
Abstract
Toxic methylmercury (CH3Hg+) is produced by microbial conversion of inorganic mercury in hypoxic environments such as rice paddy soils, and can accumulate in rice grains. Although microbial demethylation has been recognized as a crucial pathway for CH3Hg+ degradation, the identities of microbes and pathways accountable for CH3Hg+ degradation in soil remain elusive. Here, we combine 13CH3Hg+-DNA stable-isotope probing experiments with shotgun metagenomics to explore microbial taxa and associated biochemical processes involved in CH3Hg+ degradation in paddy and upland soils. We identify Pseudarthrobacter, Methylophilaceae (MM2), and Dechloromonas as the most significant taxa potentially engaged in the degradation of 13CH3Hg+ in paddy soil with high mercury contamination. We confirm that strains affiliated with two of those taxa (species Dechloromonas denitrificans and Methylovorus menthalis) can degrade CH3Hg+ in pure culture assays. Metagenomic analysis further reveals that most of these candidate 13CH3Hg+ degraders carry genes associated with the Wood-Ljungdahl pathway, dicarboxylate-hydroxybutyrate cycle, methanogenesis, and denitrification, but apparently lack the merB and merA genes involved in CH3Hg+ reductive demethylation. Finally, we estimate that microbial degradation of soil CH3Hg+ contributes to 0.08–0.64 fold decreases in CH3Hg+ accumulation in rice grains across China (hazard quotient (HQ) decrements of 0.62–13.75%). Thus, our results provide insights into microorganisms and pathways responsible for CH3Hg+ degradation in soil, with potential implications for development of bioremediation strategies.
Similar content being viewed by others

Introduction
Methylmercury (CH3Hg+) poses significant environmental concerns due to its ability to biomagnify in food chains and its profound toxicity to both humans and wildlife1,2. Even low-level dietary exposure to CH3Hg+ can cause irreversible neurological damage to humans3, reducing the intelligence quotient (IQ) of newborns4. Paddy fields are particularly concerning as they serve as hotspots for CH3Hg+ production due to their hypoxic condition favoring Hg-methylating microorganisms5. CH3Hg+ produced in the soil has a high propensity to accumulate in rice grains, and is also a crucial source of CH3Hg+ in adjacent aquatic environments5,6. In many regions, paddy and upland rotations are commonly practiced as cropping systems7, making it critical to assess the dynamics of CH3Hg+ in both soil types. Recent discoveries of CH3Hg+ production even in oxic habitats indicated that CH3Hg+ accumulation and associated environmental processes are more complex than previously understood8. Prior research disclosed that specific microorganisms have the capacity to degrade CH3Hg+ in pure culture. It was reported in the 1960s that Staphylophage aureus, originally isolated from medical waste, had the ability to demethylate CH3Hg+ to Hg0 9. Subsequently, Desulfovibrio desulfuricans LS, a sulfur-reducing bacteria, and Methanococcus maripaludis, a methanogen, were identified as potential CH3Hg+ degraders10. Recent studies have further confirmed the ability of iron-reducing bacterium Geobacter bemidjiensis Bem and methanotrophic bacterium Methylosinus trichosporium OB3b to degrade CH3Hg+ in pure culture settings11,12. Nevertheless, a significant challenge lies in the quest to identify active CH3Hg+ degraders within natural environments. This arises from the considerable variability of the merB gene, which encodes the organomercury lyase responsible for CH3Hg+ degradation among various microorganisms13,14. As a result, the development of dependable biomarkers specifically targeting CH3Hg+ degradation by microorganisms remains a largely uncharted territory14. In the context of complex soil conditions, a recent work has proposed potential microbial taxa associated with CH3Hg+ degradation in soils through ecological statistical analyses15. While these findings have offered valuable insights into microbial degradation of CH3Hg+ in soils, there is still a lack of experimental evidence pinpointing the specific microbial taxa responsible for this process.
Two predominant pathways have been identified in the microbial degradation of CH3Hg+: reductive degradation mediated by the mer operon and oxidative degradation linked to one-carbon (C1) metabolism16,17. The mer operon has garnered attention, particularly the merB gene, which encodes the alkaline mercury lyase responsible for demethylating CH3Hg+ into Hg(II), and the subsequent merA gene, which catalyzes the transformation of reducing Hg(II) into Hg(0). Oxidative degradation is believed to involve a co-metabolism of CH3Hg+ resembling the metabolism of other small organic substrates (e.g., C1 compounds) by heterotrophic bacteria, including sulfate-reducing bacteria, methanogens, and certain aerobic bacteria. Studies have demonstrated that a significant portion of putative microbial taxa associated with CH3Hg+ degradation exhibits metabolic potentials related to sulfur metabolism and nitrate reduction18,19. For instance, known microbial taxa associated with CH3Hg+ degradation through non-merB-mediated pathways often exhibit sulfate reduction abilities20,21. While previous research has emphasized the significance of microbial taxa in CH3Hg+ degradation in sediments and paddy soils20,22, there has been limited exploration of the metabolic pathways associated with CH3Hg+ degradation by soil microorganisms.
Here, we aimed to explore specific microbial taxa and metabolic pathways involved in CH3Hg+ degradation and to estimate the microbial contribution to CH3Hg+ mitigation in rice. We employed 13CH3Hg+-DNA stable-isotope probing (SIP) combined with 16S rRNA long-read sequencing to identify microbial taxa responsible for 13CH3Hg+ degradation, and subsequently validated the degradation ability of candidate taxa through pure culture assays. We further reconstructed metagenome-assembled genomes (MAGs) to explore potential metabolic pathways associated with CH3Hg+ degradation. Finally, we estimated potential changes in rice CH3Hg+ accumulation and associated health risks resulting from microbial degradation of soil CH3Hg+ in the typical paddy soils from main rice-producing areas in China.
Results
13CH3Hg+ degradation in soils
Using 13C-labeled CH3Hg+ as a metabolic tracer, we investigated microbial taxa involved in CH3Hg+ demethylation in two contrasting soils (i.e., paddy and upland soils) with historically low or high Hg contamination levels (Supplementary Table S1). Twelve microcosm experiments were conducted by incubating each soil with CH3Hg+ at a spiked concentration of 50 μg kg−1 in the dark. The CH3Hg+ concentration in sterilized soils had no discernible changes during the incubation (Fig. 1A, B). We observed consistent microbial degradation of CH3Hg+ in the eight unsterilized treatments, but the extent of degradation depended on the soil types and the initial background Hg levels (Fig. 1 and Supplementary Table S1). For the soil from high Hg-contaminated site, the mean concentrations of CH3Hg+ after 28 days of incubation were 5.16 μg kg−1 and 23.16 μg kg−1 in the paddy soil (High-P) and upland soil (High-U), respectively, and the degradation efficiencies reached 86 and 37% (Fig. 1C). Differently, the mean concentrations of CH3Hg+ were 28.05 μg kg−1 and 14.10 μg kg−1 in the paddy soil (Low-P) and upland soil (Low-U) that were from the low Hg-contaminated region, respectively, and the degradation efficiencies of CH3Hg+ were 40 and 64% (Fig. 1D). Among these treatments, CH3Hg+ concentration in the High-P and Low-U soils showed a clear decline with the spiked 13CH3Hg+, similar to the trends observed with the addition of 12CH3Hg+ in the soils (Supplementary Fig. 1). Additionally, the degradation rates of CH3Hg+ were generally positive over the entire incubation period in the 13CH3Hg+ treatments (Supplementary Fig. 2). The highest degradation rates were observed on day 14 in the High-P soil (2.95 μg kg−1 d−1) and Low-U soil (1.43 μg kg−1 d−1), whereas peak rates occurred on day 3 in the High-U soil (1.57 μg kg−1 d−1) and Low-P soil (5.30 μg kg−1 d−1). These results suggest that soil microorganisms had a strong ability to degrade the spiked CH3Hg+, with higher CH3Hg+ degradation potential in the High-P and Low-U soils compared with the High-U and Low-P soils.

A, B The dynamics of CH3Hg+ concentrations in the 13CH3Hg+ treatments of the paddy and upland soils from high and low background Hg contamination sites, respectively. C, D The biotic degradation efficiencies of CH3Hg+ in the soils. The microbial degradation efficiency of CH3Hg+ was calculated by subtracting the CH3Hg+ degradation efficiency in sterilized soils from the total CH3Hg+ degradation efficiency in unsterilized soils. Data are shown in average values with standard deviations (three biological replicates, n = 3). ** indicates statistically significant levels of p < 0.01 based on two-sided T Test in (C, D). Source data are provided as a Source Data file.
Identification of microorganisms degrading 13CH3Hg+
To identify candidate microbial taxa involved in CH3Hg+ degradation, DNA-SIP experiments were performed using 13C-labeled or unlabeled 12CH3Hg+ as substrates on the High-P and Low-U soils. Genomic DNAs from the microcosm soil with 12CH3Hg+ and 13CH3Hg+ were separated into heavy (i.e., 13C-DNA) fractions and light (i.e., 12C-DNA) fractions by CsCl gradient ultracentrifugation. The abundances of 16S rRNA genes in these fractions were then quantified using real-time quantitative PCR to assess the distribution of bacterial communities across buoyant density (BD) gradients. Results showed that 16S rRNA genes were detected at the maximum abundance in the light DNA fractions (at day 7) from both the 12CH3Hg+ and 13CH3Hg+ treatments in the examined soils, while a gradual shifting of 16S rRNA gene abundance to the heavier fractions occurred at day 14 and 28 and only in the 13CH3Hg+ treatment (Fig. 2A–F). Therefore, the fractionation of DNA subsamples reveals a promising approach to trace and identify the microorganisms degrading 13CH3Hg+.

A–F Quantitative distribution of 16S rRNA genes across CsCl gradient fractions from the 12CH3Hg+ and 13CH3Hg+ treatments in the paddy soil from high Hg contaminated site (High-P) and upland soil from low Hg contaminated site (Low-U). G, H Venn diagrams showing the shared and exclusive bacterial OTUs in the fraction of the highest 16S rRNA gene copies from 13CH3Hg+ treatment and the corresponding fraction in 12CH3Hg+ treatment at day 28 in the High-P and Low-U soils. I, J The proportion of different bacterial phylum/class incorporating 13C in the labeled microcosms from High-P and Low-U soils at 28 d. Eighteen fractions from each CsCl gradient were analyzed, and the abundance of the 16S rRNA genes in each fraction was converted to the proportion of total 16S rRNA gene abundance throughout the gradient fractions. Vertical bars in (A–F) represent standard deviations of the relative abundance (three biological replicates, n = 3) in the 12CH3Hg+ (control) and 13CH3Hg+ treatments, and horizontal error bars in (A–F) represent the standard deviations of the buoyant density of the same order fractions (three biological replicates, n = 3) in the 12CH3Hg+ (control) and 13CH3Hg+ treatments. The data in (A–F) are presented as mean values ± standard deviation. The shadings in (A–F) indicated the fraction of the highest 16S rRNA gene copies from 12CH3Hg+ and 13CH3Hg+ treatments. Source data are provided as a Source Data file.
Given that the obvious shift of the 16S rRNA gene abundance in light fractions was detected at day 28, the fractions (from nine to 14 fractions) with higher 16S rRNA gene abundance from the 12CH3Hg+ and 13CH3Hg+ treatments were selected for long-read sequencing of 16S rRNA genes on the PacBio RS II platform. Microorganisms showing 13C enrichment in the DNA fractions of the 13CH3Hg+ treatment are assumed to have the ability to assimilate CH3Hg+ and thus incorporate 13C into their DNA. By examining the abundance of 16S rRNA genes across CsCl gradient fractions in the microcosm incubation over time, the risk of potential cross-feeding of assimilated 13C within microbial communities was minimized (Fig. 2A–F). We showed significant 13C enrichments in the fraction of the highest 16S rRNA gene copies from the 13CH3Hg+ treatment of both soils, comparing with the corresponding fraction in the 12CH3Hg+ treatment (Fig. 2G–J). In brief, there were 509 OTUs and 54 OTUs significantly enriched in 13CH3Hg+-spiked soils in High-P (Fig. 2G) and Low-U (Fig. 2H), respectively. Most of the OTUs were close to Planctomycetota, Proteobacteria and Acidobacteria in the 13C-DNA from spiked-13CH3Hg+ soils (Fig. 2I, J; Supplementary Fig. 3).
Moreover, dominant families, such as Xanthomonadaceae, Xanthobacteraceae and Comamonadaceae were significantly enriched in the 13CH3Hg+ treatment in both the High-P and Low-U soils (Fig. 3A, B). We further identified 34 genera with significantly different abundances between the 13CH3Hg+ and 12CH3Hg+ treatments in the High-P soil (p < 0.05), and Pseudarthrobacter, Dechloromonas and MM2 were significantly enriched in the 13CH3Hg+ treatment (Fig. 3C, D). Furthermore, Dechloromonas and MM2 exhibited significant enrichment in both light and heavy fractions (from nine to 14 fractions) from the 13CH3Hg+ treatment compared with the 12CH3Hg+ treatment (Fig. 3D; Supplementary Fig. 4). This broad distribution prompted us to further investigate their potential roles in CH3Hg+ degradation. Pure culture assays with Dechloromonas denitrificans and Methylovorus menthalis, belonging to the candidate taxa, confirmed their ability to degrade CH3Hg+ (Supplementary Fig. 5). For the Low-U soil, the relative abundance of VadinHA49 exhibited a significant increase in the 13CH3Hg+ treatment compared to the 12CH3Hg+ treatment (Fig. 3E, F).

A, B Bacterial community composition represented by the most abundant families (top 20) in the fraction of the highest 16S rRNA gene copies from 13CH3Hg+ treatment and the corresponding fraction in 12CH3Hg+ treatment at day 28 in the SIP experiments of the paddy soil from high Hg contaminated site (High-P) and upland soil from low Hg contaminated site (Low-U), respectively. C Biomarkers of key genera in the fraction of the highest 16S rRNA gene copies from 13CH3Hg+ treatment and the corresponding fraction in 12CH3Hg+ treatment of High-P soil. D Relative abundances of enriched genera across CsCl gradient fractions at 12CH3Hg+ and 13CH3Hg+ treatments of High-P soil. E Biomarkers of key genera in the fraction of the highest 16S rRNA gene copies from 13CH3Hg+ treatment and the corresponding fraction in 12CH3Hg+ treatment of Low-U soil. F Relative abundances of enriched genera across CsCl gradient fractions at 12CH3Hg+ and 13CH3Hg+ treatments of Low-U soil. Vertical bars in (D, F) represent standard deviations of the relative abundance (three biological replicates, n = 3) in the 12CH3Hg+ (control) and 13CH3Hg+ treatments, and horizontal error bars in (D, F) represent the standard deviations of the buoyant density of the same order fractions (three biological replicates, n = 3) in the 12CH3Hg+ (control) and 13CH3Hg+ treatments. The data in (D, F) are presented as mean values ± standard deviation. Source data are provided as a Source Data file.
Putative metabolic pathways of CH3Hg+ degradation by metagenomic analyses
Combined analyses of metagenome sequencing and reconstruction of MAGs were performed to retrieve the draft genomes involved in 13C-labeled metabolic processes, and then putative metabolic pathways of CH3Hg+ degradation were proposed. Due to insufficient 13C-labeled DNA from a single extraction for shotgun metagenomic sequencing, DNA was extracted three times from the same soil sample for each replicate. The DNA from each extraction was subjected to DNA-SIP gradient fractionation. After DNA-SIP fractionation, composite samples were formed by combining the fractions with the highest 16S rRNA gene copies from the 13CH3Hg+ treatment of the High-P soil on day 28 for sequencing. At the community level in the paddy soil, the relative abundances of genes involved in Hg reduction (e.g., merA and merB) were comparable between the 13CH3Hg+ and 12CH3Hg+ treatments (Fig. 4A; Supplementary Fig. 6).

A The potential metabolic pathways linked with CH3Hg+ degradation at the community level from the high Hg-contaminated paddy soil based on the predicted genes. B Heatmaps of the genes involved in Hg reduction and metabolisms of carbon, methane and nitrogen detected in the CH3Hg+ degraders associated MAGs recovered from the 13CH3Hg+ treatment. The color gradient in (A) represents the relative abundances of major genes of metagenomic analysis in the heavy DNA fraction from the 13CH3Hg+ treatment and the corresponding fraction in the 12CH3Hg+ treatment. The square and circle in (A) represent genes in the 12CH3Hg+ and 13CH3Hg+ treatments, respectively. The definitions of genes are listed in the Source data. * indicates genes that exhibited significant enrichment in the heavy fraction of the 13C-labeled treatment compared to the corresponding fraction in the 12C control at levels of p < 0.05 based on two-sided T Test. The colors in (B) indicate the number of the gene(s) detected in the MAGs. Exact p-values and Source data are provided as a Source Data file. Figure 4A was created in BioRender (https://BioRender.com/oksd9hf).
To further explore the multifunctional metabolic characteristics related to the 13CH3Hg+ degradation, genes involved in sulfate reduction, carbon metabolism (e.g., methane metabolism) and nitrogen metabolism (e.g., nitrate reduction and denitrification) were searched within the metagenomic contigs11,19,23 (Fig. 4A; Supplementary Fig. 6). Specifically, genes associated with sulfate reduction including those encoding sulfate adenylyl transferase (sat), dissimilatory sulfite reductase (dsrAB), and adenylylsulfate reductase (aprAB) were detected, showing comparable relative abundances between the 13CH3Hg+ and 12CH3Hg+ treatments (Fig. 4A). Furthermore, several key biochemical pathways related to carbon metabolism were annotated by the presence of genes involved in tetrahydrofolate (H4F) pathway, TCA cycle, pyruvate metabolism, methane metabolism and purine metabolism. For instance, genes encoding isocitrate dehydrogenase (IDH3), 2-oxoglutarate ferredoxin oxidoreductase (korAB), tetrahydromethanopterin S-methyltransferase subunit B (mtrB), phosphoribosylaminoimidazole-succinocarboxamide synthase (purC), dimethylglycine dehydrogenase, 5-methyltetrahydrofolate homocysteine methyltransferase (metH), aconitate hydratase (acnA) and succinate dehydrogenase (sdhA) exhibited significant enrichment in the 13CH3Hg+ treatment compared with the 12CH3Hg+ treatment (Fig. 4A; Supplementary Fig. 6). Among them, the metH gene was the most abundant, accounting for 0.16% of the annotated genes. In addition, genes related to nitrogen metabolism and membrane transport, such as nitrate reduction, denitrification and ABC transporter, were also significantly enriched in the 13CH3Hg+ treatment (Fig. 4A; Supplementary Fig. 6). For instance, genes encoding glutamine synthetase (glnA), glutamate synthase (gltB), periplasmic nitrate reductase (napA and napB), nitrite reductase (nirS) and D-methionine transport system ATP-binding protein (metN) exhibited higher relative abundance in the 13CH3Hg+ treatment compared with the 12CH3Hg+ treatment.
Read assembly and binning from 13C-SIP metagenomics recovered a total of 5 fairly-complete MAGs associated with candidate CH3Hg+ degraders (completeness > 70% and contamination < 10%) (Fig. 4B; Supplementary Table S2), and the metabolic reconstruction of these MAGs revealed the potential functions of the genomes20. Among the five MAGs, 3 were identified as novel species associated with the genus Pseudarthrobacter (Supplementary Fig. 7). These Pseudarthrobacter-associated MAGs generally harbored mer operons, such as merP (encoding a periplasmic protein) and merR (encoding a regulatory protein), but none carried the merA and merB genes. Meanwhile, additional MAGs (MAG3 and MAG4) did not carry any genes associated with mer operons. Moreover, all the MAGs contained genes encoding carbon metabolism pathways, such as the Wood-Ljungdahl pathway (folD, fhs, fdhA, and metF) and dicarboxylate-hydroxybutyrate cycle (sucC, sucD, and mdh) (Fig. 4B). Furthermore, genes related to methanogenesis, denitrification and nitrate reduction were detected in all MAGs, and abundant genes involved in pyruvate metabolisms, purine metabolism, and ABC transporter were found in all MAGs consuming the spiked 13CH3Hg+, but genes involved in sulfate reduction were absent in all MAGs (Supplementary Fig. 8).
Estimation of rice CH3Hg+ and health risks through microbial degradation of soil CH3Hg+
To evaluate the mitigation of CH3Hg+ in rice through microbial degradation of CH3Hg+ in soils across the main rice-producing areas of China, we used the bioaccumulation factor (BAF) to link CH3Hg+ concentrations in grains to soils (Supplementary Table 3). Results indicated that the microbial degradation efficiencies of soil CH3Hg+ ranged from 8.16 to 64.26% in these typical soils across China, leading to decreases of 0.10–2.25 μg kg⁻¹ in rice CH3Hg+ (Supplementary Table 4). Health risks associated with dietary exposure to rice CH3Hg+ were further estimated by calculating HQ and IQ decrements (ΔIQ). According to the health risk model, the potential decreases in rice CH3Hg+ concentration equate to HQ reduction of 0.62–13.75%, corresponding to ΔIQ decreases of 0.13 to 2.84%, in response to microbial degradation of CH3Hg+ in the soils (Supplementary Table 4). These results highlight the crucial role of microbial degradation in mitigating rice CH3Hg+ and associated health risks.
Discussion
Demethylation of CH3Hg+ is a fundamental route to reduce Hg toxicity and subsequent health risks, and microbial degradation is considered one of the most promising pathways to control CH3Hg+ pollution in the environment13,22. Microbial degradation of CH3Hg+ has been observed in various habitats, such as sediments24,25, marine water26, and agriculture soils15. Our findings further revealed that Xanthomonadaceae, Comamonadaceae, and Xanthobacteraceae were shared microbial taxa participating in CH3Hg+ degradation across soils from two geographically distant regions (spanning ∼500 km) with different levels of Hg contamination (Fig. 3A, B). This observation underscores the pivotal role of these microorganisms in CH3Hg+ degradation, irrespective of differences in climate and environmental conditions. Previous studies have shown that certain taxa affiliated with Xanthomonadaceae, such as Xanthobacter autotrophicus Py2, were especially efficient in CH3Hg+ degradation with Hg-contaminated sediments27,28. The enrichment of 13C-labeled Xanthomonadaceae also supports recent findings that established a statistical association between Xanthomonadaceae (a member of Xanthomonadales) and CH3Hg+ concentrations in agricultural soils29. Moreover, in this study, Pseudarthrobacter, Dechloromonas, and MM2, which belong to Micrococcaceae, Xanthomonadaceae and Methylophilaceae, were the most significantly enriched genera degrading 13CH3Hg+ in the paddy soil from the high Hg-contaminated site (Fig. 3C). Notably, Pseudarthrobacter had been reported to exhibit the tolerance of Hg in the soil30. Moreover, Pseudarthrobacter and Dechloromonas are known for their ability to degrade organic pollutants by using them as carbon sources31,32,33, suggesting they may similarly degrade CH3Hg+ to obtain carbon for growth in the soils. These findings demonstrate the roles of Pseudarthrobacter, Dechloromonas, and MM2 as important contributors in CH3Hg+ degradation, especially in highly contaminated environments. Nevertheless, we acknowledge that the primers 27 F and 1492 R may miss some active CH3Hg+ demethylators, though they are widely used for full-length 16S rRNA gene amplification due to their broad coverage across a wide range of bacterial phyla. Moreover, we performed metagenomic sequencing, which provides additional species-level annotations and enables the identification of microbes involved in CH3Hg+ degradation. Importantly, results showed that the dominant and differential microbial taxa identified through metagenomics were generally consistent with those detected by 16S rRNA gene amplicon sequencing (Supplementary Figs. 3 and 9). This complementary approach allows for a more comprehensive identification of microbial groups involved in CH3Hg+ degradation.
Further SIP-metagenomic results indicate that multiple metabolic pathways are involved in 13CH3Hg+ degradation (Fig. 4). We reconstructed a total of five fairly complete MAGs linked to 13CH3Hg+ degradation, predominantly affiliated with Actinobacteria and Gemmatimonadota. Among these MAGs, 3 were identified as novel species associated with the genus Pseudarthrobacter (Supplementary Fig. 7). Notably, none of the 13C-labeled MAGs contained the merA and merB genes, which are pivotal for reductive demethylation via the mer operon, suggesting that mer-mediated reductive demethylation was not accountable for the dominant 13CH3Hg+ degradation pathway in these taxa. This was further supported by the similar relative abundances of mer genes between the 13CH3Hg+ and 12CH3Hg+ treatments at the community level (Supplementary Fig. 6). Instead, MAG-based analysis indicated that genes involved in carbon and nitrogen metabolism were present in the reconstructed genomes, whereas community-level analysis revealed their significant enrichments in the 13CH3Hg+ treatment. These findings suggest the involvement of mer-independent CH3Hg+ degradation, such as oxidative demethylation, potentially mediated via co-metabolic processes involving simple carbon sources (i.e., C1 compounds)13.
Interestingly, we observed that Actinobacteria and Gemmatimonadota exhibited higher relative abundances in the 13C-DNA heavy fraction of the 13CH3Hg+ treatment compared to the 12CH3Hg+ treatment (Supplementary Fig. 9), further supporting their functional involvement in 13CH3Hg+ degradation rather than being influenced by GC content bias. Functional profiling of these groups further revealed the comparable relative abundance of mer operon genes (e.g., merA and merB) in the 13CH3Hg+ and 12CH3Hg+ treatments, while several genes associated with carbon and nitrogen metabolism were significantly enriched in the 13CH3Hg+ treatment (Supplementary Fig. 10). Taken together with evidence from both the MAG and community analyses, these findings suggest that alternative pathways, such as oxidative demethylation, rather than reductive demethylation, may play a dominant role in CH3Hg+ degradation in these actively 13C-labeled microbial communities. We acknowledge that some merAB-mediated demethylators may have remained in the light fraction and were not detected due to the limited amount of 13C-methyl assimilation. Nevertheless, our 13CH3Hg+-DNA-SIP analyses provide direct evidence that at least a subset of the microbial community actively assimilated 13C from 13CH3Hg+. Future studies integrating complementary methods are needed to further elucidate the underlying metabolic mechanisms involved in CH3Hg+ degradation.
It is also noteworthy to mention that our study demonstrates the substantial benefits of microbial-mediated soil CH3Hg+ demethylation to decrease CH3Hg+ accumulation in rice grains, thereby mitigating HQ and moderating IQ losses through dietary exposure. While the global effort to reduce CH3Hg+ accumulation in food primarily focuses on controlling Hg emissions34, our results emphasize the critical role of soil microorganisms in reducing the CH3Hg+ exposure risk to humans and wildlife. We suggest promoting the in situ activities of these microorganisms by optimizing environmental factors, such as nutrient availability and oxygen levels, enabling them to exert more prominent roles in CH3Hg+ degradation within agroecosystems. Alternatively, inoculating indigenous microbial strains that are pre-adapted to contaminated environments could also promote CH3Hg+ degradation. Additionally, advancements in synthetic biology offer promising avenues to optimize their metabolic functions for more efficient CH3Hg+ degradation by designing synthetic microbial consortia35. For instance, co-culture systems combining Methylovorus menthalis with methanotrophs, such as Methylococcaceae may enhance CH3Hg+ degradation through synergistic metabolic interactions, as methanol cross-feeding between these two families has been confirmed36. Incorporating these microbial regulation strategies into existing Hg emission reduction frameworks is expected to enhance the bioremediation effectiveness and reduce health risks associated with environmental Hg contamination. Moreover, although the health risk estimates depend on several assumptions that may introduce uncertainties into the model, our sensitivity and uncertainty analyses reveal that microbial degradation efficiency and soil CH3Hg+ concentration were the most important parameters for HQ and ΔIQ, with the highest sensitivity coefficient for HQ and ΔIQ (Supplementary Tables 5–7). We acknowledge the limitations of these analyses, which assume parameter independence and do not account for joint variability or probabilistic distributions. These methods were chosen due to the limited size of our dataset, which precluded the use of more advanced probabilistic approaches like Monte Carlo simulations. Therefore, further studies expanding the dataset are warranted to reinforce and consolidate the findings presented in this study, though the field sampling and experimental analyses are challenging.
Methods
Soil sampling and characterization
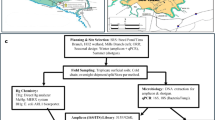
Both paddy and upland soils (0 ~ 15 cm depth) were collected from the two typical agricultural regions in Wanshan, Southwest China (27.52°N, 109.21°E), and Xianning, Center China (29.90°N, 114.23°E), in 2018 (from September to November). The mean annual precipitation and temperature in these areas ranged from 1378.7–1577.4 mm and 13.7–16.8 °C, respectively. Wanshan is known as the Hg hotspot of China, with more than 600 years of Hg mining and smelting activities, leading to elevated Hg concentrations in these soils (referred to here as high Hg-contaminated). While there are no documented Hg pollution sources in Xianning, Hg in the soil is most likely derived from parent materials and atmospheric deposition (referred to here as low Hg-contaminated). Paddy and upland rotations are used as common cropping systems in both regions7, so the presence of CH3Hg+ in the soil in both areas should not be disregarded. The collected soils were kept in an ice box during shipping and stored at 4 °C prior to incubation experiments. Details of soil geochemical properties are shown in Table S1 in the Supplementary Information.
Microcosm incubation
The 13CH3Hg+ used in this study was synthesized from 13CH4I and HgO in our laboratory37, and then dissolved in methanol. The purity of the synthesized 13CH3Hg+ compounds was evaluated by comparing the measured 13CH3Hg+ concentration with the theoretical value based on the synthesis procedure. The close match between the measured and theoretical concentrations suggests a high level of purity. In addition, we performed liquid chromatography to check for organic impurities. The conversion efficiency of 13CH3Hg+ ranged from 50.23 to 61.45%, and it could reach up to 99% after extraction and back-extraction using methylene chloride and water to remove inorganic components. The CH3Hg+ contaminated soils were prepared as follows: 10 g soil (dry weight) was spiked with CH3Hg+ solutions (both 12CH3Hg+ and 13CH3Hg+ were used in parallel for microbial comparison) to give a fixed concentration of 50 μg kg–1. As a result, the actual CH3Hg+ concentrations in the microcosms reflected the sum of the background CH3Hg+ (ranging from 0.45 to 3.48 μg kg–1; Supplementary Table 1) and the spiked CH3Hg+ (50 μg kg–1). The background CH3Hg+ concentrations may be substantial in general CH3Hg+ transformation studies; however, soil microorganisms assimilated only small amounts of 13C from 13CH3Hg+. Considering the relatively high cost of DNA-SIP, we increased the spiked CH3Hg+ concentration (50 μg kg–1) to ensure sufficient 13C enrichment for effectively tracing 13C-consuming microorganisms and identifying those involved in 13CH3Hg+ degradation. This concentration is comparable to CH3Hg+ concentrations observed in polluted farmland soils from Hg mining areas (ranging from 0.14 to 67 μg kg–1)38. A higher concentration of 13CH3Hg+ was used to obtain sufficient 13C-labeled target DNA for microbial analysis. The experiments were conducted in 50 mL serum bottles, and the microcosm experiment included the following treatments: (1) High-P, which used paddy soil from Wanshan (high Hg contaminated areas); (2) High-U, which used upland soil from Wanshan; (3) Low-P, which used paddy soil from Xianning (low Hg contaminated areas); (4) Low-U, which used upland soil from Xianning. These four treatments were all prepared using both ambient CH3Hg+ and 13C-labeled CH3Hg+. Controls were set up as follows: (5) High-SP, and (6) High-SU, representing sterilized paddy and upland soil by gamma rays from areas with high Hg-contamination, respectively; (7) Low-SP, and (8) Low-SU, representing sterilized paddy and upland soil by gamma rays from areas with low Hg-contamination, respectively. This resulted in a total of twelve treatments. During the incubation, water content was kept constant by checking the weight of the bottles regularly and adding ddH2O39. All treatments in this study were conducted in triplicate and incubated for 28 days at 25 °C in the dark. The experiment duration of 28 days was carefully selected based on literature and our pre-experiments40,41. This duration can balance both the degradation of 13CH3Hg+ and the sufficient 13C enrichment in 13CH3Hg+-degrading microorganisms.
CH3Hg+ analysis and chemical analyses
For soil CH3Hg+ analysis, ∼0.50 g dry soil of each sample was accurately weighed and then digested with 1.5 mL CuSO4 (2 mol L−1) and 7.5 mL HNO3 (25%, v/v) in an individual PE centrifuge tube. Subsequently, 10 mL dichloromethane (CH2Cl2) was added to the centrifuge tubes and closed with clean lids. These centrifuge tubes were tightly attached to an orbital shaker and shaken for 30 min at a rotational speed of 240 rpm. Then, the substrate digestion solution was transferred to a new centrifuge tube and allowed to settle in a water bath (50 °C) until all visible CH2Cl2 had completely volatilized. Finally, the extracting solution was obtained by removing any residual CH2Cl2 with high purity N2 in a water bath at 75 °C for 10 min. The concentration of CH3Hg+ in the extracting solution was measured by an automated Methyl Mercury Analytical System (Brooks Rand Labs, Seattle, WA, USA), which provides precise determination at picogram levels. CH3Hg+ concentration was measured at days 0, 3, 7, 14, and 28 in both the 13CH3Hg+ and 12CH3Hg+ treatments. However, the measured soil CH3Hg+ concentrations at day 0 (around 45 μg kg−1) did not always match the theoretical values, likely due to inevitable experimental errors, including variations in extraction efficiency and analytical recovery. According to previous studies, acceptable recoveries of CH3Hg+ from reference materials typically range from 75 to 115%, depending on soil properties15,22,42. The degradation efficiencies of CH3Hg+ were calculated based on the averaging differences of total CH3Hg+ concentrations for 28 days, using the measured CH3Hg+ concentrations at day 0 (including the background CH3Hg+) as the baseline. While we did not analytically distinguish the two isotopologues (13CH3Hg+ and 12CH3Hg+), and the individual degradation efficiencies of 13CH3Hg+ and 12CH3Hg+ were not directly quantified, the observed decline in total CH3Hg+ concentration, along with the significant 13C enrichment of microbial taxa in the heavy DNA fractions from 13CH3Hg+-DNA-SIP analyses, provide strong evidence for microbial demethylation of 13CH3Hg+ during the incubation. Moreover, soil pH was measured by a Delta pH-meter (Mettler-Toledo Instruments Co., Columbus, OH, USA) in a soil slurry with a water-to-soil ratio of 2.5:1. Dissolved C in soils was extracted with 0.5 M K2SO443. The extracted solution was used for analyses of DOC by a total organic C (TOC) analyzer (vario TOC; Elementar).
DNA extraction and SIP gradient fractionation
On the basis of the CH3Hg+ concentration changes, three typical time points (at days 7, 14, and 28) of the incubation experiment were selected for DNA extraction. Total genomic DNA was extracted using a MoBio power soil DNA isolation kit (MO BIO Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s instruction. DNA concentrations were determined using an ND-2000 UV-vis spectrophotometer (NanoDrop Technologies, Wilmington, DE). To separate 12C- and 13C-DNA, nucleic acid ultracentrifugation was conducted. Briefly, ∼2500 ng of genomic DNA was mixed with CsCl gradient in 5.1 mL Quick-Seal polyallomer tubes (Beckman Colter, Pasadena, USA) to achieve an initial BD of 1.7152 g mL−1. After balancing and sealing, density gradient centrifugation was performed at 176,770 × g for 45 h at 20 °C in an Optima XPN-100 ultracentrifuge (Beckman Colter) equipped with a VTi 65.2 vertical rotor (Beckman Colter, USA). After ultracentrifugation, DNAs were immediately fractionated into 18 gradient fractions (∼280 μL per fraction) following the published protocol44,45. Briefly, the tube was fixed in a clamp, and the bottom was carefully pierced with a needle in a smooth and controlled manner, after which the needle was discarded. A syringe pump was used to inject sterile deionized water into the top of the tube through the needle, and drops were collected from the bottom into sterile 1.5 mL tubes. The pump rate was set to yield ∼18 × 280 µL fractions within 12 minutes (425 µL min−1, ~40 s per fraction). To ensure the gradients formed correctly, the BD value of each fraction was measured using a digital refractometer (Reichert AR2000), and then CsCl was removed by glycogen-assisted ethanol precipitation46,47. Additionally, fractions were collected in a consistent manner to avoid mixing between them, and the process was performed rapidly to prevent gradient mixing.
Quantitative PCR and PacBio sequencing of the 16S rRNA genes
To quantify the 16S rRNA genes in each fraction, qPCR was performed using a CFX Connect real-time system (BioRad, CA, USA) with the 341 F (5′-CCTACGGGNGGCWGCAG-3′) and 805 R (5′-GACTACHVGGGTATCTAATCC-3′) paired primers48, which match ∼90% of all bacterial sequences and cover all phyla in the Ribosomal Database Project release 10.25. The qPCR protocol proceeded as follows: an initial step at 95 °C for 5 min, followed by 40 cycles of 94 °C for 60 s, 58 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 5 min. The standards with known copy numbers of the target sequence were diluted in a ten-fold serial dilution to achieve a concentration range spanning from 10 to 107 copies μL−1. All qPCR reactions were run in triplicate, including the blank that ran with water as a template instead of DNA.
Amplification of full-length (V1–V9) 16S rRNA gene sequences was performed on a PacBio RS II platform (Majorbio Bio-Pharm Technology, Co., Ltd., Shanghai, China). Briefly, the full-length 16S rRNA genes were amplified by PCR using the universal bacterial primer set 27 F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492 R (5′-GGTTACCTTGTTACGACTT-3′)49. The two primers contained a set of 16-nucleotide barcodes. PCR was prepared with the following reaction components: 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of Forward Primer (5 μM), 0.8 μL of Reverse Primer (5 μM), 0.4 μL of FastPfu Polymerase, 0.2 μL of BSA, 10 ng of Template DNA, and ddH2O adjusted for the sample to a 20 μL of reaction total volume. The PCR program was as follows: 95 °C for 3 min, 25 cycles of 98 °C for 20 s, 57 °C for 30 s and 72 °C for 90 s, with a final extension of 72 °C for 2 min.
The fractions (from nine to 14 fractions) with higher 16S rRNA gene abundance from the 12CH3Hg+ and 13CH3Hg+ treatments were selected for long-read sequencing of 16S rRNA genes. Moreover, we focused on analyzing the 16S rRNA gene amplicon sequences from the fraction with the highest abundance of 16S rRNA gene copies in the 13CH3Hg+ treatment and the corresponding fraction in the 12CH3Hg+ treatment50. This targeted analysis better represents the active microbial taxa directly involved in 13CH3Hg+ degradation. Sequencing of the amplicons was performed on a PacBio Sequel instrument (Pacific Biosciences, Menlo Park, CA, USA) at Shanghai Majorbio Bio-pharm Technology Co., Ltd., Shanghai, China (https://www.majorbio.com/). Raw data were retrieved and filtered with the circular consensus sequencing (CCS) software of the SMRT Link (minfullpass = 3, polish min Predicted Accuracy = 0.8, minLength = 500) to obtain the Raw CCS Reads. The barcode reads of every sample were recognized with Lima (https://github.com/PacificBiosciences/barcoding) to acquire Raw CCS, whereafter the CCS (accuracy above 99%) of each sample was tested and chimeric reads were removed with UCHIME (http://drive5.com/uchime, v8.1) to acquire quality controlled sequences. Subsequently, the representative sequences were compared using the Mothur (https://mothur.org/wiki/Classify.seqs, v1.40.0) software with Silva 132 database (classified at a bootstrap threshold of 0.9) to gain classified information.
Shotgun metagenomic sequencing
The metagenomic shotgun sequencing (2 × 150 bp) was conducted exclusively on the High-Hg contaminated paddy soil (High-P), which was selected due to its highest potential of microbial 13CH3Hg+ degradation. The sequencing was carried out on an Illumina NovaSeq 6000 platform (Shanghai Majorbio Bio-pharm Technology), which generally requires 1–5 μg of DNA for sequencing51,52. Since the DNA quantity did not meet these requirements, composite DNA samples were obtained by pooling the DNA from heavy fractions with the highest 16S rRNA gene copies from each of the triplicates of the 13CH3Hg+. The corresponding fractions from the 12CH3Hg+ treatment underwent the same procedure. A total of 10.7–18.6 Gb clean reads were obtained for each sample after quality control following the KneadData pipeline (v0.6.1), accounting for 94.3–97.9% of raw bases. De novo assembly of clean reads into contigs was performed using megahit (https://github.com/voutcn/megahit, -k-min 47 -k-max 97 -k-step 10, v1.2.9), CD-HIT (http://www.bioinformatics.org/cd-hit/, v4.6.1) and Salmon (https://github.com/COMBINE-lab/salmon, v1.10.0) were employed for subsequent open reading frames prediction53,54,55, non-redundant gene catalog clustering, and non-redundant gene quantification. Biological function annotation of the non-redundant genes was conducted on GhostKOALA annotation servers (Automatic KO assignment by GHOSTX sequence similarity search56. Binning of the assembled metagenome was computed and refined using MaxBin2 (http://sourceforge.net/projects/maxbin/, v2.2.7)57 and metaBAT2 (https://bitbucket.org/berkeleylab/metabat, v2.12.1)58 in MetaWRAP59,60. Completion and contamination estimation of all the bins were computed by CheckM (https://github.com/Ecogeno-mics/CheckM/wiki, v1.1.6)61. Taxonomic classification of MAGs was performed using GTDB-Tk v0.3.3 classify_wf command against the GTDB (http://gtdb.ecogenomic.org/, v2.3.2)62,63. Among all reconstructed MAGs, 13 were classified as medium-quality according to the Minimum Information about a Single Amplified Genome (MIMAG; >50% completeness, <10% contamination)64. Only 5 fairly complete MAGs with more than 70% completeness and less than 10% contamination were selected for further analysis65. An overview of the assembly statistics for MAGs in the selected soils was provided in Supplementary Table 2. The predicted genes of MAGs were annotated using the HMM profile database for KEGG orthology with predefined score thresholds through KofamScan (v1.3.0)66.
Validation of CH3Hg+ degradation by candidate bacterial taxa
To validate the ability of candidate microbial taxa to degrade CH3Hg+, we conducted a series of bioassays using Dechloromonas denitrificans (DSM 15892) and Methylovorus menthalis (DSM 24715), which belong to the proposed genera associated with CH3Hg+ degradation. Dechloromonas denitrificans (DSM 15892) and Methylovorus menthalis (DSM 24715) were purchased from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Both cultures were grown in the recommended media at 30 °C. Cells were harvested at the exponential growth phase and subsequently washed twice by repeated centrifugation (10 min at 1610 × g) and resuspension in phosphate buffer saline (PBS) at pH 7.2. The PBS consisted of 0.14 mol·L−1 NaCl, 3 mmol·L−1 KCl, 10 mmol·L−1 Na2HPO4, and 2 mmol·L−1 KH2PO4. The cell density was obtained by measuring optical density at 600 nm.
The CH3Hg+ degradation bioassays were conducted in sealed amber glass vials (4 mL). Each vial contained washed cells (5 × 108 cells·mL−1), supplemented once (at time zero) with 5 mmol·L−1 metal salt solution (MgCl2). The CH3Hg+ was added to the cell suspension to obtain a final concentration of 1 ng·mL−1 CH3Hg+. Experiments were performed in triplicate and included heated-killed culture controls (80 °C for 1 h) and PBS blanks. The final reaction volume was 1 mL in each vial. All vials were sealed immediately and placed on an orbital shaker and kept in the dark. At selected time points, replicate sample vials were taken out and analyzed for CH3Hg+ concentration.
Changes in rice CH3Hg+ and health risks resulting from microbial degradation of soil CH3Hg+
Exposure to CH3Hg+ through rice consumption has a prolonged effect on human health, as rice is a major source of dietary CH3Hg+ exposure in China, accounting for up to 96% of total CH3Hg+ exposure67. In our study, microorganisms may reduce the accumulation of CH3Hg+ in rice through the degradation of soil CH3Hg+. To predict the changes of CH3Hg+ accumulation in rice grains, we use the BAF to link CH3Hg+ in grains and in soils. The decreases in rice CH3Hg+ concentrations due to microbial degradation of soil CH3Hg+ are calculated using the BAFs, measured soils CH3Hg+ and microbial degradation efficiencies34:
Where soil CH3Hg+ values and microbial degradation efficiencies of CH3Hg+ were obtained from our unpublished study, in which we collected ten typical paddy soils, quantified CH3Hg+ concentrations in sterilization and unsterilization treatments, and calculated the microbial degradation efficiencies of CH3Hg+ (Supplementary Table 3). Microbial degradation efficiency (the percent of CH3Hg+ degraded over 28 days by the microbial community) was incorporated into the model to calculate changes in rice CH3Hg+ concentrations. It is important to note that the observed degradation efficiencies during the 28-day period represent net changes, accounting for both production and degradation processes. We chose microbial degradation efficiency over degradation rate as it provides a standardized index. While degradation rates vary over time, microbial degradation efficiency offers a relatively stable estimate of microbial CH3Hg⁺ degradation potential in environments.
Estimated daily intake (EDI) would be determined by calculations as described below34:
Where Ii represents the per-capita intake rate (g d−1 capita−1) in province i, Ci represents the rice CH3Hg+ (μg kg−1) while BW represents the average body weights of females (57.3 kg).
Health risks associated with dietary exposure to rice CH3Hg+ are estimated by calculating the HQ and ΔIQ in per-fetus across China. The equations for the calculation of HQ and ΔIQ are provided blow34,68:
In the formulas, HQ is used to evaluate the non-cancer health hazard, ΔEDI is the changes in daily intake, and RfD represents the reference dose for CH3Hg+, i.e., 0.1 μg kg−1·d−1 according to the USEPA (1997). Meanwhile, ΔIQ represents the change in IQ points for per-fetus due to intake of CH3Hg+. BW represents the average body weights of females, and coefficients including λ (μg Hg g-1 hair per μg Hg L−1 blood), γ (IQ points change per μg Hg g-1 hair), and β (μg Hg L−1 blood per μg Hg d−1) show the conversion from rice CH3Hg+ intake to per-fetus IQ change4,69. The values of λ, γ, and β are 0.20, 0.30, and 0.60, respectively.
We further conducted the one-at-a-time sensitivity analysis and uncertainty analysis based on interval methods to assess uncertainties in health risk estimates70,71. We identified the most important parameters for HQ and IQ decrements (ΔIQ) from microbial degradation efficiency, soil CH3Hg+ concentration, BAF, and rice ingestion rate. The results showed that microbial degradation efficiency and soil CH3Hg+ concentration were the most important, with microbial degradation efficiency having the highest sensitivity coefficients for HQ and ΔIQ (Supplementary Tables 5, 6). For instance, when microbial degradation efficiency increased from 8.16 to 64.26% under identical conditions, the reduction in HQ increased from 0.0095 to 0.0749, representing an eight-fold increase. We further conducted an uncertainty analysis based on interval methods by varying key parameters within their observed ranges (Supplementary Table 7). We quantified potential variability in HQ and ΔIQ, which ranged from 0.0027 to 0.3301 and 0.0006 to 0.0681, respectively. The results consistently demonstrated that microbial degradation efficiency is the key factor in reducing health risks.
Statistical analysis
A minimum of three independent microcosms were considered for each analysis. One-way analysis of variance followed by a two-sided Tukey post hoc test was performed by SPSS Statistics 21.0 (IBM, Chicago, IL) to detect the significant differences between treatments in the pure culture assays. LEfSe analysis (p < 0.05, LDA score > 2) was used to identify bacterial biomarkers for the 13CH3Hg+ and 12CH3Hg+ treatments on the open website (http://huttenhower.sph.harvard.edu/galaxy). A two-sided Student’s t test was conducted to identify genes or microbial species that exhibited significant enrichment in the heavy fraction of the 13C-labeled treatment compared to the corresponding fraction in the 12C control (* representing p < 0.05), and to assess differences in CH3Hg+ degradation efficiency between paddy and upland soils from both high and low Hg-contaminated sites. The phylogenetic trees were generated and visualized using a CVTree4 algorithm (https://cvtree.online/v4/prok/)72 and the Evolview v3 web application (https://www.evolgenius.info/evolview/#/treeview)73, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The full-length 16S rRNA gene amplicon sequences and metagenomic sequences generated in this study have been deposited to the NCBI SRA database under the BioProject IDs of PRJNA1183541 and PRJNA1183649, respectively. The BAF and rice ingestion rate used in this study were collected from https://doi.org/10.1038/s43016-024-00954-7 and https://doi.org/10.1038/s41467-019-09080-6, respectively. The databases used in this study include SILVA 132, Genome Taxonomy Database (http://gtdb.ecogenomic.org/) and KEGG database (https://ww.genome.ad.jp/kegg/). Source data are provided with this paper.
Code availability
All code associated with our analyses in this study is available at https://figshare.com/s/fd35576a3ffb97ea098b.
References
Wang, J. et al. Mobilization, methylation, and demethylation of mercury in a paddy soil under systematic redox changes. Environ. Sci. Technol. 55, 10133–10141 (2021).
Yang, A. et al. A review of mercury uptake, transport and bioaccumulation in rice. Water Air Soil Pollut. 234, 377 (2023).
Karagas, M. R. et al. Evidence on the human health effects of low-level methylmercury exposure. Environ. Health Perspect. 120, 799–806 (2012).
Rice, G. E., Hammitt, J. K. & Evans, J. S. A probabilistic characterization of the health benefits of reducing methyl mercury intake in the United States. Environ. Sci. Technol. 44, 5216–5224 (2010).
Liu, Y. R. et al. Unraveling microbial communities associated with methylmercury production in paddy soils. Environ. Sci. Technol. 52, 13110–13118 (2018).
Meng, B. et al. The process of methylmercury accumulation in rice (Oryza sativa L.). Environ. Sci. Technol. 45, 2711–2717 (2011).
Timsina, J. & Connor, D. J. Productivity and management of rice–wheat cropping systems: Issues and challenges. Field Crop. Res. 69, 93–132 (2001).
Xu, J. et al. Mercury methylating microbial community structure in boreal wetlands explained by local physicochemical conditions. Front. Environ. Sci. 8, 518662 (2021).
Moore, B. A new screen test and selective medium for the rapid detection of epidemic strains of Staph. Aureus. Lancet 2, 453–458 (1960).
Pak, K. R. & Bartha, R. Mercury methylation and demethylation in anoxic lake sediments and by strictly anaerobic bacteria. Appl. Environ. Microbiol. 64, 1013–1017 (1998).
Lu, X. et al. Methylmercury uptake and degradation by methanotrophs. Sci. Adv. 3, e1700041 (2017).
Lu, X. et al. Anaerobic mercury methylation and demethylation by geobacter bemidjiensis BEM. Environ. Sci. Technol. 50, 4366–4373 (2016).
Barkay, T. & Gu, B. Demethylation-the other side of the mercury methylation coin: a critical review. ACS. Environ. Au. 2, 77–97 (2022).
Yu, R. Q. & Barkay, T. Microbial mercury transformations: molecules, functions and organisms. Adv. Appl. Microbiol. 118, 31–90 (2022).
Zhou, X.-Q. et al. Microbial communities associated with methylmercury degradation in paddy soils. Environ. Sci. Technol. 54, 7952–7960 (2020).
Oremland, R.S., Miller, L.G., Dowdle, P., Connell, T. & Barkay, T. Methylmercuryoxidative degradation potentials in contaminated and pristine sediments of the Carson River, Nevada. Appl. Environ. Microbiol. 61, 2745–2753 (1995).
Barkay, T. & Wagner‐Döbler, I. Microbial transformations of mercury: potentials, challenges, and achievements in controlling mercury toxicity in the environment. Adv. Appl. Microbiol. 57, 1–52 (2005).
Liu, J. et al. Understanding of mercury and methylmercury transformation in sludge composting by metagenomic analysis. Water Res. 226, 119204 (2022).
Zheng, J. et al. Diverse methylmercury (MeHg) producers and degraders inhabit acid mine drainage sediments, but few taxa correlate with MeHg accumulation. MSystems 8, e0073622 (2023).
Oremland, R. S., Culbertson, C. W. & Winfrey, M. R. Methylmercury decomposition in sediments and bacterial cultures: Involvement of methanogens and sulfate reducers in oxidative demethylation. Appl. Environ. Microbiol. 57, 130–137 (1991).
Baldi, F., Pepi, M. & Filippelli, M. Methylmercury resistance in desulfovibrio desulfuricans strains in relation to methylmercury degradation. Appl. Environ. Microbiol. 59, 2479–2485 (1993).
Wu, Q. et al. Methanogenesis is an important process in controlling MeHg concentration in rice paddy soils affected by mining activities. Environ. Sci. Technol. 54, 13517–13526 (2020).
Gionfriddo, C. M. et al. Genome-resolved metagenomics and detailed geochemical speciation analyses yield new insights into microbial mercury cycling in geothermal springs. Appl. Environ. Microbiol. 86, e00176–00120 (2020).
Marvin-DiPasquale, M. C. & Oremland, R. S. Bacterial methylmercury degradation in Florida Everglades peat sediment. Environ. Sci. Technol. 32, 2556–2563 (1998).
Marvin-DiPasquale, M. et al. Methyl-mercury degradation pathways: a comparison among three mercury-impacted ecosystems. Environ. Sci. Technol. 34, 4908–4916 (2000).
Munson, K. M., Lamborg, C. H., Boiteau, R. M. & Saito, M. A. Dynamic mercury methylation and demethylation in oligotrophic marine water. Biogeosciences 15, 6451–6460 (2018).
Petrus, A. K., Rutner, C., Liu, S., Wang, Y. & Wiatrowski, H. A. Mercury reduction and methyl mercury degradation by the soil bacterium Xanthobacter autotrophicus Py2. Appl. Environ. Microbiol. 81, 7833–7838 (2015).
Kang-Yun, C. S. et al. Evidence for methanobactin “theft” and novel chalkophore production in methanotrophs: impact on methanotrophic-mediated methylmercury degradation. ISME. J. 16, 211–220 (2022).
Liu, Y.-R. et al. Overlooked role of putative non-hg methylators in predicting methylmercury production in paddy soils. Environ. Sci. Technol. 53, 12330–12338 (2019).
Mechirackal Balan, B., Shini, S., Krishnan, K.P. & Mohan, M. Mercurytolerance and biosorption in bacteria isolated from Ny-Ålesund, Svalbard, Arctic.J. Basic Microbiol. 58, 286–295 (2018).
Coates, J. D. et al. Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 411, 1039–1043 (2001).
Yan, Z. S. et al. Interconnection of key microbial functional genes for enhanced benzo[a]pyrene biodegradation in sediments by microbial electrochemistry. Environ. Sci. Technol. 51, 8519–8529 (2017).
Li, J. et al. Identification of an efficient phenanthrene-degrading pseudarthrobacter sp. L1sw and characterization of its metabolites and catabolic pathway. J. Hazard. Mater. 465, 133138 (2024).
Zhong, H. et al. Soil geobacteraceae are the key predictors of neurotoxic methylmercury bioaccumulation in rice. Nat. Food. 5, 301–311 (2024).
Jansson, J. K., McClure, R. & Egbert, R. G. Soil microbiome engineering for sustainability in a changing environment. Nat. Biotechnol. 41, 1716–1728 (2023).
Avila‐Nuñez, G., Saldivar, A., Ruiz‐Ruiz, P. & Revah, S. Methanol excretion by methylomonas methanica is induced by the supernatant of a methanotrophic consortium. J. Chem. Technol. Biotech. 99, 1356–1363 (2024).
Ran, X. The synthetic condition of methylmercuric chloride. China. Environ. Sci. 18, 524–526 (1998).
Tang, W. L. et al. Understanding mercury methylation in the changing environment: recent advances in assessing microbial methylators and mercury bioavailability. Sci. Total. Environ. 714, 136827 (2020).
Keighley, N., Ramwell, C., Sinclair, C. & Werner, D. Highly variable soil dissipation of metaldehyde can explain its environmental persistence and mobility. Chemosphere 283, 131165 (2021).
Jensen, S. & Jernelöv, A. Biological methylation of mercury in aquatic organisms. Nature 223, 753–754 (1969).
Rieder, S. R. & Frey, B. Methyl-mercury affects microbial activity and biomass, bacterial community structure but rarely the fungal community structure. Soil. Biol. Biochem. 64, 164–173 (2013).
Liu, J. et al. Mercury and sulfur redox cycling affect methylmercury levels in rice paddy soils across a contamination gradient. Environ. Sci. Technol. 57, 8149–8160 (2023).
Jones, D. & Willett, V. Experimental evaluation of methods to quantify dissolved organic nitrogen (DON) and dissolved organic carbon (DOC) in soil. Soil. Biol. Biochem. 38, 991–999 (2006).
Neufeld, J. D. et al. DNA stable-isotope probing. Nat. Protoc. 2, 860–866 (2007).
Sun, X. et al. Desulfurivibrio spp. Mediate sulfur-oxidation coupled to sb(v) reduction, a novel biogeochemical process. ISME. J. 16, 1547–1556 (2022).
Jiang, L., Song, M., Luo, C., Zhang, D. & Zhang, G. Novel phenanthrene-degrading bacteria identified by DNA-stable isotope probing. PloS. One. 10, e0130846 (2015).
Song, M. et al. Identification of benzo [a] pyrene-metabolizing bacteria in forest soils by using DNA-based stable-isotope probing. Appl. Environ. Microbiol. 81, 7368–7376 (2015).
Herlemann, D. P. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME. J. 5, 1571–1579 (2011).
Weisburg, W. G., Barns, S. M., Pelletier, D. A. & Lane, D. J. 16s ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703 (1991).
Sultana, N. et al. Stable isotope probing of active methane oxidizers in rice field soils from cold regions. Biol. Fert. Soils. 55, 243–250 (2019).
Liang, H. et al. Metagenomics analysis revealing the occurrence of antibiotic resistome in salt lakes. Sci. Total. Environ. 790, 148262 (2021).
Zhang, D. et al. Ecological interactions and the underlying mechanism of anammox and denitrification across the anammox enrichment with eutrophic lake sediments. Microbiome 11, 82 (2023).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015).
Sun, X. et al. Chemolithoautotropic diazotrophy dominates the nitrogen fixation process in mine tailings. Environ. Sci. Technol. 54, 6082–6093 (2020).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Parks, D. H. et al. A complete domain-to-species taxonomy for bacteria and archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., Parks, D. H. & Hancock, J. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927 (2020).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Li, Y. et al. Serratia spp. are responsible for nitrogen fixation fueled by As(III) oxidation, a novel biogeochemical process identified in mine tailings. Environ. Sci. Technol. 56, 2033–2043 (2022).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Gong, Y. et al. Bioaccessibility-corrected risk assessment of urban dietary methylmercury exposure via fish and rice consumption in China. Sci. Total. Environ. 630, 222–230 (2018).
Chen, L. et al. Trans-provincial health impacts of atmospheric mercury emissions in China. Nat. Commun. 10, 1484 (2019).
Giang, A. & Selin, N. E. Benefits of mercury controls for the United States. Proc. Natl. Acad. Sci. USA 113, 286–291 (2016).
Bastien-Olvera, B. A. & Moore, F. C. Use and non-use value of nature and the social cost of carbon. Nat. Sustain. 4, 101–108 (2020).
Tang, J., Cao, L., Mi, C., Fu, C. & Liu, Q. Interval assessments of identified parameters for uncertain structures. Eng. Comput. 38, 2905–2917 (2022).
Zuo, G. Cvtree: a parallel alignment-free phylogeny and taxonomy tool based on composition vectors of genomes. Genom. Proteom. Bioinform. 19, 662–667 (2021).
Subramanian, B., Gao, S., Lercher, M. J., Hu, S. & Chen, W. H. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 47, W270–W275 (2019).
Acknowledgements
We are grateful to Z.-C.H. for his assistance in DNA-SIP analyses. Y.-R.L. was supported by the National Natural Science Foundation of China (42425701 and 42177022) and the Fundamental Research Funds for the Central Universities (2662025PY010).
Author information
Authors and Affiliations
Contributions
Y.-R.L. developed the original idea of the analyses presented in the manuscript. Lab analyses were done by X.-Q.Z., K.-H.C., M.Y., Q.L., Y.-Y.H., H.-W.L. and Y.-R.L. Bioinformatic analyses were done by X.-Q.Z. and J.F. Statistical analyses were done by X.-Q.Z., K.-H.C. and Y.-R.L. The manuscript was written by X.-Q.Z. and K.-H.C. and edited by Y.-R.L., R.-Q.Y., J.L., W.T., Q.H. and B.G. with contributions from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Y.M., S.Z., and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, XQ., Chen, KH., Yu, RQ. et al. Microbial potential to mitigate neurotoxic methylmercury accumulation in farmlands and rice. Nat Commun 16, 5102 (2025). https://doi.org/10.1038/s41467-025-60458-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60458-1
