
Abstract
Cell-specific super-enhancers (SEs) and master transcription factors (TFs) dynamically remodel embryonic stem cell (ESC) fate, yet their regulatory interplay remains unclear. By integrating multi-omics data (H3K27ac, Hi-C, scRNA-seq) across ESC states, we identified SEs interacting with master TFs, exemplified by the Klf5-adjacent SE (K5aSE). K5aSE deletion impaired proliferation, differentiation, and Klf5 expression, partially rescued by KLF5 reintroduction. Despite phenotypic similarities between Klf5-KO and K5aSE-KO ESCs, scRNA-seq of embryoid bodies revealed distinct differentiation trajectories, suggesting K5aSE targets beyond Klf5. High-throughput 3D genome screening demonstrated K5aSE activates four distal genes via chromatin looping. CRISPRa-mediated activation of these targets rescued K5aSE-KO phenotypes and uncovered their regulatory roles. Furthermore, CTCF depletion disrupted topologically associated domains (TADs) near K5aSE, suppressing Klf5 and target gene expression, indicating CTCF-mediated TADs sustain K5aSE activity. Our study unveils a 3D genome-dependent mechanism by which SEs govern ESC identity through coordinated TF interaction and multi-gene regulation.
Similar content being viewed by others


Introduction
Precise spatiotemporal expression of genes is essential for mammalian development and cellular processes, while dysregulation of this process is closely linked to developmental abnormalities or diseases1,2,3,4. Gene expression maintenance relies on specific functional genomic elements, including promoters, enhancers, insulators, and transposons, which collectively establish transcriptional programs in a cell-type-specific manner5,6. Among these elements, super-enhancers (SEs) are a class of functional DNA elements with superior activation capacity. Compared with typical enhancers (TEs), SEs exhibit distinct and unique features, such as highly enriched H3K27ac, larger size, a greater number of transcription factor (TF) binding sites and a significant correlation with cell-type-specific transcription factors7,8,9. Recent studies also reveal that SEs play an extremely important role in mammalian development, promoting cellular activities by maintaining expression of cell-type-specific master TFs8,9,10,11,12,13,14,15. Several SEs that significantly regulate expression of pluripotent master TFs have been characterized in embryonic stem cells (ESCs), such as the Nanog proximal SE regulated by enhancer RNA (eRNA)16, a distal SE that maintains Sox2 expression through long-range chromatin interactions17, the Klf4 distal SE regulated by dynamic TF binding18, and CTCF-dependent regulation of the Prdm14 downstream SE19. Although previous studies have predicted hundreds of SEs in ESCs (ESCs) and linked some of them to transcriptional regulation of pluripotency TFs9,12,20,21,22,23, a major knowledge gap remains: few SEs have been functionally validated as direct regulators of ESC identity.
Current studies suggest that SEs in ESCs function primarily by regulating master TFs functioning in pluripotency. Interestingly, genomic regions encoding pluripotency master TFs often have an adjacent SE, with both located within the same topologically associating domain (TAD)8,9. Moreover, these master TFs also bind to their corresponding SEs, suggesting a model of circular regulation between SEs and master TFs9,21,22,23. Notably, a comparable model is reported in tumors24,25,26,27,28,29. Recent studies have revealed that three-dimensional (3D) chromatin structure plays an important role in gene expression as well as in individual development, and have also brought new perspectives on SE regulation30,31. Our previous studies indicate that TEs regulate expression of multiple target genes via long-range chromatin interactions, maintaining cell activity32,33,34,35,36,37. Also, a recent study of corneal margin stem/progenitor cells revealed that a single SE interacts with multiple genes via chromatin loops38. These findings raise a hypothesis that SEs may employ distinct 3D genomic mechanisms to globally orchestrate ESC transcriptional programs. However, the exact mechanism and function of the 3D chromatin structure in relation to the regulation of SE have not been systematically investigated.
Here, we address these gaps through an integrated analysis of SEs in ESCs. We first utilized an integrative multi-omics approach to identify a class of SEs that may coordinate ESC fate through adjacent master TFs. We show that the previously unreported ESC-specific Klf5-adjacent super-enhancer (K5aSE) is essential for ESC proliferation, EB differentiation, and Klf5 expression. However, in K5aSE-KO ESCs, KLF5 overexpression only partially rescued ESC phenotypes and gene expression profiles. Importantly, phenotypes seen in Klf5-deficient cells resembled those seen in K5aSE-KO cells, but expression profiles of genes associated with EB differentiation differed significantly between genotypes, differences validated by scRNA-seq. Moreover, combining a 3D genomic interaction screening assay with transcriptome analysis, we determined that the K5aSE regulates expression of genes in addition to Klf5 on the same chromosome by acting as a transcriptional driver. Finally, we report that CTCF-mediated TAD structure formation is necessary for K5aSE regulatory function. These findings provide an important insight into understanding ESC fate determination and regulation of SEs.
Results
Identification of a class of SEs that may coordinates cell fate through adjacent master TFs
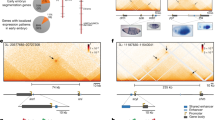
To assess how SE activities govern cell fate, we systematically analyzed SE patterns in ESCs and differentiated cells through integrating multi-omic approaches (Fig. 1a), including epigenomics (H3K27ac), transcriptomics (scRNA-seq) (Supplementary Fig. 1), and 3D genomics (Hi-C)39,40. First, we used an SE marker, namely, the activated chromatin modification H3K27ac, to predict SEs in these cells. That analysis identified 2988 SEs and revealed significant differences in the number of SEs among cell types, namely, 689 SEs in ESC-E14 cells, 388 in EB cells, 657 in mesodermal cells, 266 in NPCs, and 979 in MEFs (Fig. 1b). We then analyzed cell-specific SEs and predicted genes associated with these SEs (PSEAGs) (Fig. 1c and Supplementary Fig. 2a). GO-BP analysis revealed cell-specific PSEAGs to be significantly related to cell identity. For example, in ESC cells, we observed significant enrichment of biological processes associated with cellular responses to leukemia inhibitory factor and stem cell population maintenance. By contrast in EB cells, we observed significant enrichment in stem cell differentiation and cell differentiation processes, while in NPCs, highly enriched categories include inner ear morphogenesis and central nervous system development (Fig. 1d).

a Workflows for multi-omics analysis in ESCs and differentiated cells to identify SE patterns, including integrated epigenomics (H3K27ac), transcriptomics (scRNA sequencing) and three-dimensional genomics (Hi-C) analysis. b Analysis of SE regions based on H3K27ac ChIP-seq data. SEs were defined by signal levels above the inflection point of the curve. All H3K27ac ChIP-seq reads were aligned to the mouse genome assembly mm10 using Bowtie2, and ChIP-seq peaks were called by MACS with default parameters. The parameter “12.5 kb” was used as the maximum distance between the two regions to be sutured, and ROSE was used to distinguish SE from TSSs. H3K27ac ChIP-seq data: ESCs (GSM6911328, generated in this study), EBs (GSM1816114), NPC (GSM1603409), Mesoderm cells (GSM1163099) and MEF (GSM2912468). Publicly available ChIP-seq data have been listed in Supplementary Table 10. c Heatmap showing cell type-specific SE distribution. H3K27ac ChIP-seq signals in a ± 5 kb window for the same SE region in different cell types. d Gene ontology-biological process (GO-BP) analysis of cell-specific PSEAGs. Genes with the closest TSS to the SE (within a 50 kb window) were defined as predicted SE-associated genes (PSEAGs). GO-BP analyses were carried out using the online tool: DAVID103,104. e Gene regulatory network (GRN) analysis of SE-related TFs in ESCs. The red circle represents the master transcription factor (Seed), while the green four-sided diamonds are transcription factors (TFs) that interact with the Seed. GRN analyses were carried out using the online tool: NetworkAnalyst (https://www.networkanalyst.ca/NetworkAnalyst/home.xhtml)105,131. f UMAP embedding of single-cell RNA profiles (dots) of ESCs (green dashed line) and EBs (red dashed line). Color intensity reflects gene expression levels. g Hi-C heatmap (GSE96107), H3K27ac (GSM6911328, generated in this study) and CTCF (GSM699165) signals indicating co-localization of ESC SEs with adjacent master TFs (Klf5 and Esrrb loci). The thick red line, located at the upper part of the H3K27ac peak, delineates the SE region, whereas the thick orange line, situated at the lower part of the Hi-C heatmap, delineates the TAD region. Hi-C data are analysed and presented using online tools: 3D Genome Browser (https://3dgenome.fsm.northwestern.edu/)130. Publicly available ChIP-seq data have been listed in Supplementary Table 10. h Proposed SE function model: SE may regulate ESC properties by promoting the activity of adjacent TFs within the same TAD. Taking Pou5f1, Sox2, Esrrb and Klf5 loci as examples in the illustration. Source data are provided as a Source Data file.
To further assess the relationship between SE-associated master TFs and cell identity, we analyzed the TF regulatory network of these PSEAGs (Fig. 1e and Supplementary Fig. 2a). We identified significantly enrichment of master TFs in ESCs, including Pou5f1 (also known as Oct4), Sox2, Klf5 and Esrrb (Fig. 1f)4,41,42. Similarly, we observed key master TFs regulating cell identity in differentiated cells (Supplementary Fig. 2a, b), such as Sox9, Pax3, and Pax7 in NPCs43,44, and Runx2, Smad3, and Jun in MEFs45,46. We also observed significantly high expression of important ESC-related master TFs in ESCs, and these regulators, along with their corresponding SEs-enhancers, were located within the same TAD (Fig. 1g and Supplementary Fig. 2c). Similar features were observed in other differentiated cells, such as NPCs and MEFs (Supplementary Fig. 2d, e). These findings suggest that the identified a class of SEs may maintain cell identity by interacting with cell-specific adjacent master TFs within the same TAD (Fig. 1h and Supplementary Fig. 2f, g).
K5aSE is essential for ESC proliferation and differentiation
To assess regulatory activity of ESC-specific SEs, we focused on an unreported Klf5 adjacent super-enhancer (K5aSE, ~87.4 kb) and observed significant H3K27ac enrichment at the K5aSE locus, in addition to other chromatin-modified proteins such as H3K4me1/2/3 and BRD4 (Fig. 2a). Chromatin at the K5aSE locus also exhibited significant accessibility based on ATAC-seq analysis, indicating that K5aSE is active in ESCs. We also observed significant binding peaks of the pluripotency master transcription factors OCT4, SOX2 and NANOG (known as OSN) at the K5aSE locus47,48,49. Interestingly, KLF5 also showed significant binding to the K5aSE locus, while other ESC-essential transcription factors such as ESRRB, PRDM14, STAT3 and CTNNB1 all had significant binding peaks (Fig. 2a). These data suggest that K5aSE is an active functional element with a regulatory activity in ESCs.

a IGV screenshot showing indicated ChIP-seq signals at the K5aSE locus in ESCs and differentiated cells, including epigenetic modifications (H3K27ac, H3K4me1/2/3 and H3K27me3), chromatin accessibility (ATAC), RNA transcriptional activity (POLR2A), transcriptional regulator (BRD4) and transcription factors (OCT4, SOX2, NANOG, ESRRB, PRDM14, CTNNB1, STAT3 and KLF5). Gray-shaded area, Klf5 promoter region. Red-shaded areas, OSN binding peaks. Area within the red dashed box, K5aSE knockout region. Publicly available ChIP-seq data have been listed in Supplementary Table 10. b Bright-field and AP-staining images of WT and K5aSE-KO ESCs. Scale bar: 200 μm. c Clone size of WT and K5aSE-KO ESCs, showing significantly smaller clone size in K5aSE-KO ESCs. In WT, n = 47 clones; in KO-1#, n = 32 clones; in KO-2#, n = 17 clones; in KO-3#, n = 29 clones; two-sided unpaired t-tests. The clone size is quantified using ImageJ. d Proliferation analysis based on counting of WT and K5aSE-KO ESCs over a 4-day culture period. Analysis indicates significantly lower proliferation by K5aSE-KO ESCs. Data shown as mean ± SD, n = 3 independent replicates; two-sided unpaired t tests. e Heatmap showing DEGs between WT and K5aSE-KO ESCs (genes showing at least a twofold change and p < 0.05 were considered differentially expressed). f GO analysis of DEGs described in (e), including up- and down-regulated genes. g Bright-field images of WT and K5aSE-KO EBs at 1 and 5 days of differentiation. Scale bar: 100 μm. GO analyses were carried out using the online tool: Metascape (https://metascape.org/gp/index.html#/main/step1)130. h The sizes of both wild-type (WT) and K5aSE knockout (K5aSE-KO) embryoid bodies (EBs) were measured on differentiation days 1 and 5. K5aSE-KO EBs are significantly smaller than WT by day 5. Day 1 (left), in WT, n = 35 EBs; in KO-1#, n = 37 EBs; in KO-2#, n = 24 EBs; in KO-3#, n = 29 EBs. Day 5 (right), in WT, n = 26 EBs; in KO-1#, n = 20 EBs; in KO-2#, n = 27 EBs; in KO-3#, n = 38 EBs; two-sided unpaired t tests. The EB size is quantified using ImageJ. i Heatmap showing DEGs in K5aSE-KO compared to WT EBs at day 5 (genes showing at least a twofold change and p < 0.05 were considered differentially expressed). j GO analysis of DEGs between in K5aSE-KO relative to WT EBs on day 5, including up- and down-regulated DEGs. GO analyses were carried out using the online tool: Metascape (https://metascape.org/gp/index.html#/main/step1)130. k Heatmap illustrating the expression levels of pluripotent and lineage genes in WT and K5aSE-KO EBs on day 5 (RNA-seq, n = 3 independent replicates). The expression levels of pluripotent and lineage genes in the WT EBs were normalized to 1. Source data are provided as a Source Data file.
Next, we used CRISPR/Cas9 to knock out the core region of K5aSE in ESCs (Fig. 1a and Supplementary Fig. 3a). After screening, specific primers gDNA-PCR and Sanger sequencing-based identification, we obtained three homozygous K5aSE knockout ES cell lines (K5aSE-KO) (Supplementary Fig. 3b, c). All three showed significant clonal growth inhibition and reduced cell number compared with WT ESCs, suggesting that K5aSE is required for ESC proliferation (Fig. 2b–d). To determine K5aSE deletion effects on global gene expression, we performed transcriptome sequencing (RNA-seq) and showed significant differences in gene expression between WT and K5aSE-KO ESCs, with 255 genes up-regulated and 388 down-regulated in K5aSE-KO ESCs (Fig. 2e). GO analysis of DEGs revealed significant enrichment in biological processes related to multicellular organism development, cell differentiation, axon guidance and nervous system development in up-regulated genes, whereas processes enriched in down-regulated genes were related to regulation of angiogenesis, positive regulation of fat cell differentiation and positive regulation of cell proliferation (Fig. 2f). We also observed that differentiation genes such as T, Cdx2, Chd2 and Cebpb were significantly up-regulated in K5aSE-KO ESCs, while the pluripotency-related genes Nanog, Oct4 and Sox2 were unchanged and only Klf5 was significantly down-regulated (Supplementary Fig. 3d). Thus, we further focused on whether K5aSE regulates ESC differentiation.
To investigate potential effects on ESC differentiation, we employed the widely used in vitro EB model to compare differentiation of WT and K5aSE-KO ESCs (Supplementary Fig. 1a)50,51. Specifically, after 5 days in differentiation culture, K5aSE-KO EBs were smaller than WT EBs (Fig. 2g, h). We then performed RNA-seq of WT and K5aSE-KO EBs at the 5-day time point and identified 1497 up-regulated and 1175 down-regulated genes in K5aSE-KO relative to WT EBs (Fig. 2i). GO analysis of DEGs revealed that biological processes significantly enriched in up-regulated genes were related to embryonic organ development, cell fate commitment, axon guidance and mechanisms associated with pluripotency, whereas down-regulated genes were enriched in processes related to vasculature development, heart development and mesenchyme development (Fig. 2j), revealing overall that K5aSE deletion disrupts normal EB differentiation. To further analyze EB-specific lineage differentiation regulated by K5aSE deletion, we compared lineage gene expression in WT and K5aSE-KO EBs and observed perturbed differentiation of multiple lineages in K5aSE-KO compared to WT EBs (Fig. 2k). These results suggest that K5aSE is required to maintain proper lineage differentiation of ESCs.
KLF5 organizes regulatory networks in the context of the K5aSE
Next, we evaluated potential regulatory mechanisms of K5aSE. Based on our model of SE regulation (see Fig.1h), we hypothesized that K5aSE regulates ESCs through the adjacent master TF KLF5 (Fig. 3a, b). Also, Klf5 mRNA expression was most significantly decreased in K5aSE-KO relative to WT ESCs (Fig. 3c), suggesting that Klf5 is a K5aSE direct target gene. To test this hypothesis, we restored Klf5 expression in K5aSE-KO ESCs. RT-qPCR and WB analysis of resultant cells indicated significantly restored KLF5 mRNA and protein levels, respectively, with protein levels returning to ~80% (Fig. 3d, e). Phenotypically, Klf5 overexpression partially rescued both growth inhibition and reduced proliferation seen in ESCs following K5aSE deletion (Fig. 3f–h). RNA-seq analysis also showed that ~25% of DEGs (including Klf5) were rescued in KLF5-K5aSE-KO relative to K5aSE-KO ESCs (Fig. 3i–k). Interestingly, the promoters of some DEGs rescued by Klf5 restoration, such as Wnt3a, Klf10 and Sp5, showed significant KLF5 binding in WT ESCs (Fig. 3l), suggesting KLF5 may act as a network regulator in the context of K5aSE, directly regulating downstream genes (Fig. 3m).

a Showing rankings for K5aSE in ESC super-enhancers. b Hi-C (GSE96107) and H3K27ac ChIP-seq data (generated in this study) indicating that both K5aSE and Klf5 are located within the same TAD in ESCs. Red dashed box marking K5aSE region. Hi-C data are analyzed and presented using online tools: 3D Genome Browser (https://3dgenome.fsm.northwestern.edu/)133. c Volcano plot based on RNA-seq revealing Klf5 to be the most significant DEGs in K5aSE-KO relative to WT ESCs. Genes showing at least a twofold change and p < 0.05 were considered differentially expressed. The data showed down-regulated expression of 388 genes and up-regulated expression of 255 genes in K5aSE-KO ESCs, respectively. d RT-qPCR analysis showing relative expression of Klf5 mRNA in V-WT, V-K5aSE-KO and KLF5-K5aSE-KO ESCs. Data shown as mean ± SD, n = 3 independent replicates; two-sided unpaired t tests. e Western blot showing KLF5 protein expression in indicated ESCs (V-WT, V-K5aSE-KO and KLF5-K5aSE-KO, n = 1). KLF5 expression was normalized according to GADPH expression using ImageJ. f AP-staining of indicated ESCs (V-WT, V-K5aSE-KO and KLF5-K5aSE-KO). Scale bar: 200 μm. g Analysis of clone size in indicated ESCs (V-WT, V-K5aSE-KO and KLF5-K5aSE-KO). In V-WT, n = 34 clones; in V-K5aSE-KO, n = 96 clones; in KLF5-K5aSE-KO, n = 204 clones; two-sided unpaired t tests. The clone size is quantified using ImageJ. h Partial rescue of K5aSE-KO ESC proliferation by KLF5 overexpression. Data shown as mean ± SD, n = 3 independent replicates; two-sided unpaired t-tests. i Heatmap showing DEGs in V-K5aSE-KO compared to V-WT ESCs (FC ≥ 2, p < 0.05). j Heatmap showing rescue of DEGs shown in (i) by KLF5 overexpression (KLF5-K5aSE-KO compared to V-K5aSE-KO, FC ≥ 1, p < 0.10). k Statistical analysis of genes significantly regulated by KLF5 but aberrantly expressed in K5aSE-KO ESCs. Rescued Up-regulated genes, n = 160 genes; rescued Down-regulated genes, n = 165 genes; two-sided paired t tests. l IGV screenshots show KLF5 (GSM1208218) and H3K4me3 (GSM1871952) binding at loci of indicated genes (examples include Wnt3a, Klf10, Nek2, Sp5, Fgfbp1 and Uqcrc1) rescued by KLF5. Black arrows, direction of transcription. The relative RNA expression levels of these genes were obtained from RNA sequencing (n = 2 independent replicates). m, The schematic shows that K5aSE may regulate ESCs identity through the transcriptional regulatory network of its adjacent transcription factor KLF5. Source data are provided as a Source Data file.
Klf5 deletion promotes ESC phenotypes resembling those seen in K5aSE-KO cells
Klf5 is reportedly essential for ESC proliferation and pluripotency52,53,54,55,56,57,58, and our work reported above suggests that Klf5 is a K5aSE target gene. To investigate specific Klf5 function in the context of K5aSE under our culture system (2i/LIF), we used CRISPR/Cas9-mediated translocation mutations to knock out Klf5 in ESCs (see “Methods”) and obtained two homozygous Klf5 knockout ES cell lines (Klf5-KO) (Supplementary Fig. 4a, b). Phenotypically, Klf5-KO ESCs exhibited significantly inhibited clonal growth and reduced proliferation compared with WT cells (Supplementary Fig. 4c–e), phenotypes comparable to those seen in K5aSE-KO ESCs (Fig. 2b–d). RNA-seq analysis revealed significant differences in gene expression in Klf5-KO compared to WT ESCs, with 502 up-regulated and 210 down-regulated genes (Supplementary Fig. 4f). GO analysis of these DEGs revealed that biological processes related to gliogenesis, neuroepithelial cell differentiation and head development were significantly enriched in up-regulated genes, while down-regulated genes were significantly enriched in processes related to skin development, vasculature development and regulation of the meiotic cell cycle (Supplementary Fig. 4g). These results suggest that Klf5 is required for ESC proliferation.
Next, we examined effects of Klf5 deficiency on ESC differentiation by comparing EB differentiation of Klf5-KO and WT ESCs. After 5 days of culture in differentiation conditions, Klf5-KO EB growth was significantly inhibited relative to WT EBs (Supplementary Fig. 4h, i). Transcriptome analysis revealed significant differential gene expression in Klf5-KO compared with WT EBs, with 458 up-regulated and 1136 down-regulated genes (Supplementary Fig. 4j). GO analysis of DEGs showed that up-regulated genes were significantly enriched for processes associated with mechanisms associated with pluripotency and stem cell population maintenance, whereas down-regulated genes were significantly enriched for processes associated with connective tissue development and skin development (Supplementary Fig. 4k). When we analyzed expression levels of lineage genes in WT and Klf5-KO EBs, pluripotency genes were significantly overexpressed in Klf5-KO relative to WT EBs, while mesoderm and endoderm genes were significantly repressed (Supplementary Fig. 4l). Overall, these findings suggest that Klf5 is required for ESCs to escape pluripotency and undergo normal EB lineage differentiation and that Klf5 deletion promotes phenotypes resembling those seen in K5aSE-KO ESCs and EBs.
Functional comparison of K5aSE and Klf5 regulation of ESCs
The above findings indicate that K5aSE-KO and Klf5-KO ESCs are phenotypically similar. However, K5aSE and Klf5 may differ in terms of regulating EB differentiation: for example, K5aSE-KO EBs showed significantly enhanced ectodermal differentiation (Fig. 2k), while mesodermal differentiation was significantly inhibited in Klf5-KO EBs (Supplementary Fig. 4l). Also, when we compared differences in gene expression following K5aSE and Klf5 deletion in ESCs, both KOs shared fewer DEGs, an outcome also seen in EBs (Supplementary Fig. 5). Finally, as noted above (Fig. 3d–k), KLF5 overexpression in ESCs rescued only a subset of K5aSE-KO phenotypes and DEGs.
To further investigate these differences, we conducted 10x single-cell sequencing of WT ESCs and EBs differentiated 5 days, including EBs derived from WT, Klf5-KO, and K5aSE-KO ESCs. After data processing and cluster analysis (see Methods), we identified 9 cell subtypes (Fig. 4a, b). We observed a greater number of differentiated cell types in WT EBs relative to Klf5-KO or K5aSE-KO EBs, but also observed significant differences between Klf5-KO and K5aSE-KO EB cell subtypes. Specifically, subtypes in clusters 1 and 2 were significantly enriched in undifferentiated WT ESCs, while the proportion of subtypes in cluster 0 increased significantly upon differentiation, with K5aSE-KO cells showing the highest proportion (Fig. 4c). GO analysis of highly expressed specific genes in these cell subtypes showed that clusters 1 and 2 were significantly enriched in biological processes related to stem cells and the pluripotency markers Nanog, Sox2, Zfp42, and Bcat159,60. Cluster 0 cell subtypes in both K5aSE and Klf5 KO EBs were significantly enriched in apoptosis and cell cycle genes, such as Lgals1, Dusp1, Ddit3, Hmox1, Cdkn1c, and Cdkn1a61,62, suggesting that K5aSE and Klf5 loss decreases proliferation and may favor apoptosis of differentiated cells (Fig. 4d). Furthermore, in comparisons with WT ESC cells, we analyzed the degree of differentiation in WT, Klf5-KO, and K5aSE-KO cell populations in EBs using Monocle mode (Fig. 4e) and found that relative to WT EB differentiation processes63, Klf5-KO and K5aSE-KO cell populations exhibited incomplete differentiation processes, supporting the idea that Klf5 or K5aSE are both required for normal EB differentiation. Quantitative analyses of Monocle mode also indicated that the pseudotime progression of K5aSE-KO cells was slightly attenuated compared to Klf5-KO, but the difference was not significant (Fig. 4f).

a UMAPs of scRNA expression profiles (dots) in WT ESCs and EBs (including WT, Klf5-KO and K5aSE-KO EBs). A total of 9809 cells were enrolled in the single-cell analysis. b UMAP embeddings plots of scRNA expression profiles for different cell subsets (n = 9 clusters). Number of single cells enrolled in the analysis: WT ESCs, n = 4207 cells; WT EBs, n = 2967 cells; Klf5-KO EBs, n = 1558 cells; K5aSE-KO EBs, n = 1077 cells. c Proportion of cell subsets (using color legend shown in b) in indicated cell groups (WT ESCs, WT EBs, Klf5-KO EBs, K5aSE-KO EBs). d GO analysis of genes specifically expressed (Top100) in indicated cell subsets (n = 9 clusters). GO analyses were carried out using the online tool: DAVID103,104. e Monocle pseudotime analyses indicating cell state transitions from ESC-independent to EB states (Including WT ESCs, WT EBs, Klf5-KO EBs, K5aSE-KO EBs). Each cell is colored with pseudotime, a measure of changes occurring in each cell as differentiation proceeds; change trajectory is marked with a solid line on the UMAP. f Cell density plot of ESCs towards EB differentiation process along the pseudotime, including WT ESCs, WT EBs, Klf5-KO EBs and K5aSE-KO EBs.
Therefore, a combination of cell phenotype, transcriptome, single-cell sequencing and rescue assay analyses of K5aSE-KO and Klf5-KO suggested that K5aSE may have target genes other than Klf5 (Fig. 5a).

a Schematic illustrating that K5aSE may have target genes in addition to Klf5. b IGV screenshot showing indicated ChIP-seq signals at the K5aSE locus. Multi-colored downward-pointing arrows mark the different 4 C bait regions. Publicly available ChIP-seq data have been listed in Supplementary Table 10. c Circos plots showing candidate genes within the genome interacting with K5aSE. d GO analysis of candidate genes identified in (b). GO analyses were carried out using the online tool: DAVID103,104. e Heatmap showing expression of candidate genes (Identified in d) in WT and K5aSE-KO ESCs (RNA-seq, n = 3 independent replicates). Expressions of genes interacting with K5aSE were significantly lower in K5aSE-KO relative to WT ESCs; two-sided paired t tests. f The overlap of 4 C candidate genes with K5aSE-KO ESC DEGs confirmed five credible K5aSE target genes on chromosome 14, i.e., Klf5, Clybl, Farp1, Nkx3-1, and Tbc1d4; p-values referenced to RNA-seq data. g RT-qPCR showing the expression levels of K5aSE candidate target genes in WT and K5aSE-KO ESCs. Data shown as mean ± SD; in WT, n = 3 independent replicates; in K5aSE-KO, n = 9 independent replicates; two-sided unpaired t tests. Source data are provided as a Source Data file.
K5aSE functions as a transcriptional driver to promote target gene expression in ESCs via 3D chromatin interactions
A recent study revealed the existence of multiple facilitators within an SE that differ in regulating target gene expression64. Interestingly, we observed significant binding of proteins mediating chromatin interactions at the K5aSE locus, such as CTCF, YY1, MED1, MED12 and BRD4 (Fig. 5b), supporting the idea that Ka5SE may regulates expression of targets other than Klf5 via long-range chromatin interactions. To search for target candidates, we used 4C-seq methodology, a widely used chromatin interaction capture technique35,65,66. To comprehensively capture candidate genes interacting with K5aSE, we designed four decoy regions at binding peaks of these chromatin-interacting proteins, and after 4C-seq and genomic profiling obtained 189 genes interacting with K5aSE in ESCs (Fig. 5b-d). GO analysis of these genes revealed significant enrichment for processes associated with the cell cycle, endoderm differentiation and multicellular organism development (Fig. 5d). To identify more reliable candidates, we overlapped two replicates of the 4C-seq data to obtain 40 candidate genes and found that relative to WT ESCs, K5aSE-KO ESCs showed significantly decreased expression of these candidate genes (Fig. 5e). To narrow the candidate list, we overlapped genes with DEGs in K5aSE-KO ESCs to obtain 5 down-regulated genes located on the same chromosome, namely, Klf5 (chr14:99,296,691-99,315,412, +10 kb), Clybl (chr14:122,169,283-122,403,935, −22MB), Farp1 (chr14:121,033,200-121,285,744, −21MB), Nkx3-1 (chr14:69,190,650-69,194,722, +29MB) and Tbc1d4 (chr14:101,440,364-101,611,226, −2.2MB) (Fig. 5f). Further, based on the 4C-seq results (Supplementary Fig. 6a), we confirmed direct chromatin interactions between the K5aSE locus and these five target genes using 3C-PCR (Supplementary Fig. 6b, c). RT-qPCR results also confirmed significant down-regulation of all 5 in K5aSE-KO compared with WT ESCs (Fig. 5g). These results suggest that in ESCs, K5aSE drives expression of multiple genes on the same chromosome via 3D chromatin interactions.
Restoration of target gene expression rescues K5aSE-KO phenotypes
We next compared expression levels of our five target genes in ESCs and EBs. RT-qPCR revealed diverse expression of all five: Klf5 was highly expressed in ESCs, Clybl expression was comparable in ESCs and EBs, and Farp1, Nkx3-1 and Tbc1d4 were relatively highly expressed in EBs (Fig. 6a), suggesting that K5aSE may both promote expression of some pluripotency genes and maintain expression of select differentiation-related genes in ESCs.

a RT-qPCR showing mRNA expression levels of indicated genes (Clybl, Farp1, Nkx3-1, Tbc1d4 and Klf5) in ESCs and day 5 EBs. Two-sided unpaired t tests, n = 3 independent replicates. b Schematic diagram of the CRISPRa system used to activate target gene expression (Clybl, Farp1, Nkx3-1 and Tbc1d4). c Transcript levels of candidate K5aSE targets (Clybl, Farp1, Nkx3-1 and Tbc1d4) shown at top, as analyzed in WT ESCs, K5aSE-KO ESCs, or K5aSE-KO ESCs overexpressing the indicated target. Analysis indicates that CRISPRa restored respective target gene expression. Data shown as mean ± SD, n = 3 independent replicates; two-sided unpaired t tests. d AP-staining of indicated ESCs, including WT, K5aSE-KO, K5aSE-KO-CRISPRa-Clybl, K5aSE-KO-CRISPRa-Frap1, K5aSE-KO-CRISPRa-Nkx3-1 and K5aSE-KO-CRISPRa-Tbc1d4. Scale bar: 200 μm. e Clone size of indicated ESCs, including WT, K5aSE-KO, K5aSE-KO-CRISPRa-Clybl, K5aSE-KO-CRISPRa-Frap1, K5aSE-KO-CRISPRa-Nkx3-1 and K5aSE-KO-CRISPRa-Tbc1d4. Clone size of WT ESCs was normalized to 1. In WT, n = 71 clones; in K5aSE-KO, n = 65 clones; in K5aSE-KO-CRISPRa-Clybl, n = 57 clones; in K5aSE-KO-CRISPRa-Frap1, n = 82 clones; in K5aSE-KO-CRISPRa-Nkx3-1, n = 58 clones; in K5aSE-KO-CRISPRa-Tbc1d4, n = 83 clones; two-sided unpaired t-tests. The clone size is quantified using ImageJ. f Proliferation analysis based on counting of WT, K5aSE-KO and K5aSE-KO targets overexpressing (K5aSE-KO-CRISPRa-Clybl, K5aSE-KO-CRISPRa-Frap1, K5aSE-KO-CRISPRa-Nkx3-1 and K5aSE-KO-CRISPRa-Tbc1d4) ESCs over a 4-day culture period. The number of cells in the WT ESCs was normalized to 1. Data shown as mean ± SD, n = 4 independent replicates; two-sided unpaired t tests. g Bright-field images of indicated day 1 and day 5 EBs, including WT, K5aSE-KO, K5aSE-KO-CRISPRa-Clybl, K5aSE-KO-CRISPRa-Frap1, K5aSE-KO-CRISPRa-Nkx3-1 and K5aSE-KO-CRISPRa-Tbc1d4. Scale bar: 100 μm. h RT-qPCR analysis indicating rescue efficiency of Clybl, Farp1, Nkx3-1 and Tbc1d4 CRISPRa constructs in day 5 EBs. Data shown as mean ± SD, n = 6 independent replicates; two-sided unpaired t test. i Comparison of size of indicated EBs on days 1 (left) and 5 (right), including WT, K5aSE-KO, K5aSE-KO-CRISPRa-Clybl, K5aSE-KO-CRISPRa-Frap1, K5aSE-KO-CRISPRa-Nkx3-1 and K5aSE-KO-CRISPRa-Tbc1d4. Sizes of WT EBs were normalized to 1. EBs on days 1 (left), in WT, n = 39 clones; in K5aSE-KO, n = 69 clones; in K5aSE-KO-CRISPRa-Clybl, n = 27 clones; in K5aSE-KO-CRISPRa-Frap1, n = 49 clones; in K5aSE-KO-CRISPRa-Nkx3-1, n = 62 clones; in K5aSE-KO-CRISPRa-Tbc1d4, n = 84 clones. EBs on days 5 (right), in WT, n = 46 clones; in K5aSE-KO, n = 52 clones; in K5aSE-KO-CRISPRa-Clybl, n = 48 clones; in K5aSE-KO-CRISPRa-Frap1, n = 46 clones; in K5aSE-KO-CRISPRa-Nkx3-1, n = 49 clones; in K5aSE-KO-CRISPRa-Tbc1d4, n = 47 clones; two-sided unpaired t tests. The EB size is quantified using ImageJ. j Heatmap showing RT-qPCR analysis of pluripotency and lineage gene expression in indicated day 5 EBs. Gene expression levels in WT EBs were normalized to 1. Black asterisks, comparisons between WT and K5aSE-KO. Green asterisks, comparisons between CRISPRa groups and K5aSE-KO. Two-sided unpaired ttest, n = 3 independent replicates, * p < 0.05, ** p < 0.01, *** p < 0.001. Source data are provided as a Source Data file.
We showed above that Klf5 is a target gene of K5aSE. To determine whether the four other candidate genes have similar regulatory effects in the context of the K5aSE, we performed rescue experiments in K5aSE-KO ESCs using CRISPRa system (Fig. 6b)32,67. RT-qPCR analysis of resulting lines indicated significantly restored expression of candidate target genes, with activation efficiencies ranging from 3- to >30-fold (Supplementary Fig. 7 and Fig. 6c). Phenotypically, clonal growth and proliferation of K5aSE-KO ESCs were partially rescued by individual overexpression of each of the 4 candidate genes, with Tbc1d4 and Nkx3-1 having the most significant effect (Fig. 6d–f). Moreover, EB differentiation analysis showed that target gene expression downregulated in K5aSE-KO EBs was rescued by CRISPRa restoration of each of the 4 candidates, as was EB growth (Fig. 6g–i). RT-qPCR analysis of lineage gene expression also revealed that Farp1, Nkx3-1 or Tbc1d4 overexpression significantly blocked up-regulation of pluripotency (Sox2 and Mycn) and ectodermal (Gbx2) genes in K5aSE-KO EBs and significantly up-regulated endodermal genes (Gata6 and Foxa2) (Fig. 6j). By contrast, rescue effects of Clybl overexpression were weaker than those of Farp1, Nkx3-1 and Tbc1d4 (Fig. 6j). Overall, these results suggest that the four candidate target genes identified by 4C-seq can partially rescue ESC phenotypes promoted by K5aSE deletion and that they are the K5aSE direct targets.
ESCs regulation by Clybl, Farp1, Nkx3-1, and Tbc1d4
To investigate the regulatory role of our newly identified K5aSE target genes in ESCs, we again used the CRISPR/Cas9 to knock out these genes in ESCs (see Methods). After screening, we obtained homozygous KO lines designated Clybl-KO, Farp1-KO, Tbc1d4-KO and Nkx3-1-KO (Supplementary Fig. 8). Relative to WT ESCs, we observed no differences in morphology of any KO lines (Fig. 7a), although clonal growth was significantly inhibited in all KO lines except Clybl-KO (Fig. 7b), and proliferation was significantly decreased in all four KO lines, with Nkx3-1-KO and Tbc1d4-KO cells exhibiting the most robust phenotypes (Fig. 7c).

a Bright-field (upper) and AP-stained (lower) images of indicated ESCs, including WT, Clybl-KO, Farp1-KO, Nkx3-1-KO and Tbc1d4-KO. Scale bar: 200 μm. b Relative clone size of indicated ESCs, including WT (n = 51 clones), Clybl-KO (n = 29), Farp1-KO (n = 44 clones), Nkx3-1-KO (n = 56 clones) and Tbc1d4-KO (n = 54 clones). Clone sizes of WT ESCs were normalized to 1. Two-sided unpaired t test. The clone size is quantified using ImageJ. c Proliferation analysis based on number of indicated ESCs. The cell numbers of WT ESCs were normalized to 1. Data shown as mean ± SD, n = 6 independent replicates; two-sided unpaired t test. d Venn diagrams showing DEGs common to indicated KO relative to WT ESCs, including commonly up- and down-regulated genes. e GO analysis of DEGs evaluated in d (including co-down-regulated and co-up-regulated genes). GO analyses were carried out using the online tool: Metascape (https://metascape.org/gp/index.html#/main/step1)130. f Bright-field images of indicated day 1 and day 5 EBs. Scale bar: 200 μm. g Relative size of indicated day 1 and day 5 EBs. Sizes of WT EBs were normalized to 1. EBs on days 1 (left), in WT, n = 60 clones; in Clybl-KO, n = 62 clones; in Frap1-KO, n = 89 clones; in Nkx3-1-KO, n = 54 clones; in Tbc1d4-KO, n = 104 clones. EBs on days 5 (right), in WT, n = 106 clones; in Clybl-KO, n = 212 clones; in Frap1-KO, n = 227 clones; in Nkx3-1-KO, n = 76 clones; in Tbc1d4-KO, n = 114 clones. Two-sided unpaired t tests. The EB size is quantified using ImageJ. h Venn diagrams showing common DEGs in indicated day 5 EBs, including commonly up- and down-regulated genes. i GO analysis of commonly down-regulated genes in indicated day 5 EBs. GO analyses were carried out using the online tool: Metascape (https://metascape.org/gp/index.html#/main/step1)130. j Expression analysis of pluripotency and lineage genes in indicated day 5 EBs. Expression levels of pluripotency and lineage genes in WT EBs were normalized to 1(as shown by the gray dashed line). Two-sided paired t test. Source data are provided as a Source Data file.
To assess effects on global gene expression, we then performed transcriptome analysis of Clybl-KO, Farp1-KO, Tbc1d4-KO and Nkx3-1-KO lines. RNA-seq showed that compared with WT ESCs, there were 2078 DEGs in Clybl-KO cells, 5011 in Farp1-KO cells, 1737 in Nkkx3-1-KO cells and 2856 in Tbc1d4-KO cells (Supplementary Fig. 9a). Combined GO results of the up-regulated genes in KO cells showed significant enrichment in processes related to oxidative stress and redox pathway, glutathione metabolism and NoRC negatively regulates rRNA expression (Supplementary Fig. 9b). Combined GO results of the down-regulated genes in KO cells showed significant enrichment in processes related to cell cycle regulation, cell growth and mechanisms associated with pluripotency (Supplementary Fig. 9c). To further assess biological mechanisms regulating these four genes, we overlapped up- or down-regulated genes and observed 185 co-up-regulated and 285 co-down-regulated genes (Fig. 7d). GO analysis of co-regulated genes revealed that up-regulated genes were mainly enriched for processes related to glutathione metabolism and cholesterol metabolism, while down-regulated genes were associated with placenta development, developmental growth and stem cell population maintenance (Fig. 7e).
To determine whether loss of any of these four genes altered EB differentiation, we compared WT and gene KO EBs after 5 days of differentiation. EB growth of all candidate KO lines was significantly inhibited compared with WT EBs (Fig. 7f, g). Transcriptome analysis showed that KO of any one the four candidate genes significantly altered global gene expression relative to WT EBs, with 959 DEGs in Clybl-KO EBs, 341 in Farp1-KO EBs, 3246 in Nkx3-1-KO EBs and 313 in Tbc1d4-KO EBs (Supplementary Fig. 9d). Combined GO analysis of up-regulated genes indicated significant enrichment of processes related to the meiotic cell cycle, placenta development and differentiation of cells involved in embryonic placenta development (Supplementary Fig. 9e). GO analysis of down-regulated genes revealed significant enrichment in processes associated with heart development, neural crest differentiation, cell fate commitment and mesenchyme development (Supplementary Fig. 9f). We then overlapped DEGs to obtain 10 commonly up-regulated genes (which included Cd36, Rex2 and Cyp2j6) and 55 commonly down-regulated genes (Fig. 7h). GO analysis of commonly down-regulated genes revealed significant enrichment of processes associated with neuron projection development, embryonic morphogenesis, forebrain development and neural crest differentiation (Fig. 7i), suggesting that KO of any of these four genes disturbs EB differentiation. To assess EB lineage differentiation regulated by these four genes, we analyzed lineage gene expression in WT and KO EB lines (Fig. 7j) and observed showed significantly suppressed ectodermal and mesodermal differentiation in Clybl-KO, Farp1-KO and Tbc1d4-KO compared with WT EBs. Notably, pluripotency, endoderm and trophectoderm genes were significantly enhanced in Nkx3-1-KO compared to WT EBs. Overall, these results demonstrate that Clybl, Farp1, Nkx3-1 and Tbc1d4, which are K5aSE target genes, are required to maintain ESC proliferation and proper differentiation.
CTCF-mediated TAD formation maintains K5aSE regulatory function
Interaction of functional DNA elements with target genes requires proteins that mediate chromatin interactions and organize higher-order chromatin structures, such as CTCF, MED1/12 and YY168,69,70,71,72,73,74,75. Here, although our findings indicate that K5aSE maintains target gene expression via 3D chromatin interactions, it remained unclear which factors mediate those interactions. Interestingly, we found distinct CTCF, MED1/12 and YY1 binding peaks at the K5aSE locus (Fig. 4b), suggesting they may mediate K5aSE/target gene interactions. Given that CTCF is reportedly required to maintain higher-order chromatin structures in ESCs, we analyzed publicly available data to assess a role for CTCF in the K5aSE context76. Analysis of Hi-C data in WT ESCs indicated that K5aSE and Klf5 are located within the same TAD, while the potential K5aSE target gene, Tbc1d4, is located within the TAD boundary and separated from K5aSE by a TAD (Fig. 8a), suggestive of a relatively compact chromatin interplay state. However, Hi-C analysis of CTCF-deficient and -proficient WT ESCs showed significant loss of TAD structure in CTCF-deficient cells (Fig. 8b), likely resulting in a looser interaction between K5aSE and the Tbc1d4 locus (Fig. 8a–d). We also observed significantly lower Tbc1d4 and Klf5 expression in CTCF-depletion relative to -proficient WT ESCs (Fig. 8e). These data suggest overall that CTCF may mediate K5aSE interaction with Tbc1d4 by maintaining TAD structure, promoting both Klf5 and Tbc1d4 expression (Fig. 8f).

a Hi-C heatmap showing chromatin interactions of the K5aSE-Tbc1d4 locus in WT ESCs and in ESCs treated with auxin for 2 days to induce CTCF depletion (Hi-C data: GSE98671). Hi-C data are analyzed and presented using online tools: 3D Genome Browser (https://3dgenome.fsm.northwestern.edu/)133. b Insulation scores for the K5aSE-Tbc1d4 locus in CTCF-WT and CTCF-depletion ESCs were analyzed using Hi-C data (GSE98671) with a resolution of 25 kb. c ChIP-Seq data showing H3K27ac (GSM6911328, generated in this study) and CTCF (GSM699165) signals at the K5aSE-Tbc1d4 locus in ESCs. d ChIP-seq data showing CTCF binding at the K5aSE-Tbc1d4 locus in CTCF-WT (GSM2609185) and CTCF-depletion (GSM2609186) ESCs. e RNA-seq (GSE98671) showing significantly decreased Klf5 and Tbc1d4 expression in ESCs after CTCF depletion. f A proposed model of how the TAD structure maintained by CTCF allows remote regulation of Klf5 and Tbc1d4 by K5aSE. g Illustration of six selected CTCF binding sites (CBS1-6) within the TAD boundary region and the application of CRISPR/Cas9 technology to knockout these sites. h The mRNA expression levels of Klf5 and Tbc1d4 were detected using RT-qPCR in WT and CBS-KO ESCs. In WT, n = 16 independent replicates; in CBS1-KO, n = 12 independent replicates; in CBS2-KO, n = 12 independent replicates; in CBS3-KO, n = 8 independent replicates; in CBS4-KO, n = 4 independent replicates; in CBS5-KO, n = 8 independent replicates; in CBS6-KO, n = 12 independent replicates. Gene expression levels in WT ESCs were normalized to 1. Two-sided unpaired t test. Source data are provided as a Source Data file.
Previous studies have demonstrated that CTCF’s functional roles are context-dependent, specifically determined by its binding to particular genomic regions known as CTCF binding sites (CBS)68,77,78. Therefore, we further questioned whether the CBS at the TAD boundary of the K5aSE-Tbc1d4 genomic region is involved in regulating the expression of the K5aSE target gene (such as Tbc1d4). To test this, we identified six CBSs at these TAD boundary regions and employed CRISPR/Cas9 technology to knock out these sites (Fig. 8g). Through a series of gDNA-PCR and Sanger sequencing validations (Supplementary Fig. 10), we successfully generated homozygous knockout cell lines for these CBS sites. Further RT-qPCR analysis showed that compared to WT ESCs, the expression of Klf5 and Tbc1d4 was significantly reduced in CBS3-KO ESCs (Fig. 8h). Similarly, Klf5 was also lowly expressed in CBS2-KO ESCs, while Tbc1d4 was lowly expressed in CBS6-KO ESCs. In addition, we also found that the expression of Klf5 was reduced in CBS5-KO ESCs. These findings suggest that distinct CBSs may exhibit varied regulatory functions.
In summary, by analyzing the TAD structure and insulation scores from the Hi-C data, we observed changes in gene expression following CTCF depletion, as well as functional validation after CBS sites deletion. These results demonstrate that the CTCF-mediated formation of the TAD structure at the K5aSE-Tbc1d4 locus is involved in the regulation of K5aSE on its target genes, such as Klf5 and Tbc1d4.
Discussion
Here, we identified a class of SEs, as exemplified by K5aSE, that maintains expression of the adjacent master TF (Klf5) and promotes expression of other target genes likely through 3D chromatin interactions to ensure ESC identity (Fig. 9). These findings provide an important perspective for understanding ESC identity and SE regulatory mechanisms.

a K5aSE regulates the expression of multiple target genes located on the same chromosome in WT ESCs (left panel) through three-dimensional chromatin interactions, thereby safeguarding regular proliferation and differentiation of ESCs. In contrast, in K5aSE knockout ESCs, the expression of target genes was significantly reduced (right panel), leading to decreased proliferation and impaired differentiation of ESCs. b A proposed model of how the TAD structure maintained by CTCF allows remote regulation of Klf5 and Tbc1d4 by K5aSE.
Numerous studies show that SEs play a central role in development and disease by maintaining expression of cell- and tissue-specific genes8,9,79. Although hundreds of ESC-related SEs are predicted by bioinformatics analysis, their function or detailed regulatory mechanisms are not yet fully understood. Here, we identified K5aSE as a functional mouse ESC-specific SE through bioinformatic analyses and in vitro experiments combined with transcriptomic and phenotypic analyses, and show that K5aSE is required for ESC proliferation and EB differentiation. We also predicted numerous cell-specific SEs in this study, such as many unresearched SEs in ESCs or NPCs, but focused primarily on regulation of the ESC-specific super-enhancer K5aSE (Fig. 2). Further analysis of these SEs, such as systematic screening of these cell-specific SEs using CRISPR/Cas9 and in vivo validation of their effects on ESC differentiation, will enhance our understanding of SE function in cell growth or development. It is important to note that we characterized K5aSE in mouse ESCs, and it does not appear to function as an SE in human ESCs or other differentiated human cells, based on H3K27ac ChIP-seq and ATAC-seq data analysis. However, in this study, we observed SE clusters near the KLF5 locus in human pancreatic cancer cell lines (Supplementary Fig. 11). Bioinformatics analysis also revealed significant KLF5 overexpression in pancreatic cancer specimens and that patients with high KLF5 expression have a poor prognosis (Supplementary Fig. 12)80,81, indicating that targeting KLF5 may offer new therapeutic strategies. Interestingly, analysis of Hi-C data indicated similar high-order chromatin structures near the KLF5/Klf5 loci in human and mouse (Supplementary Fig. 13), suggesting that K5aSE is associated with ESC development in both species, although confirmation will require future analysis.
Previous studies show that SEs exert regulatory effects by maintaining expression of adjacent master TFs, which in turn bind to the SE, forming a circular SE-TF-SE regulatory model8,21,24,79,82. Several SEs reportedly function in ESCs, among them the distal SEs of Sox2 and Klf417,18, the Nanog proximal SE and the Prdm14 3’-end SE16,19. However, as part of high-order chromatin structures, SEs may not function in a simple one-to-one pattern (Fig. 1h). We previously reported that TEs regulate cell activity by promoting expression of multiple genes through long-range chromatin interactions32,33,34. Also, here, we show that K5aSE regulates Klf5 and that KLF5 also binds at the K5aSE locus (Fig. 2a), in accordance with the circular SE-TF-SE model. However, Klf5 re-introduction only partially rescued ESC phenotypes seen after K5aSE deletion, and EB differentiation gene expression patterns differed in Klf5-KO and K5aSE-KO contexts (Fig. 3, Fig. 4, Supplementary Fig. 4 and Supplementary Fig. 5), suggesting K5aSE has other targets. Moreover, our 4C-seq results reveal that K5aSE maintains expression of multiple genes through 3D chromatin interactions (Fig. 5). These results suggest that in ESCs, SEs regulate both expression of adjacent master TFs and expression of other genes through 3D chromatin interactions, and then synergistically regulate ESC activity (Fig. 9). Here, we assessed only one SE; however, as noted, ESCs harbor numerous SEs. Future use of mature chromatin interaction capture technology, such as HiChIP-seq83,84,85,86,87,88, could reveal global interactions of SEs and provide a crucial understanding of their regulatory mechanisms.
Using 4C-seq assay and rescue experiments, we identified multiple K5aSE targets, including Klf5, Clybl, Farp1, Nkx3-1 and Tbc1d4. Interestingly, others report that NKX3-1 plays a crucial role in iPSCs by promoting reprogramming in the absence of OCT489. NKX3-1 also reportedly regulates activities in prostate ductal stem cells, hematopoietic stem cells and tumor stem cells90,91,92,93. Accordingly, our deletion analysis demonstrated that Nkx3-1 is required for ESC proliferation and EB lineage differentiation, revealing a crucial role in ESCs (Fig. 7). Among the five K5aSE target genes identified here, Klf5 and Nkx3-1 are TFs. In EB differentiation, Klf5 is expressed at low levels, while Nkx3-1 is highly expressed (Fig. 6a) and likely serves as a differentiation gene. Thus, our findings suggest that in ESCs, K5aSE maintains expression of both pluripotency and differentiation genes, but how it plays such diverse roles will require further analysis.
Finally, our Hi-C analysis showed significant loss of TAD structure in ESC cells following experimental CTCF protein degradation (Fig. 8a, b). In the CTCF-depletion ESCs, we observed that the downregulation of Klf5 and Tbc1d4 is most pronounced, exhibiting specificity compared to neighboring genes such as Mzt1, Pibf1, Prr30 and Lmo7. Further experiments involving CBS deletion indicate that the TAD boundary CBS3 located downstream of K5aSE is essential for the expression of Klf5 and Tbc1d4. Accordingly, we propose that CTCF mediates K5aSE interaction with Tbc1d4 by maintaining TAD structure, which in turn promotes Klf5 and Tbc1d4 expression. Thus, CTCF deficiency would weaken these TAD-based chromatin interactions and decrease Klf5 and Tbc1d4 expression. We also observed binding peaks of OCT4, CTCF, YY1, BRD4 and MED1/12 at the K5aSE locus (Fig. 2a and Fig. 5b) in accordance with their known function in maintaining higher-order chromatin structures and their reported regulation of target gene expression via a phase-separation mechanism75,94,95,96,97,98,99. These proteins may also maintain chromatin interactions of K5aSE through a comparable phase-separation mechanism, a hypothesis to be tested in future studies.
In conclusion, our analysis of K5aSE reveals a key mechanism used by a class of SEs to maintain expression of an adjacent TF (Klf5) and enhance expression of other genes on the same chromosome through 3D chromatin interactions, ensuring ESC identity. Our findings provide an important insights into ESC fate determination and SE regulation.
Methods
Cell culture
Mouse ESCs (E14) or cells generated from this line were cultured under undifferentiated conditions37. ESCs were grown in culture dishes coated with 0.1% gelatin (Sigma) in Dulbecco’s Modified Eagle’s Medium (DMEM, CORNING, ref. 10–013-CV) supplemented with 15% fetal bovine serum (FBS, AusGeneX, ref. FBS500-S), 1x nonessential amino acids (NEAA, 100x, Gibco, ref. 11140-050), 1x L-Glutamate (100x, Gibco, ref. 25030-081), 1x Penicillin-Streptomycin (P/S, 100x, Gibco, ref. 15140-122), 50 μM β-mercaptoethanol (Sigma), 1000 U/ml leukemia inhibitory factor (LIF, ESGRO, ref. 11710035), and 2i (1 μM PD0325901, MedChemExpress; 3 μM CHIR99021, MedChemExpress). ESC medium was replaced every 1–2 days. HEK293T cells were cultured in DMEM supplemented with 10% FBS (Biological Industries, ref. 04-00101 A) and 1x P/S. HEK 293 T cells were passaged every two days using 0.25% trypsin–EDTA (Gibco, ref. 25200-072)100. All cells were maintained at 37 °C in a 5% CO2 incubator. Cultured cells undergo routine mycoplasma testing with consistently negative results.
Identification of SEs
We used H3K27ac ChIP-seq data to create SE annotations8,9. Mouse H3K27ac ChIP-seq data were downloaded from the National Center for Biotechnology Information (NCBI, www.ncbi.nlm.nih.gov/sra/). All ChIP-seq reads were aligned to the mouse genome assembly mm10 using Bowtie2101. ChIP-seq peaks were called by MACS with default parameters102. We also used the parameter “12.5 kb” as the maximum distance between two regions to be sutured. Finally, ROSE was used to separate SEs from transcription start sites (TSSs, +/- 2.5 kb). Source Data Supplementary Fig. 1b shows SEs identified in ESCs and differentiated cells.
Prediction of SE-associated genes
Methods used for Prediction of SE-Associated Genes (PSEAGs) were shown below: SEs were assigned to genes defined in the RefSeq (Mouse, GRCm38/mm10) gene annotation; to assign each SE to genes, we calculated the distance from the SE to the TSS of relevant genes (within a 50 kb window)8,9,21. PSEAGs include genes closest to the SE. Source Data Supplementary Fig. 1b shows PSEAGs in each cell type.
Gene regulatory network analysis
To further explore the transcriptional regulatory network of these PSEAGs, we first utilized the DAVID tool for GO-MF (Gene Ontology-Molecular Function) analysis to identify a class of transcription factors (TFs) with regulatory functions (i.e., transcription factor activity, sequence-specific DNA binding)103,104. Subsequently, we analyzed these TFs using the online tool NetworkAnalyst (https://www.networkanalyst.ca/NetworkAnalyst/home.xhtml) to investigate the regulatory network of a class of transcription factors associated with SEs105.
Chromatin immunoprecipitation sequencing and analysis
ChIP-seq assay was performed using a ChIP Assay Kit (Beyotime, P2078) with minor modifications. In brief, 107 ES cells were treated with 1% formaldehyde (methanol free) and cross-linked 10 min at room temperature with rotation. Glycine was added to a final concentration of 125 mM to quench the formaldehyde. Samples were washed twice in cold PBS and spun at 3000 rpm for 5 min. The supernatant was discarded and pelleted cells were harvested in SDS lysis buffer with 100× protease Inhibitor Cocktail (Merck, EMD Millipore Crop, LOT: 3446024) for 15 min on ice. 10% whole cell lysates were saved as input after genomic DNA was broken into 200– 400 bp by sonication. Samples were immunoprecipitated with 7.5 μg H3K27ac antibody (Abcam, CAT: ab4729) overnight followed by a 4 h incubation in 60 μL protein A/G agarose beads (Thermo Fisher Scientific) at 4 °C. The beads are cleaned again under the above conditions. ChIP and input DNA was de-crosslinking with Proteinase K and purified using Ampure XP beads. Precipitated DNA and input were sequenced using an Illumina sequencer. Raw sequencing reads were mapped onto the reference mouse genome mm10 using Bowtie2101. Peak calling was conducted using MACS2 software with 5% false discovery rate (FDR) cut-off values for narrow peaks102. ChIP-seq tracks were generated using IGV software or the WashU Epigenome Browser106.
Construction of sgRNA plasmids
sgRNA plasmids were constructed using reported protocols37,107. Target-specific guide RNAs (sgRNAs) were designed using an online tool (http://chopchop.cbu.uib.no/)108. sgRNAs matching the target site were selected (Mus musculus, mm10), synthesized at Sangon Biotech (Shanghai city, China) and cloned into the Cas9-puro vector (pXPR_001) using the BsmBl restriction site (NEB, ref. R0580L). Successful sgRNA plasmid construction was confirmed by Sanger sequencing at GENEWIZ (Suzhou City, China). Supplementary Table 1 shows sgRNA sequences.
CRISPR/Cas9-mediated genome editing in ESCs
Genome editing was performed according to the following methods37,107,109. ESCs were transfected with 1–2 sgRNA plasmids using Lipofectamine 3000 (Life Technologies) and 24 h later, cells were treated with 5 μM puromycin (MCE) for 24 h and then cultured in the ESC medium without puromycin for 5–7 more days. Individual colonies were selected and validated by genomic DNA PCR (gDNA-PCR), Sanger sequencing, Western Blotting or RT-qPCR. Two sgRNAs were used to knock out K5aSE, followed by gDNA-PCR using specific primers to identify homozygous K5aSE knockout lines. For Klf5 knockout, a single sgRNA was used to cause a shift mutation in the Klf5 CDS region, followed by gDNA-PCR, Sanger sequencing and Western blotting to identify Klf5 homozygous knockout lines. Two sgRNAs were used to knock out Clybl, Farp1, Nkx3-1 and Tbc1d, followed by gDNA-PCR using specific primers, Sanger sequencing and RT-qPCR to identify homozygous knockout cell lines. Supplementary Table 2 shows gDNA-PCR primers used for genotyping.
In vitro formation of EBs from ESCs
In vitro EB formation was performed as reported with minor modifications110,111. Single cell ESC suspensions were placed in AggreWell plates (STEMCELL TECHNOLOGIES) and allowed to aggregate into spheroids, which were then cultured in suspension and allowed to differentiate spontaneously. First, ~ 2 × 106 ESCs were suspended in 2 ml EB differentiation medium (ESC media without LIF and 2i) and added to wells of the AggreWell plate. The plate was then centrifuged at 100 g for 3 min to allow cells to deposit in microwells and then incubated 1 day at 37 ˚C, during which time spherical aggregates of deposited ESCs formed. EBs were then collected and transferred to a bacterial-grade culture dish, spun (at 70 rpm/min) to suspend EBs in the same medium and cultured to allow spontaneous differentiation. After 4 days, EBs were collected for photography and RNA extraction.
scRNA-seq analysis
Raw sequence read quality was assessed using FastQC. The mouse mm10 reference genome was downloaded from Ensembl (https://www.ensembl.org/). Cell Ranger software was downloaded from 10x Genomics (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/ latest) and used to process raw data, align reads to the mm10 mouse reference genome and summarize unique molecular identifier (UMI) counts against the corresponding Ensembl annotations obtained in GTF format. Empty wells were distinguished from barcoded cells using UMI count distributions. First, UMIs likely misassigned to an incorrect barcode due to sequencing index swapping were removed using DropletUtils112. The emptyDrops function from DropletUtils was then used to distinguish cells from empty droplets containing only ambient RNA, with barcodes <5% FDR retained. Also, droplet barcodes with low total UMI counts and droplets for which a high percentage of total UMIs originated from mitochondrial RNAs (>15%) were filtered out. For the remaining cells in each sample, doublet detection and filtering were performed using DoubletFinder113. Expression matrixes were loaded into R using the function Read10X in Seurat and then merged by column114. This resulted in a merged object of WT-EB samples, differentiation samples, and Klf5-KO and K5aSE-KO samples from WT (ESCs). Cell-level quality control was performed to filter out cells of which: (1) total UMI counts were no more than 1000; (2) gene numbers were <500; (3) mitochondrial gene percentages >10; and (4) ribosomal gene percentages were >25. Expression levels of each gene in each cell were normalized using the function Normalize Data with default parameters to decrease influence of the sequencing library size, converting expression values from UMI counts to ln[10,000 × UMI counts/total UMI counts in cell + 1]. Batch-effect correction for samples from different samples was performed using the harmony before clustering and visualization115. Clustering was performed using the standard Seurat clustering pipeline. Briefly, we used the following functions in this order: Find Variable Features with 2,000 genes, Scale Data, Run PCA, Find Neighbors with the first 20 harmony values and FindClusters with resolution 0.5. Otherwise, default settings were used.
To model the stem cell differentiation state, Monocle3 was applied to the minicluster expression matrix and the UMAP embedding, to better preserve local relations of cells63. Specifically, the principal components were re-computed from the mini-cluster expression matrix. Based on the function cluster_cells, mini-clusters were divided into large: confidential 10 separated groups called partitions, within each of which a principal graph was fitted using the function learn_graph. The principal graph was shown on the UMAP as “skeleton lines”, indicating differentiation trajectories. Assigning mini-clusters to the nearest principal graph nodes, the principal graph node containing the highest fraction of undifferentiation ESCs was specified as the root, and then pseudotime was calculated using function order_cells.
ESC alkaline phosphatase staining and colony growth assays
A low density of ESCs (~2 × 103 cells/cm2) was plated and cultured 4 days. Cells were then fixed and stained for alkaline phosphatase (AP) activity following the manufacturer’s instructions (SBI, Purple/Red-ColorTM AP Staining Kit). AP-positive colonies were imaged using an Olympus Inverted Fluorescence Microscope. ESC clone size was measured according to the ImageJ software scheme and normalized to values seen in control cells.
ESC proliferation assay
ESCs (5 × 104) were plated in 6-well plates at day 0. At time points of interest (Day 4), ESCs were digested to individual cells in 0.25% trypsin for counting. Relative cell numbers were calculated by normalizing the values in control cells.
RNA extraction, cDNA synthesis and RT-qPCR
Cells were lysed with Trizol reagent (Life Technologies), and total RNA was extracted based on the manufacturer’s instructions. 1 µg RNA was converted to cDNA using a PrimerScriptTM RT reagent Kit with gDNA Eraser (TaKaRa) according to the manufacturer’s instructions. To quantify gene expression, RT-qPCR was carried out on a CFX96 Real-Time PCR system (Biorad) using Hieff qPCR SYBR Green Master Mix (YEASEN, ref. 11201ES08). PCR conditions were: 95 °C for 5 min, followed by 40 three-step cycles at 95 °C for 10 s, 60 °C for 10 s and 72 °C for 30 s. Data were analyzed using the comparative Ct (ΔΔCt) method to quantitate gene expression116. Supplementary Table 3 lists RT-qPCR primers used in this study.
Stable transduction of ESC lines
Full-length Klf5 coding sequence (CDS) was synthesized at GENEWIZ (Suzhou city, China) and inserted into the FLAG-tag vector (pLCH72) using Nhe1 (NEB, ref. R3131L) and Not1 (NEB, ref. R3189L) digestion sites. ESCs were then transfected the overexpression vector using Lipofectamine 3000 (Invitrogen, ref. 2309895) and then 24 h later treated with media containing 5 μM puromycin (MCE) until stably-transduced cells were harvested. RT-qPCR and Western blotting were used to identify overexpressing lines109. Supplementary Table 4 shows full-length Klf5 CDS.
Western blotting analysis
Protein was extracted using RIPA lysis buffer (Strong, YEASEN) and electrophoresis performed using a PAGE Gel Quick Preparation Kit (10%, YEASEN) following the manufacturer’s instructions. WB was carried out with the following antibodies: (primary) KLF5 (Santa Cruz, ref. sc-398470X, 1:10000) and GAPDH (Santa Cruz, ref. sc-365062, 1:2000), and then HRP-linked secondary antibodies (Abcam, ref. ab6728). HRP activity was detected using Luminol HRP Substrate (Millipore, ref. WBKLS0500). Digital images were taken using an automatic chemiluminescence imaging analysis system (Tanon). Protein quantification was performed using the computational model of ImageJ software.
Transcriptome sequencing (RNA-seq) and data analysis
RNA-seq was performed according to the following methods36,109. ESCs were lysed with Trizol reagent (Life Technologies), and RNA was extracted based on the manufacturer’s instructions. RNA was sequenced by Novogene (Tianjin City, China). Clean reads were mapped to the Ensemble mm10 mouse genome using Hisat2 with default parameters. Gene reads were counted by Htseq117. Fold-changes (FC) were computed as a log2 ratio of normalized reads per gene using the DEseq2 R package118. Genes showing at least a twofold change and p < 0.05 were considered differentially expressed (DEGs). Heatmaps were drawn using the heatmap.2 function or Microsoft Excel 97-2003.
Circular chromosome conformation capture assay (4C-seq) and data analysis to identify K5aSE-interacting candidate genes
4C experiments were performed using the reported protocol with a few modifications35,65,66. 1 × 107 ES cells were trypsinized to single cells and resuspended in 9.4 ml DMEM/15% FBS. Cells were then cross-linked by adding 0.6 ml 16% formaldehyde for 10 min. After centrifugation at 4 degrees (2000 x g), cells were lysed in 5 ml lysis buffer (10 mM Tris-HCl [pH = 7.5]; 10 mM NaCl; 5 mM MgCl2; 0.1 mM EGTA; 1 x protease inhibitor) for 10 min (on ice with constant light shaking) and centrifuged to remove supernatants. Nuclei were then digested with DpnII (NEB, ref. R0543L) at 37 °C. After inactivation for 20 min in 1.6% SDS at 65 °C, samples were diluted in 6.125 ml of 1.15× ligation buffer and 100 U T4 ligase (NEB, ref. M0202L) and incubated 4 h at 16 °C, followed by 30 min at 25 °C. Ligated chromatin was digested by proteinase K (CWBIO, ref. 01724) and purified by phenol-chloroform extraction, and then DNA was ethanol-precipitated. The purified product was further digested with NlaIII (NEB, ref. R0125L) and then cyclized with T4 ligase. After purification, PCR reactions (TransStart FastPfu DNA Polymerase) containing 100 ng DNA were performed using bait primers listed in Supplementary Table 5. PCR products were purified by Agencourt AMPure XP paramagnetic beads (BECKMAN COULTER, ref. A63881), and purified products were sequenced at Novogene (Tianjin city, China).
4C-seq data were analyzed with reference to our previous reports35,66. Sequencing reads aligned at the 5’ end to the forward inverse PCR primer sequence were selected. The remaining selected reads, including those at the DpnII sites, were mapped to the mm10 assembly using BWA to identify ligation sites in the genome. The mapped ligated DpnII sites were subsequently compared to a reduced genome that included all DpnII site locations. For statistical analysis, we adhered to the established 4C-seq data analysis protocol119,120,121. To identify nonrandom long-range interactions, we employed a false discovery rate (FDR) of 0.05 as a threshold and compared our 4C-seq data to randomly permuted datasets.
To systematically identify potential regulatory target genes associated with K5aSE, we established a multi-level screening strategy based on 3D genomic interaction features. First, at the annotation level of genomic interaction sites, we prioritised genes whose transcription start sites (TSS) were within ±10 kb of interaction anchors. Given that K5aSE functions as an ultra-long enhancer element (>50 kb), we further optimised the 4C-seq bait design strategy. We integrated multi-omics datasets, including histone H3K27ac modification profiles, chromatin accessibility (ATAC-seq), and binding sites of chromatin interaction mediators (e.g., CTCF, BRD4, YY1, MED1, and MED12), and selected four highly active subregions enriched for transcriptional regulatory elements within K5aSE as bait regions (Fig. 5b). To ensure reproducibility, we performed two biological replicates. Captured data from the four bait regions in each replicate were pooled. Finally, high confidence target gene sets were derived from the intersection analysis of two independent experimental replicates. Source Data Supplementary Fig. 5d lists candidate genes obtained by 4C-seq.
Chromatin conformation capture (3C) assay
The 3C assay was carried out as follows33,34,122. The interactions between the K5aSE locus and the candidate target genes (Klf5, Clybl, Farp1, Nkx3-1 and Tbc1d4) were detected by primer-specific PCR experiments. Specific primers were designed upstream and downstream of the K5aSE and target gene regions, respectively, and PCR was performed using the 4C library we had generated, followed by Sanger sequencing of the PCR products to confirm the precise intercalation sites. These primer designs refer to the 4C-seq data and have been listed in Supplementary Table 6.
CRISPRa-mediated target gene activation
CRISPRa was performed as follows32,67,123. A stable ESC CRISPRa cell line was generated using lentiMPH v2 plasmid (Addgene, ref 89308). Lentiviral particles were generated in HEK293T cells using pMD2.G (Addgene, ref 12259) and psPAX2 (Addgene, ref 12260) packaging plasmids in a standard laboratory setting. ESCs were transduced 24 h and selected 5 days in 200 mg/ml Hygromycin (Invitrogen). CRISPRa sgRNAs targeting a target promoter were designed using the Genetic Perturbation Platform (GPP) Web Portal (https://portals.broadinstitute.org/gppx/crispick/public) and ordered from Sangon Biotech (Shanghai city, China)124,125. Supplementary Table 7 shows sgRNA sequences. Sequences were subsequently cloned into lentiSAM v2 plasmid (Addgene, ref 75112) using Bsmbl and packaged, and cells were transduced as described above. Transduced ESC CRISPRa cells were then selected in 3 mg/ml blasticidin S (Solarbio). Final target gene activation efficiency was verified by RT-qPCR.
Insulation score calculation and TAD boundary identification
The valid pairs (allValidPairs files) from both WT-Hi-C and CTCF-depletion-Hi-C data generated by HiC-Pro were used to create.cool files using hicpro2higlass.sh126. TADs were identified with insulation score127. Genome-wide insulation scores and boundary scores were computed using balanced interaction matrices with a 25 kb bin size and a 500 kb window size, employing the cool tools “diamond-insulation” function128. Bins with a boundary score ≥ |0.5| were considered valid boundaries. When the insulation scores for both WT and CTCF-depletion are <0, boundaries with (WT - CTCF-depletion) insulation score ≤ −0.3 were lost boundaries in CTCF-depletion cells. When the insulation scores for both WT and CTCF-depletion are > 0, boundaries with (WT - CTCF-depletion) insulation score ≥ 0.3 were lost boundaries in CTCF-depletion cells. Adjacent differential boundaries were merged.
CTCF binding sites (CBS) analysis and CRISPR/Cas9-mediated CBS deletion in ESCs
The CBS knockdown strategy refers to our previous report36. Based on TAD boundary analysis, we selected 6 CBSs for functional validation. JASPR analysis was used to confirm the precise CTCF binding sequences (https://jaspar.elixir.no/)129. sgRNAs were targeted upstream and downstream of the CBS. Homozygous CBS knockout cell lines were identified by gDNA-PCR with specific primers and Sanger sequencing. The sgRNA sequences that were used to knockout CBSs have been listed in Supplementary Table 8. Specific PCR primers used to identify CBS knockout cell lines are listed in Supplementary Table 9.
Bioinformatics analysis
Gene ontology (GO) analyses were performed using the following online tools: Metascape (http://metascape.org)130 and DAVID Functional Annotation Bioinformatics Microarray Analysis tool (https://david.ncifcrf.gov/tools.jsp)104. Briefly, DEGs (FC ≥ 2, p < 0.5) were loaded into the analysis module as instructed by the tools, and mouse was selected as the specific specie for GO analysis to obtain the final output. Transcription factor interaction networks were constructed using online tool: https://www.networkanalyst.ca/, referring to the corresponding instructions105,131. The online tool GEPIA2 was used to analyze the clinical data (http://gepia2.cancer-pku.cn/#index)132.
Statistical analysis
Data was evaluated statistically using Microsoft Excel and GraphPad Prism 9 software. Statistical tests and experimental replicates are presented in figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We thank the laboratories that generated public data used in this study. Previously published datasets used in this study are shown in Supplementary Table 10. These published data sets were downloaded and analyzed using online tools (http://cistrome.org/db/#/, http://3dgenome.fsm.northwestern.edu/ and www.ncbi.nlm.nih.gov/sra/)133,134,135. Our DNA-seq and RNA-seq data have been deposited in the NCBI’s Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE202540, GSE263578, GSM6911328, GSM6911329. Source data are provided with this paper.
References
Harmston, N. & Lenhard, B. Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res. 41, 7185–7199 (2013).
Razin, S. V. et al. Transcription factories in the context of the nuclear and genome organization. Nucleic Acids Res. 39, 9085–9092 (2011).
Stadhouders, R., Filion, G. J. & Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354 (2019).
Young, R. A. Control of the embryonic stem cell state. Cell 144, 940–954 (2011).
Kellis, M. et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 111, 6131–6138 (2014).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220 (2019).
Cook, P. R. & Marenduzzo, D. Transcription-driven genome organization: a model for chromosome structure and the regulation of gene expression tested through simulations. Nucleic Acids Res. 46, 9895–9906 (2018).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Adam, R. C. et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366–370 (2015).
Lin, L. et al. Super-enhancer-associated MEIS1 promotes transcriptional dysregulation in Ewing sarcoma in co-operation with EWS-FLI1. Nucleic acids Res. 47, 1255–1267 (2019).
Sun, X. et al. Hippo-YAP signaling controls lineage differentiation of mouse embryonic stem cells through modulating the formation of super-enhancers. Nucleic acids Res. 48, 7182–7196 (2020).
Dos Santos, M. et al. A fast Myosin super enhancer dictates muscle fiber phenotype through competitive interactions with Myosin genes. Nat. Commun. 13, 1039 (2022).
Honnell, V. et al. Identification of a modular super-enhancer in murine retinal development. Nat. Commun. 13, 253 (2022).
Kessler, S. et al. A multiple super-enhancer region establishes inter-TAD interactions and controls Hoxa function in cranial neural crest. Nat. Commun. 14, 3242 (2023).
Blinka, S., Reimer, M. H. Jr, Pulakanti, K. & Rao, S. Super-Enhancers at the Nanog Locus Differentially Regulate Neighboring Pluripotency-Associated Genes. Cell Rep. 17, 19–28 (2016).
Li, Y. et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PloS One 9, e114485 (2014).
Xie, L. et al. A dynamic interplay of enhancer elements regulates Klf4 expression in naïve pluripotency. Genes Dev. 31, 1795–1808 (2017).
Vos, E. S. M. et al. Interplay between CTCF boundaries and a super enhancer controls cohesin extrusion trajectories and gene expression. Mol. cell 81, 3082–3095.e3086 (2021).
Di Micco, R. et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Protein cell 9, 234–247 (2014).
Dowen, J. M. et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387 (2014).
Jia, R. et al. Super enhancer profiles identify key cell identity genes during differentiation from embryonic stem cells to trophoblast stem cells super enhencers in trophoblast differentiation. Front. Genet. 12, 762529 (2021).
Moorthy, S. D. et al. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 27, 246–258 (2017).
Sengupta, S. & George, R. E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 3, 269–281 (2017).
Sur, I. & Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493 (2016).
Jia, Q., Chen, S., Tan, Y., Li, Y. & Tang, F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp. Mol. Med. 52, 713–723 (2020).
Chipumuro, E. et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139 (2014).
Jiang, Y. Y. et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 66, 1358–1368 (2017).
Pelish, H. E. et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 526, 273–276 (2015).
Rowley, M. J. & Corces, V. G. Organizational principles of 3D genome architecture. Nat. Rev. Genet 19, 789–800 (2018).
Zheng, H. & Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20, 535–550 (2019).
Chen, B. et al. Long-range gene regulation network of the MGMT enhancer modulates glioma cell sensitivity to temozolomide. J. Genet. Genomics = Yi chuan xue bao 48, 946–949 (2021).
Zhang, L. et al. A HOTAIR regulatory element modulates glioma cell sensitivity to temozolomide through long-range regulation of multiple target genes. Genome Res 30, 155–163 (2020).
Qian, Y. et al. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci. Adv. 5, eaaw6710 (2019).
Wei, Z. et al. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell 13, 36–47 (2013).
Su, G. et al. CTCF-binding element regulates ESC differentiation via orchestrating long-range chromatin interaction between enhancers and HoxA. J. Biol. Chem. 296, 100413 (2021).
Su, G. et al. Enhancer architecture-dependent multilayered transcriptional regulation orchestrates RA signaling-induced early lineage differentiation of ESCs. Nucleic Acids Res 49, 11575–11595 (2021).
Li, M. et al. Comprehensive 3D epigenomic maps define limbal stem/progenitor cell function and identity. Nat. Commun. 13, 1293 (2022).
Bonev, B. et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e524 (2017).
Barutcu, A. R., Maass, P. G., Lewandowski, J. P., Weiner, C. L. & Rinn, J. L. A TAD boundary is preserved upon deletion of the CTCF-rich Firre locus. Nat. Commun. 9, 1444 (2018).
Niwa, H. The principles that govern transcription factor network functions in stem cells. Development 145, dev157420 (2018).
Pan, G. J., Chang, Z. Y., Schöler, H. R. & Pei, D. Stem cell pluripotency and transcription factor Oct4. Cell Res 12, 321–329 (2002).
Scott, C. E. et al. SOX9 induces and maintains neural stem cells. Nat. Neurosci. 13, 1181–1189 (2010).
Monsoro-Burq, A. H. PAX transcription factors in neural crest development. Semin Cell Dev. Biol. 44, 87–96 (2015).
Li, Z. et al. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci. USA 105, 13906–13911 (2008).
Sthanam, L. K. et al. Biophysical regulation of mouse embryonic stem cell fate and genomic integrity by feeder derived matrices. Biomaterials 119, 9–22 (2017).
Das, S. & Levasseur, D. Transcriptional regulatory mechanisms that govern embryonic stem cell fate. Methods Mol. Biol. 1029, 191–203 (2013).
Pan, G. & Thomson, J. A. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res 17, 42–49 (2007).
Saunders, A., Faiola, F. & Wang, J. Concise review: pursuing self-renewal and pluripotency with the stem cell factor Nanog. Stem Cells 31, 1227–1236 (2013).
Kibschull, M. Differentiating mouse embryonic stem cells into embryoid bodies in aggrewell plates. Cold Spring Harb. Protoc. 2017, pdb.prot094169 (2017).
Flamier, A., Singh, S. & Rasmussen, T. P. A standardized human embryoid body platform for the detection and analysis of teratogens. PloS one 12, e0171101 (2017).
Ai, Z. et al. Krüppel-like factor 5 rewires NANOG regulatory network to activate human naive pluripotency specific LTR7Ys and promote naive pluripotency. Sci. Adv. 40, 111240 (2022).
Aksoy, I. et al. Klf4 and Klf5 differentially inhibit mesoderm and endoderm differentiation in embryonic stem cells. Nat. Commun. 5, 3719 (2014).
Ema, M. et al. Krüppel-like factor 5 is essential for blastocyst development and the normal self-renewal of mouse ESCs. Cell Stem Cell 3, 555–567 (2008).
Hammoud, A. A. et al. Murine embryonic stem cell plasticity is regulated through Klf5 and maintained by metalloproteinase MMP1 and hypoxia. PloS one 11, e0146281 (2016).
Jiang, J. et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. cell Biol. 10, 353–360 (2008).
Parisi, S. et al. Direct targets of Klf5 transcription factor contribute to the maintenance of mouse embryonic stem cell undifferentiated state. BMC Biol. 8, 128 (2010).
Parisi, S. et al. Klf5 is involved in self-renewal of mouse embryonic stem cells. J. cell Sci. 121, 2629–2634 (2008).
Chen, S. et al. Branched-chain amino acid aminotransferase-1 regulates self-renewal and pluripotency of mouse embryonic stem cells through Ras signaling. Stem Cell Res. 49, 102097 (2020).
Raffel, S. et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388 (2017).
Choi, I. et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat. Cell Biol. 25, 963–974 (2023).
Romanov, V. S. & Rudolph, K. L. p21 shapes cancer evolution. Nat. Cell Biol. 18, 722–724 (2016).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Blayney, J. W. et al. Super-enhancers include classical enhancers and facilitators to fully activate gene expression. Cell 186, 5826–5839.e5818 (2023).
Gheldof, N., Leleu, M., Noordermeer, D., Rougemont, J. & Reymond, A. Detecting long-range chromatin interactions using the chromosome conformation capture sequencing (4C-seq) method. Methods Mol. Biol. (Clifton, N. J.) 786, 211–225 (2012).
Zhao, T. et al. lncRNA 5430416N02Rik promotes the proliferation of mouse embryonic stem cells by activating Mid1 expression through 3D chromatin architecture. Stem Cell Rep. 14, 493–505 (2020).
Joung, J. et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017).
Dehingia, B., Milewska, M., Janowski, M. & Pękowska, A. CTCF shapes chromatin structure and gene expression in health and disease. EMBO Rep. 23, e55146 (2022).
Ghirlando, R. & Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 30, 881–891 (2016).
Phillips, J. E. & Corces, V. G. CTCF: Master weaver of the genome. Cell 137, 1194–1211 (2009).
Hsieh, T. S. et al. Enhancer-promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. Nat. Genet 54, 1919–1932 (2022).
Lam, J. C. et al. YY1-controlled regulatory connectivity and transcription are influenced by the cell cycle. Nat. Genet 56, 1938–1952 (2024).
Liu, T. et al. Matrin3 mediates differentiation through stabilizing chromatin loop-domain interactions and YY1 mediated enhancer-promoter interactions. Nat. Commun. 15, 1274 (2024).
Phongbunchoo, Y. et al. YY1-mediated enhancer-promoter communication in the immunoglobulin μ locus is regulated by MSL/MOF recruitment. Cell Rep. 43, 114456 (2024).
Wang, W. et al. A histidine cluster determines YY1-compartmentalized coactivators and chromatin elements in phase-separated enhancer clusters. Nucleic Acids Res 50, 4917–4937 (2022).
Nora, E. P. et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e922 (2017).
Lee, B. K. & Iyer, V. R. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. J. Biol. Chem. 287, 30906–30913 (2012).
Bruneau, B. G. Dissecting CTCF site function in a tense HoxD locus. Genes Dev. 35, 1401–1402 (2021).
Wang, X., Cairns, M. J. & Yan, J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 47, 11481–11496 (2019).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–w102 (2017).
Li, C., Tang, Z., Zhang, W., Ye, Z. & Liu, F. GEPIA2021: integrating multiple deconvolution-based analysis into GEPIA. Nucleic acids Res. 49, W242–W246 (2021).
Jiang, Y., Jiang, Y. Y. & Lin, D. C. Super-enhancer-mediated core regulatory circuitry in human cancer. Comput. Struct. Biotechnol. J. 19, 2790–2795 (2021).
Berta, D. G. Deficient H2A.Z deposition is associated with genesis of uterine leiomyoma. Nat. Commun. 596, 398–403 (2021).
Cai, W. et al. Enhancer dependence of cell-type-specific gene expression increases with developmental age. Nucleic Acids Res. 117, 21450–21458 (2020).
Chandra, V. Promoter-interacting expression quantitative trait loci are enriched for functional genetic variants. Nature 53, 110–119 (2021).
Dai, C. et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. methods 49, 1602–1612 (2017).
Gryder, B. E. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 51, 1714–1722 (2019).
Guo, Y., Krismer, K., Closser, M., Wichterle, H. & Gifford, D. K. High resolution discovery of chromatin interactions. Nat. Genet. 47, e35 (2019).
Mai, T. et al. NKX3-1 is required for induced pluripotent stem cell reprogramming and can replace OCT4 in mouse and human iPSC induction. Nat. cell Biol. 20, 900–908 (2018).
Antao, A. M., Ramakrishna, S. & Kim, K. S. The Role of Nkx3.1 in Cancers and Stemness. Int. J. Stem cells 14, 168–179 (2021).
Xie, Q. & Wang, Z. A. Transcriptional regulation of the Nkx3.1 gene in prostate luminal stem cell specification and cancer initiation via its 3’ genomic region. J. Biol. Chem. 292, 13521–13530 (2017).
Nagel, S. et al. NKL homeobox gene activities in hematopoietic stem cells, T-cell development and T-cell leukemia. PloS One 12, e0171164 (2017).
Germann, M. et al. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. Stem Cells (Dayt., Ohio) 30, 1076–1086 (2012).
Lee, R. et al. CTCF-mediated chromatin looping provides a topological framework for the formation of phase-separated transcriptional condensates. Nucleic acids Res. 50, 207–226 (2021).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e1816 (2018).
Wang, J. et al. Phase separation of OCT4 controls TAD reorganization to promote cell fate transitions. Cell Stem Cell 28, 1868–1883.e1811 (2021).
Han, X. et al. Roles of the BRD4 short isoform in phase separation and active gene transcription. Nat. Struct. Mol. Biol. 27, 333–341 (2020).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Sci. (New York, N.Y.) 361, eaar3958 (2018).
Gibson, B. A. et al. Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e421 (2019).
Zhang, W. et al. Zscan4c activates endogenous retrovirus MERVL and cleavage embryo genes. Nucleic Acids Res. 47, 8485–8501 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–w221 (2022).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Zhou, G. et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 47, W234–w241 (2019).
Li, D. et al. WashU epigenome browser update 2022. Nucleic Acids Res 50, W774–w781 (2022).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Sci. (New York, N.Y.) 343, 84–87 (2014).
Labun, K. et al. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–w174 (2019).
Su, G. et al. A distal enhancer maintaining Hoxa1 expression orchestrates retinoic acid-induced early ESCs differentiation. Nucleic Acids Res. 47, 6737–6752 (2019).
Pedroza, M. et al. Self-patterning of human stem cells into post-implantation lineages. Nature 622, 574–583 (2023).
Weatherbee, B. A. T. et al. Pluripotent stem cell-derived model of the post-implantation human embryo. Nature 622, 584–593 (2023).
Griffiths, J. A., Richard, A. C., Bach, K., Lun, A. T. L. & Marioni, J. C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 9, 2667 (2018).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e324 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinforma. (Oxf., Engl.) 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Splinter, E., de Wit, E., van de Werken, H. J., Klous, P. & de Laat, W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods 58, 221–230 (2012).
van de Werken, H. J. et al. 4C technology: Protocols and data analysis. Methods Enzymol. 513, 89–112 (2012).
van de Werken, H. J. et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods 9, 969–972 (2012).
Hagège, H. et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 (2007).
Hua, J. T. et al. Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 174, 564–575 e518 (2018).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Sanson, K. R. et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018).
Servant, N. et al. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Hnisz, D., Day, D. S. & Young, R. A. Insulated neighborhoods: Structural and functional units of mammalian gene control. Cell 167, 1188–1200 (2016).
Abdennur, N. et al. Cooltools: Enabling high-resolution Hi-C analysis in Python. PLoS Comput Biol. 20, e1012067 (2024).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 52, D174–d182 (2024).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Xia, J., Gill, E. E. & Hancock, R. E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 10, 823–844 (2015).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 47, W556–w560 (2019).
Wang, Y. et al. The 3D genome browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Mei, S. et al. Cistrome data browser: A data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 45, D658–d662 (2017).
Zheng, R. et al. Cistrome data browser: Expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–d735 (2019).
Acknowledgements
We thank all members of the Lu Lab for their scientific discussions and helps. We also thank Dr. Fuquan Chen, Dr. Man Liu, Yaoqiang Zheng, Meng Zhang and Baoying Zhang for their helps with cell preparation in this study. We also thank Dr. Elise Lamar for revising our manuscript. The work is supported by the National Natural Science Foundation of China (Grant No.32130018 and No.32370785), the National Key R&D Program of China (No.2020YFA803700), the Natural Science Foundation of Guangdong Province (No.2024A1515010434) and the Guangzhou Youth Doctor Sailing Project (No.2024A04J4666).
Author information
Authors and Affiliations
Contributions
W.L. and G.S. conceived and designed the study. W.L., G.S., P.L., L.Z., Z.Z. and J.S. supervised the study. G.S., Q.Y., X.Z., Y.S., J.B., S.F., J.L. and D.L. performed experiments. G.S., B.C. and Q.Y. analyzed data. B.C. and W.W. analyzed sequencing data. G.S., B.C. and W.W. performed the bioinformatics analysis. G.S. and W.L. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, G., Chen, B., Song, Y. et al. Klf5-adjacent super-enhancer functions as a 3D genome structure-dependent transcriptional driver to safeguard ESC identity. Nat Commun 16, 5540 (2025). https://doi.org/10.1038/s41467-025-60389-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60389-x
