
Abstract
N-trifluoromethyl compounds, featuring a CF3 group directly attached to nitrogen, are valuable in medicinal chemistry. Despite substantial advances in their synthesis over the past decade, the efficient preparation of inherently unstable N-CF3 secondary amines remains a challenge in synthetic chemistry. Herein, we present a mild and practical method for synthesizing these compounds via oxidative fluorination of isocyanides using iodine as the oxidant, silver fluoride as the fluorinating reagent, and tert-butyldimethylsilane as the proton precursor. This approach benefits from simple workup, as all reagents and by-products can be easily removed through simple filtration and evaporation. This protocol features a broad substrate scope, good functional group tolerance, and good to excellent yields. Additionally, the resulting products can be readily converted into N-CF3 carbamoyl fluorides, valuable building blocks for the synthesis of diverse N-CF3 carbonyl derivatives.
Similar content being viewed by others
Introduction
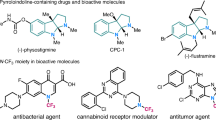
The introduction of a trifluoromethyl (CF3) group into organic molecules is a widely utilized strategy in medicinal chemistry, as its unique physicochemical properties are crucial for modulating drug lipophilicity, membrane permeability, and metabolic stability1,2,3,4,5. Consequently, substantial research efforts have been dedicated to developing trifluoromethylation reactions for the synthesis of various trifluoromethylated compounds6,7,8,9,10. In this context, N-CF3 compounds, featuring the CF3 group directly attached to the nitrogen atom, have garnered increasing interest within the medicinal and synthetic chemistry communities in recent years11,12,13,14. Some representative bioactive molecules containing the N-CF3 moiety are illustrated in Fig. 115,16,17,18,19.

N-CF3-containing compounds show great potential in drug discovery. Norfloxacin analog, an antibacterial agent.
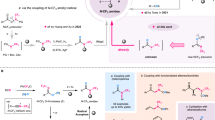
Retro-synthetic analysis reveals four primary strategies for the synthesis of N-CF₃ compounds (Fig. 2A): (a) fluorination of functionalized amine or thioamide derivatives20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38, (b) trifluoromethylation of nitrogen-containing substrates39,40,41,42,43,44,45,46,47,48,49,50,51, (c) trifluoromethylamination using appropriate N-CF3 compounds as N-trifluoromethyl transfer precursors52, and (d) derivatization of specific functionalized N-CF3 building blocks53,54,55,56,57,58,59,60. These approaches have successfully enabled the synthesis of a diverse range of N-CF3 frameworks, including N-CF3 tertiary amines20,21,22,23,24,25,26,27,28,38,39,40, amides29,30,31,32,41,42,53, and N-heterocycles33,34,35,37,43,44,45,46,47,54,55,56,57,58,59. However, the efficient synthesis of N-CF3 secondary amines—a class of N-CF3 compounds that present significant synthetic challenges yet hold promising potential—remains a formidable task in synthetic chemistry. This difficulty arises primarily from their inherent instability61,62. The nitrogen lone pair can readily promote fluoride elimination, even during purification by silica gel column chromatography (Fig. 2B).

A General strategies for the synthesis of N-CF3 compounds. B Established methods for the synthesis of N-CF3 secondary amines. C Conversion of the isocyano group into the N-CF3 moiety. D This work: synthesis of N-CF3 secondary amines via oxidative fluorination of isocyanides. Cbz carbobenzyloxy, Boc t-butyloxycarbonyl, Ts tosyl.
In early 1965, Sheppard made a pioneering contribution by reporting the desulfurization-fluorination of isothiocyanates to produce N-CF3 secondary amines using toxic mercuric fluoride as the fluorinating reagent (Fig. 2B-a)63. More recently, Schoenebeck31,53,54,55,56,57 and others27,32,37 have demonstrated that AgF serves as a highly effective fluorinating reagent for the desulfurization-fluorination of isothiocyanates. In this context, Yi and co-workers disclosed an elegant strategy for the synthesis of N-CF3 secondary amines with a broad substrate scope (Fig. 2B-a). This approach involves the in situ generation of a difluoromethyl imine intermediate via the desulfurization-fluorination of isothiocyanate with AgF, followed by an addition reaction with hydrofluoride, which is generated by the oxidation-reduction of triethylsilane with AgF64. Furthermore, the groups of Selander65 and Xu52 independently developed methodologies for the hydrogenation of prefunctionalized N-CF3 compounds, specifically (CF3)N-OH and (CF3)N-Cbz, using palladium on carbon as a catalyst under neutral conditions (Fig. 2B-b). However, these methods require the preparation of prefunctionalized N-CF3 compounds prior to their use, presenting a limitation for broader synthetic applications. In 2007, Umemoto and co-workers disclosed an alternative approach for the N-trifluoromethylation of primary amines using an electrophilic trifluoromethyl reagent (Fig. 2B-c)66. Although seemingly straightforward and ideal, this method’s reliance on a thermally unstable CF3 oxonium salt as the trifluoromethyl reagent limited its applicability and made the isolation of the desired N-CF3 secondary amines challenging, thereby reducing its utility in practical synthesis. While these examples represent significant advancements, the development of mild and practical synthetic methodologies using readily available materials to reach these intriguing N-CF3 secondary amines is still highly desirable.
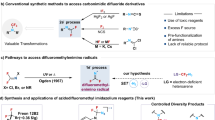
Isocyanides, featuring a unique isocyano group that resonates between zwitterionic triple bond and neutral iminocarbene structures, are valuable and versatile building blocks in synthetic chemistry67,68,69,70,71,72,73. Their distinctive structure enables participation in a wide range of transformations, including multicomponent reactions74,75,76,77,78, insertion reactions79,80,81,82,83,84, and radical-involved processes85,86,87,88. Previously, Ruppert reported the transformation of isocyanides to N-CF3 secondary amines; however, the use of deleterious fluorine gas and anhydrous hydrofluoride and the requirement for very low temperatures severely limited the broad application of this methodology (Fig. 2C-a)89. Recently, we upgraded this protocol by using N-bromosuccinimide (NBS) as oxidant and commercially available Et3N·3HF as HF source90. The reaction of the isocyano group with NBS was believed to generate an isocyanide dihalide, which then undergo facile substitution and addition reactions to form a N-CF3 funcitionality91,92. The labile N-CF3 moiety was intramolecularly captured by the in-situ-formed iminium group by using tryptamine-derived isocyanides as substrates (Fig. 2C-b). When extending this protocol to general aliphatic or aromatic isocyanides to synthesize N-CF3 secondary amines, we observed low efficiency (only a 25% 19F NMR yield for compound 2ae, Fig. 2C-c). Moreover, isolating the pure product proved challenging, as byproducts from NBS and Et3N·3HF complicated the purification process. To develop a general and practical protocol, we identified several essential requirements: (1) the reaction should proceed with high efficiency and cleanliness; (2) all reagents and solvents must be easily removable from the reaction mixture; and (3) importantly, all generated byproducts must also be easily removable. Herein, we disclose our successful synthesis of N-CF3 secondary amines via oxidative fluorination of readily accessible isocyanides using iodine as the oxidant, silver fluoride as the fluorinating reagent, and tert-butyldimethylsilane as the proton precursor (Fig. 2D). The key to the success of this method lies in the fact that all reagents and by-products can be easily removed through simple filtration and evaporation from the reaction mixture. This protocol features mild conditions, a broad substrate scope, good functional group tolerance, and good to excellent yields.
Results
Reaction optimization
Initially, ethyl 4-isocyanobenzoate (1a) was selected as the model substrate to optimize the conditions for the oxidative fluorination reaction (Table 1). The investigation commenced by screening various electrophilic halogenating agents as oxidants in the presence of triethylamine trihydrofluoride, which served as both the fluoride source and the hydrofluoride source. The use of 1,3-diiodo-5,5-dimethylhydantoin (DIH), N-iodosuccinimide (NIS), NBS, and 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) resulted in no detectable product formation (entries 1–4). Next, we evaluated the potential of iodine (I2) as the oxidant. We reasoned the excellent sublimation property of I2 may simplify post-reaction purification. To our delight, the desired N-CF3 secondary amine 2a was successfully formed, albeit in a low 5% yield (entry 5). Upon adding silver fluoride as an additional fluorinating reagent, which is also easy to remove from the reaction mixture through simple filtration, the yield was improved to 16% (entries 5–7). We then investigated alternative hydrofluoride sources. Pyridine poly-hydrofluoride complex showed no reactivity, in contrast to triethylamine trihydrofluoride (entries 7 and 8). Yi and co-workers have reported that hydrofluoride can be mildly produced via the oxidation-reduction of silane with AgF64. In our investigation, we tested three different silane sources. While triethylsilane exhibited high efficiency under Yi’s conditions, it failed to produce the desired product in our approach (entry 9). We reasoned I₂ readily reacts with HSiEt₃ to form HI, thereby consuming the oxidant in an unproductive manner93. Similarly, HSi(EtO)3 showed no reactivity (entry 10). However, tert-butyldimethylsilane (HSitBuMe2) delivered a promising 48% yield, as determined by F NMR (entry 11). Finally, a solvent screening was conducted, revealing that ether solvents were most suitable for this transformation. Using 1,4-dioxane provided an excellent 97% isolated yield (entry 14), while acetonitrile and dichloromethane (DCM) failed to produce any product (entries 12 and 13). And finally, attempts to replace AgF with other fluorine sources such as CsF, KF, tetrabutylammonium fluoride (TBAF), Selectfluor, diethylaminosulfur trifluoride (DAST), and bis(2-methoxyethyl)aminosulfur trifluoride (BAST) all failed, with no or trivial amounts of product being observed.
Substrate scope
With the optimized conditions in hand (Table 1, entry 14), we next sought to investigate the generality of this protocol. As shown in Fig. 3, this transformation proved to be remarkably general and robust across a wide range of aryl and alkyl isocyanide substrates. The scope of aromatic isocyanides was first evaluated. A diverse set of commonly encountered substituents, irrespective of their electronic properties, at different positions on the benzene ring were well tolerated, resulting in the corresponding N-CF3 secondary amines 2a–2k with generally excellent yields. Substituents such as ester (2a), cyano (2b), carbonyl (2c), sulfonyl (2d), nitro (2e), iodide (2f), benzyloxy (2i), and methylthio (2j) were valuable functional handles for potential further derivatization. Notably, substituents at the ortho position did not hamper the reactivity (2k and 2l), and multisubstituted substrates were also compatible with the reaction conditions (2l–2r). Moreover, aromatic substrates bearing polycyclic structures, such as naphthalene (2s and 2t), and fluorene (2u), as well as heteroaromatics, including dibenzofuran (2v), dibenzothiophene (2w), indole (2x), and benzofuran (2y), underwent the reaction smoothly to afford the corresponding N-CF3 secondary amines in good to excellent yields, ranging from 73 to 90%. Importantly, a drug molecule derived from acedapsone (2z)94 was also amenable to this protocol, demonstrating the potential applicability of this method in medicinal chemistry.

Unless otherwise noted, the reactions were performed with 1 (0.5 mmol) under the optimized conditions. Isolated yields are given. a2.0 equiv of HSitBuMe2 was used. bDue to the instability of this product, the yield was determined by 19F NMR analysis using PhCF3 as the internal standard.
We next explored the scope of aliphatic isocyanides. Both phenylpropyl (2aa) and benzyl-substituted isocyanides (2ab and 2ac) worked well under the optimized conditions. A glucose-derived isocyanide successfully afforded the desired product 2ad in 98% yield as a single stereoisomer, highlighting the stereospecificity of this transformation. Additionally, nitrogen-containing heterocycles were readily converted to their respective N-CF3 secondary amine products (2ae–2ag). The method also demonstrated compatibility with isocyanides derived from various α-amino acids (2ah–2ap), further underscoring the versatility of this protocol. Amantadine (1aq), quinoline (1ar) or carbazole-derived (1as) isocyanides were also tested. Unfortunately, the formation of the desired product was confirmed by ¹⁹F NMR, but isolating the pure product using our standard procedure proved challenging. Interestingly, ketone 1-isocyano-1-phenylpropan-2-one (1at) showed no reactivity, with most of the starting materials being recovered. After obtaining the N-CF3 secondary amines, we evaluated their stability under both atmospheric and inert gas conditions. The aromatic products exhibited excellent stability, remaining intact for over a week in air. In contrast, alkyl and benzyl-substituted products showed significant instability, with a storage window of less than 5 h in air, though they remained stable under argon. Interestingly, N-CF3 compounds derived from α-amino acids displayed enhanced stability compared to their alkyl and benzyl analogs.
Synthetic applications
To demonstrate the practicality and robustness of this protocol, the gram-scale synthesis was conducted (Fig. 4A). Under the standard reaction conditions, both aryl (1a) and alkyl isocyanide (1ae) underwent reaction smoothly, providing more than 1 g of the corresponding products without difficulty. Subsequently, we explored further transformations of the N-CF3 products (Fig. 4B). Upon treatment with tosyl bromide and AgOTf, the N-CF3 secondary amine 2b was successfully converted to N-CF3 sulfonamide 3 in 72% yield64. Additionally, the (CF3) N-H bond within the secondary amines were easily transformed into the N-CF3 aminoformyl fluoride motif (4 and 5) under Schoenebeck’s reaction system31. With the N-CF3 aminoformyl fluoride in hand, we performed a series of reactions with diverse nucleophiles. Treatment with N-, O-, and C- nucleophiles resulted in various N-CF3 derivatives, including N-CF3 ureas (6, 7, and 8), carbamate (9), carbamoyl azides (10, 11), and amide (12) in moderate to good yields. The reduction of N-CF3 aminoformyl fluoride with sodium borohydride provided N-CF3 formamide 13 in moderate yield. Notably, N-CF3 carbamoyl azide 11 demonstrated further synthetic utility. Using a photocatalyst and blue light to activate the azide group, we achieved a C-H bond insertion reaction, forming the cyclic N-CF3 urea 14 in a reasonable yield. Alternatively, heating 11 in a microwave reactor with water and tetrahydrofuran (THF) led to the formation of N-CF3 hydrazine 15. As a final example, we successfully synthesized N-CF3 tryptophan carbamate 16 through a three-step reaction starting from isocyanide 1ah. This series of transformations highlights the versatility of the N-CF3 secondary amines, showcasing their capacity as valuable intermediates for synthesizing various N-CF3 compounds with potential applications in medicinal chemistry.

A Gram-scale synthesis of compounds 2a and 2ae. B Further transformations of compounds 2a, 2b, and 2 g. Reaction conditions: (a) TsBr (1.5 equiv), AgOTf (1.5 equiv), Et3N (1.0 equiv), DCM, 25 °C, 15 min; (b) AgF (5.0 equiv), BTC (0.4 equiv), MeCN, 25 °C, 30 min; (c) amines (1.5 equiv), DIPEA (1.5 equiv), DMAP (0.1 equiv), DCM, 25 °C, 24 h; (d) EtOH (1.5 equiv), DIPEA (1.5 equiv), DMAP (0.1 equiv), DCM, 25 °C, 24 h; (e) NaN3 (1.2 equiv), THF, 25 °C, 16 h; (f) PhMgBr (1.2 equiv), PhMe, 25 °C, 15 min; (g) NaBH4 (2.0 equiv), DCM/tAmOH = 1:1, 25 °C, 2 h; (h) [Ir(dtbbpy)(ppy)2]PF6 (3 mol%), DCM, 25 °C, blue light for 24 h; (i) H2O/THF = 1:5, 100 °C, 3 h. BTC bis(trichloromethyl) carbonate, DIPEA N N-diisopropylethylamine, DMAP 4-dimethylaminopyridine, tAmOH tert-amylalcohol, dtbbpy 4,4’-di-tert-butyl-2,2’-bipyridyl, ppy 2-phenylpyridyl.
Stability testing
N-CF3 secondary amine compounds are prone to HF elimination and hydrolysis. To evaluate their stability, seven different products were tested in four media with varying pH values: HCl solution (pH 1.0), water (pH 7.0), phosphate-buffered saline (pH 7.4), and sodium carbonate buffer (pH 10.0) (Fig. 5). As shown in Fig. 5A–D, the aromatic products exhibited higher acid resistance compared to alkyl-substituted products, 70% of 2a, 58% of 2i and 73% of 2k still could be detected when they were dissolved in pH of 1.0 for 6 h. Under the condition of pH 7.0, three compounds were completely decomposed after 6 h, while at pH 7.4 or even 10.0, they were completely decomposed in just 2 h. In contrast, alkyl-substituted products such as 2aa and 2ae exhibited better stability under pH 7.0, 7.4, and 10.0 conditions. At pH 1.0, after 2 h, only 3% of 2aa remained, and 2ae was completely decomposed. In contrast, at pH 7.0, 7.4, and 10.0, 12–30% of both products remained after 6 h. In addition, we found that amino acid-derived product 2ak exhibited good stability under different pH conditions. N-CF3 amide product 12 was very stable at different pH values and did not decompose after 6 h. The experimental findings demonstrate a pronounced divergence in stability profiles: aromatic derivatives exhibit good stability in acidic media, whereas alkyl-substituted compounds show superior stability across neutral to alkaline conditions. Then, we selected compound 2ae to analyze the decomposition by-products under pH 1.0. By analyzing the peaks and integral values in the 19F NMR spectrum within 2 h, we observed that N-COF and HF were the primary decomposition products (Fig. 5F). To rationalize this observation, computational analysis revealed a significant pKa difference (for details, see the Supplementary Information on pages 36 and 37) between the compounds: 2i (14.4) vs. 2ae (20.7). This magnitude alkalinity disparity (ΔpKa = 6.3) dictated their stability profiles—the more basic compound 2ae underwent rapid HF elimination via acid-catalyzed decomposition. Conversely, the lower pKa of 2i rendered it to undergo base-induced deprotonation, followed by β-elimination in alkaline media.

A, B, C and D represent the percentage of remaining N-trifluoromethyl secondary and tertiary amines (relative to 0 h) after standing in a 10 mg/mL water/DMSO (4:1) solution at room temperature with a pH of 1.0, 7.0, 7.4, or 10.0 (the test times were 5 min, 15 min, 30 min, 2 h and 6 h, respectively). E Structures of testing compounds. F Analysis of decomposition by-products of compound 2ae over time in pH = 1 solution within 2 h.
Discussion
In conclusion, we have developed a mild and practical method for the oxidative fluorination of readily accessible isocyanides, providing an efficient approach to intriguing yet synthetic challenging N-CF3 secondary amines. This reaction is applicable to both aryl and alkyl isocyanides, with commercially available iodine, silver fluoride, and tert-butyldimethylsilane serving as the oxidant, fluorinating reagent, and proton precursor, respectively. The key advantage of this protocol lies in its simplicity: all reagents and by-products can be removed via simple filtration and evaporation, enhancing its practicality. This protocol features a broad substrate scope, good functional group tolerance, and good to excellent yields. Importantly, it is also suitable for the late-stage modification of complex molecules and bioactive compounds. Additionally, the resulting N-CF3 secondary amines can be converted into N-CF3 carbamoyl fluorides, which serve as valuable building blocks for synthesizing a variety of N-CF3 carbonyl derivatives.
Methods
General procedure for synthesis of N–CF3 secondary amines
A 10 mL Schlenk tube equipped with a magnetic stir bar was charged with the isocyanide (0.5 mmol), silver fluoride (444.1 mg, 3.5 mmol, 7.0 equiv), iodine (139.7 mg, 0.55 mmol, 1.1 equiv), and 1,4-dioxane (1.5 mL) in the glove box. Then tert-butyldimethylsilane (116 uL, 0.7 mmol, 1.4 equiv or 166 μL, 1.0 mmol, 2.0 equiv) was dissolved in 1,4-dioxane (1.5 mL) and added to the reaction vessel via syringe within one minute. The reaction mixture was stirred at 40 °C for 4 h. After completion, the reaction mixture was diluted with Et2O (5 mL) and stirred for 5 min. The crude mixture was filtered through a Celite pad, eluting with Et2O (10 mL). The solvents were evaporated, and the residue was redissolved in Et2O (5 mL) and filtered through a new Celite pad to remove trace salts and insoluble impurities. The purified product, the corresponding N-trifluoromethyl secondary amine, was obtained after solvent evaporation.
Data availability
The authors declare that the main data supporting the findings of this study, including experimental procedures, characterization of materials and products, general methods, and NMR spectra, are available within the article and its Supplementary Information. All data are available from the corresponding author upon request.
References
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Berger, R., Resnati, G., Metrangolo, P., Weber, E. & Hulliger, J. Organic fluorine compounds: a great opportunity for enhanced materials properties. J. Chem. Soc. Rev. 40, 3496–3508 (2011).
Zhou, Y. et al. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem. Rev. 116, 422–518 (2016).
Johnson, B. M., Shu, Y.-Z., Zhuo, X. & Meanwell, N. A. Metabolic and pharmaceutical aspects of fluorinated compounds. J. Med. Chem. 63, 6315–6386 (2020).
Zhang, C. et al. Biological utility of fluorinated compounds: from materials design to molecular imaging, therapeutics and environmental remediation. Chem. Rev. 122, 167–208 (2022).
Tomashenko, O. A. & Grushin, V. V. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 111, 4475–4521 (2011).
Tlili, A., Toulgoat, F. & Billard, T. Synthetic approaches to trifluoromethoxy-substituted compounds. Angew. Chem. Int. Ed. 55, 11726–11735 (2016).
Li, M., Xue, X.-S. & Cheng, J.-P. Establishing cation and radical donor ability scales of electrophilic F, CF3, and SCF3 transfer reagents. Acc. Chem. Res. 53, 182–197 (2020).
Xiao, H., Zhang, Z., Fang, Y., Zhu, L. & Li, C. Radical trifluoromethylation. Chem. Soc. Rev. 50, 6308–6319 (2021).
Hao, B.-Y., Han, Y.-P., Zhang, Y. & Liang, Y.-M. New synthetic approaches toward OCF3-containing compounds. Org. Biomol. Chem. 21, 4926–4954 (2023).
Milcent, T. & Crousse, B. The main and recent syntheses of the N-CF3 motifLes principales et plus récentes synthèses du motif N-CF3. C. R. Chim. 21, 771–781 (2018).
Crousse, B. Recent advances in the syntheses of N-CF3 scaffolds up to their valorization. Chem. Rec. 23, e202300011 (2023).
Lei, Z., Chang, W., Guo, H., Feng, J. & Zhang, Z. A brief review on the synthesis of the N-CF3 motif in heterocycles. Molecules 28, 3012–3038 (2023).
Yang, Y., Taponard, A., Vantourout, J. C. & Tlili, A. Synthesis of fluorinated amines: a personal account. ACS Org. Inorg. Au. 3, 364–370 (2023).
Schow, S. R. et al. Synthesis and activity of 2,6,9-trisubstituted purines. Bioorg. Med. Chem. Lett. 7, 2697–2702 (1997).
Asahina, Y. et al. Synthesis and antibacterial activity of the 4-quinolone-3-carboxylic acid derivatives having a trifluoromethyl group as a novel N-1 substituent. J. Med. Chem. 48, 3443–3446 (2005).
Sahu, K. K., Ravichandran, V., Mourya, V. K. & Agrawal, R. K. QSAR analysis of caffeoyl naphthalene sulfonamide derivatives as HIV-1 integrase inhibitors. Med. Chem. Res. 15, 418–430 (2007).
Gahman, T. C., Thomas, D. J., Lang, H. & Massari, M. E. Aminoqui nazoline cannabinoid recepetor modulators for treatment of disease. US patent WO2008157500 A1 (2008).
Kubo, O. et al. Discovery of a novel series of GPR119 agonists: Design, synthesis, and biological evaluation of N-(Piperidin-4-yl)-N-(trifluoromethyl)pyrimidin-4-amine derivatives. Bioorg. Med. Chem. Lett. 41, 116208 (2021).
Klauke, E. Preparation and properties of substances with N- or S-perhalogenomethyl groups. Angew. Chem. Int. Ed. 5, 848–848 (1966).
Lal, G. S., Lobach, E. & Evans, A. Fluorination of thiocarbonyl compounds with bis(2-methoxyethyl)aminosulfur trifluoride (deoxo-fluor reagent): a facile synthesis of gem-difluorides. J. Org. Chem. 65, 4830–4832 (2000).
Scattolin, T., Deckers, K. & Schoenebeck, F. Efficient synthesis of trifluoromethyl amines through a formal umpolung strategy from the bench-stable precursor (Me4N)SCF3. Angew. Chem. Int. Ed. 56, 221–224 (2017).
Yu, J., Lin, J.-H. & Xiao, J.-C. Reaction of thiocarbonyl fluoride generated from difluorocarbene with amines. Angew. Chem. Int. Ed. 56, 16669–16673 (2017).
Liang, S. et al. One-pot synthesis of trifluoromethyl amines and perfluoroalkyl amines with CF3SO2Na and RfSO2Na. Chem. Commun. 55, 8536–8539 (2019).
Onida, K., Vanoye, L. & Tlili, A. Direct synthesis of thiocarbamoyl fluorides and trifluoromethylamines through fluorinative desulfurization. Eur. J. Org. Chem. 35, 6106–6109 (2019).
Xu, W. et al. Thiocarbamoyl fluoride synthesis by deconstructive diversification of arylated tetrahydroisoquinolines. J. Org. Chem. 86, 12443–12451 (2021).
Yang, Y., Saffon-Merceron, N., Vantourout, J. C. & Tlili, A. Novel N(SCF3)(CF3)-amines: synthesis, scalability and stability. Chem. Sci. 14, 3893–3898 (2023).
Song, H. et al. Efficient N-trifluoromethylation of amines with carbon disulfide and silver fluoride as reagents. CCS Chem. 7, 381–391 (2024).
Fawcett, F. S., Tullock, C. W. & Coffman, D. D. The chemistry of carbonyl fluoride. I. The fluorination of organic compounds. J. Am. Chem. Soc. 84, 4275–4285 (1962).
Hagooly, Y., Gatenyo, J., Hagooly, A. & Rozen, S. Toward the synthesis of the Rare N-(Trifluoromethyl)amides and the N-(Difluoromethylene)-N-(trifluoromethyl)amines [RN(CF3)CF2R′] Using BrF3. J. Org. Chem. 74, 8578–8582 (2009).
Scattolin, T., Bouayad-Gervais, S. & Schoenebeck, F. Straightforward access to N-trifluoromethyl amides, carbamates, thiocarbamates and ureas. Nature 573, 102–107 (2019).
Liu, J. et al. Synthesis of N-trifluoromethyl amides from carboxylic acids. Chem. 7, 2245–2255 (2021).
Morimoto, K., Makino, K., Yamamoto, S. & Sakata, G. Synthesis of fluoromethyl, difluoromethyl and trifluoromethyl analogues of pyrazosulfuron-ethyl as herbicides. J. Heterocycl. Chem. 27, 807–810 (1990).
Yagupolskii, L. M. et al. N-Trihalomethyl derivatives of benzimidazole, benzotriazole and indazole. J. Fluor. Chem. 106, 181–187 (2000).
Yagupolskii, L. M. N-trifluoromethylazoles. Chem. Heterocycl. Compd. 45, 430–435 (2009).
Zhen, L., Fan, H., Wang, X. & Jiang, L. Synthesis of thiocarbamoyl fluorides and isothiocyanates using CF3SiMe3 and elemental sulfur or AgSCF3 and KBr with amines. Org. Lett. 21, 2106–2110 (2019).
Hong, J. et al. Transition-metal-catalyzed straightforward synthesis of N-trifluoromethyl indoles from 2-alkynylaryl isothiocyanates or 2-alkynylanilines. Org. Chem. Front. 11, 1720–1728 (2024).
Spennacchio, M. et al. A unified flow strategy for the preparation and use of trifluoromethyl-heteroatom anions. Science 385, 991–996 (2024).
Mamone, M., Morvan, E., Milcent, T., Ongeri, S. & Crousse, B. Electrophilic amination of fluoroalkyl groups on azodicarboxylate derivatives. J. Org. Chem. 80, 1964–1971 (2015).
Fleetwood, T. D., Kerr, W. J. & Mason, J. Copper-mediated N-trifluoromethylation of O-benzoylhydroxylamines. Chem. Eur. J. 30, e202303314 (2024).
Zhang, Z. et al. Silver-mediated N-trifluoromethylation of amides and peptides. Chin. J. Chem. 38, 924–928 (2020).
Cao, T., Retailleau, P., Milcent, T. & Crousse, B. Synthesis of N-CF3 hydrazines through radical trifluoromethylation of azodicarboxylates. Chem. Commun. 57, 10351–10354 (2021).
Niedermann, K. et al. Direct electrophilic N-trifluoromethylation of azoles by a hypervalent iodine reagent. Angew. Chem. Int. Ed. 51, 6511–6515 (2012).
Brantley, J. N., Samant, A. V. & Toste, F. D. Isolation and reactivity of trifluoromethyl iodonium salts. ACS Cent. Sci. 2, 341–350 (2016).
Engl, P. S. et al. Exploiting and understanding the selectivity of Ru-n-heterocyclic carbene metathesis catalysts for the ethenolysis of cyclic olefins to α,ω-Dienes. J. Am. Chem. Soc. 139, 13117–13125 (2017).
Zhang, R. Z., Gao, Y. F., Yu, J. X., Xu, C. & Wang, M. N-CF3 Imidoyl chlorides: scalable N-CF3 nitrilium precursors for the construction of N-CF3 compounds. Org. Lett. 26, 2641–2645 (2024).
Zheng, G., Ma, X., Li, J., Zhu, D. & Wang, M. Electrophilic N-trifluoromethylation of N–H ketimines. J. Org. Chem. 80, 8910–8915 (2015).
Xu, C. et al. Synthesis of chloro(phenyl)trifluoromethyliodane and catalyst-free electrophilic trifluoromethylations. Org. Lett. 20, 3933–3937 (2018).
Niedermann, K. et al. A ritter-type reaction: direct electrophilic trifluoromethylation at nitrogen atoms using hypervalent iodine reagents. Angew. Chem. Int. Ed. 50, 1059–1063 (2011).
Blastik, Z. E. et al. Azidoperfluoroalkanes: synthesis and application in copper(I)-catalyzed azide–alkyne cycloaddition. Angew. Chem. Int. Ed. 56, 346–349 (2017).
Teng, F., Cheng, J. & Bolm, C. Silver-mediated N-trifluoromethylation of sulfoximines. Org. Lett. 17, 3166–3169 (2015).
Liu, S., Huang, Y., Wang, J., Qing, F.-L. & Xu, X.-H. General synthesis of N-trifluoromethyl compounds with N-trifluoromethyl hydroxylamine reagents. J. Am. Chem. Soc. 144, 1962–1970 (2022).
Zivkovic, F. G., Nielsen, C. D.-T. & Schoenebeck, F. Access to N−CF3 formamides by reduction of N−CF3 carbamoyl fluorides. Angew. Chem. Int. Ed. 61, e202213829 (2022).
Bouayad-Gervais, S., Scattolin, T. & Schoenebeck, F. N-trifluoromethyl hydrazines, indoles and their derivatives. Angew. Chem. Int. Ed. 59, 11908–11912 (2020).
Nielsen, C. D. T., Zivkovic, F. G. & Schoenebeck, F. Synthesis of N-CF3 alkynamides and derivatives enabled by Ni-catalyzed alkynylation of N-CF3 carbamoyl fluorides. J. Am. Chem. Soc. 143, 13029–13033 (2021).
Bouayad-Gervais, S. et al. Access to cyclic N-trifluoromethyl ureas through photocatalytic activation of carbamoyl azides. J. Am. Chem. Soc. 144, 6100–6106 (2022).
Turksoy, A., Bouayad-Gervais, S. & Schoenebeck, F. N-CF3 Imidazolidin-2-one derivatives via photocatalytic and silver-catalyzed cyclizations. Chem. Eur. J. 28, e202201435 (2022).
Zhang, R. Z. et al. An N-trifluoromethylation/cyclization strategy for accessing diverse N-trifluoromethyl azoles from nitriles and, 3-dipoles. Angew. Chem. Int. Ed. 61, e202110749 (2022).
Gao, Y. F., Zhang, R. Z., Xu, C. & Wang, M. Controllable regioselective [3+2] cyclizations of N-CF3 imidoyl chlorides and Ph3PNNC: divergent synthesis of N-CF3 triazoles. Org. Lett. 26, 5087–5091 (2024).
Zhang, R. Z., Huang, W., Zhang, R. X., Xu, C. & Wang, M. Synthesis of N-CF3 amidines/imidates/thioimidates via N-CF3 nitrilium ions. Org. Lett. 24, 2393–2398 (2022).
Bürger, H., Pawelke, G. Difluoromethanimine, F2C=NH, a novel unstable molecule. J. Chem. Soc. Chem. Commun. 2, 105–106 (1988).
Abe, T., Joo, Y.-H., Tao, G.-H., Twamley, B. & Shreeve, J. M. Disubstituted azidotetrazoles as energetic compounds. Chem. Eur. J. 15, 4102–4110 (2009).
Sheppard, W. A. N-fluoroalkylamines. I. Difluoroazomethines. J. Am. Chem. Soc. 87, 4338–4341 (1965).
Wang, L. et al. General access to N−CF3 secondary amines and their transformation to N−CF3 sulfonamides. Angew. Chem. Int. Ed. 61, e202212115 (2022).
van der Werf, A., Hribersek, M. & Selander, N. N-trifluoromethylation of nitrosoarenes with sodium triflinate. Org. Lett. 19, 2374–2377 (2017).
Umemoto, T., Adachi, K. & Ishihara, S. CF3 oxonium salts, O-(trifluoromethyl)dibenzofuranium salts: in situ synthesis, properties, and application as a real CF3+ species reagent. J. Org. Chem. 72, 6905–6917 (2007).
Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 42, 4867–4880 (2013).
Saya, J. M. et al. Iodospirocyclization of tryptamine-derived isocyanides: formal total synthesis of aspidofractinine. Angew. Chem. Int. Ed. 57, 15232–15236 (2018).
Luo, J., Chen, G.-S., Chen, S.-J. & Liu, Y.-L. Catalyst-free formal [4+1]/[4+2] cyclization cascade sequence of isocyanides with two molecules of acylketene formed in situ from thermal-induced Wolff rearrangement of 2-diazo-1,3-diketones. Sci. Bull. 65, 670–677 (2020).
Cao, W.-B. et al. Hydrogen-bonding-promoted cascade rearrangement involving the enlargement of two rings: efficient access to polycyclic quinoline derivatives. Angew. Chem. Int. Ed. 59, 21425–21430 (2020).
Roose, T. R. et al. Transition metal-catalysed carbene- and nitrene transfer to carbon monoxide and isocyanides. Chem. Soc. Rev. 51, 5842–5877 (2022).
Cheng, S. et al. Copper/chiral phosphoric-acid-catalyzed intramolecular reductive isocyanide-alkene (1 + 2) cycloaddition: enantioselective construction of 2-azabicyclo[3.1.0]hexanes. J. Am. Chem. Soc. 146, 7956–7962 (2024).
Gao, M., Lu, S. & Xu, B. C–H functionalization enabled by multiple isocyanides. Chem. Soc. Rev. 53, 10147–10170 (2024).
Dömling, A. & Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 39, 3168–3210 (2000).
Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 106, 17–89 (2006).
Wang, Q., Wang, D.-X., Wang, M.-X. & Zhu, J. Still unconquered: enantioselective passerini and ugi multicomponent reactions. Acc. Chem. Res. 51, 1290–1300 (2018).
Cheng, S. et al. Palladium-catalyzed four-component cascade imidoyl-carbamoylation of unactivated alkenes. ACS Catal. 12, 837–845 (2022).
Liu, H., Ye, Z.-L., Cai, Z.-J. & Ji, S.-J. A multicomponent reaction of isocyanides, selenium powder and 3-aminooxetanes in pure water: green and efficient synthesis of 1,3-selenazolines. Green. Chem. 25, 4239–4243 (2023).
Qiu, G., Ding, Q. & Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 42, 5257–5269 (2013).
Vlaar, T., Ruijter, E., Maes, B. U. W. & Orru, R. V. A. Palladium-catalyzed migratory insertion of isocyanides: an emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 52, 7084–7097 (2013).
Boyarskiy, V. P., Bokach, N. A., Luzyanin, K. V. & Kukushkin, V. Y. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 115, 2698–2779 (2015).
Hong, X., Tan, Q., Liu, B. & Xu, B. Isocyanide-induced activation of copper sulfate: direct access to functionalized heteroarene sulfonic esters. Angew. Chem. Int. Ed. 56, 3961–3965 (2017).
Collet, J. W., Roose, T. R., Ruijter, E., Maes, B. U. W. & Orru, R. V. A. Base metal catalyzed isocyanide insertions. Angew. Chem. Int. Ed. 59, 540–558 (2020).
Zhang, Y. et al. Oxidative cyclization of allyl compounds and isocyanide: a facile entry to polysubstituted 2-cyanopyrroles. Chin. Chem. Lett. 35, 108836 (2024).
Ryu, I., Sonoda, N. & Curran, D. P. Tandem radical reactions of carbon monoxide, isonitriles, and other reagent equivalents of the geminal radical acceptor/radical precursor synthon. Chem. Rev. 96, 177–194 (1996).
Zhang, B. & Studer, A. Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors. Chem. Soc. Rev. 44, 3505–3521 (2015).
Chen, D. et al. Catalytic metal-enabled story of isocyanides for use as “C1N1” synthons in cyclization: beyond radical chemistry. Org. Chem. Front. 9, 4209–4220 (2022).
Jiao, Y., Shi, X., Ju, L. & Yu, S. Photoredox-catalyzed synthesis of C-benzoselenazolyl/benzothiazolyl glycosides from 2-isocyanoaryl selenoethers/thioethers and glycosyl bromides. Org. Lett. 26, 390–395 (2024).
Ruppert, I. Organylisocyaniddifluoride R=N=CF2 durch direktfluorierung von isocyaniden. Tetrahedron Lett. 21, 4893–4896 (1980).
Wu, J.-Y. et al. N-halosuccinimide enables cascade oxidative trifluorination and halogenative cyclization of tryptamine-derived isocyanides. Nat. Commun. 15, 8917 (2024).
Kühle, E., Anders, B. & Zumach, G. Syntheses of isocyanide dihalides. Angew. Chem. Int. Ed. 6, 649–665 (1967).
Kühle, E., Anders, B., Klauke, E., Tarnow, H. & Zumach, G. Reactions of isocyanide dihalides and their derivatives. Angew. Chem. Int. Ed. 8, 20–34 (1969).
Li, Y. et al. β-boron effect enables regioselective and stereospecific electrophilic addition to alkenes. J. Am. Chem. Soc. 145, 7548 (2023).
George, J. Metabolism and interactions of antileprosy drugs. Biochem. Pharmacol. 177, 113993 (2020).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22171293 to Q.L. and 22371311 to H.W.), the Guangdong Basic and Applied Basic Research Foundation (2024A1515012178 to Q.L.), the Guangdong Provincial Key Laboratory of Construction Foundation (2023B1212060022 to Q.L.), and Noncommunicable Chronic Diseases-National Science and Technology Major Project (2023ZD0507600 to H.W.). We thank Mr. Leibing Wang at Nanjing University of Science and Technology for helpful discussions. We also thank Prof. Ruibo Wu and Mr. Kangwei Xu at Sun Yat-sen University for pKa calculation.
Author information
Authors and Affiliations
Contributions
Q.L. and H.W. designed and supervised the project. J.-L.F. and J.-Y.W. performed experiments. J.-F.S. participated in the synthesis of substrates. L.-L.H. contributed to the initial investigation of the reaction. Q.L. prepared this manuscript and W.H. contributed to discussions. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, JL., Wu, JY., Shi, JF. et al. A mild and practical approach to N-CF3 secondary amines via oxidative fluorination of isocyanides. Nat Commun 16, 4873 (2025). https://doi.org/10.1038/s41467-025-60225-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60225-2
This article is cited by
-
Advances and challenges in the synthesis of N-fluoroalkyl compounds
Science China Chemistry (2026)
-
Bench-stable azidodifluoromethyl imidazolium reagents unlock the synthetic potential of carbonimidic difluorides
Nature Communications (2025)
