
Abstract
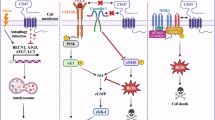
The CD47/SIRPα axis conveys a ‘don’t eat me’ signal, thereby thwarting the phagocytic clearance of tumor cells. Although blocking antibodies targeting CD47 have demonstrated promising anti-tumor effects in preclinical models, clinical trials involving human cancer patients have not yielded ideal results. Exploring the regulatory mechanisms of CD47 is imperative for devising more efficacious combinational therapies. Here, we report that inhibiting USP2 prompts CD47 degradation and reshapes the tumor microenvironment (TME), thereby enhancing anti-PD-1 immunotherapy. Mechanistically, USP2 interacts with CD47, stabilizing it through deubiquitination. USP2 inhibition destabilizes CD47, thereby boosting macrophage phagocytosis. Single-cell RNA sequencing shows USP2 inhibition reprograms TME, evidenced by increasing M1 macrophages and CD8+ T cells while reducing M2 macrophages. Combining ML364 with anti-PD-1 reduces tumor burden in mouse models. Clinically, low USP2 expression predicts a better response to anti-PD-1 treatment. Our findings uncover the regulatory mechanism of CD47 by USP2 and targeting this axis boosts anti-tumor immunity.
Similar content being viewed by others


Introduction
Immune checkpoint blockade (ICB) therapies, particularly targeting PD-1/PD-L1, have shown effective therapeutic benefits in various types of cancer patients1,2. However, the underlying mechanisms of low response rates to ICB therapies remain incompletely understood1,3,4. Recently, designing combination immunotherapies based on molecular mechanisms might be one of the major strategies to overcome resistance or enhance the efficacy of current ICB therapies5,6,7. CD47 is frequently overexpressed on tumor cells and interacts with SIRPα on macrophages, conveying a ‘don’t eat me’ signal to prevent phagocytic clearance of tumor cells8,9,10. Blocking antibodies targeting the CD47/SIRPα axis have exhibited potent anti-tumor effects in preclinical tumor models and are currently being evaluated in clinical trials for cancer therapy10,11,12,13,14. Thus, understanding the molecular mechanisms underlying CD47 regulation may aid in designing optimal combination therapies to improve anti-tumor efficacy.
Upregulation of CD47 expression has been observed in a variety of solid tumors and hematologic malignancies13,15,16,17. At the transcriptional level, Myc triggers CD47 expression via binding the promoter region of CD4718. CD47 expression is also induced by hypoxia-inducible factor 1α (HIF-1α) during hypoxic tumor immune microenvironment19. Moreover, the tumor necrosis factor (TNF) promotes the nuclear factor-κB (NF-κB) transcription factor to bind the super-enhancer of CD47 to elevate its expression20. In addition, mutant KRAS upregulates CD47 expression by relieving miR-34a-mediated repression on the 3′-UTR of CD47 mRNA21. At the post-translational level, two independent groups have demonstrated that glutaminyl-peptide cyclotransferase-like (QPCTL, also named isoQC) catalyzes the N-terminal pyroglutamate formation on CD47 protein, which is required for the binding of SIRPα and anti-human CD47 monoclonal antibody CC2C622,23,24. A recent study has reported that the EGFR signaling can promote c-Src-mediated CD47 phosphorylation, which inhibits TRIM21-dependent ubiquitination and degradation of CD47 to evade immune surveillance25. However, the deubiquitinases (DUBs) that regulate CD47 at the post-translational level remain unknown.
Ubiquitin-specific protease 2 (USP2), a member of the deubiquitinating enzyme family, is upregulated in various cancers and plays a significant role in tumorigenesis and progression26. Studies have shown that USP2 stabilizes cyclin D1 and facilitates the degradation of p53 by stabilizing MDM227,28. A recent report that USP2 inhibition sensitizes tumors to PD-1 blockade in syngeneic mouse breast tumor models largely through destabilizing VPRBP to indirectly upregulate p53 and PD-L129. Additionally, small-molecule inhibitors targeting USP2, such as ML364, have been reported to specifically inhibit USP2, leading to suppression of tumor cell proliferation30. However, the role of USP2 in tumor immunity regulation remains largely unexplored.
In this study, our findings reveal that USP2 interacts with CD47, facilitating the deubiquitination of CD47 and leading to the stabilization of the CD47 protein. USP2 inhibition by pharmacological inhibitor or genetic depletion not only reduces CD47 expression to enhance the phagocytic capacity of macrophages towards tumor cells, but also reshapes an inflamed TME, which enables tumors to overcome resistance to anti-PD-1 immunotherapy in multiple lung tumor models. Clinically, patients with low USP2 expression in tumor tissues exhibit better response to PD-1 blockade therapy, accompanied by low expression of CD47, low M2 macrophage, and high M1 macrophage and CD8+ T cells in the TME. These findings identify the USP2-CD47 axis as a critical driver of tumor immune evasion and anti-PD-1 therapy resistance. Targeting this axis may therefore enhance anti-tumor immunity and improve outcomes for cancer patients.
Results
Identifying the deubiquitinase USP2 as a positive regulator for CD47
Ubiquitination is a highly dynamic and reversible process that is governed by both ubiquitin E3 ligases and DUBs, which is a major system for controlling protein homeostasis in cells31,32. Compared to E3 ligases, more inhibitors targeting DUBs have been developed, and some of them have displayed potential therapeutic value in treating cancers33. To investigate whether the deubiquitinating enzyme regulates CD47 protein abundance, we initially generated stable H1975 and HEK293T cell lines expressing a GFP fusion in the cytoplasmic C-terminus of CD47 (CD47-GFP). Immunofluorescence experiments firstly confirmed that the GFP protein fused to the C-terminus of CD47 displays normal membrane localization in H1975 and HEK293T cells (Supplementary Fig. 1a). Then we conducted a screening of a siRNA library comprising siRNA pools targeting 96 human DUBs using these stable cell lines, as outlined in Fig. 1a. Heatmaps displaying the fluorescence intensities of the CD47-GFP signal during the screening were presented for both H1975 and HEK293T cells (Supplementary Fig. 1b, c). Additionally, we listed the top ten genes, for which their respective siRNAs noticeably reduced the median fluorescence intensity (MFI) of the CD47-GFP signal in the two cell lines (Fig. 1a). Importantly, USP2 emerged as the sole gene consistently listed in the top ten genes regulating CD47-GFP signal in both H1975 and HEK293T cell lines. This suggests that USP2 may play a crucial role in modulating CD47 expression.

a Overview of the outline for establishing H1975 and HEK293T cell lines expressing a fusion GFP gene in the C-terminus of CD47 (CD47-GFP) and subsequently identifying the deubiquitinase of CD47 through DUB siRNA library screening with two stable cell lines. DUBs deubiquitinases; FACS fluorescence-activated cell sorting; MFI mean fluorescence intensity. b Whole-cell lysates (WCL) of H1975 or PC9 cells treated with indicated deubiquitinase inhibitors (2.5 µM) or dimethyl sulfoxide (DMSO) for 12 hours (h) were prepared and subjected to immunoblotting (IB) analysis. c–e IB analysis of WCL derived from H1975 cells (c) and PC9 cells (e) treated with ML364 (1 µM and 2 µM) or DMSO for 16 h. The mRNA level of CD47 in H1975 cells (c) was measured using reverse transcription quantitative PCR (RT-qPCR) (d). f, g Immunofluorescence (IF) staining for CD47 in H1975 cells (f) and PC9 cells (g) treated with ML364 (2 µM) for 16 h. Scale bar, 25 μm. h–k IB analysis of WCL derived from H1975 (h) or PC9 (j) cells stably expressing shUSP2 or shGFP, respectively. The mRNA level of CD47 in H1975 (i) or PC9 (k) cells was measured using RT-qPCR. l, m IB analysis of Cd47 protein expression in the tissues of lung, heart, or kidney obtained from wild-type (WT) and Usp2−/− mice (l). Quantification of Cd47 protein band intensity was normalized to vinculin (m). n–p IB analysis of WCL derived from HEK293T cells co-transfected with indicated constructs (n, p). The mRNA level of CD47 was quantified by RT-qPCR (o). EV: empty vector. q, r IB analysis of WCL derived from HEK293T cells co-transfected with indicated constructs. Cells were treated with 200 μg/ml CHX for the indicated time points (q). Quantification of CD47 protein band intensity was normalized to vinculin, then compared to the t = 0 time point (r). s, t Representative images from immunohistochemical (IHC) staining of CD47 and USP2 in human lung adenocarcinoma (s). Scale bar, left panels: 100 μm; right panels: 50 μm. n = 83. Quantification of USP2 and CD47 staining intensities was performed as average optical density (AOD) [AOD = Integrated Optical Density (IOD) SUM/Area SUM] (t). For (d, i, k, m, o, and r), unpaired two-tailed Student’s t-test. Correlations were analyzed by Pearson’s test (t). Data are shown as the mean ± SD, n = 3 independent biological replicates. P < 0.05 was considered statistically significant. n = 3 biologically independent experiments for (b, c, e, h, j, l, n, p, and q). Source data are provided as the Source Data file.
To ensure that the knockdown of USP2 specifically regulates CD47 protein expression rather than merely reducing the GFP signal itself, we conducted siUSP2 transfection experiments in H1975 and HEK293T cells stably expressing either the pLenti-GFP empty vector or pLenti-CD47-GFP. The results showed that GFP MFI was only reduced in cells expressing pLenti-CD47-GFP following siUSP2 transfection, while no significant change was observed in cells expressing the pLenti-GFP empty vector (Supplementary Fig. 1d–g). To further validate this observation, we employed a set of DUB inhibitors to treat cancer cells and observed that, among the inhibitors tested, only ML364, a USP2 inhibitor, dramatically downregulated the protein abundance of CD47 in various human cancer cell lines, including H1975, PC9, and MCF7 (Fig. 1b and Supplementary Fig. 1h). ML364 treatment resulted in a dose- and time-dependent decrease in CD47 protein levels, while the CD47 mRNA levels remained unaffected (Fig. 1c–e and Supplementary Fig. 1i–m). Furthermore, immunofluorescence (IF) staining assay results demonstrated a substantial reduction in cell surface CD47 protein levels in PC9 or H1975 cells following ML364 treatment (Fig. 1f, g and Supplementary Fig. 1n, o). These findings collectively suggest that the USP2 inhibitor, ML364, robustly diminishes CD47 protein abundance in multiple cancer cell lines. Additionally, a recent study demonstrated that ML364, by targeting USP2, enhances PD-L1 protein expression in breast and lung cancer cell lines29. We treated PC9 and H460 cell lines with a gradient of ML364 concentrations and simultaneously monitored the expression of CD47, PD-L1, and p53. Our results showed that low doses of ML364 dramatically reduced CD47 levels without affecting PD-L1 and p53 expression, while higher doses increased PD-L1 and p53 expression in both cell lines (Supplementary Fig. 1p, q). These differential responses to ML364 treatment may be due to CD47 being a direct substrate of USP2, while PD-L1 and p53 are indirect downstream targets regulated through the VPRBP pathway.
To further demonstrate the role of USP2 in regulating CD47 expression, we used two independent shRNAs to knock down USP2 in multiple cancer cell lines. Our results demonstrated that knockdown of USP2 resulted in a dramatic reduction in CD47 protein abundance, without significant change in its mRNA level (Fig. 1h–k and Supplementary Fig. 1r, s). In keeping with these results, the Cd47 protein level was significantly decreased in tissues of the lung, heart, and kidney derived from Usp2−/− mice compared to wild-type (WT) mice (Fig. 1l, m). In contrast, ectopic expression WT USP2, but not the enzymatic inactivation mutant (C276A)34, markedly increased the CD47 protein abundance, but didn’t significantly change its mRNA level (Fig. 1n–p). Furthermore, we blocked protein translation by using cycloheximide (CHX) to examine the protein half-life of CD47. The results demonstrated that ectopic expression of WT USP2, rather than the USP2-C276A mutant, significantly prolonged the CD47 protein half-life, supporting the notion that USP2 stabilizes CD47 at the post-translational level in a deubiquitinating enzyme dependent manner (Fig. 1q, r and Supplementary Fig. 1t, u).
To investigate the clinical relevance of USP2 and CD47 protein expression levels in human cancer patients, the tissue microarray containing 83 cases of human lung adenocarcinoma (LUAD) tissues was immunohistochemically (IHC) stained with anti-CD47 and anti-USP2 antibodies, respectively. The IHC staining results showed that the USP2 expression was positively correlated with CD47 expression in the tissues of LUAD (Fig. 1s, t and Supplementary Fig. 1v). Taken together, these findings suggest that USP2 positively regulates CD47 protein abundance in human cancer cells and clinical specimens.
USP2 interacts with CD47 and deubiquitinates CD47
The deubiquitinating enzymes stabilize their substrates by removing ubiquitin chains on substrates to prevent the 26S proteasome-mediated degradation35. Considering USP2 as a deubiquitinating enzyme, we hypothesized that USP2 might interact with CD47 to erase the ubiquitination on CD47. To test this hypothesis, we performed co-immunoprecipitation (co-IP) assay to examine a set of DUBs interaction with CD47 in cells. The results revealed that only USP2, but not other DUBs we examined, had a strong binding affinity to CD47 (Fig. 2a and Supplementary Fig. 2a). The glutathione S-transferase (GST) pull-down assay showed that bacterially purified recombinant GST-USP2, but not GST alone, interacted with CD47 (Fig. 2b). Conversely, the recombinant GST-CD47, but not GST alone, bound to USP2 (Fig. 2c). USP2 consists of two domains, the N-terminal domain and the C-terminal USP catalytic domain26. To identify which domain of USP2 is required for binding to CD47, we generated two truncated mutants of USP2, the N-terminal domain of amino acid (aa) 1–266 and C-terminal domain of aa267–605 (Fig. 2d). The results demonstrated that the N-terminal domain, but not the C-terminal domain of USP2, interacted with CD47 (Fig. 2e–g). CD47 is a five-pass transmembrane protein and has three short cytoplasmic regions36. As USP2 mainly localizes in the cytoplasm, we speculated that USP2 might interact with CD47 through its intracellular regions. To this end, we generated three CD47 truncated mutants by deleting each of the intracellular regions, respectively (Fig. 2h). The GST pull-down assay showed that CD47-Δ3 with missing amino acid residues from 290 to 323, but not the CD47-Δ1 lacking the residues of 163–176 and Δ2 deleting residues of 229–235 in the cytoplasmic region, failed to interact with USP2 in cells, suggesting that CD47 interacts with USP2 largely through its C-terminal cytoplasmic region (Fig. 2i). Meanwhile, we observed a strong endogenous interaction between USP2 and CD47 in both PC9 and H1975 lung cancer cells (Fig. 2j, k). And colocalization of USP2 and CD47 on the cellular membrane was also observed in human lung cancer tissues (Supplementary Fig. 2b). Collectively, these results demonstrate that USP2 interacts with CD47 in cells. Furthermore, patient-derived mutations in the USP2 N-terminal domain, including H21Q, G109V, C121Y, D145N, R148L, D162Y, Q214H, R229Q, and G265S, have been identified in clinical NSCLC patients using data from The Cancer Genome Atlas (TCGA) via cBioPortal (https://www.cbioportal.org/)37. Upon ectopic expression of WT USP2 and these mutants in PC9 cells, we observed that most mutants, like USP2 WT, increased CD47 expression. However, the USP2 D145N mutant reduced CD47 protein levels (Supplementary Fig. 2c). Further investigation is needed to determine the clinical relevance of these USP2 mutations in modulating CD47 expression.

a IB analysis of HEK293T WCL and anti-HA immunoprecipitates (IPs). HEK293T cells were co-transfected with indicated constructs and treated with 10 µM MG132 for 12 h before harvesting. b IB analysis of glutathione S-transferase (GST) pull-down protein mixture from HEK293T cell lysates that overexpressed CD47-cHA incubated with bacterially purified recombinant GST or GST-USP2 protein. c IB analysis of GST pull-down products from HEK293T cell lysates that overexpressed Flag-USP2 incubated with bacterially purified recombinant GST or GST-CD47 protein. d Schematic representation of WT and truncations of USP2, including the N-terminal region of amino acid (aa) 1–266 and C-terminal domain of aa267–605. e IB analysis of WCL and anti-HA IPs obtained from HEK293T cells, which were co-transfected with indicated constructs and treated with 10 µM MG132 for 12 h before harvesting. f IB analysis of GST pull-down products derived from HEK293T cell lysates that ectopic expression of CD47-cHA incubated with bacterially purified recombinant GST, GST-USP2 WT, and GST-USP2 truncations. g IB analysis of GST pull-down products derived from HEK293T cell lysates that ectopic expression of Flag-USP2 WT and truncations incubated with bacterially purified recombinant GST-CD47 protein. h Schematic diagram of CD47 WT and its various deletion mutants. i IB analysis of GST pull-down products derived from HEK293T cell lysates that ectopic expression of CD47-cHA WT and deletion mutants incubated with bacterially purified recombinant GST-USP2 protein. j, k IB analysis of WCL and IPs derived from H1975 (j) and PC9 (k) cells. l, m IB analysis of WCL and Ni-NTA pull-down products of the in vivo ubiquitination assay in the guanidine-HCl denaturing buffer. HEK293T cells were co-transfected with the indicated constructs and treated with 10 µM MG132 for 12 h before harvesting. n IB analysis of WCL and Ni-NTA pull-down products of the in vivo ubiquitination assay in the guanidine-HCl denaturing buffer. HEK293T cells were co-transfected with the indicated constructs and treated with 2 µM ML364 for 16 h and 10 µM MG132 for 12 h before harvesting. o, p IB analysis of WCL and IPs derived from lysates of H1975 (o) and PC9 (p) cells using indicated K48-Ubi antibodies. Cells were treated with 20 µM MG132 for 6 h before harvesting. q, r IB analysis of WCL and anti-CD47 IPs derived from H1975 (q) or PC9 (r) cells stably expressing shUSP2 or shGFP, respectively. Cells were treated with 20 µM MG132 for 6 h before harvesting. n = 3 biologically independent experiments for (a, b, c, e, f, g, and i–r). Source data are provided as the Source Data file.
Next, we examined whether USP2 could serve as a deubiquitinating enzyme for removing the ubiquitination on CD47. As expected, the in vivo ubiquitination assay showed that the ectopic expression of USP2 reduced the ubiquitination on CD47 (Fig. 2l). Moreover, the enzymatically inactive mutant C276A of USP2 failed to deubiquitinate CD47, suggesting that the deubiquitinating enzyme activity of USP2 is required for removing ubiquitination on CD47 (Fig. 2m). In line with this result, the ML364 inhibitor treatment suppressed the USP2-mediated deubiquitination on CD47 in cells (Fig. 2n). To determine the specific type of ubiquitin linkage(s) assembled on CD47, we co-transfected CD47 with each of seven lysine (K)-only ubiquitin constructs, where each construct retained only the specified lysine, with the remaining six lysine residues mutated to arginine (R). The results demonstrated that CD47 is primarily modified by K48-linked ubiquitination, which can be obviously removed through ectopic expression of USP2 (Supplementary Fig. 2d–f). Additionally, K48-linked ubiquitination on endogenous CD47 was detected in both H1975 and PC9 lung cancer cell lines (Fig. 2o, p). Notably, USP2 knockdown with two different shRNAs significantly increased the levels of K48-linked ubiquitination on endogenous CD47, indicating that USP2 specifically regulates this modification (Fig. 2q, r). Taken together, these findings demonstrate that USP2 primarily stabilizes CD47 by removing its K48-linked ubiquitination through its deubiquitinating enzymatic activity in cells.
USP2 inhibition enhances macrophage phagocytosis and reshapes an inflamed tumor microenvironment
The expression level of CD47 on the tumor cell surface affects the phagocytosis of macrophages11,12. Our results above showed that USP2 inhibition reduced CD47 protein levels in tumor cells, we therefore hypothesized that USP2 inhibition may promote phagocytosis of tumor cells by macrophages. To test this hypothesis, we performed the in vitro phagocytosis assays by co-culturing THP-1-derived macrophages and cancer cells. Tumor cells (PC9 or H1975) were labeled with the fluorescent dye carboxyfluorescein diacetate succinimidyl ester (CFSE) and co-cultured with the THP-1-derived mature macrophages, which were subjected to CD11b antibody staining for flow cytometric analysis. Tumor cells pre-treated with ML364 showed a significant increase in phagocytosis rate compared with those cells treated with DMSO (Fig. 3a–d). Nevertheless, ML364 treatment on PC9 or H1975 cells with CD47 gene depletion by sgRNA didn’t enhance the phagocytic capacity of THP-1-derived macrophages (Supplementary Fig. 3a–d), indicating that USP2 inhibition by ML364 could improve the THP-1-mediated phagocytosis of tumor cells primarily through reducing CD47 protein expression on PC9 or H1975 cells. Moreover, flow cytometry analysis by 7-AAD staining showed that ML364 treatment did not significantly increase lung cancer cell death compared to the DMSO group (Supplementary Fig. 3e–j). This suggests that the enhanced phagocytosis of lung cancer cells following ML364 pretreatment is primarily due to its inhibitory effect on CD47, rather than cytotoxic effects towards cancer cells at the experimental conditions.

a–d In vitro phagocytosis assay. Representative flow cytometry plots displaying THP-1 macrophage phagocytosis of PC9 cells (a) and H1975 cells (c). Quantitative results for phagocytosis to show the percentage of CFSE+ cells (PC9 cells for b and H1975 cells for d) in CD11b+ cells (THP-1 macrophages). PC9 or H1975 cells were pre-treated with DMSO or 2 µM ML364 for 16 h and pre-stained with carboxyfluorescein diacetate succinimidyl ester (CFSE). Subsequently, PC9 or H1975 cells were co-cultured with THP-1 cells, which have been stimulated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA) for 48 h to induce THP-1 monocytes into macrophages. After 6 h of co-culture at 37 °C, cells were harvested for the flow cytometry analysis. n = 3 per group for (b, d). e A schematic treatment plan for immunocompetent C57BL/6J mice bearing LLC tumors. Mice were subcutaneously implanted with 2 × 106 LLC cells and treated with a control vehicle or USP2 inhibitor (ML364, 5 mg/kg), respectively. i.p., intraperitoneal; s.c., subcutaneous. f–h The growth of subcutaneous LLC tumors was assessed. Images of LLC tumors at the endpoint (f). Tumor volume for different treatment groups was measured with a caliper, and the tumor growth curve was plotted (g). The weight of LLC tumors was measured at the endpoint (h). i Uniform Manifold Approximation and Projection (UMAP) of single-cell RNA sequencing (scRNA-seq) data showing the myeloid lineage subsets from LLC tumors treated with ML364 (5 mg/kg) or control vehicle for 12 days. Color coding indicates the different cell types. M1, M1 macrophages; M2, M2 macrophages; M0, M0 macrophages; DCs, dendritic cells; MDSCs, myeloid-derived suppressor cells. j Dot-plot showing the selected representative gene markers enriched in myeloid lineage subsets. k Comparative abundance of the myeloid lineage subsets in LLC tumors treated with control vehicle (blue) versus ML364 (green) from the scRNA-seq data. l, m Quantification of MHCII+ (M1 macrophage) cells (l) or CD206+ (M2 macrophage) cells (m) represented as a percentage of F4/80+ macrophages in subcutaneous LLC tumors derived from mice with indicated treatments. n Quantification of Gr1+Ly6C+cells (myeloid-derived suppressor cells, MDSCs) represented as a percentage of CD11b+ monocytes in subcutaneous LLC tumors derived from mice with indicated treatments. o Quantification of CD8+ T cells represented as a percentage of CD3+ lymphocytes in subcutaneous LLC tumors derived from mice-indicated treatments. p–r Representative IF staining images of CD86 (orange) and CD206 (green) in subcutaneous LLC tumors with indicated treatments (p). Each point represents the average counts of positive cells within three high-power fields (q, r). Scale bars, 100 μm (left); zoom scale bars, 50 μm (right). For (b, d, h, l–o, q, and r), unpaired two-tailed Student’s t-test. Two-way ANOVA for (g). n = 5 mice per group for (g, h, l–o, q, and r), Data are shown as the mean ± SD. P < 0.05 was considered statistically significant. Source data are provided as the Source Data file.
To further investigate whether USP2 inhibition affects tumor immune microenvironment in vivo, we established a subcutaneous Lewis Lung Carcinoma (LLC) model in immunocompetent C57BL/6J mice, which were treated with ML364 as indicated (Fig. 3e). We observed that ML364 treatment suppressed tumor growth as measured by tumor volume and tumor weight compared with those tumors treated with control vehicle (Fig. 3f–h).
To further explore how the ML364 treatment affects the tumor-infiltrating immune cells in the TME, we performed single-cell RNA sequencing (scRNA-seq) of LLC tumors treated with ML364 or vehicle, respectively. To define the subpopulation structure of tumor cells and tumor-infiltrating immune cells, we computationally pooled data from the two groups representing a total of 39,765 cells, and clustering analysis was carried out to identify various cell populations (Supplementary Fig. 4a). Clusters were further divided into cancer cells, monocyte/macrophage, T cells, DCs, NK cells, B cells, neutrophils, stromal cells, and endothelial cells, based on the expression of marker genes (Supplementary Fig. 4b–d). Of note, the abundance of tumor-infiltrating monocyte/macrophage was the highest among immune cell types and increased in tumors with ML364 treatments (Supplementary Fig. 4e). Further analysis of myeloid lineage, we found that the proportion of M1 macrophages (anti-tumor macrophages) was increased in tumors with ML364 treatments, whereas the proportion of M2 macrophages (pro-tumor macrophages) and myeloid-derived suppressor cells (MDSCs) were dramatically decreased (Fig. 3i–k). For the analysis of the T cell population, the abundance of tumor-infiltrating effector CD8+ T cells was slightly elevated in tumors after ML364 treatment, whereas the abundance of exhausted CD8+ T cells was reduced (Supplementary Fig. 4f–h). Thus, these results demonstrated that ML364 treatment might reshape the immune cell composition of the TME from a pro-tumorigenic phenotype to an anti-tumorigenic phenotype.
Next, we utilized flow cytometry to analyze tumor infiltration of immune cells following ML364 treatment, the strategy for flow cytometry was illustrated (Supplementary Fig. 5a). The flow cytometric analysis showed that USP2 inhibition decreased CD47 protein levels in CD45− cells (mostly tumor cells) (Supplementary Fig. 5b), which is consistent with our results in vitro. In keeping with the findings of scRNA-seq, the results of flow cytometric analysis showed that ML364 treatment resulted in a significant increase in the tumor-infiltrating M1 macrophages and CD8+ T cells, accompanied by a decrease in the M2 macrophages and MDSCs in the TME (Fig. 3l–o). Moreover, effector CD8+ T cells with production of interferon-gamma (IFN-γ) and tumor necrosis factor (TNF) were significantly increased in tumors with ML364 treatment (Supplementary Fig. 5c, d). The IF staining of tumor tissues also demonstrated that upregulation of M1 macrophages, CD8+ T cells, and downregulation of M2 macrophages were observed in tumors with ML364 treatment (Fig. 3p–r and Supplementary Fig. 5e, f).
Given that anti-CD47 antibodies might elicit severe side effects such as anemia and thrombocytopenia in clinical trials or animal models38,39, we wondered whether ML364 treatment could cause hematologic or other toxicities. Of note, our results demonstrated that ML364 treatment did not cause apparent drug-related toxicities in mice. No statistically significant difference was observed in the counts of blood cells including red blood cells (RBC), white blood cells (WBC), or platelets (PLT) from the blood of mice treated with vehicle or ML364 (Supplementary Fig. 5g–i), which might account for the low gene expression level of Usp2 in RBC or PLT compared to LLC tumor (Supplementary Fig. 5j). Meanwhile, aspartate amino transferase (AST) and alanine amino transferase (ALT) were also not significantly altered in both groups of vehicle and ML364 treatment (Supplementary Fig. 5k, l). In addition, no obvious differences were observed regarding the body weight, RBC, WBC, PLT counts, and liver function (ALT, AST) between WT and Usp2−/− mice at eight weeks old (Supplementary Fig. 5m–r). Furthermore, to explore the expression levels of USP2 in different tissues, we retrieved the USP2 gene in the GTEx portal (https://www.gtexportal.org/home)40, which establishes a database and a tissue bank that analyzes gene expression in different human tissues. The result showed that USP2 expression was lower in whole blood than other organs or tissues (Supplementary Fig. 5s). Likewise, the Usp2 mRNA level was obviously lower in RBC or PLT compared to other organs such as lung, liver, colon, kidney and stomach for C57BL/6J WT mice at 8-week-old (Supplementary Fig. 5t). Therefore, the low hematologic cytotoxicity of ML364 treatment may result from the low expression of Usp2 gene in whole blood, which prevents the degradation of the CD47 protein on blood by ML364 treatment. Furthermore, we also did not observe significant organ toxicity in the colon, kidney, lung, and stomach of mice treated with ML364, likely due to the relatively low dose (5 mg/kg), when compared to the vehicle control group (Supplementary Fig. 5u). Taken together, these results suggest that pharmacological inhibition of USP2 by ML364 may reshape an anti-tumor immune microenvironment to suppress tumor growth without apparent toxic side effects.
To investigate whether the anti-tumor effect of ML364 is dependent on CD47 expression in lung cancer cells, we first established a Cd47 knockout LLC cell line and treated mice bearing subcutaneous sgControl- or sgCd47-LLC tumors (Supplementary Fig. 6a, b). The results revealed that in the sgControl group, both ML364 and CD47 mAb monotherapy significantly inhibited tumor growth compared to the vehicle group, with no substantial difference between the two treatments (Supplementary Fig. 6c, d). In contrast, in the sgCd47 group, ML364 or CD47 mAb treatment did not result in a significant further inhibition of tumor growth relative to the vehicle control group (Supplementary Fig. 6c, d). These findings indicate that the anti-tumor effect of ML364 is predominantly mediated by CD47 expression in lung cancer cells under the experimental conditions employed (Supplementary Fig. 6c, d). Furthermore, we assessed the infiltration of M1 and M2 macrophages in the tumors of treated mice. In sgControl LLC tumors, we observed that both ML364 and CD47 mAb significantly increased tumor-infiltrating M1 macrophages while reducing M2 macrophages (Supplementary Fig. 6e, f). Notably, Cd47 knockout also significantly increased M1 macrophage infiltration and decreased M2 macrophages (Supplementary Fig. 6e, f). However, ML364 or CD47 mAb did not further alter the M1 or M2 macrophage ratio in Cd47 KO tumors (Supplementary Fig. 6e, f). These findings collectively suggest that inhibition of USP2 with ML364 enhances M1 and reduces M2 macrophage infiltration in LLC tumors from C57BL/6J mice, primarily through CD47 regulation. Previous studies have demonstrated that the impact of CD47 mAb treatment on tumor-infiltrating M2 macrophages in mice has shown variability, with some reporting no effect on M2 macrophage infiltration and others observing a reduction in M2 macrophages41,42,43. These differences are likely due to variations in the cancer models, mouse strains, and the specific CD47 antibodies employed, which warrants further in-depth study.
Correspondingly, CD47 mAb led to a significant reduction in red blood cell count, but no significant blood toxicity was observed in the other treatment groups (Supplementary Fig. 6g–i). Furthermore, Cd47 knockout in LLC tumors or CD47 mAb treatment did not induce significant liver toxicity or cause notable changes in body weight in the mice, compared to the vehicle control group (Supplementary Fig. 6j–l). Overall, inhibition of USP2 by ML364 downregulates CD47 expression and remodels the tumor immune microenvironment, thereby exerting its anti-tumor effects.
USP2 inhibition by ML364 sensitizes tumors to anti-PD-1 immunotherapy in syngeneic LLC lung tumor models
Our results above demonstrate that USP2 inhibition reprograms an inflamed TME, which may set up a situation that sensitizes tumors to anti-PD-1 immunotherapy. Here, we first utilized a syngeneic mouse LLC tumor model to evaluate the effect of combined the low-dose ML364 (5 mg/kg) and anti-PD-1 therapy on tumor growth and mice survival (Fig. 4a). Compared with either single-agent or control treatment, the combination of ML364 and anti-PD-1 antibody significantly retarded the tumor growth and prolonged the overall survival of immunocompetent C57BL/6J mice bearing LLC tumors (Fig. 4b–d and Supplementary Fig. 7a). In line with the result in Supplementary Fig. 5b, ML364 monotherapy or combined therapy significantly reduced the CD47 expression on CD45− cells (mostly tumor cells) (Fig. 4e). Moreover, the combination therapy led to a significant increase in anti-tumor M1 macrophages, accompanied by a dramatical reduction in immunosuppressive M2 macrophages and MDSCs compared with monotherapy (Fig. 4f, g and Supplementary Fig. 7b). Meanwhile, the ML364 combination with anti-PD-1 therapy significantly elevated the tumor-infiltrating cytotoxic CD8+ T cells (Fig. 4h and Supplementary Fig. 7c, d). Likewise, the results of IHC staining showed that the ML364 treatment or combination therapy decreased the CD47 expression, and combination therapy increased the tumor-infiltrating macrophages and CD8+ T cells compared with ML364 or anti-PD-1 monotherapy (Supplementary Fig. 7e–h). Meanwhile, IHC staining demonstrated that treatment with ML364 alone or in combination with PD-1 mAb did not result in a significant increase in PD-L1 expression under the conditions of this experiment (Supplementary Fig. 7e, i). Additionally, we did not observe apparent toxic side effects on mice with a combination therapy of ML364 and anti-PD-1. There were no significant differences in body weights and various organs of mice among different treatment groups (Supplementary Fig. 7j, k). And no statistically significant differences in blood routine tests and liver function examination were observed (Supplementary Fig. 7l–q). These results suggest that the combination of USP2 inhibition and anti-PD-1 therapy significantly suppresses the tumor growth and does not display apparent toxic side effects in mice.

a A schematic treatment plan for immunocompetent C57BL/6J mice bearing LLC tumors. Mice were subcutaneously implanted with 2 × 106 LLC cells and treated with a control vehicle, USP2 inhibitor (ML364, 5 mg/kg, daily for 12 doses), anti-PD-1 mAb (100 μg per mouse, every three days for 4 treatments), or combined treatment, respectively. i.p., intraperitoneal; s.c., subcutaneous; MAb, monoclonal antibody. b, c Tumor growth (b) or Kaplan–Meier survival curves (c) for C57BL/6J bearing LLC tumors with indicated treatments. d The weight of LLC tumors was measured at the endpoint. e Quantification of cellular surface CD47 on CD45− cells in subcutaneous LLC tumors derived from C57BL/6J mice with indicated treatments. f–h Quantification of MHCII+ (M1 macrophage) cells (f) or CD206+ (M2 macrophage) cells (g) represented as percentage of F4/80+ macrophages, and CD8+ T cells (h) represented as percentage of CD3+ T cells in subcutaneous LLC tumors derived from C57BL/6J mice-indicated treatments. i A schematic treatment plan for the immunocompetent C57BL/6J lung tumor mouse model. Mice were injected with 1 × 106 LLC cells/per mouse via tail vein injection, followed by treatments with control vehicle, ML364 (5 mg/kg), anti-PD-1 mAb (100 μg per mouse), or combined treatment, respectively. i.p., intraperitoneal; i.v., intravenous. j–l Representative H&E-stained images of lung tissues from mice across different groups (j), tumor size was quantified by measuring the cross-sectional area of all tumors (k), and the tumor-occupied lung area (tumor area, %) was calculated (l) from three nonconsecutive sections per mouse. Scale bars, 5 mm. m LLC-GFP tumors bearing C57BL/6J mice were treated as (a). Representative images of IF staining for tumor cells (GFP, green), macrophages (F4/80, red), and nuclei (DAPI, blue). The yellow (indicated with arrows) showing colocalization of GFP (green) and F4/80 (red) double-positive cells suggested the macrophage phagocytosis of tumor cells. Each point represents the average counts of yellow cells within three high-power fields. The sections were scanned at 60× with an oil objective on a confocal microscope. Scale bars, 50 μm (left panels); zoom scale bars, 20 μm (right panels). For (d–h, k, and l), unpaired two-tailed Student’s t-test. Two-way ANOVA for (b). Log-rank test for (c). n = 5 mice per group. Data are shown as the mean ± SD. P < 0.05 was considered statistically significant. Source data are provided as the Source Data file.
Additionally, we also established a lung cancer mouse model by tail vein injection of LLC cells, and treated mice with an intraperitoneal injection of ML364, anti-PD-1, alone or combination (Fig. 4i). Hematoxylin and eosin (H&E) staining of lungs showed a combination of ML364 and anti-PD-1 antibody dramatically suppressed tumor development evidenced by tumor size and tumor area, compared with ML364 or anti-PD-1 monotherapy (Fig. 4j–l). These findings together suggest that inhibition of USP2 by ML364 treatment sensitizes tumors to anti-PD-1 immunotherapy in syngeneic LLC lung tumor models.
To evaluate the phagocytosis of tumor cells by macrophages after ML364, anti-PD-1, alone or in combination therapy in vivo, we generated a GFP-transduced LLC cell line (LLC-GFP) and subcutaneously injected LLC-GFP cells into immunocompetent C57BL/6J mice. After treatments, LLC-GFP-tumor tissues were analyzed with immunofluorescent staining. The green fluorescence represented the GFP-expressing LLC tumor cells, whereas the red fluorescence indicated the tumor-infiltrating macrophages (Fig. 4m and Supplementary Fig. 8a). Merged yellow fluorescence exhibited colocalization of green and red fluorescence, and showed stronger signals in the combined treatment group compared with ML364 or anti-PD-1 single-agent treatment group (Supplementary Fig. 8b, c). The zoom 3D stereoscopic plane diagram view along the X, Y, and Z axes demonstrate that the red macrophage fully encapsulated the green tumor cell, suggesting that LLC-GFP-tumor cells were phagocytosed in tumors with combination therapy (Fig. 4m). The findings indicate that ML364 combined with anti-PD-1 therapy potentiates the phagocytic activity of macrophages against tumor cells in vivo.
High-dose ML364 combined with anti-PD-1 therapy significantly extends the survival of mice bearing LLC tumors
Subsequently, to evaluate the long-term survival of mice, we prolonged the duration of the combination treatment. Although combination therapy significantly inhibited tumor growth in our lung cancer mouse model, survival analysis indicated that none of the mice in the combination treatment group achieved tumor cure and long-term survival, even with extended treatment of ML364 combined with PD-1 mAb therapy (Fig. 5a–c), which might be due to the low-dose ML364 (5 mg/kg) treatment. Given the strong therapeutic efficacy of higher doses of ML364 (30 mg/kg) in combination with PD-1 mAb observed in a recent mouse breast cancer study29, we administered a higher dose of ML364 (30 mg/kg) in combination with PD-1 mAb to subcutaneous LLC tumor-bearing mice. Indeed, this high-dose treatment resulted in significantly enhanced therapeutic efficacy compared to the low-dose treatment (5 mg/kg), with a notable extension in survival (Fig. 5d–f). IHC staining revealed that high-dose ML364 significantly reduced CD47 expression while increasing PD-L1 levels (Fig. 5g–i). These findings suggest that, in the highly malignant LLC mouse tumor model, high-dose ML364 treatment not only markedly decreases CD47 expression but also upregulates PD-L1, thereby potentiating the efficacy of PD-1/PD-L1 blockade therapy.

a Diagrammatic treatment plan for immunocompetent C57BL/6J mice with LLC tumors. Mice were implanted with 2 × 106 LLC cells and treated with Vehicle, ML364 (5 mg/kg daily for 18 doses), anti-PD-1 mAb (100 μg/mouse every three days for 6 doses), or combination therapy. i.p., intraperitoneal; s.c., subcutaneous. b Tumor volume in different treatment groups was assessed using calipers, and the tumor growth curve was generated. n = 8 (Vehicle); 8 (ML364); 8 (anti-PD-1 mAb) and 10 (Combined therapy) mice. c Kaplan–Meier survival curves for C57BL/6J bearing LLC tumors with indicated treatments. Log-rank test. n = 8 (Vehicle); 8 (ML364); 8 (anti-PD-1 mAb) and 10 (Combined therapy) mice. d A schematic treatment plan for immunocompetent C57BL/6J mice bearing 2 × 106 LLC tumors. Mice were treated with a control vehicle, USP2 inhibitor (ML364, 30 mg/kg for 18 doses), anti-PD-1 mAb (200 μg per mouse for 6 doses), or combined treatment, respectively. e Tumor volume in the different treatment groups was measured using calipers, and the corresponding tumor growth curve was generated. Vehicle (n = 9), ML364 (n = 8), PD-1 mAb (n = 8), ML364 & PD-1 mAb (n = 13) mice per group. f Kaplan–Meier survival curves for C57BL/6J bearing LLC tumors with indicated treatments. Vehicle (n = 9), ML364 (n = 8), PD-1 mAb (n = 8), ML364 & PD-1 mAb (n = 13) mice per group. Log-rank test. g IHC staining for CD47 and PD-L1 in LLC tumors from C57BL/6J mice treated with indicated reagents. Scale bar, 50 μm. h, i Quantification for CD47 (h) and PD-L1 (i) positive cells. Each point represents the average counts of positive cells within three high-power fields. For (h) and (i), unpaired two-tailed Student’s t-test. n = 5 mice per group. Log-rank test for (c, f). Data are shown as the mean ± SD. P < 0.05 was considered statistically significant. Source data are provided as the Source Data file.
The anti-tumor effects of ML364 combined with anti-PD-1 therapy require both macrophages and CD8+ T cells
To further investigate the role of CD47 expression in the efficacy of ML364 combined with PD-1 mAb, we treated mice bearing subcutaneous sgControl- or sgCd47-LLC tumors. The results demonstrated that, in the sgControl group, treatment with ML364 alone significantly reduced tumor growth (Fig. 6a–c). However, combination therapy with ML364 and PD-1 mAb led to even greater tumor inhibition compared to single-agent treatment, thereby reinforcing our findings. In the sgCd47 group, while Cd47 knockout resulted in reduced tumor growth, ML364 treatment did not further inhibit tumor growth, suggesting that the anti-tumor effect of ML364 may be dependent on CD47 expression under the experimental conditions employed. Moreover, no significant difference in the anti-tumor effects of ML364 & PD-1 mAb combination therapy was observed between the sgControl and sgCd47 groups, suggesting that, in the mouse lung cancer model, the anti-tumor effect of ML364 combined with PD-1 mAb is primarily driven by the USP2-CD47 axis (Fig. 6a–c). Meanwhile, no blood toxicity, liver toxicity, or body weight changes were observed in mice in the sgCd47 group compared to the sgControl group (Fig. 6d and Supplementary Fig. 8d–h).

a A schematic treatment plan for immunocompetent C57BL/6J mice bearing sgControl- or sgCd47-LLC tumors. Mice were subcutaneously implanted with 2 × 106 sgControl or sgCd47-LLC cells and treated with a control vehicle, USP2 inhibitor (ML364, 5 mg/kg, daily for 12 doses), anti-PD-1 mAb (100 μg per mouse, every three days for 4 treatments), or combined treatment, respectively. i.p., intraperitoneal; s.c., subcutaneous. b, c Tumor volume in each treatment group was measured using calipers, and the tumor growth curve was subsequently plotted (b). At the study endpoint, the weight of LLC tumors was recorded (c). n = 5 mice per group. d The body weight of C57BL/6J mice was monitored throughout the duration of the indicated treatment. n = 5 mice per group. e A schematic treatment plan for CD8 T cell or macrophage deletion experiments. Mice were randomly divided into five groups: (1) vehicle, (2) ML364 & PD-1 mAb (MP), (3) MP & CD8 deletion (αCD8), (4) MP & macrophage deletion (clodronate liposomes, CL), and (5) MP & αCD8 & CL. On day 5 post-tumor implantation, mice in MP were treated with a combination of ML364 (5 mg/kg) daily for 12 treatments and anti-PD-1 mAb (100 μg per mouse) every 3 days for 4 doses. After grouping as indicated, mice received αCD8 antibody (200 μg per mouse) every 3 days for 4 doses, starting three days before tumor implantation. Macrophage deletion by CL was administered with 200 μl per mouse every 3 days for 4 doses, starting on the day of tumor implantation. i.p., intraperitoneal; s.c., subcutaneous. f, g Tumor volume for each treatment group was measured using calipers, and the tumor growth curve was plotted (f). The weight of LLC tumors was measured at the endpoint (g). n = 5 mice per group. h, i Quantification of CD8+ represented as percentage of CD3+ T cells (h) and F4/80+ macrophages represented as percentage of CD11b+ cells (i) in subcutaneous LLC tumors derived from C57BL/6J mice treated with indicated reagents. For (c) and (g–i), unpaired two-tailed Student’s t-test. n = 5 mice per group. Two-way ANOVA for (b, d, and f). Data are shown as the mean ± SD. P < 0.05 was considered statistically significant. Source data are provided as the Source Data file.
The USP2 inhibitor ML364 downregulates CD47 protein expression in lung cancer cells and enhances macrophage-mediated phagocytosis of tumor cells in vitro. In vivo, combinatorial treatment with ML364 and anti-PD-1 antibody significantly augments macrophage-driven tumor cell clearance. Furthermore, this therapeutic regimen markedly increases intratumoral infiltration of CD8+ T cells, indicating that both macrophages and CD8+ T cells likely contribute critically to the observed anti-tumor efficacy. To determine whether the tumor growth suppression associated with ML364 combined with PD-1 mAb therapy primarily depends on CD8+ T cells and/or macrophages, we used anti-CD8α antibodies and clodronate liposomes (CL) to selectively deplete CD8+ T cells or macrophages in mice, respectively. Tumor growth analysis revealed that blockade of either CD8+ T cells or macrophages partially mitigated the tumor-suppressive effects of ML364 & PD-1 mAb therapy (Fig. 6e–i). However, when both CD8+ T cells and macrophages were blocked, the tumor-suppressive effect of the ML364 & PD-1 mAb combination therapy was completely abolished (Fig. 6e–i). These results indicate that ML364 & PD-1 mAb combination therapy inhibits tumor growth through the cooperative actions of both macrophage-mediated phagocytosis and CD8+ T cell-mediated tumor cell killing.
USP2 inhibition by ML364 or genetic depletion overcomes resistance to anti-PD-1 immunotherapy in Kras LSL-G12D/+ ; Trp53 fl/fl (KP) mouse tumor model
To further investigate the role of USP2 inhibition in regulating anti-PD-1 immunotherapy in vivo, we used a genetically engineered KrasLSL-G12D/+; Trp53fl/fl (KP) mouse tumor model, which was induced to form autochthonous lung tumors after five weeks of intranasal instillation of adenovirus expressing Cre-recombinase (Ad-Cre)44,45. KP mice bearing lung tumors were treated with an intraperitoneal injection of ML364, anti-PD-1, alone or combined (Fig. 7a). Consistent with the results of the syngeneic mouse LLC tumor model (Fig. 4), the combination of ML364 and anti-PD-1 antibody significantly inhibited tumor development in KP mice, evidenced by the reduced tumor size, and area, compared to ML364 or anti-PD-1 monotherapy (Fig. 7b–d).

a A schematic treatment plan for autochthonous lung tumors in KrasLSL-G12D/+; Trp53fl/fl (KP) mice. KP mice were administered with an intranasal injection of Ad-Cre with 2 × 106 pfu/mouse. Five weeks after induction, mice were treated as indicated with control vehicle, ML364 (5 mg/kg), anti-PD-1 mAb (100 μg per mouse), or combined treatment, respectively. i.p., intraperitoneal; i.n., intranasal. b–d Representative H&E staining images of lung tissues from mice during different groups (b), tumor size was quantified by measuring the cross-sectional area of all tumors (c), and the tumor area (%) was calculated (d) from three nonconsecutive sections per KP mouse. Scale bars, 5 mm. e A schematic treatment plan for autochthonous lung tumors in KP and KrasLSL-G12D/+; Trp53fl/fl; Usp2−/− (KPU) mice. KP and KPU mice were administered with an intranasal injection of Ad-Cre with 2 × 106 pfu/mouse. Five weeks after induction, mice were treated as indicated with a control vehicle or anti-PD-1 mAb (100 μg per mouse). i.p., intraperitoneal; i.n., intranasal. f Micro-CT images of lungs in the indicated planes from KP and KPU mice with indicated treatments. Three-dimensional rendering of micro-CT data shows lungs in gray. Scale bars, 5 mm. g The healthy lung volumes of mice in each group were quantified using computational extraction through 3D reconstruction by the Avatar program. h–j Representative images of H&E-stained lung tissues from mice in different groups (h), tumor size was quantified by measuring the cross-sectional area of all tumors (i), and the tumor area (%) was calculated (j) based on three nonconsecutive sections per KP or KPU mouse. Scale bars, 5 mm. For (c, d, g, i, and j), unpaired two-tailed Student’s t-test. n = 8 mice per group. Data are shown as the mean ± SD. P < 0.05 was considered statistically significant. Source data are provided as the Source Data file.
Next, we genetically knocked out Usp2 in KP mice and explored the impact of Usp2 deficiency on lung cancer progression and response to immunotherapy. To this end, KrasLSL-G12D/+; Trp53fl/fl; Usp2−/− (KPU) mice were generated by breeding Usp2−/− mice with KP mice (Supplementary Fig. 9a–d). KP and KPU mice bearing Ad-Cre induction of lung tumors were treated with anti-PD-1 or control IgG (Fig. 7e). We then performed CT scans of the mouse lungs and generated 3D reconstructions of the entire lung in treated mice. As the tumor developed and invaded normal lung tissue, defects in the lung morphology became apparent and expanded with tumor progression. Consequently, the volume of the reconstructed healthy lung stroma was used as a surrogate marker for spontaneous lung cancer progression. Whole lung volume analysis revealed that the KPU with PD-1 mAb treatment group had significantly larger lung volumes, with markedly reduced tumor progression compared to the remaining groups (Fig. 7f, g). Notably, H&E staining of lungs showed that anti-PD-1 treatment significantly retarded the lung tumor development in KPU mice compared to KP mice by measuring and comparing their tumor size and area in lungs (Fig. 7h–j). Together, these results reveal that suppressing USP2 by its pharmacological inhibitors, ML364, or genetic deletion, sensitizes tumors to anti-PD-1 immunotherapy in KP lung cancer models.
Cancer patients with low USP2 expression exhibit better responses to cancer immunotherapies
To evaluate the clinical correlation between USP2 expression and immune cell infiltration in tumors, we first performed multiplex immunohistochemistry (mIHC) staining for CD163+ M2 macrophages and CD8+ T cells in specimens of human lung adenocarcinoma. CD8+ cells were significantly increased in USP2 low samples as compared to those in USP2 high samples, whereas CD163+ M2 macrophages were obviously decreased (Fig. 8a, b). Then we utilized the bioinformatic method to analyze the clinical data from published databases. The results obtained from the analysis by the TIMER2.0 software46 indicated a positive correlation between USP2 expression and immunosuppressive immune cells (M2 macrophages), while demonstrating a negative correlation with immunoactive immune cells (M1 macrophages and CD8+ T cells) in the TME (Fig. 8c–e). Thus, these results from the mIHC staining and bioinformatic analysis suggest that USP2 expression may affect the infiltration of immune cells in the TME to control the clinical outcomes of cancer patients.

a, b Representative IF staining images for CD163 (green) and CD8 (red) in human lung adenocarcinoma (LUAD) tissues with the USP2 high (n = 13) and low (n = 16) expression (a). Quantifications of CD163 and CD8 positive cells in LUAD samples with USP2 high or low expression, respectively (b). Each point represents the average counts of positive cells within three high-power fields. Scale bars, 500 μm (left panels); zoom scale bars, 50 μm (right panels). c–e USP2 expression was negatively correlated with the infiltration of M1 macrophages (c) and CD8+ T cells (e), while USP2 displayed a positive correlation with the infiltration of M2 macrophages (d) in lung cancer patients based on the immune association analysis by TIMER2.0 software. f, g Representative images from IHC staining of CD47 and USP2 in responders (R) and non-responders (NR) of lung cancer patients with anti-PD-1 therapy (f). Scale bar, left panels: 100 μm; right panels: 50 μm. Association between USP2 or CD47 expression and response to anti-PD-1 therapy in lung cancer patients (g). n = 6 (R), or 6 (NR). Patients were divided into USP2 or CD47 low and high expression groups according to their IHC score mean value (f). h, i Representative images of multiplex immunohistochemistry (mIHC) staining for R and NR of lung cancer patients with anti-PD-1 immunotherapy (h). Quantifications of CD163 (white), CD8 (red), and CK (green) in each group (i). Each point represents the average counts of positive cells within three high-power fields. n = 5 per group. Scale bars, 100 μm (left panels); zoom scale bars, 50 μm (right panels). j, k Representative images from IHC staining of CD47 and USP2 in responders and non-responders of oral cancer patients with anti-PD-1 therapy (j). Scale bar, left panels: 100 μm; right panels: 50 μm. Association between USP2 or CD47 expression and response to anti-PD-1 therapy in oral cancer patients (k). n = 25 (R), or 19 (NR). Patients were divided into USP2 or CD47 low and high expression groups according to their IHC score mean value (j). l, m Representative images of mIHC staining for responders and non-responders of oral cancer patients with anti-PD-1 therapy (l). Quantifications of CD163 (white), CD8 (red), and CK (green) in each group (m). Each data point represents the average counts of positive cells within three high-power fields. n = 18 (R), or 13 (NR). Scale bars, 100 μm (left panels); zoom scale bars, 50 μm (right panels). For (b), unpaired two-tailed Student’s t-test. For (g) and (k), a two-sided χ2 test. For (i) and (m), two-tailed Non-parametric Mann–Whitney test. Data are shown as the mean ± SD P < 0.05 was considered statistically significant. Source data are provided as the Source Data file.
To explore the relationship between USP2 expression and immunotherapeutic efficacy, we performed IHC and mIHC staining of specimens from cancer patients who received immunotherapy. The results showed that samples from lung cancer patients that responded to anti-PD-1 therapy displayed low expression of USP2 and CD47, accompanied by high CD8+ T cells and low CD163+ M2 macrophages in TME (Fig. 8f–i). By contrast, specimens derived from lung cancer patients that did not respond to anti-PD-1 therapy had high expression of USP2 and CD47, as well as exhibited low tumor-infiltrating CD8+ T cells and high CD163+ M2 macrophages (Fig. 8f–i). In keeping with these results in lung cancer patients, we also observed similar results in human oral cancer patients. The results of IHC staining in oral cancer specimens demonstrated that the expression of USP2 and CD47 was lower in most responders than those non-responders to anti-PD-1 immunotherapy (Fig. 8j, k). Moreover, a similar change was observed in mIHC results that CD8+ T cells were increased and CD163+ M2 pro-tumor macrophages were decreased in immunotherapy responder specimens with low USP2 expression (Fig. 8l, m). Collectively, these results suggest that the expression levels of USP2 and CD47 determine the TME and correlate with the sensitivity to anti-PD-1 immunotherapy in human cancers.
To gain a deeper understanding of whether the upstream mechanisms regulating USP2 operate at the transcriptional or the protein level in clinical human patient samples, we collected fresh tumor and adjacent non-tumor tissues from several lung cancer patients. The analysis revealed that USP2 mRNA levels were significantly higher in tumor tissues compared to matched adjacent non-tumor tissues in four of the five pairs examined (Supplementary Fig. 10a). Correspondingly, IHC staining of the same tissue samples exhibited similar patterns of USP2 protein expression, which aligned with the mRNA expression data (Supplementary Fig. 10b, c). These results suggest that alterations in USP2 expression may predominantly occur at the transcriptional level in human clinical tumor samples. However, further validation in larger patient cohorts will be essential to confirm the underlying regulatory mechanisms responsible for the observed changes in USP2 expression.
Taken together, our data demonstrate that USP2 plays a pivotal role in remodeling the TME, primarily by modulating CD47 protein abundance and influencing the infiltration of various immune cells, thereby significantly impacting the efficacy of anti-PD-1 immunotherapy (Supplementary Fig. 10d). Specifically, targeting USP2 with ML364 increased the proportion of M1 macrophages while decreasing M2 macrophages, likely mediated through CD47 expression. Furthermore, ML364 treatment appears to enhance the recruitment of CD8+ T cells into the tumor immune microenvironment. These findings collectively suggest that inhibition of USP2 enhances the efficacy of ICB immunotherapy largely through decreasing CD47 and reshaping an immunoactive TME.
Discussion
Increasing evidence demonstrates that targeting the CD47 signaling axis can promote both innate and adaptive immune responses, and CD47 blockade has been considered as a promising strategy for cancer immunotherapy11,12,39. However, the precise molecular mechanism underlying the regulation of CD47 expression has not been completely delineated. The ubiquitination and deubiquitination process, governed by E3 ligases and DUBs, is the major system for controlling protein homeostasis and various vital biological events in cells32,33. A recent study showed that the ubiquitin E3 ligase TRIM21 can promote CD47 ubiquitination and degradation25. However, whether and which DUBs can regulate CD47 ubiquitination and homeostasis are poorly understood prior to this study. Here, through the screen, we identify that inhibition of USP2 dramatically decreases the CD47 protein abundance (Fig. 1). Mechanistically, USP2 interacts with CD47 and removes ubiquitination on CD47, leading to the stabilization of CD47 (Fig. 2). Thus, USP2 inhibition by the pharmacological inhibitor ML364, shRNA-mediated knockdown, or knockout significantly reduced CD47 protein abundance in multiple lung cancer cell lines and mouse tissues (Fig. 1). These findings advance the understanding of the regulatory mechanism for the immune checkpoint CD47.
Previous studies have demonstrated that USP2 contributes to a favorable effect for tumorigenesis and metastasis in various cancers27,28,47, suggesting that targeting USP2 may be a potential therapeutic strategy for human cancers. Moreover, Usp2 KO mice are viable, and treatment with USP2 inhibitor, ML364, does not show obvious signs of toxicity in mice29,48,49,50, further supporting the notion that USP2 may be an ideal target for cancer treatment. In keeping with these reports, our results of scRNA-seq analysis and flow cytometry assay reveal that ML364 treatment not only reduces CD47 expression on tumor cells, but also reshapes an inflamed tumor immune microenvironment, such as increasing the tumor-infiltrating M1 macrophages and CD8+ T cells as well as decreasing the immunosuppressive M2 macrophages and MDSCs cells (Fig. 3). Of note, USP2 inhibitor combination with anti-PD-1 treatment synergistically inhibits tumor growth in multiple preclinical lung cancer models (Figs. 4 and 7). Moreover, safety assessment suggests that the combination therapy does not cause significant hematological and organ system toxicity (Supplementary Fig. 7). These results are consistent with a recent report that USP2 inhibition sensitizes tumors to PD-1 blockade in syngeneic mouse breast tumor models with no obvious toxicity largely through destabilizing VPRBP to indirectly upregulate p53 and PD-L129. The activation of the p53 pathway and upregulation of PD-L1 expression mediated by USP2 inhibition29 also contribute to shaping the inflammatory TME, which aligns with our major conclusion. Together with our findings that USP2 regulates CD47 stability and tumor immune microenvironment, these studies offer a complementary molecular rationale for the combination of USP2 inhibitor with PD-1/PD-L1 blockade to improve cancer therapy.
Importantly, the results of the IF staining for lung cancer patients and bioinformatic analysis demonstrate that cancer patients with lower USP2 expression in tumor specimens were associated with more inflamed TME than those patients with higher USP2 expression (Fig. 8a–e). In particular, the results of IHC and mIHC demonstrate that the low expression of USP2 and CD47 is positively correlated with good response to anti-PD-1 immunotherapy in lung cancer and oral cancer patients, accompanied by an increase in infiltration of immunoactive immune cells and a decrease in infiltration of immunosuppressive immune cells (Fig. 8f–m). These results from clinical studies suggest that inhibition of USP2 may reshape an inflamed TME, setting up a situation that makes PD-1/PD-L1 blockade more effective. Collectively, our study not only uncovers a novel regulatory mechanism for CD47 expression but also provides a potential therapeutic strategy that combines a USP2 inhibitor with PD-1 blockade for cancer treatment.
Methods
Cell culture and transfection
HEK293T cells were provided by the laboratory of Dr. Hong-Bing Shu from Wuhan University. H1975 (CRL-5908), A549 (CCL-185), H460 (HTB-177), MCF7 (HTB-22), SCC25 (CRL-1628), CAL27 (CRL-2095), LLC (CRL-1642), and THP-1 (TIB-202) cells were purchased from the American Type Culture Collection (ATCC). PC9 cells were purchased from the European Collection of Authenticated Cell Cultures (ECACC). All cell lines were confirmed Mycoplasma-free using the MycoAlert system (Lonza). HEK293T, PC9, H1975, A549, H460, MCF7, SCC25, CAL27, and LLC were cultured in DMEM (D6429, Sigma) medium supplemented with 10% FBS (10437-028, Gibco), 100 U penicillin, and 100 mg/ml streptomycin (SV300010, Hyclone). THP-1 cells were maintained in RPMI-1640 (SH30809.01, Hyclone) medium supplemented with 10% FBS, 100 U penicillin, and 100 mg/ml streptomycin. All cells were incubated at 37 °C in an atmosphere of 5% CO2. Cells with 60-80% confluences were transfected using PEI (23966-1, Polysciences) transfection reagent in Opti-MEM medium (31985-070, Gibco) according to the manufacturer’s instructions. After 36 hours (h) of transfection, cells were collected for various assays such as immunoblotting, immunoprecipitation, and reverse transcription quantitative PCR (RT-qPCR).
Virus package and generation of stable cell lines
For the virus package, HEK293T cells were co-transfected by lentiviral constructs pLKO.1 for shRNAs together with packaging plasmids (VSVG and Δ8.9). 48 and 72 h post-transfection, the viral supernatants were collected and filtered with a 0.45 μm syringe filter. Subsequently, cells (for generating stable cell lines) with around 40%–50% confluences were incubated with viral supernatants supplemented with 4 μg/ml polybrene (107689, Sigma) for 12 h and then changed with fresh medium for another 36 h. Finally, stable cell lines were generated after selection with puromycin (1 μg/ml) for at least 3 days.
Plasmids
pEnCMV-CD47 (human)-3 × HA (P24349) was constructed and validated with sequencing by Wuhan Miaoling Biotechnology. HA-CD47 (WT and mutants: Δ163–176, Δ229–235, and Δ290–323) were subcloned into the pEnCMV vector. Flag-USP2 (WT and mutants: amino acid 1–266, 267–605, C276A, H21Q, G109V, C121Y, D145N, R148L, D162Y, Q214H, R229Q, and G265S) were subcloned into the pEF-Flag-C vector. GST-CD47 WT, GST-USP2 WT, and truncations were constructed by cloning the corresponding cDNAs into the pGEX -6p-2 vector. His-Ub and mutant constructs have been described previously44. CD47-GFP was subcloned into the pLenti-CMV vector. Flag-USP4, USP5, USP7, USP8, USP13, USP15, USP21, USP25, and USP30 were generously provided by the laboratory of Dr. Bin Wang from the Daping Hospital in Third Military Medical University. Flag-USP32, USP51, JOSD1, OTUD5, ZRANB1, YOD1, and UBR1 were purchased from Wuhan Miaoling Biotechnology. The following shRNA sequences were inserted into pLKO.1 vector. Human USP2 shRNAs: #1, 5′-CCATGCTGTTTACAACCTGTA-3′, #2, 5′-GCTCACAACATTTGTGAACTT-3′. The following sgRNA sequences were inserted into the pLenti-V2 vector. Human CD47 sgRNAs: 5′- TTGCACTACTAAAGTCAGTG-3′. Mouse Cd47 sgRNAs: #1, 5′- CACCGGATAAGCGCGATGCCATGG -3′, #2, 5′- CACCGAAATTGGGAAAATAACAATG -3′. The following siUSP2 were generated by Sangon Biotech: 5′- CCGCGCTTTGTTGGCTATAAT -3′.
Reagents
ML323 (HY-17543), ML364 (HY-100900), WP1130 (HY-13264), P22077 (HY-13865), DUB-IN-2 (HY-50737A), Spautin-1 (HY-12990), IU1 (HY-13817), LDN57444 (HY-18637), XL188 (HY-122886), PR-619 (HY-13814), EOAI3402143 (HY-111408), BAY 11-7082 (HY-13453), and MF-094 (HY-112438) were purchased from MedChemExpress. ML364 (S6748) and TCID (S7140) were purchased from Selleck. In addition, MG132 (BML-PI102-0005) was purchased from Enzo Life Science, and cycloheximide (C7698-5G) was purchased from Sigma. In vivo mAb anti-mouse PD-1 (BE0146) was purchased from Bio X Cell.
Immunoblot and immunoprecipitation
For immunoblotting, cells were collected and lysed with RIPA buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40, 10% SDS, and 10% Triton X-100) containing protease inhibitors (B14002, Bimake) and phosphatase inhibitors (B15002, Bimake). Total protein concentration was determined using the BCA Protein Quantification Kit, and samples were added with loading buffer, followed by boiling for 5 min. In this process, samples for detecting membrane protein CD47 need to avoid boiling. For immunoprecipitation assays, 1–2 mg whole-cell lysate protein was incubated with HA-/Flag-conjugated agarose or other appropriate antibodies (2 μg) in a rotating machine overnight at 4 °C. The immunocomplexes were then washed with NETN buffer (20 mM Tris, pH 8.0, 100 mM NaCl, 0.5 mM EDTA, and 0.5% NP-40) four times. Each sample was added 30 μl 3× loading buffer for boiling for 5 min.
Equal amounts of total protein were subjected to SDS-PAGE electrophoresis and electro-transferred to a PVDF membrane. After blocking with 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween (TBST) for 30 min at room temperature, the PVDF membrane was incubated with the indicated primary antibodies overnight at 4 °C. After washing three times with TBST, the PVDF membrane was incubated with secondary antibodies conjugation with horseradish peroxidase for 1 h at room temperature. Signals of the target protein were ECL-developed and exposed with X-Ray Super RX Films (Fujifilm).
Deubiquitinase screening
The Human ON-TARGETplus siRNA Library - Deubiquitinating Enzymes - SMARTpool, specifically targeting 96 human deubiquitinating enzymes (G-104705-01, Horizon Discovery), was utilized for identifying the DUB that regulates CD47 stability in this study. Firstly, we constructed the pLenti-CD47-GFP plasmid by cloning the human full-length CD47 into the pLenti-CMV-GFP vector, which co-expresses GFP and CD47 protein. Then, H1975 and HEK293T cell lines stably expressing CD47-GFP were established by lentivirus infection, and single cells were sorted by FACS selecting for high-CD47-GFP-expressing cells. After validation of the CD47-GFP positive rate of H1975 and HEK293T monoclonal cell lines by FACS, siRNA transfection was carried out using Lipofectamine 3000 (L3000015, Thermo Fisher) according to the manufacturer’s instructions. Transfection occurred when H1975 or HEK293T cells (stably expressing CD47-GFP) reached approximately 70–80% density in 24-well cell culture plates. Briefly, 6 pmol of siRNA duplex and 1 μl of Lipofectamine 3000 were separately diluted in 50 μl of serum-free Opti-MEM medium for 5 min at room temperature. Subsequently, the Opti-MEM medium containing Lipofectamine 3000 was added to each well containing the diluted siRNA. The Opti-MEM medium mixture was gently pipetted and incubated at room temperature for 15 min. After 72 h of transfection, cells were collected and FACS was performed using Beckman Cytoflex. The MFI of CD47-GFP was detected and recorded with CytExpert Software 2.3, then results were analyzed using FlowJo and GraphPad software.
Immunofluorescence staining
H1975 or PC9 cells (Fig. 1f, g), and H1975 or HEK293T cells stably expressing either the pLenti-GFP empty vector or pLenti-CD47-GFP (Supplementary Fig. 1a) were cultured on a confocal dish (801001, NEST). After ML364 treatment for 16 h in H1975 or PC9 cells, cells were washed with pre-cold PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. After membrane permeabilization with 0.05% Triton X-100 for 30 min, samples were incubated with 5% bovine serum albumin (BSA) in PBS for 1 h at room temperature. Subsequently, samples were incubated with the indicated primary antibody in the dark overnight at 4 °C. After washing three times in PBS, the secondary antibody anti-mouse IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 488 Conjugate) (4408S, Cell Signaling Technology) was incubated for 1 h in the dark. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (C0065, Solarbio) for 5 min in the dark at room temperature. Images and cells' fluorescence intensity values were obtained by the laser scanning confocal microscopy (LSCM, ZEISS).
Patient tissue samples
The tissue microarrays for lung adenocarcinoma (HLugA180Su04) for (Fig. 1s, t and Supplementary Fig. 1v) were purchased from Shanghai Outdo Biotech Co. Ltd. (Shanghai, China), and these specimens were obtained the informed consent from patients (SHYJS-CP-1610002). We performed IHC scoring and statistical analysis only on the intact spots in the tissue microarray, n = 83. The lung cancer specimens for (Fig. 8f–i and Supplementary Fig. 10a–c) were obtained from cancer patients before receiving anti‐PD-1 therapy at the Zhongnan Hospital, Wuhan University (Supplementary Table S1 for Fig. 8f–i; Supplementary Table S3 for Supplementary Fig. 10a–c). All patients provided informed consent, and ethical approval was obtained from the Medical Ethics, Zhongnan Hospital of Wuhan University (22022032K). After two cycles of anti-PD-1 immunotherapy, patients with a complete or partial response were considered as “responders”, while those still with progressive or stable disease were considered as “non-responders”. For (Fig. 8j–m), oral squamous cell carcinoma (OSCC) samples of patients who participated in a phase 2 clinical trial investigating neoadjuvant immune checkpoint blockade, were registered at ClinicalTrials.gov (NCT04649476) (Supplementary Table S2). Eligible patients, aged 18–70 years, had histologically confirmed locally advanced OSCC at clinical stage III-IVA (T1-2N1-2M0 or T3-4aN0-3M0, as per AJCC 8th edition) and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1. We obtained 44 patient tissue samples for IHC or mIHC staining. Enrollment took place between March 2021 and July 2022. All patients provided written informed consent before enrollment, authorizing the use of their tissue samples for scientific research. Ethical approval was obtained from the Ethics Committee of the School and Hospital of Stomatology, Wuhan University ([2020] Ethics No.2).
Immunohistochemical (IHC) and multiplex IHC (mIHC) staining
For the IHC staining and analysis, paraffin-embedded tissues were cut into 4-μm-thick serial sections. Sections were placed in a 60 °C oven heated 4 h for complete dewaxing, following deparaffinized in xylene, and rehydrated in 100%, 95%, and 75% ethanol for 5 min, respectively. After deparaffinizing and rehydrating, tissue antigen retrieval was carried out by heating for 15 min in 0.01 M sodium-citrate buffer (pH = 6.0) or EDTA (pH = 8.0) using a microwave oven. Subsequently, sections were incubated with 3% H2O2 for 10 min at room temperature to quench endogenous peroxidase activity, followed by incubating with 10% goat serum for 30 min at room temperature to block non-specific binding. Then, samples were incubated with the indicated primary antibodies overnight at 4 °C. Then, slides were washed three times in PBS and incubated with biotin-labeled anti-rabbit IgG for 30 min at room temperature. After washing again with PBS, peroxidase/diaminobenzidine (DAB) was added to the sections (Detection Kit rabbit/mouse G1212-200T, Servicebio) according to the manufacturer's instructions, and the staining reaction was monitored from 30 s to 5 min. At last, sections were counterstained with hematoxylin to visualize cell nuclei.
Immunohistochemical staining images were captured by an Aperio VERSA scanner and analyzed by the Leica Aperio VERSA 8 multifunctional scanner. The tissue immunohistochemical staining was quantified by Aperio Quantification software. And the IHC scoring for DAB staining was based on the average optical density (AOD) value in Fig. 1t. Following IHC staining, the AOD for each spot on the tissue microarray was quantified using the Halo v3.0.311.314 analysis software with the Indica Labs - Area Quantification v2.1.3 module, calculated as [AOD = Integrated Optical Density (IOD) SUM/Area SUM]. Patients were grouped into the low and high groups according to USP2 IHC scores relative to the mean values.
For the mIHC staining, tissue sections were performed to the same deparaffinization and rehydration processes as IHC. Tissue slices were fixed with 4% paraformaldehyde for 20 min, and antigen retrieval was carried out by heating for 15 min in EDTA (pH = 8.0) using a microwave oven. The mIHC staining was performed using the Opal 7-Color Manual IHC Kit (NEL811001KT, Akoya Biosciences) following the manufacturer’s instructions. After washing with TBST three times, sections were blocked with Opal Antibody Diluent/Block at room temperature for 12 min. The samples were incubated with the indicated primary antibodies in PBS at 37 °C for 1 h or overnight at 4 °C in the dark. After washing three times using TBST, the secondary Abs Opal Polymer HRP Ms + Rb was incubated for 10 min in the dark. After washing again in TBST, the sections were incubated with the fluorochrome diluted 1:100 in 1× Plus Amplification Diluent in the dark. Slices were subjected to the next round of heat-induced antigen retrieval, blocking, antibody, and fluorochrome incubation. Finally, nuclear staining was performed with DAPI, and images from the stained slices were acquired by Akoya Vectra3. Akoya phenoptics InForm software was used for spectral unmixing and image analysis. Specifically, images or 3D stereoscopic plane diagrams were generated by scanning the stained slides with confocal microscopy (Leica Stellaris 5 WLL) using a 60 × oil objective to obtain clearer fields of view (Fig. 4m).
Antibodies
The antibodies used for immunoblotting are listed below. Anti-CD47 (D3O7P) Rabbit mAb (63000, 1:2000), anti-p53 Rabbit mAb (9282T, 1:2000), and anti-PD-L1 (E1L3N) Rabbit mAb (13684, 1:2000) were purchased from Cell Signaling Technology. Anti-USP2 Rabbit pAb (AP2131c, 1:2000) was purchased from Abgent. Anti-Vinculin (VIN-11-5) mouse mAb (V4505, 1:100,000), anti-Flag M2 mouse mAb (F3165, 1:10,000), anti-Flag rabbit pAb (F7425, 1:10,000), and anti-HA rabbit pAb (H6908, 1:50,000) were purchased from Sigma-Aldrich. Anti-Purified anti-HA.11 Epitope Tag (16B12) mAb (MMS-101P, 1:5000) was obtained from Biolegend. Anti-CD47 Rabbit IgG (108575-T32, 1:1000) was purchased from Sino Biological. Anti-GAPDH mouse mAb (60004-1-Ig, 1:100,000) was purchased from Proteintech. Horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (BL001A, 1:5000) and HRP-conjugated anti-rabbit secondary antibody (BL003A, 1:5000) were purchased from Biosharp.
The following antibodies were used for flow cytometry analysis. Anti-mouse CD45 (557659, 1:100), anti-mouse CD3 (562600, 1:100), anti-mouse CD8α (566096, 1:100), anti-mouse CD4 (563151, 1:100), anti-mouse IFN-γ (562303, 1:100), anti-mouse TNF (554419, 1:100), fixable viability stain 700 (564997, 1:600), anti-mouse Gr-1 (563299, 1:100), anti-mouse Ly6C (560525, 1:100), anti-mouse CD11b (557396, 1:100), anti-mouse F4/80 (565411, 1:100), anti-mouse CD47 (563585, 1:100) were purchased from BD Biosciences. Anti-mouse GzmB (61-8898-82, 1:100) and anti-mouse CD206 (12-2061-82, 1:100) were purchased from eBioscience. Anti-mouse I-A/I-E(MHCII) (107614, 1:100) and anti-human CD11b (301310, 1:100) were obtained from BioLegend.
The following antibodies were used for IHC or mIHC. Anti-human CD47 (D3O7P) Rabbit mAb (63000, 1:200), anti-mouse CD8α Rabbit mAb (98941, 1:200), anti-human CD8 Rabbit pAb (85336, 1:200), and anti-human Pan-Keratin (C11) Mouse mAb (4545, 1:400) were purchased from Cell Signaling Technology. Anti-mouse CD47 Rabbit pAb (20305-1-AP, 1:800) was obtained from Proteintech. Anti-human-USP2 Rabbit pAb (AP2131c, 1:800) was purchased from Abgent. Anti-mouse F4/80 Rabbit mAb (ab111101, 1:150) and anti-mouse PD-L1 Rabbit mAb (ab213480, 1:300) were purchased from Abcam. Anti-human CD163 Mouse mAb (MAB-0869, 1:800) was purchased from MXB Biotechnologies.
The following antibodies were used for immunofluorescence. Anti-mouse F4/80 Rabbit mAb (ab111101, 1:150) and anti-mouse Mannose Receptor (CD206) Rabbit mAb (ab64693, 1:500) were purchased from Abcam. Anti-mouse CD8α Rabbit mAb (98941, 1:200) was obtained from Cell Signaling Technology. Anti-GFP (Green Fluorescent Protein) pAb (598, 1:500) was obtained from MBL Beijing Biotechnology Company. Anti-human CD47 Mouse mAb (sc-12730, 1:100) was purchased from Santa Cruz Biotechnology.
GST pull-down assay
GST-CD47, GST-USP2, and their different truncations subcloned into pGEX-6p-2 constructs were expressed in Escherichia coli BL21 and purified by glutathione-Sepharose resin (17-0756-05, GE Healthcare). For GST pull-down assays, purified GST proteins were incubated with cell lysates from HEK293T cells expressing the indicated proteins at 4 °C for 3 h. After incubation, beads were washed four times with the NETN buffer, and 30 μl 3× loading buffer was added to boil for 5 min. Samples were subjected to SDS-PAGE gel and analyzed with immunoblotting.
Protein half-life analysis
HEK293T cells co-transfected with CD47-HA Flag-USP2 were treated with the protein synthesis inhibitor cycloheximide (CHX) (200 µg/ml) (C7698, Sigma) for the indicated time courses. Cells were harvested at the indicated time post-CHX treatment for immunoblot analysis using the indicated antibodies. CD47 protein band densities were quantified by ImageJ software and normalized to vinculin and then compared to the t = 0 time point.
In vitro phagocytosis assay
Human monocyte THP-1 was maintained at 37 °C with 5% CO2 in RPMI 1640. After stimulation by phorbol 12-myristate 13-acetate (PMA) (16561-29-8, Sigma) (100 ng/ml) for 48 h for differentiation to activated macrophages. THP-1 cells (2 × 105 per well) were seeded onto a 24-well plate. Pre-treated lung cancer cells were labeled with 2.5 μM carboxyfluorescein succinimidyl ester (CFSE, C34570, Invitrogen) according to the manufacturer’s protocol. After 2 h serum starvation for macrophages, 2 × 105 CFSE-labeled lung cancer cells were added for co-incubating with macrophages. After co-culture for 6 h at 37 °C, macrophages and target cells were harvested and stained with anti-human CD11b antibody for 30 min at 4 °C. Phagocytosis was assessed via flow cytometry and calculated as the percentage of CD11b+CFSE+ cells among CD11b+ macrophages by using the FlowJo software.
Cell death assay
H1975 or PC9 cells were seeded in six-well plates and subsequently treated with 2 μM ML364 for 16 h. After treatment, the cells were harvested and stained with 7-Amino-Actinomycin D (7-AAD) (559925, 1:100, BD Biosciences). Dead cells, identified by 7-AAD positivity, were detected using a FACS flow cytometer (Cytoflex, Beckman) and analyzed using FlowJo software.
In vivo ubiquitination assay
Transfect cells at 80% confluency with His-ubiquitin (His-Ub) and the indicated constructs. After 36 h of transfection, the cells were treated overnight with 10 μM MG132. Then cells were lysed in denaturing buffer A (6 M guanidine hydrochloride, 0.1 M Na2HPO4/NaH2PO4, and 10 mM imidazole pH 8.0). After sonication, incubate the lysates with nickel-nitrilotriacetic acid (Ni-NTA) beads (QIAGEN) at room temperature for 3 h. Subsequently, wash the Ni-NTA beads twice with buffer A, twice with buffer A/TI (volume:volume = 1:3), and once with buffer TI (25 mM Tris-HCl and 20 mM imidazole pH 6.8). Add 30 μl of 2× protein loading buffer to the Ni-NTA beads. Separate the proteins by SDS-PAGE and perform immunoblotting using the appropriate antibodies.
Denatured IP for endogenous CD47 ubiquitination assay
For endogenous CD47 ubiquitination assays, H1975 or PC9 cells, treated with 20 μM MG132 for 6 h before harvesting, were lysed in a denaturing buffer containing 50 mM Tris (pH 7.5), 1% SDS, 0.5 mM EDTA, and 1 mM dithiothreitol. The lysates were incubated at 100 °C for 10 min, sonicated, and subsequently incubated with CD47 antibodies overnight at 4 °C. After added 50 μl protein A Sepharose (17078001, Cytiva), the mixture was incubated for an additional 6 h at 4 °C. Immunoprecipitates were washed four times with NETN buffer, resolved by SDS-PAGE, and analyzed by immunoblotting.
In vivo experimental therapy for mouse tumor models
All mouse studies were approved by the Institutional Animal Care and Use Committee of Wuhan University (MRI2022-LACA08, MRI2022-LACA12, and MRI2022-LAC057). Mice were bred and maintained in a specific-pathogen-free (SPF) animal facility at the Medical Research Institute of Wuhan University. Mice were provided ad libitum access to SPF Mouse Growth and Reproduction Formula Feed (24073113, KEAO XIELI) and housed in a controlled environment with a 12-h light-dark cycle, a temperature range of 20–25 °C, and humidity maintained between 30% and 70%. C57BL/6J mice were purchased from Vital River (stock number 213). KrasLSL-G12D/+; Trp53fl/fl (KP)45,51 and Usp2−/− mice50 were generously provided by the laboratory of Dr. Bo Zhong (Wuhan University). KrasLSL-G12D/+ (008179) and Trp53fl/fl (008462) were originally purchased from the Jackson Laboratory and then crossed to obtain KP mice for maintenance and experiments. The sex of the mice was not considered in the study design, as both male and female mice were included across all experimental models, including the KP and KPU tumorigenesis models as well as the C57BL/6J tumor-bearing model.
For the in vivo experimental therapy in subcutaneous mouse lung tumor models, 2 × 106 LLC tumor cells in 100 µl FBS-free DMEM were subcutaneously injected into the flank of 6-week-old C57BL/6J mice. 3 days after LLC implantation, tumor size was measured by caliper every two days, and tumor volume was calculated as length × width2 × 0.5. When the tumors reached an average volume of 30 mm3, mice bearing LLC tumors were randomly divided into the indicated two groups, vehicle or ML364 treatment (Fig. 3e). The ML364 (5 mg/kg of mouse body weight) was administered by intraperitoneal (i.p.) injection every day for a total of 12 treatments. For the treatments in (Fig. 4a), 6-week-old C57BL/6J mice bearing LLC tumors were randomly divided into the indicated four groups: (1) vehicle, (2) ML364, (3) anti-PD-1 mAb (BE0146), and (5) combined therapy of ML364 and anti-PD-1 mAb. Anti-PD-1 mAb was administered with a dosage of 100 μg per mouse every three days for a total of 4 treatments. The ML364 (5 mg/kg of mouse body weight) was administered by i.p. injection every day for a total of 12 treatments. For the treatments in (Supplementary Fig. 6b), 6-week-old mice bearing sgControl or sgCd47-LLC tumors were randomly divided into six designated groups: (1) sgControl & Vehicle, (2) sgControl & ML364, (3) sgControl & anti-CD47 mAb, (4) sgCd47 & Vehicle, (5) sgCd47 & ML364, (6) sgCd47 & anti-CD47 mAb. Anti-CD47 mAb (200 μg/mouse) was administered via i.p. injection every three days for 4 doses, and ML364 (5 mg/kg) was given daily for 12 doses. InVivoMAb anti-mouse CD47 (IAP) was purchased from BioXCell (BE0270). For the treatments in (Fig. 6a), 6-week-old mice bearing sgControl or sg Cd47-LLC tumors were randomly assigned to six designated groups, respectively: (1) sgControl & Vehicle, (2) sgControl & ML364, (3) sgControl & ML364 & anti-PD-1 mAb, (4) sgCd47 & Vehicle, (5) sgCd47 & ML364, (6) sgCd47 & ML364 & anti-PD-1 mAb. Anti-PD-1 mAb (100 μg/mouse) was administered via i.p. injection every three days for 4 doses, and ML364 (5 mg/kg) was given daily for 12 doses. For combined treatment with macrophage and CD8 T cells depletion in (Fig. 6e), 6-week-old mice randomly divided into five groups: (1) vehicle, (2) ML364 & PD-1 mAb (MP), (3) MP & CD8 blockade (αCD8), (4) MP & macrophage blockade (clodronate liposomes, CL), and (5) (MP & αCD8 & CL). Anti-CD8 mAb (200 μg/mouse) was injected i.p. every 3 days for 4 doses, starting 3 days before tumor implantation. Clodronate liposomes (200 μl/mouse) were given every 3 days for 4 doses, starting on tumor implantation day. Mice received ML364 (5 mg/kg) daily for 12 days and anti-PD-1 mAb (100 μg/mouse) every 3 days for 4 doses. Anti-mouse CD8α-InVivo (Selleck, A2102) and Clodronate liposomes (Yeasen, 40337ES) were used. At the indicated time points after treatments, mice were euthanized by carbon dioxide (CO2) inhalation, and tumors were harvested, weighed, or prepared for the other analysis.
For survival analysis, 2 × 106 LLC tumor cells were subcutaneously injected into 6-week-old C57BL/6J mice, and mice health was monitored daily. For Fig. 5d, the ML364 (30 mg/kg of mouse body weight) was administered by i.p. injection every day for 18 treatments. Anti-PD-1 mAb was administered with a dosage of 200 μg per mouse every three days for 6 treatments. Mice were euthanized by CO2 inhalation when tumor volume reached 1500 mm3 and were deemed as death. Then, Kaplan–Meier survival curves were analyzed using the log-rank test.
For treatments in (Fig. 4i), 1 × 106 LLC cells in 250 μl FBS-free DMEM were injected via the tail vein into 7-week-old C57BL/6J mice. Seven days post-injection, tumor-bearing mice were randomly assigned to 4 groups: (1) Vehicle, (2) ML364 (5 mg/kg daily for 14 doses), (3) anti-PD-1 mAb (100 μg/mouse every 3 days for 5 doses), and (4) combined ML364 and anti-PD-1 mAb. Mice were euthanized by CO2 inhalation at the indicated time points, and lung tissues were collected for H&E staining and analysis.
For the autochthonous lung tumor model, the KrasLSL-G12D/+; Trp53fl/fl (KP) and KrasLSL-G12D/+Trp53fl/fl Usp2−/− (KPU) mice were used. KPU mice were generated by mating KP mice with Usp2−/− mice. For induction of lung tumorigenesis, 7-week-old KP and KPU mice were anesthetized with 1% sodium pentobarbital and administered with intranasal instillation of 2 × 106 pfu Adenovirus-Cre (Ad-Cre) (HBAD001077OE, Hanbio biotechnology company) in 50 μl PBS per mouse. After five weeks of induction, the treatments were performed. In Fig. 7a, KP mice were pooled and randomly divided into the vehicle, ML364, anti-PD-1 mAb, and combined treatment groups. KP mice were administered by i.p. injection of anti-PD-1 mAb (100 μg per mouse) every three days for a total of 7 treatments or ML364 (5 mg/kg mouse body weight) every day for a total of 21 treatments. In Fig. 7e, KP and KPU mice were respectively divided into the vehicle and anti-PD-1 mAb treatment groups. KP or KPU mice were administered by i.p. injection of anti-PD-1 mAb (100 μg per mouse) every three days for a total of 7 treatments. 21 days after treatment, the lungs of KP and KPU mice were harvested for H&E staining and analysis.
Tumor-infiltrating cell isolation for flow cytometry analysis
All fresh tumor tissues were cut into small pieces with scissors. The tumors were mechanically dissociated by being pushed through a 70-μm cell strainer placed in a 60-mm dish using the plunger of a 5-ml syringe. After passing through another 70-μm cell strainer, the cell suspension was collected in a 15 mL conical tube and centrifuged at 500 × g for 10 min at room temperature. The supernatant was discarded, and the cell pellet was resuspended with the ACK Lysis buffer (A1049201, Thermo Fisher) for the lysis of red blood cells for 5 min at room temperature. After centrifugation, the supernatant was aspirated. Single-cell suspensions were stained with appropriate antibodies for 30 min at 4 °C in the dark to identify specific subsets of immune cells. The samples were detected by flow cytometry on FACSCelesta (BD Biosciences), and the data were processed using FlowJo software.
Cell sorting and single-cell RNA sequencing (scRNA-seq) of the LLC tumor
2 × 106 LLC tumor cells were subcutaneously implanted into 6-week-old C57BL/6J mice and treated with a control vehicle or ML364 as described in (Fig. 3e). After treatments, tumors were harvested and dissociated into single‐cell suspensions for scRNA seq. Single-cell 3′ RNA-seq libraries were prepared using the Chromium Single Cell 3′ reagent Kits v3 (10× Genomics) according to the manufacturer’s instructions. Briefly, cells were washed with DPBS containing 0.04% BSA three times and resuscitated to a concentration of 700~1200 cells/μl (viability ≥ 85%). For the preparation of single-cell transcriptome libraries, cells were loaded onto a 10× Genomics Chromium instrument and captured by a microfluidic system for generating single-cell gel beads in emulsion (GEMs). After cells were labeled by unique Cell Tag signatures and transcripts from different cell sources were distinguished, emulsions were broken, Barcoded-cDNA was purified with Dynabeads, and PCR amplification was performed. Amplified cDNA was then used for 3′ gene expression library construction. For the construction of the gene expression library, 50 ng of amplified cDNA was fragmented, end-repaired, double-size selected with SPRIselect beads, and sequenced on a NovaSeq platform (Illumia) to generate 150 bp paired-end Reads. Then, Raw data (Raw Reads) of fastq files were assembled from the Raw BCL files using Illumina’s bcl2fastq converter, following demultiplexed and mapped to the reference genome by 10× Genomics Cell Ranger pipeline using default parameters. After initial filtering with default parameters, all downstream single-cell analysis was performed.
Cells mapped <300 or >7000 genes and with >10% mitochondrial genes or >3% red blood cell genes were eliminated from downstream analysis. Genes measured >100,000 in one cell were also eliminated for the following steps. After filtering, we were working on the analysis of a total of 39,765 cells and 20,005 genes. Samples were normalized and integrated with Harmony. Principal component analysis (PCA) was used for dimensionality reduction, and Uniform Manifold Approximation and Projection (UMAP) plots were generated via the RunUMAP function. A total of 19 clusters were generated at a resolution of 0.5, and further identified into “Cancer”, “Mono/Macrophage”, “T cells”, “DCs”, “NK cells”, “B cells”, “Neutrophils”, “Endothelial”, and “Stromal cells” using SingleR (1.4.1) and based on the expression of known gene markers. Myeloid lineage was re-clustered and grouped into “M1”, “M2”, “Monocytes”, “M0”, “Neutrophiles”, “DCs”, “MDSCs”, and “Basophiles”, determined by the expression of key markers. T cells re-clustered into 14 clusters at a resolution of 2.7 and grouped into “Tn”, “CD8 Teff “, “CD8 Tcyc”, “CD8 Tpex”, “CD8 Tex”, “Tregs”, “CD4 Tcs”, and “rd Tcs”, determined by the expression of key markers. Finally, GraphPad Prism 8.0 software was used to perform statistical analysis for different cell clusters.
Tumor-infiltrated macrophage phagocytosis analysis
Green fluorescent protein (GFP)-expressing LLC cells (2 × 106) were injected subcutaneously into the right flank of 6-week-old C57BL/6J mice. After indicated treatments in Fig. 4a, all mice were euthanized by CO2 inhalation, and tumors were harvested. Excised tumors were formalin-fixed and paraffin-embedded. 2 μm tumor sections were prepared. After dewaxing and rehydration, tumor tissue slides were subjected to fix with 4% paraformaldehyde, and antigen retrieval with EDTA buffer (pH = 8.0) was performed. The tumor tissue staining was followed as the process in the method section of ‘multiplex immunohistochemical (mIHC) staining’. The primary antibodies (anti-mouse F4/80 Rabbit mAb and anti-GFP pAb) were incubated at 37 °C for 1 h or overnight at 4 °C. After washing with TBST, slides were incubated with secondary Abs and fluorochrome sequentially with the mIHC Kit. Images were acquired using Akoya Vectra3, and phagocytosis of macrophages by tumor cells was quantified by calculating the fused yellow cells merged by green GFP-tumor cells and red F4/80-macrophages in a high-power field.
Hematoxylin & eosin (H&E) staining analysis
Lung tissues from mice were fixed in 4% paraformaldehyde for at least 12 h at room temperature and embedded into paraffin blocks. The paraffin blocks were sectioned to 5 μm per slice, and the sections were stained with H&E (C0105S, Beyotime Biotech). Tissue sections were scanned and analyzed by the Aperio VERSA 8 (Leica) multifunctional scanner.
Micro-CT scanning
CT imaging was applied to scan the mice on a small animal micro-CT scanner (NEMO® Micro CT, PINGSENG Healthcare Inc.) to present lung lesions. Briefly, animals were anesthetized using 2%–3% isoflurane, set up on the thermostatically controlled CT bed, and maintained in an anesthetized state at 1%–1.5% isoflurane delivered via a nose cone. The mice were in the supine position, and a standard CT scan protocol was performed with the scanner software (Cruiser). Meanwhile, a respiratory-gated shim was used to monitor the real-time respiratory frequencies of mice, and the total scanning time was approximately 10 min per mouse. After the CT scans were complete, the data set was subsequently reconstructed and imaged following the analysis software (Avatar) program according to the manufacturer’s instructions. And the 3D reconstruction of the whole lung and the volume calculation of the reconstructed lung were performed in the Avatar program, in accordance with the instrument manufacturer’s guidelines.
Correlation analysis and gene expression analysis
The data for analyzing the correction of USP2 expression and immune cells infiltration in LUAD patients were generated by TIMER 2.0 (http://timer.cistrome.org/) in (Fig. 8c–e). The GTEx portal (https://www.gtexportal.org/home) database was utilized to display the USP2 gene expression level in different human organs and tissues (Supplementary Fig. 5s). Expression values are shown in TPM (Transcripts Per Million), calculated from a gene model with isoforms collapsed to a single gene. Box plots are shown as median and 25th and 75th percentiles.
RNA isolation and reverse transcription quantitative PCR (RT-qPCR) assay
Total RNAs from lung tissues or cultured cells were extracted using TRIzol (15596018, Invitrogen). Reverse transcription reactions were performed with the PrimeScript RT reagent kit (RR470A, TaKaRa) to generate complementary DNA (cDNA). Quantitative PCR was performed with the Bio-Rad CFX Connect Real-Time PCR Detection System (Bio-Rad) after mixing the cDNA templates with primers and PerfectStart Green qPCR SuperMix (AQ601, TransGen Biotech). Primers used for RT-qPCR are listed below. Human CD47, forward primer, 5′- AGAAGGTGAAACGATCATCGAGC -3′; reverse primer, 5′- CTCATCCATACCACCGGATCT -3′; human GAPDH, forward primer, 5′- GTCTCCTCTGACTTCAACAGCG -3′; reverse primer, 5′- ACCACCCTGTTGCTGTAGCCAA -3′; mouse Usp2, forward primer, 5′- ACACGGCCCTCATGGAAGA -3′; reverse primer, 5′- GTGGCGCATATCTCTGGATCT -3′; mouse Gapdh, forward primer, 5′- CATCACTGCCACCCAGAAGACTG -3′; reverse primer, 5′- ATGCCAGTGAGCTTCCCGTTCAG -3′.
Statistical analysis
Data statistical analyses were carried out using GraphPad Prism 8.0 (GraphPad Software) or Excel 2021. All quantitative experiments are presented as mean ± SD. with three replicates as indicated in the figure legends. Statistical analysis was performed with the unpaired, two-tailed Student’s t-test among two group differences. Pearson’s test was conducted to evaluate correlations. Statistical comparisons of tumor growth curves were made using two‐way ANOVA. For animal survival analysis, Kaplan–Meier survival curves were analyzed by log-rank test. P < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data generated from mouse tumor samples in this study have been deposited in the Gene Expression Omnibus (GEO), a public functional genomics data repository, under the accession number GSE254966. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Zappasodi, R., Merghoub, T. & Wolchok, J. D. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33, 581–598 (2018).
Bagchi, S., Yuan, R. & Engleman, E. G. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu. Rev. Pathol. 16, 223–249 (2021).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Yap, T. A. et al. Development of immunotherapy combination strategies in cancer. Cancer Discov. 11, 1368–1397 (2021).
Marin-Acevedo, J. A., Kimbrough, E. O. & Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 14, 45 (2021).
Sharma, P. & Allison, J. P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015).
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009).
Majeti, R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009).
Willingham, S. B. et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl Acad. Sci. USA 109, 6662–6667 (2012).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Veillette, A. & Chen, J. SIRPalpha-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 39, 173–184 (2018).
Jiang, Z., Sun, H., Yu, J., Tian, W. & Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 14, 180 (2021).
Weiskopf, K. et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Invest. 126, 2610–2620 (2016).
Arrieta, O. et al. Association between CD47 expression, clinical characteristics and prognosis in patients with advanced non-small cell lung cancer. Cancer Med. 9, 2390–2402 (2020).
Wang, S. et al. Blocking CD47 promotes antitumour immunity through CD103(+) dendritic cell-NK cell axis in murine hepatocellular carcinoma model. J. Hepatol. 77, 467–478 (2022).
Logtenberg, M. E. W., Scheeren, F. A. & Schumacher, T. N. The CD47-SIRPalpha immune checkpoint. Immunity 52, 742–752 (2020).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016).
Zhang, H. et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl Acad. Sci. USA 112, E6215–E6223 (2015).
Betancur, P. A. et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 8, 14802 (2017).
Hu, H. et al. Oncogenic KRAS signaling drives evasion of innate immune surveillance in lung adenocarcinoma by activating CD47. J. Clin. Invest. 133, e153470 (2023).
Logtenberg, M. E. W. et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat. Med. 25, 612–619 (2019).
Wu, Z. et al. Identification of glutaminyl cyclase isoenzyme isoQC as a regulator of SIRPalpha-CD47 axis. Cell Res. 29, 502–505 (2019).
Mair, B. et al. High-throughput genome-wide phenotypic screening via immunomagnetic cell sorting. Nat. Biomed. Eng. 3, 796–805 (2019).
Du, L. et al. EGFR-induced and c-Src-mediated CD47 phosphorylation inhibits TRIM21-dependent polyubiquitylation and degradation of CD47 to promote tumor immune evasion. Adv. Sci., 10, e2206380 (2023).
Kitamura, H. & Hashimoto, M. USP2-related cellular signaling and consequent pathophysiological outcomes. Int. J. Mol. Sci. 22, 1209 (2021).
Shan, J., Zhao, W. & Gu, W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 36, 469–476 (2009).
Stevenson, L. F. et al. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 26, 976–986 (2007).
Yi, J. et al. Targeting USP2 regulation of VPRBP-mediated degradation of p53 and PD-L1 for cancer therapy. Nat. Commun. 14, 1941 (2023).
Davis, M. I. et al. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J. Biol. Chem. 291, 24628–24640 (2016).
Sun, S. C. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 8, 501–511 (2008).
Cruz Walma, D. A., Chen, Z., Bullock, A. N. & Yamada, K. M. Ubiquitin ligases: guardians of mammalian development. Nat. Rev. Mol. Cell Biol. 23, 350–367 (2022).
Harrigan, J. A., Jacq, X., Martin, N. M. & Jackson, S. P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discov. 17, 57–78 (2018).
Graner, E. et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 5, 253–261 (2004).
Bedford, L., Lowe, J., Dick, L. R., Mayer, R. J. & Brownell, J. E. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 10, 29–46 (2011).
Fenalti, G. et al. Structure of the human marker of self 5-transmembrane receptor CD47. Nat. Commun. 12, 5218 (2021).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Advani, R. et al. CD47 blockade by Hu5F9-G4 and Rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 379, 1711–1721 (2018).
Liu, Y. et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct. Target. Ther. 8, 104 (2023).
Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Zhang, M. et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 11, e0153550 (2016).
Han, S. et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci. Adv. 9, eadg2697 (2023).
Pan, Y. et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J. Hematol. Oncol. 12, 124 (2019).
Xiong, W. et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat. Commun. 13, 1700 (2022).
Zhang, M. et al. CCL7 recruits cDC1 to promote antitumor immunity and facilitate checkpoint immunotherapy to non-small cell lung cancer. Nat. Commun. 11, 6119 (2020).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020).
Zhao, Y. et al. USP2a supports metastasis by tuning TGF-beta signaling. Cell Rep. 22, 2442–2454 (2018).
Pouly, D. et al. Mice carrying ubiquitin-specific protease 2 (Usp2) gene inactivation maintain normal sodium balance and blood pressure. Am. J. Physiol. Ren. Physiol. 305, F21–F30 (2013).
Yang, Y. et al. Regulation of behavioral circadian rhythms and clock protein PER1 by the deubiquitinating enzyme USP2. Biol. Open 1, 789–801 (2012).
An, R. et al. USP2 promotes experimental colitis and bacterial infections by inhibiting the proliferation of myeloid cells and remodeling the extracellular matrix network. Cell Insight 1, 100047 (2022).
Yang, C. et al. KRAS4B oncogenic mutants promote non-small cell lung cancer progression via the interaction of deubiquitinase USP25 with RNF31. Dev. Cell, https://doi.org/10.1016/j.devcel.2025.01.015 (2025).
Acknowledgements
We would like to thank the staff at the core facility of the Medical Research Institute at Wuhan University for their technical support. This work was supported by grants from the National Key Research and Development Program of China (2022YFC3401500 to J.Z. and D.L., 2023YFC3402100 to J.Z., 2019YFA0210500 to G.C.), the National Natural Science Foundation of China (82273062 and 31970732 to J.Z., 82341023 to G.C., 82471098 to Y.G., 82203319 to M.G., 82103149 to H.Y., 82473253 and 82471098 to C.X.), Central Government-Guided Local Science & Technology Development Project (2024ZYYD001 to C.X.), Wu Jieping Medical Foundation Clinical Research Special Grant (320.6750.2023-11-21 to C.X.), the Fundamental Research Funds for the Central Universities (2042022dx0003 to J.Z., 2042021kf0075 to H.Y.), Natural Science Foundation of Wuhan (2024040701010031 to J.Z.), the Open Projects of Hubei Key Laboratory of Tumor Biological Behavior (220172107 to J.Z.), and Translational Medicine and Interdisciplinary Research Joint Fund of Zhongnan Hospital of Wuhan University (ZNJC202312 to J.Z. and C.X.). The work was also supported in part by grants from the Regional Science and Technology Innovation Project of Hubei Province (2024EIA001 to Y.G.).
Author information
Authors and Affiliations
Contributions
J.Z., C.X., Y.G., G.C., P.D., and Y.S. designed research; P.D., Y.S., Z.H., and Y.L. performed most of the experiments with help from M.G., J.S., C.H., B.X., Y.Y., H.Y., G.X., L.K., X.X., X.W., X.Z., and W.X.; H.L. collected human oral cancer tissues. J.H. collected human lung cancer tissues. J.Z., C.X., Y.G., and G.C. guided and supervised the project. J.Z. and P.D. wrote the manuscript. D.L., B.Z., G.C., Y.G., C.X., and J.Z. gave discussion and edited the manuscript. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dai, P., Sun, Y., Huang, Z. et al. USP2 inhibition unleashes CD47-restrained phagocytosis and enhances anti-tumor immunity. Nat Commun 16, 4564 (2025). https://doi.org/10.1038/s41467-025-59621-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59621-5
This article is cited by
-
Post-translational modifications of immune checkpoints: molecular mechanisms, tumor microenvironment remodeling, and therapeutic implications
Journal of Biomedical Science (2026)
-
CDK10 suppresses nucleic acid sensors-mediated antitumor immunity
Nature Cancer (2026)
