxygen species depletion, iron ions chelation, and GPX4 ubiquitination suppression. Adv. Sci. 10, e2207216 (2023).
Zhang, Q. et al. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 13, 1182–1190 (2018).
Liu, Y. et al. Regulatory fibroblast-like synoviocytes cell membrane coated nanoparticles: a novel targeted therapy for rheumatoid arthritis. Adv. Sci. 10, e2204998 (2023).
Hasegawa, T. et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat. Immunol. 20, 1631–1643 (2019).
Acknowledgements
This study was supported by grants from The National Natural Science Foundation of China (grant nos. 82130073, 82202737, 82302722, 82273489), Shanghai Frontiers Science Center of Degeneration and Regeneration in Skeletal System, and Biomaterials and Regenerative Medicine Institute Cooperative Research Project, Shanghai Jiao Tong University School of Medicine (no. 2022LHA01).
Author information
Authors and Affiliations
Contributions
X.Y. and J.F. conceived the study and designed experiments. Y.Y. and J.F. characterized the properties of the materials. X.Y., K.R., and S.F. performed the animal experiments. X.Y., S.L., C.Z., K.X., K.Z., Y.Z., and Y.H. contributed to data analysis and interpretation. X.Y. and J.F. crafted all the figures and wrote the manuscript. All authors edited and revised the manuscript. J.Z. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Rong, K., Fu, S. et al. Engineered Spirulina platensis for treating rheumatoid arthritis and restoring bone homeostasis. Nat Commun 16, 4434 (2025). https://doi.org/10.1038/s41467-025-59579-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59579-4
