
Abstract
The proper function of lysosomes depends on their ability to store and release calcium. While several lysosomal calcium release channels have been described, how lysosomes replenish their calcium stores in placental mammals has not been determined. Using genetic depletion and overexpression techniques combined with electrophysiology and visualization of subcellular ion concentrations and their fluxes across the lysosomal membrane, we show here that TMEM165 imports calcium to the lysosomal lumen and mediates calcium-induced lysosomal proton leakage. Accordingly, TMEM165 accelerates the recovery of cells from cytosolic calcium overload thereby enhancing cell survival while causing a significant acidification of the cytosol. These data indicate that in addition to its previously identified role in the glycosylation of proteins and lipids in the Golgi, a fraction of TMEM165 localizes on the lysosomal limiting membrane, where its putative calcium/proton antiporter activity plays an essential role in the regulation of intracellular ion homeostasis and cell survival.
Similar content being viewed by others


Introduction
Calcium (Ca2+) and proton (H+) homeostasis are of crucial importance for virtually all aspects of cellular life. In mammalian cells, the free Ca2+ concentration in the cytosol [Ca2+]cyt is maintained approximately four orders of magnitude lower than that in the extracellular space and the major intracellular Ca2+ storage organelles1,2. Similarly, the concentration of protons in the cytosol [H+]cyt is up to three orders of magnitude lower than that in the lumen of lysosomes2. Such steep ion concentration gradients across cellular membranes are maintained by numerous pumps, and exchangers that remove excess Ca2+ and H+ from the cytosol to their storage organelles or the extracellular space1,3,4. Due to the effective cytosolic buffering systems for both ions, cells tolerate transient fluctuations in their concentrations. In fact, such fluctuations are essential for a wide range of physiological signaling cascades1,5. Greater and more sustained changes in ion homeostasis are, however, associated with pathological processes, and prolonged increases in either [Ca2+]cyt or [H+]cyt eventually lead to cell death and organ dysfunction6,7.
Late endosomes/lysosomes (hereafter referred to as lysosomes) are membrane-surrounded intracellular organelles originally described as sites of degradation and recycling of cellular organelles and macromolecules8,9. In recent years, lysosomes have emerged as versatile signaling organelles that regulate numerous cellular functions from metabolic pathways and cell division to malignant transformation and cell death10,11,12,13. Most, if not all, lysosomal activities depend on ion channels and transporters that establish electrolyte concentration gradients and membrane potential across the lysosomal limiting membrane2,14. Due to the low pH optimum of most lysosomal hydrolases, the catabolic function of lysosomes depends on the vacuolar-type H+-ATPase (V-ATPase), which continuously pumps protons from the cytosol to the lysosomal lumen15,16. On the other hand, TMEM175, a H+-activated and H+-permeable channel in the lysosomal limiting membrane, protects lysosomes from overacidification by leaking H+ in response to low luminal pH17,18. The significant alkalization of TMEM175-depleted lysosomes observed upon V-ATPase inhibition suggests that TMEM175 is not the only protein capable of releasing H+ from the lysosomal lumen17.
The endoplasmic reticulum (ER), Golgi, and lysosomes constitute major intracellular Ca2+ stores that can release Ca2+ to the cytosol in response to various second messengers activated by intra- and extracellular cues1,19,20. While the sarco/endoplasmic reticulum Ca2+ ATPases (SERCAs) constantly pump free cytosolic Ca2+ to the ER, and SERCA and secretory pathway Ca2+ ATPases pump it to the Golgi, the mechanisms by which mammalian lysosomes replenish their Ca2+ stores remain largely unknown. In plant and yeast cells, the Ca2+ filling of their lysosome-related organelles, vacuoles is mediated by Ca2+/H+ exchangers CAX and VCX1, respectively21,22, and closely related genes are widespread in Metazoans, including non-placental mammals23. Although strong experimental evidence suggests that a similar Ca2+/H+ exchanger activity drives lysosomal Ca2+ filling in humans and other placental mammals, neither orthologs nor known functional counterparts of CAX or VCX1 have been identified in these species23. Given that the V-ATPase constitutively pumps cytosolic H+ into the lysosomal lumen, the resulting steep H+ gradient across the lysosomal membrane could in theory provide the driving force for a putative lysosomal Ca2+/H+ exchanger. Such an exchanger could also explain the TMEM175-independent lysosomal H+ leakage discussed above and the cytosolic acidification observed in response to elevated [Ca2+]cyt observed for example in glutamate-treated rat sensory neurons24, ionomycin-treated rabbit corneal epithelial cells25, rat parotid and pancreatic acinar cells treated with various Ca2+ mobilizing agents26,27, and human cancer cells treated with cationic amphiphilic drugs (CADs) that trigger lysosomal Ca2+ release via the purinergic receptor P2X4 (P2RX4) and subsequent lysosome-dependent cancer cell death28.
In living cells, the lysosomal H+ leakage has hitherto been demonstrated only indirectly by measuring changes in the lysosomal and cytosolic pH. To detect the lysosomal H+ leakage directly, we created a spatially restricted pH sensor that allowed us to measure local pH changes in the perilysosomal area. Using this tool, we confirmed the TMEM175-mediated basal lysosomal H+ leakage in living cells and demonstrated that TMEM175-independent perilysosomal acidification occurs in response to various Ca2+ mobilizing agents. The latter triggered a search for a lysosomal Ca2+/H+ exchanger and the identification of TMEM165, which has been previously acclaimed to be a cation/H+ antiporter that exchanges Ca2+ and manganese (Mn2+) for H+ across the Golgi membrane29, as a mediator of Ca2+-induced lysosomal H+ leakage in mammalian cells. Taken together, the identification of TMEM165 as a lysosomal Ca2+/H+ exchanger provides molecular explanations for the long-sought, lysosomal Ca2+ influx and Ca2+-induced lysosomal H+ efflux pathways.
Results
Lysosomal proton efflux acidifies the perilysosomal area
To visualize lysosomal H+ efflux in living cells, we constructed a cDNA encoding a perilysosomal pH sensor consisting of 39 amino-terminal amino acids of late endosomal/lysosomal adapter, MAPK and MTOR activator 1 (LAMTOR1), which target the protein to the lysosomal surface30, and a fluorescent protein, mKeima, which exhibits dual excitation peaks (452 nm and 585 nm) that respond ratiometrically to pH31 (Fig. 1a, b). In stably transfected HeLa human cervix carcinoma cells, the obtained Lyso-mKeima sensor colocalized mainly with established lysosomal markers, Lysotracker® green (LTG) and monomeric green fluorescence (mGFP)-tagged lysosomal associated membrane protein 1 (LAMP1) but not with the Golgi resident protein RAB6A tagged with enhanced GFP (EGFP)32, ER-trackerTM Green or MitotrackerTM Green (Fig. 1a; Supplementary Fig. 1a–c). To further increase the specificity of the sensor, we performed the measurements at lysosome-specific resolution. Imaging Lyso-mKeima expressing cells with excitation at 585 and 405 nm and emission at 620 nm resulted in a steep decrease in the 585 nm/405 nm fluorescence intensity ratios when pH was increased from 6.0 to 7.5 confirming the suitability of Lyso-mKeima for perilysosomal pH analyses (Fig. 1b; Supplementary Fig. 1d, e). Thus, we combined Lyso-mKeima and either SypHer3s or pHrodoTM-Green-AM to compare perilysosomal and cytosolic pH, respectively. Notably, the average H+ concentration on the lysosomal surface of HeLa cells (pH 7.09 ± 0.02) was 2.69-fold higher than that in the cytosol (pH 7.52 ± 0.03) (Fig. 1c). Substantial acidification of the perilysosomal area was also observed in all the other human cell lines tested, i.e. in MCF7 (2.0-fold), MDA-MB-231 (1.6-fold) and MDA-MB-468 (2.1-fold) breast carcinoma cells and in noncancerous MCF10A breast epithelial cells (1.5-fold) (Fig. 1c; Supplementary Fig. 1f). Similar perilysosomal pH values were obtained in all cell lines with a single excitation wavelength at 585 nm (Supplementary Fig. 1g). Thus, we used this approach in the following studies if not otherwise indicated.

a Schematic presentation of Lyso-mKeima (top) and representative confocal images of live HeLa-Lyso-mKeima cells stained with Lysotracker® Green (bottom). r, Pearson’s colocalization coefficient (n = 20). Scale bars, 10 μm. b The standard curve for fluorescence intensity (FLI) ratios (Ex 585 nm/405 nm; Em 620 nm) of mKeima at pH ranging from 6.0 to 7.5. See Supplementary Fig. 1d for corresponding images. c Cytosolic and perilysosomal pH values of indicated cell lines. d Representative (n = 3) immunoblots of indicated proteins in HeLa cells treated with control (CTL) or TMEM175 siRNAs for 72 h. e Perilysosomal pH analyzed by Lyso-mKeima (left) and lysosomal pH analyzed by LysoSensorTM Yellow/Blue FLI (right) in HeLa cells treated with indicated siRNAs for 72 h. f Relative volumes of acidic compartments in HeLa cells treated with Con A for 1 h, stained with Lysotracker green, and analyzed by flow cytometry (left), and mKeima FLI in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with either DMSO or 10 nM Con A for 1 h (right). See Supplementary Fig. 1h for flow cytometry gating. g Representative images of live HeLa-Lyso-mKeima cells treated with DMSO or 100 μM Arachidonic acid for 15 min (left) and corresponding mKeima FLIs (right). h Relative mKeima FLIs in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with either DMSO or 100 μM Arachidonic acid for the last 20 min. Bars, SD of three independent experiments with 10 (g) 30 (b, c, e-left, f-right, h) or ≥10000 (e-right, f-left) randomly chosen cells analyzed in each sample. *P < 0.05; **P < 0.01; ***P < 0.001 as analyzed by one-way Anova with Tukey’s multiple comparisons (e, f left), two-way Anova with Dunnett’s multiple comparison (c, f right, h), or two-tailed, homoscedastic t-test (g).
Next, we tested whether the low perilysosomal pH was caused by the activity of the recently identified lysosomal H+-activated H+ channel TMEM17517,18. Indeed, the acidification of lysosomes upon siRNA-mediated depletion of TMEM175 was associated with a significant increase in the perilysosomal pH in HeLa-Lyso-mKeima cells (Fig. 1d, e). Furthermore, concanamycin A-mediated inhibition of the V-ATPase resulted in a small but significant increase in the Lyso-mKeima fluorescence in control cells but not in TMEM175-depleted cells (Fig. 1f). Even stronger increases in Lyso-mKeima fluorescence intensities were observed upon treatment of HeLa cells with arachidonic acid (Fig. 1g), which has been shown to induce a TMEM175-dependent decrease in cytosolic pH (pHcyt)17. In contrast to the reported complete inhibition of arachidonic acid-induced acidification of the cytosol in TMEM175 knockout (KO) cells17, incomplete depletion of TMEM175 by siRNAs inhibited the lysosomal H+ efflux induced by arachidonic acid only partially (Fig. 1h).
Taken together, these data introduce Lyso-mKeima as a useful tool to detect perilysosomal pH and lysosomal H+ leakage and demonstrate that TMEM175-mediated basal lysosomal H+ leakage maintains the average pH of the perilysosomal area significantly below the value of the cytosolic pH.
An increase in the free [Ca2+]cyt triggers lysosomal H+ leakage
Commonly used cationic amphiphilic drugs (CADs) including antihistamines, antidepressants and antipsychotics are emerging as potent anticancer drugs12. As basic and membrane permeable molecules, they accumulate in the lysosomal lumen, where they inhibit lysosomal function and induce lysosomal membrane permeabilization specifically in cancer cells33,34. We have previously shown that treating cancer cells with CADs increases lysosomal pH and lowers cytosolic pH hours before any signs of lysosomal membrane damage or cell death occur35. Supporting lysosomal H+ efflux as the mechanism of CAD-induced cytosolic acidification, the CAD antihistamines, ebastine and terfenadine, triggered a rapid and significant decrease in the perilysosomal pH as demonstrated by an increase in the Lyso-mKeima fluorescence intensity (Fig. 2a–c). Importantly, the enhanced Lyso-mKeima fluorescence was observed in the entire cell population and dozens of Lyso-mKeima positive puncta per cell rather than in a few dying cells or individual damaged lysosomes (Fig. 2a). Excluding the possibility that the signal originated from the accumulation of Lyso-mKeima in acidic autolysosomes, knock-out of an essential autophagy gene (ATG7) had no effect on the CAD-induced increase in the Lyso-mKeima fluorescence intensity (Supplementary Fig. 2aleft). Finally, the failure of a similar CAD treatment to alter the fluorescence intensity of mKeima-SKL directed to peroxisomal membranes by a carboxy-terminal peroxisomal targeting sequence, serine-lysine-leucine36, ensured that the early acidification was specific to the perilysosomal area (Supplementary Fig. 2aright, 2b). Taken together, these data strongly support the idea that CADs trigger an early leakage of H+ from lysosomes.

a Representative images of live HeLa-Lyso-mKeima cells treated with DMSO or ebastine (left) and corresponding mKeima FLIs (right). b Kinetics of mKeima FLI in HeLa cells treated with 6 μM terfenadine for 60 min. c Relative mKeima FLIs in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with either DMSO or 6 μM terfenadine for the last 20 or 40 min. d Relative mKeima FLIs in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with 15 μM ebastine (left) or 6 μM terfenadine (right) for the last 20, 40 or 60 min. e Relative Fluo-4-AM (left), mKeima (middle) and SypHer3 (right) FLIs in HeLa cells treated with 6 μM terfenadine, 500 nM thapsigargin, 10 μM ML-SA1 or 1 μM ATP for the last 5 min and analyzed by flow cytometry. See Supplementary Fig. 4g for gating of the cells for flow cytometry analyses. f Relative mKeima FLI in HeLa-Lyso-mKeima cells pretreated for 30 min with 20 μM BAPTA-AM and treated with 500 nM thapsigargin for the last 5 min. g Relative mKeima FLIs in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with 10 μM ML-SA1 (left) or 500 nM thapsigargin (right) for the last 5 min. Bars, SD of three independent experiments with 10 (a), 67 (b), 30 (c, d, f, g), or ≥10000 (e) randomly chosen cells analyzed in each sample. *P < 0.05; **P < 0.01; ***P < 0.001 as analyzed by two-tailed, homoscedastic t-test (a), one-way Anova with Tukey’s multiple comparisons (b) or two-way Anova with Dunnett’s multiple comparisons (c–g).
Contrary to the basal or arachidonic acid-induced lysosomal H+ efflux, CAD-induced perilysosomal acidification was insensitive to the depletion of TMEM175 (Fig. 2c). To enlighten the regulatory mechanism of CAD-induced, lysosomal H+ leakage, we first examined the role of CAD-induced rapid Ca2+ release in this process. Interestingly, siRNA-mediated depletion of P2RX4, a Ca2+ channel responsible for the CAD-induced lysosomal Ca2+ release28, effectively blocked ebastine- and terfenadine-induced perilysosomal acidification in HeLa-Lyso-mKeima cells (Fig. 2d; Supplementary Fig. 2c), while neither the slightly acidic pH on the surface of lysosomes in control siRNA-treated cells (Supplementary Fig. 2d) nor its further acidification by maximal inhibition of the V-ATPase depended on P2RX4 (Supplementary Fig. 2e).
The reliance of CAD-induced lysosomal H+ leakage on the P2RX4 Ca2+ channel inspired us to further explore the link between lysosomal Ca2+ release and perilysosomal acidification. Given that the elevation in the [Ca2+]cyt controls diverse cellular functions and that lysosomal Ca2+ uptake has been reported to depend on acidic lysosomal pH21,37, we investigated the possibility that lysosomes export H+ in exchange for importing excessive cytosolic Ca2+. For this purpose, we treated the Lyso-mKeima-expressing HeLa cells with three stimuli that increase the free [Ca2+]cyt via different means: (i) thapsigargin, which inhibits the ER and Golgi resident SERCA Ca2+ pumps38, (ii) ATP, which stimulates P2 purinoreceptors to generate inositol 1,4,5-triphosphate (IP3), which in turn triggers the release of Ca2+ from the ER through IP3 receptor-regulated channels39, and (iii) ML-SA1, an agonist of the lysosomal Ca2+ release channel, mucolipin TRP cation channel 1 (TRPML1)40. Following rapid and transient (20 – 180 s) increases in the [Ca2+]cyt (Fig. 2e, left; Supplementary Fig. 2f), all three Ca2+ mobilizing compounds induced significant decreases in the perilysosomal pH already 4–5 min after the treatment (Fig. 2e, middle). Supporting lysosomal H+ efflux as the cause of perilysosomal acidification, neither thapsigargin nor terfenadine induced detectable changes in the cytosolic pH in this time frame (Fig. 2e, right). In contrast to the transient [Ca2+]cyt peak induced by thapsigargin, ATP, and ML-SA1, the increase in the [Ca2+]cyt induced by CADs was weaker but longer lasting (Fig. 2e, left)28. Accordingly, it was followed by a more substantial and persistent decrease in perilysosomal pH (Fig. 2e, middle; Supplementary Fig. 2g). Further corroborating Ca2+ as the trigger of lysosomal H+ efflux, the thapsigargin-induced increase in the Lyso-mKeima signal was effectively inhibited by pretreatment of cells with an intracellular Ca2+ chelator, BAPTA-AM (Fig. 2f). Like CADs, thapsigargin and ML-SA1 induced TMEM175 independent lysosomal H+ effluxes (Fig. 2g).
Taken together, the data presented above reveal that an increase in the free [Ca2+]cyt serves as a trigger for lysosomal H+ leakage that is independent of TMEM175.
TMEM165 mediates Ca2+-induced lysosomal proton leakage
In our search for a protein mediating Ca2+ influx into lysosomes, transmembrane protein 165 (TMEM165), a putative divalent cation/H+ exchanger with a well-described role in manganese homeostasis and glycosylation of proteins and lipids in the Golgi network29,41,42,43, attracted our attention. In addition to its presence in the Golgi apparatus, TMEM165 has been detected in the lysosomal compartment of HeLa cells, where its depletion has been reported to increase the volume of the acidic compartment29,44. Supporting the role of TMEM165 in CAD-induced, lysosomal H+ leakage, its partial depletion by two independent siRNAs effectively reduced ebastine- and terfenadine-induced lysosomal H+ leakage and subsequent acidification of the cytosol in HeLa cells (Fig. 3a–c). The strong TMEM165 dependence of CAD-induced lysosomal H+ leakage and cytosolic acidification was also observed in MDA-MB-468 and MCF7 cells, respectively (Supplementary Fig. 3a–d). Likewise, the depletion of TMEM165 in HeLa and MDA-MB-468 cells inhibited lysosomal H+ leakage induced by other Ca2+-mobilizing agents, ML-SA1 and thapsigargin (Fig. 3d; Supplementary Fig. 3e), while TMEM165 was dispensable for the perilysosomal acidification induced by arachidonic acid and concanamycin A (Supplementary Fig. 3f, g). Notably, siRNA-mediated depletion of TMEM165 did not alter the perilysosomal pH in untreated HeLa cells (Fig. 3e), and even complete CRISPR-mediated depletion of TMEM165, which effectively inhibited CAD-induced cytosolic acidification, altered neither the lysosomal nor the cytosolic pH in unstimulated HeLa or MDA-MB-468 cells (Fig. 3f–h; Supplementary Fig. 3h–j). However, the overexpression of either TMEM165-Flag or TMEM165-mCherry in HeLa cells significantly altered the intracellular pH homeostasis by increasing the lysosomal pH and acidifying not only the perilysosomal area but the entire cytosol (Fig. 3e–i). While the manipulation of TMEM165 levels had significant effects on the lysosomal regulation of intracellular pH, neither TMEM165 depletion nor overexpression altered the average diameter of LAMP-2 positive lysosomes (Supplementary Fig. 3k). On the other hand, both TMEM165 depletion and overexpression increased the average number of lysosomes per cross-section. Due to a large intercellular variation, these increases were, however, not statistically significant (Supplementary Fig. 3k).

a Representative (n = 3) immunoblots of indicated proteins in HeLa cells treated with indicated siRNAs for 72 h. b Relative mKeima FLI in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with 15 μM ebastine (left) or 6 μM terfenadine (right) for the last 20, 40 or 60 min. c Cytosolic pH analyzed by pHrodo-AM FLI in HeLa cells treated with indicated siRNAs for 72 h and with 15 μM ebastine or 6 μM terfenadine for 2 h. d Relative mKeima FLI in HeLa-Lyso-mKeima cells treated for 72 h with indicated siRNAs and with 10 μM ML-SA1 (left) or 500 nM thapsigargin (right) for the last 0–300 sec. e Perilysosomal pH analyzed by Lyso-mKeima FLI ratio (585/405 nm) in HeLa cells treated with indicated siRNAs for 72 h (left) or in TMEM165 overexpressed cells (right). f Representative (n = 3) immunoblots of indicated proteins in wild type (WT), TMEM165 overexpressed cell lines (top) and TMEM165-KO cell lines (bottom). g Lysosomal pH analyzed by LysoSensorTM Yellow/Blue FLI in HeLa TMEM165-KO cells (left) and TMEM165 overexpressed cells (right). h Cytosolic pH analyzed by pHrodo-AM FLI in HeLa TMEM165-KO cells (left) and TMEM165 overexpressed cells (right). i Representative (n = 3) immunoblots of indicated proteins in mCherry- and TMEM165-mCherry-trasfected HeLa clones. Bars, SD of three independent experiments with 30 (b–e, h) or ≥15000 (g) randomly chosen cells analyzed in each sample. *P < 0.05; **P < 0.01; ***P < 0.001 as analyzed by one-way Anova with Tukey’s multiple comparison (e-left, h-right), two-way Anova with Dunnett’s multiple comparison (b–d) or two-tailed, homoscedastic t-test (e-right, g, h-left).
These data indicate that TMEM165 is required for the lysosomal H+ leakage in response to elevations in the free [Ca2+]cyt, and that its overexpression disturbs cellular pH homeostasis.
TMEM165 replenishes lysosomal Ca2+ stores
Next, we investigated the effect of TMEM165 on the cellular Ca2+ homeostasis. To determine the effect of TMEM165 on the capacity of lysosomes to store Ca2+, we emptied lysosomal Ca2+ stores by glycyl-L-phenylalanine-beta-naphthylamide (GPN), a lysosomotrope that permeabilizes lysosomal membranes in a cathepsin C-dependent manner45,46, and followed the changes in the free [Ca2+]cyt using Fluo-4-AM as a cytosolic Ca2+ sensor. Suggesting a role for TMEM165 in lysosomal Ca2+ storage, the GPN-induced increase in the free [Ca2+]cyt was significantly reduced and increased in TMEM165-depleted and TMEM165-Flag expressing HeLa cells, respectively (Fig. 4a, left and middle). Similarly, GPN-mediated permeabilization of lysosomes increased the free [Ca2+]cyt significantly less in MDA-MB-468-TMEM165-KO cells than in parental MDA-MB-486 cells (Fig. 4a, right). Similar results were obtained in both cell lines when measuring the free [Ca2+]cyt with a ratiometric Ca2+ indicator Fura-2-acetoxymethyl ester (AM) and correcting the Fura-2-AM FLIs according to the changes in cytosolic pH measured by a cell permeant pH indicator, SNARFTM-1-AM (Fig. 4b; Supplementary Fig. 4a–d). Further corroborating the ability of TMEM165 to fill lysosomal Ca2+ stores, the transient expression of TMEM165-mCherry in HEK293 human kidney epithelial cells increased the release of lysosomal Ca2+ upon ML-SA1-mediated activation of the lysosomal TRPML1 Ca2+ channel while siRNA-mediated depletion of TMEM165 reduced it as determined by the fluorescence intensity of the co-expressed TRPML1-GCaMP6s Ca2+ sensor (Fig. 4c, d; Supplementary Fig. 4e, f). Notably, the TMEM165-R126C mutant, which has been reported to increase the lysosomal localization of TMEM165 without affecting its activity42, increased the capacity of lysosomes to release Ca2+ in response to ML-SA1 even more than the wild-type TMEM165 (Fig. 4c). As expected, both the wild-type and the mutant TMEM165-mCherry co-localized with the lysosomal markers, LysoTracker Deep Red and LAMP1, and their localization on the lysosomal limiting membrane was confirmed by imaging lysosomes enlarged by a pretreatment with a PIKfyve inhibitor apilimod (Fig. 4e). Importantly, the transient expression of neither wild type nor mutated TMEM165 altered the expression levels of the TRPML1-GCaMP6s Ca2+ sensor significantly (Supplementary Fig. 4e).

Relative Fluo-4-AM FLIs (a) and pH-corrected Fura-2-AM FLI ratios (b, see Supplementary Fig. 4a, b for the basis of pH corrections) in indicated HeLa and MDA-MB-468 cells treated with 300 μM GPN for 300 sec. siRNA transfection was performed 72 h before the treatment (a). Bars, SD of three independent experiments with ≥10000 (a) or ≥15000 (b) cells analyzed in each sample. Ca2+ signals in HEK293 cells transiently transfected with TRPML1-GCaMP6s and mCherry (control, n = 19), TMEM165-WT-mCherry (n = 17) or TMEM165-R126C-mCherry (n = 17) (c) or indicated siRNAs (n = 4) from a representative experiment (d). Thin and bold lines represent Ca2+ signal curves in response to 10 μM ML-SA1 in individual cells and the mean values of all cells analyzed, respectively. The bar charts represent mean values for the area under the curve corresponding to relative fluorescence unit (RFU) x seconds. Bars, SEM of 17–19 (c) and 4 (d) independent experiments with 3–10 cells in each sample. e Representative images of live HEK293 cells transfected as in d and stained with CellMaskTM green plasma membrane stain, LysoTrackerTM Deep Red and Hoechst-33342 (left) and same cells treated with 1 µM apilimod for 16 h, fixed, and stained with LAMP1 antibodies and DAPI (right). Asterisks indicate colocalization of LysoTracker/LAMP1 and TMEM165. Scale bars, 10 µm. f Schematic presentation of Lyso-GCaMP8s. g Relative Lyso-GCaMP8s FLIs in indicated HeLa (left and middle) and MDA-MB-468 (right) cells. siRNA transfection was performed 72 h before the analyses (left). Bars, SD of three independent experiments with ≥10000 cells analyzed in each sample. h Lyso-GCaMP8s (top) and Lyso-mKeima FLIs in HeLa-WT (left) and HeLa-TMEM165-KO (right) cells transfected with Lyso-GCaMP8s/Lyso-mKeima 48 h earlier and treated three times with 10 µM ML-SA1 for 5 min. Cells were allowed to recover in fresh medium for 7 min between the treatments. Mean values of 30 cells in a representative (n = 3) experiment are shown. i Mean heights of ML-SA1-induced perilysosomal Ca2+ peaks shown in (h). Bars, SD of three independent experiments with 30 randomly chosen cells analyzed in each sample. *P < 0.05; **P < 0.01; ***P < 0.001 as analyzed by two-way Anova with Dunnet’s (a, b, g, i) or Tukey’s (c, d) multiple comparison.
To study the perilysosomal free [Ca2+] without disturbing the cellular Ca2+ homeostasis by the overexpression of a lysosomal Ca2+ channel, we constructed a novel perilysosomal Ca2+ sensor, Lyso-GCaMP8s, by fusing the lysosomal membrane-targeting amino terminus of LAMTOR1 to the amino terminus of the GCaMP8s Ca2+ sensor (Fig. 4f). Supporting the role of TMEM165 in lysosomal Ca2+ influx, siRNA-mediated depletion of TMEM165 increased and TMEM165-mCherry expression reduced the perilysosomal free [Ca2+] in HeLa cells, and the reintroduction of TMEM165 into MDA-MB-468-TMEM165-KO cells completely reverted the high perilysosomal free [Ca2+] phenotype of the TMEM165-KO cells (Fig. 4g; Supplementary Fig. 4g). To further validate the role of TMEM165 in refilling lysosomal Ca2+ stores, we investigated the ability of repetitive 5 min treatments with 10 µM ML-SA1 to release Ca2+ from the lysosomes in cells expressing Lyso-GCaMP8s. As expected, the treatment of parental HeLa cells with 10 µM ML-SA1 resulted in a rapid increase in the perilysosomal free [Ca2+], which returned to the basal levels by the end of the 5 min treatment (Fig. 4h top, left). After 7 min recovery in the ML-SA1-free medium, the parental cells were fully recovered and responded to the second 5 min ML-SA1 stimulation like to the first one with a rapid and transient increase in the perilysosomal free [Ca2+], after which they again recovered fully before the third treatment that triggered even higher lysosomal Ca2+ release than the previous treatments (Fig. 4h top, left). As expected, ML-SA1-induced peaks in perilysosomal free [Ca2+] were followed by transient drops in perilysosomal pH as analyzed by Lyso-mKeima FLI (Fig. 4h bottom, left). Contrary to the parental HeLa cells, ML-SA1 triggered a relatively small increase in the perilysosomal free [Ca2+] in HeLa-TMEM165-KO cells, and the recovery to the basal [Ca2+]cyt was significantly delayed in the absence of TMEM165 (Fig. 4h top, right). Moreover, the second and third ML-SA1 treatments triggered significantly smaller increases in Lyso-GCaMP8s fluorescence intensity than the first treatment (Fig. 4h top, right, 4i). In line with the function of TMEM165 as a Ca2+/H+ exchanger, ML-SA1-induced perilysosomal Ca2+ peaks were not followed by lysosomal proton leakage in TMEM165-KO cells (Fig. 4h bottom, right).
The data presented above strongly support an important role for TMEM165 in transporting perilysosomal Ca2+ into the lysosomal lumen and thereby maintaining lysosomal Ca2+ stores.
TMEM165 protects cells against cytosolic Ca2+ overload
Prompted by the ability of TMEM165 to import perilysosomal Ca2+ into the lysosomal lumen, we investigated its role in clearing excess cytosolic Ca2+ using cytosolic Ca2+ sensors Fluo-4-AM and Fura-2-AM. While the deficiency of TMEM165 had no effect on the basal free [Ca2+]cyt in unstimulated MDA-MB-468 cells, it delayed significantly the clearance of the excess cytosolic Ca2+ following ML-SA1-induced activation of lysosomal TRPML1 Ca2+ channels, thapsigargin-mediated inhibition of ER Ca2+ pumps and ebastine-induced prolonged activation of lysosomal P2RX4 Ca2+ channels (Fig. 5a–c; Supplementary Fig. 5a, b). TMEM165 was, however, dispensable for the removal of cytosolic Ca2+ after the ATP-induced activation of IP3 receptor-regulated Ca2+ channels in the ER (Supplementary Fig. 5c), possibly due to the effective SERCA-mediated re-uptake of Ca2+ to the ER.

Cytosolic calcium concentrations analyzed by Fluo-4-AM in WT and TMEM165-KO MDA-MB-468 clones treated with 10 μM ML-SA1 (a) or 500 nM thapsigargin (b) for 0–300 sec or with 15 μM ebastine for 1 h (c). Death of indicated MDA-MB-468 cell clones treated with indicated concentrations of thapsigargin (d), ebastine (e, left) or terfenadine (e, right) for 24 h. Cells were stained with propidium iodide (dead cells) and Hoechst-33342 (total cells) and cell death was analyzed by Celigo Imaging Cytometer. f Cytosolic [Ca2+] in WT and TMEM165-KO HeLa clones treated with DMSO or 15 µM ebastine for 1 h and analyzed by Fluo-4-AM. g Death of indicated HeLa clones treated with 0–15 µM ebastine for 24 h and analyzed as in (d). h Cytosolic [Ca2+] in indicated HeLa clones treated with DMSO or 15 µM ebastine for 1 h and analyzed by Fluo-4-AM. i Death of indicated HeLa cell clones treated with 6 µM terfenadine or 15 µM ebastine for 24 h. Cells were stained with SYTOX Green (dead cells) and Hoechst-33342 (total cells) and cell death was analyzed by Celigo Imaging Cytometer. j Galectin 1 puncta (leaky lysosomes) in indicated HeLa clones treated with DMSO or 15 μM ebastine for the last 16 h. Bars, SD of three independent experiments with 30 (a, b), ≥5000 (c, f, h) or ≥15000 (d, e, i) cells analyzed in each sample. *P < 0.05; **P < 0.01; ***P < 0.001 as analyzed by two-way Anova with Dunnett’s multiple comparisons.
Underlining the importance of TMEM165 in regaining cellular Ca2+ homeostasis after Ca2+ stress, the prolonged thapsigargin-induced cytosolic Ca2+ overload was associated with significantly increased cell death in TMEM165-deficient MDA-MB-468 cells (Fig. 5d). Prompted by these results, we investigated the effect of TMEM165 on CAD-induced cancer cell death, which depends on the lysosomal Ca2+ release and activation of Ca2+-dependent cyclic AMP synthesis28. In line with the almost 3-fold increase in CAD-induced free [Ca2+]cyt caused by TMEM165 deficiency (Fig. 5c), TMEM165-KO cells were significantly sensitized to the subsequent cell death induced by either ebastine or terfenadine (Fig. 5e). The crucial role of TMEM165 in protecting cancer cells from CAD-induced cytosolic Ca2+ overload was further confirmed in HeLa cells, where TMEM165 deficiency also enhanced the CAD-induced increase in the free [Ca2+]cyt and the subsequent cell death while the ectopic expression of TMEM165-mCherry effectively inhibited both events (Fig. 5f–i; Supplementary Fig. 5d). Notably, TMEM165-mCherry-mediated protection against CAD-induced cell death was associated with a nearly complete inhibition of CAD-induced formation of galectin 1 puncta, indicative of the inhibition of CAD-induced lysosomal membrane permeabilization (Fig. 5j)47.
Taken together, these data reveal that TMEM165 contributes to the removal of excess Ca2+ from the cytosol and protects cells against cytosolic Ca2+ overload and subsequent lysosomal membrane permeabilization and cell death.
A fraction of TMEM165 localizes to the lysosomal limiting membrane
TMEM165 was originally characterized as a Golgi resident protein, whose mutations cause congenital disorders of glycosylation41. In line with the presence of a tyrosine-based, putative lysosomal targeting signal 124YNRL127 in the human TMEM165 protein, visualization of GFP- or RFP-tagged TMEM165 has revealed that TMEM165 traffics to lysosomes via the plasma membrane44. To ensure that the TMEM165-mCherry used in this study also localized to lysosomal membranes, we analyzed its localization in HeLa and MDA-MBA-468 cells. In HeLa cells, the overexpressed TMEM165-mCherry formed strongly fluorescent punctuate structures colocalizing with the lysosomal markers Lyso-GFP (r = 0.93 ± 0.05) and LysotrackerTM Green (r = 0.83 ± 0.05) and larger, perinuclear sheet-like structures with weaker fluorescence intensity and colocalization with a trans-Golgi marker EGFP-RAB6 (r = 0.42 ± 0.08) (Fig. 6a; Supplementary Fig. 6a). Indicating that the observed lysosomal localization was not caused by the overexpression, TMEM165-mCherry, which was expressed in MDA-MB-468-TMEM165-KO cells at a level similar to that of the endogenous TMEM165 protein (Supplementary Fig. 3h), exhibited a similar distribution pattern with high intensity punctuate structures colocalizing with LysotrackerTM Green (r = 0.85 ± 0.02) and sheet-like weaker intensity structures colocalizing with EGFP-RAB6 (r = 0.57 ± 0.12) (Supplementary Fig. 6b). Importantly, the formation of larger lysosomes upon inhibition of PIKfyve by vacuolin-1 revealed that also the stably expressed TMEM165-mCherry decorated lysosomal limiting membranes (Fig. 6a, bottom)48, strongly suggesting that TMEM165-mCherry is a lysosomal membrane protein rather than a lysosomal cargo. This was further supported by the similar lysosomal localization of TMEM165-mCherry in cells where autophagy was either pharmacologically (3-methyladenine) or genetically (ATG7-KO) inhibited (Fig. 6b).

a Representative (n = 3) confocal images of live HeLa-TMEM165-mCherry cells transfected with a Golgi marker EGFP-RAB6 (top), or stably expressing Lyso-GFP (middle and bottom). Yellow arrows point to vacuolin-enlarged lysosomes with both Lyso-GFP and TMEM165-mCherry on limiting membranes. DNA is visualized by Hoechst staining. White squares mark the area shown in enlarged images. r, Pearson’s colocalization coefficient (n = 20). Scale bar, 10 μm. b Representative (n = 3) confocal images of live HeLa-TMEM165-mCherry-Lyso-EGFP cells with indicated treatments or genetic alteration. r, Pearson’s colocalization coefficient (n = 20). Scale bars, 10 μm. c Representative (n = 3) immunoblots of indicated proteins from purified lysosomes (Lys), light membrane fractions (LMF) and whole cell lysates (WC) of HeLa cells. When indicated, 10 nM concanamycin A (ConA) was added to all buffers used for the extraction. d Representative (n = 3) immunoblots of indicated proteins in WT and TMEM165-KO MDA-MB-468 clones treated with 1 μM MnCl2 for indicated times (left), and relative quantification of fully glycosylated LAMP2 (right). e Cytosolic Ca2+ concentration analyzed by Fluo-4-AM FLI in WT and TMEM165- KO MDA-MB-468 clones treated with DMSO or 15 μM ebastine for the last 1 h of the 48 h treatment with 1 μM MnCl2. Bars, SD of three independent experiments with at least 5000 cells analyzed in each sample. f Cytosolic pH analyzed by pHrodo-AM FLI in WT and TMEM165- KO MDA-MB-468 clones treated with DMSO, 6 μM terfenadine, or 15 μM ebastine for the last 1 h of the 48 h treatment with 1 μM MnCl2. Bars, SD of three independent experiments with 30 randomly chosen cells analyzed in each sample. ***P < 0.001 as analyzed by one-way Anova with Tukey’s multiple comparison (d right) or two-way Anova with Dunnett’s multiple comparison (e, f).
In line with previous reports41,44, immunostaining failed to reveal a significant colocalization of either endogenous or overexpressed TMEM165 with lysosomal markers (Supplementary Fig. 6c, d). To address this discrepancy, we realized that the localization of TMEM165-mCherry in HeLa cells dramatically altered from punctuate and mainly lysosomal structures in live or fixed cells to sheet-like and mainly Golgi-localized (colocalization with N-acetylgalactosaminyltransferase-EGFP (Golgi-EGFP, trans-Golgi) structures following the permeabilization of fixed cells (Fig. 6a; Supplementary Fig. 6a, b, e). Due to this phenomenon, we were not able to demonstrate the lysosomal localization of the endogenous TMEM165 by immunostaining. Instead, we purified iron-dextran (FeDex)-loaded lysosomes from HeLa cells by magnetic capture and analyzed the proteins in the obtained eluates by immunoblotting. A small fraction of the endogenous TMEM165 was detected in the purified LAMP2-rich lysosomes, which did not contain detectable proteins from endosomes (early endosome antigen 1 (EEA1) and cation-independent mannose 6 phosphate receptor (CI-M6PR)), the Golgi (receptor-binding cancer antigen expressed on SiSo cells (RCAS1), Golgi reassembly stacking protein 1 (GORASP1), and CI-M6PR), the ER (protein disulfide isomerase (PDI)) or the plasma membrane (Na+/K+-ATPase) (Fig. 6c). However, the purified lysosomal fraction contained a small amount of mitochondrial import receptor subunit TOM20 homolog (TOM20) possibly due to copurified mitochondria attached to lysosomes or mitophagy (Fig. 6c). Notably, the inhibition of the V-ATPase by concanamycin A during the lysosome purification procedure, which inhibits the degradation of cargo proteins in the lysosomal lumen, did not significantly alter the level of TMEM165 in purified lysosomes, while the putative cargo proteins Na+/K+-ATPase and CI-M6PR became detectable after concanamycin A treatment (Fig. 6c).
Because the TMEM165-mediated maintenance of the Golgi Mn2+ homeostasis is essential for the proper N-glycosylation of several membrane proteins42, we next asked whether the lysosomal phenotype observed in TMEM165 depleted cells could be a result of the defective glycosylation of the normally highly glycosylated lysosomal membrane proteins. Treatment with 1 µM MnCl2 for 48 h completely reversed the hypoglycosylation phenotype in MDA-MB-468-TMEM165-KO cells as demonstrated by the MnCl2-induced reversal of the increased mobility of LAMP2 in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Fig. 6d). However, the MnCl2 supplementation rescued neither the higher cytosolic Ca2+ peak nor the defective cytosolic acidification in CAD treated MDA-MB-468-TMEM165-KO cells (Fig. 6e, f).
These data support the idea that a small fraction of TMEM165 localizes to the lysosomal limiting membrane where it regulates lysosomal ion homeostasis.
TMEM165 mediates H+ and Ca2+ dependent currents
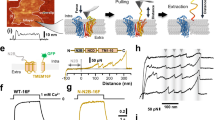
The data presented above strongly suggest that TMEM165 plays a role in Ca2+ influx to the lysosome, and H+ efflux from the lysosome to the cytosol. To test this hypothesis, we performed patch-clamp experiments using HEK293 cells transfected with wild-type TMEM165-mCherry, the R126C mutant of TMEM165-mCherry, or the mCherry vector as a control. We applied a step protocol from −100 mV to +60 mV with 20 mV increments in the whole-cell mode with an asymmetrical pH (pipette 7.5/bath 4.6) and [Ca2+] (pipette 1 µM/bath 10 nM) across the membrane mimicking the conditions in the cytosol (pipette) and the lysosomal lumen (bath) (Fig. 7a). In cells expressing wild-type TMEM165-mCherry, we detected significant inward current activity (most likely the net current of H+ inward and Ca2+ outward) in a physiologically relevant voltage range that was absent in control cells (Fig. 7b, c). Here, TMEM165-R126C mutant currents were considerably weaker than wild-type currents (Fig. 7b, c), while in the lysosomal Ca2+ imaging experiments, the mutant protein had stronger effect on Ca2+ release than the wild-type protein (Fig. 4c). This difference is presumably due to the increased lysosomal and decreased plasma membrane localization of the mutant protein (Figs. 4e and 7d)44. Thus, we focused on wild-type TMEM165-mCherry and increased the bath pH from 4.6 to 7.5 with the result that the measured inward current was almost completely reduced to zero (Fig. 7e, f, left). Additionally, we reduced the Ca2+ concentration in the pipette from 1 µM to 10 nM, resulting in a similar, almost complete loss of the current (Fig. 7e, f, middle). However, when omitting only Ca2+ we noticed a comparably small reduction in the current (Fig. 7e, f, right), leading us to speculate that this may be the result of the presence of 1 mM Mg2+ both in the pipette and the bath. We therefore next omitted both Ca2+ and Mg2+ from the bath, now resulting in a complete loss of the current (Fig. 7g, left). When only Mg2+ was omitted, the current decreased less (Fig. 7g, right) and remained significant in the TMEM165-expressing cells when compared to control cells (Supplementary Fig. 7) or TMEM165-expressing cells with both Mg2+ and Ca2+ omitted (Fig. 7h). These data suggest that the omission of either Ca2+ (in the absence of Mg2+) or H+ significantly affects the observed current.

a Voltage step protocol employed for current recordings. b Examples of macroscopic current recordings conducted for TMEM165-WT-mCherry (green), TMEM165-R126C-mCherry mutant (R126C, red), and mCherry control (black) transiently expressed in HEK293 cells. Measurements were performed using indicated pH and Ca2+ concentrations in the pipette/bath. c Current densities plotted against respective voltages for TMEM165-WT-mCherry (left, green squares, n = 6) and TMEM165-R126C-mCherry (right, red triangles, n = 6) and the control (black circles, n = 12). d Representative (n = 3) live images of HEK293 cells transiently transfected with TMEM165-WT- or TMEM165-R126C-mCherry. Arrows, plasma membrane; scale bars, 10 µm. e Examples of macroscopic current recordings conducted for TMEM165-WT-mCherry using indicated pH and Ca2+ concentrations in the pipette/bath. f Quantitative analyses of current densities (I/Cm) plotted against respective voltages for TMEM165-WT-mCherry at indicated pH and Ca2+ concentrations in the pipette/bath solution with asymmetrical Ca2+ and pH (green squares, n = 6) or asymmetrical Ca2+ and symmetrical pH (blue reverted triangles, n = 4), symmetrical Ca2+ and symmetrical pH (magenta circles, n = 3), and symmetrical Ca2+ and asymmetrical pH (mustard circles, n = 4). g, h Current densities plotted against respective voltages for TMEM165-WT-mCherry at indicated pH and Ca2+ concentrations in the pipette/bath solution with asymmetrical Ca2+ and pH with (green squares, n = 6) and without (mustard circles, n = 9, right) Mg2+ or symmetrical Ca2+ and asymmetrical pH without Mg2+ (cyan circles n = 7, left). Bars, mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001 as analyzed by two-tailed, homoscedastic t-test using GraphPad Prism 10.2.3.
Although PIKfyve inhibitors enlarged TMEM165-mCherry-positive lysosomes to some degree (Figs. 6a and 4e), our efforts to detect TMEM165 currents across the lysosomal limiting membrane were unfortunately hindered by the failure of PIKfyve inhibitors to trigger the formation of large enough TMEM165-positive giant lysosomes for patch clamp, possibly due to TMEM165-induced disturbances in the lysosomal Ca2+ homeostasis.
These data indicate that TMEM165 expression results in the voltage-dependent development of H+- and Ca2+-dependent currents, which are not present in controls.
Discussion
Lysosomal fitness depends on tightly regulated pH and Ca2+ concentration gradients across the limiting membrane2,14. While a low pH is essential for the activity of most lysosomal hydrolases, the ligand-activated Ca2+ release through TRPML1, two pore channels, and P2RX4 governs endolysosomal membrane trafficking and initiates numerous signaling cascades controlling for example lysosomal biogenesis, lysosomal exocytosis, cellular metabolism and lysosomal membrane stability28,49,50,51,52. Despite the crucial role of lysosomal Ca2+ stores in the maintenance of lysosomal and cellular health, the identity of the protein(s) responsible for filling mammalian lysosomes with Ca2+ remains unknown. The data presented above identify TMEM165 not only as a protein contributing to the maintenance of lysosomal Ca2+ stores but also as a mediator of Ca2+-induced lysosomal H+ leakage in mammalian cells. The role of TMEM165 in maintaining lysosomal Ca2+ stores is supported by ample data showing i) a significant reduction in the amount of Ca2+ released to the cytosol upon GPN-mediated lysosomal membrane permeabilization in TMEM165-depleted HeLa and MDA-MB-468 cells; ii) increased capacity of lysosomes to release Ca2+ in TMEM165 overexpressing HeLa and HEK293-TRPLM1 cells upon treatment with GPN and ML-SA1, respectively; iii) reduced capacity of MDA-MB-468-TMEM165-KO cells to refill their Ca2+ stores shortly after a ML-SA1 stimulation; iv) TMEM165-dependent recovery from the cytosolic Ca2+ overload in thapsigargin-, ML-SA1- and ebastine-treated MDA-MB-468 cells and ebastine-treated HeLa cells; and finally, v) the presence of voltage-dependent H+ and Ca2+ currents observed when patch-clamping TMEM165 overexpressing HEK293 cells. It should be noted that while our data demonstrate an essential role for TMEM165 in filling lysosomal Ca2+ stores, TMEM165-KO cells still have a higher [Ca2+] than the cytosol indicating the presence of additional mechanisms.
Previous studies have demonstrated the dependence of lysosomal Ca2+ influx on ER Ca2+ stores, ER-lysosome contact sites and acidic lysosomal pH9,53,54,55. The reported delay in the inhibition of lysosomal Ca2+ influx following the disruption of the lysosomal pH gradient has, however, suggested that the pH dependence of lysosomal Ca2+ influx observed in living cells is caused by the disruption of ER-lysosome contact sites rather than the loss of the lysosomal pH gradient56. Based on our data, TMEM165-mediated Ca2+ influx is dependent on the pH gradient, whose removal abolished the TMEM165-mediated currents almost completely. This is further supported by recently published data from Krishnan and coworkers, who introduced TMEM165 as a proton-activated lysosomal Ca2+ importer57. As mentioned above, lysosomal Ca2+ stores are not completely depleted in TMEM165-KO cells. Thus, other, yet unidentified proteins may drive lysosomal Ca2+ influx in a pH independent manner.
In addition to demonstrating the ability of TMEM165 to import Ca2+ from the cytosol to the lysosome, our data also highlight the ability of TMEM165 to protect mammalian cells against Ca2+ overload. This finding is in line with the reported sensitivity of the yeast strain depleted of the TMEM165 ortholog Gdt1p (gdt1Δ) to high external Ca2+ concentrations, and the ability of the amino-terminally truncated human TMEM165 to partially restore the growth of the gdt1Δ strain under Ca2+ stress29. While the mammalian TMEM165 may play only a moderate role in the clearance of cytosolic Ca2+ in general, its role in removing Ca2+ locally from the perilysosomal area is likely to be of greater importance. This is clearly demonstrated by the substantial sensitization to and inhibition of CAD-induced lysosome-dependent cancer cell death upon TMEM165 depletion and overexpression, respectively. These data emphasize the role of P2RX4-mediated lysosomal Ca2+ release in CAD-induced cancer cell death and are in line with our previous identification of Ca2+-dependent adenylate cyclase 1 as a mediator of CAD-induced lysosomal membrane permeabilization and cell death28,35. Providing further support for a link between the lysosomal P2RX4 Ca2+ channel and TMEM165-mediated lysosomal H+ leakage, a recent report describes a significant, P2RX4-dependent increase in the lysosomal pH in metastatic breast cancer cells58, and the expression of both P2RX4 and TMEM165 has been linked to the metastatic potential of breast cancer cells58,59.
To study the molecular basis of the lysosomal H+ leakage conductance, we created a fluorescent pH sensor, Lyso-mKeima, that detects the local pH on the surface of lysosomes. In the meanwhile, Xu and colleagues identified TMEM175 as the long-sought channel responsible for the H+-activated lysosomal H+ leakage, which maintains lysosomal pH at approximately 4.517. Using cytosolic pH indicators and Lyso-mKeima to measure cytosolic and perilysosomal pH, respectively, we demonstrated a substantial TMEM175-dependent acidification of the perilysosomal area in unstimulated HeLa cells and a further, TMEM175-independent acidification in response to Ca2+-mobilizing agents. The latter was effectively inhibited by the chelation of cytosolic free Ca2+ by BAPTA-AM and by depletion of TMEM165, strongly suggesting that the TMEM165-mediated lysosomal Ca2+ uptake triggers a TMEM165-dependent lysosomal H+ leakage. This hypothesis is strongly supported by our electrophysiological experiments, which demonstrated the ability of TMEM165 to mediate voltage-dependent H+- and Ca2+-dependent currents within the physiological voltage range (−100 mV to −20 mV). The inward current, likely representing the sum of H+ export from the lysosome and Ca2+ import to the lysosome, suggests a putative antiporter mechanism. In line with this, the omission of either Ca2+/Mg2+ or H+ almost completely abolished the inward current. Based on our results and published data, we propose that TMEM165 can transport Mg2+ cations in addition to Ca2+, Ba2+, and Mn2+60. Importantly, the measured current density of TMEM165 as a function of divalent cations varies as follows: Ca2+ > Ba2+ ~ Mg2+, with Ca2+ current density being at least seven times higher than that for Mg2+57. Consequence of this is that even if the Ca2+ concentration is much lower than that of Mg2+, the Ca2+ influx through TMEM165 will still be sufficient to accumulate Ca2+ in the lysosomal lumen.
Recently, Krishnan and coworkers postulated that TMEM165 could be a lysosomal Ca2+ uniporter57. However, they measured currents within a very broad voltage range (−100 mV to 100 mV) using a ramp protocol instead of a step protocol as used in this study, the latter being more suitable for detecting slowly developing currents. Consequently, they measured strong outward currents in the voltage range of +60 mV to +100 mV and only very small, non-significant inward currents, possibly due to the ramp protocol used. It should be noted here that the assumed lysosomal resting membrane potential is around −30 mV9, and recent studies using either newly developed DNA-based voltage sensors or optical methods argue that the values might even reach −114 mV or −56.7 mV, respectively61,62. Importantly, our measurements were performed in this physiological voltage range.
In sum, while Krishnan and coworkers proposed a uniporter mechanism, our electrophysiological and cell biological data suggest an antiporter mechanism for TMEM165. Additional work is, however, needed, to clarify the exact mechanism by which TMEM165 controls lysosomal Ca2+ import and H+ efflux.
Our Ca2+ imaging experiments showed that the depletion of TMEM165 reduced lysosomal Ca2+ and increased perilysosomal Ca2+, while not affecting cytosolic Ca2+. These data suggest that TMEM165 is to some extent constitutively active under resting conditions and that transient cytosolic Ca2+ signals activate TMEM165 to facilitate lysosomal Ca2+ reloading resulting in the observed accumulation of Ca2+ in the lysosomal lumen while the accompanying transient changes in the lysosomal and perilysosomal pH remain undetectable possibly due to rapid adjustments by the V-ATPase activity.
The observed low pH on the lysosomal surface in unstimulated cells, and especially its further, TMEM165-dependent acidification induced by Ca2+ mobilizing agents is highly intriguing and argues against the idea that variations in the cytosolic pH are insignificant, because cellular buffers quickly stabilize them. Whereas the number of protons pumped into lysosomes is likely to be equal to that “leaked” out in cellular level, our data suggest that this is not the case for individual lysosomes, whose pH varies from 4–6. Since TMEM175 is activated when the capacity of a lysosome to store protons is exceeded, it is possible that the acidic lysosomes leak more protons than the V-ATPase pumps in, while lysosomes with higher pH do the opposite. This could be achieved, for example, by the regulation of the V-ATPase activity by the luminal pH. In this case, the average perilysosomal pH would be lower than the cytosolic pH. Alternatively, the low pH at the lysosomal surface within a nanometer scale could reflect the number of protons in transit from TMEM175-mediated export to V-ATPase-mediated import.
Low pH values observed in the perilysosomal area during Ca2+ overload are known to alter the protonation status and function of several proteins, for example phosphofructokinase, whose glycolytic activity regulates AMP-dependent protein kinase (AMPK) on the lysosomal membrane63,64, and the GTP-binding protein RHEB, which controls the activity of mammalian target of rapamycin complex 1 (mTORC1) by recruiting it to the lysosomal membrane65. Thus, it is tempting to speculate that Ca2+-induced acidification of the perilysosomal area could contribute to the Ca2+ overload-induced shift from anabolic to catabolic metabolism by altering the activity of AMPK, mTORC1 or other metabolic regulators residing on the lysosomal membrane12,64,66.
As discussed above, the maintenance of lysosomal H+ and Ca2+ homeostasis is essential for cellular fitness. Accordingly, their disturbances are associated with a wide range of pathologies ranging from degenerative disorders to cancer3,4,12,16,20,67. Thus, the identification of TMEM165 as a regulator of lysosomal H+ efflux and Ca2+ influx opens new avenues for the development of therapeutics for the treatment of these diseases. In particular, TMEM165 antagonists and their combination with CADs or other Ca2+ mobilizing cancer drugs could prove highly effective in the future cancer treatment by increasing and prolonging the cytosolic Ca2+ overload and promoting the subsequent lysosomal membrane permeabilization, while TMEM165 agonists could reverse defective lysosomal Ca2+ stores in lysosomal storage disorders and assist the recovery of neurons from the cytosolic Ca2+ overload and reduced degradative activity of lysosomes in Parkinson’s disease.
Methods
Cell lines
HeLa cervical, human, female carcinoma cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC, 93021013). The TNF-sensitive S1 subclone of human MCF7 female breast cancer cells68 and HeLa-ATG7-KO cells69 have been described previously. MDA-MB-231 (HTB-26) and MDA-MB-468 (HTB-132) human, female breast cancer cell lines, MCF10A (CRL-10317) human female breast epithelial cells, and human kidney HEK-293 (CRL-1573) cells originating from a female fetus were obtained from the American Type Culture Collection (ATCC). TMEM165-KO cell lines were generated using CRISPR/Cas9-mediated gene editing by transfecting single-guide RNA targeting TMEM165 (PXPR001TMEM165) into HeLa and MDA-MB-468 cells, and after 2 weeks clones were picked up and validated by immunoblotting and sequencing. HeLa cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific, 31966021) supplemented with 10% heat-inactivated fetal calf serum (Thermo Fisher Scientific, 10270-106) and penicillin/ streptomycin (Gibco, 15140122). MDA-MB-231 and MDA-MB-468 cells were cultured in DMEM supplemented with 2 mM Glutamine, 1x MEM nonessential amino acids solution (Thermo Fisher Scientific, 11140035), 10% heat-inactivated fetal calf serum and penicillin/ streptomycin. MCF-7 cells were cultured in RPMI 1640 (Thermo Fisher Scientific, 61870010) supplemented with 10% heat-inactivated fetal calf serum and penicillin/ streptomycin. MCF-10A cells were cultured in DMEM/ F12 (Thermo Fisher Scientific, 31330038) supplemented with 5% horse serum (Thermo Fisher Scientific, 16050-122), 20 ng/ ml epidermal growth factor (Sigma-Aldrich, e-4127), 0.5 µg/ ml hydrocortisone (Sigma-Aldrich, 3867), 100 ng/ ml choleratoxin (Sigma-Aldrich, c-8052), 10 µg/ ml insulin (Sigma-Aldrich, I9278) and penicillin/ streptomycin. HEK293 cells were cultured in DMEM supplemented with penicillin/ streptomycin and 10% fetal bovine serum. All cells were cultured at 37 °C in a humidified chamber with 95% air and 5% CO2 and tested regularly for mycoplasma.
Plasmids
Primers used for cloning are listed in Supplementary Table 1. The Lyso-mKeima expression construct was generated by replacing Lyso-GFP in pLV-Lyso-GFP-puro (kindly provided by Dr. Shawn M. Ferguson, Yale University, New Haven, CT) with the first 39 amino acids of LAMTOR1 and the GGSGGS linker (Lyso) using restriction sites EcoRI and BamHI. Then the mKeima was inserted into pLV-Lyso-puro using the restriction sites BamHI and XbaI. The TMEM165-mCherry expression construct was generated by inserting mCherry into pLentiCMVie-IRES-BlastR (Addgene #119863) using the restriction sites XhoI and BamHI. Then, the TMEM165 cDNA was amplified from pCDNA3.1NEGFP-TMEM165 (Genscript) and inserted into pLentiCMVie-mCherry-BlastR via NheI and XhoI sites. The Lyso-GCaMP8s expression construct was generated by inserting Lyso into pLentiCMVie-IRES-BlastR (Addgene #119863) via the EcoRI and EcoRV sites. Then, the GCaMP8s was amplified from pGP-CMV-jGCaMP8s (Addgene #162371) and inserted into pLentiCMVie-lyso-BlastR using the restriction sites EcoRV and BamHI. The TMEM165-flag expression construct was generated by inserting TMEM165-flag into pLentiCMVie-IRES-BlastR via the NheI and XhoI sites. The TRPML1-GCaMP6s expression construct was created by subcloning the human TRPML1 insert from the human TRPML1-EYFP plasmid70 into the pGP backbone harboring GCaMP6s (Addgene #40753) using restriction sites BglII (N-terminal) and SalI (C-terminal). PXPR001TMEM165 was generated by using the gRNA: GCAGC CGGGG CGCCG AUGCG. Golgi-EGFP (#79809), EGFP-RAB6A (#49469) and LAMP1-mGFP (#34831) were purchased from Addgene.
pCDH-EF1-mKeima-SKL was kindly provided by Dr. Katrine Hartfeld (Danish Cancer Institute, Copenhagen, Denmark), and pcDNA3.1-mCherry by Dr. Christian Wahl-Schott (Biomedical Center, LMU, Munich, Germany).
siRNAs
P2X4: CAAGUCGUGCAUUUAUGAUtt/AUCAUAAAUGCACGACUUGtt, GUCCUCUACUGCAUGAAGAtt/UCUUCAUGCAGUAGAGGACtt. TMEM165: GUAUCUGAAUUG GGUGAUAtt/UAUCACCCAAUUCAGAUACaa,CAGGGUCUAUACAUACUAUtt/AUAGUAUGUAUAGACCCUGaa. TMEM175: GCCUUACACGUUUUCGUUAtt/ UAACGAAAACGUGUAAGGCag, CUGCA UGAUG ACCAU CACC aa/GGTG AUGGT CAUCA UGCAGag.
Antibodies
Anti-α-Tubulin (ab40742), anti-β-Actin (ab20272), anti-EEA1 (ab2900), anti-Galectin 1 (ab25138), anti-GFP (Ab290), anti-Lamin B1 (ab194109), anti-M6PR (ab124767), anti-P4HB (ab137110), and anti-sodium potassium ATPase (ab76020) were purchased from Abcam; anti-cofilin (3318S), anti-GAPDH (5174S), anti-RCAS1 (12290), and anti-VDAC (12454S) from Cell Signaling Technology; anti-LAMP2 (L0668-200UL) from Sigma Aldrich; anti-LAMP1 (sc20011) and anti-Tom20 (sc-11415) from Santa Cruz; anti-TMEM175 (PA5-100355), anti-mouse IgG Alexa Fluor 488 (A-11001), anti-rabbit IgG Alexa Fluor 594 (A-21207), and anti-rabbit IgG Alexa Fluor 488 (A-21206) from Thermo Fisher Scientific; anti-TMEM165 (20485-1-AP) from Proteintech Europe; anti-P2RX4 (APR-002) from Labome; and goat anti-rabbit IgG (PI-1000) from Vector Laboratories.
Chemicals and reagents
Ammonium hydroxide solution (28.0-30.0%) (221228), D-(+)-glucose (G7021), Hoechst-33342 (B2261), iron (II) chloride (372870), iron (III) chloride (157740), NMDG (66930), propidium iodide (P4864), terfenadine (T9652), and valinomycin (V0627) were purchased from Sigma Aldrich. BAPTA-AM (B1205), CellLight™ peroxisome GFP (C10604), CellMask™ Green (C37608), ER-Tracker™ Green (E34251), Fluo-4-AM (F14201), Fura-2-AM (F1221), Hanks balanced salt solution (HBSS; 14025050), HEPES (BP310), Live Cell Imaging Solution (A14291DJ), LysoTracker™ deep red (L12492), LysoTracker™ green DND-26 (L7526), MitoTracker™ green (M7514), Nigericin (N1495), pHrodo™ Green AM (P-35373) PluronicTM F127 (P3000MP), PowerLoadTM concentrate (10429993), probenecid (P36400), SNARFTM-1-AM (C1272), and SYTOX™ green, nucleic acid stain (S7020) were obtained from Thermo Fisher Scientific. ML-SA1 (4746) and thapsigarin (1138) were from Tocris, ebastine (15372) from Caymann, arachidonic acid (HY-109590) from MedchemExpress, concanamycin A (sc-202111) from Santa Cruz, dextran 40 BioChemica (A2249) from PanReac AppliChem, hydrochloric acid (VWRC30024.290) from VWR, MES (02195309-CF) from MP Biomedicals, and EGTA (324626) from Merck.
Transfections
Plasmid transfections were performed by using TurboFectin 8.0 (Origene, TF81001), Lipofectamine 3000 transfection agent (Thermo Fisher Scientific, L3000008) or TurbofectTM (Thermo Fisher, R0531) according to the manufacturer’s instructions. siRNA transfections were performed by using Lipofectamine RNAiMax (Thermo Fisher Scientific, 13778075) according to the manufacturer’s instructions.
Western blot
Cell lysates were prepared in Laemmli sample buffer (125 mM Tris, pH 6.7; 20% glycerol; 140 mM SDS) supplemented with complete protease inhibitor cocktail (Roche, 04693159001) and PhosSTOP™ (Roche, 04906837001), and 0.05 M dithiothreitol and then loaded onto a 4%–20% gradient SDS-polyacrylamide gel for electrophoresis. Proteins were transferred onto polyvinylidene difluoride membranes by using a Bio-Rad Trans-Blot Turbo system. The membranes were blocked with phosphate-buffered saline (PBS) containing 5% milk and 0.1% Tween-20 and incubated with the indicated primary antibodies and corresponding peroxidase-conjugated secondary antibodies. The signal was detected with Clarity Western ECL Substrate and Luminescent Image Reader, and quantified by densitometry with Image Studio Lite software.
Immunostaining
Cells were grown on coverslips and fixed in 4% paraformaldehyde in Dulbecco PBS (DPBS; Thermo Fisher Scientific, 14190094) for 20 min were permeabilized with 0.1% saponin in DPBS for 10 min and blocked in 5% goat serum in DPBS for 10 min. Sequential incubation of the cells with the indicated primary antibodies and corresponding Alexa Fluor 488- or Alexa Fluor 594-coupled secondary antibodies was performed to label the protein of interest. Nuclei were labeled either with 5 mg/ml Hoechst-33342 or DAPI in the Prolong Gold Antifade Mounting medium. Unless otherwise indicated, images were acquired with an LSM700 microscope with a Plan-Apochromat 63×/1.40 Oil DIC M27 objective and Zen 2010 software (all equipment and software from Carl Zeiss, Jena, Germany) and analyzed with Image J (Fiji) software.
For live imaging, cells were treated for 10 min with CellMaskTM Green dye (1:1000) at 37 °C, then treated with LysoTrackerTM DeepRed for 30 min at 37 °C, washed three times in PBS, treated with a Hoechst-33342 and then processed for live imaging with an Opera Phenix High Content Confocal Imaging System (Revvity).
Cytosolic Ca2+
For Ca2+ imaging with Fluo-4-AM, cells were stained with 3 mM Fluo-4-AM for 25 min in DMEM (Thermo Fisher, 10567014). After two washes with DPBS, the cells were resuspended in DPS supplemented with 20 mM HEPES. Further incubation with the indicated treatments was performed at 37 °C. Relative quantification and comparison of Fluo-4-AM fluorescence were performed using a BD FACSVerse flow cytometer.
To measure the absolute [Ca2+]cyt, cells were grown in 96-well plates and stained with 3 mM Fluo-4-AM in medium for 25 min. After the indicated treatments, we changed the medium to HBSS and imaged the cells on the ImageXpress Pico platform with excitation at 467 nm and emission at 520 nm. The absolute [Ca2+]cyt was calculated according to
Where F is the desired fluorescence intensity measured by the ImageXpress Pico platform; Fmax is the fluorescence intensity at saturating free Ca2+ concentration ([Ca2+] free); Fmin is the fluorescence intensity at zero [Ca2+]free; To determine Kd,, we stained the cells with 3 mM Fluo-4-AM and incubated them with a series of Ca2+ calibration buffers ([Ca2+]free = 0, 0.0717, 0.1613, and 39.8 μM) supplemented with 10 µM valinomycin, 10 µM nigericin and 3 µM ionomycin for 5 min. Thereafter, the images of the cells were taken with the ImageXpress Pico platform. The obtained fluorescence intensities of Fluo-4-AM were used to plot Log [(F-Fmin)/(Fmax-F)] vs. Log [Ca2+]. The X-intercept from the linear plot is the Log Kd. For the preparation of calcium calibration buffer, the [Ca2+]free was estimated using the equation:
where KdEGTA is the dissociation constant of CaEGTA at a given temperature (https://biotium.com/wp-content/uploads/2013/07/PI-59100.pdf), ionic strength and pH and [CaEGTA/K2EGTA] is the molar ratio of CaEGTA to K2EGTA in the solution.
For Ca2+ imaging with Fura-2-AM, cells were grown in 96-well plates and stained with 5 µM Fura-2-AM and 0.05% Pluronic F-127 for 1 h in HEPES buffered imaging solution (HBIS, 145 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose). Then cells were incubated with 2.5 mM probenecid for 20 min, washed twice with HBIS, and treated as indicated before the fluorescence intensities of Fura-2-AM were measured by SpectraMax iD3 with excitation/emission at 340 nm/510 nm and 380 nm/510 nm.
pH corrected cytosolic Ca2+ measurement
Cells were grown in 96-well plates and stained with 5 µM fura-2-AM and 5 µM SNARFTM-1-AM for 45 min in HEPES buffer (145 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose). Then cells were washed twice with HEPES buffer, and treated as indicated. The fluorescence intensities of fura-2-AM and SNARFTM-1-AM were measured with excitation/emission at 340 nm/510 nm, 380 nm/510 nm and 514 nm/580 nm, 514 nm/640 nm respectively by SpectraMax iD3. Standard curves of SNARFTM-1-AM used to estimate cytosolic pH were created by a similar analysis of cells incubated with a series of pH calibration buffers (pH 6.5, 7, 7.5 and 8) supplemented with 10 µM valinomycin and 10 µM nigericin for 5 min. To establish the standard curve between Kd of fura-2-AM and pH, we stained cells with 5 µM fura-2-AM for 45 min and incubated them with a series of Ca2+ calibration buffers ([Ca2+]free = 0, 0.0717, 0.225, 0.602 and 39.8 μM) at different pH (pH 6.0, 6.5, 7.0 and 7.5) supplemented with 10 µM valinomycin, 10 µM nigericin and 3 µM ionomycin for 5 min. Thereafter, the fluorescence intensities were measured with excitation/emission at 340 nm/510 nm and 380 nm/510 nm. The obtained fluorescence intensities of fura-2-AM in each pH condition were used to plot Log [(F-Fmin)/(Fmax-F)] vs. Log [Ca2+]. The X-intercept from the linear plot is the Log Kd at the given pH.
Perilysosomal Ca2+
HEK293 cells were transfected with 1.5 μg of TRPML1-GCaMP6s or 1 μg of the respective control plasmid or TMEM165-mCherry-carrying plasmid, 4 μL of TurbofectTM, and 200 μl of serum-free DMEM per well in a 6-well plate. Ca2+ imaging was performed 24 h later using an inverted Leica DMi8 live cell microscope. First, transfected cells were washed with DMEM and Ca2+-free buffer was used to carefully rinse the wells before placing the glass coverslips into the imaging chamber. All GCaMP6s experiments were conducted in Ca2+-free buffer comprising 138 mM NaCl, 6 mM KCl, 1 mM MgCl2, 10 mM HEPES, 5.5 mM D-glucose monohydrate and 2 mM EGTA (adjusted to pH = 7.4 with NaOH). Then, 450 μl Ca2+-free buffer was slowly added to the chamber to prevent the cells being washed away. The osmolarity of the Ca2+-free buffer was adjusted to 300 mOsmol/L. GCaMP6s was excited at 470 nm (GFP excitation wavelength) and the emitted fluorescence was captured with a 515 nm longpass filter. mCherry was excited at 568 nm and the emitted fluorescence was captured with a 590 nm filter. Images were obtained every 2.671 sec with a 63x objective. Regions of interest were drawn around each cell coexpressing the GCaMP6s and mCherry constructs. Background areas without cells were selected for manual background subtraction. The fluorescence intensity was calculated using LAS X 5.1.0 software. Baseline values were acquired by averaging fluorescence intensity values from a 30 s recording before the addition of 10 μM ML-SA1. The area under the curve was calculated using GraphPad Prism 9.0.1 software. The mean values per experiment containing 4–12 cells were calculated and used for plotting.
HeLa cells grown in 6-well plates were transfected with Lyso-GCaMP8s by using TurboFectin 8.0. After 48 h, cells were transferred to 96-well plates and 24 h later the medium was changed to a HEPES-buffered imaging solution (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM glucose, 1 mM EGTA and 15 mM HEPES, pH 7.4) and cells were treated 3 ×5 min with 10 μM ML-SA1 with 7 min recovery in fresh imaging solution between the treatments. The fluorescence intensity was measured by the ImageXpress high-content platform with excitation at 467 nm and emission at 520 nm.
Cytosolic pH measurement
Stably transfected HeLa SypHer3S cells were grown on 96-well plates. After the indicated treatments, the medium was changes to Live Cell Imaging Solution and images were taken with the ImageXpress high-content platform. Standard curves used to estimate cytosolic pH were created by a similar analysis of cells incubated with a series of pH calibration buffers (pH 6, 6.5, 7, and 7.5) supplemented with 10 µM valinomycin and 10 µM nigericin for 5 min.
Alternatively, the cells were incubated for 30 min at 37 °C in the medium containing an 1:1.000 dilution of pHrodo™ Green AM Intracellular pH Indicator and an 1:100 dilution of PowerLoad™ concentrate. The cells were subsequently washed with Live Cell Imaging Solution and analyzed on an ImageXpress high-content platform.
Perilysosomal pH measurement
Lyso-mKeima expressing cells were grown in 96-well plate and treated as indicated. The imaging of sub confluent cells was performed in Live Cell Imaging Solution (Thermo Fisher Scientific, A14291DJ) employing the ImageXpress high content microconfocal platform with excitation at 585 nm and emission at 620 nm. For the ratiometric measurements of Lyso-mKeima, cells were additionally imaged with excitation at 405 nm and emission at 620 nm, which results in a pH-independent signal. The subsequent analyses were done by the Molecular Devices MetaXpress software. Individual lysosomes were masked and selected according to the threshold of Lyso-mKeima fluorescence and lysosomal size. Subsequently, the mean intensity of Lyso-mKeima for all pixels in selected areas (lysosomes) was calculated by the software. Background was subtracted prior to calculation of the intensity using the MetaXpress software. Standard curves used to estimate perilysosomal pH were created by a similar analysis of cells incubated with a series of pH calibration buffers (pH 4.5 − 7.5; 140 mM NaCl; 2.5 mM KCl; 1.8 mM CaCl2; 1 mM MgCl2; and 20 mM HEPES) supplemented with 10 µM valinomycin and 10 µM nigericin for 5 min. The calibration was repeated for each experiment by creating standard curves for each cell line individually.
Lysosomal pH measurement
To calculate the absolute lysosomal pH, we plated cells in 96-well plates. The next day, the cells were stained with 1 μM LysoSensor Yellow /Blue DND-160 (Thermo Fisher Scientific, L-7545) for 5 min, washed twice with DPBS. Then we changed the medium to phenol-red-free DMEM supplemented with high glucose and HEPES (Thermo Fisher Scientific, 21063029) and measured the fluorescence of the cells in the SpectraMax iD3 at Ex 329 nm/ Em 440 nm and Ex 380 nm/ Em 540 nm. Standard curves used to estimate lysosomal pH were created by a similar analysis of cells incubated with a series of pH calibration buffers (pH 4, 4.5, 5, and 5.5, 15 mM HEPES, 130 mM KCl, 1 mM MgCl2) supplemented with 10 µM valinomycin and 10 µM nigericin for 5 min.
Volume of acidic compartment
Cells were treated as indicated and stained with 75 nM Lysotracker Green for 5 min in DMEM (Thermo Fisher, 31966021). After two washes with DPBS, cells were resuspended in PBS supplemented with 5% serum. Then the fluorescence was measured by the BD FACSVerse flow cytometer.
Whole-cell electrophysiology
HEK293 cells were seeded on 12 mm2 glass coverslips in standard 24-well tissue culture plates and were allowed to grow for 24-48 h before transfection. Transfection involved the use of 0.6 μg of total DNA, 2 μL of TurbofectTM, and 100 μL of serum-free DMEM per well. Manual patch-clamp recordings of transiently transfected HEK293 cells were carried out in whole-cell configuration. The extracellular/bath solution contained 140 mM NMDG, 10 mM HEPES, 10 mM MES, 5 mM EGTA, 5 mM glucose, and 10 nM free [Ca2+] CaCl2 with or without 1 mM MgCl2. The solution was adjusted to either pH 4.6 or 7.5 with MSA. The intracellular/pipette solution contained 140 mM NMDG, 10 mM HEPES, 5 mM EGTA with or without 1 mM MgCl2, and variable concentrations of CaCl2 to reach either 10 nM or 1 μM free [Ca2+]. The pH was adjusted to 7.5 with MSA. The total calcium concentration at each pH was calculated to maintain the indicated amount of free calcium using https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/CaMgATPEGTA-TS.htm. Recording glass pipettes, with resistances in the range of 5–10 MΩ were pulled and polished. Electrophysiological recordings were performed using an EPC10 patch-clamp amplifier (HEKA, Lambrecht, Germany), controlled by PatchMaster software (HEKA Elektronik). The compensation circuit of the EPC-10 amplifier canceled fast and slow capacitive transients. In all the experiments, a step protocol was used: 400 ms voltage steps from −100 to +60 mV in 20 mV increments were applied every 5 s for a total of 13 steps, and the holding potential was 0 mV. Digitized and filtered (40 kHz and low-pass filter frequency of 2.9 kHz) current amplitudes at the end (last 5%) of each step, recording from −100 to +60 mV were extracted and normalized to the cell size. All the experiments were conducted at room temperature (22–23 °C) and the data were analyzed with GraphPad Prism 10.0.2.
Cell death and lysosomal membrane permeabilization
Cells (5000 cells/well) were plated on 96-well plates, and 24 h later, they were treated as indicated. After indicated times, the cells were incubated with 0.2 µg/ml propidium iodide or 170 nM SYTOX™ Green Nucleic Acid Stain and 2.5 µg/ml Hoechst-33342 for 10 min at 37°C, and cell death was analyzed using a Celigo Imaging Cytometer (Nexcelom Bioscience) according to the manufacturer’s instructions. Leaky lysosomes were detected by Galectin 1 puncta quantification essentially as described previously47,71. microscope with a Plan-Apochromat 63×/1.40 Oil DIC M27 objective and Zen 2010 software.
Purification of iron dextran (FeDex) loaded lysosomes
To prepare the FeDex solution, FeCl2 (0.38 g) and FeCl3 (0.73 g) were dissolved in 2.5 ml H2O each. After the two obtained solutions were mixed, 2.5 ml of 30% (v/v) NH4OH and 25 ml of 5% (v/v) NH4OH were added sequentially while a magnetic stirrer was used to stir the solution for 30 min. After the iron particles had settled, the supernatant was discarded and the iron particles were washed twice with 25 ml H2O and resuspended in 20 ml 0.3 M HCl with a magnetic stirrer for 30 min. Finally, we added 1 g of Dextran 40, stirred the mor for another 30 min and dialyzed the iron dextran solution with H2O for 24 h at 4 °C.
To load lysosomes with FeDex, cells grown on 15 cm petri dishes were incubated in 20 ml medium containing 100 µl of FeDex solution for 18 h. After a 3 h chase in fresh medium, the cells were harvested in PBS and washed three times in SuMa buffer (10 mM HEPES; 0.21 M mannitol; 0.07 M sucrose; pH 7.5). The cells were then resuspended in SuMa4 buffer (SuMa 2, 1 µl Benzonase, protease inhibitor) at a concentration of 30×106 cells/ml and homogenized by a gentleMACS Dissociator machine or syringe with a 25 G needle. The resulting samples were pelleted by 2×10 min centrifugation at 1500 g. The supernatant was collected as the light membrane fraction (LMF). To further enrich lysosomes containing iron dextran particle, the LMF were passed through MS columns (Miltenyi Biotec, 130-042-201) and the columns were washed three times (once with SuMa4 and twice with SuMa2 (SuMa, 0.5 mM DTT, 0.5% fatty acid-free BSA in SuMa buffer)). Finally, the lysosomal fractions were eluted with 600 μl of SuMa2 from the columns, which were detached from the magnetic stand.
Statistical analysis
Unless otherwise indicated, the statistical analyses were performed using GraphPad Prism 9 or GraphPad Prism 10 software. Data are shown as means ± SD unless stated otherwise. The specific statistical tests applied are given in the respective figure legends, where statistical significance is given by *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data needed to evaluate the conclusions in the paper are present in the paper and Supplementary Information. Cell lines, plasmids and data sets generated during the current study will be available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Berridge, M. J., Bootman, M. D. & Roderick, H. L. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 (2003).
Li, P., Gu, M. & Xu, H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem. Sci. 44, 110–124 (2019).
White, K. A., Grillo-Hill, B. K. & Barber, D. L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 130, 663–669 (2017).
Chen, R., Jäättelä, M. & Liu, B. Lysosome as a central hub for rewiring PH homeostasis in tumors. Cancers 12, 2437 (2020).
Flinck, M., Kramer, S. H. & Pedersen, S. F. Roles of pH in control of cell proliferation. Acta Physiol. 223, e13068 (2018).
Orrenius, S., Zhivotovsky, B. & Nicotera, P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565 (2003).
Lagadic-Gossmann, D., Huc, L. & Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death Differ. 11, 953–961 (2004).
De Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R. & Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 60, 604–617 (1955).
Xu, H. & Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80 (2015).
Holland, L. K. K., Nielsen, I. O., Maeda, K. & Jaattela, M. SnapShot: lysosomal functions. Cell 181, 748–748.e741 (2020).
Ballabio, A. & Bonifacino, J. S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118 (2020).
Ellegaard, A.-M., Bach, P. & Jäättelä, M. Targeting cancer lysosomes with good old cationic amphiphilic drugs. Rev. Physiol. Biochem. Pharm. 185, 107–152 (2023).
Stahl-Meyer, J., Stahl-Meyer, K. & Jaattela, M. Control of mitosis, inflammation, and cell motility by limited leakage of lysosomes. Curr. Opin. Cell Biol. 71, 29–37 (2021).
Sterea, A. M., Almasi, S. & El Hiani, Y. The hidden potential of lysosomal ion channels: a new era of oncogenes. Cell Calcium 72, 91–103 (2018).
Maxson, M. E. & Grinstein, S. The vacuolar-type H(+)-ATPase at a glance - more than a proton pump. J. Cell Sci. 127, 4987–4993 (2014).
Stransky, L., Cotter, K. & Forgac, M. The function of V-ATPases in cancer. Physiol. Rev. 96, 1071–1091 (2016).
Hu, M. et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell 185, 2292–2308.e2220 (2022).
Zheng, W. et al. pH regulates potassium conductance and drives a constitutive proton current in human TMEM175. Sci. Adv. 8, eabm1568 (2022).
Dolman, N. J. & Tepikin, A. V. Calcium gradients and the Golgi. Cell Calcium 40, 505–512 (2006).
Lloyd-Evans, E. & Waller-Evans, H. Lysosomal Ca(2+) homeostasis and signaling in health and disease. Cold Spring Harb. Perspect. Biol. 12, a035311 (2020).
Morgan, A. J., Platt, F. M., Lloyd-Evans, E. & Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 439, 349–374 (2011).
Pittman, J. K. Vacuolar Ca(2+) uptake. Cell Calcium 50, 139–146 (2011).
Melchionda, M., Pittman, J. K., Mayor, R. & Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 212, 803–813 (2016).
Hwang, S. M. et al. Intracellular acidification is associated with changes in free cytosolic calcium and inhibition of action potentials in rat trigeminal ganglion. J. Biol. Chem. 286, 1719–1729 (2011).
Grant, R. L. & Acosta, D. Interactions of intracellular pH and intracellular calcium in primary cultures of rabbit corneal epithelial cells. Vitr. Cell Dev. Biol. Anim. 32, 38–45 (1996).
Tanimura, A., Tojyo, Y. & Matsumoto, Y. The cytosolic acidification in rat parotid cells is associated with an increase in cytosolic Ca2+ concentration. Jpn J. Pharm. 61, 357–360 (1993).
Gonzalez, A., Camello, P. J., Pariente, J. A. & Salido, G. M. Free cytosolic calcium levels modify intracellular pH in rat pancreatic acini. Biochem. Biophys. Res. Commun. 230, 652–656 (1997).
Anand, A. et al. Cell death induced by cationic amphiphilic drugs depends on lysosomal Ca(2+) release and cyclic AMP. Mol. Cancer Ther. 18, 1602–1614 (2019).
Demaegd, D. et al. Newly characterized Golgi-localized family of proteins is involved in calcium and pH homeostasis in yeast and human cells. Proc. Natl Acad. Sci. USA 110, 6859–6864 (2013).
Amick, J., Tharkeshwar, A. K., Talaia, G. & Ferguson, S. M. PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J. Cell Biol. 219, e201906076 (2020).
Tantama, M., Hung, Y. P. & Yellen, G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J. Am. Chem. Soc. 133, 10034–10037 (2011).
Tie, H. C. et al. A novel imaging method for quantitative Golgi localization reveals differential intra-Golgi trafficking of secretory cargoes. Mol. Biol. Cell 27, 848–861 (2016).
Ellegaard, A. M. et al. Repurposing cationic amphiphilic antihistamines for cancer treatment. EBioMedicine 9, 130–139 (2016).
Petersen, N. H. et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 24, 379–393 (2013).
Liu, B. et al. Cationic amphiphilic antihistamines inhibit STAT3 via Ca 2+ -dependent lysosomal H+ efflux. Cell Rep. 42, 112137 (2023).
Wolins, N. E. & Donaldson, R. P. Binding of the peroxisomal targeting sequence SKL is specified by a low-affinity site in castor bean glyoxysomal membranes. A domain next to the SKL binds to a high-affinity site. Plant Physiol. 113, 943–949 (1997).
Wu, Y., Huang, P. & Dong, X. P. Lysosomal calcium channels in autophagy and cancer. Cancers 13, 1299 (2021).
Lytton, J., Westlin, M. & Hanley, M. R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 266, 17067–17071 (1991).
Dixon, C. J. et al. Extracellular nucleotides stimulate proliferation in MCF-7 breast cancer cells via P2-purinoceptors. Br. J. Cancer 75, 34–39 (1997).
Shen, D. et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 3, 731 (2012).
Foulquier, F. et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 91, 15–26 (2012).
Potelle, S. et al. Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis. Hum. Mol. Genet. 25, 1489–1500 (2016).
Morelle, W. et al. Galactose supplementation in patients with TMEM165-CDG rescues the glycosylation defects. J. Clin. Endocrinol. Metab. 102, 1375–1386 (2017).
Rosnoblet, C. et al. Impact of disease-causing mutations on TMEM165 subcellular localization, a recently identified protein involved in CDG-II. Hum. Mol. Genet. 22, 2914–2928 (2013).
Haller, T., Dietl, P., Deetjen, P. & Volkl, H. The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+ release. Cell Calcium 19, 157–165 (1996).
Yuan, Y. et al. The lysosomotrope GPN mobilises Ca(2+) from acidic organelles. J. Cell Sci. 134, jcs256578 (2021).
Aits, S. et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11, 1408–1424 (2015).
Sano, O. et al. Vacuolin-1 inhibits autophagy by impairing lysosomal maturation via PIKfyve inhibition. FEBS Lett. 590, 1576–1585 (2016).
Ruas, M. et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr. Biol. 20, 703–709 (2010).
Scotto Rosato, A. et al. TPC2 rescues lysosomal storage in mucolipidosis type IV, Niemann-Pick type C1, and Batten disease. EMBO Mol. Med. 14, e15377 (2022).
Cao, Q. et al. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 209, 879–894 (2015).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Christensen, K. A., Myers, J. T. & Swanson, J. A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 115, 599–607 (2002).
Lopez-Sanjurjo, C. I., Tovey, S. C., Prole, D. L. & Taylor, C. W. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 126, 289–300 (2013).
Garrity, A. G. et al. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife 5, e15887 (2016).
Atakpa, P., Thillaiappan, N. B., Mataragka, S., Prole, D. L. & Taylor, C. W. IP(3) receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca(2+) to lysosomes. Cell Rep. 25, 3180–3193.e3187 (2018).
Zajac, M. et al. A mechanism of lysosomal calcium entry. Sci. Adv. 10, eadk2317 (2024).
Chadet, S. et al. P2x4 receptor promotes mammary cancer progression by sustaining autophagy and associated mesenchymal transition. Oncogene41, 2920–2931 (2022).
Murali, P. et al. Novel role for the Golgi membrane protein TMEM165 in control of migration and invasion for breast carcinoma. Oncotarget 11, 2747–2762 (2020).
Jankauskas, S. S. et al. Insights into molecular and cellular functions of the Golgi calcium/manganese-proton antiporter TMEM165. J. Biol. Chem. 300, 107567 (2024).
Matamala, E., Castillo, C., Vivar, J. P., Rojas, P. A. & Brauchi, S. E. Imaging the electrical activity of organelles in living cells. Commun. Biol. 4, 389 (2021).
Saminathan, A. et al. A DNA-based voltmeter for organelles. Nat. Nanotechnol. 16, 96–103 (2021).
Srivastava, J., Barber, D. L. & Jacobson, M. P. Intracellular pH sensors: design principles and functional significance. Physiology22, 30–39 (2007).
Zhang, C. S. et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 (2017).
Takahara, T., Amemiya, Y., Sugiyama, R., Maki, M. & Shibata, H. Amino acid-dependent control of mTORC1 signaling: a variety of regulatory modes. J. Biomed. Sci. 27, 87 (2020).
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Zaichick, S. V., McGrath, K. M. & Caraveo, G. The role of Ca(2+) signaling in Parkinson’s disease. Dis. Model Mech. 10, 519–535 (2017).
Jaattela, M., Benedict, M., Tewari, M., Shayman, J. A. & Dixit, V. M. Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene 10, 2297–2305 (1995).
Vanzo, R. et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 27, 1134–1153 (2020).
Grimm, C. et al. Small molecule activators of TRPML3. Chem. Biol. 17, 135–148 (2010).
Aits, S., Jäättelä, M. & Nylandsted, J. Methods for the quantification of lysosomal membrane permeabilization: a hallmark of lysosomal cell death. Methods Cell Biol. 126, 261–285 (2015).
Acknowledgements
We thank Shawn M. Ferguson, Katrine Hartfeld and Christian Wahl-Schott for providing valuable reagents, and Dianna Skousborg Larsen, Louise Vanderfox and Tiina Naumanen Dietrich for the technical assistance. This work was supported by the Danish National Research Foundation grant DNRF125 to M.J., the Danish Cancer Society grants R269-A15695 and R352-A20587 to M.J., the Novo Nordisk Foundation grant NNF19OC0054296 to M.J., and DFG GR4315/2-2, DFG GR4315/7-1, SFB/TRR152 P04, and SFB1328 A21 to C.G.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.L. and M.J.; Methodology: B.L., K.M., and M.J.; Investigation: R.C., B.L., D.J., L.K., V.K., B.W., Y.E.K., A.P., and S.P.C.; Visualization: R.C., B.L., D.J., L.K., V.K., B.W., Y.E.K., A.P., and S.P.C.; Supervision: B.L., K.M., C.G., and M.J.; Writing—original draft: B.L. and M.J.; Writing—review & editing: R.C., B.L., D.J., L.K., V.K., K.M., C.G., and M.J.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, R., Liu, B., Jaślan, D. et al. Lysosomal TMEM165 controls cellular ion homeostasis and survival by mediating lysosomal Ca2+ import and H+ efflux. Nat Commun 16, 5209 (2025). https://doi.org/10.1038/s41467-025-60349-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60349-5
