
Abstract
Peptidylarginine deiminases (PAD1-4) are calcium dependent enzymes responsible for protein citrullination, a post-translational modification converting arginine residues to citrulline. Elevated levels of citrullinated proteins have been associated with rheumatoid arthritis, neurodegenerative diseases, and cancers. Though highly selective PAD4 inhibitors have been described, inhibitors to the broader family currently are limited to covalent substrate analogs. Herein, we describe an allosteric binding pocket common to PAD1-4 suitable for the identification of potent, non-covalent enzyme inhibitors. A ligand-based virtual screen is utilized to identify a PAD4 inhibitor for which surface plasmon resonance confirms target binding but non-competitively with a known PAD4 ligand. We further show through co-crystal structure analysis that the ligand binds PAD4 at an allosteric pocket resulting in stabilization of a catalytically inactive, calcium-deficient enzyme conformation. A ligand designed based on this site potently inhibits all four PAD isozymes and prevents protein citrullination in neutrophils with a broader protein repertoire than observed with a PAD4-selective inhibitor.
Similar content being viewed by others


Introduction
Protein citrullination is a post-translational modification catalyzed by the peptidylarginine deiminase (PAD) enzyme family in which arginine residues are converted to citrulline. There are five human PAD isozymes, including the catalytically active members PAD1-4 and PAD6, which is reported to be enzymatically inactive1,2,3. The PADs are calcium (Ca2+)-dependent enzymes with either four (PAD1), five (PAD3 and PAD4), or six (PAD2) Ca2+ ion binding sites (designated as Ca1-6) identified from protein crystal structure analysis4,5,6,7. As a consequence, PAD functional activity is tightly controlled by local Ca2+ ion concentration. All the calcium binding sites are required to be occupied in order to induce a conformational change in the protein fold, leading to the holoenzyme with the active site cysteine (Cys645, PAD4) in the proper orientation for the enzymatic reaction8. The expression profile of the PAD family is broad and isozyme dependent, with localization to the epidermis and uterus (PAD1), hematopoietic cells (PAD2 and PAD4), central nervous system (PAD2), hair follicles (PAD3), and oocytes and embryos (PAD6)9. All of the PADs may be found in the cytoplasm of cells; however, PAD4 is additionally located in the nucleus10. PAD2 and PAD4 are also commonly observed in extracellular compartments as a consequence of release during certain cell death mechanisms11.
Protein citrullination mediated by the PAD enzyme family has a profound impact on the electrostatics of substrate proteins since a positively charged arginine residue is replaced by a neutral citrulline. This change may have consequences on protein structure, protein-protein interactions, and protein stability, leading to altered signaling networks and cellular responses. The scope of known citrullination sites has recently been estimated at over 14,000, covering more than 4000 proteins12. Elevated levels of citrullinated proteins or their corresponding autoantibodies have been associated with disorders including autoimmunity13,14,15,16, neurodegenerative disease17,18, and cancers19. Of particular note is the role that protein citrullination plays in neutrophils and its possible linkage to the progression of rheumatoid arthritis (RA). Neutrophils may undergo NETosis, a form of programmed cell death that releases neutrophil extracellular traps (NETs)20. During this event, citrullinated proteins and PAD enzymes are released into the surrounding tissue, such as the joint synovium, where inflammation occurs during RA disease11,21. Indeed, peptides derived from protein citrullination by PAD2 and PAD4 have been isolated from synovial fluid samples of RA patients, and the presence of anti-citrullinated protein antibodies (ACPAs) is diagnostic of RA progression22,23. In addition, a strong genetic association has been identified between the PADI4 gene, responsible for encoding PAD4, and RA incidence24. Therefore, significant interest has emerged to identify inhibitors to the PAD enzyme family. The presence of a catalytic cysteine residue within the highly conserved active site of PAD1-4 has led to the development of substrate-based, irreversible covalent inhibitors utilizing a Cl-amidine or F-amidine covalent reactive group with varying specificity within the PAD family25,26,27. Lewis et al. have described the identification of PAD4-specific inhibitors by screening of a DNA-encoded small molecule library28. The reported inhibitors bind a calcium-deficient isoform of the enzyme at a pocket resulting from a ligand-induced reorganization of the active site.
In this work, we describe an allosteric binding pocket common to PAD1-4 suitable for the identification of potent, non-covalent enzyme inhibitors. A ligand-based virtual screen is utilized to identify a PAD4 inhibitor for which surface plasmon resonance confirms target binding. Unexpectedly, the ligand displays non-competitive binding with known PAD4 ligands, suggesting a differentiated mechanism of action. We further show through co-crystal structure analysis that the ligand binds PAD4 at an allosteric pocket, resulting in stabilization of a catalytically inactive, calcium-deficient enzyme conformation. Further supporting this mechanism, the allosteric ligand, in combination with a known inhibitor binding to a different site, synergistically inhibits PAD4 enzyme function. An optimized ligand designed based on the binding site identified in this study potently inhibits all four PAD isozymes and prevents protein citrullination in neutrophils with a broader protein repertoire than observed with a PAD4-selective inhibitor.
Results
PAD4 inhibitor identified through ligand-based virtual screen
A virtual screen was conducted of the Pfizer compound library (4.3 M compounds) using a 2D fingerprint with data fusion29 methodology to identify compounds similar to the PAD4-selective ligand GSK14728 (Fig. 1a). The top scoring 1250 compounds were screened for inhibition of PAD4-mediated deimination of N-α-benzoyl-L-arginine ethyl ester (BAEE) through fluorescent detection of ammonia release at a single dose of 40 µM. Compound PAD-PF1 (Fig. 1a) was identified as a hit (56% inhibition) in this screen. Inhibition of PAD4 by PAD-PF1 was confirmed utilizing an orthogonal fluorescence quenching (FQ) assay employing a fluorescently labeled arginine-containing peptide substrate30. A Ca2+ ion concentration of 0.4 mM was employed in this assay to approximate the concentration yielding half-maximum activity (K0.5)31. Under these conditions, PAD-PF1 inhibited PAD4 in a dose-dependent manner with an IC50 of 15.9 µM (Fig. 1b). The binding affinity and kinetics of PAD-PF1 were measured using a surface plasmon resonance (SPR) binding assay employing immobilized bap-tagged human PAD4 enzyme. PAD-PF1 demonstrated rapid on/off kinetics of binding to PAD4 with a KD of 2.82 µM (Fig. 1c). Since PAD-PF1 was identified based on its 2D similarity with GSK147, the assumption was that PAD-PF1 was binding to the same pocket previously described28 and would demonstrate binding competitive with GSK147. In order to confirm this assumption, the binding affinity of GSK147 was determined by an SPR binding assay utilizing the standard running buffer (Fig. 1d) and where PAD-PF1 (10 µM) was added to the running buffer (Fig. 1e). Surprisingly, the affinity of GSK147 did not significantly change in the presence or absence of PAD-PF1 (KD = 0.380 µM and 0.469 µM, respectively) (p = 0.0754 student t test). This unexpected result suggested that PAD-PF1 was binding to a different binding site than occupied by GSK147 and that both ligands might bind simultaneously.

a Chemical structures of PAD-PF1, GSK147, and PAD-PF2. b PAD-PF1 inhibits PAD4-mediated citrullination of peptide substrate Atto655-(Ser-Arg-Gly-Ala)3 in a fluorescence quenching (FQ) assay in the presence of 0.4 mM Ca2+. Data points and error bars represent the mean and SD of quadruplicate determinations of a representative experiment. IC50 = 15.9 µM, pIC50 = 4.80 (0.26), mean (SD), n = 29 biologically independent experiments. c–e Representative surface plasmon resonance (SPR) traces characterizing binding of respective ligands to Bap-tagged PAD4 captured on streptavidine sensor. The red line represents the fitted curve. c PAD-PF1. KD = 2.82 µM, pKD = 5.55 (0.12), mean (SD), n = 4 technical replicates. The highest concentration of PAD-PF1 tested was 10 µM, with five more 3-fold dilutions. d, GSK147. KD = 0.469 µM, pKD = 6.33 (0.016), mean (SD), n = 4 technical replicates. The highest concentration of GSK147 tested was 3.33 µM with five more 3-fold dilutions. e Same concentration series of GSK147 in the presence of 10 µM PAD-PF1. KD = 0.380 µM, pKD = 6.42 (0.071), mean (SD), n = 4 technical replicates. Source data are provided as a Source Data file.
PAD-PF1 binds to an allosteric site distinct from substrate and GSK147
In order to understand the binding mode of PAD-PF1 and its relationship to the GSK147 binding site, a crystal structure of the tertiary complex between PAD4, PAD-PF1, and GSK147 was determined to a resolution of 2.41 Å. The resulting structure confirmed that PAD-PF1 and GSK147 can bind PAD4 simultaneously at different sites (Fig. 2a). The overall protein structure closely resembled the reported binary structure between PAD4 and GSK14728 (Supplementary Fig. 1a). As anticipated, GSK147 bound to a cryptic site immediately adjacent to the active site of PAD4 (Fig. 2b and Supplementary Fig. 2a, c). As described previously by Lewis28, the central benzimidazole ring of GSK147 packs tightly against Phe634 and Val643, while the amino hydroxypiperidine inserts into the active site where it engages residues Asp473, Asp350, and His471 involved in guanidine binding of the substrate. Consistent with the SPR results, PAD-PF1 binds at a remote site on the interface between the second immunoglobulin domain (IG2, residues 122–296) and the catalytic domain (CD, residues 297–666) adjacent to the active site (Fig. 2c and Supplementary Fig. 2b, d). The bromophenyl group of PAD-PF1 inserts between hydrophobic residues Phe368, Phe407, and Leu447, which all move in a coordinated manner to accommodate it (Supplementary Fig 2b). The pyrrolopyrimidine ring stacks against Phe450 while also contributing to the shift in Phe368 via steric clash with the methyl substituents. The resulting rotamer of Phe368 is likely stabilized in part by a π-stacking interaction with Trp296. The pyrrolopyrimidine ring also displaces the strand Val392-Pro396, forming a stabilizing interaction with the hydrophobic portion of the side chain of Arg394. The glycine amide distal terminus of PAD-PF1 is more solvent exposed, forming hydrogen bonds with IG2 residues Leu254 (CO, water mediated) and Phe256 (NH). The terminal pyrrolidine ring sits on a hydrophobic surface formed by Val392, Val152, and Phe256. The opposite face of the proline ring is largely solvent-exposed and ringed by carbonyl oxygens of Asn153 and Leu254. Owing to the low resolution of the structure, no calcium ions could be reliably modeled, and the two enantiomers of racemic PAD-PF1 could not be differentiated. The (S)-enantiomer is depicted as supported by subsequent higher resolution structures in the series (vide infra).

a Crystal structure of human full-length recombinant PAD4 simultaneously bound to GSK147 (orange carbon sticks and van der Waals surface) and PAD-PF1 (green carbon sticks and van der Waals surface). Protein backbone displayed as a ribbon diagram. b PAD4 binding pocket of GSK147 (orange carbon sticks). c PAD4 binding pocket of PAD-PF1 (green carbon sticks). In (b, c), side chains interacting with the small molecules are shown as white carbon sticks, hydrogen bonds are dotted lines, and water molecules are represented as red spheres. Key protein residues with hydrophobic interactions with the ligand are labeled in red.
Potent inhibition of PAD1-4 achievable by targeting allosteric site
Utilizing information gained from the bound conformation of PAD-PF1 and its interactions in the PAD4 binding pocket, iterative ligand design led to an inhibitor with significantly improved potency. The pyrrolopyrimidine ring was replaced by an isoquinoline ring as docking studies suggested it would lead to a better trajectory for the aryl substituent into the hydrophobic pocket and remove the potentially unfavorable polar ring nitrogen atom in proximity to the hydrophobic Leu447 residue. The isoquinoline ring was also expected to engage the hydrophobic surface of the pocket more efficiently than the dimethyl substituents found in PAD-PF1. Orthogonality of the aryl substituent was viewed as critical for high potency by maximizing hydrophobic interactions at the binding site formed by residues Phe368, Phe407, and Leu447. Therefore, ortho-substituted aromatics were evaluated for replacement of the 4-bromophenyl group. In line with our structural hypothesis, a 3,5-dichloropyridine group was found to be one of the 4-bromophenyl group replacements that demonstrated improved potency, resulting in the identification of PAD-PF2 (Fig. 1a).
PAD-PF2 was found to be a significantly more potent inhibitor than PAD-PF1 of PAD4 mediated citrullination in the fluorescent quenching assay with an IC50 of 42.7 nM (Fig. 3a). The enantiomer of PAD-PF2 (ent-PAD-PF2) was found to be significantly less potent (IC50 = 26.0 µM) suggesting that the amine substituent plays a critical role in interactions with the binding pocket. Given the high level of homology between PAD1-4 for the amino acid residues bordering the binding pocket identified in this study (Supplementary Fig. 3a), we wondered whether PAD-PF2 could also inhibit other PAD isozymes. PAD-PF2 was found to stabilize the PAD2 protein against thermal denaturation with a similar EC50 to that of PAD4 (EC50 = 2.04 µM and 1.95 µM, respectively), suggesting the potential to bind PAD2 (Fig. 3b). The ability of PAD-PF2 to functionally inhibit other PAD isozymes was assessed using a glutamate dehydrogenase (GDH)-enzyme coupled assay (Fig. 3c–f). In contrast to GSK147, which is reported to be PAD4-specific, PAD-PF2 was found to inhibit all four catalytically active PAD isozymes (PAD1-4) with comparable potency under conditions of low (0.25 mM) calcium. Given the critical role calcium ions play in the activation mechanism of the PADs, the dependence of Ca2+ concentration on the inhibition of both PAD2 and PAD4 by PAD-PF2 was further evaluated (Fig. 3g, h, respectively). For this purpose, the Ca2+ concentrations in the assay were set to 0.25 mM and 1.5 mM, which correspond to values near or above the half-maximal concentrations (K0.5) reported for these enzymes (0.2 mM PAD2 and 0.5 mM PAD4). The IC50 value for inhibition of PAD2 and PAD4 by PAD-PF2 increased 111-fold and 35-fold, respectively, at the higher Ca2+ concentration compared to the low Ca2+ conditions.

a PAD-PF2 (open circles) inhibits PAD4-mediated citrullination of peptide substrate Atto655-(Ser-Arg-Gly-Ala)3 employing a fluorescence quenching (FQ) assay in the presence of 0.4 mM Ca2+. Data points and error bars represent the mean and SD of three independent experiments in duplicate. IC50 = 42.7 nM, pIC50 = 7.37 (0.03), mean (SD), n = 3 independent experiments. The enantiomer of PAD-PF2 (ent-PAD-PF2, open squares) was less potent. IC50 = 26.0 µM, pIC50 = 4.59 (0.45), mean (SD), n = 3 independent experiments. b PAD-PF2 induces a thermal stabilization of PAD2 (open circles, EC50 = 2.04 µM) and PAD4 (open squares, EC50 = 1.95 µM) protein. Individual data points shown for a duplicate determination. c–f Inhibition of the respective PAD isoform by PAD-PF2 determined using the GDH-enzyme coupled assay in the presence of 0.25 mM Ca2+. Individual data points shown for a duplicate determination along with the fitted IC50 curves (line). c PAD1, IC50 = 109 nM. d PAD2, IC50 = 28.5 nM. e PAD3, IC50 = 106 nM. f PAD4, IC50 = 24.0 nM. g, h Calcium dependence of PAD2 or PAD4 inhibition by PAD-PF2. Inhibition was determined using the GDH enzyme-coupled assay. Data points from duplicate experiments performed at high (open squares) and low (open circles) Ca2+ concentrations are plotted together, and the fitted IC50 curves are shown (line). g The determined IC50 values for PAD2 inhibition were 27.9 nM and 3.09 µM (0.25 and 1.5 mM Ca2+, respectively). h The determined IC50 values for PAD4 inhibition were 20.1 nM and 694 nM (0.25 and 1.5 mM Ca2+, respectively). Source data are provided as a Source Data file.
The functional consequences from simultaneous binding of PAD-PF2 and GSK147 to PAD4, and specifically the potential for synergy was explored by the method of Loewe32. For this experiment, enzyme kinetics was measured using the GDH-coupled assay at different combinations of the two inhibitors in the presence of 1.5 mM Ca2+. IC50 values for either PAD-PF2 or GSK147 were determined at fixed concentrations of the other compound by nonlinear regression fitting of the initial velocity data. This analysis, Fig. 4, shows coordinates (x, y), where x and y are normalized doses of the two inhibitors calculated from the IC50 values of each inhibitor determined at a fixed concentration of the other. For compounds showing an additive effect, these coordinates fall on a line called the isobole, satisfying the equation: (x/IC50x) + (y/IC50y) = 1, where x and y are the combination of doses of the two compounds and IC50x and IC50y are the IC50 values of each compound alone. Coordinates that fall either above or below the line demonstrate either antagonism or synergy, respectively, between the two compounds. As shown in Fig. 4, PAD-PF2 and GSK147 show strong synergistic inhibition of PAD4 under the conditions tested.

The method of Loewe32 was used to assess synergy between PAD-PF2 and GSK147 towards inhibition of PAD4. Serial dilutions of the inhibitors (0–3.6 μM) were tested in all combinations (two-by-two matrix) under the PAD4 GDH-coupled assay condition with 1.5 mM CaCl2. Plotted are the normalized doses of the two compounds that were observed to produce half-maximal activity. For circles, x = [GSK147]/GSK147 IC50 in the absence of PAD-PF2, y = PAD-PF2 IC50/PAD-PF2 IC50 in the absence of GSK147. For squares, x = GSK147 IC50/GSK147 IC50 in the absence of PAD-PF2, y = [PAD-PF2]/PAD-PF2 IC50 in the absence of GSK147. Isobole represented by the dotted line. Source data are provided as a Source Data file.
Common binding mode observed for PAD2 and PAD4 isozymes
Structures of PAD-PF2 bound to PAD4 and PAD2 were determined to 2.44 Å and 1.77 Å, respectively (Fig. 5 and Supplementary Fig 4). As anticipated, the isoquinoline ring of PAD-PF2 makes extensive hydrophobic interactions with PAD4 residues Phe368, Phe450, and Leu447 (Fig. 5a and Supplementary Fig. 4a, c). The benzo portion of the ring occupies the volume filled by the two methyl groups of PAD-PF1. The dichloropyridine substituent of PAD-PF2 occupies the same space as the bromophenyl group of PAD-PF1, with the ortho-chloro substituent situated within van der Waals distance of the CD2 atom of Phe450, suggesting a potential halogen bond to the polar aromatic proton. All other PAD-PF2 interactions with PAD4 are identical to those seen in the PAD-PF1 structure, although the water molecule mediating the hydrogen bond between the amine and Leu254 is missing from the latter owing to the low resolution.

a Crystal structure of human full-length recombinant PAD4 bound to PAD-PF2 (green carbon sticks). The disordered side chains of Asp369 and Ser370 were constructed using the program ADDS43. b Crystal structure of human full-length recombinant PAD2 bound to PAD-PF2 (green carbon sticks). In a, b, side chains interacting with the small molecules are shown as white carbon sticks, hydrogen bonds are dotted lines, and water molecules are represented as red spheres. Key protein residues with hydrophobic interactions with the ligand are labeled in red.
The structure of PAD-PF2 bound to PAD2 reveals nearly identical interactions between the compound and the corresponding residues from PAD4 (Fig. 5b and Supplementary Fig. 4b, d). The isoquinoline ring stacks against PAD2 residues Phe450 and Leu369, which is equivalent to Phe368 in PAD4. The loop comprising Tyr392-Leu398 is well defined, and the hydrophobic side chain of Arg395 packs against the isoquinoline ring as seen in the PAD4:PAD-PF1:GSK147 structure. This conformation of Arg395 is further stabilized by electrostatic interactions between the guanidinium of Arg395 and the side chains of Asp406 and Ser371. The placement of the positively charged guanidinium 3.6 Å from the dichloropyridine nitrogen suggests a further direct electrostatic stabilization of the complex. The distal end of PAD-PF2 forms identical interactions to those observed in the PAD4 complex, including the water mediated hydrogen bond between the amine group and Leu255 (CO, water A) and a direct hydrogen bond between the amide oxygen of PAD-PF2 and Phe257 (NH). Additional water mediated hydrogen bonds are visible between the amine of PAD-PF2 to Phe257 (CO) and Asp259 through water B, as well as to Glu396 through water C. All other interactions are similar to those seen in the PAD4 structure. Unlike the lower resolution structures of PAD4, the PAD2:PAD-PF2 structure allowed for confident placement of four of the six calcium ions noted in previous structures7, including Ca3, Ca4, and Ca5 in IG2, and Ca6 (Supplementary Fig. 5).
PAD-PF2 inhibits PAD-mediated citrullination in neutrophils
To quantify the cellular potency of PAD-PF2, human isolated neutrophils were stimulated with 5 µM ionomycin in the presence of increasing concentrations of the compound. Protein citrullination was detected by flow cytometry using either the monoclonal IgM F95 antibody, which detects a broad range of citrullinated proteins, or the polyclonal IgG antibody that specifically detects citrullinated histone H3 (citH3). Ionomycin induced robust protein citrullination in human isolated neutrophils as detected by both antibodies. GSK147 and PAD-PF2 fully inhibited CitH3 detected protein citrullination (Fig. 6a) with comparable IC50 values (1.1 and 1.0 µM, respectively). PAD-PF2 was also able to fully inhibit F95 detected protein citrullination (IC50 = 1.5 µM); however, GSK147 only afforded a partial inhibition (39%) of the F95 detected protein citrullination (IC50 = 1.1 µM) (Fig. 6b). These data are indicative of the broader PAD isozyme inhibition of PAD-PF2 compared with GSK147.

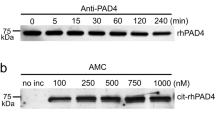
a, b Dose-dependent inhibition of ionomycin-induced protein citrullination was observed for both PAD-PF2 (open circles) and GSK147 (open squares) in human isolated neutrophils. Data points represent mean ± S.D. from four separate experiments. Determination of citrullinated proteins performed by flow cytometry employing detection by either (a) anti-CitH3 antibody: PAD-PF2: IC50 = 1.0 µM, pIC50 = 5.98 (0.20); GSK147: IC50 = 1.1 µM, pIC50 = 5.97 (0.11) (mean (SD), n = 4 biologically independent experiments) or (b), F95 antibody: PAD-PF2: IC50 = 1.5 µM, pIC50 = 5.83 (0.15); GSK147: IC50 = 1.1 µM, pIC50 = 5.98 (0.14) (mean (SD), n = 4 biologically independent experiments). c Western blot analysis of human isolated neutrophil lysates, either unstimulated or stimulated with 5 µM ionomycin in the absence or presence of three concentrations of PAD-PF2 or GSK147. Citrullinated proteins were detected with either F95 (green) or CitH3 (red) antibodies. Control proteins include CitH3 (Bands 6 and 7) and GAPDH (Band 4). d, e Western blot densitometry of (d) Band 1 and (e) Band 6 representing citrullinated histone H3 as a ratio of GAPDH protein loading control. Bars represent the mean from two biologically independent experiments. Source data are provided as a Source Data file.
Western blot analysis (Fig. 6c) of ionomycin-stimulated human neutrophil lysates demonstrated the broad detection of citrullinated proteins by the F95 antibody (Bands 1–3 and 5) and the specific protein detection by the CitH3 antibody (Bands 6 and 7). The detection of two bands by the CitH3 antibody has been observed by others33. Qualitatively, PAD-PF2 inhibits the citrullination of a greater number of detectable proteins than the PAD4-specific inhibitor GSK147. Through quantification of the F95-specific band 1 (Fig. 6d) and the CitH3-specific band 6 (Fig. 6e), these data corroborate the flow cytometry results. PAD-PF2 demonstrated a dose-dependent inhibition of band 1 protein citrullination as detected by the F95 antibody, while GSK147 failed to inhibit citrullination even at a concentration of 10 µM. On the other hand, both PAD-PF2 and GSK147 were able to inhibit citrullination of band 6 protein corresponding with CitH3. Therefore, broader PAD isozyme inhibition by PAD-PF2 translates into suppression of citrullination events on a greater number of proteins in stimulated human neutrophils compared with the PAD4-specific inhibitor GSK147.
The target specificity of PAD-PF2 was evaluated by profiling the compound against a diverse panel of receptors, enzymes, kinases, transporters, and ion channels at a dose of 10 µM concentration (Supplementary Tables 1 and 2). PAD-PF2 showed > 80-fold selectivity relative to the PAD4 enzymatic IC50 value. Among the targets tested above, PAD-PF2 was found to be a weak agonist of the κ-opioid receptor (EC50 = 7.55 µM, 177-fold) and weak antagonist of the M1 muscarinic acetylcholine receptor (IC50 = 12.3 µM, 288-fold). Also, weak binding affinity was observed to the choline transporter (Ki = 3.45 µM, 81-fold) and the L-type calcium ion channel (Ki = 11.1 µM, 260-fold).
Discussion
Ligand-based virtual screening is an established method to identify small-molecule binders to a wide range of protein classes. However, chemical structure similarity of the hit with the originating seed molecule alone does not imply both ligands will demonstrate a common mechanism of action. For example, the two ligands may bind to the same site but elicit different functional pharmacology, such as agonism versus antagonism. In the case described above, the two ligands elicit the same functional pharmacology but do so by interacting with the protein in a different manner. The serendipitous nature to discovering ligands of the latter outcome must be admitted.
PAD-PF1 shares many of the same structural features with GSK147, including a primary amine, a secondary amide, and two aromatic ring systems. Both compounds were found to inhibit the functional activity of PAD4. Nonetheless, through careful characterization by SPR binding and crystallography studies, it was deduced that the two molecules inhibit the enzyme through binding to two different binding sites. PAD-PF1 and PAD-PF2 bind to PAD4 at a site adjacent to the active site in the interface between the IG2 and catalytic domains, making specific hydrophobic, aromatic, and hydrogen-bond mediated contacts with the protein. A notable differentiation between the two series of compounds is their functional specificity among the PAD isozymes (PAD1-4). GSK147 is reported to be a PAD4-specific inhibitor28. PAD-PF2 was found to be a potent inhibitor of PAD1-3 in addition to PAD4. Binding of PAD-PF2 to PAD2 and PAD4 was confirmed by thermal shift assay, and co-crystal structures demonstrating occupancy at a similar binding pocket in both proteins. The protein sequences for PAD1-4 show a high level of homology in residues surrounding the binding site (Supplementary Fig. 3a). Of the four PAD isozymes, inhibition by PAD-PF2 was weakest against PAD1 and PAD3. Both PAD1 and PAD3 have a hydroxylated amino acid residue (Thr and Ser, respectively) proximal to the amine substituent (residue 255, PAD4 numbering) as opposed to hydrophobic residues in PAD2 and PAD4 (Cys and Ala, respectively) (Supplementary Fig. 3b). The polar nature of the residue may induce a repulsive polar-polar interaction or may interfere with the water hydrogen-bond network surrounding the amine. Although PAD6 is reported to be enzymatically inactive, the significant homology differences in key residues interacting with PAD-PF2 in the case of the other PAD isozymes would suggest the compound may have low binding affinity for PAD6.
The structures of PAD-PF2 bound to PAD2 and PAD4 suggest that modulation of the protein calcium site occupancy plays a critical role in the inhibitor mechanism of action. Our structural analysis focuses on PAD2 given the higher resolution nature of the structures, but we have confidence that these conclusions would also hold for PAD4. A comparison of the PAD2 structure bound to PAD-PF2 with the PAD2 F221/222 A holoenzyme structure (4N2C)7 reveals extensive reorganization of the catalytic domain (Supplementary Fig. 1b). Importantly, Arg347 is positioned such as to “cap” the substrate binding pocket and Cys647 is directed out of the active site suggesting a catalytically inactive enzyme form (Fig. 7a and Supplementary Fig. 6a) as opposed to the accessible active site seen in the holoenzyme (Fig. 7c). In the holoenzyme, the Ca2 binding site comprises the side chain carboxylates of Asp370, Glu352, and Asp374, as well as the carbonyl oxygen of Ser371 (Fig. 7d). PAD-PF2 was found to bind adjacent the Ca2 binding site such that the dichloropyridyl group would be only 2.6 Å from Pro372 as found in the holoenzyme structure (Fig. 7b and Supplementary Fig. 6b). This proximity forces the proline residue to flip upwards into the Ca2 binding site in the PAD-PF2 bound structure resulting in disruption of the metal binding site. Since Ca2 binding represents the ultimate step in PAD enzyme activation7, critical relocations of Trp348, Arg347, and Cys647 needed to form the active site are not observed in its absence. Therefore, binding of PAD-PF2 results in stabilization of a calcium-deficient enzyme form lacking Ca2 and incapable of forming the catalytically competent active site. Interestingly, although GSK147 and PAD-PF2 bind at different sites, they both induce a structurally similar inactive form of PAD4 (Supplementary Fig. 1c).

a, b Crystal structure of human full-length recombinant PAD2 bound to PAD-PF2 (PDB 9DOL). a Active site pocket with residues in catalytically inactive orientations. b Putative calcium ion (Ca2) binding domain with residue D374 disordered. PAD-PF2 (green carbon sticks). c, d, Crystal structure of holoenzyme PAD2 F221/222 A mutant (PDB 4N2C)7. c Active site pocket with calcium ion (Ca1, purple). d Calcium ion (Ca2, yellow) binding domain overlayed with position of PAD-PF2 (green carbon sticks) from PAD2:PAD-PF2 structure demonstrating potential clash with Pro372.
Note that since our PAD2 construct contains the native Phe221/222 sequence, we observe interactions between Phe222 of a symmetry related PAD2 monomer with amino acids Phe310, Phe355, Leu385, and Phe390 (Supplementary Fig. 6c). These interactions are distinct but reminiscent of those seen by Slade et al. 7, in which those authors saw interactions between residues Phe221 and Phe222 with symmetry related residues Phe310, Val337, Cys338, Trp348, Glue384, and Leu 385. Slade et al. 7 demonstrated through site-directed mutagenesis that this symmetry-related interaction precluded calcium binding at site Ca2. Although the symmetry interactions between Phe222 and its neighboring monomer in our structure are fewer and more removed from the Ca2 site, we cannot unequivocally decouple the effect of PAD-PF2 disruption of the Ca2 site from that of a symmetry contact. However, owing to the steric clashes induced by PAD-PF2 binding, the structure presented does predict that PAD-PF2 and calcium would bind mutually exclusive of one another at the Ca2 site. The sensitivity of inhibition to calcium ion concentration supports this hypothesis.
PAD4 has received considerable attention for its potential role in neutrophils to initiate neutrophil extracellular trap formation through citrullination of CitH320. However, it is now clear that a wide array of proteins are subject to post-translational citrullination, though their roles in physiology and human disease are not fully understood. All four PAD isozymes, either specifically or redundantly, likely plays a role in orchestrating the resulting citrullinated protein array. As shown here, stimulation by ionomycin leads to citrullination of multiple proteins in primary neutrophils. When an antibody with broad specificity was employed to detect citrullinated proteins, a PAD4-specific inhibitor was unable to fully inhibit citrullination. On the other hand, PAD-PF2 with a broader PAD isozyme specificity was able to inhibit protein citrullination more fully, and we identified specific proteins for which PAD-PF2, but not a PAD4-specific inhibitor, could block their citrullination. The ability of the compound tools described here to potently inhibit citrullination by all four of the catalytically active PAD family members will help to further expand our knowledge of the role protein citrullination plays in biology, including human disease.
Methods
Ethics Statement
Primary human blood samples were obtained with informed written consent from healthy human adult donors in accordance with the Pfizer Inc. Global Colleague Wellness Research Support Program (protocol CW RDP-02, IRB number: 08-33390) approved by the Advarra Institutional Review Board.
PAD proteins for assays
Human full-length recombinant N-terminal His-tagged PAD1 and PAD3 were purchased from Cayman Chemical (10784 and 10786, respectively). The Bac-to-Bac system (Invitrogen) was used to express recombinant PAD2 protein. Full-length PAD2 (amino acids 1–665) containing N-terminal BAP, 6X-His affinity, and FLAG tags, followed by a PreScission Protease cleavage site, was cloned into the pFastBac vector (Invitrogen). The plasmid was transformed into DH10Bac (Gibco) competent cells to produce bacmid. The bacmid was then purified and transfected into Sf9 cells using Cellfectin II (Invitrogen) at an MOI of 0.1. Sf9 cells were grown at 27 °C for 68 hours, followed by centrifugation. The resulting cell pellets were stored at − 80 °C. Cell paste was lysed in Lysis Buffer (50 mM Tris, pH 7.6, 400 mM NaCl, 1 mM TCEP, 1% Triton-X 100, 1.5 mM benzamidine) supplemented with EDTA-free complete Protease inhibitor tablets (Roche) and DNase. The resuspended cells were lysed in a microfluidizer at 18,000 psi and clarified by centrifugation. The clarified lysate was diluted to 1000 mL in lysis buffer and loaded onto a FLAG-resin column. Following elution using FLAG peptide, the PAD2-containing fractions were pooled and concentrated for loading onto a Superdex 200 size exclusion column equilibrated in 50 mM Tris, pH7.6, 10% glycerol, 500 mM NaCl, 1 mM TCEP, EDTA-free protease inhibitor cocktail, 1.5 mM benzamidine. PAD2-containing fractions were pooled, concentrated, and dialyzed overnight at 4 °C against 50 mM Tris, pH 7.6, 10% glycerol, 500 mM NaCl, 1 mM TCEP. This concentrated sample was aliquoted into single-use aliquots and flash frozen in liquid nitrogen prior to storage at − 80 °C.
A codon-optimized sequence encoding full-length PAD4 with an N-terminal fusion of Avitag-His6-FLAG tags, followed by a PreScission Protease site, was cloned into pET28b(+) by Genscript. For the production of biotinylated protein, the PAD4 plasmid was co-transformed with a plasmid expressing E.coli BirA. Plasmid DNA was used to transform BL21(DE3) (New England Biolabs) chemically competent cells, which were grown in LB media until they reached an OD600 of 0.8, and protein expression was induced by the addition of 0.3 mM IPTG (Sigma). Induction was continued for 19 h at 16 °C followed by centrifugation and storage of the cell pellets at − 80 °C. Cell pellets were resuspended in 50 mM Tris-HCl pH 8.0, 400 mM NaCl, 10% glycerol, and 1 mM TCEP with the addition of a Complete EDTA-free proteinase inhibitor tablet (Roche) and benzonase nuclease. Cells were lysed via microfluidizer at 18,000 psi and clarified via centrifugation at 30,000 g for 1 h. Supernatants were bound to a Fast Flow Histrap (Cytiva) column, washed with 20 column volumes of Buffer A (50 mM Tris-HCl pH 8.0, 400 mM NaCl, 10% glycerol, and 1 mM TCEP), followed by washing of the resin with 12% Buffer B (50 mM Tris-HCl pH 8.0, 400 mM NaCl, 10% glycerol, 1 mM TCEP, and 250 mM imidazole). The protein was then eluted with 100% Buffer B and injected onto a HiLoad s200 16/60 size exclusion column equilibrated with 50 mM Tris pH 8.0, 200 mM NaCl, 1 mM TCEP, and 10% Glycerol. Fractions containing PAD4 were pooled and concentrated to 50 µM using an Amicon 10 kDa NMWL filter (Millipore Sigma) and snap frozen in liquid N2 for storage at − 80 °C.
Compounds
PAD-PF1, PAD-PF2, and ent-PAD-PF2 were prepared by Pfizer Inc. For synthetic procedures and analytical, characterization see Supplementary Note 1. GSK-147 was prepared as described by Lewis et al. 28.
PAD4 Ammonia release assay
A PAD4 inhibitor screening assay kit (Cayman Chemical, 700560) was employed. Human recombinant PAD4 (Cayman Chemical) in Ammonia assay buffer (50 mM borate, pH 8.0, 5 mM DTT, and 10 mM CaCl2) was preincubated with various concentrations of compound or DMSO (1% final) in a Corning 3820 microplate. Following 10 min preincubation at room temperature, the reaction was initiated by adding substrate N-α-benzoyl-L-arginine ethyl ester (BAEE) at 250 µM and incubated for 1 h at room temperature. The reaction was terminated by incubation with Stop/Detection buffer for 1 h at room temperature before measuring fluorescence on an Envision microplate reader with excitation at 415 nm and emission at 475 nm. The fluorescence intensity data was normalized as a percentage of inhibition using 0 % and 100 % inhibition controls.
PAD4 fluorescence quenching (FQ) assay
Recombinant full-length human PAD4 was diluted to a final concentration of 2.4 nM in FQ assay buffer (20 mM HEPES pH 7.4, 140 mM NaCl, 0.01% Triton X-100, 0.4 mM CaCl2, and 5 mM DTT). The enzyme was preincubated with various concentrations of compound or DMSO control (1% final) in a 384-well black flat-bottom microplate (Corning 3820) for 10 min at room temperature. The enzymatic reaction was started with the addition of 8 µM of substrate Atto655 - (Ser-Arg-Gly-Ala)3 (CPC Scientific Inc.) in assay buffer. After 2 h, the reaction was stopped by adding stop reagent (10 mM N-ethyl maleimide in assay buffer) and followed by quenching with 60 µM Acid Green 27 (TCI America) for overnight incubation at room temperature. The fluorescence was read in an Envision microplate reader (excitation 620 nm; emission 685 nm). The fluorescence intensity data was normalized as a percentage of inhibition using 0% and 100% inhibition controls.
Glutamate dehydrogenase (GDH) enzyme-coupled assay for PAD enzyme activity
The enzyme activity of each of the PAD isoforms was assayed by detection of the product, ammonia, via coupled enzyme detection. GDH catalyzes the conversion of ammonium, α-ketogluterate, NADH, and H+ to NAD+, glutamate, and water, which is monitored spectrophotometrically at 340 nm34. The PAD selectivity assay conditions were: 100 mM HESPES, pH 7.5, 50 mM NaCl, 0.25 mM CaCl2, 1 mM substrate BAEE, 2 mM DTT, 0.01% Triton X-100, 40 μ/mL GDH (Sigma, G2626), 8.5 mM α-ketogluterate, and 0.2 mM NADH; the enzyme concentrations used were: 10 nM PAD1, 6 nM PAD2, 20 nM PAD3, or 4 nM PAD4. Serial dilutions of the compound were prepared in DMSO at 100x concentrations, then diluted into buffer to prepare a 4x stock solution immediately prior to assay. Reactions (40 μL) were prepared by combining 10 μL of [4x] BAEE, 10 μL of [4x] compound, and [2x] assay solution that contained all other reagents. Assays were performed in black/clear-bottom 384 well plates (Corning #3544), absorbance (340 nm) data were collected on an EnVision 2103 plate reader (PerkinElmer), and reactions were monitored at 140 s intervals for 50 plate repeats. An apparent molar absorptivity for NADH under the conditions of the assay was determined from a standard solution of NADH to be 4.13 mM-1. IC50 values were determined via nonlinear regression by fitting the rate data to the equation: f(x) = a/(1 + (x/b)), where f(x) is the measured enzyme rate at inhibitor concentration, x; a is the maximum observed velocity at x = 0 (i.e., in the absence of inhibitor); and b is the IC50. Enzyme inhibition kinetic data are graphically reported as % inhibition: 100*(1-(f(x)/a)).
Surface plasmon resonance binding
The binding affinity and kinetics of binding were measured using a surface plasmon resonance based binding assay. These experiments were carried out on a Biacore T200 instrument (Cytiva). All the samples were prepared in buffer consisting of 10 mM HEPES, pH 7.5, 150 mM NaCl, 0.25 mM TCEP, 0.01% Tween-20, and 2% DMSO. The same buffer was used during the capture of the protein and as the running buffer during the binding experiments. Bap-tagged PAD4 protein at 10 µg/mL was injected at 5 µL/min on a Streptavidin-coated sensor chip to achieve about 6,500 RUs of surface density. Compound samples were injected at a flow rate of 30 µL/min for 120 seconds of association time, followed by at least 300 s of dissociation period. For data fitting purpose, only 300 seconds of the dissociation phase was used but, actual time elapsed between the injections was more than 600 seconds. Regeneration was not used between the injections. The compounds were tested in a concentration series consisting of 6 samples made with 3-fold dilution. The concentration series tested for GSK147 consisted of 13.7, 41.2, 123.5, 370.3, 1111, and 3333 nM samples of GSK147. The concentration series tested for PAD-PF1 consisted of 41.2, 123.5, 370.3, 1111, 3333, and 10000 nM samples of PAD-PF1. For the competition experiment, the same concentration series for GSK147 was tested in the presence of 10 µM PAD-PF1. Multiple blank injections were run before and after each compound series to allow double reference subtraction during data processing and analysis. A DMSO curve was run during each experiment to properly correct for excluded volume. The data were processed and analyzed using Scrubber and Biaeval software to calculate binding affinities and kinetics by fitting the data to the Langmuir 1:1 binding model.
Thermal shift assay
A fluorescence-based thermal shift assay using recombinant human PAD2 or PAD4 proteins and firefly luciferase (fLuc) as a control was employed. Serial dilutions of PAD-PF2 in DMSO were prepared and then further diluted in assay buffer containing 20 mM HEPES pH 7.4, 140 mM NaCl, 0.4 mM CaCl2, 2 mM GSH, and 0.01% Triton X-100. Aliquots of each compound concentration (5 µL) were added to a 384-well PCR plate, followed by 10 µL of either 2x PAD4, 2x PAD2, or 2x fLuc (0.1 mg/mL final concentration). A 5 µL aliquot of 4x Sypro Orange dye (Invitrogen) was added to each well, and the plate was sealed with optical adhesive film. The plate was centrifuged briefly at 1000 rpm and loaded into a QuantStudio 6 Flex real-time PCR system (Applied Biosystems). A melt curve protocol was performed with the following settings: 25 °C for 2 min, followed by a gradual increase from 25 °C to 99 °C at 0.1 °C/s. The data was analyzed using Protein Thermal Shift Software (Applied Biosystems), and the melting temperature (Tm) of each sample was calculated based on the Boltzmann derivative.
PAD2 crystal construct and expression
The Bac-to-Bac system (Invitrogen) was used to express recombinant PAD2 protein for crystallography. Full-length PAD2 (amino acids 1–665) containing N-terminal BAP, 6X-His affinity, and FLAG tags, followed by a PreScission Protease cleavage site, was cloned into the pFastBac vector (Invitrogen) and transformed into DH10Bac (Gibco) competent cells to produce bacmid. The bacmid was then purified and transfected into Sf9 cells using Cellfectin II (Invitrogen) at an MOI of 0.1. Sf9 cells were grown at 27 °C for 65 h, followed by centrifugation. PAD2 cells were lysed in Lysis Buffer (50 mM Tris pH 7.6, 400 mM NaCl, 0.5 mM TCEP, 5 mM imidazole, 1% Triton-X 100) supplemented with protease inhibitor tablets (Roche) and nuclease (ThermoFisher Pierce). The resuspended cells were lysed in a microfluidizer at 18,000 psi and clarified by centrifugation. Clarified lysate was loaded onto a Ni-NTA column equilibrated in Buffer A (50 mM Tris pH 7.6, 10% glycerol, 400 mM NaCl, 0.5 mM TCEP, and 5 mM imidazole). PAD2 protein was washed on the column with Buffer B (50 mM Tris pH 7.6, 10% glycerol, 400 mM NaCl, 0.5 mM TCEP, and 100 mM imidazole) and eluted using Buffer C (50 mM Tris pH 7.6, 10% glycerol, 400 mM NaCl, 0.5 mM TCEP, and 250 mM imidazole). PAD2-containing fractions were pooled and put over a Superdex 200 16/600 (Cytiva) column equilibrated in 20 mM Tris-HCl pH 8.0, 10% glycerol, 500 mM NaCl, 0.5 mM TCEP, and 1 mM EDTA. PAD2-containing fractions were pooled, aliquoted in single-use aliquots, and stored at −80 °C.
PAD4 crystal construct and expression
Full-length PAD4 (amino acids 1–663) was expressed containing point mutations Gly55Ser, Val82Ala, and Gly112Ala, which were previously shown to be amenable for crystallization35. The sequence contained an N-terminal 6X-His tag and FLAG tag, followed by a PreScission Protease cleavage site, and was expressed in E. coli using a kanamycin resistance marker. PAD4 cells were lysed in Lysis Buffer (50 mM Tris pH 8.0, 10% glycerol, 500 mM NaCl, 1 mM TCEP, and 20 mM imidazole) supplemented with protease inhibitor tablets (Roche) and nuclease (ThermoFisher Pierce). The resuspended cells were lysed in a microfluidizer at 18,000 psi and clarified by centrifugation. Clarified lysate was loaded onto a Ni-NTA column equilibrated in Buffer A (50 mM Tris pH 7.6, 10% glycerol, 500 mM NaCl, 1 mM TCEP, and 20 mM imidazole). PAD4 protein was washed on the column with 10 column volumes of Buffer A and eluted using Buffer B (50 mM Tris pH 7.6, 10% glycerol, 500 mM NaCl, 1 mM TCEP, and 250 mM imidazole). PreScission protease was added to the peak fractions and simultaneously dialyzed overnight back into Buffer A. The sample was repassed over a Ni-NTA column equilibrated in Buffer A to remove the cleaved tag and protease. The flowthrough was collected. The PAD4 sample was put over a Superdex 200 16/600 (Cytiva) column equilibrated in 10 mM Tris pH 8.5, 500 mM NaCl, 1 mM DTT, and 1 mM EDTA. PAD4-containing fractions were pooled, aliquoted in single-use aliquots, and stored at − 80 °C.
Crystallization and structure determination
PAD2 was crystallized by the sitting drop vapor diffusion method, with the protein at 8 mg/mL in buffer 20 mM Tris pH 7.5, 10% glycerol, 500 mM NaCl, 0.5 mM TCEP, and 1 mM EDTA, and well solution composed of 0.1 M magnesium acetate, 50 mM MES pH 5.5, and 9-16% 2-methyl 2,4-pentane diol. PAD2 protein was mixed with an equal volume of the well solution and equilibrated against 75 µL of the well solution. Crystals typically appeared within 1 day. Diffraction quality crystals suitable for compound soaking were produced by microseeding into fresh crystallization droplets. Crystals were transferred to a soaking/cryoprotectant solution consisting of 0.1 M magnesium acetate, 50 mM MES pH 5.5, 100 µM CaCl2, 20% 2-methyl 2,4-pentane diol, and 20% PEG400. Compounds were diluted into this solution to a final concentration of 1–30 mM. Crystals were soaked for 1-2 hours before flash cooling in liquid nitrogen.
PAD4 was crystallized by the sitting drop vapor diffusion method, with the protein at 4 mg/mL in buffer 10 mM Tris pH 8.5, 500 mM NaCl, 1 mM DTT, and 1 mM EDTA, and well solution composed of 0.2 M sodium malonate pH 7 and 6% PEG 3350. PAD4 protein was mixed with an equal volume of the well solution and equilibrated against 75 µL of the well solution. Crystals typically appeared within 1 day. Crystals were transferred to a soaking/cryoprotectant solution consisting of 0.2 M sodium malonate, pH 7, 10% PEG 3350, and 20% glycerol. Compounds were diluted into this solution to a final concentration of 1–30 mM. Crystals were soaked for 1-2 hours before flash cooling in liquid nitrogen.
Crystallographic data were collected on each complex at Beamline 17ID of the Industrial Macromolecular Crystallographic Association Collaborative Access Team (IMCA-CAT, Advanced Photon Source, Argonne National Laboratory, Lemont, IL, USA), outfitted with a Dectris Eiger 9 M direct photon detector. Data processing was performed using AUTOPROC36. All subsequent data processing and handling operations were performed using the CCP4 suite of programs37. The structures of PAD2 and PAD4 were solved with the molecular replacement method, using program Phaser38 and PDB entries 4N2B and 3APM as starting models, respectively. Compounds were fit into Fo-Fc difference density maps using the program RHOFIT39. Manual rebuilding was conducted in COOT40, and automated refinement against the data was performed using Phenix41. The final refined coordinates have been deposited at http://www.rcsb.org42. Data and structural quality metrics are reported in Supplementary Table 3.
Human Neutrophil Isolation
Neutrophils were isolated from the peripheral whole blood from human blood samples via negative magnetic selection (Miltenyi, 130-104-434) followed by removal of residual erythrocytes (Miltenyi, 130-098-196). Neutrophils were then washed and resuspended in RPMI-1640 without phenol red (Gibco, 11835) for assessment of ionomycin induced citrullination.
Flow cytometry-based measurement of neutrophil citrullination
Neutrophils were plated at a density of 2 × 105 cells per well in a 96-deep well plate and preincubated with various concentrations of compounds or DMSO (0.2% final DMSO content) for 30 minutes prior to activation with 5 µM ionomycin (Sigma, I0634). Incubations were done at 37 °C, 5% CO2. After 30 minutes of activation, the assay was terminated with an equal volume of 2x Fixation/Permeabilization buffer (Invitrogen, 00-5523-00). Neutrophil fixation was done at room temperature for 20 min. After fixation, cells were washed one time using 1x permeabilization buffer (Invitrogen, 00-5523-00) and one additional time using 1x blocking buffer (1x permeabilization buffer, 2% BSA, 0.05% Tween-20, 100 µg/mL single-stranded DNA (Sigma, D7656)). Cells were washed by centrifugation at 600 × g for 5 min. After washing, neutrophils were resuspended in 1x blocking buffer and incubated for 1 h at room temperature. Neutrophils were then incubated with primary antibody cocktail (1:400 F95 (Millipore, MABN328), 1:1000 Hoechst 33342 (BD Bioscience, 561908) final) for 1 h at room temperature. Neutrophils were then washed using PBS (0.1% BSA) and resuspended in 1x blocking buffer prior to incubation with secondary antibody cocktail (1:100 anti-mouse IgM FITC (Invitrogen, 31992), 1:500 anti-citH3-AF647 (Abcam, Ab5103) final) for 1 h at room temperature. CitH3 antibody was conjugated using Alexa Fluor 647 microscale protein labeling kit (Invitrogen, A30009). Neutrophils were then washed one final time using PBS (0.1% BSA) and resuspended in this same buffer for assessment by flow cytometry. Gating for flow cytometry analysis (Supplementary Fig. 7) was as follows: (1) FSC-A vs SSC-A to gate on neutrophils, (2) FSC-A vs FSC-H to gate on single cells, (3) Hoechst + to identify nucleated neutrophils, (4a) AF647 + for neutrophils expressing citH3, and (4b) FITC + for neutrophils expressing additional citrullinated proteins detectable using F95. Integrated MFI (iMFI) was then calculated for both AF647 + and FITC + populations by multiplying the relative frequency (percent positive) of cells by the MFI of that population. IC50 curves were then generated using the iMFI values plotted against compound concentrations and fitting a 4-parameter non-linear regression using GraphPad Prism.
Western blot analysis of ionomycin stimulated human isolated neutrophils
Neutrophils were plated at a density of 5 × 106 cells per well in a deep well 96-well plate and preincubated with various concentrations of compounds or DMSO (0.2% final DMSO content) for 30 min prior to activation with 5 µM ionomycin (Sigma, I0634). Incubations were done at 37 °C, 5% CO2. After 20 minutes of activation, neutrophils were pelleted through centrifugation at 600 × g for 5 min and lysed in a final volume of 200 µL with shaking for 45 min using EPIgeneous Lysis buffer A, Part 1 (4x) (CisBio, 62EL1FDD) supplemented with 1x Phospho and Total Protein Blocking Agent (CisBio, 64KB1AAC) and 5 mM EDTA. After incubation, 50 µL of EPIgeneous Lysis Buffer A, Part 2 (5x) was added to each sample to complete the reaction. Protein concentrations were then determined using the Protein Assay Kit II with BSA standard (Bio-Rad, 5000002). Samples were then prepared for western blot analysis by adding protein lysates to appropriate volumes of 4x LDS Sample Loading Buffer (Invitrogen, NP0007) and 10x Sample Reducing Agent (Invitrogen, NP0004) so that ~ 3-4 µg of total protein can be loaded per lane. Samples were then heated to 95 °C for 10 min using a thermocycler and then placed on ice. Once cooled, 5 µL of SeeBlue Plus2 pre-stained protein standard (Invitrogen, LC5925) or 20 µL of sample was loaded into separate lanes of a 10-well 4-12% Bis-Tris gel (Invitrogen, NP0321BOX) and run at a constant 170 V using 1x MES Running Buffer (Invitrogen, NP0002). Proteins were transferred to a nitrocellulose membrane (Invitrogen, IB23001) using the iBlot 2, after which the membrane was blocked using Intercept PBS Blocking Buffer (Li-cor, 927-70001) for 1 hour at room temperature with gentle rocking. The membrane was incubated with primary antibody cocktail made up in Intercept P20 Antibody Diluent (1:250 F95 antibody, 1:1000 citH3 antibody, 1:1000 GAPDH antibody (Cell Signaling, 2118)) for 1 h at room temperature with rocking. Membrane was then washed 3x for 5 min using PBS (0.05% Tween-20) followed by incubation with secondary antibody cocktail made up in Intercept P20 Antibody Diluent (1:5000 goat anti-mouse IgM 800CW (Li-cor, 926-32280), 1:5000 goat anti-rabbit IgG 680RD (Li-cor, 926-68071)) for 1 h at room temperature with rocking. The membrane was further washed 3x for 5 minutes using PBS (0.05% Tween-20). Images for each membrane were then captured using the Li-Cor Odyssey Imaging System, and densitometry was performed. Band intensities were normalized to GAPDH level within the same lane.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates for crystal structures have been deposited in the Protein Data Bank (PDB) under the accession codes 9DPZ for PAD4:PAD-PF1:GSK147 complex; 9DOP for PAD4:PAD-PF2 complex; 9DOL for PAD2:PAD-PF2 complex, 4N2B for PAD2 10 mM Ca2+, and 4N2C for PAD2 F221/222 A holoenzyme. Source data for Figs. 1b–e, 3a–h, 4, and 6a–e are provided in the Source Data file accompanying this paper. Source data are provided in this paper.
References
Vossenaar, E. R. et al. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. BioEssays 25, 1106–1118 (2003).
Taki, H. et al. Purification of enzymatically inactive peptidylarginine deiminase type 6 from mouse ovary that reveals hexameric structure different from other dimeric isoforms. Adv. Biosci. Biotechnol. 2, 304–310 (2011).
Ranaivoson, F. M. et al. Crystal structure of human peptidylarginine deiminase type VI (PAD6) provides insights into its inactivity. IUCrJ 11, 395–404 (2024).
Saijo, S. et al. Monomeric form of peptidylarginine deiminase type I revealed by x-ray crystallography and small-angle x-ray scattering. J. Mol. Biol. 428, 3058–3073 (2016).
Funabashi, K. et al. Structures of human peptidylarginine deiminase type III provide insights into substrate recognition and inhibitor design. Arch. Biochem. Biophys. 708, 108911 (2021).
Arita, K. et al. Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 11, 777–783 (2004).
Slade, D. J. et al. Protein arginine deiminase 2 binds calcium in an ordered fashion: implications for inhibitor design. ACS Chem. Biol. 10, 1043–1053 (2015).
Alghamdi, M. et al. An interplay of structure and intrinsic disorder in the functionality of peptidylarginine deiminases, a family of key autoimmunity-related enzymes. Cell. Mol. Life Sci. 76, 4635–4662 (2019).
Yu, K. & Proost, P. Insights into peptidylarginine deiminase expression and citrullination pathways. Trends Cell Biol. 9, 746–761 (2022).
Nakashima, K., Hagiwara, T. & Yamada, M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J. Biol. Chem. 277, 49562–49568 (2002).
Spengler, J. et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. 67, 3135–3145 (2015).
Rebak, A. S. et al. A quantitative and site-specific atlas of the citrullinome reveals widespread existence of citrullination and insights into PADI4 substrates. Nat. Struct. Mol. Biol. 31, 977–995 (2024).
Tilvawala, R. et al. The rheumatoid arthritis-associated citrullinome. Cell Chem. Biol. 25, 691–704 (2018).
Mishra, N. et al. Cutting edge: Protein arginine deiminase 2 and 4 regulate NLRP3 inflammasome–dependent IL-1β maturation and ASC speck formation in macrophages. J. Immunol. 203, 795–800 (2019).
Liu, Y. et al. Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7–dependent lupus. JCI Insight 3, e124729 (2018).
Sun, B. et al. Reciprocal regulation of Th2 and Th17 cells by PAD2-mediated citrullination. JCI Insight 4, e129687 (2019).
Yang, L., Tan, D. & Piao, H. Myelin basic protein citrullination in multiple sclerosis: A potential therapeutic target for the pathology. Neurochem. Res. 41, 1845–1856 (2016).
Yusuf, I. O. et al. Protein citrullination marks myelin protein aggregation and disease progression in mouse ALS models. Acta Neuropathol. Commun. 10, 135 (2022).
Yuzhalin, A. E. Citrullination in cancer. Cancer Res. 79, 1274–1284 (2019).
Thiam, H. R. et al. Cellular mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 36, 191–218 (2020).
Foulquier, C. et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 56, 3541–3553 (2007).
Wang, F. et al. Identification of citrullinated peptides in the synovial fluid of patients with rheumatoid arthritis using LC-MALDI-TOF/TOF. Clin. Rheumatol. 35, 2185–2194 (2016).
Nielen, M. M. J. et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 50, 380–386 (2004).
Suzuki, A. et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 34, 395–402 (2003).
Knuckley, B. et al. Haloacetamidine-based inactivators of protein arginine deiminase 4 (PAD4): Evidence that general acid catalysis promotes efficient inactivation. ChemBioChem 11, 161–165 (2010).
Causey, C. P. et al. The development of N-α-(2-carboxyl)benzoyl-N5-(2-fluoro-1-iminoethyl)-L-ornithine amide (o-F-amidine) and N-α-(2-carboxyl)benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide (o-Cl-amidine) as second generation protein arginine deiminase (PAD) Inhibitors. J. Med. Chem. 54, 6919–6935 (2011).
Subramanian, V. et al. Design, synthesis, and biological evaluation of tetrazole analogs of Cl-amidine as protein arginine deiminase inhibitors. J. Med. Chem. 58, 1337–1344 (2015).
Lewis, H. D. et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 11, 189–191 (2015).
Sciabola, S. et al. Critical assessment of state-of-the-art ligand-based virtual screening methods. Mol. Inf. 41, 2200103 (2022).
Wang, Q. et al. Monitoring of protein arginine deiminase activity by using fluorescence quenching: Multicolor visualization of citrullination. Angew. Chem. Int. Ed. 52, 2323–2325 (2013).
Kearney, P. L. et al. Kinetic characterization of protein arginine deiminase 4: A transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 44, 10570–10582 (2005).
Tallarida, R. J. Quantitative methods for assessing drug synergism. Genes Cancer 2, 1003–1008 (2011).
Konig, M. F. & Andrade, F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front. Immunol. 7, 461 (2016).
Liu, Y. L. et al. Functional role of dimerization of human peptidylarginine deiminase 4 (PAD4). PLoS ONE 6, e21314 (2011).
Horikoshi, N. et al. Structural and biochemical analyses of the human PAD4 variant encoded by a functional haplotype gene. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 112–118 (2011).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 293–302 (2011).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242 (2011).
Grosse-Kunstleve, R. W. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Smart, O. et al. RHOFIT. 1.2.4 (Global Phasing Ltd., Cambridge, UK, 2014).
Emsley, P. et al. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 75, 861–877 (2019).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Gehlhaar, D. K. et al. The Pfizer crystal structure database: An essential tool for structure-based design at Pfizer. J. Comput. Chem. 43, 1053–1062 (2022).
Acknowledgements
We thank Xueming Liu, Bing Hu, and Zhan Tian of WuXi AppTec for compound synthesis, and Justyna Kilewski and Kristen Ford for compound screening. This research used resources at the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) beamline 17-ID, supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research also used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
L.X. performed the virtual screen. J.H. and F.F.V. supervised all structural and biophysical studies, determined the structures, and analyzed the results. S.L. analyzed the structural data. W.D. and P.V.S. performed SPR experiments. S.R. and F.V. supervised pharmacology experiments. J.W.P. performed neutrophil experiments. M.E.B. performed PAD4 FQ enzyme inhibition experiments. P.B. performed PAD1-4 NH3 release enzyme inhibition and calcium dependence experiments. W.Y.C. performed thermal shift experiments. J.S.C., J.A.N. and K.F.F. expressed and purified the protein. S.W.W., S.K. and J.I.T. supervised chemical compound synthesis. S.E.D. and J.M.C. synthesized final chemical compounds and intermediates. L.A.D., J.E.D., F.L., K.F. and K.L.L. designed inhibitor compounds. J.S.C. and J.A.N. prepared protein reagents. M.E.S. and F.F.V. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors were employees of Pfizer Inc. at the time this work was performed.
Peer review
Peer review information
Nature Communications thanks Kyohei Arita, Feliciana Real Fernandez, Paolo Rovero, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dakin, L.A., Xing, L., Hall, J. et al. Inhibiting peptidylarginine deiminases (PAD1-4) by targeting a Ca2+ dependent allosteric binding site. Nat Commun 16, 4579 (2025). https://doi.org/10.1038/s41467-025-59919-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59919-4
