
Abstract
Nickel photocatalysis has recently become vital to organic synthesis, but how the Ni(II)X2L pre-catalyst (X = Cl, Br; L = bidentate ligand) becomes activated to Ni(I)XL has remained puzzling and is typically addressed on a case-by-case basis. Here, we reveal a general mechanism where light induces photolysis of the Ni(II)-X bond, either via direct excitation or triplet energy transfer. Photolysis produces Ni(I)XL and a halogen radical, X•. Subsequent hydrogen atom abstraction, often from the solvent, produces a C(sp3) radical, R•, that recombines with Ni(I) to form organonickel(II) complexes, Ni(II)XRL. Rather than acting as a loss pathway, Ni(II)XRL behaves as a light-activated reservoir of Ni(I) via photolysis of the Ni(II)-C bond. These results explain the role of the solvent in protecting the catalyst from off-cycle dimerization, demonstrate that two photons are often required to drive the reaction, and show how tuning the ligand can control the concentration of active Ni(I) species.
Similar content being viewed by others

Introduction
NiX2L is widely used as a pre-catalyst (commonly, L = dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, X = Cl, Br) in both singly Ni-catalyzed1 and dual Ni/photosensitizer (PS) catalyzed2,3,4,5,6,7 synthetic methodologies that have seen extremely rapid growth over the last decade. Its advantages over traditional Pd-catalyzed methods have led to adoption at scale in the pharmaceutical industry8,9,10,11, and include: mild conditions (e.g., weak bases, room temperature operation), different scope due to the increased propensity of Ni towards radical reactivity, and the abundance and low cost of Ni sources. Until now, a central mystery has been how Ni(II) pre-catalysts such as NiX2L are activated to form the Ni(I) or Ni(0) species generally proposed as the active state of the catalyst. Past experiments have supported varying electron12,13,14,15 and energy transfer (EnT) type1,16,17,18,19 initiation mechanisms in specific methodologies, giving rise to a broad consensus that the mechanism is unique to each set of reaction conditions.
However, we show herein that a general mechanism operates in the case of many methodologies where pre-catalyst activation occurs through energy transfer from a photosensitizer (PS) or via direct photoexcitation. Indeed, the same initiation step of Ni(II)-X bond photolysis (Fig. 1A) has recently been proposed across diverse methodologies (e.g., C-H activations, cross electrophile couplings, and C-heteroatom couplings)20,21,22,23,24,25,26,27, yet no study has elucidated the full photolysis mechanism through both rigorous detection of the photo-products, Ni(I)XL and the halogen radical (X•), and determination of their downstream chemistry (especially the fate of X•).

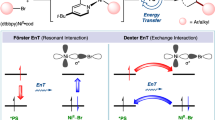
A Hypothesis 1: energy transfer quenching (left) or direct excitation (right) of Ni(II) results in Ni-X photolysis to produce Ni(I) and a halogen (X = Cl, Br) radical. The 8 refs. 20,21,22,23,24,25,26,27. B Hypothesis 2: halogen radical engages in hydrogen atom abstraction with an organic solvent or reagent, R-H. The resulting carbon-centered radical reacts with Ni(I) to produce NiXRL, shown for the specific case of L = dtbbpy. C Reaction where hypothesized steps in (A) and (B) are directly relevant and product forming. For example, see refs. 20,48,50. D Reaction where above steps are off-cycle, leading to a solvent-coupled side product. For example, see refs. 21,28,35.
Herein, we discover that X• engages in hydrogen atom transfer with any suitable partner (often the solvent, e.g., ethereal or benzylic compounds) to form a carbon-centered radical, R•, that adds to Ni(I) to form organometallic Ni(II)XRL intermediates (See Fig. 1B), where X = Br, Cl and L is a bidentate N,N′ type ligand, commonly dtbbpy. Despite significant recent mechanistic advances20 and direct incorporation of the R group into the product in C-H activations (Fig. 1C), a Ni(II)XRL intermediate has never been proposed in this reaction class. In cross-electrophile coupling reactions, solvent-derived byproducts have been detected (Fig. 1D)28, yet the mechanism of their formation has remained elusive until now. Herein, we also find that Ni(II)XRL can be further photolyzed (Fig. 1B), serving as a visible-light absorbing reservoir state which releases Ni(I)XL prior to oxidative addition of aryl halide substrates.
We substantiate the full photolysis initiation mechanism using a comprehensive suite of techniques, including X-ray and optical transient absorption spectroscopies (XTA, OTA) and electrochemical methods. Further, we follow the photolysis of NiX2L resulting from energy transfer or direct excitation of NiX2L in the absence of a PS, finding a wavelength dependence where UV (360 nm) excitation results in formation of a greater proportion of photo-products than violet (400 nm) light for the model case of L = dtbbpy, X = Cl. Two photo-products are detected, a major one (λmax = 500 nm) and a minor one (λmax = 420, 660 nm). Ni(I)Cl(dtbbpy) is assigned as the minor photo-product through a combination of techniques, including steady state ultraviolet-visible spectroscopy (UV-vis), electron paramagnetic resonance spectroscopy (EPR), and pulse radiolysis (PR).
The major photo-product (λmax = 500 nm) is assigned as Ni(II)XRL, formed via downstream dark reactivity involving the Ni(I) and X radical photo-products, as in Fig. 1B. A series of highly water sensitive, orange to red to pink colored Ni(II)XRL complexes are detected with UV-vis, where L = dtbbpy, 4,4′-ditrifluoromethyl-2,2′-bipyridine (CF3bpy), 6,6′-dimethyl-2,2′-bipyridine (66Mebpy), or 2,9-Dimethyl-1,10-phenanthroline (29Mephen), X = Cl or Br, and R = an organometallic ligand derived from 1,2-dimethoxyethane (DME), tetrahydrofuran (THF), 1,4-dioxane (Diox), toluene (Tol), or 4-fluorotoluene (4FTol). For NiCl(CDME)(dtbbpy), further characterization by nuclear magnetic resonance (NMR), X-ray absorption spectroscopy (XAS), PR, EPR, and density functional theory calculations (DFT) is consistent with its assigned square planar structure and singlet ground state. Importantly, NiCl(C4FTol)(dtbbpy) is independently synthesized, fully characterized (including by Single Crystal X-ray Diffraction, SCXRD), and found to spectroscopically match in situ experiments producing it photochemically from 4FTol. Further, NiCl(CDME)(dtbbpy) is found to convert to \({{\mbox{NiBr}} }({{\rm{C}}}_{{\mbox{BzCF}}_{3}})({{\mbox{dtbbpy}}})\) in the presence of aryl halide and light, showing that Ni(II)XRL also serves as a reservoir for catalytically active Ni(I) species.
Results
Defining main hypotheses and a model system
In the following sections, we examine two main mechanistic hypotheses outlined in Fig. 1A, B. First, the hypothesis that both energy transfer from a PS and direct excitation of NiX2L result in the formation of Ni(I)XL and X•. Second, that a carbon-centered radical, R•, formed via X• engaging in hydrogen atom abstraction with R-H, undergoes radical addition to Ni(I)XL to form a Ni(II)XRL reservoir state. We employ NiCl2(dtbbpy) (X = Cl, L = dtbbpy) dissolved in DME (R-H = DME) as a model system, with the optional photosensitizer [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, referred to from here on as “Ir PS”, where dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine. In addition, we optionally include aryl bromide 4-bromobenzotrifluoride (ArBr) and the base 2,6-lutidine (lut). These Ni source, PS, base, and substrate were selected as they are utilized in pivotal methodologies and mechanistic studies20,23,28.
Importantly, this choice of model system also results in a homogeneous solution with comparatively simple Ni speciation. Many other solvents commonly used in Ni catalysis (e.g., N,N-dimethylacetamide [DMAc], N,N-dimethylformamide [DMF], acetonitrile [MeCN]) bind to Ni, creating a complex mixture of species15,16,29. Here, the speciation of commercially purchased, isolated NiCl2(dtbbpy) upon dissolution in DME matches that of the 1:1 molar ratio mixture of NiCl2(DME) and dtbbpy (See SI, Fig. S6). As such, a ligand exchange equilibrium must exist where dtbbpy displaces DME. From UV-vis titration experiments where the dtbbpy concentration is increased while the total [Ni] is held constant, the 1:1 mixture is found to contain both NiCl2(DME) and NiCl2(dtbbpy), with the latter as the dominant species (See SI, Figs. S2–3). For the following experiments, either a 1:1 ratio solution or a 1:3 ratio solution will be employed, as the former is most commonly used in catalysis and the latter predominantly contains NiCl2(dtbbpy). The common solutions employed throughout are defined here:
Steady-state evidence for photolysis of Ni(II)Cl2(dtbbpy) to Ni(I)Cl(dtbbpy)
As one of the immediate photo-products of Ni(II)-Cl photolysis, detecting Ni(I)Cl(dtbbpy) is vital to testing the first main hypothesis of this work. Since Ni(I)Cl(dtbbpy) has never been isolated, we produced it in situ via two distinct methods, Ni(0/II) comproportionation and Ni(II/I) reduction by pulse radiolysis, both of which are consistent with its reported EPR and UV-vis (i.e., λ = 420, 660 nm maxima) spectroscopic handles (For a detailed discussion, See SI, Section 17). The Ni/Ir mixture in DME was used to test the hypothesis that energy transfer to Ni(II)Cl2(dtbbpy) from the Ir PS results in Ni(I) formation (See Table 1 for component details). Support for the assignment of the quenching mechanism as EnT will be detailed in the next section below. Upon steady state irradiation of this mixture with a 405 nm LED (216 mW cm−2), EnT results in appearance of a new absorption difference feature centered at 500 nm (See SI, Figs. S13–14). Introducing 2,6-lutidine23,28 to the reaction mixture results in a 6-fold increase in the steady state concentration of the 500 nm photo-product. The feature, due to the major photo-product absorbing at 500 nm, does not match the UV-vis spectrum for Ni(I)Cl(dtbbpy), but its enhancement by addition of a base is consistent with the mechanism in Fig. 1C, where HX is a reaction product. A shift in lutidine 1H NMR resonances (See SI, Fig. S71) upon irradiation is observed, consistent with formation of the lutadinium-Cl salt. In support of this explanation, addition of HCl to lutidine in the absence of light, Ni, or Ir results in chemical shift changes of the same magnitude and direction (See SI, Fig. S72). We thus infer that the 500 nm absorption peak can be assigned to Ni(II)Cl(CDME)(dtbbpy), which we substantiate extensively in the following sections.
In addition to enhancement of the major photo-product buildup, an additional minor photo-product appears as a growth of additional spectral features at λ = 420, 660 nm at later irradiation times (See SI, Figs. S50–51). These can be most readily deconvoluted through spectral subtraction. The double difference spectrum (Fig. 2B) calculated via subtraction of the final two time points reveals the diagnostic features of Ni(I)Cl(dtbbpy) which match those produced independently via comproportionation or pulse radiolysis (See SI, Figs. S8, S100–105). Furthermore, if the same mixture is prepared in an air-tight EPR tube and irradiated at the same LED power, EPR analysis reveals diagnostic features of Ni(I)Cl(dtbbpy) (Fig. 2C). Fitting the irradiated difference EPR spectrum reveals g-values of gx = 2.056, gy = 2.086, and gz = 2.278, which compare well with the values gx = 2.050, gy = 2.070, and gz = 2.248 reported by Hadt and colleagues30. Together, these results strongly support the formation of Ni(I)Cl(dtbbpy) through photolysis of the Ni(II)-Cl bond. While the photolysis of Ni(III)-X bonds is well studied31,32, these findings represent direct evidence of Ni(II)-X photolysis via energy transfer from a PS. In the next section, we present evidence to support assignment of the role of the Ir PS as an energy transfer donor.

A Scheme describing Ni(II)-Cl bond photolysis resulting from energy transfer from the Ir PS excited state. Downstream reactivity of the Ni(I) and Cl• photo-products will be elucidated in depth in a later section of this work. B Double difference UV-vis spectrum of the Ni/Ir/lut mixture after illumination with a 405 nm LED (216 mW cm−2) (left axis). Ni(I)Cl(dtbbpy) as produced by comproportionation (right axis, see SI, Fig. S8, for further details). C Continuous wave EPR difference spectrum (t = 0 subtracted) for the Ni/Ir/lut mixture at t = 30 min irradiation with a 405 nm LED (128 mW cm−2). EPR conditions: temperature T = 5 K, microwave power P = 0.1 mW, microwave frequency F = 9.372 GHz. For EPR experimental details and additional time points, See SI, Figs. S92–93.
Time-resolved evidence for photolysis of Ni(II)Cl2(dtbbpy) to Ni(I)Cl(dtbbpy)
In this section, we continue probing the hypothesis that energy transfer to NiCl2(dtbbpy) results in Ni(I) formation through a combination of transient optical and X-ray absorption techniques (OTA and XTA). Time-resolved methods, in conjunction with techniques to aid in spectral assignment, enable the unambiguous assignment of photochemical reaction mechanisms through the detection (or lack thereof) of specific photo-products33. Figure 3A shows a ns-μs OTA experiment we use to assign the nature of the photoreaction between the Ir PS and Ni, while Fig. 3B, C show a photolysis scheme and kinetics for fs-ns timescale OTA experiments conducted in flow, where we directly excite Ni(II)Cl2(dtbbpy) at both UV (360 nm) and visible (400 nm) pump wavelengths in the absence of the Ir PS.

A Transient absorption spectral traces at the indicated time delays of a mixture of the Ir PS (134 μM) and NiCl2(dtbbpy), saturated, filtered and diluted (0.9:1) in DME. Dotted lines indicate selected probe wavelengths for which kinetic traces are shown (inset) along with a global fit (black lines). B Scheme explaining rationale for the observed wavelength dependence. A higher lying excited state with increased quantum yield (ϕ) for photolysis can be accessed with a 360 nm photon but not with 400 nm. C Fluence matched kinetic slices at λprobe = 660 nm of the NiCl2(DME): dtbbpy 1:3 mixture excited at λpump = 360 or 400 nm without a PS. A persistent signal decays to the baseline with a 338 ps lifetime. For additional details, See SI, Optical Transient Absorption section.
OTA on the ns-μs timescale of the Ir/Ni mixture in Fig. 3A reveals a significantly shortened decay compared to Ir alone (1.78 μs to 0.44 μs from global analysis fitting, See SI, Figs. S113–118), indicating quenching of the excited Ir PS. In Fig. 3A: inset, the kinetics and global fit clearly show persistent features that survive beyond the experimental window (45 μs). The datasets with/without Ni are both globally fit with models involving monoexponential decay of the Ir PS triplet metal to ligand charge transfer (3MLCT) state; however, an additional infinite component was added to the Ir/Ni model to account for the persistent features. The species associated spectrum (SAS) obtained from the global fit for the infinite component (See SI, Figs. S119–120) is assigned as a mixture of the Ni(I)Cl(dtbbpy) and Ni(II)Cl(CDME)(dtbbpy) photo-products.
The SAS of the persistent component aids in assignment of the quenching mechanism as energy transfer, as it does not match the spectrum of either the electrochemically oxidized or reduced Ir PS (See SI, Fig. S63) that would be the product of photoinduced electron transfer quenching. Specifically, we note that reductive quenching would result in formation of the singly reduced Ir PS, and its diagnostic sharp features (λ = 480–530 nm, See SI, Fig. S63) are not present in either the spectral evolution shown in in Fig. 3A or the SAS (Fig. S120). In addition, cyclic voltammetry data in DME (See SI, Fig. S62) does not show an oxidation wave for NiCl2(dtbbpy) accessible with the known excited state oxidation potential of the Ir PS (E*ox = 1.21 V vs. SCE)34, suggesting that the reductive quenching pathway is unlikely on thermodynamic grounds. Importantly, XTA experiments at the Ir L3 edge do not show a persistent component, as is consistent with that component arising from Ni-based photo-products in the OTA experiment, instead exhibiting complete recovery of the ground state of the Ir PS along with a quenched 3MLCT lifetime (See SI, Fig. S149). This complete recovery is inconsistent with the presence of Ir(IV), which would be the product of oxidative quenching. Stern-Volmer quenching studies (See SI, Fig. S58) with increasing [Ni] concentration allow extraction of a rate constant for this process of kq = (3.41 ± 0.31) * 109 M−1 s−1, while quenching experiments including reaction components used in one relevant methodology, cross electrophile coupling of aryl and alkyl halides35, reveal the Ni catalyst as the dominant quencher in that system (See SI, Fig. S57).
These observations together strongly support a catalytically relevant energy transfer quenching mechanism, in agreement with our recent work in a closely related acetonitrile-based system21. Notably, reported OTA data in that work employs the same Ni/Ir mixture in acetonitrile where a persistent signal was not observed, consistent with our DME solvent-dependent hypothesis outlined in Fig. 1C. Although excitation of the Ir PS in the Ir/Ni mixture produces long-lived photo-products (Fig. 3A), it is not a suitable experiment for resolving the photolysis dynamics. EnT does not proceed instantaneously, which limits the maximum available concentration of the excited Ni products, as they are spread out in time. Considering that the longest-lived excited-state lifetime of Ni(II)Cl2(dtbbpy) has been reported to be less than 60 ps36, it cannot be expected to appear at any significant concentration during the ns timescale EnT. Therefore, we investigated the photolysis step with femtosecond OTA.
Figure 3C shows kinetic traces from OTA experiments conducted in flow at 2 pump wavelengths to test the hypothesis of ultrafast Ni(II)-Cl bond photolysis. We find that photolysis of Ni(II)Cl2(dtbbpy) occurs if λpump = 360 nm (estimated ϕphotolysis = 0.13) as indicated by a persistent signal at λprobe = 660 nm (i.e., the λmax of Ni(I)Cl(dtbbpy)), while employing λpump = 400 nm with matched fluence results in minimal photolysis (estimated ϕphotolysis = 0.01; See SI, Optical Transient Absorption section for details). We emphasize that this estimation describes the quantum yield for the photolysis step without influence from downstream steps such as recombination or cage escape that typically contribute to quantum yields measured by steady-state methods.
The spectral shape and evolution differ between the two datasets even at sub-picosecond times (For spectral traces, 2D maps, and global fitting see SI, Figs. S127–137), with the λpump = 360 nm dataset showing greater absorption at wavelengths longer than 510 nm. There is complete baseline recovery within 25 ps (Fig. 3C) for 400 nm excitation, while for 360 nm excitation the signal persists until 800 ps at 660 nm as shown by the 660 nm dynamics in Fig. 3C. The λpump = 400 nm data is globally fit with a model that includes a 2 component decay to the ground state with τ0 = 0.25 ps and τ1 = 1.88 ps, which we assign as nonradiative decay processes occurring after photoexcitation into the lower energy absorption feature (λshoulder = 380 nm, ϵ = 330.7 M−1 cm−1; See SI, Fig. S4). The λpump = 360 nm data, in contrast, involves photoexcitation into a higher energy feature (λshoulder = 330 nm, ϵ = 1118.1 M−1 cm−1; See SI, Fig. S4).
The λpump = 360 nm dataset requires the inclusion of an additional species component and kinetic parameter to capture the appearance and subsequent decay of the persistent features. The first two components decay with lifetimes τ0 = 0.11 ps and τ1 = 1.94 ps similar to the λpump = 400 nm case and consistent with the same nonradiative decay pathways being present. In a parallel pathway, photolysis from the higher lying excited state occurs within the instrument response (≃250 fs), producing photolysis products which recombine to recover the ground state with a lifetime of τ2 = 338 ps. We note that the photolysis products SAS does not match the spectrum of Ni(I)Cl(dtbbpy) (See SI, Figs. S136–137), though it is consistent with Ni(I)Cl(dtbbpy) being present as a major component.
This stark wavelength dependence in the quantum yield for photolysis (i.e., ϕ360 = 0.13 and ϕ400 = 0.01) constitutes an “anti-Kasha”37 photochemical event in that it can be explained through accessing a higher lying electronic transition with 360 nm light that is inaccessible with 400 nm light (Fig. 3B). This higher state could have greater activity for photolysis if it involves ligand-to-metal charge transfer (i.e., chloride to Ni) character, as suggested by reported time-dependent density functional theory calculations36. In a related recent study, the rate of Ni(II)-C photolysis in NiCl(dtbbpy)(o-tolyl) was found to exhibit a similar wavelength dependence wherein increased reactivity was observed approaching shorter wavelengths from 450–350 nm38. Importantly, these data support that direct photoexcitation of Ni(II)Cl2(dtbbpy) results in ultrafast photolysis of the Ni-Cl bond. In another study and consistent with this observation, photolysis of Cu(II)-Cl bonds has been observed to occur on an ultrafast timescale39.
Identifying NiCl(dtbbpy)(CDME) as the major photolysis product
The second main hypothesis of this work involves the fate of initially produced Ni(I)XL. The proposal put forward in Fig. 1C is that radical addition of R• to Ni(I)XL forms Ni(II)XRL, also shown in Fig. 4A for the specific case of Ni(I)Cl(dtbbpy).

A Scheme describing R• addition to NiCl(dtbbpy) to form NiClR(dtbbpy). B Pulse radiolysis spectral slices at the indicated time delays of the comproportionation mixture, 2.5 mM total [Ni] in DME (left axis). Overlay (right axis): UV-vis difference spectrum of the Ni/Ir/lut mixture after 405 nm LED irradiation for t = 3 min with the t = 0 spectrum subtracted. The 500 nm absorption is assigned as NiCl(CDME)(dtbbpy). C XANES spectrum of the Ni/Ir (air headspace) pre-irradiated mixture zoomed in on the pre-edge region. NiCl(o-tolyl)(TMEDA) XANES is overlaid as a square planar reference compound. TMEDA = N,N,\({{{{\rm{N}}}}}^{{\prime} },{{{{\rm{N}}}}}^{{\prime} }\)-Tetramethylethane-1,2-diamine. D 1H NMR experiments with varying irradiation time of the Ni/Ir/lut mixture in benzene-d6 containing DME or 4-fluorotoluene (4FTol) in a 10:1 ratio. Photoinduced signals are assigned to the tert-butyl protons of dtbbpy in each complex, where observation of 2 peaks corresponds to the 2 regioisomers of NiCl(CDME)(dtbbpy) and 1 peak to NiCl(C4FTol)(dtbbpy). E UV-vis of independently synthesized NiCl(C4FTol)(dtbbpy) overlaid with the UV-vis difference spectrum of the Ni/Ir mixture in 4FTol after 405 nm LED irradiation for t = 5 min with the t = 0 spectrum subtracted. The crystal structure (50% thermal ellipsoids) of isolated NiCl(C4FTol)(dtbbpy) is overlaid. See SI, NMR, PR, XAS, SCXRD, and Synthesis Sections for further experimental details.
Upon irradiation of the Ni/Ir/lut mixture, it was noted above that a species with visible λmax = 500 nm forms as the dominant photo-product after steady state illumination with 405 nm light, associated with a color change in the mixture from yellow to orange-red (See SI, Fig. S51). The same feature was also detected under UV LED irradiation (365 nm, 340 nm) without the Ir PS, consistent with the proposed EnT pathway (See SI, Figs. S9–10) and the evidence of ultrafast photolysis of Ni(II)Cl2(dtbbpy) with a 360 nm pump. Consistent with the wavelength dependence found in the previous section, repeating the same experiment with 405 nm light does not produce any significant photo-products (See SI, Figs. S11–12).
Here, we substantiate our spectral assignment of the 500 nm absorption peak to Ni(II)Cl(CDME)(dtbbpy). We employ pulse radiolysis (PR) experiments to make Ni(II)Cl(CDME)(dtbbpy) via an entirely different route, conduct Ni K-edge XAS experiments on concentrated solutions of the photo-product to understand its coordination environment, and use proton NMR to follow its production. Finally, we repeat the UV-vis and NMR experiments with 4-fluorotoluene (4FTol) instead of DME to produce a related organonickel complex, Ni(II)Cl(C4FTol)(dtbbpy), which we also synthesized independently, isolated, and crystallographically characterized. Additional circumstantial evidence for this assignment is that the λmax = 500 nm species is quickly decomposed upon exposure to water vapor in the air or in a wet nitrogen stream (See SI, Figs. S31–34). Unexpectedly, when the Ni/Ir mixture is prepared under air and sealed with a limited headspace, a greater amount of the 500 nm absorbing species is observed after an induction period (See SI, Figs. S35–37). We take advantage of this finding for XAS measurements below.
Pulse radiolysis is an exceptionally powerful tool for studying radical chemistry because it allows exogeneous production of radical intermediates, breaking reaction steps down into their elementary components. Here we use it to produce Ni(II)Cl(CDME)(dtbbpy) in situ without photolysis of Ni(II)Cl2(dtbbpy) (Fig. 4A, R = DME). Notably, PR has advantages over traditional methods of probing radical involvement, such as radical or spin trapping, in that no additional species (e.g., traps) need to be added that may interfere with the process under study. Indeed, spin trapping was unsuccessful here since introduction of the trap changed the quenching mechanism, resulting in detection of the reduced Ir PS by EPR (See SI, Fig. S97). PR in DME creates the DME-derived radical via electron ejection followed by fragmentation of the radical cation to a neutral radical and a solvated proton. In THF, this process is known to occur with a 500 fs time constant40, suggesting it is plausible for DME radicals to form in the PR experiment with sufficient concentration for addition to Ni(I)Cl(dtbbpy), forming NiCl(CDME)(dtbbpy) (See SI, Fig. S106 for a scheme with additional details). Ni(I)Cl(dtbbpy) is supplied in high concentration using the Ni(0/II) comproportionation mixture.
Figure 4B shows the pulse radiolysis spectrum of the comproportionation mixture as a function of delay time, compared with the difference spectrum obtained earlier by photolysis of Ni(II)Cl2(dtbbpy). Evidently, pulse radiolysis of this mixture results in growth of the same 500 nm absorption feature as in photolysis of Ni(II)Cl2(dtbbpy) simultaneous with loss of the 660 and 420 nm peaks associated with Ni(I)Cl(dtbbpy) in Fig. 2A (See SI, Fig. S111 for an overlaid plot). This data strongly supports production of Ni(II)Cl(CDME)(dtbbpy) on the 100s of ns to μs timescale (Fig. 4B; for additional time points, see SI, Fig. S109). Formation of Ni(II)Cl(CDME)(dtbbpy) occurs to a lesser extent at lower concentrations of total [Ni], becoming the dominant process at 2.5 mM. At 5.0 mM [Ni], Ni(II)Cl(CDME)(dtbbpy) forms with increased rate (See SI, Figs. S107–110). Finally, the absorption spectrum of Ni(II)Cl(CDME)(dtbbpy) persists in UV-vis after conclusion of the PR experiment, consistent with its long timescale stability (See SI, Fig. S112). This experiment allows observation of C(sp3) radical addition to Ni(I) in real time, an important step proposed across the field of Ni photocatalysis, for example, in C(sp3)-C(sp2) carboxylations or alkyne hydroalkylations41,42. Further investigation of this step utilizing this comproportionation platform will be explored in future work.
X-ray absorption (XAS) experiments can explicitly probe the coordination environment around metal atoms, and we use it here to test the hypothesis that radical addition of DME• to NiCl(dtbbpy) forms a square planar Ni(II)Cl(CDME)(dtbbpy) complex, as shown in Fig. 4A. Samples were prepared by irradiating both the Ni/Ir/lut (under N2) and Ni/Ir (under air) mixtures and subsequently concentrating them under vacuum on a Schlenk line (See SI, section 13 for further details). Ni K-edge X-ray absorption near edge structure (XANES) spectra of both mixtures revealed the growth of the pre-edge absorption at 8333.7 eV and the appearance of a new near-edge absorption feature at 8338 eV (See SI, Fig. 4C, Fig. S146). The presence and energies of these features are consistent with either a planar or square pyramidal geometry of Ni(II). Based on literature precedent, they are assigned to 1s → 3d (8333.7 eV) and 1s → 4pz (8338 eV) transitions43. The new absorption features are compared to those of reported four-coordinate Ni(II)(Caryl)Ln species44, as well as a commercially available square planar nickel reference compound, Ni(II)(TMEDA)(o-Tol)Cl (TMEDA = N,N,N′,N′-Tetramethylethane-1,2-diamine, o-tol = ortho-tolyl). The XAS spectrum of Ni(TMEDA)(o-Tol)Cl is plotted in 4C, highlighting the similarity with the species formed upon irradiation of the Ni/Ir (air) mixture. Compared to NiCl2(dtbbpy), both species demonstrate increased intensity and blue-shifted energy of the 1s → 3d peak, as well as the appearance of 1s → 4pz features of similar energies. Time-dependent DFT (TD-DFT) calculations carried out on Ni(II)Cl(CDME)(dtbbpy) using Ni(TMEDA)(o-Tol)Cl as an energy shift reference show good agreement between experimental and theoretical NiCl(CDME)(dtbbpy) X-ray absorption maxima energies (See SI, Fig. S165).
The low intensity of the 1s → 4pz peak of the irradiated Ni/Ir (air) sample is attributed to the presence of a mixture of species in solution. Because of this, quantitative peak area analysis that might be correlated to coordination number was not possible, and a five-coordinate square pyramidal geometry cannot be ruled out based on the XANES data alone. However, we favor four-coordinate singlet Ni(II)Cl(CDME)(dtbbpy) as the source of the observed pre-edge XAS features and the 500 nm optical absorption, as there are no known strongly coordinating L type ligands in the mixture that could occupy a fifth coordination site. Efforts to optimize singlet Ni(II)Cl(CDME)(dtbbpy) with either H2O or κ1-DME in the axial coordination site resulted in the exogenous ligand dissociating in silico. A trigonal planar Ni(I) species could also impart intensity to both sets of transitions, although a reference compound of this type shows that the 1s → 3d peak energy is red shifted by about 3 eV compared to that measured here45. Consideration of additional species that could contribute to the XANES spectra of the Ni/Ir (air) mixture can be found in the SI, Section 13. The XANES data, consistent with square planar geometry at Ni(II), in conjunction with DFT computations of alternative possible structures and configurations (See SI, Section 14), strongly supports assignment of the 500 nm absorbing species as NiCl(CDME)(dtbbpy) with a singlet ground state configuration.
The major photo-product, Ni(II)Cl(CDME)(dtbbpy), can exist as 2 isomers (Fig. 4D, depending on whether a DME radical is formed through methylene or methyl C-H activation. We used 1H NMR to probe the Ni/Ir/lut mixture in a 1:10 DME:benzene-d6 mixed solvent system, irradiated in a custom tube photoreactor setup (For setup details, See SI, Section 8). Upon irradiation, a pair of 1H NMR signals appear that vary in ratio with increasing irradiation time (Fig. 4D and SI, Figs. S65–70). We assign these peaks to the Ni-bound, dtbbpy tert-butyl groups (18H) of each regioisomer. If this assignment is correct, only a single photoinduced signal should be observed if DME is replaced with 4FTol, as 4FTol is only susceptible to H-atom abstraction in the benzylic position and thus cannot form organonickel regioisomers. Indeed, only 1 peak is observed at a similar chemical shift in Fig. 4D (i.e., DME: δ = 0.952/0.946; 4FTol: δ = 0.912)(See SI, Figs. S78–80). Further, these NMR assignments are supported by the independent synthesis and full characterization of Ni(II)Cl(C4FTol)(dtbbpy) (See SI, Section 3 for characterization and stability details; Section 15 for SCXRD).
Notably, the UV-vis spectrum of the isolated deep red solid dissolved in 4-fluorotoluene exactly matches that generated through energy transfer induced photolysis (See Fig. 4E). In crystalline form, Ni(II)Cl(C4FTol)(dtbbpy) exhibits distorted square planar geometry and a Ni-C bond distance of 1.958(4) Å consistent with the small subset of known organonickel(II) complexes featuring ligands derived from toluene (1.964(2) Å) or 2-methylnaphthalene (1.958(2) Å) (For SCXRD details, See SI, Section 15)42,46. Comparatively, Ni-Caryl complexes have significantly shorter Ni-C bond lengths, for example, 1.896(4) Å for Ni(II)Cl(Cp-Tol)(dtbbpy)47 and 1.873(5) Å for Ni(II)Cl(Co-Tol)(dtbbpy)36. Overall, these experiments provide strong support for the identities of the square planar organonickel intermediates, Ni(II)Cl(C4FTol)(dtbbpy) and Ni(II)Cl(CDME)(dtbbpy), and their formation via the steps detailed in Fig. 1A–B. Further, these observations offer insight into the regioselectivity of product formation where multiple C-H activation possibilities exist, for example, in catalytic reactions with DME28,48. The ratio of regioisomers observed in Ni(II)Cl(CDME)(dtbbpy) here agrees well with the reported product regiochemistry (i.e., 1.35:1) for the coupling of DME with 4-chloroacetophenone48.
In addition, while the formation and role of Ni(II)-Caryl intermediates in catalysis are well-studied49, comparatively little is known of Ni-C(sp3) species. In a recent advance, a Ni(II) complex possessing two organometallic ligands, a C(sp3) neosilyl ligand and a C(sp2) o-tolyl ligand, was reported20 and found to undergo light-driven reductive elimination of a C(sp2)-C(sp3) product. While interesting, this type of intermediate was not proposed to be on-cycle for C-H activation. Thus, further investigation of Ni(II)XRL, which lacks the organometallic aryl ligand, is crucial to achieve mechanistic understanding in these and other diverse methodologies where these intermediates form.
The roles of NiCl(CDME)(dtbbpy) in catalysis and mechanism generality
To this point, we have presented substantial evidence that the ultimate product of photolyzing Ni(II)Cl2(dtbbpy) in DME is Ni(II)Cl(CDME)(dtbbpy), whether through direct excitation or energy transfer from an iridium sensitizer. We have also suggested in Fig. 1C that Ni(II)Cl(CDME)(dtbbpy) can itself be photolyzed to re-form the catalytically active Ni(I)Cl(dtbbpy), making it a potentially productive reservoir species that protects the Ni(I)Cl(dtbbpy) from dimerization and deactivation. In support of this idea, the double difference spectrum in Fig. 2B also shows a negative feature at λ = 500 nm, indicating loss of some NiCl(CDME)(dtbbpy) concomitant with Ni(I)Cl(dtbbpy) formation. Here, we directly assess the participation of Ni(II)Cl(CDME)(dtbbpy) in catalysis.
The structure of Ni(II)Cl(CDME)(dtbbpy) suggests several hypotheses concerning its role in 2 prominent C(sp2)-C(sp3) cross-coupling reactions that have recently been studied in significant mechanistic detail (Fig. 1C, D)20,28. In both reactions, the Ni-Caryl complex \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\) has been identified as the organometallic Ni intermediate present in the highest concentration during the reaction. As such, we hypothesized that Ni(II)Cl(CDME)(dtbbpy) may convert to \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\) under photochemical conditions in the presence of an aryl bromide substrate (Fig. 5A).

A Scheme describing hypothesis for conversion of Ni(II)Cl(CDME)(dtbbpy) to \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\) via energy transfer induced photolysis. OA = oxidative addition; comp. = comproportionation. B Left axis: UV-vis difference spectra (t = 0 subtracted) of the Ni/Ir/ArBr mixture after increasing irradiation times with a 405 nm LED (216 mW cm−2). Right axis: the overlaid (dashed black trace) UV-vis spectrum of separately synthesized \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\). Arrows indicate an evolution from Ni(II)Cl(CDME)(dtbbpy) to \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\) (C) The scope of R-H. Normalized UV-vis difference spectra of Ni/Ir (N2) mixtures irradiated with a 405 nm LED (216 mW cm−2) in Diox, THF, DME, 4FTol, or Tol as solvent.
Photolysis of the Ni-CDME bond, analogous to the known Ni-Caryl photolysis pathway in Ni-Caryl complexes, would result in formation of Ni(I)Cl(dtbbpy), which is known to undergo oxidative addition of aryl halides30. Subsequent comproportionation of the resulting Ni(III) with Ni(I)Cl(dtbbpy) would form the \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\) intermediate and one equivalent of Ni(II)Cl2(dtbbpy) (Fig. 5A). To test this hypothesis, the Ni/Ir/ArBr mixture was prepared and irradiated with 405 nm light.
At early irradiation times, the 500 nm feature diagnostic of Ni(II)Cl(CDME)(dtbbpy) forms first (Fig. 5B), suggesting that the hydrogen atom abstraction step and radical addition to Ni(I) are faster than oxidative addition of the aryl bromide, quite consistent with the relative concentrations of DME (solvent) and aryl bromide (reagent). However, as the irradiation progresses, the Ni(II)Cl(CDME)(dtbbpy) is converted to \({{\mbox{NiBr}} }({{\rm{C}}}_{{{\mbox{BzCF}}}_{3}})({{\mbox{dtbbpy}}})\) as evidenced by the formation of a bimodal blue-shifted feature that matches that in the known absorption spectrum of \({{\mbox{NiBr}} }({{\rm{C}}}_{{\mbox{BzCF}}_{3}})({{\mbox{dtbbpy}}})\)28 in DME, which was also supported by independent synthesis (See SI, Fig. S53).
These observations suggest that NiCl(CDME)(dtbbpy) is catalytically relevant in two ways. First, it can be converted back to Ni(I) and undergo oxidative addition. Second, even with aryl halide present in ten-fold excess, oxidative addition is too slow to prevent formation of NiCl(CDME)(dtbbpy), suggesting it continues to form in low concentration throughout the reaction. It is likely that its relative concentration would increase as the rate of oxidative addition decreases, for example, if aryl chlorides were used instead of bromides. As such, it is plausible that in the general case Ni(II)XRL could serve as on-cycle intermediates in many reported C-H activation reactions23,24,48,50, where trapping of an aryl radical produced via photolysis of Ni(II)X(Caryl)L30,38,47 would form the desired product following reductive elimination from Ni(III). We note that NiCl(CDME)(dtbbpy) may additionally be susceptible to photo-oxidation by the Ir PS, a possibility that could be explored in future studies.
Importantly, the reactivity observed in DME to form NiCl(CDME)(dtbbpy) is not limited to that solvent, nor specifically to chloride. Rather, consistent with the scope of ethers and benzylic C-H containing compounds shown in C-H activation methods in the literature23,48, related but distinct UV-vis features form when the solvent in the Ni/Ir mixture is replaced with toluene, 4-fluorotoluene, 1,4-dioxane, or tetrahydrofuran (Fig. 5C). Interestingly, in support of the broad applicability of these findings across Ni photocatalysis, the toluene-derived complex, NiCl(CTol)(dtbbpy), has been isolated and was proposed in recent work as an on-cycle intermediate for the Markovnikov-type alkylation of alkynes42. Similarly, if Ni(II)Br2(dtbbpy) is used in place of Ni(II)Cl2(dtbbpy), a similar spectral evolution is observed (See SI, Figs. S15–16). These results show that the observed reactivity applies generally to other Ni(II)-halide salts and to other solvents with C-H bonds susceptible to abstraction (Fig. 5C). Benzene proved significantly less reactive, consistent with the comparatively higher bond dissociation energy of the aryl C(sp2)-H bond (See SI, Fig. S32)51. If a mixed solvent system contains two species, for example DME and toluene, Ni complexes derived from both solvents are observed, with a greater contribution from the most reactive species (See SI, Figs. S30–31).
Further, the same general mechanism remains at play if dtbbpy is replaced with CF3bpy, resulting in the formation of both Ni(I)Cl(CF3bpy) and NiCl(CDME)(CF3bpy) photo-products at red-shifted wavelengths, although the Ni(I) is observed in a greater ratio relative to Ni(II) (See SI, Figs. S40–42). We hypothesized that the prevalence of the Ni(I) photo-product could be further increased if the bpy ligand were modified through methylation and/or changing the core to phenanthroline. Indeed, Ni(I) becomes the dominant photo-product if dtbbpy is replaced with 66Mebpy (See SI, Figs. S34–35) or 29Mephen (See SI, Figs. S37–38). In both cases, the NiCl(CDME)L photo-product is detected initially, after which Ni(I) becomes dominant with continued irradiation (See SI, Figs. S36, S39). Several effects may be involved to explain these observations, including a less negative Ni(II)/(I) reduction potential upon methylation ortho to the N-atoms of the ligand52, an increase in Ni(II)-C photolysis efficiency, or enhanced stability of the Ni(I) towards dimerization due to increased steric bulk around the Ni center. Overall, these observations can enable rational ligand selection to control the concentration of Ni(I) during catalysis.
A general initiation mechanism
The above experiments provide substantial evidence for a general initiation mechanism defined as follows (Fig. 6).

The simplest system consists of NiX2(dtbbpy) dissolved in R-H with no additional reagents. Grayed out segments become active if additional reagents (i.e., PS, base, or substrates) are present in the mixture. CEC = cross-electrophile coupling. ArBr = aryl bromide.
A Ni(II)X2(dtbbpy), where X = Cl or Br, excited state forms through either photoexcitation or energy transfer and undergoes photolysis of the Ni-X bond. The resulting X• abstracts a H-atom from R-H to form R• and HX. The HX can be intercepted by a base (if present) to form the base-halide salt, which serves to suppress the protonation that limits Ni(II)XR(dtbbpy) buildup. R• recombines with Ni(I)Cl(dtbbpy) to form Ni(II)XR(dtbbpy), which is also susceptible to photolysis induced by photoexcitation or energy transfer. As such, Ni(II)XR(dtbbpy) serves as a reservoir of active Ni(I) species that protects the Ni(I), limiting its concentration and thus its propensity to undergo off-cycle dimerization. Further, the rapid formation of Ni(II)XR(dtbbpy) suggests the possibility that, through reacting with an aryl radical produced via photolysis of Ni-Caryl species, NiXR(dtbbpy) could lead to product formation directly in C-H activation C(sp2)-(Csp3) coupling reactions. Lastly, changing L = dtbbpy to CF3bpy, 66Mebpy, or 29Mephen increases the ratio of Ni(I)XL to Ni(II)XRL photo-products, thus providing a handle to control the buildup of these reactive intermediates to suit a particular catalytic system.
Discussion
In this work, we have investigated how Ni-mediated cross-coupling reactions are initiated when the starting Ni source is the pre-catalyst Ni(II)X2(dtbbpy). Spectroscopic evidence supports an initiation mechanism involving the photolysis of Ni(II)-X bonds (X = Br or Cl) to form Ni(I)X(dtbbpy) and NiXR(dtbbpy) as the long-lived photo-products. Importantly, this mechanistic paradigm explains how Ni(I) species active for oxidative addition of aryl halides form through either direct photolysis with UV light or through energy transfer from a suitable photosensitizer. This result is relevant to a number of pivotal methodologies used to create cross-coupled products through C-H activation20,23,24,48,50,53, cross-electrophile coupling26,28,35, or C-heteroatom coupling22,24,25. Femtosecond transient absorption measurements in flow suggest that the efficiency of photolysis through direct excitation increases at wavelengths shorter than 400 nm (i.e., ϕ360 = 0.13 and ϕ400 = 0.01).
C-H activation by X• produced from photolysis results in the formation of R• and HX from any available H-atom source, R-H (e.g., ethers, toluene). Radical addition of R• to Ni(I) results in formation of a rarely studied class of organonickel(II) complexes, NiXR(dtbbpy), (R is derived from toluene, 4-fluorotoluene, THF, DME, or dioxane and X = Cl, Br) that are determined here to possess square planar geometry by XAS and are further supported by multiple spectroscopic techniques, as well as the synthesis of Ni(II)Cl(C4FTol)(dtbbpy). Replacement of dtbbpy with CF3bpy, 66Mebpy, or 29Mephen ligands leads to increasing steady-state Ni(I) buildup, in that order, which can enable control of the Ni(I) concentration during catalysis. Importantly, UV-vis experiments show that these complexes are re-photolyzed to Ni(I)Cl(dtbbpy), which reacts to form NiX(Caryl)(dtbbpy) in the presence of aryl halide coupling partners, complexes that are well-studied and have been identified as the dominant Ni species during catalysis in both C-H activation and cross-electrophile coupling20,28. As such, NiXR(dtbbpy) can be thought of as a light-activated reservoir of active Ni(I), protecting it from pathways (e.g., dimerization) that lead to off-cycle Ni species. The photochemical initiation mechanism presented here could find applicability beyond the reaction systems considered in this work, as NiXRL complexes have been proposed to be on-cycle for light-driven carboxylation41, decarboxylative cross-coupling46, and alkyne hydroalkylation reactions42.
Methods
Representative method for in situ generation of NiCl(CDME)(dtbbpy)
To make and convert the Ni/Ir/lut solution: in a N2-filled glovebox, NiCl2(dtbbpy)(6.0 mg, 0.015 mmol) was weighed into a 25 mL volumetric flask to which 25 mL DME was added to the line. A stir bar was then added, and the solution was stirred for 24 h and syringe filtered (0.2 μm) into two 20 mL scintillation vials. An Ir PS stock solution was separately prepared by weighing the Ir PS (1.5 mg, 0.00625 mmol) into a vial and dissolving it in 1 mL DME. To a quartz 1 cm path length screw cap cuvette, 2.7 mL of the filtered NiCl2(dtbbpy) solution and 0.3 mL of the Ir PS stock solution were combined, along with 2.1 μL of 2,6-lutidine. Liquid handling was done with gas-tight glass syringes. The cuvette was sealed, removed from the glovebox, and irradiated in a custom photoreactor setup with a 405 nm LED (216 mW cm−2 at the sample position). A typical irradiation time at this light flux to observe the color change from pale yellow to orange, indicating formation of NiCl(CDME)(dtbbpy), is 20 min.
For additional methods to prepare spectroscopic solutions and additional supporting experimental data, see the Supplementary Information file.
Data availability
All data are available from the corresponding author upon request. The single crystal X-ray crystallography data generated in this study for Ni(II)Cl(C4FTol)(dtbbpy) have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under deposition number CCDC 2449925. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The data used to produce all main text and Supplementary Information figures are available free of charge for download from the NREL Data Catalog via https://data.nrel.gov/submissions/292/.
Code availability
All code written for data analysis and figure plotting is available in the NREL Data Catalog, organized by Figure number.
References
Wenger, O. S. Photoactive nickel complexes in cross-coupling catalysis. Chem. A Eur. J. 27, 2270–2278 (2021).
Chan, A. Y. et al. Metallaphotoredox: the merger of photoredox and transition metal catalysis. Chem. Rev. 122, 1485–1542 (2022).
Mantry, L., Maayuri, R., Kumar, V. & Gandeepan, P. Photoredox catalysis in nickel-catalyzed C-H functionalization. Beilstein J. Org. Chem. 17, 2209–2259 (2021).
Marchi, M. et al. The nickel age in synthetic dual photocatalysis: a bright trip toward materials science. ChemSusChem 15, e202201094 (2022).
Milligan, J. A., Phelan, J. P., Badir, S. O. & Molander, G. A. Alkyl carbon-carbon bond formation by nickel/photoredox cross-coupling. Angew. Chem. Int. Ed. 58, 6152–6163 (2019).
Twilton, J. et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1, 52–71 (2017).
Zhu, C., Yue, H., Chu, L. & Rueping, M. Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chem. Sci. 11, 4051–4064 (2020).
Hsieh, H.-W., Coley, C. W., Baumgartner, L. M., Jensen, K. F. & Robinson, R. I. Photoredox iridium-nickel dual-catalyzed decarboxylative arylation cross-coupling: from batch to continuous flow via self-optimizing segmented flow reactor. Org. Process Res. Dev. 22, 542–550 (2018).
Gesmundo, N. J., Rago, A. J., Young, J. M., Keess, S. & Wang, Y. At the speed of light: the systematic implementation of photoredox cross-coupling reactions for medicinal chemistry research. J. Org. Chem. 89, 16070–16092 (2024).
Grimm, I. et al. Upscaling photoredox cross-coupling reactions in batch using immersion-well reactors. Org. Process Res. Dev. 24, 1185–1193 (2020).
Duvadie, R. et al. Photoredox iridium-nickel dual catalyzed cross-electrophile coupling: from a batch to a continuous stirred-tank reactor via an automated segmented flow reactor. Org. Process Res. Dev. 25, 2323–2330 (2021).
Sun, R. et al. Elucidation of a redox-mediated reaction cycle for nickel-catalyzed cross coupling. J. Am. Chem. Soc. 141, 89–93 (2019).
Qin, Y., Sun, R., Gianoulis, N. P. & Nocera, D. G. Photoredox nickel-catalyzed c-s cross-coupling: mechanism, kinetics, and generalization. J. Am. Chem. Soc. 143, 2005–2015 (2021).
Till, N. A., Tian, L., Dong, Z., Scholes, G. D. & MacMillan, D. W. C. Mechanistic analysis of metallaphotoredox c-n coupling: photocatalysis initiates and perpetuates ni(i)/ni(iii) coupling activity. J. Am. Chem. Soc. 142, 15830–15841 (2020).
Chrisman, C. H. et al. Halide noninnocence and direct photoreduction of Ni (II) enables coupling of aryl chlorides in dual catalytic, carbon-heteroatom bond-forming reactions. J. Am. Chem. Soc. 145, 12293–12304 (2023).
Kudisch, M., Lim, C.-H., Thordarson, P. & Miyake, G. M. Energy transfer to ni-amine complexes in dual catalytic, light-driven c-n cross-coupling reactions. J. Am. Chem. Soc. 141, 19479–19486 (2019).
Kancherla, R. et al. Mechanistic insights into photochemical nickel-catalyzed cross-couplings enabled by energy transfer. Nat. Commun. 13, 2737–2746 (2022).
Tian, L., Till, N. A., Kudisch, B., MacMillan, D. W. C. & Scholes, G. D. Transient absorption spectroscopy offers mechanistic insights for an iridium/nickel-catalyzed c-o coupling. J. Am. Chem. Soc. 142, 4555–4559 (2020).
Fan, Y., Kang, D. W., Labalme, S., Li, J. & Lin, W. Enhanced energy transfer in a pi-conjugated covalent organic framework facilitates excited-state nickel catalysis. Angew. Chem. Int. Ed. 62, e202218908 (2023).
Cusumano, A. Q., Chaffin, B. C. & Doyle, A. G. Mechanism of Ni-catalyzed photochemical halogen atom-mediated c(sp3)-h arylation. J. Am. Chem. Soc. 146, 15331–15344 (2024).
DiLuzio, S. et al. Reconceptualizing the IrIII role in metallaphotoredox catalysis: from strong photooxidant to potent energy donor. ACS Catalysis 11378–11388 https://doi.org/10.1021/acscatal.4c03350 (2024).
Song, G. et al. Ni-catalyzed photochemical C-N coupling of amides with (hetero)aryl chlorides. Chem. A Eur. J. 29, e202300458 (2023).
Ishida, N., Masuda, Y., Ishikawa, N. & Murakami, M. Cooperation of a nickel-bipyridine complex with light for benzylic c-h arylation of toluene derivatives. Asian J. Org. Chem. 6, 669–672 (2017).
Li, J. et al. The multiple roles of bipyridine-nickel(II) complex in versatile photoredox c(sp2)-c(sp3) cross-coupling. ACS Catalysis 3328–3338 https://doi.org/10.1021/acscatal.4c07605 (2025).
Cavedon, C. et al. Intraligand charge transfer enables visible-light-mediated nickel-catalyzed cross-coupling reactions**. Angew. Chem. Int. Ed. 61, e202211433 (2022)..
Anghileri, L. et al. Evidence for a unifying NiI/NiIII mechanism in light-mediated cross-coupling catalysis. J. Am. Chem. Soc. 147, 13169–13179 (2025).
Na, H. & Mirica, L. M. Deciphering the mechanism of the ni-photocatalyzed c-o cross-coupling reaction using a tridentate pyridinophane ligand. Nat. Commun. 13, 1313 (2022).
Ben-Tal, Y. & Lloyd-Jones, G. C. Kinetics of a ni/ir-photocatalyzed coupling of arbr with rbr: Intermediacy of arniii(l)br and rate/selectivity factors. J. Am. Chem. Soc. 144, 15372–15382 (2022).
McNicholas, B. J. et al. Electronic structures of nickel(II)-bis(indanyloxazoline)-dihalide catalysts: Understanding ligand field contributions that promote c(sp2)-c(sp3) cross-coupling. Inorg. Chem. 62, 14010–14027 (2023).
Cagan, D. A. et al. Photogenerated ni(i)-bipyridine halide complexes: Structure-function relationships for competitive c(sp2)-cl oxidative addition and dimerization reactivity pathways. Inorg. Chem. 62, 9538–9551 (2023).
Hwang, S. J. et al. Trap-free halogen photoelimination from mononuclear ni(III) complexes. J. Am. Chem. Soc. 137, 6472–6475 (2015).
Na, H., Watson, M. B., Tang, F., Rath, N. P. & Mirica, L. M. Photoreductive chlorine elimination from a ni(iii)cl2 complex supported by a tetradentate pyridinophane ligand. Chem. Commun. 57, 7264–7267 (2021).
Arias-Rotondo, D. M. & McCusker, J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 45, 5803–5820 (2016).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Zhang, P., Le, C. C. & MacMillan, D. W. Silyl radical activation of alkyl halides in metallaphotoredox catalysis: a unique pathway for cross-electrophile coupling. J. Am. Chem. Soc. 138, 8084–8087 (2016).
Shields, B. J., Kudisch, B., Scholes, G. D. & Doyle, A. G. Long-lived charge-transfer states of nickel(ii) aryl halide complexes facilitate bimolecular photoinduced electron transfer. J. Am. Chem. Soc. 140, 3035–3039 (2018).
Demchenko, A. P., Tomin, V. I. & Chou, P.-T. Breaking the kasha rule for more efficient photochemistry. Chem. Rev. 117, 13353–13381 (2017).
Cagan, D. A. et al. Elucidating the mechanism of excited-state bond homolysis in nickel-bipyridine photoredox catalysts. J. Am. Chem. Soc. 144, 6516–6531 (2022).
Fayad, R. et al. Direct evidence of visible light-induced homolysis in chlorobis(2,9-dimethyl-1,10-phenanthroline)copper(II). J. Phys. Chem. Lett. 11, 5345–5349 (2020).
Cook, A. R. Sub-picosecond production of solute radical cations in tetrahydrofuran after radiolysis. J. Phys. Chem. A 125, 10189–10197 (2021).
Davies, J. et al. Kinetically-controlled ni-catalyzed direct carboxylation of unactivated secondary alkyl bromides without chain walking. J. Am. Chem. Soc. 146, 1753–1759 (2024).
Zhang, Y., Tanabe, Y., Kuriyama, S. & Nishibayashi, Y. Photoredox- and nickel-catalyzed hydroalkylation of alkynes with 4-alkyl-1,4-dihydropyridines: Ligand-controlled regioselectivity. Chem. A Eur. J. 28, e202200727 (2022).
Colpas, G. J. et al. X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases. Inorg. Chem. 30, 920–928 (1991).
Feth, M. P., Klein, A. & Bertagnolli, H. Investigation of the ligand exchange behavior of square-planar nickel(II) complexes by x-ray absorption spectroscopy and x-ray diffraction. Eur. J. Inorg. Chem. 2003, 839–852 (2003).
Wilson, D. W. N. et al. Three-coordinate nickel and metal-metal interactions in a heterometallic iron-sulfur cluster. J. Am. Chem. Soc. 146, 4013–4025 (2024).
Nsouli, R. et al. Decarboxylative cross-coupling enabled by Fe and Ni metallaphotoredox catalysis. J. Am. Chem. Soc. 146, 29551–29559 (2024).
Westawker, L. P., Bouley, B. S., Vura-Weis, J. & Mirica, L. M. Photochemistry of Ni (II) tolyl chlorides supported by bidentate ligand frameworks https://doi.org/10.1021/jacs.5c03770 (2025).
Shields, B. J. & Doyle, A. G. Direct c(sp3)-h cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc. 138, 12719–12722 (2016).
Cagan, D. A., Bím, D., Kazmierczak, N. P. & Hadt, R. G. Mechanisms of photoredox catalysis featuring nickel-bipyridine complexes. ACS Catal. 9055–9076 https://doi.org/10.1021/acscatal.4c02036 (2024).
Heitz, D. R., Tellis, J. C. & Molander, G. A. Photochemical nickel-catalyzed c-h arylation: Synthetic scope and mechanistic investigations. J. Am. Chem. Soc. 138, 12715–12718 (2016).
Xue, X.-S., Ji, P., Zhou, B. & Cheng, J.-P. The essential role of bond energetics in C-H activation/functionalization. Chem. Rev. 117, 8622–8648 (2017).
Huang, H., Alvarez-Hernandez, J. L., Hazari, N., Mercado, B. Q. & Uehling, M. R. Effect of 6,\({6}^{{\prime} }\)-substituents on bipyridine-ligated Ni catalysts for cross-electrophile coupling. ACS Catal. 14, 6897–6914 (2024).
Kariofillis, S. K. & Doyle, A. G. Synthetic and mechanistic implications of chlorine photoelimination in nickel/photoredox c(sp3)-h cross-coupling. Acc. Chem. Res. 54, 988–1000 (2021).
Acknowledgements
This work was authored in part by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy under Contract No. DE-AC36-08GO28308. Funding was provided by the US Department of Energy (DOE), Office of Science, as part of the Bio-Inspired Light-Escalated Chemistry Energy Frontier Research Center under grant DE-SC0019370, awarded to O.G.R., G.R., H.S., A.A.C., and M.J.B. EPR measurements were conducted using the NREL Advanced Spin Resonance Facility. We thank Steven Rowland for assistance with FT-ICR-MS measurements and data analysis. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Use of the Laser Electron Accelerator Facility (LEAF) of the BNL Accelerator Center for Energy Research (ACER) was supported by US Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences & Bioscience through contract DE-SC0012704. This research used resources of the Advanced Photon Source; a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank Stanford University and the Stanford Research Computing Center for providing computational resources that contributed to these research results. We thank Kacie Nelson for preparing and measuring the XANES spectrum of the reference nickel TMEDA complex. The views expressed in the article do not necessarily represent the views of the DOE or the US Government. The US Government retains and the publisher, by accepting the article for publication, acknowledges that the US Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for US Government purposes.
Author information
Authors and Affiliations
Contributions
R.X.H. performed XAS experiments at SLAC with assistance from M.K. and analyzed the XAS data. L.K.V. performed PR experiments at BNL, and L.K.V. and M.J.B. analyzed the PR data. J.D.E., O.G.R. and J.Y. performed XAS experiments at ANL, and J.D.E. and O.G.R. analyzed the data. J.D.E. performed TRPL experiments and analysis. A.Z. performed quenching and spectroelectrochemistry experiments at NREL, analyzed the data, and conceived of initial project direction. R.W.S. and M.K. performed single crystal X-ray diffraction experiments at NREL. R.W.S. analyzed the diffraction data, including crystal structure solution and refinement. M.K. performed all other experiments and analysis at NREL. R.X.H. performed the DFT computations. S.D., X.Z., A.A.C., H.S., M.J.B. and G.R. provided intellectual contributions. M.K. and O.G.R. wrote the manuscript incorporating feedback and contributions from the other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interest: the provisional patent application 63/686,695 was filed with the Alliance for Sustainable Energy, LLC as applicant and with M.K., G.R., and O.G.R. listed as inventors. The patent covers preparation and use of NiXRL complexes as catalysts (as described in this work) for processes not described in this work. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kudisch, M., Hooper, R.X., Valloli, L.K. et al. Photolytic activation of Ni(II)X2L explains how Ni-mediated cross coupling begins. Nat Commun 16, 5530 (2025). https://doi.org/10.1038/s41467-025-60729-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60729-x
