
Abstract
Ankylosing spondylitis (AS) is a chronic inflammatory arthritis that primarily affects the enthesis and may culminate in bony ankylosis of the spine. Despite TNF inhibitor (TNFi) being foundational in managing active inflammation, 30-40% of patients with AS remain non-responsive. Through longitudinal and multi-omics profiling of peripheral blood mononuclear cells from TNFi-receiving patients with AS, here we reveal that elevated type I IFN signatures at baseline are associated with poor TNFi response, leading to a paradoxical enhancement of IFN signatures and Th17 responses following TNFi therapy. Among type I IFN-related genes, we identify and validate AIF-1 as a predictive biomarker reflecting the inherent IFN signature that differentiates responders from non-responders. AIF-1 also contributes to an inflammatory cycle by increasing IFNα receptor expression and Th17 responses. In summary, our findings advocate for a personalized approach to managing AS by considering individual variations in AIF-1 levels and IFN signatures.
Similar content being viewed by others


Introduction
Spondyloarthritis (SpA) is an inflammatory disease affecting the axial skeleton and peripheral joints1,2,3. It is characterized by enthesitis, inflammation at the sites where tendons and ligaments attach to the bone4. SpA encompasses several subtypes, including ankylosing spondylitis (AS), a chronic inflammatory condition characterized by inflammation in the spine along with extra-articular manifestations, such as uveitis, psoriasis, inflammatory bowel disease, and aortitis5. However, the mechanisms by which pathological immune cells are aberrantly activated in patients with AS remain unclear. Effective disease-modifying treatments for AS became available with the advent of TNF inhibitors (TNFi)6,7,8. Nearly half of active patients with AS do not experience significant improvement with initial NSAID treatment9 and are candidates for treatment with TNFi. However, only 50-60% of TNFi users achieve >50% improvement in disease activity6,7. Considering the limited response rate, high cost, and safety issues associated with prolonged TNFi use10,11, risk stratification is required based on the mechanisms of TNFi failure rather than the current ‘try-and-fail’ approach.
The role of monocytes in AS and their responses to TNFα and TNFi treatment have been of particular interest. TNFα has been shown to induce significant changes in monocyte differentiation and function, promoting an inflammatory phenotype characterized by increased production of pro-inflammatory cytokines, such as IL-1β, IL-6, and TNFα12,13. In addition, TNFα can induce a state of tolerance in monocytes, in which pre-exposure to TNFα reduces their sensitivity to subsequent stimulation with this cytokine14,15. Conversely, TNFi treatment has been reported to suppress these inflammatory pathways12. However, the impact of TNFi on monocyte subsets and their role in mediating the response or non-response to treatment in patients with AS remains poorly understood.
Recent research has highlighted the role of IL-17-producing CD4+ T cells, known as T helper 17 (Th17) cells, in the pathogenesis of AS. The therapeutic efficacy of IL-17A inhibitor (IL-17i) highlights the importance of the Th17 axis in AS16,17. Previous studies have shown that TNF promotes Th17 differentiation by stimulating monocytes to produce IL-6 and IL-1β18, whereas TNFi suppresses Th17 cell functions19. However, the clinical response to TNFi is heterogeneous among patients with rheumatoid arthritis (RA) or AS. For example, paradoxical psoriasis, characterized by a strong Th17 response, can be induced by TNFi in patients with AS rather than RA20,21,22. In addition, a paradoxically enhanced Th17 response has been observed in patients with a non-response to TNFi21,22. The mechanisms underlying this enhancement of the Th17 response remain unresolved.
Previous studies have proposed various serum molecules and composite markers, such as C-reactive protein (CRP), vascular endothelial growth factor, serum amyloid A, and tumor necrosis factor superfamily member 14, as potential predictors of the TNFi response in AS23,24,25,26. A few attributes have also been linked to a favorable response, including younger age, shorter disease duration, male gender, and HLA-B27 positivity25,27. Despite these findings, none of these markers or models have been widely adopted in clinical practice.
In this study, we aim to clarify immunological mechanisms behind TNFi non-response in AS and identify biomarkers for predicting therapeutic outcomes. Utilizing single-cell RNA sequencing, we identify transcriptional signatures linked to poor TNFi responses, particularly highlighting type I IFN-related pathways. Our findings support personalized therapeutic strategies and provide insights that may guide improved patient management and treatment optimization.
Results
Comparative immune landscape of PBMCs from TNFi-treated patients with AS highlights significant cellular differences between responders and non-responders
To elucidate the mechanisms differentiating TNFi responders from NRs among patients with AS, we collected PBMCs from 225 patients both before and after treatment with either TNFi or IL-17 inhibitor (IL-17i). This included a discovery cohort (n = 22) and three validation cohorts of TNFi users: cohort 1 (n = 18), 2 (n = 117), and 3 (n = 58) (Fig. 1a, Supplementary Data 1).

a Schematic of the study design, showing blood sample collection from 167 patients with ankylosing spondylitis (AS) before and after TNFi treatment. This includes a discovery cohort (n = 32), of which 22 patients (responders: R_Pre, R_Post, n = 12; non-responders: NR_Pre, NR_Post, n = 10) underwent scRNA-seq analysis, as well as validation cohort 1 (n = 18) and validation cohort 2 (n = 117). Figure created with BioRender. Created in BioRender. https://BioRender.com/q70l192. b UMAP of single-cell RNA sequencing data from PBMCs (n = 199,926), revealing 10 major cell clusters colored according to cell type. c Comparisons between pre- and post-treatment within the same group (R, NR) were performed using the Wilcoxon matched-pairs signed rank test or a paired t-test, while comparisons between different groups (R_Pre vs. NR_Pre, R_Post vs. NR_Post) used a two-sided Mann-Whitney U test or an unpaired t-test, based on normality. In the cluster names, “CD14 M” and “CD16 M” refer to CD14⁺ and CD16⁺ monocytes, respectively, and “prolif” indicates proliferating cells. Bar plots indicate means, and error bars represent standard deviation (SD). Exact p-values for CD14 Monocyte and NK cell comparisons within responders are 0.00012 and 0.00029, respectively (Wilcoxon matched-pairs signed rank test, two-sided). d Milo analysis comparing PBMC neighborhoods (k = 45) between NR and R baselines. Red indicates neighborhoods more abundant in the NR group, and blue indicates neighborhoods more abundant in the R group. Nhood size corresponds to circle size, and overlap size corresponds to line thickness. e Neighborhood function that uses community detection to partition neighborhoods, showing automatic grouping of 15 Nhood groups in different colors. f Visualization of differential abundance (DA) fold changes in different neighborhoods, focusing on the distribution in PBMCs: cytotoxic (CTX), proliferating (PRF), central memory (CM), and effector memory (EM). Statistical testing for differential abundance utilized the quasi-likelihood F-test implemented in edgeR (two-sided), with spatial FDR controlled using a weighted Benjamini–Hochberg method. g Pearson correlation coefficient analysis of TNFi-induced changes. Responder pairs (n = 12), non-responder pairs (n = 10); error bars indicate standard error. Statistical significance was tested using the Wilcoxon matched-pairs signed rank test (two-sided). Source data are provided as a Source Data file.
Among the discovery cohort, we performed 3′ scRNA-seq on PBMCs collected from responders (n = 12) and non-responders (n = 10) before (R_Pre and NR_Pre) and 1 month after TNFi treatment (R_Post and NR_Post); 19 patients were male (Fig. 1a). Louvain clustering followed by UMAP visualization revealed 10 major cell clusters characterized by canonical gene expression (n = 199,926; Fig. 1b, Supplementary Fig. 1a, b and Supplementary Data 2). From the perspective of cell proportions in the 10 clusters across the four conditions, a distinct responsive pattern to TNFi was observed between responders and NRs. Specifically, a TNFi response was associated with a marked decrease in CD14+ monocytes and an increase in B cells, as reported previously (Fig. 1c)28. Conversely, the NRs exhibited an increase in NK cells and plasma cells with TNFi treatment. At baseline, the NR group had higher proportions of T cells and proliferating cells compared to responders, both before and after TNFi treatment. We additionally summarized the more detailed subtypes and assessed their statistical significance (Supplementary Fig. 1c, d).
To further investigate baseline differences, we employed the neighborhood-based Milo analysis, which provides a differential abundance analysis on k-nearest neighbor (KNN) graphs29 (Fig. 1d, e and Supplementary Data 3). The results showed an increased abundance of cytotoxic T cells and proliferating cells (Nhoodgroup 10) and NK cells (Nhoodgroup 14) at baseline among the NRs, whereas certain monocyte neighborhoods were also more prevalent (Fig. 1f). In contrast, responders exhibited higher abundance of naive (Nhoodgroup 4) or effector memory T cells (Nhoodgroup 13) at baseline. This suggests that the increased T-cell proliferation in the NR group may be related to stimulation by autoantigens or an inflammatory milieu.
To further elucidate the cell populations responsible for the alterations induced by TNFi, we assessed the Pearson correlation coefficients depicting the amount of interval change with treatment within individual clusters (Fig. 1g and Supplementary Fig. 1d). The greatest differences between responders and NRs were observed in monocytes (P = 0.0596) and B cells (P = 0.0533). B cells underwent fewer transcriptome changes in NRs than in responders, whereas monocytes underwent the most prominent transcriptome changes of all the cells in both the responder and NR groups, prompting us to focus on monocyte analysis in subsequent investigations.
Monocyte heterogeneity highlighting interferon-related signatures distinguishes TNFi non-responders
We performed subclustering (n = 52,218) in subsets of CD14+ and CD16+ monocytes, revealing three main populations: classical monocytes (cMo), intermediate monocytes (intMo), and non-classical monocytes (ncMo) (Fig. 2a and Supplementary Data 4). Within these populations, specific subclusters were identified, including those with high expression of IL1B (inflammatory cytokine), interferon-stimulated genes (ISGs) (e.g., HERC5), EGR1, and CXCL10 (Fig. 2b). CXCL10, a ligand for CXCR3, defines a pathogenic monocyte in the central nervous system during autoimmune neuroinflammation and systemic lupus erythematosus (SLE)30. This cluster also presents elevated GBP1 and SLAMF7 expression, both of which are induced by interferon (IFN)31. SLAMF7 serves as a significant marker of hyperinflammation in RA patients. In addition, ncMo subclusters were distinguished by their differing levels of ISG expression (Fig. 2b).

a UMAP of CD14+ or CD16+ monocytes (n = 52,218), with 13 clusters identified and color-coded, including classical (cMo), intermediate (intMo), and non-classical (ncMo) monocytes. b Dot plot showing the average expression of marker genes for each monocyte cluster. c Milo analysis UMAP comparing non-responders (NRs) and responders (Rs), with red indicating higher abundance in NRs and blue indicating a higher abundance in Rs. Neighborhoods with a spatial FDR < 0.5 are plotted in the figure. Cells are depicted as circles proportional in size to the number of cells contained. d Beeswarm and box plots illustrating log2-fold differences across neighborhoods. Boxplot center lines represent medians, boxes show interquartile ranges (IQR), whiskers extend to the smallest and largest values within 1.5 × IQR from the box edges, and points beyond whiskers indicate outliers. e Boxplots comparing baseline proportions between groups (R_Pre, n = 12; NR_Pre, n = 10). Statistical tests were two-sided t-test or Wilcoxon tests, depending on normality. Boxplot definitions as in panel d. Data are presented as mean ± standard deviation. f Radar plot showing -log(adjusted P value) of the top 5 cytokines from Immune Response Enrichment Analysis for EGR1hi cMo and CXCL10hi cMo. FDR-adjusted p-value from two-sided Wilcoxon rank-sum test. g K-means clustering of DEGs in monocytes, showing the relative expression under different conditions. Significant modules are highlighted in red. h Pathway analysis of each module using MSigDB HallMark 2020, showing the top 3 significant pathways per module. Red intensity indicates the significance, and circle size represents the enrichment score. Statistical analysis using MAST (two-sided), BH-adjusted. i, Gene Ontology analysis of DEGs between R_Pre and NR_Pre. Blue represents R_Pre, and red represents NR_Pre enriched ontology, BH-adjusted. j Correlation analysis of the IFN\(\alpha \) response score with CXCL10hi cMo cluster and delta ASDAS. Spearman correlation was used for the cluster score and Pearson correlation for delta ASDAS, both two-sided. Shaded bands represent 95% confidence intervals. k Bar graphs showing the average IFN\(\alpha \) score (paired samples: R n = 12, NR n = 10, IL-17i n = 10) per sample, compared across conditions. Source data are provided as a Source Data file.
Neighborhood-based Milo analysis of monocytes showed that the CXCL10hi cMo, transitional monocyte, and ISGhi ncMo populations were significantly more abundant in the NR group (Fig. 2c, d). A cluster-based comparison reaffirmed that CXCL10hi cMo and ISGhi ncMo populations were more abundant in the NR group (Fig. 2e, Supplementary Fig. 2a). Conversely, EGR1hi monocytes were more prevalent in the R_Pre samples, reflecting that EGR1 is a gatekeeper for inflammation32.
To identify cytokine combinations that influence these transitions and activations, we conducted immune response enrichment analysis (IREA)33 (Fig. 2f). IREA employs statistical tests to evaluate the enrichment of cell polarization or cytokine signatures in transcriptomes. This analysis revealed that EGR1hi monocytes are influenced by a variety of cytokines, including IL-1a and IL-3, whereas CXCL10hi monocytes are influenced by IFN\(\alpha \), IFN\(\gamma \), and IL-15. This observation was further supported by a comparison of the expression levels of IFN receptors, with CXCL10hi cMo demonstrating elevated expression levels for both IFNAR1 and IFNAR2, in contrast to the EGR1hi cMo, which exhibited the least expression (Supplementary Fig. 2b).
Next, we used k-means clustering to identify differentially expressed genes (DEGs) across conditions, yielding eight modules with significant pathway associations (Fig. 2g, h). Module 7, which prominently features specific type 1 IFN response genes, such as STAT2 and OAS1, was highly elevated in the NR group both pre- and post-TNFi treatment. Interestingly, module 8, which includes CXCL10 and STAT1, showed an increase in response to TNFi treatment, suggesting an intensified IFN response. Other modules displayed diverse patterns; module 4 (TNF and mTOR1-related genes) decreased across both groups, and module 1 (IFN response genes) increased in NR_Post but decreased in R_Post.
These findings underscore the elevated IFN signature in the monocytes of NRs at baseline, prompting us to perform gene set enrichment analysis (GSEA) of baseline DEGs (Fig. 2i). NR_Pre samples were enriched for type 1 IFN modules, whereas R_Pre samples were enriched in neutrophil migration and activation ontologies34. This elevated IFN signature is dependent on phosphorylated ISGF3 (P-ISGF3) which is mediated by phosphorylation at the Tyr690 of STAT2 and IRF9, highly expressed in two clusters that were enriched in NR (Supplementary Fig. 2c, d)35,36. These findings indicate that IFN signaling was continuously active up to the time of sampling.
We subsequently examined the specific cell types within PBMCs that exhibited distinct scores for the IFN\(\alpha \) response37 (Supplementary Fig. 2e). As expected, the most substantial discrepancy in the IFN response score between responders and NRs occurred in monocytes. Remarkably, this module showed a strong positive correlation with the CXCL10hi cMo population, which was previously abundant in NRs, and a negative correlation with the treatment response (delta ASDAS), supporting its predictive value for the response to TNFi (Fig. 2j). Finally, we compared the IFN\(\alpha \) response score (signature) not only pre- and post-TNFi, but also in the IL-17i-treated group, as IL-17i can be an alternative for TNFi-refractory patients with AS38. Because our IL-17i-treated group had a history of TNFi-refractoriness, the IL-17i_Pre group has a significantly higher IFN\(\alpha \) signature than the R_Pre group (Fig. 2k). Meanwhile, the IFNγ response score did not show as marked a difference between responders and NRs (Supplementary Fig. 2f). In contrast to the tendency of the IFN\(\alpha \) signature to increase in the NR group with TNFi treatment, it tended to decrease after IL-17i treatment (P = 0.084).
Specific monocyte populations and AIF-1 levels at baseline strongly correlate with a TNFi non-response, identifying AIF-1 as a potential predictive biomarker
To deepen our understanding of baseline differences and identify potential markers, we focused on the cluster that increased in the NR_Pre group. The ISGhi ncMo population, which was enriched in the NR_Pre group, showed a significant positive correlation with the IFN\(\alpha \) signature (Fig. 3a and Supplementary Fig. 3a). Notably, the slope of the correlation was steeper for NRs than for responders, suggesting a stronger correlation. Furthermore, similar to the CXCL10hi cMo population, the proportion of this cluster negatively correlated with delta ASDAS, suggesting clinical relevance. Therefore, we explored specific markers distinguishing the ISGhi ncMo population from typical ncMo (Fig. 3b). Among various markers, AIF1 emerged with the strongest significance. Given its high expression in infiltrating monocytes and macrophages in RA induced by IFN39,40 and its known status as a soluble protein, we hypothesized that AIF-1 could serve as a predictive marker for TNFi non-responders. Therefore, we designed experiments to validate AIF-1 as a biomarker and determine its function (Fig. 3c).

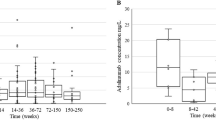
a Correlation of ISGhi ncMo proportion with IFN\(\alpha \) score and delta ASDAS. P values were calculated using Pearson correlation (two-sided), shaded bands represent 95% confidence intervals. Left, the black line indicates the overall correlation slope, and the gray lines indicate the NR (steeper slope) and R groups. b Volcano plot comparing ISGhi ncMo and ncMo. Thresholds were set at absolute log2 fold change of 0.25 and adjusted P value of 0.05 (MAST (two-sided, BH adjustment)). c Experimental design to validate AIF-1 as a predictive biomarker of the response to treatment with TNF inhibitor (TNFi). Figure created with BioRender.com. Created in BioRender. https://BioRender.com/q70l192. d Gene expression of AIF1 by qRT-PCR compared between Rs (n = 10) and NRs (n = 8) using PBMCs from TNFi-treated SpA patients in the validation cohort 1 at baseline (Welch two-sample t-test (two-sided)). Data are presented as mean ± standard deviation. e Baseline serum level of AIF-1 measured by ELISA compared between Rs (n = 69) and NRs (n = 48), two-sided Mann-Whitney test. Three different criteria (BASDAI50, ASAS40, and ASAS20) were used to determine the clinical response at week 14. Data are presented as mean ± standard deviation. f, g ROC curve of baseline serum AIF-1 levels for predicting a non-response at week 14 (f), odds ratio analysis of serum AIF-1 levels for TNFi response prediction. The gray diamond represents the unadjusted odds ratio, while the red diamond represents the adjusted odds ratio, accounting for age, initial BASDAI, CRP, and ESR in the logistic regression model. After adjustment, the odds ratio remained significant at 6.96 (95% CI). Error bars indicate 95% confidence intervals, with center points representing odds ratios (unadjusted, gray; adjusted, red) (g). h Validation of baseline serum AIF-1 levels in an independent cohort (Validation cohort 3, n = 58). Data shown as mean ± standard error; statistical significance assessed by Wilcoxon rank sum test. Exact P-value = 3.64e-06. i Change in serum AIF-1 between week 0 (baseline) and week 14 after initiating TNFi (R n = 59, NR n = 39), two-sided t-test. Source data are provided as a Source Data file.
The demographic and clinical characteristics of the responder and NR groups were not significantly different in the validation cohort 1 (Supplementary Data 1). Significant upregulation of AIF1 in NRs was confirmed by qRT-PCR (n = 18; Fig. 3d). In validation cohort 2, which was derived from an independent clinical trial evaluating TNFi biosimilar CT-P13, the primary endpoints included pharmacokinetic parameters as well as efficacy endpoints such as ASAS20 and ASAS40 response rates41. In this cohort, the serum concentration of AIF-1 was consistently higher in non-responders compared to responders across all three evaluated response criteria (n = 117; Fig. 3e).
The AUC analysis provided an optimal cut-off value of 63.5 pg/ml for the serum AIF-1 concentration to differentiate NRs from responders (Fig. 3f). After a multivariate adjustment, the patients with AS and high AIF-1 ( > 63.5 pg/ml) had a 6.96-times higher risk of a non-response than the low AIF-1 group (Fig. 3g). Additionally, this serum cut-off level was further validated in the independent cohort (validation cohort 3, n = 58), the sensitivity and specificity were 89.5% and 82.1, respectively (Fig. 3h and Supplementary Fig. 3b–d). In validation cohort 3, serum levels of IFNγ did not differ significantly between responders and non-responders, and type 1 interferons (IFNα and IFNβ1) were predominantly below 1 pg/ml without a clear trend (Supplementary Fig. 3e).
Serum AIF-1 levels correlated positively with the initial BASDAI score. However, they did not correlate with the blood levels of common inflammatory markers, such as CRP and erythrocyte sedimentation rate (Supplementary Fig. 3f). Serum AIF-1 levels did not change significantly with TNFi treatment when compared between weeks 0 and 14 (Fig. 3i). Taken together, these results show that serum AIF-1 levels were higher in patients with AS who were not responsive to TNFi, and can be used as a stable predictive biomarker that is not affected by TNFi use.
AIF-1 increases monocyte populations with the footprint of non-responders to TNFi
To understand the role of elevated AIF-1 in the NR group, we focused on its impact on monocytes, the population most affected by TNFi (Fig. 1g). First, we treated PBMCs from healthy donors with AIF-1 to evaluate its direct and indirect interactions in human immune cells and performed scRNA-seq analysis (Fig. 4a, Supplementary Fig. 4a–d, and Supplementary Data 5, 6). Second, we isolated CD14+ monocytes of PBMCs from healthy donors using CD14 microbeads to compare the direct stimulatory effect of AIF-1 in vitro with the transcriptome level.

a Schematic of the experimental design. PBMCs from healthy donors (n = 4) were treated with AIF-1 or control (Ctl) and subjected to scRNA-seq. In addition, CD14+ monocytes were isolated from PBMCs using CD14 Microbeads and treated with AIF-1 or Ctl, followed by bulk RNA sequencing. Created in BioRender. https://BioRender.com/q70l192. b UMAP of monocytes from AIF-1-treated PBMCs, revealing 10 clusters (n = 2245). c Dot plot showing the average expression of key markers in each cluster from AIF-1-treated UMAP. d Violin plot of log-normalized expression values for SLAMF7, GBP1, IRF7, and ISG20. e Bar plots showing the proportion of each cluster in AIF-1-treated versus Ctl PBMCs, represented as the AIF-1/Ctl ratio. f Dot plot comparing the expression of IFNAR1 and IFNGR1 between AIF-1 treated monocytes and control monocytes. The accompanying table summarizes the average log2 fold change (avg_log2FC), Bonferroni-adjusted p-value (adj p-value), and expression levels (exp) in AIF-1 treated versus control cells. (Wilcoxon rank sum test, two-sided). g Heatmap showing the expression of inflammatory cytokines (e.g., IL1B, IL6, IL-23) in monocytes treated with AIF-1 versus Ctl as determined by bulk RNA sequencing. h GSEA plot of DEGs from bulk RNA-seq, highlighting monocyte-derived dendritic cells (Mo-DCs) and BDCA1+ DC markers. i Schematic overview of the experimental design. THP-1 monocytes were transfected with siRNA targeting AIF1 (siAIF1) or scrambled control siRNA via liposomal carrier-based transfection. Created in BioRender. https://BioRender.com/q70l192. j Bulk RNA-seq analysis (n = 4) comparing the expression of AIF1 and Th17-promoting cytokines (IL1B, IL6, IL23A) between control and AIF1 knockdown (KD) THP-1 cells. Statistical comparisons performed t-test and Wilcoxon test (two-sided). Exact p-values: AIF1 = 2.68e-5, IL6 = 1.37e-5, IL23A = 7.30e-5. k FACS analysis comparing the proportion of IL-17A+ and IFNγ+ populations within memory CD4+ T cells between control and siAIF1-treated conditions (n = 5, biological replicates). Statistical significance assessed by paired t-test (two-sided). Figure 4a and i created with BioRender. Source data are provided as a Source Data file.
In the scRNA-seq data, we analyzed monocytes and re-clustered them (n = 2245), identifying 10 distinct clusters (Fig. 4b). Importantly, we identified clusters mirroring the CXCL10hi cMo and EGR1hi cMo populations found in the TNFi-treated groups depicted in Figs. 2a, 4c). SLAMF7 and GBP1, critical markers identified earlier, were highly expressed in the CXCL10hi cluster which exhibited elevated ISG expression (Fig. 4d and Supplementary Fig. 4e). Conversely, the EGR1hi cluster had low expression of SLAMF7 and GBP1, consistent with our previous patient data analysis. These findings suggest that AIF-1-treated monocytes share characteristics with those observed in NRs.
We further investigated how these monocytes transitioned in response to AIF-1 treatment (Fig. 4e). Inflammatory monocytes significantly increased, followed by the CXCL10hi cluster, indicating that AIF-1 promotes these specific monocyte subpopulations similar to those seen in the NRs. Conversely, the control group had higher proportions of clusters associated with anti-inflammatory markers, such as CD163, and the EGR1hi cluster, which was more prevalent in the responder group (Fig. 4e).
Next, we sought to understand the mechanism by which AIF-1 contributes to the IFNα signature. AIF-1 was found to upregulate IFNAR1, thereby increasing the sensitivity to IFNα, while downregulating IFNGR1, highlighting differential regulation of interferon receptors (Fig. 4f). Through gene ontology analysis of type 1 IFN-related pathways, we confirmed that monocytes with heightened sensitivity due to AIF-1 are more susceptible to IFNα-driven responses (Supplementary Fig. 4f).
The bulk RNA-seq results revealed that AIF-1 treatment increased the expression of inflammatory cytokines, such as IL-1β and IL-6, as well as IL-23, which can promote Th17 cells42,43,44,45 (Fig. 4g). ELISA confirmed that these cytokines were secreted by AIF-1 stimulation in monocytes (Supplementary Fig. 4g). GSEA of DEGs from bulk RNA-sequencing revealed that AIF-1-treated monocytes exhibited characteristics of monocyte-derived dendritic cells (Mo-DCs), which have strong Th17 driving features compared with other monocyte or macrophage populations46 (Fig. 4h).
We performed AIF-1 knockdown experiments using the human monocyte cell line THP-1, transfecting cells with either siRNA targeting AIF1 (siAIF1) or scrambled control siRNA via a liposomal carrier-based method (Fig. 4i). Successful knockdown of AIF-1 significantly reduced critical inflammatory mediators (IL1B, IL6, IL23A, IL12B), known to enhance Th17 responses (Fig. 4j). Additionally, primary CD4+ T cells cultured with conditioned media from siAIF1-treated monocytes showed substantially reduced Th17 responses compared to cells cultured with control-conditioned media (Fig. 4k). These results indicate that AIF-1-induced monocyte transitions reflect the state of NRs and AIF-1 is a critical factor contributing to this phenotype.
TNFi treatment paradoxically enhances IFN response in non-responders, highlighting distinct gene expression patterns that differentiate these patients from responders
Having established that an IFN signature marked by AIF-1 is a critical factor determining non-response to TNFi at baseline, we focused on the interval changes induced by TNFi in the responder and NR groups (Fig. 5a, b). Analyzing the distribution of significantly altered genes in each quadrant, we observed that most genes in the responder group were located in the third quadrant. In contrast, genes changed only in the NR group predominantly occupied the first quadrant, indicating an upregulation by TNFi (Fig. 5b). Interestingly, key TFs and response elements of the type 1 IFN response, such as IRF1, GBP1, and STAT1, exhibited significant upregulation regardless of the TNFi response. Furthermore, CXCL10 and SLAMF7, which are critical for monocyte activation in autoimmune diseases, paradoxically increased with TNFi treatment only in NRs. We also identified genes that decreased in responders but increased in NRs, such as DDIT4 and MX1, as well as genes primarily heightened in NRs (e.g., IFIT1) that correspond to unphosphorylated ISGF3 (U-ISGF3). Conversely, the anti-inflammatory genes CTNNB1 and ATF3 are associated with WNT signaling, with ATF3 activating WNT signaling by increasing tenascin-C47,48 and SGK1 expression, reducing the amount of inflammatory cytokines, such as TNF\(\alpha \). All three of these were reduced in NRs but significantly increased in the responders49.

a A plot of DEGs in monocytes for responders (Rs) and non-responders (NRs) before and after treatment with TNF inhibitor (TNFi). Each point represents a gene, colored by gene type as indicated in the lower right legend. b Bar graphs showing the proportion of genes in each quadrant, specifically highlighting R_only and NR_only genes. c Dot plot of Gene Ontology analysis for significant genes based on their response pattern, showing the top 5 terms per group, with red indicating higher significance. (Wilcoxon rank sum test (BH adjustment, two-sided)). d Schematic of in vitro experiments to assess the effect of adalimumab (ADA). Human PBMCs from four individuals were treated with TNFα for 12 h, followed by IgG1 control or ADA for 6 h prior to scRNA-seq. e The proportion change in the CXCL10hi cMo cluster in response to ADA treatment, with each dot representing one sample (n = 4, biological replicates). f Violin plots of normalized expression of SLAMF7 and JUN across four conditions. g A volcano plot of DEGs between ADA and control (IgG1), with red indicating downregulated genes and blue indicating upregulated genes (BH-adjusted P value). h Boxplots comparing the average expression of ADA-upregulated gene modules in Rs and NRs. Boxplot comparison (R n = 10, NR n = 12), two-sided t-test. i A 3D plot of baseline patient status defined by three variables, with arrows indicating the Pearson correlation between IFN\(\alpha \) score and delta ASDAS (upper left) and between the score and ADA-upregulated module (lower right). Each point represents a patient (blue for R, red for NR). j Plots of regulon specificity scores (RSS) based on the AUC in each condition. The top 10 regulons with the highest RSS are indicated by red circles. k Heatmap with binarized regulon activity. Regulons that showed significant differences between pre- and post-TNFi in the NR group are highlighted with red boxes. l Motif logos of the highest normalized enrichment score (NES) motifs for key transcription factors in the R and NR groups. m A PCA plot illustrating the contribution of different programs to the separation of Rs and NRs and their changes post-TNFi treatment, with ADA-up representing in vitro ADA-upregulated modules. Statistical analyses were performed using the unpaired t-test. Source data are provided as a Source Data file.
We classified the genes based on their expression patterns and performed ontology analysis to identify associated biological processes. Genes that increased only in the NR group were significantly associated with viral defense and IFN response pathways. Interestingly, genes that increased in both responders and NRs due to TNFi were also linked to the IFN response (Fig. 5c).
To verify whether these responses were TNFi-dependent, we conducted in vitro experiments in which we treated PBMCs from healthy donors with TNF and adalimumab (ADA) and performed scRNA-seq to consider all cellular interactions (Fig. 5d, Supplementary Fig. 5a, b, and Supplementary Data 7). The CXCL10hi cMo cluster, which was enriched in NRs, decreased with TNFi treatment across all samples (Fig. 5e). Specifically, critical markers, such as CXCL10 and SLAMF7, which increased in NRs treated with TNFi, significantly decreased with in vitro ADA treatment (Fig. 5f, g). In addition, genes such as STAT1 and IRF5, which are important for the type 1 IFN response50, increased with ADA treatment, and MS4A6A, which is co-expressed with CD163 in monocytes, and EGR1, which was enriched in the responders, also increased. We then compared the ADA-upregulated features in both groups to examine whether these features were increased in the pre-treatment state. The ADA-upregulated module was significantly higher in the NR_Pre group, and it positively correlated with the IFN\(\alpha \) response (Fig. 5h, i). Thus, the TNFi response in patients with AS is influenced by a specific biological pathway (i.e., IFN\(\alpha \) signature), drug characteristics (i.e., ADA), and clinical outcomes (i.e., ASDAS), separating patients into two distinct groups (Fig. 5i).
To delve into the mechanisms regulating each group and identify key regulatory factors, we analyzed the upstream regulons of these programs using pySCENIC for TF network analysis (Fig. 5j). In the responder group, EGR1 maintained its influence pre- and post-TNFi, acting as a gatekeeper, with ELK4 expression emerging post-TNFi to activate anti-inflammatory programs51. EGR1 inhibits the activation of the inflammatory enhancer, which in turn suppresses the IFN response and the more commonly known IL-632,52. In contrast, the NR group lacked the influence of EGR1; instead, IRF5, a central driver of the IFN\(\alpha \) signature and known for its role in SLE53,54, dominated pre-treatment. Post-TNFi, NRs exhibited increased regulation by STAT2 and IRF9, key regulators of IFN\(\alpha \) responses. Notably, ETS2, which was recently associated with AS and inflammatory bowel disease, emerged as a top regulon, indicating its role in worsening conditions by amplifying inflammatory responses55 (Fig. 5j). Motif analysis indicated that IRF9 interacts with STAT, which possesses a high NES motif (Fig. 5k, l, Supplementary Fig. 5e). These changes were unique to NRs, highlighting the distinct IFN burden.
To explore other detrimental effects of TNFi in the NRs, we examined the changes in TNF\(\alpha \)-induced tolerized genes15. TNF\(\alpha \) typically epigenetically desensitizes genes with NF-kB and partial ISRE binding motifs, but IFN\(\alpha \) can reverse this effect. We hypothesized that reduced cross-tolerance by TNFi would heighten IFN\(\alpha \)-induced disruption of tolerization. Analyzing our samples, we found that tolerized genes (Class 2) and type 1 IFN-related genes (Class 3) were expressed at significantly higher levels in NRs (Supplementary Fig. 5c). Furthermore, the differences between responders and NRs broadened after TNFi treatment, with significant ISRE motif enrichment in genes showing extreme differences. Class 2 genes exhibit a strong positive correlation with the NOD-like receptor (NLR) pathway, which is recognized for mediating type 1 IFN signaling through IRF756. Notably, CXCL10hi cMo demonstrated the most pronounced signal within this correlation (Supplementary Fig. 5d). This suggests that NR exacerbation post-TNFi is linked to epigenetic regulatory mechanisms, as evidenced by the CXCL10hi cluster proportions.
Finally, principal component analysis (PCA) analysis of responders and NRs highlighted that PC1 (69.58% explained variance) distinctly separated patients into two groups. The primary forces contributing to non-response and worsening conditions post-TNFi in NRs were pathways related to IFN, NLR, and JAK-STAT34,57,58 (Fig. 5m). In summary, the paradoxical effect of TNFi in NRs manifests through both direct and indirect mechanisms. Commonly upregulated ISGs and NR-specific U-ISGF3 highlight why TNFi are ineffective in patients with a high IFN burden.
IFNα synergizes with TNFα to enhance inflammatory gene expression in NR model monocytes, reflecting in vitro validation
We aimed to validate whether the presence of IFNα can reproduce the NR model observed in AS patients. Human CD14+ monocytes from healthy donors were isolated and cultured under six different conditions with combinations of cytokines and ADA. Cytokines were administered for 24 h, with ADA or IgG1 isotype control added 12 h after the initial cytokine treatment for an additional 12 h. The R model was represented by conditions T and T + ADA, whereas the NR model was represented by conditions I + T and I + T + ADA (Fig. 6a and Supplementary Fig. 6a).

a In vitro bulk RNA-seq experimental design. CD14+ monocytes were isolated from human PBMCs and treated with combinations of cytokines and adalimumab (ADA) to create six conditions before harvest and library construction. ‘I’ denotes IFNα and ‘T’ denotes TNFα. b Heatmap of DEGs identified through k-means clustering across six conditions. Modules are named and characterized on the left. Statistical confirmation was performed using the MAST algorithm (Seurat implementation), with significance determined by Bonferroni-adjusted P < 0.05 and a log2FC > 0.1. c Heatmap showing the relative comparison of modules scored against 11 CD14+ monocyte clusters from patients with AS. Columns are ordered based on the Euclidean distance. d Bar plots showing the pathway analysis and TF analysis for module 4, with the top three P value terms highlighted. Terms with an adjusted P < 0.05 are in green; non-significant terms are in gray. Combined scores were calculated by multiplying the Fisher exact test log(P value) by the z-score of the deviation from the expected rank. (BH adjustment, two-sided) e Venn diagram of ADA-upregulated genes in the R and NR models. Roman numeral I denotes R model-specific genes, II denotes genes upregulated in both models, and III denotes NR model-specific genes, with counts provided. Bar graphs show the counts and proportion of genes upregulated by IFNα added to TNFα or to control within each category.f Line graph of deconvoluted proportions (n = 3) from in vitro bulk RNA-seq data using patient data as the reference. Key clusters from patient data are in bold. Error bars represent the standard error of the mean (SEM). g Box plot comparing the cosine similarity of deconvoluted data for the key clusters in bold from the I + T condition between R_Pre and NR_Pre samples (R n = 12, NR n = 10) and analyzed using the Wilcoxon rank sum exact test. Boxplot definitions as in Fig. 2d. Source data are provided as a Source Data file.
To assess the expression patterns across these conditions, we performed k-means clustering and identified seven significant modules (Fig. 6b). Modules 6 and 7 exhibited a synergistic effect of IFNα and TNFα, particularly module 7, which exhibited unique behavior not observed in the R model. Notably, the CXCL10hi cMo population prominently expressed this feature in the AS patient data (Fig. 6c). In addition, two modules (3 and 4) were upregulated by ADA, with module 4 being specific to the NR model. Pathway analysis of this module revealed enrichment in the IFN response genes, and TF analysis identified IRF8 as being significantly involved (Fig. 6d). We further explored the impact of ADA on gene expression by analyzing ADA-upregulated genes in both the R and NR models. As expected, ADA did not increase TNFα-induced genes, but it did upregulate IFNα response genes (IFNα signature) in both models. In particular, the NR model-specific genes had a high proportion (48.6%) of IFNα response genes, compared to only 2.9% in the R model (Fig. 6e, Supplementary Fig. 6b, c). Thus, we observed an increase in some IFN responses due to TNFi, as seen in the patient data.
Next, we utilized CIBERSORTx to deconvolute our in vitro data59 (Fig. 6f). A striking observation was an absence of the EGR1hi cluster in all conditions containing IFNα, indicating that IFNα significantly reduces this cluster. This finding aligns with the increased presence of the CXCL10hi cluster and decreased EGR1hi cluster in the NR group, demonstrating the causal role of IFNα in shaping the NR environment.
Finally, we evaluated the cosine similarity of deconvoluted proportions between in vitro conditions and patient data. We found that the I + T condition more accurately reflects the NR group (P = 0.0090; Fig. 6g). Thus, our in vitro experiments confirmed that the combined presence of IFNα and TNFα simulates the NR conditions.
Non-responders exhibit increased activated Th17 cells with enhanced effector functions post-TNFi treatment
Our study, along with others, has emphasized the importance of Th17 cells in the pathophysiology of AS60,61. Given that T cells likely reflect long-term patient-specific differences, and considering our previous data showing that, in NRs, monocytes exhibit Mo-DC features and likely interact with CD4+ T cells, we analyzed memory CD4 + T cells (Figs. 2i, 7a, Supplementary Fig. 7a). Memory CD4+ T cells were categorized into eight distinct clusters, the most notable being the activated Th17 cluster (Supplementary Data 8). This cluster highly expressed TNFRSF4, NR4A1, and FURIN (activation marker)62. Another notable cluster was the dysfunctional MAFlo Treg cluster63. The activated Th17 cluster and MAFlo Treg cluster were both enriched in the NR group (Fig. 7b).

a UMAP of memory CD4+ T cells (n = 17,777) from TNF inhibitor (TNFi)-treated patients, displaying eight distinct clusters. b Boxplots (R n = 12, NR n = 10) comparing effector memory (EM, Wilcoxon two-sided), activated Th17, and MAFlo Treg clusters (two-sided t-test) in the R and NR baselines. Boxplot definitions as in Fig. 2d. c Violin plots showing normalized expression levels of RORA and CD69 in activated Th17 cells. (Wilcoxon rank-sum test, two-sided, BH adjustment) d Plots showing the correlation between CXCL10hi cMo and the activated Th17 and MAFlo Treg clusters. Pearson and Spearman correlations (two-sided); shaded bands represent 95% confidence intervals. e, f Serum AIF-1 concentrations higher or lower than the cut-off of 63.5 pg/ml were denoted as ‘High’ (n = 12 patients with AS) or ‘Low’ (n = 16 patients with AS), respectively. PBMCs were stimulated by anti-CD3 antibody and anti-CD28 antibody for 6 h, and the CD4+CD45RA- population was considered memory CD4+ T cells. e Proportions of IL-17A+, IFNγ+, and IL-17A+IFNγ+ cells among memory CD4+ T cells were measured by flow cytometry after intracellular cytokine staining (ICS). Two contour plots show representative cases of AIF-1 high (AIF-1hi) and low (AIF-1lo) among patients with AS. Data are presented as mean ± standard deviation. (Two-sided t-test) f Serum cytokine changes compared between AIF-1hi (above cut-off) and AIF-1lo groups (AIF1 low, n = 4; AIF1 high, n = 8; two-sided t-test) among TNFi-treated patients with AS. Changes from baseline to week 14 are shown as percentages and are presented as mean ± standard deviation. g TNFi was added to AIF-1-stimulated and control PBMCs from active patients with AS (n = 10) stimulated with anti-CD3 antibody and anti-CD28 antibody for 6 h. Data are presented as mean ± standard deviation. (two-sided t-test) h Summary of the clinical history of the patient with AS who experienced paradoxical psoriasis after initiating TNFi and improvement with IL-17 inhibitor secukinumab, including the immunophenotype of memory CD4+ T cells (left contour plot), clinical photos, and serum AIF-1 level. Source data are provided as a Source Data file.
Upon examining the changes induced by TNFi in both groups, we found a significant increase in RORA expression, the gene product of which is critical for maintaining Th17 effector function long term64, and increased expression of the activation marker CD69 in the NR group with TNFi treatment (Fig. 7c, Supplementary Fig. 7b, c). We also observed that both the activated Th17 cluster and MAFlo Treg cluster had a significant positive correlation with the previously identified CXCL10hi cMo cluster (Fig. 7d).
To verify whether the characteristics of these CD4+ T cells are reproduced in individuals with high levels of AIF-1 at the protein level, we conducted immunophenotyping between the responder and NR groups. Patients with high AIF-1 levels had an increased proportion of Th17 cells, which is consistent with the effect of AIF-1 treatment in the same patients (Fig. 7e, Supplementary Fig. 8a, b, 9a–d). Furthermore, an increase in Th17 features induced by TNFi was clearly observed in AIF-1hi patients. Similar results were validated at the protein level when the same patients were treated with AIF-1 followed by TNFi (Fig. 7f, g). The soluble protein level of IL-17A had a significant positive correlation with AIF-1 (Supplementary Fig. 8c). Therefore, AIF-1 relatively increases Th17 features.
Because TNFi has efficacy to treat psoriasis, psoriasis that newly develops after initiating TNFi is termed ‘paradoxical psoriasis’20. We examined one AS patient with severe paradoxical psoriasis and found high serum AIF-1 (147.8 pg/ml) and a high frequency of IL-17A+ cells at the moment of psoriasis development (Fig. 7h). After psoriasis development, TNFi was discontinued and the patient switched to secukinumab, an IL-17i, with dose escalation. The patient improved with IL-17i treatment, indicating that the strong Th17 activity was responsible for the development of paradoxical psoriasis in this patient. Notably, serum AIF-1 levels remained high (132.3 pg/ml) even after clinical improvement, underscoring AIF-1 as a potentially stable biomarker reflecting the IFN signature of TNFi non-responses.
Discussion
This study revealed that type I IFN signatures differentiate clinical response to TNFi among patients with AS. Although TNFi is considered an effective drug, the variability in patient responses remains poorly understood65. A common assumption is that higher TNFα levels result in less neutralization due to pharmacokinetic differences. However, TNFi treatment can affect the immune system beyond neutralization66,67. Our findings indicate that elevated type I IFN signatures at baseline are associated with poor TNFi response, contributing to the paradoxical enhancement of the IFN signatures and Th17 responses in NR patients. One of the type I IFN-related gene, AIF-1, was validated as a predicting biomarker, and may actively contribute to the vicious inflammatory cycle by elevating the expression of IFNα receptor and Th17 responses. Previous research has shown that TNF and IFNα can reprogram the epigenome of macrophages, breaking TNF-induced tolerization by type I IFN15. Further, our results elucidate how TNF inhibition by drugs can have divergent effects on the overall human immune system depending on the state of type I IFN response.
Single cell transcriptome analysis reveals important contrasting subsets of monocytes that help explain these divergent features between responders and NRs. Regulon analysis indicated that EGR1 consistently regulated monocytes in the responder group. EGR1 is known to suppress type 1 IFN pathways and LPS signaling via the NuRD corepressor complex32. In contrast, ETS2 emerged as the top regulon in the NR group post-TNFi. Recent studies have shown that ETS2 is a central regulator of inflammatory macrophages and is associated with inflammatory diseases, such as inflammatory bowel disease and AS55. By investigating an intergenic haplotype on chr21q22 that is independently linked to several inflammatory diseases, ETS2 was identified as a key gene amplifying inflammatory responses.
CXCL10hi cMo, a monocyte subset that tends to increase in NRs, is an inflammatory cell type differentiated in response to AIF-1 stimulation and is known to play a significant role in autoimmune pathology30,68,69. Identification of the CXCL10hi cluster, which also expressed GBP1 and SLAMF7, is consistent with earlier studies highlighting sensitivity to IFNα stimulation31. For example, the presence of CXCL10hi monocytes as a pathogenic subset in autoimmune neuroinflammation and their correlation with serum IFNα and CXCL10 levels in SLE provide a parallel to the situation in NRs70. Thus, this evidence helps explain why TNF inhibitors not only failed to demonstrate efficacy in clinical trials involving SLE patients but also why cases of new-onset SLE during TNF inhibitor use have been reported71,72,73.
Typically, the most representative source of type I IFN is the plasmacytoid dendritic cell (pDC). It has been reported that pDCs increase in the enthesis, which is crucial for the pathology of AS, due to infections, and this increase may contribute to the exacerbation of psoriatic arthritis74. Furthermore, there is evidence suggesting that TNFi treatment may influence pDC maturation, leading to increased IFNα secretion75. This phenomenon has been reported in cases of paradoxical psoriasis76 and may also contribute to the heightened IFN response observed in the NR group in the present study.
It is intriguing that the characteristics of NR, represented by AIF-1 or ISG, are associated with increased Th17 responses after treatment. Given that TNF inhibitors were originally developed for RA, a condition with a different immunological background compared to ankylosing spondylitis AS, which is characterized by a high burden of Th17 cells, such phenomena may not have been anticipated initially77. If we can predict NR to TNF inhibitors using biomarkers associated with baseline features presented in our study, we could consider treatments that directly target the IFN-signature contributing to treatment resistance. Among targeted agents, JAK inhibitors might be promising candidates. Given that JAK1 and TYK2 are essential signaling proteins directly associated with the IFNα receptor, upadacitinib, with its pronounced selectivity for JAK1 inhibition, may offer advantages over tofacitinib78. Clinical trials focusing on TNF inhibitor non-responders have demonstrated that upadacitinib, a selective JAK1 inhibitor, achieves a response rate approximately 10-20% higher than that of tofacitinib79,80. Future prospective trials using IFN-signature or AIF-1 as biomarkers to select patients could help validate this approach.
The strengths of our study include the longitudinal multi-omics approach, which combined single-cell and bulk RNA sequencing with protein-level validation in the well-controlled cohort. This allowed us to dissect the complex interactions between different immune cell populations and their responses to TNFi treatment. However, we were unable to determine the specific causes of the increased type I IFN response or whether it is unique to AS patients. While it is well known that viral infections generally cause transient exacerbations of autoimmune diseases, understanding how these infections regulate responses to TNFi is challenging within human cohorts. Additionally, we did not observe changes in immune cells within tissues, which is a limitation of our study. Although we validated AIF-1 as a biomarker associated with ISGs and found that it can enhance ISGs and increase Th17 levels, we do not yet know the exact receptor involved.
In conclusion, our study demonstrated that elevated type I IFN signatures marked by AIF-1 are associated with a non-response to TNFi treatment in patients with AS, leading to increased Th17 activity and clinical deterioration in NRs. These findings highlight the need for personalized treatment strategies based on baseline IFN signatures to improve clinical outcomes in AS.
Methods
Patients
Patients fulfilling the 2009 Assessment of SpondyloArthritis International Society (ASAS) classification criteria for axial SpA and treated with TNFi were enrolled. Responders were defined clinically at week 12 or 14 as having a ≥ 50% reduction in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI 50) from baseline81. Additionally, we enrolled patients refractory to TNFi treatment who subsequently received IL-17 inhibitors (Supplementary Data 1).
The discovery cohort was enrolled consecutively from the Rheumatology Clinic at Seoul National University Hospital (SNUH), and PBMCs were collected before and 4 weeks after TNFi treatment. The blood draws for all cohorts were consistently performed before 10:00 AM, in accordance with standard clinical practice. Given the relatively smaller number of NR patients, we matched the R group to the NR group by age and sex to minimize potential bias. For the validation cohort 1, PBMCs were collected before and after 14 weeks after TNFi treatment to measure the gene expression level of AIF-1. For the validation cohort 2, serum samples of the participants from the PLANETAS study and the SNUH cohort were used to measure serum concentration of AIF-182. For the validation cohort 3, serum samples were obtained from patients at the Rheumatology Clinic at SNUH.
Ethical approval was obtained from the ethics committees of Seoul National University Hospital (C-1202-055-398 and H-1511-094-721), adhering to the Declaration of Helsinki and Good Clinical Practice guidelines. All participants provided written informed consent.
Single-cell RNA sequencing
PBMCs were isolated from peripheral venous blood via standard Ficoll-Paque (GE Healthcare, Uppsala, Sweden) density gradient centrifugation, frozen in freezing media, and stored in liquid nitrogen until use. The scRNA-seq libraries were generated using the Chromium Single Cell 3′ Library & Gel Bead Kit v3 (10× Genomics, Pleasanton, CA) following the manufacturer’s instructions. A total of 17 batch runs were performed, with 2–4 samples multiplexed per run. Single Nucleotide Polymorphism (SNP)-based demultiplexing was then applied, in accordance with the 10× Genomics standard protocols. We generated single-cell libraries using the Chromium Single Cell 3′ Library & Gel Bead Kit v3 (10× Genomics, Pleasanton, CA) according to the manufacturer’s instructions. A total of 17 batch runs were performed, with 2–4 samples multiplexed per run. Single Nucleotide Polymorphism (SNP)-based demultiplexing was then applied, in accordance with the 10× Genomics standard protocols. Libraries were sequenced on an Illumina NovaSeq platform, using a paired-end strategy at approximately 50,000 reads per cell. The read length was set to 2 × 75 bp to cover both cell barcodes and the captured transcript fragments.
scRNA-seq data processing
The sequenced data were demultiplexed using mkfastq (Cell Ranger 10× Genomics, v3.0.2) to generate fastq files. After demultiplexing, the reads were aligned to the human reference genome (GRCh38; 10× Cell Ranger reference GRCh38 v3.0.0), feature-barcode matrices were generated using the cellranger count, and then aggregated by cellranger aggr using default parameters. The subsequent analysis was performed using Seurat 4 with R 4.3.0.1 (Satija Lab, New York, NY, USA)83. After generating the feature-barcode matrix, cells expressing <200 genes or >10% mitochondrial genes were discarded. To exclude low-quality cells from our data, we filtered out the cells in which the expression of mitochondrial genes was >10% of their total gene expression84. To align the cells originating from different samples, 3000 highly variable genes from each sample were identified by sctransform (SCT; v2 from CRAN, sctransform v0.3.3 + )85. The SCT approach leverages the Pearson residuals derived from negative binomial regression as the initial data for conventional methods of dimensional reduction.
Unless otherwise noted, data were scaled and transformed and variable genes identified using the SCTransform() function, and linear regression was performed to remove unwanted variation due to cell quality (% mitochondrial reads and feature counts). PCA was performed using the highly variable genes, and the first 30 PCs were used to perform UMAP to embed the dataset into two dimensions. Next, the first 30 PCs were used to construct an SNN graph using FindNeighbors(), and this SNN was used to cluster the dataset in FindClusters(). Although upstream quality control removed many dead or low-quality cells, if any clusters were identified that were defined by few canonical cell lineage markers and enriched for genes of mitochondrial or ribosomal origin, these clusters were removed from further analysis.
Differential abundance analysis using scRNA-seq data
We performed differential abundance analysis of patients with AS using scRNA-seq data. We used miloR (version 3.19) to detect sets of cells that are differentially abundant in various conditions by modeling counts of cells in the neighborhoods of a KNN graph29. We first used the buildGraph function to construct a KNN graph based on pre-computed supervised PCA with k = 45 and using 30 PCs (d = 30). Next, we used the makeNhoods function to assign cells into neighborhoods based on their connectivity over the KNN graph. To test for differential abundance, Milo fit an NB GLM to the counts for each neighborhood, accounting for different numbers of cells across samples using TMM normalization. The log2 fold change in the number of cells between two conditions in each neighborhood was used for visualization. Each dot in Fig. 1f corresponds to an individual neighborhood, colored by statistical significance (adjusted p < 0.05), reflecting log2-fold changes in abundance between responder and non-responder groups
K-means clustering analysis
As shown in Fig. 2g, we performed K-means clustering of DEGs among all pairs of patients with AS. The log2-transformed relative gene expression of DEGs compared with each condition was subjected to K-means clustering (k = 8). Here, we used up-regulated DEGs in at least one condition.
Identification of DEGs using MAST
To analyze DEGs following TNFi treatment, we used the MAST algorithm in Seurat’s implementation based on a Bonferroni-adjusted P < 0.05 and a log2 fold change > 0.1.
In vitro adalimumab-treated PBMCs
PBMCs were isolated from four healthy donors and subjected to four different treatment conditions. For the control condition, cells were incubated for 18 h without any treatment. In the TNFα condition, cells were treated with TNFα (PeproTech, #300-01 A) for 18 h. For the TNFα_IgG condition, cells were treated with TNFα for 12 h, followed by human IgG1 (BD Biosciences, #569605) for the remaining 6 h. In the TNFα_ADA condition, cells were treated with TNFα for 12 h and then with ADA (BD Pharmingen™, #569601) for 6 h. For each condition, 0.5 million PBMCs were cultured in 100 μl of medium per well in a 96-well U-bottom plate, and all experiments were performed in triplicate. Following treatment, single-cell libraries were prepared using a standard single-cell RNA sequencing protocol, and sequencing was performed to analyze the gene expression profiles.
Monocyte regulon analysis
Regulon analysis of monocytes was performed using the pySCENIC package86. The Seurat object containing the single-cell RNA sequencing data was converted to the h5ad file format for compatibility with the pySCENIC workflow. Gene regulatory network inference was carried out using GRNBoost2, and the area under the curve (AUC) matrix, representing the activity scores of TFs for each cell, was calculated to determine the regulon specificity scores for each condition. Logo analysis, identifying the most significant motif for each TF, was also conducted using pySCENIC.
The TF combinations used in the analysis were obtained from the ‘Lambert2018.txt’ file available at aertslab GitHub repository. For ranking databases, the following resources were utilized from the aertslab cistarget folder: ‘hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.genes_vs_motifs.rankings.feather’ and ‘hg38__refseq-r80__500bp_up_and_100bp_down_tss.mc9nr.genes_vs_motifs.rankings.feather’. Motif annotation was conducted using the ‘motifs-v9-nr.hgnc-m0.001-o0.0.tbl’ file. The regulon prediction was executed through a two-step process involving network inference based on GRNBoost2 from CLI, followed by regulon prediction using cisTarget from CLI.
Bulk RNA-Seq deconvolution
The annotated single-cell RNA-Seq data was uploaded to CIBERSORTx (http://cibersortx.stanford.edu) to create the novel signature matrix with default parameters59. Quantile normalization was disabled. CIBERSORTx utilizes a linear model to infer cell type abundance from bulk tissue expression profiles by modeling the mixture samples as a system of linear equations. Specifically, M represents an n × k matrix with n genes and k mixture gene expression profiles (GEPs), and B is a subset of the cell type expression matrix H containing discriminatory marker genes for each cell subset. The system can be solved to impute F, the c × k fractional abundance matrix, representing cell type proportions. CIBERSORT, which implements ν-support vector regression, was used to estimate F, providing robust imputation against noise, unknown mixture content, and collinearity among cell type reference profiles.
Study design for biomarker discovery
The overall experimental flow is summarized in Fig. 3c. AIF-1 was selected from the scRNA-seq data. Blood samples were taken before and 14 weeks after TNFi treatment. We used serum samples from the participants in the PLANETAS study, a previous phase I clinical study of CT-P13 (Clinicaltrials.gov; NCT01220518)82.
qRT-PCR
Biomarker discovery was confirmed using PBMCs from the validation cohort (11 responders and 7 non-responders) for qRT-PCR. To validate the difference in AIF1 expression levels, we used the TaqMan® Gene Expression Assay (AIF1, Hs00610419_g1; Applied Biosystems). Gene expression was normalized to an endogenous reference gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Hs02786624_g1), and compared in responders and NRs using the ΔΔCт method.
ELISA and cytometric bead array
Serum AIF-1 levels were measured using ELISA kits (LSbio, Seattle, WA, #LS-F6826-1) according to the manufacturer’s instructions. Other cytokines, including IL-17A, IL-17F, IFNγ, IL-1β, IL-6, GM-CSF, and TNF, were quantified using BD Biosciences ELISA kits or BD Cytometric Bead Array (CBA) Flex Sets (BD Biosciences). Specifically, IL-17A was measured using CBA Human IL-17A Flex Set (BD, #560383), IL-17F (BD, #562151), IFNγ (BD, #555142), IL-1β (BD, #557953), IL-6 (BD, #555220), GM-CSF (BD, #555126), and TNF (BD, #555212). We evaluated a total of 79 serum samples from the SNUH cohort and 38 serum samples from the PLANETAS study41, a previous phase I clinical study of CT-P13 (Clinicaltrials.gov; NCT01220518).
Isolation of CD4+ T cells and CD14+ monocytes
CD14⁺ monocytes from healthy donors were isolated from PBMCs using CD14 MicroBeads (Miltenyi Biotec, #130-050-201) following the manufacturer’s protocol. Flow-through cells from this isolation were used to purify CD4⁺ T cells using the CD4⁺ T Cell Isolation Kit (Miltenyi Biotec, #130-097-048). After isolation, cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Thermo Fisher Scientific, #15140122). For short-term cultures (12–24 h) in non-tissue-culture–treated plates, cells are only weakly adherent and thus easily collected by gentle pipetting. If any residual adherence was noted, we used a standard cell scraper or gentle enzymatic dissociation (EDTA) to detach them, followed by washing with PBS.
In vitro stimulation of T cells and intracellular cytokine staining
Cryopreserved PBMCs were thawed, resuspended in RPMI 1640 containing 5% fetal bovine serum and 2 mmol/L L-glutamine, and rested for 12 h at 37 °C. The PBMCs were then cultured in the presence or absence of 100 ng/ml AIF-1 (Cloud-Clone Corp., Katy, TX, #RPC288Hu01) for 3 days and stimulated with anti-CD3 antibody (0.1 mg/ml OKT-3; eBioscience) and anti-CD28 antibody (1 mg/ml; BD Biosciences). When isolated CD4+ T cells were stimulated, Dynabeads Human T-Activator CD3/CD28 (ThermoFisher Scientific, Waltham, MA) was used with a 4:1 cell-to-bead ratio. Brefeldin A (GolgiPlug, BD Biosciences) and monesin (GolgiStop, BD Biosciences) were added 1 h later. After another 5 h of incubation, cells were stained using Live/Dead fixable cell stain to exclude dead cells, and then stained with fluorochrome-conjugated antibodies against surface markers, including anti-CD3 (BV605; BD Biosciences, #742623; All antibodies used for FACS were diluted 1:100), anti-CD4 (BV510; BD Biosciences, #562970), anti-CD14 (PE-eFluor610; eBioscience, #61-0149-42), anti-CD19 (PE-eFluor610; eBioscience, #61-0199-42), anti-CD45RA (APC-Cy7; Biolegend, #304128), anti-CCR6 (BV421; BD Biosciences, #565925), and anti-IL-1R1 (PE; R&D, # FAB269P). Stained cells were permeabilized using the Foxp3 staining buffer kit and further stained with anti-RORγt (Alexa Fluor 488; eBioscience, #53-6981-82), anti-T-bet (PE-Cy7; eBioscience, #25-5825-82), anti-TNF (APC; eBioscience, #17-7349-82), anti-IFN-γ (Alexa Fluor 488; eBioscience or APC-R700; BD Biosciences, #564981), and anti-IL-17A (BV711; Biolegend, #512328). Flow cytometry was performed on an LSR II instrument using FACSDiva software (BD Biosciences) and the data analyzed using FlowJo software (Treestar, San Carlos, CA).
For in vitro experiments, to assess the effect of TNFα inhibition, cells were subjected to four different treatment conditions. Each condition was performed in a 96-well U-bottom plate, with 0.5 × 10^6 PBMCs per well, in RPMI-based media. TNFα (50 ng/ml, PeproTech, #300-01 A) was added to PBMC cultures, with or without 10ug/ml adalimumab (ADA). ADA treatment was performed using adalimumab biosimilar (BD Pharmingen™, #569601). To assess cytokine responses, PBMCs were stimulated with 100 ng/ml AIF-1 (Cloud-Clone Corp., Katy, TX) for 3 days, followed by activation with anti-CD3/CD28 antibodies (Gibco, #11161D). To evaluate the effect of TNFi, ADA (InvivoGen, Hong Kong, China) was added to cultures 1 h before AIF-1 stimulation, and IgG1 isotype control (BD Biosciences, #569605) was included as a negative control. All experiments were performed using three biological replicates, each from a different healthy donor. Technical replicates were included in selected assays to ensure reproducibility.
RNA-seq and data analysis
CD4+ T cells were stimulated for 72 h, and CD14+ monocytes for 24 h, with 100 ng/ml of recombinant human AIF-1. Regarding the experiment in Fig. 6, CD14+ monocytes were isolated from human PBMCs using MACS with CD14 Microbeads. The isolated monocytes were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated defined FBS, P/S, L-glutamine, and M-CSF (20 ng/ml, Peprotech, #300-25-10UG). The cells were then treated with 50 ng/ml TNFα (PeproTech, #300-01 A), 125 ng/ml IFNα (Peprotech, #300-02AA), 10 μg/ml ADA (BD, #569601), or human IgG1 isotype control (BD, #569605) according to the designated conditions. Cytokines were added for 24 h, and ADA or IgG1 isotype control was added 12 h after the cytokines. After 24 h of culture, RNA was isolated using standard protocols and sequenced to analyze the differential gene expression profiles under each condition. The process of RNA-sequencing is described in the online supplementary information.
Statistical analysis
Statistical analyses were performed using R 4.3.0.1 or Prism software version 5.0 (GraphPad, La Jolla, CA). Detailed information on statistical analyses can be found in each figure legend. P values ≤ 0.05 were considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
For all data analyses, we used publicly available software. All raw and processed data for single-cell sequencing are deposited in the GEO (GSE277117 and GSE277791). All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
Code availability
Scripts used to analyze the AS data were generated by R 4.1.3 and Seurat 4.2.0 or 5.0.3, and have been deposited with the link [https://github.com/swooju/TNFi-refractorness-in-AS].
References
Rudwaleit, M. et al. The development of assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann. Rheum. Dis. 68, 777–783 (2009).
Rudwaleit, M. et al. The Assessment of SpondyloArthritis International Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann. Rheum. Dis. 70, 25–31 (2011).
Sieper, J. et al. Axial spondyloarthritis. Nat. Rev. Dis. Prim. 1, 15013 (2015).
Kehl, A. S., Corr, M. & Weisman, M. H. Review: Enthesitis: New Insights Into Pathogenesis, Diagnostic Modalities, and Treatment. Arthritis Rheumatol. 68, 312–322 (2016).
Taurog, J. D., Chhabra, A. & Colbert, R. A. Ankylosing Spondylitis and Axial Spondyloarthritis. N. Engl. J. Med 374, 2563–2574 (2016).
Braun, J. et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet 359, 1187–1193 (2002).
Gorman, J. D., Sack, K. E. & Davis, J. C. Jr Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N. Engl. J. Med 346, 1349–1356 (2002).
Haroon, N. et al. The impact of tumor necrosis factor α inhibitors on radiographic progression in ankylosing spondylitis. Arthritis Rheum. 65, 2645–2654 (2013).
Dougados, M. et al. Efficacy of celecoxib, a cyclooxygenase 2-specific inhibitor, in the treatment of ankylosing spondylitis: a six-week controlled study with comparison against placebo and against a conventional nonsteroidal antiinflammatory drug. Arthritis Rheum. 44, 180–185 (2001).
Boonen, A. A review of work-participation, cost-of-illness and cost-effectiveness studies in ankylosing spondylitis. Nat. Clin. Pr. Rheumatol. 2, 546–553 (2006).
Winthrop, K. L. Risk and prevention of tuberculosis and other serious opportunistic infections associated with the inhibition of tumor necrosis factor. Nat. Clin. Pr. Rheumatol. 2, 602–610 (2006).
Gane, J. M., Stockley, R. A. & Sapey, E. TNF-α Autocrine Feedback Loops in Human Monocytes: the pro- and anti-inflammatory roles of the tnf-α receptors support the concept of selective TNFR1 blockade in vivo. J. Immunol. Res 2016, 1079851 (2016).
Iwamoto, S. et al. TNF-alpha drives human CD14+ monocytes to differentiate into CD70+ dendritic cells evoking Th1 and Th17 responses. J. Immunol. 179, 1449–1457 (2007).
Huber, R. et al. TNF tolerance in monocytes and macrophages: characteristics and molecular mechanisms. J. Immunol. Res 2017, 9570129 (2017).
Park, S. H. et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 18, 1104–1116 (2017).
Baeten, D. et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med 373, 2534–2548 (2015).
Blanco, F. J. et al. Secukinumab in active rheumatoid arthritis: a phase iii randomized, double-blind, active comparator- and placebo-controlled study. Arthritis Rheumatol. 69, 1144–1153 (2017).
Zheng, Y. et al. TNFα promotes Th17 cell differentiation through IL-6 and IL-1β produced by monocytes in rheumatoid arthritis. J. Immunol. Res 2014, 385352 (2014).
Lin, Y. C. et al. The immunomodulatory effects of TNF-α inhibitors on human Th17 cells via RORγt histone acetylation. Oncotarget 8, 7559–7571 (2017).
Bae, J. M. et al. Paradoxical psoriasis following anti-TNF therapy in ankylosing spondylitis: A population-based cohort study. J. Allergy Clin. Immunol. 142, 1001–1003.e2 (2018).
Chen, D. Y. et al. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res Ther. 13, R126 (2011).
Alzabin, S. et al. Incomplete response of inflammatory arthritis to TNFα blockade is associated with the Th17 pathway. Ann. Rheum. Dis. 71, 1741–1748 (2012).
Haroon, N. et al. From gene expression to serum proteins: biomarker discovery in ankylosing spondylitis. Ann. Rheum. Dis. 69, 297–300 (2010).
de Vries, M. K. et al. Erythrocyte sedimentation rate, C-reactive protein level, and serum amyloid a protein for patient selection and monitoring of anti-tumor necrosis factor treatment in ankylosing spondylitis. Arthritis Rheum. 61, 1484–1490 (2009).
Baraliakos, X. et al. Predictors of clinical remission under anti-tumor necrosis factor treatment in patients with ankylosing spondylitis: pooled analysis from large randomized clinical trials. J. Rheumatol. 42, 1418–1426 (2015).
Hwang, M. et al. Quantitative proteomic screening uncovers candidate diagnostic and monitoring serum biomarkers of ankylosing spondylitis. Arthritis Res. Ther. 25, 57 (2023).
Wang, R., Dasgupta, A. & Ward, M. M. Predicting probability of response to tumor necrosis factor inhibitors for individual patients with ankylosing spondylitis. JAMA Netw. Open 5, e222312 (2022).
Aeberli, D. et al. Regulation of peripheral classical and non-classical monocytes on infliximab treatment in patients with rheumatoid arthritis and ankylosing spondylitis. RMD Open 2, e000079 (2016).
Dann, E. et al. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Giladi, A. et al. Cxcl10+ monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nat. Immunol. 21, 525–534 (2020).
Simmons, D. P. et al. SLAMF7 engagement superactivates macrophages in acute and chronic inflammation. Sci. Immunol. 7, eabf2846 (2022).
Trizzino, M. et al. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci. Adv. 7, eaaz8836 (2021).
Cui, A. et al. Dictionary of immune responses to cytokines at single-cell resolution. Nature 625, 377–384 (2024).
Consortium, T. G. O. et al. The gene ontology knowledgebase in 2023. Genetics 224, iyad031 (2023).
Michalska, A. et al. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type i and type ii ifn responses. Front Immunol. 9, 1135 (2018).
Cheon, H. et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. Embo j. 32, 2751–2763 (2013).
Rouillard, A. D. et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, baw100 (2016).
Baeten, D. et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet 382, 1705–1713 (2013).
Pawlik, A. et al. Expression of allograft inflammatory factor-1 in peripheral blood monocytes and synovial membranes in patients with rheumatoid arthritis. Hum. Immunol. 77, 131–136 (2016).
Elizondo, D. M. et al. Allograft inflammatory factor-1 in myeloid cells drives autoimmunity in type 1 diabetes. JCI Insight 5, e136092 (2020).
Park, W. et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 18, 25 (2016).
Teng, M. W. L. et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 21, 719–729 (2015).
Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Schnell, A. et al. Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 184, 6281–6298.e23 (2021).
Schnell, A., Littman, D. R. & Kuchroo, V. K. TH17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 24, 19–29 (2023).
Segura, E. et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 38, 336–348 (2013).
Sha, H. et al. ATF3 promotes migration and M1/M2 polarization of macrophages by activating tenascin‑C via Wnt/β‑catenin pathway. Mol. Med Rep. 16, 3641–3647 (2017).
Labzin, L. I. et al. ATF3 is a key regulator of macrophage IFN responses. J. Immunol. 195, 4446–4455 (2015).
Han, X. et al. SGK1 negatively regulates inflammatory immune responses and protects against alveolar bone loss through modulation of TRAF3 activity. J. Biol. Chem. 298, 102036 (2022).
Ban, T. et al. Genetic and chemical inhibition of IRF5 suppresses pre-existing mouse lupus-like disease. Nat. Commun. 12, 4379 (2021).
Zheng, L. et al. ELK4 promotes the development of gastric cancer by inducing M2 polarization of macrophages through regulation of the KDM5A-PJA2-KSR1 axis. J. Transl. Med 19, 342 (2021).
Rui, L. Oncogenic Cooperation between EGR1 and BRD4 in Diffuse Large B Cell Lymphoma. Blood 134, 1508–1508 (2019).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 (2011).
Cham, C. M., Ko, K. & Niewold, T. B. Interferon regulatory factor 5 in the pathogenesis of systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 780436 (2012).
Stankey, C. T. et al. A disease-associated gene desert directs macrophage inflammation through ETS2. Nature 630, 447–456 (2024).
Watanabe, T. et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Invest 120, 1645–1662 (2010).
Huang, R. et al. The NCATS bioplanet - an integrated platform for exploring the universe of cellular signaling pathways for toxicology, systems biology, and chemical genomics. Front Pharm. 10, 445 (2019).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 51, D587–d592 (2023).
Steen, C. B. et al. Profiling cell type abundance and expression in bulk tissues with CIBERSORTx. Methods Mol. Biol. 2117, 135–157 (2020).
Yi, K. et al. Analysis of single-cell transcriptome and surface protein expression in ankylosing spondylitis identifies OX40-positive and glucocorticoid-induced tumor necrosis factor receptor-positive pathogenic Th17 Cells. Arthritis Rheumatol. 75, 1176–1186 (2023).
Burton, P. R. et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337 (2007).
Ortutay, Z. et al. Proprotein convertase FURIN regulates T cell receptor-induced transactivation. J. Leukoc. Biol. 98, 73–83 (2015).
Neumann, C. et al. c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host–microbiota homeostasis. Nat. Immunol. 20, 471–481 (2019).
Hall, J. A. et al. Transcription factor RORα enforces stability of the Th17 cell effector program by binding to a Rorc cis-regulatory element. Immunity 55, 2027–2043.e9 (2022).
Emery, P. Optimizing outcomes in patients with rheumatoid arthritis and an inadequate response to anti-TNF treatment. Rheumatology 51, v22–v30 (2012).
Askling, J. et al. Cancer risk in patients with rheumatoid arthritis treated with anti-tumor necrosis factor alpha therapies: does the risk change with the time since start of treatment?. Arthritis Rheum. 60, 3180–3189 (2009).
Gertel, S. et al. Ex vivo cell-based assay for assessment of response to TNF inhibitors in patients with rheumatic diseases. Rheumatology 64, 2233–2241 (2024).
Lee, E. Y., Lee, Z. H. & Song, Y. W. CXCL10 and autoimmune diseases. Autoimmun. Rev. 8, 379–383 (2009).
Zhao, Q. et al. A novel function of CXCL10 in mediating monocyte production of proinflammatory cytokines. J. Leukoc. Biol. 102, 1271–1280 (2017).
Enocsson, H. et al. Comparison of surrogate markers of the type i interferon response and their ability to mirror disease activity in systemic lupus erythematosus. Front Immunol. 12, 688753 (2021).
Shakoor, N. et al. Drug-induced systemic lupus erythematosus associated with etanercept therapy. Lancet 359, 579–580 (2002).
De Bandt, M. et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 7, R545–R551 (2005).
Aringer, M. et al. Adverse events and efficacy of TNF-alpha blockade with infliximab in patients with systemic lupus erythematosus: long-term follow-up of 13 patients. Rheumatol. (Oxf.) 48, 1451–1454 (2009).
Zhou, Q. et al. SARS-CoV-2 infection induces psoriatic arthritis flares and enthesis resident plasmacytoid dendritic cell type-1 interferon inhibition by JAK antagonism offer novel spondyloarthritis pathogenesis insights. Front Immunol. 12, 635018 (2021).
Psarras, A. et al. TNF-α regulates human plasmacytoid dendritic cells by suppressing ifn-α production and enhancing T cell activation. J. Immunol. 206, 785–796 (2021).
Conrad, C. et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 9, 25 (2018).
Monaco, C. et al. Anti-TNF therapy: past, present and future. Int Immunol. 27, 55–62 (2015).
Bonelli, M. et al. Selectivity, efficacy and safety of JAKinibs: new evidence for a still evolving story. Ann. Rheum. Dis. 83, 139–160 (2024).
van der Heijde, D. et al. Upadacitinib in active ankylosing spondylitis: results of the 2-year, double-blind, placebo-controlled SELECT-AXIS 1 study and open-label extension. RMD Open 8, e002280 (2022).
Deodhar, A. et al. Tofacitinib efficacy and safety in patients with ankylosing spondylitis by prior biologic disease-modifying antirheumatic drug use: a post hoc analysis. ACR Open Rheumatol. 5, 632–643 (2023).
Garrett, S. et al. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J. Rheumatol. 21, 2286–2291 (1994).
Park, W. et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann. Rheum. Dis. 72, 1605–1612 (2013).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Wei, K. et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 582, 259–264 (2020).
Choudhary, S. & Satija, R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 23, 27 (2022).
Van de Sande, B., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protocols, 15, 2247–2276 (2020).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT 800-20240195; 2022M3A9D3016848; RS-2024-00356421; RS-2023-000279801; RS-2024-00440930; RS-2023-00262527), by Yuhan Corporation (SNU grant No. 800-20230067), and by the Korea Health Industry Development Institute (RS-2023-00265820).
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: W.S., E.E.L., S.P., I.J., J.S.L. and E.Y.L.; Methodology: W.S., S.P., B.C., M.-G.K., S.Y.B., S.R.C., J.Y.K., S.U.K., I.J., J.S.L., and E.Y.L.; Investigation: W.S., E.E.L., S.P., I.J., J.S.L., and E.Y.L.; Resources: J.-I.K., E.-C.S., I.J., J.S.L., and E.Y.L.; Writing:; Review & Editing: W.S., E.E.L., S.P., I.J., J.S.L., and E.Y.L.; Supervision: I.J., J.S.L., and E.Y.L.; Funding: J.-I.K., I.J., J.S.L., and E.Y.L.
Corresponding authors
Ethics declarations
Competing interests
Eun Young Lee is a consultant for Samsung Bioepis and IMBiologics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Paul Bowness and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, W., Lee, E.E., Park, S. et al. Type 1 interferon signature and allograft inflammatory factor-1 contribute to refractoriness to TNF inhibition in ankylosing spondylitis. Nat Commun 16, 5531 (2025). https://doi.org/10.1038/s41467-025-60445-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60445-6
