
Abstract
Focal gene amplification serves as an oncogenic driver during tumorigenesis and is a hallmark of many forms of cancer. Oncogene amplifications promote genomic instability, which is integral to cancer cell survival and evolution. However, focal gene amplification potentially affords an opportunity for therapeutic exploitation. As a proof-of-concept, we leverage CRISPR-Cas9 nickase to selectively promote cancer cell death in MYCN-amplified neuroblastoma in a gene amplification-dependent manner. Our analysis demonstrates that CRISPR-Cas9 nickase can generate a lethal number of highly toxic, replication-dependent double-strand breaks in cells harboring amplified loci. Furthermore, we demonstrate that Cas9 nickase—mediated toxicity can be modulated in combination with small molecule inhibitors targeting key regulators of the DNA-damage response or cell death pathways. Importantly, our findings in MYCN-amplified neuroblastoma translate to other cancer types with distinct oncogene amplifications.
Similar content being viewed by others
Introduction
Neuroblastoma is an aggressive pediatric malignancy accounting for approximately 15% of childhood cancer mortality1,2,3. Due to a complex genetic landscape, clinical manifestations of neuroblastoma demonstrate a high degree of variability leading to a risk-based stratification scheme – low, intermediate, and high4,5. Currently, both low and intermediate-risk groups maintain excellent prognoses with an overall survival rate of >90%6,7. Despite recent advancements in cancer therapeutics, however, high-risk neuroblastomas remain a significant challenge to treat with an overall survival rate of ~50%5. The aggressive behavior demonstrated by high-risk neuroblastoma may be ascribed to segmental chromosomal aberrations, such as amplification of the MYCN oncogene4, which occurs in ~20% of neuroblastomas8. MYCN amplifications are highly heterogenous ranging anywhere from ~10 to >1000 copies, and may extend from 350 kb to 8 Mb depending on the site and stage of disease9,10,11,12.
High-risk neuroblastomas often respond to conventional therapeutic modalities such as dose-intensive chemotherapy, surgery, radiotherapy, myeloablative chemotherapy with subsequent hematopoietic stem-cell rescue, and anti-GD2 immunotherapy2,4. Regrettably, this intensive therapeutic regimen is frequently insufficient with relapse occurring in ≥50% of affected individuals with no curative salvage therapy currently available4,5. Of those that remain in remission, most experience significant treatment-associated morbidity: secondary malignancy, impaired growth and development, hearing loss, and infertility among others3,13,14,15,16. As such, the modest efficacy and detrimental side effects associated with conventional chemoradiotherapies in the context of high-risk neuroblastoma constitutes an urgent unmet medical need for novel therapeutic interventions.
Clustered regularly interspaced short palindromic repeats (CRISPR)-based genome editing systems have become an increasingly popular platform for the development of novel therapeutics17,18. CRISPR-based nuclease systems, such as Cas9 or Cas12a, are commonly utilized for the inactivation of genes or regulatory elements by means of the mutagenic, non-homologous end joining (NHEJ) DNA repair pathway19. While nuclease-based genome editing systems have shown promise for some therapeutic applications20,21,22, double-strand breaks (DSBs) can elicit unintended consequences such as large deletions, chromosomal truncations, translocations, or chromothripsis among other complex genomic rearrangements that may hinder its potential use for certain therapeutic applications23,24,25. Notably, Cas9 nuclease—induced cytotoxicity was observed in cancer cell lines when targeting amplified sequences in the context of large-scale gene inactivation screens26,27,28, which informed efforts to implement Cas9 nucleases for selective cell-killing by targeting cancer-specific sequence amplifications29,30.
Cas9 nickases, which are catalytically inactivated at either of the two cleavage domains (HNH or RuvC), have been implemented for the targeted generation of single-strand breaks (SSBs)31. Single-strand breaks are largely innocuous in post-mitotic cells, however persistent SSBs within proliferating cells can impose significant cellular toxicity32,33. In actively dividing cells, the conversion of SSBs into potentially toxic single-ended or double-ended DSBs (seDSB or deDSB, respectively) depending on encounter with leading or lagging strand synthesis during DNA replication can impose replication stress, replication fork collapse, and ultimately cell death if not adequately repaired34,35,36. For example, Cas9 nickases have been observed to promote cellular toxicity when targeted to endogenous, repetitive elements in the mammalian genome33,37.
Here, we demonstrate that targeting highly amplified regions within the genome, such as MYCN in neuroblastoma cells, with the RuvC mutant SpyCas9D10A nickase affords an opportunity to exploit genomic vulnerabilities to induce cell death in a gene amplification-dependent manner. We show that Cas9 nickase—mediated SSBs within amplified loci are subsequently converted to seDSB or deDSBs during DNA replication (Fig. 1A). Thus, only actively dividing, MYCN-amplified neuroblasts are exposed to a high level of genotoxicity, sparing copy-normal cells in the process. Additionally, we find that this nickase-based, genotype-specific toxicity is not limited to MYCN-amplified neuroblastoma; we also demonstrate its efficacy in additional malignancies that exhibit gene amplifications such as ERBB2 (HER2)-amplified breast cancer, MYC-amplified non-small cell lung cancer, and MYC-amplified colorectal cancer.

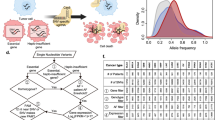
A Top pathway: Transient single-strand breaks (SSBs) are largely innocuous and quickly repaired in non-dividing cells. Bottom pathway: Persistent SSBs at high copy number loci within the genome in proliferating cells can induce cellular toxicity through their conversion into single-ended double-strand breaks (seDSBs) during DNA replication. Created in BioRender. Wolfe, S. (2025) https://BioRender.com/ttl9vxt. B Average MYCN genome copy number across various neuroblastoma cell lines obtained by qPCR (n = ≥ 3 biological replicates). For each box plot the horizontal line represents the median, the top and bottom of the box represent the upper and lower quartiles, respectively, and the top and bottom whiskers represent the maximum and minimum values, respectively. C A schematic of piggyBac-integrated sgRNA expression cassettes used to generate stable sgRNA-expressing cell lines. D A schematic of in vitro transcribed Cas9 nickase – mRNA construct variants. Created in BioRender. Wolfe, S. (2025) https://BioRender.com/tgt62po. Source data are provided as a Source Data file.
Results
Cas9D10A eliminates neuroblastoma cells in a gene amplification dependent manner
Given the high degree of heterogeneity found within neuroblastomas with respect to MYCN genome copy number38,39, a diverse cohort of neuroblastoma cell lines was selected for screening (Fig. 1B). The average MYCN genome copy number present in each cell line was validated by real-time quantitative PCR (qPCR; Fig. 1B) and was found to be in relative agreement with previously reported values40. Of the cell lines selected, CHP-212 is known to harbor MYCN amplification on extrachromosomal DNA (ecDNA)41 and SH-SY5Y serves as a MYCN non-amplified neuroblastoma control.
As previous work has shown, Cas9 nickases induce cellular toxicity when targeted to human long interspersed nuclear element (LINE1)33, an endogenous transposable element exceeding >5000 copies in human cell lines42. As such, Cas9 nickase targeting LINE-1 served as a positive control for induced cellular toxicity. Cas9 nickase targeting PPP1R12C – AAVS1, a non-amplified safe harbor locus in the human genome43, served as a negative control. Empirically, we observed no appreciable difference in cell viability when targeting AAVS1 compared to a panel of non-targeting or mock treatment controls (Supplementary Fig. 1). As a therapeutic target, we selected a non-coding region ~700 bp downstream of the annotated MYCN coding sequence that is still maintained within the MYCN amplicon to avoid any potential disruption of MYCN expression by means of coding-sequence alteration, as Cas9 nickases can introduce mutations at a low frequency44,45. To optimize screening, each cell line was modified to individually express one of the three guide RNAs (sgLINE-1, sgMYCN-1, or sgAAVS1) from a U6 promoter integrated as a stable transgene (Fig. 1C).
For the initial assessment of Cas9 nickase mediated toxicity, each cell line was electroporated with a dose of in vitro transcribed Cas9D10A – mRNA (Fig. 1D) ranging from 7.5 to 60 nM. LINE-1 and MYCN targeted cells were subsequently assessed for changes in cell viability relative to AAVS1 targeted cells after 3 days of incubation. Consistent with previous reports33, targeting of LINE-1 with Cas9D10A was toxic to all cell lines in a dose-dependent manner (Fig. 2A). Likewise, targeting a non-coding region adjacent to MYCN with Cas9D10A led to the rapid depletion of MYCN-amplified neuroblastoma cell lines (SK-N-BE(2 C), KELLY, NGP, CHP-212, and IMR-32) in a dose-dependent manner (Fig. 2B). Conversely, targeting MYCN with Cas9D10A was well-tolerated in MYCN non-amplified (SH-SY5Y) and MYCN non-amplified, non-neuroblastoma (HEK293T) cell lines (Fig. 2B).

A Positive control for Cas9D10A nickase – mediated cell-killing. Long interspersed nuclear element 1 (LINE-1) is a widespread endogenous transposable element where there are >5000 canonical LINE-1 repeats in the reference human genome. Dose dependent nickase toxicity is observed in all cell lines expressing LINE-1 targeting sgRNA (sgLINE-1) at 3 days post-treatment (n = 3 biological replicates). BMYCN-amplified neuroblastoma cells expressing MYCN targeting sgRNA (sgMYCN-1) are rapidly depleted in a dose-dependent manner whereas MYCN non-amplified (SH-SY5Y) and non-neuroblastoma (HEK293T) cells expressing sgMYCN-1 are not appreciably affected at 3 days post-treatment (n = 3 biological replicates). Focal MYCN amplifications present as a homogeneously staining region (HSR) or ecDNA produce toxicity when targeted by Cas9 nickase. Data in panels (A and B are presented as mean ± s.d. and normalized relative to viability of cells expressing AAVS1 targeting sgRNA treated with Cas9D10A. C MYCN copy number variance determined by real-time qPCR demonstrates a reduction in MYCN copy number 3 days following treatment with Cas9D10A-mRNA (30 nM) targeting MYCN in MYCN-amplified cell lines but no change in MYCN non-amplified SH-SY5Y cells (n = 3 biological replicates with technical duplicates). D Western blot of N-MYC protein expression levels from SK-N-BE(2)C or NGP cells expressing MYCN or AAVS1 sgRNA at 24 h post-treatment with Cas9D10A. E, F MYCN expression analysis by qRT-PCR demonstrates a modest reduction in MYCN expression in SK-N-BE(2)C or NGP MYCN-amplified neuroblastoma cells up to 72-h post-treatment with Cas9D10A-mRNA (30 nM) when targeting LINE-1 or MYCN (n = 4 biological replicates with technical duplicates). G, H MCM7 expression analysis by qRT-PCR demonstrates an increase in MCM7 expression in SK-N-BE(2)C or NGP MYCN-amplified neuroblastoma cells up to 72 h post-treatment with Cas9D10A-mRNA (30 nM) when targeting LINE-1 or MYCN (n = 4 biological replicates with technical duplicates). Data are presented as individual data points around the mean ± s.d. and normalized to untreated cells as a baseline control. Data analyzed by multiple unpaired t-tests (two-tailed); ns, P > 0.05; *, P ≤ 0.05; ** P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001 relative to untreated cells. Source data are provided as a Source Data file.
Given these observations, all subsequent applications of Cas9D10A – mRNA were carried out at a 30 nM dose, where a high therapeutic index for each of the target cell populations was observed. Next, to provide an orthogonal measure of Cas9D10A toxicity when targeting amplified sequences, we assessed cell population growth by quantitative image-based cytometry (QIBC). Population dynamics were monitored over a 3 day period post-Cas9D10A treatment (Supplementary Fig. 2). When targeting LINE-1 with Cas9D10A, all cell lines demonstrated population collapse, whereas only MYCN-amplified neuroblastoma cell lines demonstrated population collapse when targeting MYCN. We did not observe any appreciable changes in population dynamics when targeting AAVS1 relative to an untreated control.
Next, we sought to verify that the decrease in cell viability was not due to alterations in N-Myc levels. Despite targeting Cas9D10A to a non-coding sequence downstream of the MYCN 3’UTR, we questioned whether Cas9D10A – mediated DNA damage within MYCN amplifications could result in genomic or transcriptomic alterations within the target locus of the surviving cell population. Interestingly, MYCN copy number was observed to have decreased modestly in surviving MYCN targeted SK-N-BE(2)C and NGP cells ( ~ 25% and ~17%, respectively; Fig. 2C). Likewise, MYCN mRNA levels decreased when targeting MYCN, although similar reductions were observed in all Cas9D10A treatment groups at early time points (Fig. 2E, F).
Despite the observed decrease in MYCN gene and transcript copy number when targeting MYCN with Cas9D10A in SK-N-BE(2)C and NGP cells, no substantial differences in N-Myc levels were observed when targeting MYCN 48-h post-treatment with Cas9D10A relative to AAVS1 targeted or untreated cells (Fig. 2D). To assess N-Myc activity post-treatment with Cas9D10A, we assessed the expression of MCM7, a direct transcriptional target of N-Myc46 that functions to maintain genomic stability during S-phase47, at 1-, 2-, and 3 days post-treatment with Cas9D10A (Fig. 2G, H). Consistent with the notion that Cas9D10A – induced toxicity is associated with replication of DNA during S-phase, a substantial enrichment in MCM7 expression was observed at 24-h post-treatment with Cas9D10A when targeting LINE-1 or MYCN, but not AAVS1 (Fig. 2G, H). Expression of MCM7 remained elevated, though decreased progressively at 48-, and 72-h. These observations suggest that targeting the amplified MYCN region does not appreciably affect N-Myc expression or activity.
Next, we asked whether the efficacy of Cas9D10A could be target site-dependent with respect to the MYCN locus. As such, MYCN-amplified SK-N-BE(2)C and NGP cell lines were modified for the stable expression of sgRNAs targeting different regions within the MYCN locus: exon 2/intron (sgMYCN-2; ENST00000281043.4, ENST00000638417.1), exon 2 (sgMYCN-3) or the 3’-UTR (sgMYCN-4) (Supplementary Fig. 3A). No substantial position-specific variation in the efficacy of Cas9D10A—mediated cell-killing was observed in any MYCN sgRNA expressing cell line, supporting the notion that the observed cytotoxic effects conferred by Cas9D10A are largely dependent on the number of available target sites and not the position of the target sequence within an amplified genomic region (Supplementary Fig. 3B).
We also examined whether the cellular toxicity induced by Cas9D10A nickase when targeting an amplified gene could be replicated by the HNH mutant Cas9H840A nickase31. As such, SK-N-BE(2)C, NGP, and SH-SY5Y cells were treated with Cas9D10A or Cas9H840A—mRNA (30 nM) targeting LINE-1, MYCN, or AAVS1 and assessed for changes in cell viability 3 days post-treatment. As anticipated, we observed that both Cas9D10A and Cas9H840A reduced cell viability when targeting LINE-1 or MYCN in MYCN-amplified neuroblastoma cell lines (Supplementary Fig. 4). However, the reduction of cell viability was significantly greater in cells treated with Cas9D10A nickase. The disproportionate efficacy between nickase variants may largely be due to superior cleavage activity of Cas9D10A compared to Cas9H840A48 or a longer residence time of Cas9D10A at the cleavage site, which may occlude the nick and prevent repair, due to preferential recognition and removal of Cas9H840A intermediate by HLTF35,49.
Cas9D10A generates DSBs when targeted to amplified regions
We posited that the accumulation of SSBs generated by Cas9D10A, when intersecting with an active replication fork during S-phase, may lead to the formation of seDSBs or deDSBs as a function of the number of available Cas9D10A target sites within the genome35,36. As such, the presence of DNA damage induced by Cas9D10A within treated cells was assessed with single-cell resolution using the comet assay (Fig. 3; Supplementary Fig. 5)50.

Cumulative DNA damage (alkaline) and DSBs (neutral) quantified by single cell gel electrophoresis (comet assay). Alkaline conditions (pH > 13) relax the supercoiled structure DNA allowing both SSBs and DSBs to be detected. Neutral conditions restrict detection to fragmented DNA from DSBs. A–C SK-N-BE(2)C, NGP, or SH-SY5Y cells expressing LINE-1, MYCN or AAVS1 targeting sgRNA were electroporated with Cas9D10A-mRNA (30 nM). Individual cells were assessed for DNA damage at 3 days post-treatment by alkaline and neutral comet assay (n = 150). Representative images of Cas9D10A-treated cell comets are shown below each graph, respectively. Data are presented as individual data points around the median (black line) and were analyzed using a one-way ANOVA; ns, P > 0.05; *, P ≤ 0.05; ** P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Significance was determined using untreated cells as a baseline control. Source data are provided as a Source Data file.
Cumulative DNA damage (SSBs & DSBs) was assessed by comet assay under alkaline conditions at 3 days post-treatment with Cas9D10A – mRNA (30 nM) in various neuroblastoma cell lines, as well as a non-neuroblastoma control cell line (HEK293T) that had been previously modified to express either LINE-1, MYCN, or AAVS1 targeting sgRNAs. SK-N-BE(2)C, KELLY, NGP, CHP-212, SH-SY5Y and HEK293T cells demonstrated a dramatic increase in DNA damage (719–2677-fold) when targeting LINE-1 (Fig. 3A; Supplementary Fig. 5A). Cell lines harboring MYCN amplification (SK-N-BE(2)C, KELLY, NGP and CHP-212) demonstrated a substantial increase in DNA damage (122 to 746-fold) when targeting MYCN, whereas cell lines without MYCN amplification (SH-SY5Y and HEK293T) demonstrated only a modest increase in DNA damage (2.1 to 2.7-fold) when targeting MYCN (Fig. 3B; Supplementary Fig. 5B), which was similar to the increase in DNA damage (1.1 to 2.4-fold) when targeting AAVS1 (Fig. 3C; Supplementary Fig. 5C).
Double-strand breaks were specifically assessed by comet assay under neutral conditions for the same treatment groups (Fig. 3). Under these conditions, cells targeted at LINE-1 demonstrated a substantial increase in DSBs (83 – 646-fold; Fig. 3A; Supplementary Fig. 5A). Likewise, MYCN-amplified cell lines demonstrated a similar increase in DSBs (102 to 381-fold) when targeting MYCN, whereas cell lines without MYCN amplification (SH-SY5Y and HEK293T) demonstrated a modest increase in DSBs (0.3 – 0.6-fold) when targeting MYCN (Fig. 3B; Supplementary Fig. 5B) similar to the increase in DSBs (0.2 to 1.3-fold) when targeting AAVS1 relative to a mock/untreated control (Fig. 3C; Supplementary Fig. 5C).
Cas9D10A – mediated DSBs induce replication stress and G2/M cell-cycle arrest
Given that the conversion of Cas9D10A-generated SSBs to DSBs is contingent on active replication of the genome, we anticipated cell cycle aberrations to occur in S-phase. Consistent with our expectations, cell cycle analysis of MYCN-amplified SK-N-BE(2)C cells revealed a substantial accumulation of cells in S-phase at 24-h post-treatment with Cas9D10A when targeting LINE-1 or MYCN (2.2 & 2-fold, respectively; Fig. 4A, B).

A–D Flow cytometric cell cycle analysis of SK-N-BE(2)C cells expressing LINE-1, MYCN or AAVS1 targeting sgRNA at 1-, 2-, and 3 days post-treatment with Cas9D10A mRNA (30 nM). LINE-1 and MYCN targeted cells display an extension of S-phase at 1 day post-treatment and eventual arrest in G2/M at days 2 and 3 (n = 3 biological replicates). AAVS1 targeted cells demonstrate no apparent alteration in cell cycle progression. Data in panels (B–D) are presented as mean ± s.d. E Western blot of DNA damage markers from SK-N-BE(2)C cells expressing LINE-1, MYCN or AAVS1 sgRNA at 3 days post-treatment with Cas9D10A. LINE-1 and MYCN targeted cells demonstrate a substantial elevation in DNA damage markers. F, G Cell viability assessment for SK-N-BE(2)C, or SK-N-BE(2)C-RPA(123) cells expressing (F) LINE-1 or (G) MYCN targeting sgRNA treated with Cas9D10A, Cas9D10A + siRNA, or siRNA only. Cells treated with both Cas9D10A and siRNA were pre-incubated with siRNA for 24 h prior to the delivery of Cas9D10A – mRNA (30 nM). For each condition, cell viability was assessed at 3 days post-treatment (n = 3 biological replicates). Data are presented as mean ± s.d. normalized relative to viability of cells expressing AAVS1 targeting sgRNA treated with Cas9D10A. Data were analyzed multiple unpaired t-tests (two-tailed); ns, P > 0.05; *, P ≤ 0.05; ** P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. Source data are provided as a Source Data file.
Replication stress was demonstratively enriched in all cell lines targeted at LINE-1 and in MYCN-amplified cell lines targeted at MYCN 24 h post-treatment with Cas9D10A, as assessed by elevated of cytosolic calcium (Ca2+) levels (Supplementary Fig. 6A–C)51. Additional indicators of replication stress that are congruent with fork-stalling as a means to delay cell cycle progression and mitigate fork breakage within S-phase observed in these Cas9D10A-treated cell populations, include both the elevation of reactive oxygen species (ROS; Supplementary Fig. 6 D-F)52,53,54, and the increase in ATR-mediated phosphorylation of RPA2 (pS33) and CHK1 (pS345; Fig. 4E)55,56,57. Moreover, the production of DSBs during replication when Cas9D10A targets LINE-1 or MYCN in SK-N-BE(2) cells is evident by elevated levels of the canonical biomarker γ-H2AX (Fig. 4E)58,59,60.
Excessive DNA resection by EXO1 is known to propagate genomic instability and toxicity by hindering DSB repair61,62. Phosphorylation of EXO1 (pS746) by the ATR-CHK1 or CMKK2-AMPK pathways in response to replication stress protects DNA ends exposed by DSBs by preventing DNA resection51. Following Cas9D10A treatment, we observed an increase in phosphorylated EXO1 (pS746) in SK-N-BE(2)C cells when targeting LINE-1 or MYCN but not AAVS1 (Fig. 4E). DNA2 is partially redundant with EXO1 for DNA resection during S-phase DNA damage repair63. We examined whether EXO1 or DNA2 activity may be contributing to the observed toxicity conferred by Cas9D10A when targeting amplified loci. To investigate this, SK-N-BE(2)C cells expressing LINE-1, MYCN, or AAVS1 targeting sgRNAs were pre-treated with EXO1 (siEXO1), DNA2 (siDNA2), or a non-targeting (siNT) siRNA and recovered for 24-h before delivering Cas9D10A. Notably, the downregulation of EXO1, but not DNA2 was observed to increase cell viability by 3 to 4-fold for Cas9D10A targeting LINE-1 or MYCN (Fig. 4F, G; Supplementary Fig. 7). Likewise, the downregulation of EXO1 was shown to have a modest positive effect on cell proliferation for Cas9D10A targeting LINE-1 or MYCN in SK-N-BE(2)C cells (Supplementary Fig. 8A–C).
In the absence of Cas9D10A, cells treated with siEXO1 displayed appreciable stalling in S-phase of the cell cycle after 48-h (Supplementary Fig. 8D–F). Conceivably, the increase observed in cell viability in SK-N-BE(2)C cells treated with Cas9D10A targeting LINE-1 or MYCN may be due to slower rates of cell proliferation or a decrease in extensive 5’ DNA resection due to the reduction of EXO1 activity. As such, we reasoned these siEXO1-treated cells may display an alteration in target site sequence modifications due to changes in their ability to resolve DNA damage induced by Cas9D10A during replication. Evaluation of the MYCN target site in Cas9D10A-treated, siEXO1+ SK-N-BE(2)C cells by amplicon-sequencing revealed ~15% editing rate, whereas Cas9D10A, siDNA2+ SK-N-BE(2)C cells revealed a modest ~1% editing rate, relative to an untreated control (Supplementary Fig. 9). Our observations suggest that EXO1 activity may play a contributing role in Cas9D10A-mediated cell-killing.
Overexpression of RPA has been shown to fortify cells from the catastrophic impacts of replication stress by modulating exonuclease activity57,64,65. Previous work indicated that a free RPA reservoir is critical for the protection of replication forks against breakage during replication stress57, exhaustion of which promotes and accelerates fork collapse. As such, we suspected that the extensive number of Cas9D10A-mediated SSBs may deplete the free RPA reservoir, potentially exacerbating Cas9D10A-induced cellular toxicity. To investigate this, SK-N-BE(2)C cells expressing LINE-1 or MYCN targeting sgRNA were modified to overexpress human RPA1, RPA2, and RPA3 from a synthetic CAG promoter as a stable transgene (Supplementary Fig. 10) and subsequently treated with Cas9D10A. Interestingly, the overexpression of RPA increased survival in Cas9D10A – treated, SK-N-BE(2)C cells when targeting either LINE-1 or MYCN (4 to 5-fold) but remained insufficient to completely abolish cellular toxicity (Fig. 4F, G).
Given the partial increase in cell viability observed by the downregulation of EXO1 or the overexpression of RPA independently, we reasoned combining these approaches may increase resistance against Cas9D10A-mediated DNA damage in an additive manner. To test this, RPA-overexpressing SK-N-BE(2)C cells expressing LINE-1 or MYCN targeting sgRNA were pre-treated with siEXO1 24 h prior to Cas9D10A delivery. Surprisingly, no additional improvement in cell viability was observed (Fig. 4F, G), suggesting that these two perturbations (EXO1 knockdown and RPA overexpression) are impacting the same cytotoxic DNA repair intermediate associated with excessive DNA resection.
Furthermore, we observed stalling in S-phase to be only a transient state in response to Cas9D10A-mediated nicking at LINE-1 or MYCN, as evident by the progression into, and subsequent arrest of SK-N-BE(2)C cells in G2/M-phase of the cell cycle at days 2 and 3 post-treatment, with a failure to repopulate G1 (Fig. 4A, C and D). As such, we speculated that cell death is likely occurring shortly after cell division. Flow cytometric analysis of surviving SK-N-BE(2)C cells 3 days post-treatment with Cas9D10A targeting LINE-1 or MYCN revealed a substantial increase in cells harboring micronuclei (4.9 and 4.6-fold, respectively), whereas AAVS1 targeted cells showed only a modest increase (1.3-fold) relative to a mock/untreated control (Supplementary Fig. 11A, B).
Provided that micronuclei formation is contingent on the mitotic entry of cells harboring unrepaired DNA damage66, we asked whether Cas9D10A-mediated cell-killing is dependent on cell division. As such, SK-N-BE(2)C, NGP, and SH-SY5Y cells treated with Cas9D10A targeting LINE-1, MYCN, and AAVS1 were supplemented with the AURKA inhibitor (AURKAi), alisertib (0.5 µM), known to promote mitotic arrest67. Remarkably, an AURKAi significantly attenuated the cytotoxic effects conferred by Cas9D10A-mediated DNA damage (Supplementary Fig. 11C, D). These findings suggest that cell death in Cas9D10A-treated cells may occur subsequent to a mitotic catastrophe, which is consistent with the observed arrest of Cas9D10A-treated cells in the G2/M phase of the cell cycle, and a failure to repopulate G1.
Cas9D10A-induced DNA damage promotes PARP1 hyperactivation and necrotic cell death in MYCN-amplified neuroblastoma cells
The vast majority of primary neuroblastomas express wild type (WT) p53, with less than 2% harboring a p53 inactivating mutation68,69,70. In neuroblastoma, MYCN-driven tumorigenesis is contingent upon inhibition of p53-mediated apoptosis through the upregulation of MDM270. To assess the potential role of core apoptosis factors in cell death resulting from Cas9D10A-induced DNA damage, we screened two p53-WT neuroblastoma cell lines (MYCN-amplified NGP and MYCN non-amplified SH-SY5Y)71 for caspase 3 activation, a hallmark of intrinsic apoptosis72,73. Caspase 3 cleavage was evaluated by Western blot in both NGP and SH-SY5Y cells 3 days post-treatment with Cas9D10A and targeting LINE-1, MYCN, or AAVS1 (Supplementary Fig. 12A). As a positive control, SH-SY5Y cells were co-incubated with staurosporine (STS; 1 µM), a known inducer of caspase activation74,75. Not surprisingly, caspase 3 activation was readily apparent in STS-treated SH-SY5Y cells, whereas no obvious caspase 3 activation was detected in either cell line when targeting LINE-1, MYCN, or AAVS1 with Cas9D10A. To further validate our observations, we co-treated MYCN-amplified SK-N-BE(2)C (p53-mutant) and NGP (p53-WT) cells with Cas9D10A targeting either LINE-1, MYCN, or AAVS1 and Z-DEVD-FMK (18 µM), a known caspase 3 inhibitor, or pifithrin-α (PFT-α; 20 µM), a presumed inhibitor of p53 activity and apoptosis (Supplementary Fig. 12B, C)76,77. No appreciable difference in the efficacy of Cas9D10A – mediated cell-killing was observed.
Continued investigation of cell death pathways activated by Cas9D10A – mediated DNA damage was carried out in the p53-deficient, highly MYCN-amplified neuroblastoma cell line, SK-N-BE(2)C. Poly [ADP-ribose] polymerase 1 (PARP1) cleavage is a common occurrence observed across various cell death pathways, displaying distinct cleavage patterns by which proteases involved in each pathway can be identified78. As such, PARP1 cleavage was used as a benchmark to provide mechanistic insight into the cell death pathway(s) stimulated by Cas9D10A. As expected, when targeting LINE-1 and MYCN in SK-N-BE(2)C cells with Cas9D10A we observed substantially more PARP1 cleavage compared to targeting AAVS1 (Supplementary Fig. 12D). Multiple cleavage fragments ranging from 40 – 70 kDa were present, a pattern indicative of calpain-mediated PARP1 cleavage with continued proteolysis by cathepsin proteases79,80 and consistent with necrotic cell death.
Calpains are a family of Ca2+-activated cysteine proteases81. Their activation would be consistent with the elevation in cytosolic Ca2+ observed upon treatment of cells with Cas9D10A when targeting amplified loci (See, Supplementary Fig. 6A–C). Calpain activation, as marked by autoproteolytic cleavage82,83, was readily apparent in both LINE-1 and MYCN targeted SK-N-BE(2)C cell lysates (Supplementary Fig. 13A). Expanding on this observation, we assessed the level of calpain activity in both MYCN-amplified, SK-N-BE(2)C and NGP LINE-1, MYCN, or AAVS1 sgRNA expressing cells at 3 days post-treatment with Cas9D10A-mRNA (30 nM) relative to a mock/untreated control. As a positive control, calpain activity was induced in both SK-N-BE(2)C and NGP cells by preincubation in growth media supplemented with CaCl2 (2 mM). Consistent with the autoproteolytic cleavage observed previously, cells targeted by Cas9D10A at either LINE-1 or MYCN demonstrated an increase in calpain activity in both SK-N-BE(2)C (2.7 & 2.4—fold) and NGP (2.5 & 2.2—fold) cells (Supplementary Fig. 13B). Calpain activity was not observed to be appreciably altered in either SK-N-BE(2)C or NGP cells when targeting AAVS1 (1.0 & 1.1—fold).
Given the observed activation of calpains post-treatment with Cas9D10A, we reasoned that calpain inhibition may promote cell survival. As such, SK-N-BE(2)C, NGP, and SH-SY5Y cells treated with Cas9D10A targeting LINE-1, MYCN or AAVS1 were supplemented with calpastatin (CAST; 20 nM) an endogenous calpain inhibitor84. CAST supplementation in the presence of Cas9D10A significantly promoted cell survival (Supplementary Figs. 13C, D; 14A, B), while CAST supplementation in the absence of Cas9D10A had no effect on cell proliferation (See, Supplementary Fig. 15). These observations implicate calpains as a key protease involved in the execution of cell death post-treatment with Cas9D10A targeting amplified loci.
Ca2+ and ROS are key factors governing both the induction, and execution of necrotic cell death85,86,87. Furthermore, the influx of Ca2+ and ROS are known to elicit PARP1 hyperactivation subsequent to DNA damage88,89,90. PARP1 hyperactivation is marked by the enrichment of poly-ADP ribosylation (PARylation) of endogenous proteins and segments of damaged DNA, resulting in the depletion of intracellular ATP and NAD+ pools and the promotion of necrotic cell death78,91,92,93,94,95,96,97. As such, we observed a stark elevation in PARylation in SK-N-BE(2)C cells when targeting LINE-1 or MYCN with Cas9D10A (Supplementary Fig. 12D). Moreover, the depletion of ATP and NAD+ levels were observed across all tested neuroblastoma cell lines (SK-N-BE(2)C, NGP, and SH-SY5Y) when targeting LINE-1 or when targeting MYCN in MYCN-amplified neuroblastoma cell lines (SK-N-BE(2)C, and NGP; Supplementary Fig. 16).
Given these observations, we investigated the effect of PARP inhibition on Cas9D10A-mediated cell-killing in MYCN-amplified (SK-N-BE(2)C and NGP) and MYCN non-amplified (SH-SY5Y) neuroblastoma cells. Treatment with the PARP1 inhibitor (PARPi) rucaparib (10 µM) significantly attenuated ATP and NAD+ depletion (Supplementary Fig. 16) and promoted cell survival in the presence of Cas9D10A when targeting LINE-1 or MYCN (Supplementary Figs. 17A, B; 14C, D). In the absence of Cas9D10A, cell proliferation was only modestly affected by rucaparib treatment (See, Supplementary Fig. 12). Similar inhibition of Cas9D10A cell killing was observed with a second PARPi, olaparib (10 µM), which has a different allosteric effect on PARP1 recognition of DNA lesions98 (Supplementary Fig. 17C, D). Consistent with the modest change in cell cycle progression in PARPi-treated cells, no substantial reduction in the DNA damaging activity was observed for Cas9D10A targeting amplified loci (Supplementary Fig. 17E). Additionally, we observed only a modest increase in the fraction of cells in the G2/M phase of the cell cycle after 24-h of incubation with either rucaparib or olaparib relative to an untreated control (Supplementary Fig. 18). As such, these data suggest that the increase in cell survival observed when PARPi are combined with Cas9D10A targeting amplified loci is likely due to a reduction in PARP1 activity. These observations are consistent with the extensive Cas9D10A-mediated DNA damage occurring during DNA replication leading to PARP1 hyperactivation followed by necrotic cell death.
Release of cellular contents, such as the cytosolic enzyme lactate dehydrogenase (LDH), through permeabilization of the plasma membrane is a hallmark of necrotic cell death99. As such, we assessed the amount of LDH present in the cell culture media in both SK-N-BE(2)C and NGP LINE-1, MYCN, or AAVS1 sgRNA expressing cells at 3 days post-treatment with Cas9D10A – mRNA (30 nM; Supplementary Fig. 19). Consistent with plasma membrane disruption, LDH activity was increased when targeting LINE-1 or MYCN in SK-N-BE(2)C and NGP cells by ~3-fold, whereas LDH activity was not appreciable altered when targeting AAVS1 relative to an untreated control. Collectively, these observations suggest necrosis as the principal mode of cell death following Cas9D10A—mediated DNA damage at amplified loci within the genome.
Surviving Cas9D10A-treated neuroblastoma cells display markers of neuronal differentiation
Though Cas9D10A demonstrates robust cell-killing efficiency across a variety of neuroblastoma cell lines, a small percentage of cells manage to survive. As such, we were interested in expanding the surviving cell population as a means of testing their susceptibility to retreatment with Cas9D10A. However, the outgrowth of the surviving cell population failed on numerous occasions. Moreover, the surviving cells displayed morphological changes, such as de novo neurite formation and extension, consistent with neuronal differentiation100,101 (Supplementary Fig. 20A). DNA damage—induced cell differentiation has previously be reported to occur in stem cells102,103,104. Given the stem cell-like properties associated with neuroblastomas105,106, we explored whether Cas9D10A—induced DNA damage prompted the surviving cell population to undergo neuronal differentiation.
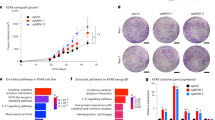
Here, we treated the MYCN-amplified, SK-N-BE(2)C cell line with Cas9D10A – mRNA (30 nM) targeting MYCN or AAVS1. Cells were assessed by immunocytochemistry for canonical biomarkers of neuronal differentiation, such as the elevation of class III β-tubulin (TUBB3) and a reduction in the proliferation marker, Ki-67107 at 5 days post-treatment (Supplementary Fig. 20B, C). SK-N-BE(2)C cells undergo neuronal differentiation when treated with retinoic acid107. Thus, untreated SK-N-BE(2)C cells were co-incubated with all-trans-retinoic acid (ATRA; 10 µM) as a positive control. Consistent with neuronal differentiation, SK-N-BE(2)C cells treated with MYCN targeting Cas9D10A displayed an enrichment in TUBB3 expression ( ~ 6.2-fold) and decrease in Ki-67 ( ~ 2.9-fold), whereas no appreciable differences were observed when targeting AAVS1 relative to an untreated control (Supplementary Fig. 20B, C). These results indicate that a fraction of the cell population may exit the cell cycle following treatment with Cas9D10A targeting the amplified MYCN locus to promote cell survival during a period of high cellular stress108. Whether this is a transient or terminal induction of neuronal differentiation is unclear.
Cas9D10A-mediated cell-killing can be augmented in combination with DDR pathway inhibitors or multiplex targeting
ATR and CHK1 inhibitors are commonly employed to potentiate the effectiveness of genotoxic chemotherapeutics by promoting continued origin firing and G2/M checkpoint escape109,110,111,112. The ATR-CHK1 DNA-damage response axis plays a critical role in restraining DNA replication in the context of replication stress by halting new origin firing and restricting EXO1-mediated DNA resection activity45,51. Therefore, we reasoned that the inhibition of ATR or CHK1 may yield a cellular environment that is more permissive to the accumulation of Cas9D10A-mediated DNA damage during S-phase, bypassing the G2/M-checkpoint, and aberrantly allowing for mitotic entry with unrepaired DNA damage.
To test this combination approach, we supplemented Cas9D10A treatment with a sub-lethal concentration (≤IC50) of the CHK1 inhibitor (CHK1i), MK8776 (500 nM) or the ATR inhibitor (ATRi), berzosertib (M6620, VX-970, VE-822; 20 nM)112,113 and assessed changes in cell viability 3 days post-treatment. Combining Cas9D10A and CHK1i or ATRi was shown to potentiate cell killing across all cell lines when targeting LINE-1, and significantly improved efficacy when targeting MYCN within MYCN-amplified cell lines at lower concentrations of Cas9D10A—mRNA (Fig. 5A, B; Supplementary Figs. 21, 22). Notably, only a modest reduction in cell viability was observed when targeting MYCN within the MYCN non-amplified SH-SY5Y cell line in the presence of CHK1i or ATRi which may be ascribed to the toxicity of either small molecule inhibitor in the context of dysregulated cellular proliferation in neuroblastoma cell lines (See, Supplementary Fig. 15).

A, B SK-N-BE(2)C, NGP, and SH-SY5Y cells expressing (A) LINE-1 or (B) MYCN targeting sgRNA were treated with Cas9D10A-mRNA at increasing doses in the absence or presence of a CHK1 inhibitor, MK8776 (500 nM). Co-treatment with CHK1i potentiated Cas9D10A-mediated cell-killing across cell lines at 3 days post-treatment (n = 3 biological replicates). Data were analyzed by multiple unpaired t-tests (two-tailed); ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. C Average MYCN and ALK genome copy number in IMR-32 cells determined by qPCR (n = 4 biological replicates). Data are presented normalized to HFF cells as a baseline control where for each box plot the horizontal line represents the median, the top and bottom of the box represent the upper and lower quartiles, respectively, and the top and bottom whiskers represent the maximum and minimum values, respectively. D Targeting of LINE-1, MYCN, ALK, or MYCN and ALK in IMR-32 cells (2 × 105) with Cas9D10A demonstrates a dose-dependent cytotoxic effect. Multiplex targeting of MYCN and ALK increases Cas9D10A-mediated cell-killing relative to targeting MYCN or ALK individually (n = 3 biological replicates). Data were analyzed using a two-way ANOVA with a Tukey’s multiple comparison test; ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. E No additional cellular toxicity is observed for multiplex targeting of an amplified (MYCN or ALK) locus with a non-amplified (AAVS1) locus in IMR-32 cells with Cas9D10A-mRNA (15 nM; n = 3 biological replicates). Data were analyzed by multiple unpaired t-tests (two-tailed); ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. F Multiplex targeting of non-amplified loci in SH-SY5Y cells with Cas9D10A-mRNA (30 nM) resulted in similar rates of cell viability to targeting individual loci. Cells were assessed for changes in cell viability at 3 days post-treatment with Cas9D10A-mRNA (n = 3). Data in panels (A, B, and E, F) is presented as mean ± s.d. and normalized relative to viability of cells expressing AAVS1 targeting sgRNA treated with Cas9D10A. Source data are provided as a Source Data file.
Co-amplification of oncogenes with proximal non-coding regulatory regions, such as enhancers, or even additional genes or gene segments is a phenomenon readily observed across tumor types114. In addition, amplification of distant genes can also occur. For example, focal amplifications of the receptor tyrosine kinase gene, ALK, located ~14 Mb from MYCN (Fig. 5C), have been observed in a subset of MYCN-amplified neuroblastomas115. As such, we reasoned gene amplifications from distinct genomic regions could be targeted simultaneously to further enhance Cas9D10A-mediated cell-killing by increasing the amount of associated DNA damage. To test this hypothesis, we utilized the IMR-32 neuroblastoma cell line harboring both MYCN and ALK gene amplification, which contains ~80 genome copies of each gene40 (Fig. 5C). We reasoned that targeting both MYCN and ALK amplifications simultaneously, which would double the number of Cas9D10A target sites, should have an additive effect. IMR-32 cells were modified in the same manner as previously described to individually express one of four guide RNAs (sgLINE-1, sgMYCN-1, sgALK or sgAAVS1), or co-express both sgMYCN-1 and sgALK simultaneously. Like sgMYCN-1, sgALK was designed to target a non-coding region neighboring the 3’ UTR of ALK.
Similar to other Cas9D10A-treated neuroblastoma cell lines, the efficacy of Cas9D10A-mediated cell-killing in IMR-32 cells increased in a dose-dependent manner (Fig. 5D). Given the similarity in MYCN and ALK gene copy number in IMR-32 cells, we observed a comparable decrease in cell viability when targeting MYCN or ALK at each of the tested concentrations of Cas9D10A-mRNA (Fig. 5D). However, when targeting MYCN and ALK simultaneously we observed an appreciable increase in the efficacy of Cas9D10A-mediated cell-killing at each of the tested concentrations of Cas9D10A-mRNA with respect to targeting MYCN (1.7 to 2.6-fold) or ALK (1.7 to 2.1-fold) individually (Fig. 5D). Notably, the observed fold increase in Cas9D10A-mediated cell-killing efficacy when targeting both gene amplifications in IMR-32 cells is consistent with the approximate doubling of sites targetable by Cas9D10A. We did not observe any substantial increase in toxicity when targeting MYCN or ALK in combination with AAVS1 in IMR-32 cells (Fig. 5E), or when targeting MYCN and ALK simultaneously in SH-SY5Y cells (Fig. 5F). These observations provide further evidence that the toxicity conferred by Cas9D10A is dependent on the number of targetable sites in the genome.
Cas9D10A demonstrates comparable cell killing efficacy to Cas9 nuclease with better selectivity
As noted previously, observations of Cas9 nuclease (Cas9WT)-induced cellular toxicity during large-scale loss of function screens26,27,28 proved foundational for the development of some Cas9WT-based cancer cell-killing technologies29,30. Unlike Cas9D10A, however, DSBs generated by Cas9WTare not replication dependent, and the repair of Cas9WT DSBs is considerably more mutagenic19,23,29,30. Consequently, we speculate that a cancer therapeutic based on Cas9WT could encounter potential problems including: 1) target site depletion by means of sequence alteration due to imprecise DSB repair116,117,118; and 2) DSB-related toxicity in post-mitotic119,120 or labile cell types, such as hematopoietic stem and progenitor cells (HSPCs)121.
In theory, employing Cas9D10A instead of Cas9WT would circumvent some of these issues. As such, we sought to directly compare Cas9D10A and Cas9WT with regards to selective cell killing. First, we validated Cas9WT-mediated cell-killing in MYCN-amplified, SK-N-BE(2)C cells and assessed its efficacy when delivered as mRNA at the same concentration (30 nM) as was previously employed for Cas9D10A (Supplementary Fig. 23). As anticipated, Cas9WT displayed efficacious cell-killing when targeting either LINE-1 or MYCN. Surprisingly, however, Cas9WT induced an appreciable degree of toxicity (0.5-fold) when targeting AAVS1, a safe-harbor locus, an outcome not observed when employing Cas9D10A (Supplementary Fig. 23A). Upon further investigation, SK-N-BE(2)C cells targeted at AAVS1 with Cas9WT demonstrated an ~2-fold median enrichment in DNA damage, as would be expected of a non-amplified locus, but an ~105-fold mean enrichment in DNA damage potentially as a result of off-target activity in a subset of cells within the population that received higher levels of Cas9WT mRNA (Supplementary Fig. 23B, C).
Next, we assessed SK-N-BE(2)C cells for sequence alterations at the MYCN target site post-treatment with either Cas9WT or Cas9D10A. Target site variations were assessed by amplicon-sequencing using genomic DNA isolated from surviving SK-N-BE(2)C cells at 3 days post-treatment with Cas9WT or Cas9D10A. Amplicon-sequencing analyses revealed ~84% of target sequences were modified in SK-N-BE(2)C cells treated with Cas9WT (Supplementary Fig. 18A), whereas only ~3.7% were modified when treated with Cas9D10A (Supplementary Fig. 24B). Moreover, sequences modified by Cas9WT-mediated DSBs demonstrate mixed imprecise repair outcomes with the majority being a single-nucleotide insertion, consistent with NHEJ (Supplementary Fig. 24C). In contrast, sequences modified by Cas9D10A, though present in low abundance, predominantly exhibit deletions, consistent with exonuclease-mediated end-resection and repair during S-phase of the cell cycle (Supplementary Fig. 24D).
Given the ostensible restriction of Cas9D10A-induced toxicity to proliferating cells, we sought to validate this notion by assessing cytotoxic effects in post-mitotic cells. We chose to target LINE-1 as it provides a challenging test given the large number of sites present within the genome. For a post-mitotic cell system, we induced neuronal differentiation in SK-N-BE(2)C cells expressing LINE-1 or AAVS1 targeting sgRNA. Neuronal SK-N-BE(2)C cells were transfected with either Cas9WT or Cas9D10A-mRNA and assessed for changes in confluency with crystal violet stain at 3 days post-treatment relative to an untreated control as a measure of cellular toxicity (Supplementary Fig. 25A, B). Cas9WT proved highly toxic when targeting LINE-1, and to our surprise, it was also toxic at AAVS1 in neuronal cells, suggesting a high level of mRNA delivery with substantial off-target activity. In contrast, no significant toxicity (P » 0.05) was observed when either site was targeted with Cas9D10A at the same mRNA dose.
Lastly, we sought to assess unwanted activity in HSPCs, as they are labile cells where levels of N-MYC and c-MYC are critical for stem cell maintenance and proper hematopoietic differentiation122. Moreover, the damage and depletion of HSPCs is an unfortunate consequence of more conventional chemotherapies4. Here, primary human cord blood CD34+ cells were electroporated with Cas9WT-mRNA (30 nM) and sgMYCN-1 (50 µM) or sgAAVS1 (50 µM), Cas9D10A-mRNA (30 nM) and sgMYCN-1 (50 µM), GFP-mRNA only (30 nM) or sgRNA only (50 µM) and assessed for changes in cell viability at 3 days post-treatment relative to a mock control (Supplementary Fig. 25C). Modest toxicity was observed in Cas9WT-treated HSPCs when targeting MYCN (0.7-fold) or AAVS1 (0.8-fold), whereas Cas9D10A-treated HSPCs maintained a similar level of viability comparable to other controls.
Next, we assessed target site mutagenesis in surviving HSPCs. Genomic DNA was isolated at 3 days post-treatment with Cas9WT or Cas9D10A and assessed for editing at the sgMYCN-1 target site. Despite the appreciable level of activity at the sgAAVS1 target site, Cas9WT demonstrated modest editing at the sgMYCN-1 target site as assessed by Sanger sequencing (Supplementary Fig. 25D). An assessment of Cas9WT-mediated editing at the MYCN and AAVS1 loci in MYCN non-amplified SH-SY5Y resulted in similar editing activity and viability as observed in HSPCs (Supplementary Fig. 25E). Importantly, Cas9D10A editing levels at the MYCN locus in HSPCs were below the level of detection by Sanger sequencing. These observations suggest that Cas9D10A editing activity at the MYCN locus in HSPCs is minimal.
Cas9D10A eliminates other cancer cell types in a gene amplification dependent manner
Given the success of Cas9D10A in the selective depletion of MYCN-amplified neuroblastoma cells, we asked whether our observations would translate to other cancer types exhibiting gene amplification. Consequently, we targeted ERBB2 (HER2), which is amplified ( ~ 50 copies) in the breast cancer cell line BT-474, and MYC, which is amplified ( > 400 copies as ecDNA)123,124 in non-small cell lung cancer cell line NCI-H2170 and the colorectal cancer cell line NCI-H716 (Fig. 6A, B). Each cell line was modified to express an sgRNA targeting a non-coding sequence downstream of the annotated coding sequence of the amplified gene, LINE-1 or AAVS1. Cellular toxicity was observed when targeting LINE-1 with Cas9D10A in each cell line, whereas targeting AAVS1 with Cas9D10A had minimal effect on cell viability or proliferation (Fig. 6C and Supplementary Fig. 15). Moreover, cellular toxicity was observed in BT-474, NCI-H716, and NCI-H2170 cells when targeting gene amplified loci (ERBB2 (HER2) or MYC) with Cas9D10A consistent with observations in the MYCN-amplified cell lines (Fig. 6D). Cas9D10A-mediated cell killing was dose responsive in BT-474 and NCI-H716 cell lines when targeting amplified loci. However, NCI-H2170 cells demonstrated high sensitivity to Cas9D10A at all concentrations when targeting amplified loci (Fig. 6C, D).

A, B qPCR analysis of gene copy number variance in (A) ERBB2 (HER2)-amplified breast cancer cell line, BT-474 and (B) MYC-amplified non-small-cell lung cancer and MYC-amplified colorectal cancer cell lines, NCI-H2170 and NCI-H716, respectively (n = 3 biological replicates with technical duplicates). Data are presented normalized to non-amplified human foreskin fibroblast (HFF) where for each box plot the horizontal line represents the median, the top and bottom of the box represent the upper and lower quartiles, respectively, and the top and bottom whiskers represent the maximum and minimum values, respectively. C, D Targeting of (C) LINE-1 or (D) ERBB2 (HER2) locus in BT-474 or MYC locus in NCI-H716 or NCI-H2170 cells demonstrates a cytotoxic effect similar to that observed when targeting the MYCN locus in MYCN-amplified neuroblastoma cells. All cells were assessed for changes in cell viability at 3 days post-treatment with Cas9D10A-mRNA (n = 3 biological replicates). Data are presented as mean ± s.d. and normalized relative to viability of cells expressing AAVS1 targeting sgRNA treated with Cas9D10A. Source data are provided as a Source Data file.
Next, we evaluated Cas9D10A-mediated cell killing in the presence of a CHK1 or ATR inhibitor (Supplementary Fig. 26). Similar to our observations in the MYCN-amplified neuroblastoma cell lines, delivery of Cas9D10A targeting an amplified loci in combination with CHK1i appreciably improved the cell-killing efficacy of Cas9D10A at lower concentrations for BT474 and NCI-H716 cells (Supplementary Fig. 26A–C) whereas delivery of Cas9D10A in combination with ATRi was more variable in its effect on cell killing (Supplementary Fig. 26D–F).
Finally, we assessed whether the cellular toxicity of Cas9D10A-induced DNA damage could be attenuated in the same manner as the MYCN-amplified neuroblastoma cell lines by treating BT-474, NCI-H716, or NCI-H2170 cells with either rucaparib (PARPi) or calpastatin (CAST) in the presence of Cas9D10A (Supplementary Fig. 27). Consistent with our observations in MYCN-amplified neuroblastoma cells, the cytotoxic effects of Cas9D10A targeting an amplified locus were significantly reduced at many doses in the presence of PARP inhibitors (Supplementary Fig. 27A–C) or calpastatin (Supplementary Fig. 27D–F). Overall, our observations suggest a conserved mechanism of amplification-dependent Cas9D10A-mediated cell-killing across four distinct cancer types harboring four distinct oncogene amplifications.
Discussion
Here we have demonstrated that CRISPR-Cas9 nickases can be employed for the selective depletion of cancer cells by targeting disease-specific gene amplifications. Gene amplification is associated with >50% of common cancer types and is more prevalent in recurrent or metastatic cancers125 suggesting that Cas9 nickase could have broad utility. A previous report has leveraged Cas9 nucleases to promote selective cancer cell death by generating DSBs in sequence-divergent, repetitive elements within the non-coding genome as a potential therapeutic approach for cancer30. However, targeting extensively amplified loci using Cas9D10A affords an opportunity to exploit this genomic vulnerability using reagents that do not require optimization to patient-specific sequence variations. In our survey of the MYCN locus, the position of the Cas9D10A target sequence did not appreciably influence the degree of toxicity conferred by the nickase suggesting a vast array of target sites can be utilized provided that they are amplified. This attribute of Cas9 nickase-induced toxicity in the context of high-copy number genomic sequences has important implications for the development of reagents tailored to specific cancer types that avoids potential damage to functional sequence elements within the genome of normal cells, thereby minimizing collateral damage and associated systemic toxicity.
One critical advantage of nickase-mediated cell killing over nuclease-based systems is the low indel rates induced at the target site by the nickase45. The low indel frequencies associated with a nickase minimize the chance of unintended sequence alterations at the target locus in normal cells (e.g. MYCN in HSPCs). Avoiding the use of a nuclease also minimizes the occurrence of other unwanted DNA repair outcomes, such as large deletions at the target site, that are associated with DSB repair in normal cells23,126. Low indel frequencies at the Cas9D10A target site within a cancer cell harboring a gene amplification minimizes the opportunity for resistance by means of target sequence-alteration, a pitfall that has been observed with nuclease-based systems for some therapeutic applications116,117,118. In addition, Cas9D10A can be used for multiplex targeting in instances of dual gene amplification (MYCN and ALK in IMR-32 cells), demonstrating the ability to harness multiple guides to increase the potency of the Cas9D10A system.
Our working model for cellular toxicity mediated by Cas9D10A nickase treatment is the replication-dependent formation of DSBs34 followed by the aberrant entry of cells into mitosis with extensive, unrepaired DNA damage. As it stands, there are many other genetic alterations (TP53 status, DNA repair factor inactivation, etc.) that may influence the efficiency of Cas9D10A-mediated cell-killing. Our observations suggest that the toxicity conferred by Cas9D10A is unlikely to be caused by changes in target locus copy number or gene expression level, as the observed changes for MYCN were relatively modest in Cas9D10A-treated MYCN-amplified neuroblastoma cell lines relative to the distribution of MYCN-amplification levels across different neuroblastoma cell lines tested in our experiments. These results suggest that the mechanism underlying Cas9D10A-mediated cell-killing is independent of gene function. However, the dependence of Cas9 nickase toxicity on DNA replication is a potential limitation for this approach, as quiescent cancer cells will not be subjected to the generation of DSBs. However, this challenge is also faced by other effective chemotherapies, such as topotecan and etoposide, where genotoxicity is associated with DNA replication127.
Furthermore, we have demonstrated that Cas9D10A can be used in combination with existing cytotoxic agents, such as CHK1 inhibitors, to augment the potency of Cas9D10A-mediated cell-killing. Thus, Cas9D10A treatment could potentially serve as a means of increasing the sensitivity of cancer cells harboring gene amplifications to a variety of conventional therapeutics128,129,130. Surprisingly, ATR inhibition was less effective than CHK1 inhibition when used in combination with Cas9D10A to augment cell-killing. In some cell lines, ATR inhibition was modestly protective when paired with a high concentration of Cas9D10A. These observations suggest that stimulation of the ATR-mediated DDR pathway could play a positive or negative role in Cas9D10A-mediated cell-killing depending on the cellular environment. For example, NCI-H2170 cells harbor a missense mutation (p.R2190C) within ATR, located between the FAT (FRAP-ATM-TRRAP) and kinase domains, which may be responsible for the attenuated response to ATR and CHK1 inhibitors131,132. As such, more comprehensive analyses of the DDR and cell death pathway(s) elicited by targeting Cas9D10A to high copy number sites may provide insights into additional cell-signaling pathway perturbations for augmenting nickase-induced cellular toxicity.
Finally, despite the favorable activity observed when applying Cas9D10A—mediated cell-killing in vitro, its utility for therapeutic application is contingent on the feasibility of delivery to the tumor in vivo. As such, directing future efforts into the development and optimization of a suitable carrier system is paramount for the therapeutic functionalization of this promising Cas9 nickase–based approach.
Methods
Ethics statement
Human Umbilical Cord Blood (UCB) used to isolate CD34+ HSPCs was obtained in accordance with the Committee for the Protection of Human Subjects in Research guidelines of the University of Massachusetts Chan Medical School. UCB was provided by the medical staff of the University of Massachusetts Memorial Umbilical Cord Blood Donation Program. The UBC core does the verbal consenting. The UCB are provided to us a deidentified specimens from the UCB Core at UMass: https://www.umassmed.edu/research/cores/cord-blood/. Consequently, sex and/or gender were not considered in the study design.
Mammalian cell culture
SK-N-BE(2)C [CRL-2268], CHP-212 [CRL-2273], IMR-32 [CCL-127], SH-SY5Y [CRL-2266], BT-474 [HBT-20], NCI-H2170 [CRL-5928], NCI-H716 [CCL-251], and HEK293T [CRL-3216] cells were sourced through the American Type Culture Collection (ATCC). KELLY [ACC 355] and NGP [ACC 676] cells were sourced through the Leibniz Institute (DSMZ). All mammalian cell cultures were maintained at 37 °C, 5% CO2. SK-N-BE(2)C, NGP, CHP-212, IMR-32 and SH-SY5Y neuroblastoma cell lines were grown and maintained in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12, GlutaMAX™ supplement (DMEM/F12, GlutaMAX™ supplement; Gibco, #10565018), supplemented with 10% fetal bovine serum (FBS; R&D Systems, #S12450H) and 1% penicillin-streptomycin (PS; 10,000 U/mL; Gibco, #15140122). KELLY neuroblastoma cells were grown and maintained in RPMI 1640, 1X with L-glutamine (Corning, #10-040-CV), supplemented with 15% FBS (R&D Systems, #S12450H) and 1% PS (10,000 U/mL; Gibco, #15140122). BT-474 breast cancer cells were grown and maintained in RPMI 1640, 1X with L-glutamine (Corning, #10-040-CV), supplemented with 10% FBS (R&D Systems, #S12450H) and 1% PS (10,000 U/mL; Gibco, #15140122). HEK293T human embryonic kidney cells were grown and maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, #11965092) supplemented with 10% FBS (R&D Systems, #S12450H) and 1% PS (10,000 U/mL; Gibco, #15140122). NCI-H2170 non-small cell lung cancer cells and NCI-H716 colorectal cancer cells were grown and maintained in RPMI 1640, 1X with L-glutamine (Corning, #10-040-CV), supplemented with 10% FBS (R&D Systems, #S12450H) and 1% PS (10,000 U/mL; Gibco, #15140122). All sgRNA expression cell lines were grown and maintained in the same media as previously described, further supplemented with Geneticin™ (1 mg/mL) (G418 Sulfate; 50 mg/mL; Gibco, #10131027). HEK293T sgRNA expression cell lines and SK-N-BE(2)C RPA overexpressing cell lines were grown and maintained in the same media as previously described, further supplemented with blasticidin S HCl (10 µg/mL) (Gibco, #A1113903). Primary CD34+ hematopoietic stem and progenitor cells (HSPCs) isolated from human cord blood were obtained as gift from Dr. Michael Brehm (University of Massachusetts Chan Medical School). HSPCs were maintained in StemPro®—34 SFM Complete Medium (Gibco, #10639-011) supplemented with L-glutamine (2 mM; Gibco, #21051-024), and cytokines SCF (MedChemExpress, #HY-P70781), IL-3 (MedChemExpress, #HY-P70576), and GM-CSF (MedChemExpress, #HY-P7016A\CS-P7016A) according to the manufacturer’s instructions. All cell lines were grown and maintained in 25 cm2 (Celltreat, #229331) or 75 cm2 (Celltreat, #229341) tissue culture flasks. All cell lines were tested for mycoplasma contamination at regular intervals using the e-MycoTM VALiD mycoplasma PCR detection kit (LiliF Diagnostics, #25239).
Expression vectors
Cas9 nickase in vitro transcription vectors contain T7 RNA polymerase promoter for the production of synthetic Cas9D10A or Cas9H840A mRNA. Additional elements include a 5’UTR derived from human hemoglobin alpha (hHBα), Kozak sequence, and 3’UTR derived from amino-terminal enhancer of split (AES) and mitochondrially encoded 12S rRNA (mt-RNR1) motifs133,134,135, and poly-A150. Additionally, all coding sequences were codon optimized to deplete the corresponding mRNA transcript of any unnecessary uridine bases to reduce the stimulation of innate immune sensors, with all necessary uridine bases being replaced with the modified nucleobase N1-methylpseudouridine (m1Ψ) during mRNA production136.
Guide RNA expression donor vectors contained a G418 sulfate selection marker (NeoR/KanR), or a blasticidin selection marker under the transcriptional control of a murine phosphoglycerate kinase (mPGK) promoter, and the desired sgRNA sequence under the transcriptional control of a U6 promoter. The expression cassette was placed between piggyBac left and right inverted terminal repeats (ITRs) to facilitate integration by piggyBac transposition137. The RPA1, RPA2, and RPA3 expression donor vector was derived from p11d-tRPA(123) (Addgene, #102613; a gift from Marc Wold)138 and placed into a piggyBac donor backbone containing a blasticidin selection marker under the transcriptional control of an EF1α promoter. RPA was expressed under the transcriptional control of a CAG promoter and separated by 2 A self-cleavable peptide linkers139, RPA1-T2A-RPA2-P2A-RPA3. Expression of the RPA transgene was verified by Western blot.
Cloning of sgRNAs
Oligonucleotides to construct sgRNA inserts were ordered from GENEWIZ (Azenta Life Sciences, Inc., USA). Vector backbone was digested overnight with BbsI-HF (New England Biolabs, #R539L) and supplemented with Quick CIP (New England Biolabs, #M0525L) to prevent recircularization. Forward and reverse oligonucleotides corresponding to each sgRNA were resuspended to a concentration of 20 µM then mixed; 5 µL forward oligo, 5 µL reverse oligo, 5 µL T4 polynucleotide kinase reaction buffer (10x; New England Biolabs, #B0201S), and 35 µL ddH2O. Mixture was incubated at 95 °C for 4 min and left to cool to room temperature. After cooling, ATP (10 mM) and T4 polynucleotide kinase (T4 PNK, New England Biolabs, #M0201L) were added directly to the mixture and incubated at 37 °C for 30 min. Annealed oligos (2 µL) and digested backbone (20 ng) were then ligated overnight at 16 °C with T4 DNA ligase, and used to transform NEB Stable (New England Biolabs, #C3040) cells under ampicillin selection (Thermo Scientific, #J63807-06). Successful insertion of sgRNA sequences verified by Sanger sequencing at GENEWIZ (Azenta Life Sciences, Inc., USA).
Stable transfection for the production of sgRNA and RPA(123) expressing cell lines
Neuroblastoma cell lines, BT-474 breast cancer cells, NCI-H716 colorectal cancer cells or NCI-H2170 non-small cell lung cancer cells ( ~ 2 × 106) were transfected with 0.5 µg of Super PiggyBac transposase expression vector (System Biosciences, #PB210PA-1) and 2 µg of sgRNA donor vector corresponding to either sgLINE-1 (5’ – ATTCTACCAGAGGTACAAGG – 3’); sgMYCN-1 (5’ – CAATGGAGACCCCATATGGG – 3’); sgMYCN-2 (5’ – CAGCTCCGAGCCCCCGAGCT – 3’); sgMYCN-3 (5’ – GGACCCAGGGCTGCGTTCTT – 3’); sgMYCN-4 (5’ – CACCAGTGTGGGGTTCTGCT – 3’); sgHER2-1 (5’ – GAGATCGAGTCGCGCCTCGG – 3’); sgMYCC (5’ – GATTCCTACCGTCGTCTGAG – 3’); sgALK (5’ – ACAGGCCGCTGGTGGTCCTA – 3’); or sgAAVS1 (5’ – GTCCCCTCCACCCCACAGTG – 3’) using Lipofectamine 3000 (Invitrogen, #L3000001) and incubated for 6 h before replacing the media. Cells were allowed to recover for 3 days prior to initiating antibiotic selection with either Geneticin™ (1 mg/mL) (G418 Sulfate; 50 mg/mL; Gibco, #10131027), blasticidin S HCl (10 µg/mL) (Gibco, #A1113903), or both when two differentially marked donor vectors were used to select for the integration of two different sgRNA expression cassettes. Cells were cultured under antibiotic selection for 3–5 days and then allowed to recover for 3 days without antibiotic selection to facilitate outgrowth and dilution of unincorporated donor vector prior to initiating an additional round antibiotic selection. The method used for the generation of SK-N-BE(2)C-RPA(123) is identical to that described for the sgRNA transgenes but employed an alternate selection marker to allow the selection of cells harboring both sgRNA and RPA(123) expression cassettes.
in vitro transcription (IVT)
Cas9D10A, Cas9H840A, or Cas9WT IVT vectors were constructed and subsequently digested overnight with Esp3I (New England Biolabs, #R0734L) to ensure complete linearization of the DNA template. Linearized template was subsequently purified using a DNA clean & concentrator – 5 spin-column (Zymo Research, #D4013) and eluted with nuclease-free H2O (NF- H2O). Individual IVT reactions (20 µL) were carried out with 1 µg of linearized template using the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs, #E2040S), exchanging UTP with N1-methylpseudouridine-5’-triphosphate (100 mM; TriLink, #N-1081-5) and further supplementing the reaction with CleanCap Reagent AG (3’ OMe) (4 mM; TriLink, #N-7413), RNasin Plus ribonuclease inhibitor (40 U; Promega, #N2615), and yeast inorganic pyrophosphatase (New England Biolabs, #M2403S). Reaction mixture was incubated at 37 °C for 2 h before increasing the reaction volume to 50 µL with nuclease free—H2O and treating the mixture with DNase I (New England Biolabs, #M0303L) at 37 °C for 15 min to remove residual DNA template. The final IVT product was purified using the Monarch RNA Clean-up Kit (500 µg; New England Biolabs, #T2057L) according to the manufacturer’s instructions. Typical yields obtained from the method described were in excess of 100 µg per reaction, as quantified by a NanoDrop UV/Vis spectrophotometer (Thermo Scientific, #ND-ONE-W). IVT products were assessed by denaturing 1 µg of IVT-produced mRNA in RNA loading dye (2X; New England Biolabs, #B0363S) for 10 min at 70 °C and running the product on a non-denaturing agarose gel (1%). Verified mRNA product was subsequently aliquoted at a concentration of 1 µg/µl and stored at −80 °C for downstream applications.
Electroporation of cells for the delivery of Cas9D10A, Cas9H840A, or Cas9WT mRNAs in vitro
Cas9D10A, Cas9H840A, or Cas9WT mRNAs were delivered by electroporation to each of the cell lines using the Neon™ Transfection System 10 µL kit (Invitrogen, #MPK1096) or Neon™ NxT Electroporation System 10 µL kit (Invitrogen, #N1096). Electroporation parameters were determined empirically for the following cell lines; SK-N-BE(2)C, IMR-32, and SH-SY5Y were electroporated with a 1300 V pulse voltage, 20 ms pulse width, 3 pulses; KELLY, NGP, and CHP-212 with a 1650 V pulse voltage, 10 ms pulse width, 3 pulses; HEK293T cells with a 1200 V pulse voltage, 20 ms pulse width, 2 pulses; and BT-474, NCI-H716, and NCI-H2170 cells with a 1450 V pulse voltage, 20 ms pulse width, 2 pulses. CD34+ HSPCs were electroporated a 1600 V pulse voltage, 10 ms pulse width, and 3 pulses according to the manufacturer’s instructions. All non-primary cells (2 × 105 per condition) were early to mid-passage ranging from passage number 5–10. Primary HSPCs were cultured for 24 h after thawing prior to electroporation. All electroporated cells demonstrated >90% viability at the time of electroporation as determined by trypan blue exclusion (Gibco, #15250061) using a TC20 automated cell counter (BioRad, #1450102). Cas9 nickase or Cas9 nuclease mRNA was prepared in resuspension buffer R supplemented with RNasin Plus (40 U, Promega, #N2615) at the desired concentration in which to resuspend the cells for electroporation. Co-delivery of Cas9D10A mRNA (30 nM) and synthetic sgRNA (300 pmol) (Integrated DNA Technologies, Inc., USA) was carried out in the same manner as previously described.
Cell viability and proliferation assays
Changes in cell viability and cytotoxicity were assessed using the RealTime-Glo™ MT Cell Viability Assay (Promega, #G9713), a nonlytic ATP-independent bioluminescent assay that measures the reduction potential of cells. Given the semi-adherent nature of many of the neuroblastoma cell lines, minimal manipulation of cells post-electroporation was critical. Cell viability was determined in an end-point manner at 3 days post-treatment with either Cas9D10A, Cas9H840A, or Cas9WT mRNAs by measuring the relative luminescence units (RLUs) using a GloMax® Explorer Multimode Microplate Reader (Promega, #GM3500). Changes in cell viability were normalized to AAVS1 targeted cells as no significant difference in viability was observed between AAVS1 and mock/untreated controls (Supplementary Fig. 15). Cell proliferation was monitored at 1-, 2-, and 3 days post-treatment with Cas9 nickase mRNA or by QIBC using the Celigo Image Cytometer (Nexcelcom Bioscience, USA) to measure the cell population following staining cell with Hoecsht 33342 (1 µg/mL; Invitrogen, #H1399) at 37 °C for 30 min. Where applicable, cells were supplemented with various reagents: in the presence of MK8776 (MedChemExpress, #HY-15532), rucaparib (MedChemExpress, #HY-10617A), PDD00017273 (MedChemExpress, # HY-108360), berzosertib (Selleck, #S7102), calpastatin (Millipore Sigma, #208902), alisertib (MedChemExpress, #HY-10971), etoposide (MedChemExpress, #HY-13629R), or topotecan (MedChemExpress, #HY-13768A) at concentrations ≤IC50 to prevent confounding toxicity.
Comet assay
Single cell gel electrophoresis was carried out on SK-N-BE(2)C, KELLY, NGP, CHP-212, SH-SY5Y, and HEK293T cells expressing different sgRNAs 3 days post-electroporation with Cas9D10A (30 nM) or Cas9H840A (30 nM) using the Comet Assay Electrophoresis Kit (R&D Systems, #4250-050-ESK) according to the manufacturer’s instructions. Cells were stained with Hoecsht 33342 (1 µg/mL; Invitrogen, #H1399) for 1 h at room temperature. Cells were visualized and imaged using the EVOS™ FL Imaging System (Invitrogen, #AMF4300, LED, Ex: 357/44 Em: 447/60, 4/10 PH, Sony ICX285AQ color CCD, 2/3 in 1360 × 1024, 1.4 Megapixels). Cells used for analysis were imaged at 10X magnification, representative cell images taken at 20X magnification. Tail moments were quantified using an ImageJ (NIH) Comet Assay macro (developed by Herbert M. Geller, NIH, 1997, and further modified by Robert Bagnell, 2011, Department of Pathology and Laboratory Medicine, UNC-CH).
Cell cycle analysis
SK-N-BE(2)C cells (2 × 105 cells per condition) expressing the LINE-1, MYCN or AAVS1 sgRNA were electroporated with Cas9D10A mRNA (30 nM) and collected at 1-, 2-, and 3 days post-treatment to assess changes in cell cycle progression. Harvested cells were fixed in 95% ethanol overnight at 4 °C, washed twice with PBS (1X), and resuspended in propidium iodide (Thermo Scientific, #J66764-MC) and RNase A (Invitrogen, #AM2271) solution. Cells were stained at 37 °C for 30 min and subsequently analyzed using the MACSQuant® Analyzer 10 Flow Cytometer (Miltenyi Biotec), and the resulting cell populations were characterized using FlowJo analysis software.
Metabolism assays
SK-N-BE(2)C, NGP, and SH-SY5Y cells (2 × 105) expressing LINE-1, MYCN or AAVS1 targeting sgRNA were electroporated with Cas9D10A mRNA (30 nM) and monitored for changes in intracellular calcium flux at 1-, 2-, and 3 days post treatment using the calcium indicator dye, Fluo-4 AM (Invitrogen, #F14201). Fluo-4 AM was reconstituted in DMSO and added to cells at a final concentration of 3 µM in PBS (1X) and incubated at room temperature for 1 h prior to analysis. ROS accumulation was monitored at 1-, 2-, and 3 days post treatment using the Total Reactive Oxygen Species (ROS) Assay Kit, 520 nm (Invitrogen, #88-5930-74) according to the manufacturer’s instructions. Mean integrated fluorescence intensities were acquired by QIBC using the Celigo Image Cytometer (Nexcelcom Bioscience, USA). Changes in total cellular ATP were assessed at 3 days post-treatment with Cas9D10A using CellTiter-Glo® Luminescent Cell Viability Assay (Promega, #G7572) according to the manufacturer’s instructions. Changes in NAD+ were assessed at 3 days post-treatment with Cas9D10A using NAD/NADH-Glo™ (Promega, #G9072). Changes in ATP or NAD+ were determined by measuring the relative luminescence units (RLUs) using a GloMax® Explorer Multimode Microplate Reader (Promega, #GM3500).
Western blotting
SK-N-BE(2)C, NGP, and SH-SY5Y cells ( ~ 2 × 106) expressing the desired sgRNA were electroporated with Cas9D10A mRNA (30 nM) for protein extraction 3 days post-treatment. Cells washed twice with ice-cold PBS (1X) and lysed with RIPA buffer (Thermo Scientific, #89900) containing Halt™ Protease and Phosphatase Inhibitor (Thermo Scientific, #78440) on ice for 30 min. Lysates were centrifuged at >15,000 g for 30 min at 4 °C to pellet cell debris and collect the supernatant. Protein concentration was measured by bicinchoninic acid (BCA) assay (Thermo Scientific, #23225). SDS-PAGE gel electrophoresis was carried out using 10 – 20% tricine gels (Invitrogen, #EC6625BOX) in tricine SDS running buffer (1X; Invitrogen, #LC1675) at 130 V constant voltage for 1.5 h at room temperature. Gels were transferred to PVDF membranes using the iBlot® Dry Blotting System, and subsequently blocked for 1 h at room temperature in SuperBlock™ Blocking Buffer (0.05% Tween-20; Thermo Scientific, #37535) before overnight primary antibody incubation at 4 °C.
Primary antibodies used include; N-Myc monoclonal antibody (NCM-II 100; 1:500, Invitrogen, #MA1-170); PARP1 polyclonal antibody (1:1000, Invitrogen, #PA5-34803); Poly ADP-ribose antibody, clone 10H (1:1000, Millipore Sigma, #MABC547); Caspase 3 polyclonal antibody (1:1000, Invitrogen, #PA5-77887); Mu-Calpain polyclonal antibody (1:1000, Invitrogen, #PA5-17547); Calpain 2 polyclonal antibody (1:1000, Invitrogen, #PA5-17494); phospho-EXO1(Ser746) (1:1000, Millipore Sigma, #ABE1066); anti-gamma-H2AX polyclonal antibody (1:10,000, Bethyl Laboratories, #A300-081A); phospho-CHK1 (Ser345) polyclonal antibody (1:1000, Invitrogen, #PA5-34625); phospho-RPA32 (Ser33) polyclonal antibody (1:1000, Invitrogen, #PA5-39809); RPA2 monoclonal antibody (9H8) (1:500, Invitrogen, #MA1-26418); β-actin (8H10D10) mouse monoclonal antibody (1:2000, Cell Signaling Technology, #3700). Blots were washed 3–5 times in TBST prior to secondary antibody incubation for 1.5 h at room temperature with either anti-rabbit IgG, HRP-conjugated antibody (1:3000, Cell Signaling Technology, #7074) or anti-mouse IgG, HRP-conjugated antibody (1:3000, Cell Signaling Technology, #7076). Blots were incubated in ECL substrate (Thermo Scientific, #32106) for 1–2 min before visualization using ChemiDoc™ Touch Imaging System (BioRad). Uncropped blots are present in the Source Data file.
Assessing apoptotic and necrotic cell death activity in Cas9D10A—treated neuroblastoma cells
Cleavage of caspase 3 was assessed in NGP and SH-SY5Y LINE-1, MYCN, or AAVS1 sgRNA expressing cells by Western blot at 3 days post-treatment with Cas9D10A (See Western blotting section). Staurosporine (1 µM; MedChemExpress, #HY-15141) was used as a positive control for caspase 3 activation in SH-SY5Y cells and harvested for protein extraction following 3-h of incubation under normal culture conditions (See mammalian cell culture section). SK-N-BE(2)C and NGP LINE-1, MYCN, or AAVS1 sgRNA expressing cells ( ~ 2 × 105) were electroporated with Cas9D10A – mRNA (30 nM) and incubated with Z-DEVD-FMK (18 µM; MedChemExpress, # HY-12466) or PFT-α (20 µM; Millipore Sigma, #P4359). Changes in cell viability and cytotoxicity were assessed at 3 days post-treatment using the RealTime-Glo™ MT Cell Viability Assay (Promega, #G9713). Calpain autoproteolytic cleavage was assessed in SK-N-BE(2)C LINE-1, MYCN, or AAVS1 sgRNA expressing cells at 3 days post-treatment with Cas9D10A (See Western blotting section). Calpain activity was assessed in both SK-N-BE(2)C and NGP LINE-1, MYCN, or AAVS1 sgRNA expressing cells ( ~ 2 × 105) electroporated with Cas9D10A – mRNA (30 nM). Calpain activity was measured at 3 days post-treatment with Cas9D10A using the Calpain-Glo™ Protease Assay (Promega, #G8501) according to the manufacturer’s instructions. Induction of calpain activation was achieved following 30 min of incubation in growth media supplemented with CaCl2 (2 mM). Lactate dehydrogenase activity was assessed in both SK-N-BE(2)C and NGP LINE-1, MYCN, or AAVS1 sgRNA expressing cells ( ~ 2 × 105) electroporated with Cas9D10A – mRNA (30 nM). Lactate dehydrogenase activity was assessed at 3-days post-treatment with Cas9D10A using the CyQUANT™ LDH Cytotoxicity Assay Kit (Invitrogen, #C20301) according to the manufacturer’s instructions.
Assessment of neuronal differentiation by immunocytochemistry post treatment with Cas9D10A
Surviving SK-N-BE(2)C cells expressing the designed sgRNAs electroporated with Cas9D10A – mRNA (30 nM) or incubated with ATRA (10 µM; MedChemExpress, # HY-14649) were assessed at 5 days post-treatment. Cells were fixed with ice-cold methanol (100%) for 15 min at 4 °C, washed briefly with PBS (1X), and permeabilized with PBST (0.5%) for 5 min at room temperature. Following permeabilization, cells were washed briefly with PBS (1X) and blocked with SuperBlock™ Blocking Buffer (0.05% Tween-20; Thermo Scientific, #37535) for 30 min at room temperature. Primary antibody incubation performed overnight at 4 °C. Primary antibody was removed by washing 3x with PBS (1X). Secondary antibody incubation performed for 1 h at room temperature. TUBB3 was probed with beta-3 tubulin polyclonal antibody (1:50; Invitrogen, #PA5-25655) and goat anti-rabbit IgG (H + L) AF488 (1:250; Invitrogen, #A32731). Ki-67 was probed with Ki-67 (SP6) recombinant monoclonal antibody (1:200; Invitrogen, #MA5-14520) and goat anti-rabbit IgG (H + L) AF594 (1:250; Invitrogen, #A11012). Expression analysis of TUBB3 and Ki-67 was achieved by QIBC using the Celigo Image Cytometer (Nexcelcom Bioscience, USA). Representative images were taken using the EVOS™ FL Imaging System (Invitrogen, #AMF4300, LED, Ex: 357/44 Em: 447/60, Ex: 482/25 Em: 524/24, 4/10 PH, Sony ICX285AQ color CCD, 2/3 in 1360 × 1024, 1.4 Megapixels). Image processing conducted with Fiji (ImageJ)140.
Transfection and assessment of cellular toxicity in post-mitotic neuronal cells
SK-N-BE(2)C cells expressing either LINE-1 or AAVS1 targeting sgRNA were cultured in DMEM/F12, GlutaMAX™ supplement (Gibco, #10565018) supplemented with ATRA (10 µM; MedChemExpress, # HY-14649) in 6-well plates (Celltreat, #229106). ATRA-containing growth media was exchanged every 3 days with gradual serum deprivation (10–2%). Cells were maintained in differentiation media until the halting of proliferation and demonstration of distinct morphological changes consistent with neuronal differentiation. Neuronal SK-N-BE(2)C cells expressing either LINE-1 or AAVS1 targeting sgRNA were subsequently transfected with Cas9WT or Cas9D10A-mRNA (5 µg) using Lipofectamine MessengerMAX Reagent (Invitrogen, #LMRNA008) and assessed for cytotoxic effects at 3 days post-treatment. Cellular toxicity was determined by assessing changes in cell confluency using crystal violet staining (Millipore Sigma, #V5265). Representative images were taken using the EVOS™ FL Imaging System #AMF4300, LED, Sony ICX285AQ color CCD, 2/3 in 1360 × 1024, 1.4 Megapixels) at 4X magnification. Differences in cell confluency determined with ImageJ140.
Micronuclei detection assay
Surviving SK-N-BE(2)C cells expressing the designed sgRNAs were harvested 3 days following electroporation with Cas9D10A mRNA (30 nM) and washed twice in PBS (1X) to remove dead cells and debris before being stained with Hoecsht 33342 (1 µg/mL; Invitrogen, #H1399) for 30 min at room temperature. Cells were pelleted at 1,000 x g and resuspended in Nuclei Prep Buffer (New England Biolabs, #T3052) containing RNase A (Invitrogen, #AM2271) and incubated at room temperature for 5 min. Nuclei Prep Buffer is an incomplete lysis buffer disrupting the cell membrane while leaving nuclei intact. SK-N-BE(2)C nuclei from each condition were analyzed using the MACSQuant® Analyzer 10 Flow Cytometer (Miltenyi Biotec), and further characterized using FlowJo analysis software. Cells were visualized and imaged using the EVOS™ FL Imaging System (Invitrogen, #AMF4300, LED, Ex: 357/44 Em: 447/60, 4/10 PH, Sony ICX285AQ color CCD, 2/3 in 1360 ×1024, 1.4 Megapixels) at 20X magnification.
Genome copy number variance assay
Genomic DNA was extracted from each of the cell lines using the DNeasy Blood & Tissue Kit according to the manufacturer’s instructions (Qiagen, #69504). DNA concentration was obtained using the Qubit™ dsDNA quantitation high-sensitivity kit (Invitrogen, #Q32851). Genome copy number variance was determined by performing a TaqMan copy number variance real-time qPCR assay using the QuantStudio3 (Applied Biosystems, USA) according to the manufacturer’s instructions using RPPH1 (RNase P) as an internal control. MYCN was amplified using forward (5’ – ACTCTGTCCCTTAAGGAGCAACC – 3’) and reverse (5’ – CCAACCAGGATTGTACAGGTGC – 3’) primers. MYC was amplified using forward (5’ – CACAAAGACTCATCCACATGC – 3’) and reverse (5’ – CTCCTGTACTCACAAGCAGC – 3’) primers. ERBB2 (HER2) was amplified using forward (5‘ – CCACATTCCCAGACTGGACG – 3’) and reverse (5’ – GGTAATACCTGTCTGATGCCAC – 3’) primers. ALK was amplified using forward (5’ – CGTTTCAGTCATGGAATGAACTACC – 3’) and reverse (5’ – CATGAGGAAGGGAGAGAAAACGC – 3’) primers. Average gene copy number for each cell line was determined by comparative Ct. Genome copy numbers were normalized to normal human foreskin fibroblast cells (HFF) as a control.
MYCN and MCM7 expression analysis by qRT-PCR
Total RNA was extracted from SK-N-BE(2)C or NGP cells using the Monarch® Total RNA Miniprep Kit according to the manufacturer’s instructions (New England Biolabs, #T2010S). Total RNA concentration was obtained using a NanoDrop UV/Vis spectrophotometer (Thermo Scientific, #ND-ONE-W). Reverse transcription was performed using the SuperScript™ III First-strand Synthesis kit according to the manufacturer’s instructions (Invitrogen™, #11752050). Quantitative PCR was performed using PowerTrack™ SYBR Green Master Mix (Applied Biosystems™, #A46012) and the QuantStudio3 (Applied Biosystems, USA) according to the manufacturer’s instructions.
Site-specific cDNA was amplified using the following forward (MYCN: 5’ – CACTGAGTATGTCCACTCCCTCC – 3’; MCM7: 5’ – CCAAGTCTCAGCTCCTGTCATAC – 3’) and reverse (MYCN: 5’ – GTGGCAGTGACTGTCCAGTTTTG – 3’; MCM7: 5’ – CCTCTAAGGTCAGTTCTCCACTC – 3’) primers. Expression values were normalized to GAPDH which was amplified using the following forward (5’ – GTCTCCTCTGACTTCAACAGCG – 3’) and reverse (5’ – ACCACCCTGTTGCTGTAGCCAA – 3’) primers. Changes in MYCN or MCM7 expression were determined by comparative Ct relative to a mock/untreated control.
EXO1/DNA2 knockdown
Pre-validated Silencer® Select siRNAs targeting EXO1 or DNA2 (Invitrogen, #AM16708), or Silencer® Select Negative Control siRNA (Invitrogen, #4390843) were delivered to SK-N-BE(2)C cells expressing LINE-1, MYCN, or AAVS1 targeting sgRNA using the Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, #13778075) in accordance with the manufacturer’s instructions. Transfected cells were recovered for 24 h prior to downstream application.
Indel analysis
Genomic DNA was extracted from each of the cell lines using the DNeasy Blood & Tissue Kit (Qiagen, #69504) 3 days post-treatment with Cas9D10A mRNA. DNA concentration was obtained using the Qubit™ dsDNA quantitation high-sensitivity kit (Invitrogen, #Q32851). The sgMYCN-1 target site was amplified using forward (5’ – GTAGAGCATAGTTTGTCTCC – 3’) and reverse (5’ – CTATCTCCTTAACACACAAG – 3’) primers. Amplicon-Seq was performed GENEWIZ (Azenta Life Science, Inc., USA). Raw data was analyzed using the CRISPResso2 deep sequencing analysis webtool141.
Statistics and Reproducibility
No statistical method was used to predetermine sample size. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment. No data were excluded from the analyses. All statistical analyses were carried out using GraphPad Prism (10.2.3) software. Quantitative data, apart from where individual values are demonstrated, represent the mean ± standard deviation. Comparative statistical analyses were conducted using multiple unpaired t-tests or two-way ANOVA with a Tukey’s multiple comparison test; ns, P > 0.05; *, P ≤ 0.05; ** P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
A Reporting Summary for this article is available as a supplementary information file. Illumina sequencing data has been submitted to the Sequence Read Archive accession code PRJNA1251614. Source data are provided as a Source Data file. Source data are provided with this paper.
References
Smith, V. & Foster, J. High-risk neuroblastoma treatment review. Children 5, 114 (2018).
Krystal, J. & Foster, J. H. Treatment of high-risk neuroblastoma. Children 10, 1302 (2023).
Matthay, K. K. et al. Neuroblastoma. Nat. Rev. Dis. Prim. 2, 16078 (2016).
Maris, J. M. Recent advances in neuroblastoma. N. Engl. J. Med 362, 2202–2211 (2010).
Irwin, M. S. et al. Revised neuroblastoma risk classification system: a report from the children’s oncology group. J. Clin. Oncol. 39, 3229–3241 (2021).
Strother, D. R. et al. Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of children’s oncology group study P9641. JCO 30, 1842–1848 (2012).
Iehara, T. et al. Successful treatment of infants with localized neuroblastoma based on their MYCN status. Int. J. Clin. Oncol. 18, 389–395 (2013).
Louis, C. U. & Shohet, J. M. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev. Med. 66, 49–63 (2015).
Gundem, G. et al. Clonal evolution during metastatic spread in high-risk neuroblastoma. Nat. Genet. 55, 1022–1033 (2023).
Akiyama, K. et al. Megabase-scale analysis of the origin of N-myc amplicons in human neuroblastomas. Nucleic Acids Res. 22, 187–193 (1994).
Łastowska, M. et al. Identification of candidate genes involved in neuroblastoma progression by combining genomic and expression microarrays with survival data. Oncogene 26, 7432–7444 (2007).
Beheshti, B. et al. Chromosomal localization of DNA amplifications in neuroblastoma tumors using cDNA microarray comparative genomic hybridization. Neoplasia 5, 53–62 (2003).
Qiu, B. & Matthay, K. K. Advancing therapy for neuroblastoma. Nat. Rev. Clin. Oncol. 19, 515–533 (2022).
Cohen, L. E. et al. Late effects in children treated with intensive multimodal therapy for high-risk neuroblastoma: High incidence of endocrine and growth problems. Bone Marrow Transpl. 49, 502–508 (2014).
Gurney, J. G. et al. Hearing loss, quality of life, and academic problems in long-term neuroblastoma survivors: a report from the children’s oncology group. Pediatrics 120, e1229–e1236 (2007).
Applebaum, M. A. et al. Second malignancies in patients with neuroblastoma: the effects of risk-based therapy. Pediatr. Blood Cancer 62, 128–133 (2015).
Zhao, Z., Shang, P., Mohanraju, P. & Geijsen, N. Prime editing: advances and therapeutic applications. Trends Biotechnol. 41, 1000–1012 (2023).
Doudna, J. A. The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Fu, B. et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia. Nat. Med. 28, 1573–1580 (2022).
Frangoul, H. et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 384, 252–260 (2021).
Gillmore, J. D. et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Leibowitz, M. L. et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet 53, 895–905 (2021).
Frock, R. L. et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186 (2015).
Munoz, D. M. et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 6, 900–913 (2016).
Aguirre, A. J. et al. genomic copy number dictates a gene-independent cell response to crispr/cas9 targeting. Cancer Discov. 6, 914–929 (2016).
Wang, T. et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015).
Kwon, T. et al. Precision targeting tumor cells using cancer-specific InDel mutations with CRISPR-Cas9. Proc. Natl. Acad. Sci. USA. 119, e2103532119 (2022).
Tan, I.-L. et al. Targeting the non-coding genome and temozolomide signature enables CRISPR-mediated glioma oncolysis. Cell Rep. 42, 113339 (2023).
Jinek, M. et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA. 98, 8241–8246 (2001).
Smith, C. J. et al. Enabling large-scale genome editing at repetitive elements by reducing DNA nicking. Nucleic Acids Res. 48, 5183–5195 (2020).
Caldecott, K. W. Causes and consequences of DNA single-strand breaks. Trends Biochem. Sci. 49, 68–78 (2024).
Pavani, R. et al. Structure and repair of replication-coupled DNA breaks. Science 385, eado3867 (2024).
Kimble, M. T. et al. Repair of replication-dependent double-strand breaks differs between the leading and lagging strands Mol. Cell 85, 61–77.e6 (2025)
Koeppel, J. et al. Randomizing the human genome by engineering recombination between repeat elements. Science 387, eado3979 (2025).
Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E. & Bishop, J. M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121–1124 (1984).
Pugh, T. J. et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet 45, 279–284 (2013).
Lodrini, M. et al. Using droplet digital PCR to analyze MYCN and ALK copy number in plasma from patients with neuroblastoma. Oncotarget 8, 85234–85251 (2017).
Helmsauer, K. et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 11, 5823 (2020).
Coufal, N. G. et al. L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131 (2009).
Papapetrou, E. P. & Schambach, A. gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 24, 678–684 (2016).
Cho, S. W. et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132–141 (2014).
Rees, H. A., Yeh, W.-H. & Liu, D. R. Development of hRad51-Cas9 nickase fusions that mediate HDR without double-stranded breaks. Nat. Commun. 10, 2212 (2019).
Shohet, J. M. et al. Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer Res 62, 1123–1128 (2002).
Bailis, J. M. & Forsburg, S. L. MCM proteins: DNA damage, mutagenesis and repair. Curr. Opin. Genet. Dev. 14, 17–21 (2004).
Gopalappa, R., Suresh, B., Ramakrishna, S. & Kim, H.(H. enry) Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res. 46, e71–e71 (2018).
Reginato, G. et al. HLTF disrupts Cas9-DNA post-cleavage complexes to allow DNA break processing. Nat. Commun. 15, 5789 (2024).
Olive, P. L. & Banáth, J. P. The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 1, 23–29 (2006).
Li, S. et al. Ca2 + -stimulated AMPK-dependent phosphorylation of Exo1 protects stressed replication forks from aberrant resection. Mol. Cell 74, 1123–1137.e6 (2019).
Ragu, S. et al. A noncanonical response to replication stress protects genome stability through ROS production, in an adaptive manner. Cell Death Differ. 30, 1349–1365 (2023).
Andrs, M. et al. Excessive reactive oxygen species induce transcription-dependent replication stress. Nat. Commun. 14, 1791 (2023).
Somyajit, K. et al. Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science 358, 797–802 (2017).
Friedel, A. M., Pike, B. L. & Gasser, S. M. ATR/Mec1: coordinating fork stability and repair. Curr. Opin. Cell Biol. 21, 237–244 (2009).
Sorensen, C. S. & Syljuasen, R. G. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 40, 477–486 (2012).
Toledo, L. I. et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013).
Strumberg, D. et al. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5’-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell Biol. 20, 3977–3987 (2000).
Xu, Y. & Her, C. inhibition of topoisomerase (DNA) i (top1): DNA damage repair and anticancer therapy. Biomolecules 5, 1652–1670 (2015).
Kuo, L. J. & Yang, L.-X. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. Vivo 22, 305–309 (2008).
Tomimatsu, N. et al. DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J. Biol. Chem. 292, 10779–10790 (2017).
Chen, X. et al. 14-3-3 proteins restrain the Exo1 nuclease to prevent overresection. J. Biol. Chem. 290, 12300–12312 (2015).
Cejka, P. & Symington, L. S. DNA end resection: mechanism and control. Annu Rev. Genet 55, 285–307 (2021).
Zhao, F., Kim, W., Kloeber, J. A. & Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 52, 1705–1714 (2020).
Myler, L. R. et al. Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. Proc. Natl. Acad. Sci. USA 113, E1170-9 (2016).
Kalsbeek, D. & Golsteyn, R. M. G2/M-phase checkpoint adaptation and micronuclei formation as mechanisms that contribute to genomic instability in human cells. Int J. Mol. Sci. 18, 2344 (2017).
Manfredi, M. G. et al. characterization of alisertib (MLN8237), an investigational small-molecule inhibitor of aurora a kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 17, 7614–7624 (2011).
Hosoi, G. et al. Low frequency of the p53 gene mutations in neuroblastoma. Cancer 73, 3087–3093 (1994).
Vogan, K. et al. Absence of p53 gene mutations in primary neuroblastomas. Cancer Res 53, 5269–5273 (1993).
Slack, A. et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. USA. 102, 731–736 (2005).
Chen, L. et al. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 6, 10207–10221 (2015).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417 (2020).
Mookherjee, P. et al. Mitochondrial-targeted active Akt protects SH-SY5Y neuroblastoma cells from staurosporine-induced apoptotic cell death. J. Cell Biochem 102, 196–210 (2007).
Boix, J., Llecha, N., Yuste, V. J. & Comella, J. X. Characterization of the cell death process induced by staurosporine in human neuroblastoma cell lines. Neuropharmacology 36, 811–821 (1997).
Dhani, S., Zhao, Y. & Zhivotovsky, B. A long way to go: caspase inhibitors in clinical use. Cell Death Dis. 12, 949 (2021).
Gary, R. K. & Jensen, D. A. The p53 inhibitor pifithrin-alpha forms a sparingly soluble derivative via intramolecular cyclization under physiological conditions. Mol. Pharm. 2, 462–474 (2005).
Chaitanya, G. V., Alexander, J. S. & Babu, P. P. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal 8, 31 (2010).
McGinnis, K. M., Gnegy, M. E., Park, Y. H., Mukerjee, N. & Wang, K. K. W. Procaspase-3 and poly(ADP)ribose polymerase (PARP) are calpain substrates. Biochem. Biophys. Res. Commun. 263, 94–99 (1999).
Gobeil, S., Boucher, C. C., Nadeau, D. & Poirier, G. G. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 8, 588–594 (2001).
Huang, Y. & Wang, K. K. The calpain family and human disease. Trends Mol. Med 7, 355–362 (2001).
Baki, A., Tompa, P., Alexa, A., Molnár, O. & Friedrich, P. Autolysis parallels activation of mu-calpain. Biochem. J. 318, 897–901 (1996).
Croall, D. E. Proteolytic modification of calcium-dependent protease 1 in erythrocytes treated with ionomycin and calcium. Biochemistry 28, 6882–6888 (1989).
Wang, K. K. Calpain and caspase: can you tell the difference?. Trends Neurosci. 23, 20–26 (2000).
Görlach, A., Bertram, K., Hudecova, S. & Krizanova, O. Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271 (2015).
Festjens, N., Vanden Berghe, T. & Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochimica. Biophysica. Acta (BBA) - Bioenerg. 1757, 1371–1387 (2006).
Yan, Y., Wei, C., Zhang, W., Cheng, H. & Liu, J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol. Sin. 27, 821–826 (2006).
Fatokun, A. A., Dawson, V. L. & Dawson, T. M. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016 (2014).
Baxter, P., Chen, Y., Xu, Y. & Swanson, R. A. mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl. Stroke Res. 5, 136–144 (2014).
Bentle, M. S., Reinicke, K. E., Bey, E. A., Spitz, D. R. & Boothman, D. A. calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and dna repair. J. Biol. Chem. 281, 33684–33696 (2006).
Murata, M. M. et al. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 30, 2584–2597 (2019).
Wang, Y. et al. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J. Neurochem 110, 687–696 (2009).
Leist, M., Single, B., Castoldi, A. F., Kühnle, S. & Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185, 1481–1486 (1997).
Ferrari, D., Stepczynska, A., Los, M., Wesselborg, S. & Schulze-Osthoff, K. Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during CD95- and anticancer drug–induced apoptosis. J. Exp. Med. 188, 979–984 (1998).
Eguchi, Y., Shimizu, S. & Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 57, 1835–1840 (1997).
Zamaraeva, M. V. et al. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ. 12, 1390–1397 (2005).
Chappidi, N. et al. PARP1-DNA co-condensation drives DNA repair site assembly to prevent disjunction of broken DNA ends. Cell 187, 945–961.e18 (2024).
Zandarashvili, L. et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 368, eaax6367 (2020).
Chan, F. K.-M., Moriwaki, K. & De Rosa, M. J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 979, 65–70 (2013).
Sidell, N., Altman, A., Haussler, M. R. & Seeger, R. C. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Exp. Cell Res. 148, 21–30 (1983).
Reynolds, C. P., Matthay, K. K., Villablanca, J. G. & Maurer, B. J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 197, 185–192 (2003).
Vitale, I., Manic, G., De Maria, R., Kroemer, G. & Galluzzi, L. DNA damage in stem cells. Mol. Cell 66, 306–319 (2017).
Sherman, M. H., Bassing, C. H. & Teitell, M. A. Regulation of cell differentiation by the DNA damage response. Trends Cell Biol. 21, 312–319 (2011).
Sjakste, N. & Riekstiņa, U. DNA damage and repair in differentiation of stem cells and cells of connective cell lineages: a trigger or a complication?. Eur. J. Histochem. 65, 3236 (2021).
Walton, J. D. et al. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 6, 838–845 (2004).
Kamijo, T. Role of stemness-related molecules in neuroblastoma. Pediatr. Res. 71, 511–515 (2012).
Zimmerman, M. W. et al. Retinoic acid rewires the adrenergic core regulatory circuitry of childhood neuroblastoma. Sci. Adv. 7, eabe0834 (2021).
Parker, A. L., Turner, N., McCarroll, J. A. & Kavallaris, M. βIII-Tubulin alters glucose metabolism and stress response signaling to promote cell survival and proliferation in glucose-starved non-small cell lung cancer cells. CARCIN 37, 787–798 (2016).
Middleton, M. R. et al. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 125, 510–519 (2021).
Burris, H. A. et al. A phase I study of ATR inhibitor gartisertib (M4344) as a single agent and in combination with carboplatin in patients with advanced solid tumours. Br. J. Cancer 130, 1131–1140 (2024).
Qiu, Z., Oleinick, N. L. & Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 126, 450–464 (2018).
Russell, M. R. et al. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res 73, 776–784 (2013).
Guzi, T. J. et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 10, 591–602 (2011).
Morton, A. R. et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell 179, 1330–1341.e13 (2019).
Bellini, A. et al. SIOPEN) High-risk neuroblastoma trial (HR-NBL1). J. Clin. Oncol. 39, 3377–3390 (2021).
Wang, G., Zhao, N., Berkhout, B. & Das, A. T. CRISPR-Cas9 Can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 24, 522–526 (2016).
Wang, Z. et al. CRISPR/Cas9-Derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 15, 481–489 (2016).
Yoder, K. E. & Bundschuh, R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci. Rep. 6, 29530 (2016).
Lombard, D. B. et al. DNA repair, genome stability, and aging. Cell 120, 497–512 (2005).
Shanbhag, N. M. et al. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 7, 77 (2019).
Lattanzi, A. et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol. Ther. 27, 137–150 (2019).
Laurenti, E. et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 3, 611–624 (2008).
Fessler, J. et al. CytoCellDB: a comprehensive resource for exploring extrachromosomal DNA in cancer cell lines. NAR Cancer 6, zcae035 (2024).
Camps, J. et al. Integrative genomics reveals mechanisms of copy number alterations responsible for transcriptional deregulation in colorectal cancer. Genes Chromosomes Cancer 48, 1002–1017 (2009).
Kim, H. et al. Mapping extrachromosomal DNA amplifications during cancer progression. Nat. Genet 56, 2447–2454 (2024).
Park, S. H., Cao, M. & Bao, G. Detection and quantification of unintended large on-target gene modifications due to CRISPR/Cas9 editing. Curr. Opin. Biomed. Eng. 28, 100478 (2023).
Yakkala, P. A., Penumallu, N. R., Shafi, S. & Kamal, A. Prospects of topoisomerase inhibitors as promising anti-cancer Agents. Pharm. (Basel) 16, 1456 (2023).
Smith, G., Alholm, Z., Coleman, R. L. & Monk, B. J. DNA Damage repair inhibitors-combination therapies. Cancer J. 27, 501–505 (2021).
Grunblatt, E. et al. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev. 34, 1210–1226 (2020).
Casinelli, G. et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2, 16082 (2016).
DepMap, B. DepMap 23Q4 Public. 21888030738 Bytes. https://depmap.org/portal/data_page/?tab=allData&releasename=DepMap+Public+23Q4&filename=CRISPRGeneEffect.csv (2023).
Maréchal, A. & Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5, a012716 (2013).
Truong, B. et al. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 116, 21150–21159 (2019).
Holcik, M. & Liebhaber, S. A. Four highly stable eukaryotic mRNAs assemble 3’ untranslated region RNA-protein complexes sharing cis and trans components. Proc. Natl. Acad. Sci. USA 94, 2410–2414 (1997).
Chen, R. et al. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 41, 262–272 (2023).
Karikó, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Wilson, M. H., Coates, C. J. & George, A. L. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 15, 139–145 (2007).
Henricksen, L. A., Umbricht, C. B. & Wold, M. S. Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 269, 11121–11132 (1994).
Liu, Z. et al. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 7, 2193 (2017).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Acknowledgements
We thank members of the Shohet and Wolfe laboratories and colleagues within the Molecular, Cell and Cancer Biology Department for helpful suggestions and discussions. We thank Michael Brehm, his laboratory and the UMCMS Humanized Mouse Core for providing CD34+ HSPCs for the studies. We are grateful to UMass Chan’s BRIDGE Innovation & Business Development Fund and NIH R01CA275945 (Wen Xue – UMass Chan) for supporting this research.
Author information
Authors and Affiliations
Contributions
M.B.H. and S.A.W. conceptualized the project, M.B.H., J.M.S. and S.A.W. designed the experiments, M.B.H. conducted the experiments and M.B.H. and S.A.W. interpreted the data and wrote the manuscript with input from J.M.S.
Corresponding author
Ethics declarations
Competing interests
S.A.W. is a consultant for Editas Medicine. M.B.H. and S.A.W. have submitted a patent application to the US patent office pertaining to aspects of this work (application number PCT/US2025/019912). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hanlon, M.B., Shohet, J.M. & Wolfe, S.A. Selective targeting of genome amplifications and repeat elements by CRISPR-Cas9 nickases to promote cancer cell death. Nat Commun 16, 5126 (2025). https://doi.org/10.1038/s41467-025-60160-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60160-2
