
Abstract
In the phase II NIMBUS trial, patients with human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer (MBC) and high tumor mutational burden (TMB ≥ 9 mut/Mb) received nivolumab (3 mg/kg biweekly) and low-dose ipilimumab (1 mg/kg every 6 weeks) for 2 years or until progression. The primary endpoint was objective response rate (ORR) per RECIST 1.1 criteria. Among 30 patients enrolled, the median TMB was 10.9 mut/Mb (range: 9–110) and the confirmed objective response rate was 20%. Secondary endpoints included progression-free survival, overall survival, clinical benefit rate, and safety and tolerability, including immune-related adverse events (irAEs). A prespecified correlative outcome was to evaluate the ORR in patients with a TMB ≥ 14 mut/Mb. Patients with TMB ≥ 14 mut/Mb (n = 6) experienced higher response rates (60% vs 12%; p = 0.041) and showed a trend towards improved progression-free survival and overall survival compared to patients with TMB < 14 mut/Mb. Exploratory genomic analyses suggested that ESR1 and PTEN mutations may be associated with poor response, while clinical benefit was associated with a decrease or no change in tumor fraction by serial circulating tumor DNA during treatment. Stool microbiome analysis revealed that baseline blood TMB, PD-L1 positivity, and immune-related diarrhea are associated with distinct taxonomic profiles. In summary, some patients with hypermutated HER2-negative MBC experience extended clinical benefit with a dual immunotherapy regimen; a higher TMB, and additional genomic and microbiome biomarkers may optimize patient selection for therapy with nivolumab plus low-dose ipilimumab. (Funded by Bristol Myers Squibb; ClinicalTrials.gov identifier, NCT03789110).
Similar content being viewed by others

Introduction
Despite treatment advances, metastatic breast cancer (MBC) is still considered incurable1, underscoring the need for new therapeutic approaches. Currently, the use of immune checkpoint inhibitors (ICI) with chemotherapy is the preferred first line of systemic treatment for patients with PD-L1 positive, de novo or recurrent metastatic triple-negative breast cancer (mTNBC)2,3,4. However, for the vast majority of patients with MBC, including PD-L1 negative TNBC, those with hormone receptor-positive (HR+) disease or human epidermal growth factor receptor 2-positive (HER2+) disease, ICIs have not proven useful5,6,7. This limited benefit of immunotherapy in MBC underscores the need to define the exact molecular processes that drive cancer immune evasion in these tumors. Additional biomarkers besides PD-L1 status are essential to broaden the indication for ICIs among MBC patients.
Different studies have shown that tumor mutational burden (TMB) is associated with markers of antitumor immunity, including tumor neoantigen burden, tumor infiltrating lymphocytes (TILs), and efficacy outcomes to ICIs8,9,10,11,12,13,14. Results of the phase II KEYNOTE-158 study demonstrated an objective response rate (ORR) of 29% (95% confidence interval [CI]: 21%–39%) to pembrolizumab monotherapy among patients with advanced solid tumors and TMB ≥ 10 mutations per megabase of DNA (mut/Mb). In contrast, the ORR was 6% (95% CI: 5%–8%) in patients with TMB < 10 mut/Mb15. While these findings supported the approval of pembrolizumab to treat patients with TMB ≥ 10 mut/Mb (i.e., TMB-high status per the FoundationOne®CDx assay [F1CDx]), no patients with MBC were included in the KEYNOTE-158 study16.
Previously, our group observed that high TMB was associated with longer progression-free survival (PFS) and overall survival (OS) among mTNBC patients treated with ICIs, regardless of clinical factors and PD-L1 status17. Exploratory analyses from KEYNOTE-11918 and IMpassion13019 also suggested that mTNBC patients with high TMB derived significant survival benefit from pembrolizumab monotherapy and atezolizumab plus nab-paclitaxel, respectively. In both studies, these associations were not observed in patients treated with chemotherapy alone. Furthermore, the phase II TAPUR basket study included a cohort of 28 MBC patients with TMB ≥ 9 mut/Mb. The patients were treated with pembrolizumab monotherapy, which demonstrated an unconfirmed ORR of 21% (95% CI: 8%–41%)20.
Several lines of evidence suggest that tumors with high TMB might respond better to dual immune checkpoint blockade. Both preclinical and clinical data suggest that a combination of nivolumab (anti-PD-1) with ipilimumab (anti-CTLA-4) could activate complementary mechanisms that promote T cell antitumor activity21,22,23. The CHECKMATE-142 study showed that patients with DNA microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer had a numerically higher response rate when treated with a combination of ipilimumab and nivolumab compared to those treated with nivolumab alone24,25. Nivolumab in combination with low-dose ipilimumab is approved by the U.S. Food and Drug Administration as a first-line treatment for patients with microsatellite instability-high metastatic colorectal cancer, as well as for patients with metastatic non-small cell lung cancer with PD-L1 expression of 1% or more, as measured by an approved companion diagnostic assay26,27. Based on these findings, we hypothesized that patients with MBC with high TMB could derive benefit from a combination of nivolumab plus ipilimumab.
Here, we report the clinical and translational results of the NIMBUS trial, an open-label, single-arm, multicenter phase II trial of nivolumab plus low-dose ipilimumab in patients with hypermutated (>9 mut/Mb) human epidermal growth factor receptor 2 (HER2)-negative MBC. The primary endpoint was ORR per RECIST 1.1 criteria28. Secondary endpoints included PFS, OS, clinical benefit rate (CBR), and safety and tolerability, including immune-related adverse events (irAEs). A prespecified correlative outcome was to evaluate the ORR in patients with a TMB ≥ 14 mut/Mb. To evaluate genomic profiling between responders and non-responders, we conducted genomic analyses on tumor tissue taken before treatment and on liquid biopsy samples taken at baseline and on treatment from patients run centrally using the Foundation Medicine, Inc. (Boston, MA) panels. Furthermore, based on the emerging data that the gut microbiome diversity and composition can impact the outcome of immune checkpoint blockade by influencing the tumor immune microenvironment29,30,31,32, stool samples were collected at baseline, and metagenomic data were analyzed for exploring possible differences in taxonomic profiling according to tumor characteristics, clinical benefit to immunotherapy, and irAEs.
Results
Patients
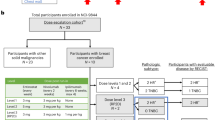
Between 2/19/2019 and 3/6/2024, 30 patients were enrolled at 3 cancer centers (Dana-Farber Cancer Institute [DFCI] [n = 25]; University of Texas Southwestern [n = 4]; University of Pittsburgh Medical Center [n = 1]). The trial is now complete. The study design is shown in Supplemental Fig. 1. The sex of all patients was female, and the median age was 63 years (range 36–72). A total of 21 patients (70%) had HR+ breast cancer, and the remaining 30% had TNBC. Patients received a median of 1.5 prior lines of chemotherapy in the metastatic setting (range: 0–3). PD-L1 status was positive in 5 patients (16.7%), negative in 24 (80%), and not determined in 1 (3.3%). Among patients with TNBC and PD-L1 status available, 33.3% (3 out of 9) were positive for PD-L1. A total of 27 patients were included in NIMBUS based on results from a tissue-based panel (21 OncoPanel, 5 F1CDx, 1 CARIS), and 3 patients used data from blood-based panels (2 Guardant and 1 FoundationOne® Liquid CDx [F1LCDx]). Overall, the median TMB was 10.9 mut/Mb (range: 9–110); 25 patients (83.3%) had TMB ≥ 9–<14 mut/Mb, two patients (6.7%) had TMB ≥ 14–<20 mut/Mb, and three patients (10%) had TMB ≥ 20 mut/Mb (Table 1).
Efficacy
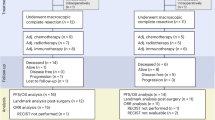
After a median follow-up of 30.6 months (interquartile range: 18.4–35.3), no patients had a complete response, and 6 patients (20%) had a partial response, for a confirmed ORR of 20% (95% CI: 7.7–38.6%). Stable disease was the best response in 5 patients (16.7%) (Fig. 1A). The CBR, defined as a best response of complete response or partial response or stable disease ≥24 weeks, was 20% (95% CI: 7.7–38.6%), as no patients with stable disease achieved this time point of 24 weeks (Table 2).

A Waterfall plot showing change in diameter of target lesions in the full patient population after a median of 30.6 months of follow-up, excluding three unevaluable patients (N = 27). B Kaplan–Meier curves showing progression-free survival in the full population (N = 30), and D among patients with TMB < 14 mut/Mb and patients with TMB ≥ 14 mut/Mb; (N = 30); C Kaplan–Meier curves showing overall survival in the full population (N = 30), and E among patients with TMB < 14 mut/Mb and patients with TMB ≥ 14 mut/Mb (N = 30). P-values are from Log-Rank tests. F Swimmer plot showing time to response and duration of response among the full patient population (N = 30). CI confidence interval, HR hazard ratio, m months, mOS median overall survival, mPFS median progression-free survival, mut/Mb mutations per megabase of DNA, NA not applicable, NE not evaluable, OS overall survival, PD progressive disease, PFS progression-free survival, PR partial response, SD stable disease, TMB tumor mutational burden.
Exploratory analyses did not reveal any differences in response rate based on hormone receptor status (HR+ vs. TNBC), PD-L1 status (positive vs. negative), or stromal TIL percentage (<10% vs. ≥10%). However, patients with TMB < 14 mut/Mb had an ORR of 12%, compared to 60% for patients with TMB ≥ 14 mut/Mb (p = 0.041, two-sided Fisher exact test) (Table 3).
The median PFS was 1.4 months (95% CI: 1.3–4.6) (Fig. 1B), and the median OS was 16.9 months (95% CI: 7.1–Not Reached) (Fig. 1C). Median PFS and OS were not statistically different according to HR+ status (Supplemental Fig. 2). Among patients with TMB ≥ 14 mut/Mb, the median PFS was 9.5 months (95% CI: 4.6–Not Reached), compared to 1.4 months (95% CI: 1.3–2.6) among patients with TMB < 14 mut/Mb (Fig. 1D). The median OS was not reached (95% CI: 18.6–Not Reached) in patients with TMB ≥ 14 mut/Mb compared to 8.2 months (95% CI: 5.1–Not Reached) in patients with TMB < 14 mut/Mb (Fig. 1E). Among the 6 responders, the median time to response was 1.9 months and the median duration of response was 10.2 months (Fig. 1F). Importantly, there were two patients who remained progression-free for more than two years since starting therapy (subject #1: 43 months; subject #11: 30.6 months). Patients who experienced a partial response to therapy had a 12-month OS of 100% (Supplemental Fig. 3).
Safety
The median number of cycles of nivolumab and ipilimumab administered was one. Disease progression was the most common reason for treatment discontinuation. Two responders discontinued treatment due to toxicity: one due to myocarditis and the other due to fatigue.
The most common adverse events of grade 2 or higher included fatigue (33%) and diarrhea (13.3%). Grade 3 adverse events included fatigue (6.7%) and elevated aspartate aminotransferase (6.7%). No grade 4 or 5 adverse events were seen (Supplemental Table 1). IrAEs of grade 2 or higher occurred in 30% of patients. Grade 2 irAEs occurred in 20% of patients, and grade 3 irAEs occurred in 10%, including one patient who developed grade 3 adrenal insufficiency and grade 3 myocarditis, and two patients with grade 3 hepatitis (Supplemental Table 2).
Pre-trial genomic profiling and clinical outcomes
A total of 29 patients (6 responders and 23 non-responders) were subjected to at least one pretreatment genomic panel (centrally performed): 16 were subjected to both, 5 had pretreatment F1CDx only, and 8 had F1LCDx panel only. Analysis of clinical samples from the most recent genomic panel available (including 24 baseline blood and 5 baseline tissue samples), revealed that the most frequently mutated genes (>30%) in this population were TP53 (65.52%, n = 19/29 patients; all with a pathogenic/likely pathogenic mutation), PIK3CA (44.83%, n = 13/29 patients; all with a pathogenic/likely pathogenic mutation), ESR1 (34.48% n = 10/29 patients; all with a pathogenic/likely pathogenic mutation), and CDH1 (34.48%, n = 9/29; 20.69%, n = 6/29 with a pathogenic/likely pathogenic mutation) (Fig. 2A). Additional mutational frequencies were computed and visualized for tissue samples, baseline blood, on trial (C1D15 or C2D1) blood, and end of treatment (EOT) blood separately (Supplemental Figs. 4–7).

A The mutational landscape of the full patient population for the top 25 mutated genes (N = 29). B Kaplan–Meier curve of the overall survival (OS) outcomes for 20 HR+ patients with or without ESR1 oncogenic mutations showing a significant decrease in survivability. C Progression-free survival (PFS) Kaplan–Meier curve for PTEN showing a negative correlation between PTEN oncogenic mutations and PFS. D Kaplan–Meier curve of OS outcomes for 29 patients showing a negative correlation between PTEN oncogenic mutations with OS. E, F Kaplan–Meier survival curves for the composite immuno-oncology score and TMB ≥ 14 mut/Mb, PFS (E) and OS (F) for high or low composite immuno-oncology (IO) score. PFS (G) and OS (H) for TMB ≥ 14 mut/Mb or TMB < 14 mut/Mb. 21 patients with tissue F1CDx testing are included. P-values are Log-Rank tests of the variables in each Kaplan–Meier curve. Pathogenic/likely pathogenic variants are shown with a black line around the box denoting the variant, while variants of unknown significance are not. HR hormone receptor, NE not evaluable, PD progressive disease, PR partial response, SD stable disease.
Analysis of the top 10 mutated genes with oncogenic (pathogenic/likely pathogenic) mutations revealed a trend towards correlation between oncogenic ESR1 mutations and a decrease in OS among patients with HR+ disease (n = 20, log-rank test p = 0.10, HR = 2.5) (Fig. 2B). Among all 29 patients, there was a trend toward negative correlation between oncogenic PTEN mutations and PFS and OS (PFS: [n = 29, log-rank test p = 0.17, HR = 2.4], OS: [n = 29, log-rank test p = 0.13, HR = 2.4],) (Fig. 2C-D). A composite immuno-oncology (IO) score that integrates TMB, genomic features associated with ICI, and PD-L1 status33,34 identified 5 patients with a high IO score, 4/5 of which intersected with patients with TMB ≥ 14 mut/Mb. High IO score and TMB ≥ 14 mut/Mb status each had statistically significant correlation with PFS (n = 21, IO score: [log-rank test p = 0.014, HR = 0.18], TMB ≥ 14 mut/Mb: [log-rank test p = 0.0024, HR = 0.071]) (Fig. 2E–H).
Baseline blood TMB and clinical outcomes
Blood TMB (bTMB) was evaluable for 24 patients at baseline, 5 who achieved an objective response, and 19 without response. Overall, the median bTMB was 10.12 mut/Mb (interquartile range [IQR] 6.32–21.49; range 2.53–113.79). The median bTMB was 41.72 mut/Mb (IQR 12.64–78.39; range 2.53–113.79) among those with objective response and 8.85 mut/Mb (IQR 6.32–11.38; range 5.06–44.25) among those without response.
bTMB ≥ 14 mut/Mb was found in 60% (n = 3/5) of patients with objective response and in 21% (n = 4/19) of patients without response (Supplemental Table 3). Comparing bTMB vs tissue F1CDx TMB in 16 patients with both assays based on their objective response (4 patients with objective response, 12 without objective response), TMB ≥ 14 mut/Mb was found in 75% (n = 3/4, bTMB) vs 100% (n = 4/4, tissue TMB) of patients with objective response and in 25% (n = 3/12, bTMB) vs 8% (n = 1/12, tissue TMB) of non-responders (Supplemental Table 3).
bTMB levels at baseline were compared with values obtained using the DFCI-NGS Panel (OncoPanel) and with tissue F1CDx (Fig. 3A). Twelve patients had paired values for all three panels, which includes 3 patients with clinical benefit (patients 1, 11, and 14). Between 15 and 17 patients had paired values for at least two panels (Fig. 3B, C). bTMB was highly correlated with tissue F1CDx (r = 0.95, two-sided Pearson correlation) and with OncoPanel (r = 0.91, two-sided Pearson correlation; Fig. 3B–D). For tissue F1CDx or OncoPanel samples with TMB > 9 mut/Mb, bTMB samples that differed by 50% or more often had a higher TMB (3/3 for blood vs OncoPanel and 5/7 for blood vs tissue F1CDx) (Fig. 3A, C, D).

A Distribution of the 12 paired tissue and baseline blood samples showing the tissue, blood, and OncoPanel TMB or bTMB, respectively. B Correlation between the tissue TMB and baseline bTMB showing the significant positive correlation with bTMB. C The correlation between baseline bTMB and OncoPanel TMB reveals a significantly higher bTMB. D The correlation between tissue and OncoPanel TMB shows a significant positive correlation between them, and the strongest correlation among the different TMB panels. P-values in (B–D) are two-sided Student t tests for the Pearson correlation coefficient r. E Changes in tumor fraction from seven paired baseline and end of treatment patients reveal a significant correlation based on clinical benefit status. P-value is an unpaired two-sided Wilcoxon rank-sum test between the clinical benefit groups using paired tumor fraction changes per patient. Patients 1 and 7 had similar changes in tumor fraction, and they overlap in the trajectory plot.
Tumor fraction estimates from 7 matched baseline and EOT samples revealed that all patients with clinical benefit had a decrease in tumor fraction or a baseline of zero, compared to an increase in patients with no clinical benefit (n = 7, p = 0.034, Wilcoxon two-sided test) (Fig. 3E, F).
Distinct taxonomic profiles are associated with blood TMB and with the occurrence of immune-related diarrhea
With regards to the gut microbiome, first, we applied a filter requiring features to have at least 0.01% relative abundance in 10% of samples prior to analyses unless noted, giving 1209 species genomic bins (SGBs) before filtering and 416 SGBs after. Then we examined if compositional taxonomic differences in the gut microbiota of patients would be associated with specific baseline features or with efficacy outcomes. The alpha diversity (within-sample diversity) of the fecal microbiome at baseline was not different among patients with distinct baseline characteristics or clinical outcomes. Notably, no patient included in this study reported use of antibiotics in the last 60 days before starting the experimental therapy. Each clinical covariate and outcome was tested for association with overall microbiome structure in baseline stool samples. Beta diversity (between-sample diversity) did not differ between responders and non-responders. This parameter significantly differed only between patients harboring baseline bTMB high versus low (Supplemental Table 4), Q = 0.078.
Next, we investigated differences in species relative abundance between different groups. The top 20 species by mean relative abundance for each patient baseline sample are presented in Supplemental Fig. 8. We found an enrichment of Clostridium scindens in TMB low patients (p = 0.0011). PD-L1 positive status was associated with members of the Lachnospiraceae family (Supplemental Table 5). Gut microbiota composition did not significantly differ between patients with or without objective response, and we found no significant correlation with either OS or PFS.
Although gut microbiota composition did not differ regarding the overall incidence of irAEs, we found significant hits associated with immune-related diarrhea, including members of the Clostridia class (Clostridiaceae SGB14839 and Clostridiales SGB29342 order) (Supplemental Table 6). Interestingly, stool from patients without irAEs had higher functional potential of SpoIVB peptidase (Supplemental Table 7).
Discussion
The NIMBUS study demonstrated that patients with HER2-negative MBC and a high TMB (≥9 mut/Mb) who were treated with a combination of nivolumab and low-dose ipilimumab had a confirmed ORR of 20%, thereby meeting the trial’s primary endpoint. No new toxicities were observed with this dual ICI regimen, and there were no reported grade 4 or 5 adverse events. Additionally, while we did not observe any association between clinical response and hormone receptor status, PD-L1 expression, or TIL percentage, the analysis of a prespecified correlative outcome revealed that patients with TMB ≥ 14 mut/Mb had an ORR of 60%, while those with TMB < 14 mut/Mb had an ORR of 12%. Furthermore, patients with TMB ≥ 14 mut/Mb showed a trend toward longer PFS and OS compared to those with lower TMB.
Genomic exploratory analyses suggested that ESR1 and PTEN mutations may be associated with a lack of benefit from treatment. Furthermore, all patients with clinical benefit had a decrease in ctDNA tumor fraction from baseline to end of treatment, or a baseline tumor fraction of zero, compared to an increase in patients with no clinical benefit. An IO score that integrated TMB, genomic alterations, and biomarkers associated with ICI response identified similar patients as identified by TMB ≥ 14 mut/Mb alone. Furthermore, stool microbiome analysis revealed that baseline bTMB, PD-L1 positivity, and occurrence of immune-related diarrhea are associated with distinct taxonomic profiles.
Currently, in the metastatic setting, only patients with PD-L1-positive mTNBC are eligible for treatment with ICIs in combination with chemotherapy4. The continued development of ICIs in breast cancer requires the identification of additional biomarkers that can predict benefit to this therapy, both for sparing patients from an ineffective therapy potentially associated with severe adverse events, as well as trying to expand their use among patients with other breast cancer subtypes, such as HR+ breast cancer. It is also worthwhile to explore whether patients with likely immunogenic biologic features could be treated with a chemotherapy-free ICI-based regimen.
High TMB (cutoff of ≥10 mut/Mb) is not rare among MBC. Our group and others have shown that approximately 10% of all patients with MBC have high TMB35,36,37. Interestingly, in our prior work using whole-exome sequencing data from 3969 publicly available breast cancer samples, we found that the proportion of breast tumors with high TMB does not differ among breast cancer subtypes35. However, there is a significant enrichment of high TMB among metastatic invasive lobular carcinoma compared with invasive ductal tumors, a finding also described by Sokol and colleagues35,38. Interestingly, APOBEC dysregulation has been characterized as the most frequent mutational process among MBC with high TMB35,37,39,40.
It is also important to note that the nivolumab and low-dose ipilimumab regimen used in the NIMBUS study was feasible, and the toxicity profile was consistent with other studies using this combination. Grade 3 irAEs occurred in 3 (10%) patients, and no grade 4 or 5 irAEs were reported. This regimen has also been used in metastatic non-small cell lung cancer26 and DNA microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer27, with high objective response rates and good tolerability.
Outside of NIMBUS, the only other study to prospectively evaluate the use of ICIs in patients with MBC and high TMB was the phase II TAPUR basket study. In the TAPUR study, a cohort of 28 patients with MBC and TMB ≥ 9 mut/Mb received pembrolizumab, with an unconfirmed ORR of 21%20. Notably, at the time we designed NIMBUS, there was no approval for immunotherapy based on a specific agnostic TMB cutoff, and we decided to use as an inclusion criteria the same TMB cutoff of ≥9 mut/Mb used in TAPUR. Additionally, as there were emerging data derived from studies in lung cancer suggesting that a TMB cutoff of 10 mut/Mb would not be ideal41, we decided to explore a higher cutoff as a prespecified secondary objective. Thus, we found that patients with TMB ≥ 14 mut/Mb had an ORR of 60% versus 12% among patients with TMB ≥ 9 and <14 mut/Mb. Acknowledging that there were only 5 patients with TMB ≥ 14, data from NIMBUS suggest that further work is needed to investigate the optimal TMB cutoff for selecting patients with MBC for receiving ICI. Consistent with our findings, the results of the phase IIa multi-basket MyPathway study showed that atezolizumab monotherapy demonstrated promising and durable clinical activity, with an ORR of 38.1% among patients diagnosed with different advanced solid tumor types and high TMB (TMB ≥ 16 mut/Mb) versus a limited activity (ORR of 2%) among patients whose tumors had a TMB between 10 and 16 mut/Mb42.
Importantly, given that both the TAPUR and NIMBUS studies met their primary endpoints and have similar ORR, an important question is whether the addition of ipilimumab adds benefit. It is important to note the differences between the TAPUR and NIMBUS study populations. In NIMBUS, 70% of patients had HR + MBC, and 14% of patients had PD-L1-positive tumors, whereas in TAPUR, only 43% of patients had HR + MBC, and the rate of PD-L1 positivity is unknown. It is reasonable to hypothesize that such differences can be explained by the fact that ICIs were not yet approved for mTNBC when the TAPUR study was enrolling, but atezolizumab had been approved in MBC prior to patient enrollment for the NIMBUS study. This might have skewed enrollment in the NIMBUS trial toward MBC patients who did not meet the criteria for treatment with atezolizumab plus chemotherapy.
Notably, two patients enrolled in the NIMBUS study have remained progression-free more than two years after the end of treatment. It is plausible to hypothesize that these durable responses may be attributable to the use of dual checkpoint inhibition. In preclinical studies using mouse solid tumor models, dual blockade of PD-1 and CTLA-4 resulted in enhanced proliferation, intratumoral infiltration, and antitumor activity of antigen-specific CD8+ and CD4+ T cells, along with a decrease in intratumoral regulatory T cells (Tregs)43,44,45. These preclinical findings are supported by two additional clinical studies that have reported data on the efficacy of a dual ICI regimen in MBC: one study46 included only patients with metaplastic MBC (ORR = 18%) and used nivolumab plus ipilimumab; the other study47 included patients with HER2-negative MBC and treated them with durvalumab and tremelimumab (ORR = 17%). In addition to dual checkpoint inhibition, the phase Ib ETCTN-9844 trial evaluated the safety and ORR of a triplet regimen consisting of the class I histone deacetylase inhibitor etinostat given weekly at a dose of 3 mg in combination with biweekly nivolumab 3 mg/kg plus ipilimumab 1 mg/kg every 6 weeks48. The ORR for this triplet was 25%. Importantly, among the 20 evaluable patients, 55% had TNBC, 71% had PD-L1-positive tumors, and only one patient (5%) had TMB > 10 mut/Mb. The combination was well tolerated48.
Similar to our previous findings from a retrospective cohort study from DFCI17, we found a trend between the presence of PTEN oncogenic mutations and reduced PFS and OS in the NIMBUS study. Several groups have shown that PTEN loss is associated with immune evasion in different animal models49,50. In addition, we observed a trend between ESR1 mutations and decreased OS among HR+ patients. This is also consistent with our previous studies, in which we showed that higher ER signaling is associated with decreased antigen presentation and response to ICI treatment51,52.
In certain tumor types, particularly lung cancer, the application of a composite IO score that is composed of multiple predictive biomarkers has been shown to be better associated with outcomes after ICI treatment than any individual biomarker33,34. Here we found that patients with a high composite IO score had a large overlap with those with TMB ≥ 14 mut/Mb (4/5 patients), thus resulting in similar predictive power of the IO score and TMB ≥ 14 mut/Mb for PFS and OS. In this study, we assessed the IO score in patients with MBC, and further studies in larger cohorts are needed to assess the utility and optimal components of a composite IO score in the breast cancer field.
Another interesting finding in this study was that 60% of patients (n = 3/5) with objective responses had bTMB ≥14 mut/Mb, while 21% of those without response (n = 4/19) had a bTMB ≥14 mut/Mb. In addition, tumor fraction estimates from 7 paired biopsies from baseline to EOT revealed that all patients with a clinical benefit had a decrease or no change in tumor fraction compared to an increase in patients with no clinical benefit.
By using preclinical models, different groups have shown that the composition of the gut microbiota impacts the response of hosts to ICI therapy. Additionally, the gut microbiome profile has been identified as a predictor of response to ICI therapy in patients with melanoma, kidney cancer, and lung cancer29,30,31,32.
In breast cancer, few data are available on the structure and composition of the intestinal microbiome53,54, and its role as a predictive biomarker of response to immunotherapy is not established. Although we did not find an association between alpha diversity with patient characteristics or clinical outcomes, nor beta diversity or significant hits with clinical outcomes, we found an enrichment of Clostridium scindens, implicated in bile acid metabolism and carcinogenesis, in patients with TMB low55,56. Further, patients with PD-L1-positive tumors harbor members of the Lachnospiraceae family associated with benefit in cancer immunotherapy57. The finding that different hits were associated with immune-related diarrhea is notable (Clostridiaceae SGB14839 and Clostridiales SGB29342 order). Interestingly, members of the Clostridia class were already reported as related to colitis and treatment-related toxicity in patients with breast cancer54,58,59. Furthermore, we observed higher functional potential of SpoIVB peptidase in patients without irAEs, often necessary in spore formation. Bacillus spore-forming bacteria have a protective effect in autoimmune colitis60,61.
This study has several limitations, including the small sample size. Furthermore, while the results of the NIMBUS study corroborate the existing evidence to support the utilization of ICIs for MBC patients with high TMB, it does not address the important issue of whether dual checkpoint inhibition is more effective than pembrolizumab alone. Another limitation is that different genomic panels could be used to determine whether a patient had high TMB for study eligibility, and central testing was not performed to meet the inclusion criteria. However, 21 out of 30 patients had TMB determined using the same test, a DFCI in-house panel called OncoPanel62.
In summary, the NIMBUS study reached its primary endpoint and supports the use of ICIs for MBC patients with high TMB63. The study corroborates other available evidence that TMB can serve as a clinical biomarker to broaden access to these agents for MBC patients with breast cancer subtypes beyond PD-L1-positive TNBC. Additionally, we can hypothesize that patients with PD-L1-positive metastatic TNBC and high TMB could be treated solely with an immunotherapy-based regimen, thereby avoiding the need for concurrent cytotoxic drugs. Moreover, in the NIMBUS trial, we found that a higher cutoff of TMB (≥14 mut/Mb) is more adequate to select responders to nivolumab plus low-dose ipilimumab. Additionally, there was a trend between the presence of oncogenic ESR1 and PTEN mutations and a lack of benefit. Finally, distinct taxonomic profiling was found in stool samples from patients with and without immune-related diarrhea. If validated, these results could help tailor the use of ICI among patients with TMB-high MBC.
Methods
Compliance with ethical standards
The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice Standards and the Declaration of Helsinki. Institutional review board (IRB) approval was obtained at Dana-Farber/Harvard Cancer Center (DF/HCC). The DF/HCC Data and Safety Monitoring Committee (DSMC), which is composed of clinical specialists with experience in oncology and who had no direct relationship with the study, reviewed and monitored toxicity and accrual data from the study. Information that raised questions or concerns was addressed with the overall PI and study team. Participants provided written informed consent prior to the performance of any protocol-specific procedures or assessments.
Study design and patient population
The study enrolled patients with MBC that was HER2-negative per American Society of Clinical Oncology/College of American Pathologists guidelines. Participants with HR+ breast cancer must have progressed on at least one prior line of endocrine therapy in the metastatic setting or had disease recurrence while on adjuvant endocrine therapy. Participants were required to have TMB ≥ 9 mut/Mb as assessed by a cancer-gene panel (tissue or blood-based test) evaluating >300 genes and performed in a CLIA-certified laboratory. The cutoff of TMB ≥ 9 mut/Mb was chosen to be consistent with the cutoff used in the phase II TAPUR basket study of pembrolizumab20. We use TMB data mainly from 3 distinct targeted next-generation sequencing panels, which were used both for trial eligibility and correlative genomic analyses: OncoPanel62, F1CDx, and F1LCDx64. Two additional commercial panels (Guardant and CARIS) were used for a total of 3 patients for trial eligibility. These panels, being targeted sequencing panels, are designed to only sequence genomic regions associated with a set of cancer-associated genes that vary between panels (e.g., 287–462 genes for OncoPanel and 324 genes for F1CDx and F1LCDx). These panels differ in the type of material used for sequencing: tissue (OncoPanel and F1CDx) or blood (F1LCDx, Guardant, and CARIS). These panels are tumor-only, which means that alterations identified can be of somatic or germline origin, and the lack of a paired germline sample makes it challenging to reliably distinguish somatic from germline, and rely on the use of known germline mutation databases and additional algorithms65 to distinguish them.
Additional eligibility criteria included measurable disease by RECIST 1.1 criteria28 and no prior treatment with ICIs. Participants may have received 0–3 prior lines of chemotherapy in the metastatic setting and must have been off chemotherapy for at least 14 days prior to the initiation of protocol therapy. All participants were at least 18 years old with an Eastern Cooperative Oncology Group performance status of 0 or 1. Patients with a medical condition requiring chronic systemic steroid therapy or on any other form of immunosuppressive medication were excluded.
Procedures
Participants were treated with nivolumab 3 mg/kg given intravenously every 2 weeks in combination with ipilimumab 1 mg/kg given intravenously every 6 weeks, for each 6-week cycle. Participants were treated until disease progression, unacceptable toxicity, or up to 24 months (whichever occurred first). Participants were required to undergo a research biopsy at baseline and at cycle 1 day 29 if the tumor was safely accessible. Patients also underwent an additional optional research biopsy at the EOT. Stool samples were required at baseline, before cycle 2, and at EOT.
Assessments
The primary endpoint was ORR per RECIST 1.1 criteria28. Secondary endpoints included PFS, OS, CBR, and safety and tolerability, including irAEs. A prespecified correlative outcome was to evaluate the ORR in patients with a TMB ≥ 14 mut/Mb.
Exploratory analysis evaluated the association between ORR and PD-L1 status, TILs, and CD8+ TILs. All tests were performed centrally and retrospectively. PD-L1 testing was assessed by the DAKO/AGILENT assay using the 22C3 antibody66. PD-L1 positivity was defined according to manufacturer instructions for calculating the combined positive score (CPS), which is a method for evaluating the number of PD-L1 staining cells in relation to the total number of viable tumor cells in breast cancer: all PD-L1+ cells/total # viable tumor cells ×100; any membrane staining in tumor cells, any membrane or cytoplasmic staining in immune cells considered as positive cells. Particularly for patients with TNBC, a tumor with CPS ≥ 10 is classified as PD-L1-positive. Stromal TILs were assessed according to the International TIL Working Group guidelines67. CD8+ TILs were determined as the proportion of the area of TILs that scored only on the CD8+ component by IHC.
Genomic, TMB, and ctDNA tumor fraction analysis
Genomic analyses were performed on the tissue obtained before registering the trial (archival samples) and on liquid biopsy samples obtained during the trial from the patients run centrally on the F1CDx and F1LCDx targeted DNA sequencing panels, respectively. The genomic analysis focused on mutations, including single-nucleotide variants (SNVs) and insertions/deletions (INDELS). Mutations were annotated as oncogenic (pathogenic/likely pathogenic) based on their presence in multiple public (dbSNP, 1000 Genomes Project, ExAC, and COSMIC) and private databases, and whether they had been previously characterized as pathogenic (e.g., literature or truncating mutations in tumor suppressor genes), as previously described68. Mutations were annotated with gnomAD v2.1.1, and those with a gnomAD allele frequency ≥0.01% were considered putative germline mutations. Mutations in CHIP genes (DNMT3A, ATM, CHEK2, and MLL2) were also removed. The genomic analysis was performed with putative somatic mutations (those not annotated as germline and those not in CHIP genes) and germline mutations classified as pathogenic/likely pathogenic.
The remaining variants were plotted using the CoMut69 library to visualize the mutational landscape from the tissue, baseline, on trial (C1D15 or C2D1), and EOT timepoints. The top mutated genes and clinical variables were used to perform survival analysis. Survival analysis on PFS and OS was done using a cox proportional hazards model, and the significance of the Kaplan–Meier curves was determined using log-rank P-values, which was done using the survival 3.5–7 and ggsurvfit 1.0.0R packages (R 4.3.2).
TMB was obtained from OncoPanel (a tumor-only Dana-Farber Brigham Cancer Center-NGS Panel), tissue (F1CDx), and liquid biopsies (F1LCDx). Correlation between OncoPanel TMB, tissue TMB, and baseline ctDNA bTMB paired per patient was assessed using Pearson correlation using the cor.test function in the stats 4.3.2 package (R 4.3.2) and the SciPy 1.7.1 stats Python library (Python 3.917).
ctDNA tumor fraction from Foundation Medicine, Inc., was analyzed for longitudinal changes during trial treatment. Samples with low tumor fraction in which the tumor fraction could not be estimated were assigned a tumor fraction of 0%. To quantify the statistical significance between clinical benefit groups of tumor fraction changes per patient, an unpaired two-sided Wilcoxon rank-sum test of the paired tumor fraction changes per patient was used.
A composite IO signature that uses genomic and biomarker data from tissue was assessed on all 21 patients who received F1CDx testing. The signature was comprised of genomic features previously established as independent predictors of ICI response among patients with advanced squamous cell lung cancer33. For each patient, the score was increased for each positive predictive biomarker detected (+2 for TMB ≥ 20, +1 for TMB ≥ 10 and <20, +1 for PD-L1-positive), +1 for any known or likely pathogenic ARID1A alteration, and +1 for any known or likely pathogenic alteration in any DNA damage response gene (MLH1, MSH2, MSH6, PMS2, ATM, BRCA1, BRCA2, PALB2, BRIP1, or BARD1)70 and decreased for each negative predictive biomarker detected (−1 for any known or likely pathogenic alteration detected in either KEAP1 or NFE2L2, −1 for any known or likely pathogenic alteration detected in STK11, and −1 for homozygous deletion of CDKN2A). All patients in this study had a score between 0 and 3. Patients with a score of either 0 or 1 were categorized as having a low composite IO score, and patients with a score of either 2 or 3 were categorized as having a high composite IO score.
Metagenomics analysis
Patients self-collected stool specimens into OMNIgene•GUT kit(DNA Genotek, OMR-200) tubes. Stabilized stool samples were either shipped via USPS prepaid standard shipment/postage or brought back to their doctor’s appointment and were processed within 60 days from the date of collection. In preparation for DNA extraction, OMNIgene® Liquefaction Reagent (OM-LQR) was used following the manufacturer's protocol to render each stool sample collected in OMNIgene•GUT pipettable, and subsequently stored at −80 °C until DNA extraction. All samples were stored and processed in the same laboratory. A total of 41 stool samples were collected across 3 timepoints, with 23 baseline samples for 23 patients. In this study, we analyzed only the baseline samples.
Genomic DNA was extracted using the PowerSoil Pro HT following the manufacturer’s instructions. Libraries were constructed using the Illumina DNA Prep (formerly known as Illumina DNA Flex). Each sample was barcoded using a kit appropriate for Unique Dual Index adapter sets (IDT for Illumina UD indexes). Completed libraries were QC’d using a combination of PicoGreen (Thermo), Qubit (Thermo), Fragment Analyzer (Agilent), and Tapestation (Agilent) to assess concentration and fragment size distribution. Sequencing MTG pooled libraries was carried out via the Illumina NovaSeq6000 platform, using the 2 × 150 bp paired-end protocol. The provided metagenomic data, sequenced at the Baylor College of Medicine Sequencing Facility, was processed using the bioBakery meta’omics workflow71 following the standard pipeline with default parameters. In short, this comprises three fundamental steps: data were first passed through KneadData v0.12.0 for read-level quality control, then taxonomic profiling was performed using MetaPhlAn v4.0.372, and functional potential profiling was performed with HUMAnN v3.6.171. The median final read count was 89520455, with a minimum of 36312577, therefore, no samples were considered to be a QC failure by this metric.
Statistical analyses were performed in the R 4.3.0 environment. In particular, vegan (https://CRAN.R-project.org/package=vegan) was used for measurements of alpha diversity (within-sample diversity) and beta diversity (between-sample diversity), tests, and visualizations. MaAsLin273 was used as a linear modeling framework on total sum scaling, normalized, log-transformed relative abundances. We applied a filter requiring features to have at least 0.01% relative abundance in 10% of samples prior to analyses, unless noted, giving 1209 species genomic bins (SGBs) before filtering and 416 SGBs after. Among each of the comparison types generated, multiple comparisons are adjusted using a Benjamini and Hochberg procedure (BH) using the p.adjust function in R, and FDR-adjusted p-values of 0.25 or lower are reported as significant.
Statistics & reproducibility
The study followed a Simon’s two-stage design with 14 patients enrolled in the first stage. If at least one patient had an objective response (complete or partial response) in the first stage, the study would enroll an additional 16 patients in the second stage, for a total of 30 patients. If at least four out of 30 patients had an objective response, the regimen would be declared worthy of further study. Based on recent trials of anti-PD-1/PD-L1 agents used as monotherapy in patients with MBC and considering that patients in the present study would not be selected based on PD-L1 status, a true ORR of 5% or less would not be of clinical interest. The study had 90% power to distinguish between an ORR of 5% and 25%. ORR and CBR were reported with 95% confidence intervals. PFS and OS were analyzed using the Kaplan–Meier estimation method. Fisher’s exact test was used to assess the associations between response rate and the following factors: estrogen receptor (ER) status (HR+ vs. TNBC), PD-L1 status (positive vs. negative), and stromal TIL percentage (<10% vs. ≥10%). All analyses of clinical efficacy endpoints were performed using SAS 9.4 and R 4.0.2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
In accordance with the Health Insurance Portability and Accountability Act, we do not have IRB approval or patient consent to share individualized patient genomic data, which contains potentially identifying or sensitive patient information and cannot be reported in a public data repository. Foundation Medicine is committed to collaborative data analysis and has well-established and widely used mechanisms by which qualified researchers can query our core genomic database of >500,000 de-identified sequenced cancers. More information and mechanisms for data access can be obtained by contacting the corresponding author or the Foundation Medicine Data Governance Council at data.governance.council@foundationmedicine.com. Metagenomic data for stool samples have been deposited at SRA (https://www.ncbi.nlm.nih.gov/bioproject/1222800) under BioProject PRJNA1222800. The source data to generate Figs. 2, 3 and Supplemental Figs. 4–8 is available in GitHub (https://github.com/Breast-Oncology-Computational-Group/NIMBUS/). The remaining source data are available within the Source Data file. Source data for confidential variables is not included. Requests for additional raw and processed data and materials will be reviewed by the senior authors to determine whether the request is subject to any intellectual property or confidentiality obligations. These additional data and materials may be subject to patient confidentiality and might require a material transfer agreement. Source data are provided with this paper.
Code availability
All software and pipelines for genomic and microbiome data generation are described in detail in the “Methods” section. The scripts used to generate Figs. 2–3 and Supplemental Figures 4–8 are available on GitHub (https://github.com/Breast-Oncology-Computational-Group/NIMBUS/). Scripts that require confidential data are not included in the GitHub repository. The scripts used to generate the rest of the figures are available upon request from the corresponding author.
References
Caswell-Jin, J. L. et al. Change in survival in metastatic breast cancer with treatment advances: meta-analysis and systematic review. JNCI Cancer Spectr. 2, pky062 (2018).
Schmid, P. et al. Atezolizumab and Nab-Paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 (2018).
Schmid, P. et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 21, 44–59 (2020).
Cortes, J. et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 396, 1817–1828 (2020).
Dirix, L. Y. et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN solid tumor study. Breast Cancer Res. Treat. 167, 671–686 (2018).
Rugo, H. et al. Abstract S5-07: Preliminary efficacy and safety of pembrolizumab (MK-3475) in patients with PD-L1–positive, estrogen receptor-positive (ER+)/HER2-negative advanced breast cancer enrolled in KEYNOTE-028. Cancer Res. 76, S5-07–S05-07 (2016).
Tolaney, S. M. et al. Effect of eribulin with or without pembrolizumab on progression-free survival for patients with hormone receptor-positive, ERBB2-negative metastatic breast cancer: a randomized clinical trial. JAMA Oncol. 6, 1598–1605 (2020).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015).
Giannakis, M. et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 17, 1206 (2016).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Hellmann, M. D. et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378, 2093–2104 (2018).
Campesato, L. F. et al. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 6, 34221–34227 (2015).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020).
Barroso-Sousa, R., Pacifico, J. P., Sammons, S. & Tolaney, S. M. Tumor mutational burden in breast cancer: current evidence, challenges, and opportunities. Cancers https://doi.org/10.3390/cancers15153997 (2023).
Barroso-Sousa, R. et al. Tumor mutational burden and PTEN alterations as molecular correlates of response to PD-1/L1 blockade in metastatic triple-negative breast cancer. Clin. Cancer Res. 26, 2565–2572 (2020).
Winer, E. P. et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): a randomised, open-label, phase 3 trial. Lancet Oncol. 22, 499–511 (2021).
Emens, L. A. et al. 296P Tumour mutational burden and clinical outcomes with first-line atezolizumab and nab-paclitaxel in triple-negative breast cancer: exploratory analysis of the phase III IMpassion130 trial. Ann. Oncol. 31, S360–S361 (2020).
Alva, A. S. et al. Pembrolizumab in patients with metastatic breast cancer with high tumor mutational burden: results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J. Clin. Oncol. 39, 2443–2451 (2021).
Curran, M. A., Montalvo, W., Yagita, H. & Allison, J. P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 107, 4275–4280 (2010).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Motzer, R. J. et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 378, 1277–1290 (2018).
Overman, M. J. et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 36, 773–779 (2018).
Overman, M. J. et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 18, 1182–1191 (2017).
Ready, N. et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 37, 992–1000 (2019).
Lenz, H. J. et al. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the Phase II CheckMate 142 study. J. Clin. Oncol. 40, 161–170 (2022).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Matson, V. et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018).
Derosa, L. et al. Intestinal Akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat. Med. 28, 315–324 (2022).
Gandara, D. et al. 29 Lung-MAP composite signature for immune checkpoint inhibitor (ICI) efficacy in advanced squamous cell lung cancer (SCC). J. Immunother. Cancer 10, A31 (2022).
Tolba, K. et al. 584 Evaluation of a composite immunotherapy signature in non-small cell lung cancer patients treated with atezolizumab. J. Immunother. Cancer 10, A611 (2022).
Barroso-Sousa, R. et al. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 31, 387–394 (2020).
Sammons, S. et al. Genomic evaluation of tumor mutational burden-high (TMB-H) versus TMB-low (TMB-L) metastatic breast cancer to reveal unique mutational features. J. Clin. Oncol. 39, 1091–1091 (2021).
Sammons, S. et al. Abstract P2-08-08: concurrent predictors of an immune responsive tumor microenvironment within tumor mutational burden-high breast cancer. Cancer Res. 82, P2-08-08–P02-08-08 (2022).
Sokol, E. S. et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann. Oncol. 30, 115–123 (2019).
Chumsri, S. et al. Durable complete response with immune checkpoint inhibitor in breast cancer with high tumor mutational burden and APOBEC signature. J. Natl Compr. Canc Netw. 18, 517–521 (2020).
Chumsri, S. et al. Abstract PD14-09: APOBEC signature, clinical characteristics, and outcome in hormone receptor-positive (HR+) HER2-negative (HER2-) breast cancer (BC) patients (pts) in real-world data (RWD). Cancer Res. 82, PD14-09–PD14-09 (2022).
Gandara, D. R. et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 24, 1441–1448 (2018).
Friedman, C. F. et al. Atezolizumab treatment of tumors with high tumor mutational burden from MyPathway, a multicenter, open-label, phase IIa multiple basket study. Cancer Discov. 12, 654–669 (2021).
Duraiswamy, J., Kaluza, K. M., Freeman, G. J. & Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 73, 3591–3603 (2013).
Beavis, P. A. et al. Dual PD-1 and CTLA-4 checkpoint blockade promotes antitumor immune responses through CD4(+)Foxp3(-) cell-mediated modulation of CD103(+) dendritic cells. Cancer Immunol. Res. 6, 1069–1081 (2018).
Shi, L. Z. et al. Interdependent IL-7 and IFN-gamma signalling in T-cell controls tumour eradication by combined alpha-CTLA-4+alpha-PD-1 therapy. Nat. Commun. 7, 12335 (2016).
Adams, S. et al. A multicenter phase II trial of ipilimumab and nivolumab in unresectable or metastatic metaplastic breast cancer: cohort 36 of Dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART, SWOG S1609). Clin. Cancer Res. 28, 271–278 (2022).
Santa-Maria, C. A. et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget 9, 18985–18996 (2018).
Roussos Torres, E. T. et al. Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: a phase Ib trial. Nat. Cancer 5, 866–879 (2024).
Bergholz, J. S. et al. PI3Kbeta controls immune evasion in PTEN-deficient breast tumours. Nature 617, 139–146 (2023).
Trujillo, J. A. et al. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or PTEN loss in metastatic melanoma. J. Immunother. Cancer 7, 295 (2019).
Hermida-Prado, F. et al. Endocrine therapy synergizes with SMAC mimetics to potentiate antigen presentation and tumor regression in hormone receptor-positive breast cancer. Cancer Res. 83, 3284–3304 (2023).
Keenan, T. E. et al. Molecular correlates of response to eribulin and pembrolizumab in hormone receptor-positive metastatic breast cancer. Nat. Commun. 12, 5563 (2021).
Barroso-Sousa, R. & Thommen Teles, L. Gut microbiome and breast cancer in the era of cancer immunotherapy. Curr. Breast Cancer Rep. 11, 272–276 (2019).
Terrisse, S. et al. Intestinal microbiota influences clinical outcome and side effects of early breast cancer treatment. Cell Death Differ. 28, 2778–2796 (2021).
Cai, J., Sun, L. & Gonzalez, F. J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 30, 289–300 (2022).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science https://doi.org/10.1126/science.aan5931 (2018).
Derosa, L. et al. Microbiota-centered interventions: the next breakthrough in immuno-oncology? Cancer Discov. 11, 2396–2412 (2021).
Wang, Y. et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat. Med. 24, 1804–1808 (2018).
Forbes, J. D., Van Domselaar, G. & Bernstein, C. N. The gut microbiota in immune-mediated inflammatory diseases. Front. Microbiol. 7, 1081 (2016).
Hoa, N. T., Brannigan, J. A. & Cutting, S. M. The Bacillus subtilis signaling protein SpoIVB defines a new family of serine peptidases. J. Bacteriol. 184, 191–199 (2002).
Catinean, A. et al. Probiotic Bacillus spores together with amino acids and immunoglobulins exert protective effects on a rat model of ulcerative colitis. Nutrients 12, 3607 (2020).
Garcia, E. P. et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med. 141, 751–758 (2017).
Weis, L. N., Tolaney, S. M., Barrios, C. H. & Barroso-Sousa, R. Tissue-agnostic drug approvals: how does this apply to patients with breast cancer? NPJ Breast Cancer 7, 120 (2021).
Woodhouse, R. et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 15, e0237802 (2020).
Sun, J. X. et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput. Biol. 14, e1005965 (2018).
Merck & Co., Inc. PD-L1 testing and scoring. https://www.keytrudahcp.com/biomarker-testing/pd-l1-expression-testing/ (2022).
Salgado, R. et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann. Oncol. 26, 259–271 (2015).
Lee, J. K. et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precis. Oncol. 6, 91 (2022).
Crowdis, J., He, M. X., Reardon, B. & Van Allen, E. M. CoMut: visualizing integrated molecular information with comutation plots. Bioinformatics 36, 4348–4349 (2020).
Ricciuti, B. et al. Impact of DNA damage response and repair (DDR) gene mutations on efficacy of PD-(L)1 immune checkpoint inhibition in non-small cell lung cancer. Clin. Cancer Res. 26, 4135–4142 (2020).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife https://doi.org/10.7554/eLife.65088 (2021).
Blanco-Miguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
Acknowledgements
The authors acknowledge receiving institutional support for this study from Bristol Myers Squibb, The Department of Defense Physician Research Award (W81XWH-21-1-0084, PC200150) and Idea Development Award—Early-Career Investigator (KC210042/W81XWH-22-1-0455) (S.H.A.), National Comprehensive Cancer Network Oncology Research Program - Pfizer Enhancing Academic-Community-Patient Partnerships in Metastatic Breast Cancer Care. E.A.M. acknowledges support as the Rob and Karen Hale Distinguished Chair in Surgical Oncology. The funders did not have a role in the study design, data collection and analysis, or manuscript writing. The authors would like to acknowledge Timothy K. Erick, PhD, for writing and editorial assistance, and Kate Bifolck for editorial and submission assistance in the preparation of the manuscript. They are full-time employees of Dana-Farber Cancer Institute and were not compensated beyond their regular salaries. These data were presented in part at the 2021 San Antonio Breast Cancer Symposium (San Antonio, TX, USA) and the 2023 San Antonio Breast Cancer Symposium (San Antonio, TX, USA).
Author information
Authors and Affiliations
Contributions
Conceptualization: R.B.-S. and S.M.T. Data curation: J.G.T.Z., L.A.E., T.M.K., M.M., T.L., and N.T. Formal analysis: J.G.T.Z., T.L., T.M.K., M.M., N.T., and R.J. Funding acquisition: R.B.-S. and S.M.T. Investigation: R.B.-S., S.M.R., L.A.E., B.O., N.U.L., and S.M.T. Methodology: J.G.T.Z., T.L., T.M.K., M.M., N.T., and R.J. Project administration: P.L., M.K.D., J.K., M.E.H., V.A., and A.B. Resources: R.B.-S., S.M.R., L.A.E., B.O., N.U.L., and S.M.T. Supervision: S.M.T. Validation: J.G.T.Z., T.L., T.M.K., and M.M. Visualization: J.G.T.Z., T.L., T.M.K., and M.M. Writing – original draft: R.B.-S. and S.M.T. Writing – review & editing: R.B.-S., J.G.T.Z., T.L., S.M.R., L.A.E., T.M.K., C.A.C.S., S.H.A., H.C., B.O., P.L., M.K.D., M.M., J.K., M.E.H., V.A., A.B., N.U.L., N.T., R.J., E.A.M., and S.M.T.
Corresponding author
Ethics declarations
Competing interests
R.B-S. reported receiving speaker bureau fees from AstraZeneca, Daichi-Sankyo, Eli Lilly, Pfizer, Novartis, Merck, and Roche. He has also served as a consultant/advisor for AstraZeneca, Eli Lilly, Libbs, Roche, Merck, and has received support for attending medical conferences from AstraZeneca, Roche, Eli Lilly, Daichi-Sankyo, and Merck. Has received institutional research funding from AstraZeneca and Daichi-Sankyo and owns stocks in the educational company RD Educacao Medica LTDA. J.G. owns stocks in the biotechnology exchange-traded funds CNCR, IDNA, IBB, and XBI, owns stocks in Novo Nordisk, and owned stocks in Adaptive Biotechnologies, 2seventy bio, and bluebird bio. M.M. is an employee of Foundation Medicine and owns stock in Roche Holding AG. S.M.R. has received institutional research funding from Eli Lilly, Celldex Therapeutics, Bristol Myers Squibb, Tvardi Therapeutics, and Silverback Therapeutics. She has served as a scientific advisor for Pfizer, Eli Lilly, and Silverback Therapeutics. V.A. reports current employment at Olema Pharmaceuticals and stock ownership in OLMA. N.U.L. reports institutional research support from Genentech (and Zion Pharmaceutical as part of GNE), Pfizer, Merck, Seattle Genetics (now Pfizer), Olema Pharmaceuticals, and AstraZeneca; consulting honoraria from Puma, Seattle Genetics, Daiichi-Sankyo, AstraZeneca, Olema Pharmaceuticals, Janssen, Blueprint Medicines, Stemline/Menarini, Artera Inc., and Eisai; royalties from UpToDate; and travel support from Olema Pharmaceuticals and AstraZeneca. R.J. received institutional research support from Pfizer and Lilly. Consulting honoraria from Lilly, Pfizer, Novartis, and Carrick Therapeutics. E.A.M. reports compensated service on scientific advisory boards for AstraZeneca, BioNTech, Merck, and Moderna; uncompensated service on steering committees for Bristol Myers Squibb and Roche/Genentech; speakers honoraria and travel support from Merck Sharp & Dohme; and institutional research support from Roche/Genentech (via SU2C grant) and Gilead. E.A.M. also reports research funding from Susan Komen for the Cure for which she serves as a Scientific Advisor, and uncompensated participation as a member of the American Society of Clinical Oncology Board of Directors. S.M.T. receives institutional research funding from Genentech/Roche, Merck, Exelixis, Pfizer, Lilly, Novartis, Bristol Myers Squibb, Eisai, AstraZeneca, Gilead, NanoString Technologies, Seattle Genetics, OncoPep, Daiichi-Sankyo, Menarini/Stemline, and Jazz Pharmaceuticals; has served as an advisor/consultant for Novartis, Pfizer (SeaGen), Merck, Eli Lilly, AstraZeneca, Genentech/Roche, Eisai, Sanofi, Bristol Myers Squibb, CytomX Therapeutics, Daiichi-Sankyo, Gilead, Zymeworks, Zentalis, Blueprint Medicines, Reveal Genomics, Sumitovant Biopharma, Umoja Biopharma, Artios Pharma, Menarini/Stemline, Aadi Bio, Bayer, Incyte Corp, Jazz Pharmaceuticals, Natera, Tango Therapeutics, Systimmune, eFFECTOR, Hengrui USA, Cullinan Oncology, Circle Pharma, Arvinas, BioNTech, Johnson&Johnson/Ambrx, Launch Therapeutics, Zuellig Pharma, Bicycle Therapeutics, BeiGene Therapeutics, Mersana, and Summitt Therapeutics; and receives travel support from Eli Lilly, Sanofi, Gilead, Jazz Pharmaceuticals, Pfizer, and Arvinas. C.A.C.S. reported receiving speaker bureau fees from Bristol Myers Squibb. The remaining authors declare no financial or non-financial competing interests.
Peer review
Peer review information
Nature Communications thanks Ludmila Danilova, Alberto Montero, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Barroso-Sousa, R., Zanudo, J.G.T., Li, T. et al. Nivolumab plus low-dose ipilimumab in hypermutated HER2-negative metastatic breast cancer: a phase II trial (NIMBUS). Nat Commun 16, 4430 (2025). https://doi.org/10.1038/s41467-025-59695-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59695-1
This article is cited by
-
Long non-coding RNAs and signaling networks in non-small cell lung cancer: mechanistic insights into tumor pathogenesis
Cancer Gene Therapy (2025)
-
The Potential Role of Immunotherapy in ER-positive Breast Cancer
Current Breast Cancer Reports (2025)
