
Abstract
Differences in transmission and ploidy between sex chromosomes and autosomes drive divergent evolutionary trajectories, with sex chromosomes generally evolving faster. Because sex-linked genes are transmitted less frequently, they are under less efficient selection. Conversely, exposure of recessive mutations on haploid sex chromosomes creates more efficient selection. In most systems, these effects occur simultaneously and are confounded. The fly families Sciaridae (fungus gnats) and Cecidomyiidae (gall midges) have X0 sex determination, but males transmit only maternally inherited chromosomes. This phenomenon results in equal transmission of the X and autosomes, allowing the effect of haploid selection to be studied in isolation. We discover that, unlike well-studied systems, X chromosomes diverge more slowly than autosomes in these flies. Using population genomic and expression data, we show that despite the X evolving more adaptively, stronger purifying selection explains slower divergence. Our findings demonstrate the utility of non-Mendelian inheritance systems for understanding fundamental evolutionary processes.
Similar content being viewed by others
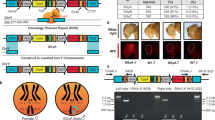
Introduction
In most sexually reproducing organisms, an individual’s sex is determined by sex chromosomes1. Aside from their role in sex determination, sex chromosomes are distinct from autosomes in other ways that have important consequences for their evolutionary fates. Sex chromosomes typically evolve from a pair of homologous autosomes on which a sex-determining locus emerges. Recombination becomes suppressed around the locus, expands along the chromosome, and degeneration ensues for the non-recombining region, which is found only in the heterogametic sex (the Y and W in XY and ZW systems, respectively); sometimes the sex-limited chromosome is lost completely2. This process leads to sex-linked genes being expressed and transmitted differently to those on autosomes (Fig. 1a). The patterns of evolution that result from this are influenced by a complex interaction of evolutionary processes3. Understanding how sex chromosomes and autosomes evolve differently, and the processes that drive these differences, can contribute to a more comprehensive understanding of how genomes evolve.

a In classic heterogametic systems, the presence of a sex-limited chromosome (e.g., the Y in XX/XY systems) results in the X being haploid in males and transmitted less frequently than autosomes. b In systems with paternal genome elimination (PGE), the males are also haploid for the X but eliminate paternally inherited chromosomes during spermatogenesis, so that every sperm contains the maternal haploid genome and the X and autosomes are transmitted equally. c In systems with PGE, sex is determined after fertilisation, establishing female (XX) and male (X0) karyotypes. Note that in fungus gnats, sperm have two duplicate copies of the maternally inherited X, and fertilised embryos are therefore XXX. Male and female embryos, therefore, lose 2 and 1 Xs, respectively. Figure created in BioRender (http://BioRender.com/ajxef9e).
Genomes are shaped by both selective and neutral processes, as well as the rate, effect, and dominance of new mutations4. Where recessive alleles are hemizygous (i.e., X- or Z-linked in the heterogametic sex), they are always exposed to selection. Mutations should therefore be swiftly purged if deleterious or invade and become fixed more quickly if beneficial. X- or Z-linked genes are indeed often found to diverge faster between species (i.e., have higher dN/dS) than autosomal genes5,6,7,8,9,10,11,12, known as the ‘faster-X’ effect3,13. Conversely, because sex chromosomes are only transmitted to half of offspring by the heterogametic sex and thus have a smaller effective population size (Ne) than the autosomes (NeX/NeA = ¾), selective constraints on many new mutations should be relaxed on the sex chromosomes, allowing a greater number of slightly deleterious alleles to behave neutrally and fix through drift (for the sake of brevity, we will refer to these opposing forces as ‘selection’ and ‘drift’).
Faster-X evolution may therefore be driven by a combination of positive selection on recessive beneficial mutations (adaptive faster-X) and stronger drift on weakly deleterious mutations (nonadaptive faster-X)3. Although numerous studies across a range of taxa have found evidence for accelerated rates of X/Z chromosome evolution compared to autosomes, support in some lineages is mixed9,14,15,16 and the precise causes can be difficult to disentangle. Some studies find support for drift as the predominant driver9,12,17, others, adaptive evolution6,10,18. A key limiting factor in systems with regular chromosome inheritance is that these two effects are naturally confounded: X-linked genes are simultaneously hemizygous in males and have a smaller Ne than do autosomal genes. As a result, it is often unclear what drives the different patterns of genetic diversity we see on sex chromosomes and autosomes.
Dark-winged fungus gnats (Diptera: Sciaridae) and gall midges (Diptera: Cecidomyiidae) are two distantly related families of flies that have independently evolved an unusual form of haplodiploidy known as germline paternal genome elimination19. They have X0 sex determination, but all paternally inherited homologues are eliminated during the first meiotic division during spermatogenesis. Males therefore inherit but do not transmit their fathers’ genes, instead passing on only the maternally inherited genome, including an X chromosome to every offspring (Fig. 1b). The result of this is that both X chromosomes and autosomes are transmitted in a haploid state through males, and thus NeX should equal NeA20. However, elimination of X chromosomes in somatic cells during embryogenesis, which establishes the sex of an individual, produces X0 males and XX females (Fig. 1c). As such, the X is haploid in males but diploid in females, while the autosomes are diploid in both sexes. Thus, while in other systems the cause of a lower Ne for the X arises from the difference in ploidy relative to the autosomes, under PGE, this is not the case due to non-Mendelian transmission of the chromosomes.
PGE, therefore, negates the effects of X and autosomal differences in Ne when it comes to comparisons between sex chromosomes and autosomes. Moreover, this uniform transmission of the genome makes the system invariant to many processes that affect the NeX/NeA ratio, such as sex-differences in variance in reproductive success21, demography22, and fluctuations in population size23. Recombination rates and mutation rates, which usually differ between the X and autosomes due to differences in transmission24,25, should also be equal under PGE. Many of these factors can differ between populations of closely related species26, which may explain why faster-X results from Drosophila are difficult to interpret and often seem contradictory5,14,18,27. Under PGE, hemizygosity of the X should be the sole driver of differences in autosome and X chromosome evolution20. This may be particularly useful when investigating, for example, patterns of neutral genetic diversity, which are influenced by multiple processes22,28,29, as well as whether dominant versus recessive favourable mutations are more prevalent, which is difficult to estimate a priori3,30.
These PGE systems have a further idiosyncrasy in that some species have unorthodox reproductive biology associated with unusual genomic architecture. Some fungus gnats and gall midges have genetically distinct male-producing and female-producing females, a phenomenon known as ‘monogenic’ reproduction31. Female producers are heterozygous for large inversions that are X-linked (X’) in sciarids and autosomal (A’) in cecidomyiids32,33, and these appear to be recently evolved in at least one case33. Other species in the families are ‘digenic’, do not have inversions, and produce mixed-sex broods. Therefore, in addition to being a good model for studying faster-X, these systems provide opportunities to explore the consequences of recent recombination suppression on both X chromosomes and autosomes.
Here, we investigate patterns of evolution in fungus gnats and gall midges and find that, in contrast to most previous studies on systems with regular Mendelian inheritance, X chromosomes consistently diverge more slowly than autosomes under PGE. In some of these species, we use population resequencing and gene expression data to examine polymorphism, selection, and the contribution of differentially expressed genes. We find that, although X-linked genes evolve more adaptively than autosomal genes, purifying selection on weakly deleterious mutations being stronger than positive selection on beneficial mutations causes the slower-X divergence we observe. We find that this purifying selection is likely driven by a combination of hemizygous expression of the X in males, supplemented by higher overall expression of X-linked compared to autosomal genes in both sexes. We also find that recent loss of recombination has the potential to drastically reduce the levels of neutral diversity within recombination-suppressed regions. Our results demonstrate the value of using systems with unusual genetics in understanding complex evolutionary processes.
Results
Effective population size, patterns of neutral diversity, and unusual genomic architecture
Under PGE, autosomes and X chromosomes should have equal effective population sizes (NeX/NeA = 1)20. One method of testing this prediction is by comparing neutral diversity, i.e., diversity at sites that should be evolving in the absence of selection, such as fourfold degenerate sites17. Many factors contribute to driving different patterns of neutral diversity on autosomes and X chromosomes, such as variance in male reproductive success and X/A differences in recombination and mutation rates21,24,25. PGE systems should be invariant to such processes because they rely on differences in autosome and X transmission. We used population resequencing data to estimate neutral diversity (as a proxy for NeX/NeA) in the species Bradysia (formerly Sciara) coprophila, Lycoriella ingenua (both fungus gnats), and Mayetiola destructor (a gall midge), by calculating nucleotide diversity (θ) at fourfold degenerate sites. For L. ingenua and M. destructor, θX/θA was 0.77 and 0.86, respectively. A value lower than 1 could be caused by background selection removing synonymous polymorphism at linked nonsynonymous sites34, which is likely to be higher on the X due to haploid selection.
Surprisingly, we found θX/θA to be only 0.28 for B. coprophila. In this species, half of females (the female-producers) are heterozygous for a large inversion-based supergene that comprises ~ 80% of the X chromosome and formed < 0.5 mya33 (Fig. 2a). One consequence of this is an overall reduction in the rate of recombination for the X: in addition to male meiosis being achiasmatic35, most of the X does not recombine in half of females. This should enhance the effects of linked selection, including background selection, hitchhiking, and selective interference, which in turn should reduce polymorphism34. To examine whether lower θX/θA in B. coprophila is likely to be a consequence of these processes, we performed a sliding window analysis of heterozygosity along the X in wild-caught XX and X’X females. We found strongly reduced heterozygosity in the region of recombination suppression in B. coprophila, suggesting that lower recombination is indeed responsible for purging neutral diversity. In contrast, L. ingenua, in which females are always XX, did not show a pronounced reduction (Fig. 2b). While this unusual genomic architecture may affect X versus autosomal evolution in B. coprophila, this is unlikely to be true of L. ingenua and other species without X-linked supergenes.

a Schematic of the X chromosomes of B. coprophila. Female-producing females are heterozygous for a large X-linked supergene composed of paracentric inversions on the long arm of the chromosome. b The supergene region is visible as raised heterozygosity in X’X females, and causes reduced diversity in XX females. In L. ingenua, a species which does not have a supergene, females are all XX and heterozygosity is similar between the autosomes and X chromosome.
X chromosomes diverge more slowly under PGE
We sought to explore the consequences of PGE for rates of X chromosome divergence. To estimate whether X chromosomes evolve faster than autosomes, studies often examine whether X-linked genes accumulate more nonsynonymous substitutions than do autosomal genes by measuring the scaled rate of divergence, i.e., dN/dS3. We calculated dN/dS from single-copy orthologs between 8 species pairs within the Dipteran infraorder Bibionomorpha: 4 pairs of fungus gnats (Sciaridae), 3 of gall midges (Cecidomyiidae), and one of fever flies (Bibionidae). Fever flies are a non-PGE outgroup with male heterogamety and differentiated X and Y chromosomes36, thus serving as a control (Fig. 3a). Surprisingly, in all PGE pairs studied, we found significantly lower dN/dS for the X, indicating slower divergence of X-linked genes relative to autosomal genes (Fig. 3b and Table 1). This result places PGE in stark contrast with the faster-X divergence seen in many classic reproductive systems. For the non-PGE outgroup, dN/dS was around 20% greater on the X chromosome compared to autosomes, in-line with the faster-X divergence seen in many comparisons of sex chromosomes and autosomes in other systems5,6,7,8,9,10,11,12.

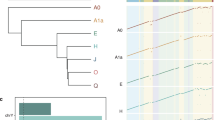
a BUSCO-based phylogenetic tree showing all species analysed (see Supplementary Fig. 1 for separate X and autosome trees). Values at branches represent branch length; bold values at nodes represent ultrafast bootstrap support (%). b Comparisons of dN/dS for autosomal (grey) and X-linked (blue) genes between each species pair (outliers not shown). For a version with medians emphasised, see Supplementary Fig. 2. Numbers on x-axes show the number of autosomal and X-linked genes single-copy orthologs tested. Asterisks represent significance levels for two-sided Mann-Whitney-Wilcoxon tests between the X and autosomes (**P < 0.01, ***P < 0.001; see Supplementary Table 1 for full statistical results). Box plots show median (central line), IQR (25th and 75th percentiles, box limits); whiskers extend to 1.5 x IQR. Outliers not shown. Images were obtained from https://www.inaturalist.org/, credit: Sciaridae (Odontosciara nigra), J Gallagher; Cecidomyiidae (Contarinia pseudotsugae), G San Martin; Bibionidae (Dilophus febrilis), P Le Mao.
Purifying selection slows rates of X chromosome divergence despite adaptive evolution
One limitation of dN/dS as a measure of faster-X evolution is that, although it is informative about rates of sequence divergence, it cannot distinguish between adaptive and non-adaptive causes of divergence3. For example, positive, hemizygous selection on the X can increase the rate of non-synonymous substitutions by fixing recessive beneficial mutations (e.g., monarch butterflies10), but relaxed purifying selection due to the smaller effective population size of the X (in XX/XY systems without PGE), which allows more weakly deleterious mutations to fix through drift, also contributes to sequence divergence (e.g., in Timema stick insects37). As such, faster-X evolution does not always imply faster-X adaptative evolution, as studies of the latter generally require evidence of a higher fixation rate of new beneficial mutations. Since X chromosomes in PGE systems are diverging more slowly, this implies that purifying selection dominates over positive selection, potentially reflecting a distribution of fitness effects biased away from beneficial recessive mutations. However, factors such as sex-specific mutation rates and differences in the distribution of dominance and selection coefficients of new mutations could also contribute towards patterns we observe24.
We used population resequencing data in two fungus gnat species, B. coprophila and L. ingenua, and one gall midge, M. destructor, to examine the impact of different types of selection on polymorphism. Notably, our estimates of nonsynonymous (pN) and scaled rates (pN/pS) of polymorphism were significantly lower on the X chromosome compared to the autosomes in all three species (Fig. 4a, b, full statistical results in Supplementary Table 2), indicating stronger purifying selection on X-linked genes. In B. coprophila and M. destructor, pS was also lower on the X, which could be explained by associated background selection. Strangely, we found pS to be higher on the X in L. ingenua (Fig. 4c). Higher pS can be caused by a higher mutation rate or a lower recombination rate (and reduced background selection), but because of equal transmission across the genome we do not expect these to differ between the X and autosomes under PGE. However, it is possible that particular genomic architecture on the autosomes could affect pS, similar to how the X’ supergene in B. coprophila affects neutral diversity through recombination suppression. We checked heterozygosity along the genome in the resequenced individuals in this species and did not find evidence of recombination suppression on the autosomes, but we did find that some individuals had runs of homozygosity that may result from cross-overs where parents were siblings, which likely explains this finding, and may also reflect a degree of inbreeding in the population that we sampled which could explain why both pN and pS were lower for L. ingenua compared to the other two species (Supplementary Fig. 2).

Scaled rates of a polymorphism (pN/pS), b nonsynonymous polymorphism (pN), and c synonymous polymorphism (pS) are shown. d Per-gene α. The result shown for B. coprophila is after removing polymorphisms with frequencies lower than 0.2 (see Supplementary Notes 1). e Aggregate α. Error bars in e represent 95% confidence intervals obtained by parametric bootstrapping. Asterisks represent significance levels for two-sided Mann-Whitney-Wilcoxon tests between the X and autosomes (*P < 0.05, **P < 0.01, ***P < 0.001, NS = not significant). Box plots show median (central line), IQR (25th and 75th percentiles, box limits); whiskers extend to 1.5 x IQR. Outliers in A-C are not shown. See Supplementary Table 2 for full statistical results, including numbers of genes in each comparison.
Although purifying selection appears to be predominant on the X, this does not preclude the X from evolving more adaptively relative to the autosomes, because haploid selection should cause recessive beneficial mutations to fix more quickly. To examine the contribution of adaptive evolution, we combined polymorphism and divergence in B. coprophila, L. ingenua, and M. destructor to calculate α, the proportion of substitutions driven by positive selection38. We calculated α via two methods: (i) separately for each gene, and (ii) from summed variants across the X and autosomes (i.e., aggregated α) to increase power to detect signatures of selection. Per-gene α was significantly higher for the X relative to autosomes in L. ingenua and M. destructor, suggesting faster adaptation on the X. This was originally not the case for B. coprophila, where α was lower on the X. Since an excess of segregating weakly deleterious mutations can reduce α values39, we removed low-frequency polymorphisms in a manner similar to an asymptotic McDonald-Kreitman test40 prior to calculating α (Supplementary Notes 1), after which we were able to recover positive α for the X (Fig. 4d). For aggregated α, the X was higher than the autosomes in L. ingenua and M. destructor, but not significantly so. Only in B. coprophila was there a significant difference, but this species again had lower α on the X. We hypothesise that this may be due to recombination suppression and the reduced Ne of the B. coprophila X, resulting in less efficient positive selection as well as less efficient purging of the segregating weakly deleterious variants that drive down α. Together, these findings suggest that the X may still be adapting faster under PGE, but that in B. coprophila, recombination suppression may reduce the adaptability of this chromosome. Overall, our results suggest that the predominance of purifying selection on X-linked genes causes the slower-X divergence we observe under PGE.
Sex-biased gene expression and hemizygous selection
Since genes are only likely exposed to selection if they are expressed, and the X is only hemizygous in males, the effect of selection on genes should increase with male-biased expression. Moreover, because sex chromosomes spend disproportionately more time in one sex, they should accumulate more genes with sex-biased expression than autosomes41,42. Accordingly, X and Z chromosomes are often found to have an excess of female-biased (feminisation) or male-biased genes (masculinisation), respectively43,44,45,46, a pattern which tends to be driven primarily by sexually dimorphic expression in reproductive tissues47.
We analysed differential gene expression (DGE) in bodies and reproductive tissues of B. coprophila and L. ingenua, and we did not find a consistent distribution of sex-biased genes: for genes expressed in the body, the X appeared to have an excess of male-biased genes and a dearth of female-biased genes relative to the autosomes in L. ingenua and B. coprophila, and we found the opposite pattern for reproductive tissues (residuals in Supplementary Table 3). In flies, the X is generally found to harbour an excess of female-biased genes and a deficit of male-biased genes, and strong sex-biased gene expression is largely limited to gonadal tissue48. Interestingly, we found extreme patterns of sex-biased gene expression that was not limited to the gonads, with 72–80% of genes showing sex-biased expression in the body and 73–81% in the reproductive tissues (Fig. 5a). Such extreme patterns of sex-biased gene expression are often associated with sexual dimorphism49 or resolution of intragenomic conflicts50,51. Extreme sex-biased expression also appears to be associated with PGE in scale insects52, which could hint at an association with a history of intragenomic conflict. We extended our DGE analysis to three other species: the fungus gnat B. odoriphaga and the gall midges M. destructor and Sitodiplosis mosellana, and found similarly extreme patterns (Fig. 5b), further suggesting that these results could be associated with PGE rather than being an idiosyncrasy of the sciarid clade. Previous work suggests that PGE creates strongly asymmetric conditions for male- and female-beneficial variants to invade53, which may explain an accumulation of genes with strongly male- and female-biased expression. The inconsistent patterns of sex-biased gene content that we found on the X and autosomes could be indicative of turnover in sex-biased gene content, but a causal link requires further exploration.

a Proportions of genes with strongly sex-biased (> 90% in one sex), sex-biased (> 70% in one sex), and unbiased (30–70% in one sex) expression on the autosomes (A) and X chromosome in the body and somatic reproductive tissues of the fungus gnats Bradysia coprophila and Lycoriella ingenua (3 RNAseq replicates each). b Same as above for Bradysia odoriphaga (3 replicates) and the gall midges Sitodiplosis mosellana (2 replicates) and Mayetiola destructor (1 replicate). Asterisks represent significance differences in sex-biased composition using two-sided χ2 tests (***P < 0.001, NS = not significant). Significance test results are not shown for S. mosellana or M. destructor as the comparisons were based on fewer than 3 biological replicates. See Supplementary Table 3 for all residuals and P-values.
We calculated dN/dS and pN/pS for male-biased, female-biased, and unbiased genes in B. coprophila and L. ingenua to determine whether the slower-X divergence we observed is primarily driven by haploid selection in males (full statistical results in Supplementary Table 4). We found that genes with sex-biased expression generally evolve more quickly than those with unbiased expression, in line with findings from previous studies on other organisms7,10,54. Consistent with haploid selection in males, male-biased X-linked genes had lower dN/dS than male-biased autosomal genes (Fig. 6a). In B. coprophila and L. ingenua, this result was not limited to gonadal tissues, which was surprising given that sexually dimorphic tissues are often the most rapidly evolving traits55. Similarly, Heliconius shows no particular correlation between faster-evolving Z-linked genes and expression in the ovaries16. Surprisingly, we found a similar pattern for female-biased genes (Fig. 6a). Such a finding could be due to background selection from selection on male-biased genes, but it may also suggest that some effect other than haploid selection is driving slower-X divergence.

Genes are designated as body- or gonad-expressed if they are predominantly expressed in one tissue (i.e., > 50% of a gene’s expression). Note that we excluded germline (testes and ovaries) from reproductive tissues as germline and somatic karyotypes differ (see methods). a Scaled rates of divergence (dN/dS); (b) scaled rates of polymorphism (pN/pS) for autosomal (grey) and X-linked (blue) genes. MB = male-biased, UB = unbiased, FB = female-biased. Asterisks represent significance levels for two-sided Mann-Whitney-Wilcoxon tests between the X and autosomes (*P < 0.05, **P < 0.01, ***P < 0.001, NS = not significant). Box plots show median (central line), IQR (25th and 75th percentiles, box limits); whiskers extend to 1.5 x IQR. Outliers not shown. See Supplementary Table 4 for full statistical test results, including numbers of genes in each comparison.
Due to haploid selection, we also expect to see the patterns of pN/pS observed above to apply primarily to genes with male-biased expression. However, with the exception of male-biased body-expressed genes in L. ingenua, we did not to recover significant differences in pN/pS between the X and autosomes. In some cases, this may be because relatively few genes were being tested (Fig. 6b and Supplementary Table 4).
Differences in X and autosome expression may contribute to slower X divergence
We investigated differences in expression levels between autosomal and X-linked genes, which could influence the exposure of alleles to selection: genes with higher expression are likely to have a larger phenotypic effect and should, therefore, be under stronger purifying selection56,57,58. For example, in the pea aphid system, in which parthenogenetic sex determination also predicts that NeX/NeA should be approximately equal59,60, X-linked genes have much lower expression than autosomal genes and show correspondingly higher dN/dS7.
Across all species in which we assayed gene expression levels, we found that X-linked genes were generally more highly expressed than autosomal genes in both sexes (Fig. 7). Only in L. ingenua was the difference in autosomal versus X-linked expression not significant in every tissue. That we found similar patterns of autosomal and X expression in both sexes confirms previous findings of dosage compensation in fungus gnats61,62, and suggests that the X may also be dosage balanced in gall midges. Moreover, the fact that we find higher expression of the X may reflect possible dosage overcompensation, a theoretically predicted but rarely observed phenomenon in sex chromosome evolution63. The X chromosomes of fungus gnats are evolutionarily independent from those of gall midges: the former are comprised of Muller Elements A and E, while the latter are mainly C, D, and F64. That both families show higher expression of X-linked genes, therefore suggests a potential evolutionary explanation for this pattern, which remains to be explored.

X chromosomes generally show higher expression than autosomes in the fungus gnats Bradysia coprophila, Lycoriella ingenua, B. odoriphaga, and the gall midges Sitodiplosis mosellana and Mayetiola destructor. Asterisks represent significance levels for two-sided Mann-Whitney-Wilcoxon tests, Bonferroni-corrected for multiple tests, between the X and autosomes (*P < 0.05, ***P < 0.001, NS = not significant). Box plots show median (central line), IQR (25th and 75th percentiles, box limits); whiskers extend to 1.5 x IQR. Outliers not shown. See Supplementary Table 5 for all residuals and P-values, including numbers of genes in each comparison.
Discussion
Most species in which evolutionary rates of autosomes and sex chromosomes have been compared show faster rates of divergence for X- or Z-linked genes, and this is often attributed to faster adaptation or stronger drift acting on sex-linked genes. However, results are often mixed, and the causes of faster divergence are a challenge to disentangle. We found that under PGE in fungus gnats and gall midges, where NeX/NeA = 1, X chromosomes diverge more slowly than do autosomes. The slower-X divergence appears to be driven by stronger purifying selection of X-linked genes, resulting from a combination of haploid selection in males and purifying selection on the more highly expressed X chromosome.
Interestingly, the unusual genomic architecture of B. coprophila, where inversions suppress recombination, also results in drastically reduced diversity on the X. While this could theoretically contribute to the slower-X divergence we see, it is unlikely to explain our observations across PGE species for two reasons. Firstly, L. ingenua and many other fungus gnats lack inversions on their X chromosomes yet show similar patterns of X versus autosomal divergence. Secondly, gall midges can also be monogenic with much smaller associated inversions32, but these are autosomal and should, therefore, have the opposite effect to that which we see.
More broadly, our findings suggest that the consistent observations of higher dN/dS for sex chromosomes relative to autosomes5,6,7,8,9,10,11,12 may be driven primarily by the smaller effective population size of the X chromosomes, and, therefore, that faster divergence of X chromosomes is primarily a result of more weakly deleterious mutations fixing through drift rather than beneficial mutations through selection. Thus, while evidence suggests that X chromosomes often indeed adapt faster than autosomes, faster-X divergence is not necessarily adaptive. Overall, our findings demonstrate how species with unusual inheritance mechanisms are uniquely placed to provide insights into complex questions surrounding sex chromosome evolution.
Methods
Genome assembly, annotation, and sex-linkage assignment for species pairs
We used publicly available genomes for A.aphidimyza, B. coprophila, B. odoriphaga, C. nasturtii, D. febrilis, L. ingenua, M. destructor, P. flavipes, and S. moselllana (see Supplementary Table 6 for all accessions). For all remaining species, we generated de novo genome assemblies using WGS Illumina data. For D. femoratus, C. rumicis, and M. hordei, we used publicly available data. We collected and generated WGS data for L. agraria, B. pectoralis, B. confinis, and B. desolata. B. confinis, B. desolata, and B. pectoralis were collected from Cerová Vrchovina forest, Slovakia. L. agraria was collected from Whytham Woods, Oxford, UK. All collected specimens were identified from genital clasper morphology and amplification and Sanger sequencing of the COI barcode sequence using the primer pair LCO1490/HCO219865.
Genomic DNA was extracted from collected flies using a protocol modified to obtain high-molecular-weight DNA with low-input material, developed by C Laumer at the Wellcome Sanger Institute (dx.doi.org/10.17504/protocols.io.bypypvpw). Quantification and quality control was performed with Qubit and Nanodrop (ThermoFisher). Samples were sequenced for 150 bp paired-end Illumina reads on the Novoseq 6000 platform (Supplementary Table 7 for per-sample coverage). Raw reads were trimmed with fastp v0.12.466. We generated de novo short read assemblies for B confinis, B. desolata, and B. pectoralis using SPAdes v3.14.167 and used Blobtools v168 to identify and remove contaminant (non-metazoan), low-coverage (< 5x), and short (< 500 bp) contigs. Assembly quality was assessed with BUSCO v569 (Supplementary Fig. 4). We constructed a phylogenetic tree to determine species pairs using BUSCO Phylogenomics (github.com/jamiemcg/BUSCO_phylogenomics) and IQTree v2.2.570, using FigTree v1.4.471 to plot the trees.
For B. coprophila, B. odoriphaga, and C. nasturtii, RefSeq gene predictions were publicly available (Supplementary Table 6). For all other species, we generated de novo predictions. The genomes were soft-masked with Red v272 and annotated with BRAKER273,74,75,76,77 using the OrthoDB v10 Diptera protein sequences78. The L. ingenua genome annotation was generated at a later date using BRAKER379 and the OrthoDB v11 Dipteran protein sequences, along with all RNAseq libraries generated in this study (see below), which were mapped to the genome with HISAT80.
We identified autosomal and X-linked genes based on read depth from the homogametic sex (males), as autosomal sequences should have diploid (2n) coverage while X-linked sequences should have haploid (1n) coverage. Where male reads were available for both members of a species pair, we aligned them with BWA-MEM81 and used per-scaffold read depth, computed with SAMtools v1.1482, to identify autosomal and X-linked sequences. For 3 species pairs (B. coprophila/B. odoriphaga, L. ingenua/L. agararia, M. destructor/M. hordei), we inferred X-linkage from only one species since male reads were not available for both. For one species pair (C. nasturtii/C. rumicis), only female reads were available, so we inferred X-linkage based on alignments, using Minimap v2.17-r94183, to S. mosellana, which was the closest chromosome-level outgroup for which sex linkage information was available (for more information on sex-linkage assignments see Supplementary Notes 2).
Analysis of neutral diversity
We calculated nucleotide diversity (θ) at fourfold degenerate (i.e., putatively neutral) sites to test our prediction that under PGE, NeX/NeA should equal 1. To this end, we used alignments of resequencing datasets for B. coprophila, L. ingenua, and M. destructor (described below), and the population genomic tool ANGSD v0.94084 to calculate θ for each scaffold.
We analysed θ across sliding windows of the X chromosome in B. coprophila and L. ingenua individuals using resequencing datasets (see below). We called genotypes at all sites with GATK-485,86,87 using the output mode EMIT_ALL_CONFIDENT_SITES with the option -include-non-variant-sites when genotyping VCFs, in order to call all (variant and invariant) genotypes at every site. The scripts parseVCF.py and popgenWindows.py (https://github.com/simonhmartin/genomics_general) were used to calculate θ in 100 kb sliding windows across the X in XX and X’X individuals.
Analysis of between-species divergence
To calculate dN/dS we used alignments of WGS data of one species to the genome of another. For each pair, the species with the better quality (more contiguous) genome was chosen as the ingroup to ensure better mapping rates and reduce mapping times (Table 1; see Supplementary Table 8 for assembly statistics). Outgroups were aligned to their respective ingroup species with BWA-MEM81 using default parameters. PicardTools (http://broadinstitute.github.io/picard/) was used to sort alignments, mark and remove duplicates, and add read groups. Variants were called using the GATK-4 best practises pipeline85,86,87, and were filtered for Quality by Depth > 2, Fisher Strand-bias < 60, and Mapping Quality > 40. Variant annotations were generated using SnpEff v5.1d88 and SnpSift v5.1d89. The script partitioncds.py90 was used to annotate degeneracy for all genic sites. Finally, a custom R script91 was used to calculate scaled divergence (dN/dS), i.e., the ratio between non-synonymous variants per non-synonymous site (dN) and synonymous variants per synonymous site (dS), for single-copy orthologs within each species pair. We calculated dN/dS only for single-copy orthologs between species pairs, which we identified by running Orthofinder v1.4.292 on the longest predicted isoforms of the predicted genes.
We calculated DXY between species by aligning whole genomes with Minimap v2.17-r94183 with the option -x asm20 to allow for 20% sequence divergence and –secondary = no to avoid secondary alignments. We also repeated this for the X and autosomes separately. DXY was calculated as 1 – percentage identity (i.e., the number of mismatches divided / total length of aligned sequence).
Analysis of within-species polymorphism and selection
To estimate scaled rates of polymorphism (i.e., pN/pS), we utilised population resequencing data for B. coprophila, L. ingenua, and M. destructor. We collected 11 B. coprophila females and 5 L. ingenua XX females for resequencing. In taxonomic literature, B. coprophila is traditionally named B. tilicola, but we use the former name here to maintain consistency with evolutionary literature. Females of B. coprophila were collected from houseplants in different apartments in Edinburgh, as well as from the Royal Botanic Gardens, Edinburgh, UK. Because B. coprophila females can either be X’X (heterozygous for an X-linked supergene) or XX, and we identified XX and X’X females based on distributions of heterozygosity using a sliding window analysis (Supplementary Notes 3). We identified 3 individuals as having the XX genotype and the remaining 8 as having the X’X genotype. Since we were interested in polymorphisms on the standard X chromosome, we analysed only the 3 XX individuals. We collected L. ingenua from a single mushroom straw log obtained from Mycobee Mushroom Farm, North Berwick, from which thousands of individuals were emerging, and may, therefore, have been descended from relatively fewer individuals, which may explain why we observed lower polymorphism in this species (see main text). DNA extractions and sequencing were performed as described above, for ~20x coverage per individual. For M. destructor, we used 5 publicly available resequencing libraries from female individuals93. We calculated pN/pS using the same method described above for dN/dS but aligning resequencing libraries to their respective genomes and combining VCFs with GATK-4 combineGVCFs prior to genotyping.
To assess the extent of positive (adaptive) selection on the X versus the autosomes, we calculated α for each gene using a calculation of the Neutrality Index (1 – NI)38. A higher α value indicates a higher proportion of fixed non-synonymous differences that are driven by positive selection. We calculated α both per-gene as well as aggregated within a chromosome (i.e., by summing dN, dS, pN, and pS across autosomal and X-linked genes prior to calculating α). Because some genes contain very few variants, the latter method arguably allows for a more powerful test of positive selection38. Many of the per-gene α values were negative, as was aggregate α for the B. coprophila X and L. ingenua X and autosomes. Since an excess of weakly deleterious segregating variants can cause negative α, we applied a sliding cut-off of both synonymous and non-synonymous polymorphisms, akin to an asymptotic McDonald-Kreitman test40. We provide all α estimates in Supplementary Notes 1, and present original estimates in the main text, except for B. coprophila per-gene α where we present the estimate after removing polymorphisms with frequencies < 0.2.
Differential gene expression
We mass-reared lab cultures of B. coprophila94 and L. ingenua and performed dissections to separate gonadal tissue from the remainder of the body (Supplementary Fig. 5). Germline (i.e., testes or ovaries) and somatic reproductive tissues were separated and sequenced separately, since male Sciaridae germ cells are XX95. Adult flies were anaesthetised on ice, dissected in 1 × phosphate buffered saline on a glass slide under a Leica EZ4 microscope, and flash-frozen at − 70 °C. 20–60 individuals were pooled per replicate, and 3 replicates were collected for each sex; for B. coprophila, we used male-producing (XX) females, which lack the female-limited X’ supergene. RNA was extracted using a modified version of the PureLink RNA purification kit (ThermoFisher) with a TRIzol solubilisation step. All samples were quantified and quality-checked using the Qubit and Nandrop (ThermoFisher) and were sequenced for 30 M 150 bp paired-end reads on the NovaSeq 6000 platform, for 9 G data per sample.
To analyse sex-biased gene expression and classify genes as differentially expressed, we used Kallisto v0.46.196. Indices were generated from the B. coprophila33 and L. ingenua gene predictions, against which RNAseq libraries were quantified. Data quality control (PCA and comparing transcript per million (TPM) distributions, Supplementary Fig. 6) and quantile-normalisation of transcript counts were performed in R. Sex-biased expression was assessed using the specificity metric (SPM) for male versus female expression by dividing the squared mean counts for each gene in females by the sum of squared mean male and female counts97, such that male-limited genes have an SPM of 0 and female-limited genes an SPM of 1. Genes with SPM < 0.3 were assigned as male-biased and SPM > 0.7 as female-biased, corresponding to a difference of 1.5x in expression between the sexes. Genes with SPM < 0.1 and > 0.9 were assigned as strongly male-biased and strongly female-biased, respectively, though when analysing dN/dS and pN/pS of sex-biased genes we combined strongly-biased and biased genes into one category. Prior to calculating SPM, we excluded genes with normalised counts < 4 to ensure that our results were not driven by potentially unreliable assignment of bias in genes with very low expression. Because sexually dimorphic reproductive tissues generally contribute disproportionately to sex-biased gene expression, we categorised genes into those that were predominantly (> 50%) expressed in the body (i.e., non-sexually dimorphic tissues) and those predominantly expressed in the somatic reproductive tissues (i.e., sexually dimorphic tissues). For analyses of dN/dS and pN/pS for differentially expressed genes, we only included somatic gonadal tissue (i.e., excluding testes and germline) since germ cells are XX in both sexes and should thus not be subject to haploid selection.
We also performed a complementary differential gene expression analysis on counts with DESeq298, which is more conservative in defining sex-biased genes compared to using SPM. We used an adjusted P-value cut-off of < 0.05 to define expression as significant and a Log2 fold-change of > 0.6 to categorise genes as sex-biased, again corresponding to a difference in expression between the sexes of approximately 1.5x. DESeq2 assigned fewer genes as sex-biased compared to SPM, but there were no major inconsistencies between the two methods (i.e., no genes were categorised as male-biased by one method but female-biased by the other, Supplementary Table 9).
Statistical analyses
Significant differences in dN/dS, pN/pS, and α were tested with Mann-Whitney-Wilcoxon tests for comparisons between X chromosomes and autosomes. Sex-biased composition of chromosomes was assessed using Chi-squared (χ2) tests. To test for significance differences in α between the X and autosomes, we used a permutation test framework10,12,99. We calculated point estimates of α for the X and autosomes and used the absolute value of the difference between the two estimates as the permutation test statistic. From the two gene sets being tested, we randomly sampled individual genes to create two permuted sets of equal size to the true sets, without replacement. We then calculated absolute differences in α between the two sampled gene sets for 10,000 permutations and constructed a distribution of differences in point estimates for α. These distributions were then compared to the true value, and P-values were calculated as the proportion of times the observed value was smaller than the values in the permuted distribution.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing reads generated for this study are available in SRA, and genome assemblies are available in GenBank, under NCBI BioProject accession PRJNA1109384. Accessions for already-available data analysed in this study are provided in Supplementary Table 6. Gene predictions generated in this study, and tables containing all per-gene metrics, are available in a public GitHub repository (https://github.com/RossLab/PGE_slowerX, https://doi.org/10.5281/zenodo.15212538). Source data is available as a Source data file. Source data are provided with this paper.
Code availability
The walkthrough code to reproduce the analyses in this study are detailed in a public GitHub repository (https://github.com/RossLab/PGE_slowerX).
References
Bachtrog, D. et al. Sex determination: Why so many ways of doing it?. PLoS Biol. 12, e1001899 (2014).
Charlesworth, D., Charlesworth, B. & Marais, G. Steps in the evolution of heteromorphic sex chromosomes. Heredity 95, 118–128 (2005).
Meisel, R. P. & Connallon, T. The faster-X effect: integrating theory and data. Trends Genet. 29, 537–544 (2013).
Ohta, T. The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23, 263–286 (1992).
Ávila, V. et al. Faster-X Effects in two drosophila lineages. Genome Biol. Evol. 6, 2968–2982 (2014).
Kousathanas, A., Halligan, D. L. & Keightley, P. D. Faster-X adaptive protein evolution in house mice. Genetics 196, 1131–1143 (2014).
Jaquiéry, J. et al. Disentangling the causes for faster-X evolution in aphids. Genome Biol. Evol. 10, 507–520 (2018).
Bechsgaard, J. et al. Evidence for faster X chromosome evolution in spiders. Mol. Biol. Evol. 36, 1281–1293 (2019).
Hayes, K., Barton, H. J. & Zeng, K. A study of faster-Z evolution in the great tit (Parus major). Genome Biol. Evol. 12, 210–222 (2020).
Mongue, A. J., Hansen, M. E. & Walters, J. R. Support for faster and more adaptive Z chromosome evolution in two divergent lepidopteran lineages*. Evolution 76, 332–345 (2022).
Chase, M. A., Vilcot, M. & Mugal, C. F. Evidence that genetic drift not adaptation drives fast-Z and large-Z effects in Ficedula flycatchers. Mol. Ecol. e17262, (2024).
Mongue, A. J. & Baird, R. B. Genetic drift drives faster-Z evolution in the salmon louse Lepeophtheirus salmonis. Evolution qpae090 (2024).
Charlesworth, B., Coyne, J. A. & Barton, N. H. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130, 113–146 (1987).
Thornton, K., Bachtrog, D. & Andolfatto, P. X chromosomes and autosomes evolve at similar rates in Drosophila: no evidence for faster-X protein evolution. Genome Res. 16, 498–504 (2006).
Rousselle, M., Faivre, N., Ballenghien, M., Galtier, N. & Nabholz, B. Hemizygosity enhances purifying selection: lack of fast-Z evolution in two satyrine butterflies. Genome Biol. Evol. 8, 3108–3119 (2016).
Pinharanda, A. et al. Sexually dimorphic gene expression and transcriptome evolution provide mixed evidence for a fast-Z effect in Heliconius. J. Evol. Biol. 32, 194–204 (2019).
Mank, J. E., Nam, K. & Ellegren, H. Faster-Z evolution Is predominantly due to genetic drift. Mol. Biol. Evol. 27, 661–670 (2010).
Charlesworth, B., Campos, J. L. & Jackson, B. C. Faster-X evolution: Theory and evidence from Drosophila. Mol. Ecol. 27, 3753–3771 (2018).
Herbette, M. & Ross, L. Paternal genome elimination: patterns and mechanisms of drive and silencing. Curr. Opin. Genet. Dev. 81, 102065 (2023).
Hitchcock, T. J., Baird, R. B., Gardner, A. & Ross, L. Paternal genome elimination, monogenic reproduction, and the evolutionary genetics of atypical sex chromosome systems. Preprint at https://doi.org/10.1101/2024.10.23.619029 (2024).
Vicoso, B. & Charlesworth, B. Effective population size and the faster-x effect: an extended model. Evolution 63, 2413–2426 (2009).
Charlesworth, B. Patterns of age-specific means and genetic variances of mortality rates predicted by the mutation-accumulation theory of ageing. J. Theor. Biol. 210, 47–65 (2001).
Pool, J. E. & Nielsen, R. Population size changes reshape genomic patterns of diversity. Evolution 61, 3001–3006 (2007).
Vicoso, B. & Charlesworth, B. Evolution on the X chromosome: unusual patterns and processes. Nat. Rev. Genet. 7, 645–653 (2006).
Vicoso, B. & Charlesworth, B. Recombination rates may affect the ratio of X to autosomal noncoding polymorphism in African populations of Drosophila melanogaster. Genetics 181, 1699–1701 (2009).
Hutter, S., Li, H., Beisswanger, S., De Lorenzo, D. & Stephan, W. Distinctly different sex ratios in African and European populations of Drosophila melanogaster inferred from chromosomewide single nucleotide polymorphism data. Genetics 177, 469–480 (2007).
Garrigan, D., Kingan, S. B., Geneva, A. J., Vedanayagam, J. P. & Presgraves, D. C. Genome diversity and divergence in Drosophila mauritiana: multiple signatures of faster X evolution. Genome Biol. Evol. 6, 2444–2458 (2014).
Kirkpatrick, M., Guerrero, R. F. & Scarpino, S. V. Patterns of neutral genetic variation on recombining sex chromosomes. Genetics 184, 1141–1152 (2010).
Sayres, M. A. W. Genetic diversity on the sex chromosomes. Genome Biol. Evol. 10, 1064 (2018).
Lasne, C., Sgrò, C. M. & Connallon, T. The relative contributions of the X chromosome and autosomes to local adaptation. Genetics 205, 1285–1304 (2017).
Baird, R. B., Mongue, A. J. & Ross, L. Why put all your eggs in one basket? Evolutionary perspectives on the origins of monogenic reproduction. Heredity 131, 87–95 (2023).
Benatti, T. R. et al. A neo-sex chromosome that drives postzygotic sex determination in the Hessian Fly (Mayetiola destructor). Genetics 184, 769–777 (2010).
Baird, R. B. et al. Recent evolution of a maternally acting sex-determining supergene in a fly with single-sex broods. Mol. Biol. Evol. 40, msad148 (2023).
Charlesworth, B. & Jensen, J. D. Effects of selection at linked sites on patterns of genetic variability. Annu. Rev. Ecol. Evol. Syst. 52, 177–197 (2021).
Amabis, J. M., Reinach, F. C. & Andrews, N. Spermatogenesis in Trichosa Pubescens (Diptera: Sciaridae). J. Cell Sci. 63, 199–213 (1979).
White, M. J. D. Animal Cytology and Evolution. (Cambridge University Press, London, 1973).
Parker, D. J., Jaron, K. S., Dumas, Z., Robinson-Rechavi, M. & Schwander, T. X chromosomes show relaxed selection and complete somatic dosage compensation across Timema stick insect species. J. Evol. Biol. 35, 1734–1750 (2022).
Stoletzki, N. & Eyre-Walker, A. Estimation of the neutrality index. Mol. Biol. Evol. 28, 63–70 (2011).
Charlesworth, J. & Eyre-Walker, A. The McDonald–Kreitman test and slightly deleterious mutations. Mol. Biol. Evol. 25, 1007–1015 (2008).
Messer, P. W. & Petrov, D. A. Frequent adaptation and the McDonald–Kreitman test. Proc. Natl. Acad. Sci. USA 110, 8615–8620 (2013).
Rice, W. R. Sex chromosomes and the evolution of sexual dimorphism. Evolution 38, 735–742 (1984).
Klein, K., Kokko, H. & Ten Brink, H. Disentangling verbal arguments: intralocus sexual conflict in haplodiploids. Am. Nat. 198, 678–693 (2021).
Wright, A. E., Moghadam, H. K. & Mank, J. E. Trade-off between selection for dosage compensation and masculinization on the avian Z chromosome. Genetics 192, 1433–1445 (2012).
Allen, S. L., Bonduriansky, R. & Chenoweth, S. F. The genomic distribution of sex-biased genes in Drosophila serrata: X chromosome demasculinization, feminization, and hyperexpression in both sexes. Genome Biol. Evol. 5, 1986–1994 (2013).
Albritton, S. E. et al. Sex-biased gene expression and evolution of the x chromosome in nematodes. Genetics 197, 865–883 (2014).
Mongue, A. J. & Walters, J. R. The Z chromosome is enriched for sperm proteins in two divergent species of Lepidoptera. Genome 61, 248–253 (2018).
Grath, S. & Parsch, J. Sex-biased gene expression. Annu. Rev. Genet. 50, 29–44 (2016).
Vicoso, B. & Bachtrog, D. Numerous Transitions of Sex Chromosomes in Diptera. PLoS Biol. 13, e1002078 (2015).
Harrison, P. W. et al. Sexual selection drives evolution and rapid turnover of male gene expression. Proc. Natl. Acad. Sci. USA 112, 4393–4398 (2015).
Gallach, M., Domingues, S. & Betrán, E. Gene duplication and the genome distribution of sex-biased genes. Int. J. Evol. Biol. 2011, https://doi.org/10.4061/2011/989438 (2011).
Wright, A. E. et al. Male-biased gene expression resolves sexual conflict through the evolution of sex-specific genetic architecture. Evol. Lett. 2, 52–61 (2018).
Bain, S. A. et al. Sex-specific expression and DNA methylation in a species with extreme sexual dimorphism and paternal genome elimination. Mol. Ecol. 30, 5687–5703 (2021).
Hitchcock, T. J., Gardner, A. & Ross, L. Sexual antagonism in haplodiploids. Evolution 76, 292–309 (2022).
Yang, L., Zhang, Z. & He, S. Both male-biased and female-biased genes evolve faster in fish genomes. Genome Biol. Evol. 8, 3433–3445 (2016).
Kopp, A., Duncan, I. & Carroll, S. B. Genetic control and evolution of sexually dimorphic characters in Drosophila. Nature 408, 553–559 (2000).
Krylov, D. M., Wolf, Y. I., Rogozin, I. B. & Koonin, E. V. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 13, 2229–2235 (2003).
Drummond, D. A., Bloom, J. D., Adami, C., Wilke, C. O. & Arnold, F. H. Why highly expressed proteins evolve slowly. Proc. Natl Acad. Sci. 102, 14338–14343 (2005).
Bédard, C., Cisneros, A. F., Jordan, D. & Landry, C. R. Correlation between protein abundance and sequence conservation: what do recent experiments say?. Curr. Opin. Genet. Dev. 77, 101984 (2022).
Jaquiéry, J. et al. Accelerated evolution of sex chromosomes in aphids, an X0 system. Mol. Biol. Evol. 29, 837–847 (2012).
Hitchcock, T. J. Effective population size of X chromosomes and haplodiploids under cyclical parthenogenesis. Preprint at https://doi.org/10.1101/2024.04.05.586733 (2024).
da Cunha, P. R., Granadino, B., Perondini, A. L. & Sánchez, L. Dosage compensation in sciarids is achieved by hypertranscription of the single X chromosome in males. Genetics 138, 787–790 (1994).
Urban, J. M. et al. High contiguity de novo genome assembly and DNA modification analyses for the fungus fly, Sciara coprophila, using single-molecule sequencing. BMC Genomics 22, 643 (2021).
Charlesworth, B. Model for evolution of Y chromosomes and dosage compensation. Proc. Natl. Acad. Sci. USA 75, 5618–5622 (1978).
Anderson, N. et al. Gene-rich X chromosomes implicate intragenomic conflict in the evolution of bizarre genetic systems. Proc. Natl. Acad. Sci. USA 119, e2122580119 (2022).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol., (1994).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Laetsch, D. R. & Blaxter, M. L. BlobTools: Interrogation of genome assemblies. F1000Res. 6, 1287 (2017).
Seppey, M., Manni, M. & Zdobnov, E. M. in Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 227–245 (Springer New York, New York, NY, 2019).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Rambaut, A. FigTree v1.3.1. (2010).
Girgis, H. Z. Red: an intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinform. 16, 1–19 (2015).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119–e119 (2014).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2016).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-genome annotation with BRAKER. Gene Predict. Methods Protoc. 65, 95 (2019).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinform. 3, lqaa108 (2021).
Kriventseva, E. V. et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2018).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. https://doi.org/10.1101/gr.278090.123 (2024).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinforma. 15, 1–13 (2014).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. (O’Reilly Media, 2020).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012).
Cingolani, P. et al. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 3, https://doi.org/10.3389/fgene.2012.00035 (2012).
Mackintosh, A. et al. The genome sequence of the scarce swallowtail, Iphiclides podalirius. G312, jkac193 (2022).
R Core Team. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, Vienna, Austria, 2023).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Navarro-Escalante, L., Zhao, C., Shukle, R. & Stuart, J. BSA-Seq discovery and functional analysis of candidate hessian fly (Mayetiola destructor) avirulence genes. Front. Plant Sci. 11, 956 (2020).
Metz, C. W. Genetic evidence of a selective segregation of chromosomes in Sciara (Diptera). Proc. Natl. Acad. Sci. USA 12, 690–692 (1926).
Gerbi, S. A. Non-random chromosome segregation and chromosome eliminations in the fly Bradysia (Sciara). Chromosome Res. 30, 273–288 (2022).
Bray, N., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal RNA-Seq quantification with kallisto. Nat. Biotechnol. 34, 525–527 (2016).
Kryuchkova-Mostacci, N. & Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 18, 205–214 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Mongue, A. J., Hansen, M. E., Gu, L., Sorenson, C. E. & Walters, J. R. Nonfertilizing sperm in Lepidoptera show little evidence for recurrent positive selection. Mol. Ecol. 28, 2517–2530 (2019).
Acknowledgements
The authors would like to thank Kamil Jaron, Christina Hodson, and Aleksandra Bliznina for providing early access to the Lycoriella ingenua genome. We thank Bea Herliczka at Mycobee Mushroom Farm and Gunnar Ovstebo at the Royal Botanic Garden Edinburgh for providing access to collect fly specimens, and Reuben Nowell, Priscilla Chin, and Réka Jenei for allowing the collection of flies from their houseplants. We also thank Maria Shlyakonova for helping to mass-rear flies for dissections. Finally, we thank Simon Martin, Deborah and Brian Charlesworth, Darren Obbard, Scott Roy, Konrad Lohse, Yannick Wurm, Alexander Mackintosh, and members of the Ross Lab for useful discussions throughout the project. Finally, we would like to thank three anonymous reviewers for their suggestions and help in improving the manuscript. This work was funded by a European Research Council starting grant (PGErepro, to L.R.), the RIKEN Special Postdoctoral Researchers Programme (to T.J.H.), a RIKEN Incentive Grant (to T.J.H.), and a European Research Council Consolidator Grant (no. 771387, to A.G.).
Author information
Authors and Affiliations
Contributions
R.B.B. conceptualised this study with T.J.H., A.G., L.R. and A.J.M. R.B.B. and J.S. collected and identified species. R.B.B. and K.M.M. performed species barcoding and DNA extractions. R.B.B. collected population and expression data and carried out all analyses with guidance from L.R. and A.J.M. R.B.B. wrote the manuscript, which all authors reviewed. L.R. and A.J.M. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Baird, R.B., Hitchcock, T.J., Ševčík, J. et al. Faster adaptation but slower divergence of X chromosomes under paternal genome elimination. Nat Commun 16, 5288 (2025). https://doi.org/10.1038/s41467-025-60114-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60114-8
