
Abstract
Alzheimer’s disease (AD), the leading cause of senile dementia, lacks effective therapies. While microglia are central to AD pathology, key therapeutic targets remain unclear. Here we identify microglial connexin43 (Cx43) hemichannels as a regulator of microglial reactivity in AD, positioning them as a promising therapeutic target. Post-mortem AD patient tissue showed elevated Cx43 levels in periplaque microglia. In the APPswe/PS1dE9 (APP/PS1) mouse model of amyloidosis, we demonstrated that microglial Cx43 hemichannels correlated with microglial malfunction, which in turn exacerbated β-amyloid pathology. Ablation of microglial Cx43 hemichannels by genetic knockout shifts microglia to a neuroprotective phenotype, enhancing the microglia-plaque interaction while suppressing neurotoxicity, thereby mitigating the progression of AD-like pathology. We developed TAT-Cx43@LNPs, a Cx43 hemichannel-targeting peptide delivered by a lipid nanoparticle system, which effectively delayed and rescued β-amyloid-related neuropathology and cognitive impairment in APP/PS1 mice. This study provides evidence for advancing Cx43 hemichannel targeting therapy into clinical trials.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative and cognitive disorder among the elderly, with projections indicating a doubling of AD-related dementia cases in Europe and tripling globally by 20501. Decades of extensive research on AD were unable to fully reveal its complex and enigmatic pathophysiology2. Ongoing new drug development efforts have been mainly guided by the widely accepted amyloid cascade hypothesis, which led to a staggering 98% failure rate in clinical trials3,4. The recent β-amyloid (Aβ) antibody therapies are only suitable for AD patients at the very early stages, yet still failing to halt its progression5. To address the stalemate in treating this irreversible disease, we must identify new targets to tackle pathophysiological mechanisms beyond Aβ accumulation1.
Among an array of cell types involved in early-stage AD6, our attention is on microglia, which are the first responders to the central nervous system (CNS) pathology, including AD7. Microglial cells undergo significant and complex pathological changes at the pre-plaque stage in AD mice8. These changes are also observed in patients with mild cognitive impairment that manifest the initial stages of AD9,10,11. Such microglia was called disease-associated microglia12, characterized by overexpression of genes (such as Trem2, Apoe, and Grn) associated with AD1,13,14. Recent studies highlighted the dual role of microglia in AD. Microglial cells establish close interactions with Aβ plaques, forming a core-shell structure15 to restrict the damage to the neighboring neuropil and actively participate in Aβ clearance13. However, reactive microglia also contribute to neurodegeneration by engaging in aberrant synapse removal and inducing a pro-inflammatory, neurotoxic microenvironment13,16,17. Although extensively studied, targeting microglia remains an uncharted area due to their remarkable flexibility and multiple roles during disease progression. Nevertheless, any cell-specific approach to shift microglia towards a neuroprotective state may represent a new approach in AD treatment.
A unique feature of pathological microglia during AD progression is associated with connexin43 (Cx43). An upregulation of Cx43 in periplaque microglia in AD mice was demonstrated by in situ single-cell transcriptome mapping15. Elevated Cx43 proteins were also confirmed by proteomic analysis of microglia isolated from AD mice18. Connexins form gap junction channels that are fundamental for cell-to-cell communication, and hemichannels for autocrine or paracrine signaling. Connexin expression represents a distinctive feature of macroglia in the CNS, connecting astroglia and oligodendroglia into panglial syncytia19. Recent studies suggested elevated Cx43 expression and altered channel functions as the critical elements in AD pathology20,21,22,23,24,25,26, while Aβ exposure has been found to activate microglial Cx43 hemichannels in vitro27. However, the detailed contribution of microglial Cx43 in AD pathology and the potential of microglial Cx43 as a therapeutic target remain largely unexplored.
Our study explores a cell-specific treatment for AD by targeting Cx43 hemichannels. Our in-depth examination of post-mortem human AD brains confirmed an abnormal increase in microglial Cx43. Using the APPswe/PS1dE9 (APP/PS1) mouse model of Aβ pathology, we further identified microglial Cx43-related pathogenetic mechanisms in AD pathogenesis, demonstrating that ablation of microglial Cx43 hemichannels shifted microglia to a neuroprotective reactive state. We translated this finding into a lipid nanoparticle-based peptide drug delivery system, which inhibits microglial Cx43 hemichannels to arrest and reverse AD progression in the pre-clinical trial.
Results
Increased microglial Cx43 in AD patient brains
We first analyzed the global changes in Cx43 expression in AD patient brains from online transcriptome and proteome databases. Analysis of the ROSMAP-DLPFC bulk RNA-sequencing data28 showed that Cx43 RNA expression is significantly increased in AD patients and positively correlates with the Braak stage (Fig. 1a and Supplementary Fig. 1a). Similarly, data from a multi-center consortium proteomics study (ACT-BLSA-Banner-MSSB)29 revealed a positive correlation between Cx43 protein level and the Braak stage, indicating a significant elevation of Cx43 in AD patients compared to the healthy controls and asymptomatic AD patients (defined as cognitively normal individuals with AD neuropathological hallmarks30) (Fig. 1b). However, the cell-specific distribution of elevated Cx43 remained unknown.

a Cx43 (gene name: GJA1) expression from bulk RNA-sequencing of the human dorsolateral prefrontal cortex in the ROSMAP cohort was plotted against the Braak score, revealing a positive correlation. Right: expression of Cx43 in healthy (N = 172), mild cognitive impairment (MCI, N = 138), and AD (N = 219) groups. Statistical test: one-way ANOVA. b Cx43 protein level from a proteomic study of BLSA-Banner-MSSB-ACT multicenter cohort was plotted against the Braak score. Right: Cx43 protein level in control (N = 72), asymptomatic AD (asymAD, N = 75), and AD (N = 179) groups. Statistical test: one-way ANOVA. c Representative IBA1 and Aβ immunostaining of healthy controls and AD patients, scale bar: 10 μm. Right: Quantification of IBA1+ microglia number. N = 5 subjects. Statistical test: two-tailed t test. d Representative IBA1, GFAP, and Aβ immunostaining of AD patients, scale bar: 50 μm. Right: Distribution of microglia and astrocytes at different distances from Aβ plaques (N = 30 plaques). e Cx43 and Aβ immunostaining, scale bar: 10 μm. C x 43 intensity at different distances from plaques (n = 91) was quantified and compared to the area distal to the plaque (55–100 μm). Statistical test: two-tailed t test. f Representative Cx43, IBA1, and Aβ immunostaining of healthy controls and AD patients, scale bar: 10 μm. The white boxed area was 3D rendered to display Cx43 signal in IBA1+ domain. Arrowhead: periplaque microglia. g Quantification of Cx43 + area within IBA1+ domain in normal controls and AD patients, normalized to normal controls. N = 5 subjects, n = 153 (control) and 206 (AD) cells. Statistical test: two-tailed t test. h Quantification of Cx43 + area within IBA1+ domain in periplaque microglia and distal microglia in AD patients, normalized to normal controls. N = 5 subjects, n = 85 (periplaque) and 121 (distal) cells. Statistical test: two-tailed t test. Dot plots show mean ± SEM, each data point, and p-value. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
Thus, we performed immunostaining on five post-mortem human AD brains and five age- and sex-matched healthy control brains (Supplementary Fig. 1b). We found a significant increase in microglia numbers in the AD brain, while microglial density was the highest in the vicinity of Aβ plaques (Fig. 1c, d). We further analyzed the Cx43 protein level in relation to Aβ plaques, and found that Cx43 expression was also significantly higher in the periplaque area (Fig. 1e). Subsequently, we examined variations in Cx43 expression around four different classes of Aβ plaques (dense-cored plaque, cotton-wool plaque, neuritic plaque with AT8+ Tau neurofilament tangle (NFT), and diffuse plaque with AT8+ Tau NFT), as well as to neuronal Tau NFT alone (Supplementary Fig. 1c). Elevated periplaque Cx43 expression was observed around all four classes of Aβ plaques; cotton-wool plaques and plaques with AT8+ NFT showed higher periplaque Cx43 expression compared to the dense-cored plaques (Supplementary Fig. 1d). In contrast, Cx43 expression was not elevated around AT8+ Tau NFT residing in the neuronal cell body (Supplementary Fig. 1d).
To evaluate the changes in microglial Cx43 levels, we quantified the colocalization of Cx43 with the microglial marker IBA1 using super-resolution microscopy. We found that the Cx43+ area associated with microglia was increased by 182.1 ± 18.8% (N = 153 and 217 cells, p < 0.0001) in the AD patient brains when compared to the healthy controls (Fig. 1f, g). It was suggested that microglia at the adjacent areas of Aβ plaques constitute a subset of disease-associated microglia with distinctive morphological and transcriptional changes, which may play a significant role in Aβ plaque formation, containment, and neurodegeneration12,15. We therefore classified microglia into two groups: the periplaque microglia with their cell body located within 50 μm off the plaque center and in contact with Aβ plaque, and the distal microglia located > 50 μm to the plaque center. We found that the level of Cx43 was 61.9 ± 12.4% (N = 85 and 121 cells, p < 0.0001) greater in periplaque microglia when compared to the distal microglia (Fig. 1f, h).
Pannexin 1 (Panx1) is a plasmalemmal channel distantly related to the connexin family; Panx1 is expressed in microglia and can be affected by neurodegeneration31. We analyzed Panx1 in post-mortem brain samples and found that Panx1 expression was low in microglia and was not significantly altered in AD brains (Supplementary Fig. 1e). As a reference, we also quantified Cx43 changes in the ALDH1L1+ astrocytes in controls and AD patients. Astrocytic Cx43 level was significantly increased in AD patient brains, but the degree of the change (AD vs control: 52.4 ± 8.1% increase, N = 321 and 129 cells, p < 0.0001; periplaque vs distal: 51.6 ± 6.6% increase, N = 124 and 197 cells, p < 0.0001) was lower compared to that of microglia (Supplementary Fig. 1f).
Taken together, these observations suggest that a marked increase in Cx43 is one of the features characterizing pathological microglia in AD.
Increased microglial Cx43 and hemichannel activity correlates with Aβ pathology in a mouse model of AD
We used the APP/PS1 mouse model, known to resemble human AD-related Aβ pathology in distinct disease stages (2 months: pre-onset; 4 months: onset of Aβ pathology; 9 months: profound Aβ pathology and measurable cognitive decline)32,33 (Supplementary Fig. 2a). In APP/PS1 mice, microglial density was higher in the periplaque area than in the area distal to the plaques; in contrast, astrocytes, oligodendrocyte precursor cells (OPC) or oligodendrocytes (OL) were not as enriched at the periplaque area (Supplementary Fig. 2b). We first accessed the immunofluorescence staining of Cx43 in the hippocampus, the brain region known to regulate cognitive function and is impaired by AD34,35. There were no significant changes in Cx43 levels in the hippocampus of 2- and 4-month-old APP/PS1 mice. However, a marked increase in Cx43 immunoreactivity was evident in 9-month-old APP/PS1 mice (Fig. 2a). Further analysis revealed an increase in the Cx43 immunoreactivity in the periplaque areas in both 4 and 9-month-old mice (Fig. 2b). A similar stage and position-dependent Cx43 increase was also observed in the cortex of APP/PS1 mice (Supplementary Fig. 2c, d). Hence, an increase in Cx43 levels is closely associated with Aβ plaques.

a Representative Cx43 and Aβ immunostaining in 9-month-old wild type (WT) and APP/PS1 mice, scale bar: 100 μm. Right: Quantification of Cx43 intensity in the hippocampus of WT and APP/PS1 mice at 2-, 4-, and 9-month-old. N = 5 mice. Statistical test: two-tailed t test. b Representative Cx43 and Aβ immunostaining in 4- and 9-month-old APP/PS1 mice, scale bar: 10 μm. Cx43 staining intensity at different distances from Aβ plaques (n = 20, 25) was quantified and compared to the area distal to the plaque (50–55 μm). Statistical test: two-tailed t test. c Representative super-resolution image of Cx43, IBA1, and Aβ staining in 4- and 9-month-old WT and APP/PS1 mice, accompanied by 3D reconstruction of Cx43 signal within IBA1+ volume. Scale bar: 10 μm. d Quantification of Cx43 + area within IBA1+ domain in WT and APP/PS1 mice. Cx43+ volume was normalized to the cell domain volume, display in percentage. N = 97, 92, 79, 79, 90, 95 cells, respectively, from 5 mice. Statistical test: one-way ANOVA. e Comparison of Cx43+ area within IBA1+ domain between periplaque and distal microglia. N = 40, 39, 61, 34 cells from 5 mice. Statistical test: one-way ANOVA. f Representative Ethidium bromide (EtBr) uptake by 9-month-old APP/PS1 acute brain slices and counterstained with IBA1. Cx43-specific channel blocker GAP26 and pan gap junction/hemichannel blocker CBX were added to 9-month-old APP/PS1 acute brain slices. Scale bar: 10 μm. g Quantification of EtBr dye intensity in dye uptake experiment on 2-, 4- and 9-month-old WT and APP/PS1 acute brain slices. For 2-month-old, N = 4 experiments; for 4-month-old, N = 5 experiments; for 9-month-old, N = 5 experiments (GAP26) or 10 experiments (other groups). Statistical test: two-tailed t test (2-month-old) or one-way ANOVA (4- and 9-month-old). Dot plots show mean ± SEM, each data point, and p-value. Box plots show minima, maxima, 25 ~ 75% percentile, medium, each data point, and p-value. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
To assess changes in microglia-specific Cx43 levels, we quantified the colocalization between Cx43 and IBA1 staining. Cx43+ area co-localized with IBA1+ microglia was not significantly altered in APP/PS1 mice at 2 months of age, but increased at 4 and 9 months of age (by 276 ± 52% at 4 months, and by 354 ± 48% at 9 months, N > 80 cells from 5 mice, p < 0.0001 Fig. 2c, d). Similarly, Cx43 levels in periplaque microglia at both 4 and 9 months of age were significantly increased (Fig. 2c, e). Increased microglial Cx43 expression was also observed in APP/PS1 mouse cortex (Supplementary Fig. 2e). These results indicate a correlation between increased Cx43 levels in microglia and the progression of Aβ pathology. We further analyzed the contribution of astrocytes, OPCs, and OLs to the periplaque Cx43 changes. At 9-month of age, astrocytic Cx43 level was elevated in APP/PS1 mice compared to WT, and in periplaque astrocytes compared to distal ones; however, Cx43 level in OPCs/OLs was not significantly changed in APP/PS1 animals (Supplementary Fig. 2f). This suggests that periplaque Cx43 elevation is driven by microglia and astrocytes, with minimal involvement of cells of oligodendroglial lineage. We also examined the changes in Panx1 and Cx30 levels in microglia. While Panx1 was increased in APP/PS1 mice, the change was not age-dependent, nor was it significantly different between periplaque and distal microglia (Supplementary Fig. 2g). Microglial Cx30 was not significantly changed in APP/PS1 mice compared to WT (Supplementary Fig. 2h).
Altered Cx43 function was reported in AD24, and it correlated with Aβ pathology27. Connexins in reactive microglia have been reported to possess both hemichannel and gap junction functions in vitro31. However, no functional gap junction was found in surveilling or reactive microglia in vivo36. To investigate microglial Cx43 hemichannel activity in APP/PS1 mice, we monitored ethidium bromide (EtBr) uptake in acute brain slices. At 2 months of age, no change in dye uptake by microglia from APP/PS1 mice compared to WT control was detected (Fig. 2g). Starting from 4 months of age, we observed a significant increase in dye accumulation in the periplaque microglia (Fig. 2g). At 9 months of age, markedly increased accumulation of EtBr was observed in both the periplaque and the distal microglia in APP/PS1 mice compared to WT mice (83.2 ± 9.2% and 42.5 ± 8.4%, N ≥ 5 experiments, p < 0.0001, Fig. 2f, g). Addition of carbenoxolone (CBX), a non-selective hemichannel blocker, prior to dye uptake experiment decreased the dye uptake by microglia from APP/PS1 mice to the WT level (Fig. 2g). Because dye uptake into microglia could be mediated not only by Cx43 but also by Panx1 channels31, we further used a more selective Cx43 channel blocker Gap2637, which decreased microglial dye uptake in APP/PS1 mice brain (reduced by 39.9 ± 8.3% in periplaque microglia, N ≥ 5 experiments, p = 0.0002), almost to the levels of WT and the CBX-treated group (Fig. 2g). Thus, we can attribute an increased dye uptake to an elevated Cx43 hemichannels activity in the periplaque microglia from APP/PS1 mice.
In summary, we observed a significant elevation in microglial Cx43 levels and related hemichannels activity during disease progression in APP/PS1 mice, particularly in the periplaque microglia. Our findings from AD patients and the mouse model indicate that microglial Cx43 may contribute to progressive pathogenesis of AD.
Depletion of microglial Cx43 alleviates AD pathology and cognitive impairment
To understand the pathological role of microglial Cx43 in AD, we crossed the APP/PS1 mice with microglia-specific Cx3cr1-CreERT mice and Cx43-flox mice to achieve microglia-specific conditional Cx43 knockout in amyloidosis mouse model brains (APP/PS1:Cx43mgl-cKO) (Fig. 3a). We confirmed the significant down-regulation of Cx43 in microglia in the APP/PS1:Cx43mgl-cKO mice (Supplementary Fig. 3a). The changes in brain histology and cognitive performance were analyzed at 9 months of age. APP/PS1:Cx43mgl-cKO mice demonstrated no changes in either the area percentage or the number of Aβ plaques. However, Cx43 conditional knockout increased the percentage of compact plaques (plaques with a condensed core) as opposed to filamentous plaques (Fig. 3b). Cx43 conditional knockout may thus reduce the neurotoxicity of the plaques, as filamentous plaques are suggested to be more neurotoxic38,39,40.

a Mating strategy to achieve Cx43 conditional knockout in microglia (Cx43mgl-cKO). b Representative Methoxy X04 staining in 9-month-old APP/PS1 and APP/PS1:Cx43mgl-cKO mouse hippocampus. Scale bar, 50 μm. The number of plaques, percentage of Methoxy X04+ area, the compact plaque percentage, and plaque core area ratio were quantified. N = 5 mice. Statistical test: two-tailed t test. c Representative Aβ (6E10) and dystrophic neurites (RTN3). Scale bar: 10 μm. The number of RTN3+ puncta at different distances from the plaque and the sum number were quantified. N = 32 plaques. Statistical test: two-way ANOVA (intersection curves), and two-tailed t test (sum intersection). d Representative Aβ (Methoxy X04) and dystrophic neurites (LAMP1). Scale bar: 10 μm. LAMP1+ area % at different distances from the plaque and the total area per plaque were quantified. N = 32 plaques. Statistical test: two-way ANOVA (area curves) and two-tailed t test (area per plaque). e Left: Lamp1+ area around compact and fibrillar plaques. N = 33, 27, 46, and 27 plaques. Right: Lamp1 + area around small (0 ~ 300 μm2), medium (300 ~ 1000 μm2) and large (> 1000 μm2) plaques. N = 21, 18, and 21 plaques (APP/PS1), and 27, 29, and 17 plaques (APP/PS1:Cx43mgl-cKO). Statistical test: two-way ANOVA. f Representative Aβ and Homer1 staining. Scale bar: 10 μm. Homer1+ area percentage at different distances from the plaque was quantified. N = 32 plaques. Statistical test: two-way ANOVA. g MitoSox staining of hippocampal CA1 neuron. Scale bar, 50 μm. MitoSox intensity was quantified and normalized to the WT control. N = 5 mice. Statistical test: two-sided t test. h Novel object recognition test was performed at 9-month-old to examine cognitive function. Statistical test: two-sided t test. i Open field test was performed at 9-month-old to examine the anxiety (Distance in center) and activity traits (total distance). Statistical test: two-sided t test. For behavioral tests, N = 21 (WT), 12 (Cx43mgl-cKO), 13 (APP/PS1), or 8 (APP/PS1:Cx43mgl-cKO) mice. Dot plots show mean ± SEM, each data point, and p-value. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
To confirm this, we used reticulon-3 (RTN3) and lysosomal-associated membrane protein 1 (LAMP1) antibodies to assess periplaque neurite dystrophy; both proteins are commonly used as dystrophic neurite markers in AD brains and animal models. LAMP1, a lysosomal marker, is highly enriched in the inner layer of periplaque dystrophic neurites41, while RTN3, a tubular endoplasmic reticulum marker, is enriched in the outer layer of periplaque dystrophic neurites42. APP/PS1:Cx43mgl-cKO mice showed less RTN3+ or LAMP1+ dystrophic neurites surrounding the plaques compared to APP/PS1 mice (Fig. 3c, d). After we sub-classified plaques according to the types or size, we found that microglial Cx43 ablation significantly reduced the dystrophic neurites around filamentous plaques but not the compact plaques, and around large plaques (> 1000 μm2) but not medium (300 ~ 1000 μm2) or small (0 ~ 300 μm2) ones (Fig. 3e). We also found an increased level of the postsynaptic marker Homer1 in the periplaque area (Fig. 3f), suggesting alleviated synaptic loss. While microglial Cx43 ablation may not further reduce periplaque neurite dystrophy near compact/small plaques (already low in dystrophy), its role in promoting overall plaque compaction likely decreases dystrophic neurites by limiting diffuse, damaging plaques.
In addition, microglial Cx43 knockout attenuated neuronal oxidative stress in APP/PS1 mice, as indicated by reduced MitoSox staining intensity in the acute brain slices, reflecting reduced superoxide production (Fig. 3g). Neuronal loss is one of the leading pathologies in humans with AD2. To evaluate neuronal death, we performed cleaved-caspase3 (c-Casp3) staining and Tunel assay in the brain sections of WT, APP/PS1, and APP/PS1:Cx43mgl-cKO mice. We did not observe positive staining c-Casp3+ or Tunel+ cells in the brains of APP/PS1 mice (Supplementary Fig. 3b, c). We also performed NeuN staining to quantify neuronal number in the hippocampus, which was not significantly changed among the three experimental groups (Supplementary Fig. 3d). Our findings are consistent with the current consensus that there is no neuronal loss in the APP/PS1 mouse model43.
We further investigated whether microglial Cx43 knockout affected other manifestations of AD-related pathology. In particular, using immunostaining, we found decreased myelin loss (as assessed by MBP immunostaining) in APP/PS1 mice with microglial Cx43 ablation (Supplementary Fig. 3e). Downregulation of microglial Cx43, however, did not affect the number or the area of GFAP+ astrocytes (Supplementary Fig. 3f).
Subsequently, we examined the behavior of APP/PS1 mice with microglial Cx43 ablation. Conditional knockout of microglial Cx43 in APP/PS1 mice significantly improved cognitive performance as evidenced by the discrimination index in the novel object recognition test (Fig. 3h). Microglial Cx43 ablation on the wildtype (WT) background, however, did not additionally improve the cognitive function compared to the WT control (Fig. 3h). Previous studies identified anxiety or hyperactivity symptom in APP/PS1, similar to the psychiatric manifestations in some AD patients44,45; however, evidence demonstrating these behavioral deficits in APP/PS1 mice is inconsistent45. In our hand, open field test showed no anxiety-like (distance in the central area) or hyperactivity (total distance) behaviors in APP/PS1 mice compared to WT (Fig. 3i). Microglial Cx43 knockout in APP/PS1 mice or WT mice did not cause abnormality in these behavioral traits, either (Fig. 3i).
Together, these findings suggest that ablation of Cx43 in pathological microglia mitigates neuropathology and cognitive decline during AD progression.
Turning microglia neuroprotective by microglial-specific Cx43 knockout in AD mice
To understand the mechanisms underlying the beneficial effects of suppressing microglial Cx43 on neuropathology and cognitive impairment, we compared the cell densities and cellular morphology of hippocampal microglia in APP/PS1:Cx43mgl-cKO, APP/PS1, and WT mice. While APP/PS1 mice displayed a significant increase in microglia numbers compared to the WT controls, the number of microglia was even higher in APP/PS1:Cx43mgl-cKO mice (Fig. 4a, b). Increased microglial density in APP/PS1:Cx43mgl-cKO mice was due to an increased population of periplaque microglia, while the number of microglia distal to plaques did not significantly differ from the APP/PS1 mice (Fig. 4b). Arguably, periplaque microglia engage closely with the Aβ plaques to limit the damage to the adjacent neuropil39,40,46. Given the increased compactness of Aβ plaques and reduced plaque-associated neural damage observed in APP/PS1:Cx43mgl-cKO mice, we speculated that the microglial barrier surrounding the plaques was strengthened by Cx43 knockout. Upon closer examination of individual plaques, it became evident that APP/PS1:Cx43mgl-cKO mice exhibited a higher number of periplaque microglia compared to APP/PS1 mice when normalized to the plaque perimeter (Fig. 4c and Supplementary Fig. 4a). Quantification of Aβ plaque volume within the microglial domain using Imaris software also showed that microglia with Cx43 knockout displayed increased interaction with Aβ plaques (Fig. 4d and Supplementary Fig. 4b), which reflected an increased number of periplaque microglia; individual microglia phagocytosis, however, was not affected, as the amount of Aβ engulfed per microglia was not changed (Supplementary Fig. 4c). Nonetheless, such increased microglia-plaque interaction might promote the compactness of Aβ plaque and reduce periplaque neurite dystrophy, as suggested above (Fig. 3b). We also evaluated the morphology of microglia using Sholl analysis. Both periplaque and distal microglia in APP/PS1 showed reduced process complexity compared to WT microglia; the distal microglia in APP/PS1:Cx43mgl-cKO extend more numerous, complex processes compared to that in APP/PS1 mice, though the morphology of periplaque microglia was unchanged (Supplementary Fig. 4d).

a Representative image of IBA1, Aβ and DAPI staining of mouse hippocampus. Scale bar: 20 μm. b The total number of IBA1+ microglia, periplaque microglia, and distal microglia was quantified and normalized to the area. N = 5 mice. Statistical test: two-sided t test. c Representative image of IBA1, Aβ and DAPI staining. Scale bar: 10 μm. For each plaque, the number of periplaque microglia was quantified and normalized to the plaque perimeter. N = 5 mice. Statistical test: two-sided t test. d 3D reconstruction image of panel c displaying Aβ plaque and the fraction of Aβ plaque within IBA1+ microglia domain. Scale bar: 10 μm. The ratio between plaque within microglia and total plaque volume was quantified. N = 5 mice. Statistical test: two-sided t test. e Dye uptake experiment was performed on acute brain slices and counterstained with IBA1 to analyze the microglial hemichannel opening. Scale bar: 10 μm. EtBr intensity was quantified and normalized to the WT group for each experiment. N = 5 mice. Statistical test: paired t test. f ELISA analysis of TNFα and IL1β cytokine levels in acute brain slices normalized to protein content. N = 4 mice. Statistical test: paired t test. g Bulk RNA-seq was performed on microglia isolated from WT, APP/PS1, and APP/PS1:Cx43mgl-cKO mouse cortex and hippocampus. h Volcano plot highlighting the differentially expressed genes between APP/PS1:Cx43mgl-cKO microglia and APP/PS1. i Heatmap shows expression of disease-associated microglia 3 (DAM3) enriched genes, which were split into 5 clusters according to their expression change in APP/PS1:Cx43mgl-cKO microglia compared to APP/PS1. Left: normalized gene expression level of 4 clusters. Right: functional annotation and representative genes of 4 clusters. j, k Heatmap shows differentially expressed genes that are associated with Aβ recognition, as well as the neuroprotective or neurotoxic signature of microglia. Dot plots show mean ± SEM, each data point, and p-value. Box plots show minima, maxima, 25 ~ 75% percentile, and medium. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
Consistent with our observation of microglial Cx43 operating as hemichannels (as shown in Fig. 2f-g), Cx43 knockout in microglia significantly reduced the hemichannels activity, as indicated by the reduced EtBr uptake in IBA1+ microglia in the brain slice (Fig. 4e). Cx43 hemichannels opening was proposed to aggravate proinflammatory response by an autocrine mechanism that activates inflammasome47,48. Microglial Cx43 may play a similar role in Aβ pathology. Indeed, treatment with minocycline, an anti-inflammatory agent49, suppressed the microglial hemichannels opening in acute brain slices from APP/PS1 mice (Supplementary Fig. 4e), whereas knockout of microglial Cx43 significantly reduced the levels of proinflammatory cytokines TNFα and IL1β in acute brain slices (Fig. 4f). This suggests that opening of the microglial Cx43 hemichannels contributes to an inflammatory milieu in AD and may drive microgliosis to a neurotoxic pro-inflammatory phenotype.
To further understand how Cx43 ablation affects the microglial reactive landscape, we performed RNA sequencing of acutely isolated microglia from APP/PS1, APP/PS1:Cx43mgl-cKO, and age-matched WT control mice (Fig. 4g). Differential expression analysis showed that Cx43 knockout in microglia promoted a distinctive neuroprotective reactive phenotype (Fig. 4h–k and Supplementary Fig. 4f). Specifically, we analyzed the expression of previously reported genes enriched in the so-called disease-associated microglia (DAM)12 in WT, APP/PS1, and APP/PS1:Cx43mgl-cKO mice (Fig. 4i). Our RNA-sequencing data confirmed the overexpression of DAM-associated genes in the APP/PS1 mice compared to WT mice (Fig. 4i). We then compared the expression of these genes between APP/PS1 and APP/PS1:Cx43mgl-cKO mice and identified 5 clusters according to the differential expression (Fig. 4i). Ablation of microglial Cx43 upregulated or maintained a set of DAM genes which are associated with Aβ binding and microglia attraction to Aβ, such as Tlr250, Scarb151, Apoe52 (Fig. 4i, j), thus consistent with the histological observation that Cx43 knockout microglia interact more closely with the Aβ plaques. Lysosome- and phagosome-related genes were not prominently altered by Cx43 knockout (Fig. 4i), suggesting an unaffected phagocytosis capacity. We also observed a reduced expression of genes involved in the ribosome-, oxidative phosphorylation-, and AD-related pathways in Cx43 knockout microglia (Fig. 4i). In addition, the downregulation of genes linked to TNF production and inflammatory-related pathways (Fig. 4i), as well as IL1β (Fig. 4k), are consistent with our ELISA data.
RNA-sequencing analysis provides another aspect of how microglial Cx43 knockout-mediated phenotypic shift could ameliorate Aβ pathology. We observed that the neuroprotective reactive microglial signature was upregulated in APP/PS1:Cx43mgl-cKO mice, including Arg1, Chil3, and Grn, while a majority of the neurotoxic signature genes, such as Il1b, Ifng, and Itgax were downregulated (Fig. 4k). Concomitantly, immunostaining showed increased levels of ARG1 in periplaque microglia of Cx43 knockout mice compared to APP/PS1 control (Supplementary Fig. 4g). Consistently, Cx43 knockout in microglia upregulated several genes which have been shown to be neuroprotective in Aβ pathology, including Grn53 and Tspo54,55; and several neurotoxic or pro-inflammatory genes related to Aβ pathology such as Clec7a56, Ch25h57, and Tyrobp58 were reduced by Cx43 ablation (Fig. 4i). We further spatially validated the selected differential expressed genes, Clec7a and Grn, using immunostaining. Expression of Clec7a showed only a trend of downregulation in periplaque microglia, but was significantly downregulated in distal microglia (Supplementary Fig. 4h). In contrast, GRN was significantly upregulated in periplaque microglia, but only showed trends of increase in distal microglia (Supplementary Fig. 4i).
Proinflammatory cytokines from reactive microglia may activate astrocytic connexin hemichannels59. Given that the knockout of microglial Cx43 suppressed the proinflammatory profile, we wondered whether the astrocytic connexin hemichannel activation was also affected. EtBr uptake experiment of acute brain slices indicated that astrocyte connexin hemichannel activity was also reduced in APP/PS1:Cx43mgl-cKO mice (Supplementary Fig. 4j). Opening of astrocytic connexin hemichannel can mediate release of gliotransmitter such as D-serine60. We further measured the level of D-serine in the conditional medium from acute brain slices and found that D-serine level in APP/PS1:Cx43mgl-cKO group was also significantly downregulated compared to that of APP/PS1, possibly reflecting reduced astrocytic connexin hemichannel activity (Supplementary Fig. 4k). Considering our previous study that activation of astrocytic connexin hemichannels contributes to neuronal damage24, the reduced astrocytic hemichannel activity in microglial Cx43 cKO can also ameliorate neuronal damage in Aβ pathology.
Thus, ablation of microglial Cx43 promotes a transformation of reactive microglia towards neuroprotective phenotype in AD; these microglia establish a closer interaction with Aβ plaques to limit the toxicity of the latter, while also reducing the presence of neurotoxic factors, which, we suggest, is due to the inhibition of Cx43 hemichannels activity.
A lipid nanoparticle-based drug delivery strategy inhibits Cx43 hemichannels
Our finding that the neuroprotective state of microglia can be promoted by Cx43 knockout, combined with our previous observation that inhibition of astroglial Cx43 hemichannels can protect neurons in APP/PS1 mice24, supports the idea that targeting Cx43 hemichannels is a promising therapeutic approach for AD treatment.
We, therefore, targeted Cx43 hemichannels using a mimetic peptide-based approach. TAT-Cx43266–283 peptide was designed to mimic the SH3-domain of the Cx43 C-terminus61 (Fig. 5a). Treatment with TAT-Cx43266–283 inhibits Cx43 hemichannels but not the gap junction channels in vitro62. In addition, TAT-conjugated peptide readily crosses the blood-brain barrier (BBB)63, supporting potential in vivo efficacy, which, however, has not been tested previously.

a Illustration of Cx43 structure highlighting the 266–283 region. TAT-Cx43266-283 is the mimetic peptide of this region conjugated to a cell-penetrating peptide TAT. b Dye uptake experiment was performed on primary microglia in combination with TAT-Cx43266-283 treatment at different concentrations. The addition of 1.8 mM Ca2+, which inhibits hemichannel opening64 serves as a negative control. Scale bar, 10 μm. EtBr signal was quantified, and a non-linear regression analysis was performed to determine the IC50 value. N = 3 coverslips. c EtBr uptake experiment on acute brain slices followed by IBA1 counter-immunostaining. Scale bar, 10 μm. EtBr signal intensity in microglia was quantified and normalized to the TAT-treated control. N = 4 experiments. Statistical test: one-way ANOVA. d Schematic of TAT-Cx43@LNP construction. e Electronic microscopy of lipid nanoparticles with different compositions. Scale bar, 100 nm. f Lipid nanoparticle size measurement. N = 3 experiments. g Measurement of zeta potential of lipid nanoparticles. N = 3 experiments. h The size of lipid nanoparticles at different days post-production incubated in saline control (0.9% NaCl) or DMEM supplemented with 10% fetal bovine serum (DMEM:10% FBS). N = 3 experiments. i Measurement of the peptide release rate at 37 °C of lipid nanoparticles in vitro. N = 3 experiments. Statistical test: two-way ANOVA. j Cy5.5-labeled TAT-Cx43266-283 peptide packaged with DOTAP nanolipid gel (DOTAP-NLG) was i.p. injected into mice, followed by analysis at different time points. k Representative biofluorescence imaging at days 0, 1, and 7 post-injection, revealing the retainment of peptides in the brain. l Microplate reader analysis of Cy5.5 intensity of brain lysate and serum collected at day 0, 1, 3, 7, 14 post-injection of TAT-Cx43266-283 peptide with or without DOTAP-NLG packaging, as well as Cx43266-283 peptide without packaging. N = 5 (TAT-Cx43266-283@LNP), 3 (TAT-Cx43266-283), and 3 (Cx43266-283) experiments. The area under the curve were calculated for statistical analysis (two-sided t test). Dot plots show mean ± SEM, each data point, and p-value. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
We first confirmed the capability of the TAT-Cx43266–283 to inhibit Cx43 hemichannels. In the primary microglial culture, TAT-Cx43266–283 effectively suppressed hemichannels opening as assessed by the EtBr uptake assay (IC50 = 0.5641 μM, 95% confidence interval = 0.2625 ~ 1.271 μM, n = 3 experiments) (Fig. 5b). In acute brain slices from APP/PS1 mice, treatment with 50 μM of TAT-Cx43266–283 was sufficient to inhibit microglial Cx43 hemichannels opening (assessed by EtBr uptake) with an efficacy similar to that of 200 μM CBX; both reduced hemichannels activation to ~ 60% of the control level (Fig. 5c). In primary microglia treated with Aβ25-35, exposure to 50 μM of TAT-Cx43266-283 modified microglial reactive state, as indicated by the increased ARG1 and reduced iNOS staining (Supplementary Fig. 5a, b). Furthermore, as Cx43 is also expressed in astrocytes forming both hemichannels and gap junction channels64, we examined the effect of TAT-CX43266-283 on astrocytes. In acute brain slices, TAT-Cx43266-283 inhibited astrocytic Cx43 hemichannels opening (shown by EtBr uptake assay), but did not affect gap junctions as determined by gap-FRAP assay, which reveals the extent of syncytial coupling (Supplementary Fig. 5c, d). Considering that activation of astrocytic connexin hemichannels contributes to neuronal damage in APP/PS1 mice24, and the critical contribution of astrocytic gap junctions towards neuronal function maintenance64, these observations are further in favor of the use of TAT-CX43266-283 in the treatment of AD. To verify the direct interaction of TAT-Cx43266-283 peptide with Cx43 protein, we conjugated the TAT-Cx43266-283 peptide to the Cy5.5 fluorophore, applied the conjugated peptide to acute brain slices, and counterstained with Cx43 antibody. We found that the Cy5.5 signal was enriched at Cx43+ puncta, indicating that TAT-Cx43266-283 directly binds to Cx43 protein (Supplementary Fig. 5e, f). In contrast, Cy5.5 conjugated TAT peptide increased the overall fluorescence intensity of Cy5.5, but did not colocalize with Cx43+ puncta (Supplementary Fig. 5e, f), suggesting non-specific distribution. These data support the notion that TAT-Cx43266-283 can effectively and specifically inhibit Cx43 hemichannels and modulate microglial reactivity without affecting Cx43 gap junction functions.
However, peptide proteins normally have a short half-life in vivo, which limits their bioavailability65. To address this limitation, we adopted a liposome-based peptide drug delivery system. We packaged the peptide with the 1,2-dioleoyl-3-trimethylammonium propane (DOTAP)-based, chitosan-methacrylate (CMA)-crosslinked, nanolipid gel (Fig. 5d), initially designed for siRNA delivery66. We named the packaged TAT-Cx43266-283 formulation ‘TAT-Cx43@LNPs’. Electron microscopy revealed that the addition of DOTAP and/or CMA crosslinking slightly increased the size of the nanolipid particle compared to the basic egg phosphatidylcholine (EPC) particle, while the addition of DOTAP significantly increased the surface potential of the nanolipid particles. (Fig. 5e–g). The size of the nanolipid gel remained stable in vitro for 7 days, in either saline buffers or serum supplemented buffers (Fig. 5h and Supplementary Fig. 5j), and could achieve sustained, stable release of peptides for 14 days (Fig. 5i). Next, we examined the capacity of the sustained release of the lipid nanoparticle in vivo. The peptide was labeled with fluorophore Cy5.5 before being packaged with lipid nanoparticles and was subsequently intraperitoneally injected into mice (Fig. 5j). Biofluorescence imaging detected Cy5.5 signal in the brain for up to 7 days post injection (Fig. 5k). To further confirm the tissue retainment of the peptide drug, we isolated tissues post-injection and homogenized them to analyze the fluorescence intensity. When TAT-Cx43266-283 peptides were injected without LNP packaging, they peaked on day 1 post-injection but were absent on the 7th day post-injection in the brain lysate and serum (Fig. 5l). However, when LNP-packaged, significantly longer stability of peptides was observed in vivo. On the 14th day post-injection, LNP-packaged peptides were maintained at a relatively high level in the brain lysates (42.9 ± 17.1% compared to day 1) but dropped to a low level (9.7 ± 13.4% compared to day 1) in serum (Fig. 5l). In addition, the unpackaged Cx43266-283 peptide failed to enter the brain parenchyma after i.p. injection (Fig. 5l), consistent with the literature data indicating that the TAT peptide can facilitate cargo across BBB delivery63,67. We also verified the entry of LNP-packaged peptide into the brain parenchyma by imaging of brain slices on day 1 post-injection: TAT-Cx43-Cy5.5@LNP injected mice displayed higher Cy5.5 signal intensity in the brain parenchyma compared to the control. Consistent with the ex vivo treatment, Cy5.5 signal was also higher at Cx43 + puncta (Supplementary Fig. 5g, h). We also observed colocalization of Cy5.5 signal with Cx43 within the periplaque IBA1+ microglia domain (Supplementary Fig. 5i). Our LNP packaging method moderately elevated the peptide level in the brain, but could not achieve exclusive brain delivery, as Cy5.5-peptides were also found in the heart, liver, and kidney (Supplementary Fig. 5k). Nonetheless, these data show that TAT-Cx43@LNPs can achieve sustained drug release and brain retention, substantiating their feasibility for in vivo application.
We further examined the biosafety of the TAT-Cx43@LNPs. Injection of TAT@LNPs or TAT-Cx43@LNPs did not significantly change the mouse weight upon consecutive weekly injections for 6 weeks (Supplementary Fig. 5l). Cx43 is highly expressed in several vital organs, especially in the heart, where Cx43 contributes to action potential propagation68. To investigate potential adverse effects on cardiac function, we performed electrocardiograms on mice before and after the treatment. We found that TAT-Cx43@LNPs altered neither the heart rate nor the propagation of electrical signal across the heart, demonstrated by the unchanged duration of P wave, P-R interval, QRS complex, and Q-T interval, compared to the baseline measurement or the TAT@LNP treated control mice (Supplementary Fig. 5m). We also performed the serum biochemistry test before and after treatment to examine the impact on the biomarkers of vital organ functions, including the heart, liver, kidney, muscle, and pancreas. It appeared that LNP treatment did not significantly affect serum biomarkers reflecting all these organ functions (Supplementary Fig. 5n). In summary, our nanocarrier-based TAT-Cx43266-283 delivery strategy appears to be safe without apparent off-target effects.
Nanocarrier-delivered Cx43 blocker rescues neuropathology and cognitive decline in AD mice
To test whether TAT-Cx43@LNPs can affect AD progression, we treated WT and APP/PS1 mice with TAT-Cx43@LNPs once per week (120 μg/g, peptide 8.2 nmol/g, i.p.) starting from 8 months of age. After 6 weeks of treatment, mice were subjected to behavioral tests and histological analysis (Fig. 6a). The TAT@LNPs treated mice acted as control (which did not affect the cognitive outcome as indicated by the novel object test; Supplementary Fig. 6a). In the Barnes Maze test, TAT-Cx43@LNPs treated APP/PS1 mice spent more time in the target quadrant in the probe trial compared to the TAT@LNPs treated APP/PS1 mice (Fig. 6b, c and Supplementary Fig. 6b, c), suggesting an improved spatial memory. In the novel object recognition test, TAT-Cx43@LNPs treated APP/PS1 mice showed a higher preference for the new object compared to the TAT-treated APP/PS1 mice, indicating an improved short-term memory (Fig. 6d). However, TAT-Cx43@LNPs treatment did not affect the locomotive activity or anxiety-like behavior in the open field test (Fig. 6e; Supplementary Fig. 6d). TAT-Cx43@LNPs treatment in the WT mice affected neither cognitive function, nor locomotive activity, nor anxiety-like behavior (Fig. 6b–e and Supplementary Fig. 6b–d). In summary, these results indicate that TAT-Cx43@LNPs restored the cognitive functions of APP/PS1 mice.

a Experimental protocol. b Barnes’ maze-acquisition trial. The total latency to the target hole, primary latency, and primary exploratory error were quantified. c Barnes’ maze-probe trial. Time spent in the target quadrant and the total distance were quantified. Statistical test: two-sided t test. d Novel object recognition test. Discrimination index and exploration time were quantified. Statistical test: two-sided t test. e Open field test. Distance and time spent in the center zone and the total distance were quantified. Statistical test: two-sided t test. For behavioral tests, N = 9 (WT TAT and WT TAT-CX43) or 13 (APP/PS1 TAT and APP/PS1 TAT-CX43) mice. f EtBr uptake of acute brain slices with IBA1 counterstaining. Microglial EtBr intensity quantified was normalized to the TAT@LNP control. Scale bar, 20 μm. N = 5 mice. Statistical test: one-way ANOVA. g D-serine level in acute brain slice supernatant normalized to TAT@LNP control. N = 5 mice. Statistical test: two-sided t test. h IL1β and TNFα levels in acute brain slices. N = 5 mice. Statistical test: two-sided t test. i IBA1 and Methoxy X04 staining. Scale bar, 50 μm. Hippocampal microglia number and the periplaque microglia number were quantified. N = 5 mice. Statistical test: two-sided t test. j Methoxy X04 staining in the hippocampus. Scale bar, 50 μm. Aβ plaque number and compact plaque percentage were quantified. N = 5 mice. Statistical test: two-sided t test. k RTN3 and Aβ staining. Scale bar, 10 μm. RTN3 + puncta number at different distances from the plaque was quantified. N = 45 and 42 plaques. Statistical test: two-way ANOVA. l MitoSox Red staining of hippocampal CA1 neuron. Scale bar, 50 μm. Quantified MitoSox intensity was normalized to WT. N = 5 mice. Statistical test: two-sided t test. m LTP experiment on acute hippocampal slices. n Representative field excitatory post-synaptic potential (fEPSP) before and after LTP induction. o fEPSP slope during recording. Statistical test: two-way ANOVA. p Normalized fEPSP slope post-LTP induction. For the LTP experiment, N = 8 experiments from 4 mice. Statistical test: two-sided t test. Dot plots show mean ± SEM, each data point, and p-value. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
TAT-Cx43@LNPs suppressed Cx43 hemichannels in both microglia (Fig. 6f) and astrocytes (Supplementary Fig. 6g) as determined by the dye uptake assay in the acute brain slices, but did not affect Cx43 protein levels (Supplementary Fig. 6e, f). Gap junctional communication of astrocytes was not affected either, as determined by the gapFRAP experiment (Supplementary Fig. 6h). Furthermore, TAT-Cx43@LNPs reduced the extracellular level of D-serine (Fig. 6g). TAT-Cx43@LNPs also made microglia less pro-inflammatory, as reflected by the reduced production of proinflammatory cytokine TNF-α and IL1-β (Fig. 6h), as well as an increased ARG1 expression (Supplementary Fig. 6i). Similarly to the APP/PS1:Cx43mgl-cKO mice, TAT-Cx43@LNPs treatment increased microglial numbers, especially of the periplaque microglial population (Fig. 6i and Supplementary Fig. 6j, k), increased microglia-plaque interaction (Supplementary Fig. 6l), and slightly increased microglial ramification (Supplementary Fig. 6m). These observations are consistent with those in the APP/PS1:Cx43mgl-cKO mice, supporting the notion that the inhibition of microglia Cx43 hemichannels promotes a neuroprotective microglia reactive state.
We next examined whether TAT-Cx43@LNPs rescued the neuropathology in APP/PS1 mice. Immunohistology showed that while TAT-Cx43@LNPs did not significantly alter the total area or the number of Aβ plaques, instead, it increased their compactness (Fig. 6j and Supplementary Fig. 6n), which was accompanied by reduced plaque-associated neurite dystrophy (Fig. 6k). TAT-Cx43@LNPs treatment mitigated the neuronal oxidative stress reflected by MitoSox staining of brain slices (Fig. 6l). In addition, TAT-Cx43@LNPs treatment ameliorated the myelin loss, as reflected by the elevated MBP+ area (Supplementary Fig. 6o). Finally, TAT-Cx43@LNPs treatment rescued the reduced long-term potentiation (LTP) in APP/PS1 mice. (Fig. 6m–p).
Collectively, our findings demonstrate that TAT-Cx43@LNPs can be delivered into the CNS to effectively inhibit microglial and astrocytic Cx43 hemichannels, mitigate neuropathology, and restore impaired neuronal functions and cognitive deficits.
TAT-Cx43@LNPs prevent neuropathology at the early stage of Aβ pathology
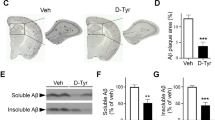
We demonstrated that ablation of microglial Cx43 or pharmacological suppression of Cx43 hemichannels did not alter the plaque load. It is known, however, that microglia differentially affect the plaque load at different stages of Aβ pathology40. To investigate whether suppression of Cx43 hemichannels by TAT-Cx43@LNPs at the early stage of Aβ pathology can arrest or delay disease progression, we treated APP/PS1 mice with TAT-Cx43@LNPs weekly from 3.5 to 5.5 months of age (Fig. 7a). We first verified that TAT-Cx43@LNPs treatment effectively inhibited Cx43 hemichannels in microglia in APP/PS1 mice at 5.5 months using the EtBr uptake experiment (Supplementary Fig. 7a). MitoSox staining of the acute brain slices revealed that TAT-Cx43@LNPs reduced the oxidative stress in hippocampal neurons compared to the LNP-TAT treated control (Fig. 7b). Finally, TAT-Cx43@LNPs alleviated neurite dystrophy as revealed by a reduced RTN3 staining and increased Homer1 staining intensities (Fig. 7c, d), suggesting that administration of TAT-Cx43@LNPs at early ages can delay the neuropathological progression in APP/PS1 mice.

a 3.5-month-old WT and APP/PS1 mice were treated with TAT-Cx43@LNP or with TAT@LNP as control weekly for 8 weeks, before being subjected to histology analysis. b MitoSox Red staining assay was performed on acute brain slices to analyze hippocampal CA1 neuronal oxidative stress. Scale bar, 50 μm. MitoSox intensity was quantified and normalized to the WT control. N = 5 mice. Statistical test: two-sided t test. c Representative images of RTN3 and Aβ staining in the hippocampus. Scale bar, 10 μm. RTN3 + puncta number at different distances from the plaque was quantified. N = 26 (TAT) and 29 (TAT-Cx43) plaques. Statistical test: two-way ANOVA. d Representative images of Aβ and postsynaptic element labeling by Homer1. Scale bar: 10 μm. Homer1 + puncta intensity at different distances from the plaque was quantified. N = 10 (TAT) and 15 (TAT-Cx43) plaques. Statistical test: two-way ANOVA. e Representative images of IBA1 and Methoxy X04 staining. Scale bar, 50 μm. f Quantification of Aβ plaque number. N = 5 mice. Statistical test: two-sided t test. g Higher resolution images of IBA1 and Methoxy X04 staining. Scale bar, 10 μm. h Quantification of the percentage of compact plaque. N = 5 mice. Statistical test: two-sided t test. i For each plaque, the number of periplaque microglia was quantified and normalized to the plaque perimeter. N = 5 mice. Statistical test: two-sided t test. j 3D reconstruction image of the panel g displaying Aβ plaque and the fraction of Aβ plaque within the IBA1 + microglia domain. Scale bar, 10 μm. k The ratio between plaque within microglia and total plaque volume was quantified. N = 5 mice. Statistical test: two-sided t test. Dot plots show mean ± SEM, each data point, and p-value. Significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Source data are provided as a Source Data file.
In contrast to the treatment at the late stage of AD, early TAT-Cx43@LNPs exposure significantly reduced the number of Aβ plaques (41.2 ± 13.2% reduction, N = 5 mice) as shown by the Methoxy-X04 staining (Fig. 7e, f). Early TAT-Cx43@LNPs treatment also increased the compactness of Aβ plaques, similar to the late-stage treatment (Fig. 7g, h and Supplementary Fig. 7c). Alleviated Aβ pathology by the TAT-Cx43@LNPs was associated with the enhanced microglia-plaque interactions, as shown by the increased number of periplaque microglia, as well as the microglia-interacting fraction per plaque (Fig. 7i–k). In addition, TAT-Cx43@LNPs increased the ramification of microglia distal to plaques (Supplementary Fig. 7d). Our findings highlight the potential therapeutic value of suppressing Cx43 hemichannel activity for both the prevention and treatment of AD.
Discussion
This study develops a cell-targeted treatment strategy for AD, based on switching microglia to a neuroprotective phenotype by targeting microglial Cx43 hemichannels. We provide comprehensive evidence to support the role of microglial Cx43 hemichannels in the pathogenesis of AD in patients and in a mouse model of Aβ pathology. We found an increased presence of microglial Cx43 in both AD patient brains and in APP/PS1 mice, suggesting the relevance to human disease. We subsequently demonstrated that microglial Cx43 operates as a hemichannel and provided in vivo evidence that inhibition of microglial Cx43 hemichannels alleviates AD progression by inducing a neuroprotective microglial state. The neuroprotective microglia established a closer interaction with and promoted the compactness of Aβ plaques, as well as reduced the expression of neurotoxic factors. We further provided preclinical evidence that targeting Cx43 hemichannels using our formulation TAT-Cx43@LNPs represents a promising strategy for AD management through shifting reactive microglia towards a neuroprotective state, leading to (i) more compacted Aβ plaque morphology and reduced plaques number when treated at the early disease stage; (ii) reduced periplaque neurite dystrophy and synaptic loss; (iii) decreased neuronal oxidative stress; (iv) rescued myelin loss; (v) ameliorated hippocampal LTP deficit; and last but not least, (vi) alleviated the cognitive decline. Our discoveries highlight the potential of microglial Cx43 as a therapeutic target for AD therapy.
Although the role of microglial Cx43 in neuropathology has been suggested by in vitro studies27, there is a lack of in vivo evidence; similarly, the impact of Cx43 on microglia reactive state remained unexplored. In the healthy brain, microglia express low levels of Cx43, which, however, is increased by lipopolysaccharide (LPS) or proinflammatory factors69,70,71. Increased microglial Cx43 was also found in vivo around stab wounds72. The suggested gap junction function of microglia connexins was only based on in vitro observations and was never confirmed in vivo36. Treatment of primary cultured microglia with Aβ increases Cx43 hemichannel activity, triggering the release of glutamate and ATP, and inducing excitotoxicity27,73,74. These observations consolidated the notion of changes in Cx43 expression and function associated with reactive microgliosis. In this study, we revealed how Cx43 hemichannels modulate reactive microgliosis in the context of AD. We observed a significant increase in microglial Cx43 hemichannel activity, especially in the periplaque microglia. We provided evidence showing that conditional knockout of microglial Cx43 significantly reduces hemichannel activity, alters microglial reactivity, increases periplaque microglia population, and consequently alleviates neuropathology and cognitive impairments. Moreover, manipulating microglial Cx43 leads to marked transcriptomic changes in microglia in APP/PS1 mice that are instrumental in promoting neuroprotective reactive status. Further research is needed to reveal the molecular mechanism by which Cx43 hemichannels regulate gene expression.
To date, most of the AD drug development is focused on disease-modifying therapies aimed at Aβ5,75. The Aβ cascade hypothesis, however, is insufficient to explain many facets of AD progression, leading to a 98% failure rate of clinical trials3,4; hitherto, no disease-modifying therapy exists5. Microglia-targeting AD therapy has gained increasing attention as a growing body of evidence indicates that microglia engage in the pathological progression of AD76. However, previous attempts at suppressing reactive microgliosis using minocycline have proven ineffective in clinical trials77, suggesting the need for new strategies to modulate microglia state. Our study illustrates the role of microglial Cx43 hemichannels in regulating the microglia response to Aβ pathology. Moreover, genetic deletion or pharmacological inhibition of microglial Cx43 alleviates AD progression. In CNS, Cx43 is highly expressed by astrocytes, forming gap junctions that mediate cell-to-cell communication within the CNS. In our previous study, we demonstrated that Cx43 hemichannels in astrocytes disrupt the microenvironment homeostasis and contribute to AD progression24, while the deletion of astroglial Cx43 in APP/PS1 blocked hemichannels opening, leading to reduced neuronal damage in the hippocampus24. Notably, despite short intervention windows (inducible microglial Cx43 knockout: 8.5 ~ 9.5-month-old; TAT-Cx43@LNP: 8 ~ 9.5-month-old or 3.5 ~ 5.5-month-old), we achieved robust rescue of cognitive deficits and neuropathology. Together, these results highlight the potential of targeting Cx43 hemichannels as a preventive and disease-modifying strategy of AD, suggesting a promising avenue for therapeutic intervention.
However, there are two major obstacles to be circumvented in targeting Cx43 hemichannels for AD treatment. Firstly, given that appropriate Cx43 gap junctions and hemichannels are essential for the function of multiple organs, any therapeutic strategy targeting Cx43 needs to be carefully controlled to minimize side effects. Our previous studies have shown that an alkaloid compound, Boldine, that can cross the BBB, alleviates the neuropathology of AD by blocking Cx43 hemichannels78. Boldine has recently been under clinical trial for treating overactive bladder in women79. However, its off-target effects remain unknown. There are other Cx43-targeting agents in clinical trials or preclinical tests80; among them, mefloquine, a potent Cx channel blocker, combined with donepezil (THN201), is under preclinical testing for cognitive deficit linked to AD80. However, mefloquine has a higher efficiency in targeting Cx36 compared with Cx43. Several other Cx43 channel blockers were used ex vivo or in various animal models. CBX, for instance, is widely used to inhibit connexin hemichannels in cell cultures or in acute or organotypic brain slices. However, CBX is unspecific (and targets other protein channels such as Panx1 channel), potently inhibits gap junction channels, and could not cross BBB81. Gap26 and Gap27 are Cx43 mimetic peptides that target the extracellular loop of Cx4382, which not only block Cx43 hemichannels but also interfere with gap junctions83, as well as with other connexin channels82. D4 is a newly developed connexin channel blocker that showed high efficacy in suppressing reactive gliosis in an epilepsy model84, but it does not discriminate between Cx26, Cx30, Cx43, and Cx45 hemichannels, which may lead to significant side effects.
TAT-Cx43266-283 peptide specifically inhibits the Cx43 hemichannels in astrocytes62. Here, we demonstrated that TAT-Cx43266-283 peptide blocks Cx43 hemichannels in both microglia and astrocytes without measurable effects on astrocytic gap junctions. The safety profile of the TAT-Cx43266-283 peptide in our preclinical study also supports its clinical translation, showing that it does not interfere with the physiological function of vital organs expressing Cx43, such as the heart, liver, and kidney. Secondly, peptide drugs have a short half-life in serum due to proteolytic digestion65, an obstacle that needs to be addressed for the management of chronic diseases such as AD. To this end, we developed a lipid nanoparticle packaging system to formulate the TAT-Cx43@LNPs drug, which improves the bioavailability of TAT-Cx43266-283 peptide in the CNS, achieves long-term glial Cx43 hemichannels inhibition, and more importantly, not only prevents and rescues the Aβ-related neuropathology but also alleviates the cognitive decline. One limitation of our lipid nanoparticle packaging system is the lack of brain-specific targeting, which may lower the drug availability in the brain and increase the risk of side effects, which awaits further iteration to introduce the active brain targeting strategy85. While we provide solid evidence that the efficacy of TAT-Cx43@LNP in alleviating cognitive deficits and Aβ pathology in APP/PS1 mice is centrally mediated, peripheral contributions remain a potential adjunct mechanism, which requires future studies to follow up. Nonetheless, our data demonstrate that targeting Cx43 hemichannels by a lipid nanoparticle-based mimetic peptide delivery has promising clinical value for AD therapy and prevention.
Another limitation of our study is associated with the animal model. APP/PS1 mice were constructed to model severe Aβ pathology in familial AD with APP and PSEN1 mutation, and lacks several critical histopathological hallmarks of human disease, including tau pathology, neuronal death, etc.43,86, rendering it unable to determine whether Cx43 hemichannel inhibition would impact on these pathologies. Regarding neuronal death, however, in vitro evidence suggests that inhibition of microglial or astrocytic Cx43 hemichannel protects against Aβ-induced primary neuronal death27. In addition, aging is a critical factor contributing to AD. However, current AD mouse models developed pathological change at a relatively early life stage. APP/PS1 mice, for instance, develop Aβ deposition at 4 months old (this age is comparable to 20 ~ 30 years old in humans87), and cognitive decline before 10 months old (comparable to ~ 40 years old in humans87). In this aspect, APP/PS1 mice, along with other similar mouse models, are unsuitable for studying the interaction of aging and AD pathogenesis, or the efficacy of Cx43 hemichannel inhibition in aged AD conditions. Taken together, to further confirm the usefulness of Cx43 hemichannel inhibitors in AD, they need to be tested in other models, including patient-derived organoids, Tau pathology models, or inducible, aging-dependent, AD mouse models.
In conclusion, our study provides insight into the early cellular changes in AD pathology and introduces a cell-specific strategy with significant therapeutic potential. Future work is needed to fully realize the treatment potential of our strategies for patients with AD in clinical trials.
Methods
Study ethics and approval
This study complies with all relevant ethical regulations. Human post-mortem brain tissues were provided by the Human Brain Bank, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China, under the ethical approval of the Ethics Committee of the Chinese Academy of Medical Sciences (NO. 009-2014). Informed consent was obtained by participants. All animals-related procedures were performed under the ethical approval of the Sun Yat-Sen University Institutional Animal Care and Use Committee (NO. SYSU-IACUC-2024-000452).
Human samples
Human post-mortem brain tissues were provided by the Human Brain Bank, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China, under the ethical approval of the Ethics Committee of the Chinese Academy of Medical Sciences (NO. 009-2014). Informed consent was obtained by participants. For immunostaining, the paraffin-embedded sections were deparaffinized, treated with 3% H2O2 and processed for citric acid antigen retrieval. The sections were blocked with 2% BSA in TBST and incubated with primary antibody for 2 h at 37 °C and then overnight at 4 °C, followed by serial rinsing and incubation with fluorophore-conjugated secondary antibody. The detailed information of healthy controls and AD patients was listed in Supplementary Fig. 1b. Primary antibodies used are Rabbit-anti-Cx43 (1:200, Merck, C6219), Rabbit-anti-PANX1 (Thermo, 487900, 1:200), Goat-anti-IBA1 (Abcam, ab5076, 1:400), Rat-anti-GFAP (Abcam, ab279291, 1:400), Mouse-anti-pTAU (AT8, Thermo, MN1020, 1:1000), Rat-anti-ALDH1L1 (1:200, Oasis biofarm, OB-PRT023).
Animals
All animals used in the study were maintained in a PC2 pathogen-free animal facility in a 12 h/12 h light/dark cycle with a temperature of ~ 20 °C and ~ 50% humidity. All procedures were performed under the ethical approval of the Sun Yat-Sen University Institutional Animal Care and Use Committee (NO. SYSU-IACUC-2024-000452).
Wild type (WT) or APPswe/PS1dE9 (APP/PS1) mice with a C57BL/6j background were obtained from GemPharmatech Co., Ltd. Cx3cr1-CreERT mice, as previously reported, were obtained from the Jackson Lab (Strain # 21160). They were mated with Cx43-flox (Strain # 008039) mice and APP/PS1 mice to generate APP/PS1: Cx3cr1-CreERT: Cx43-floxed mice (APP/PS1:Cx43mgl-cKO). It is worth noting that APP/PS1 transgene and Cx3cr1-CreERT knockin/knockout are both located at chromosome 9. In this study, APP/PS1 heterozygote littermates were used as a control for behavioral tests, histological analysis, acute brain slice experiments, and RNA-sequencing experiments. To induce Cx43 knockout, 30 mg/mL tamoxifen in 1:9 ethanol/corn oil mixture was given to mice via gavage at 50 μL each day for 5 consecutive days. The induction started at 8.5-month-old, and the behavioral and histological at 9.5-month-old. For TAT-Cx43@LNP treatment, mice received TAT@LNP or TAT-CX43@LNP (15 mg/mL, 8 μL/g) injection once per week (i.p., equivalent to 120 μg LNPs/g, and 8.2 nmol peptides/g). Two experiment schemes were included: (1) treatment from 8-month-old to 9.5-month-old, to examine the rescue effect; (2) treatment from 3.5-month-old to 5.5-month-old, to examine the prevention effect.
Mice of both sexes were used for most experiments. The number of mice in different sex used in different experiments are listed below as (male: female): In the conditional knockout of microglial Cx43, for behavioral tests, wild type (8:13), Cx43mgl-cKO (5:7), APP/PS1 (5:9), APP/PS1:Cx43mgl-cKO (3:5); for acute brain slice and histology experiments, (2:3) for all experiment groups (wild type, APP/PS1, and APP/PS1:Cx43mgl-cKO); for RNA-seq experiment (1:2) for all animal groups (wild type, APP/PS1, and APP/PS1:Cx43mgl-cKO). In the TAT-Cx43@LNP treatment experiments at 9.5 months, for behavioral tests, WT TAT@LNP (4:5), WT TAT-CX43@LNP (4:5), APP/PS1 TAT@LNP (5:8), APP/PS1 TAT-CX43@LNP (6:7); for acute brain slice and histology experiments, (2:3) for all experiment groups, for electrophysiology experiment, (2:2) for all experiment groups. In the TAT-Cx43@LNP treatment experiments at 5 months, (2:3) for all experiment groups.
Behavioral tests
Before being subjected to behavioral tests, mice were handled for 3 consecutive days until they appeared calm on the handler’s hand without displaying stressful behaviors, such as jumping, biting, defecating, or urinating. Tests were conducted in the following sequence: open field test, novel object recognition, and Barnes’ maze test for the TAT-CX43@LNP experiment. Videotaping and data analysis were conducted using the VisuTrack software and hardware (Xinrun, Shanghai).
Open field test
was used to determine the anxiety trait and locomotor function. Mice were placed in the center of a 50 × 50 × 40 (height) cm3 white box and were allowed to explore for 20 min. The floor was divided into 5 × 5 squares, and the center 9 squares were considered as center zone.
Novel object recognition test
was used to determine the cognitive function. In the familiarization session, mice were placed in a 25 × 25 × 40 (height) cm3 white box with 2 rectangular plastic objects placed 8 cm away from the walls and allowed to explore for 5 min. The test session took place two hours later, where one of the rectangular objects was replaced by a cylinder (the novel object) before the mouse was re-introduced into the apparatus. The discrimination index calculated as (Timenew-Timeold) / (Timenew-Timeold) was used to determine the exploratory preference toward the novel object.
Barnes’ Maze test
was used to determine spatial memory and cognitive functions and performed according to a previous publication88 with minor adjustments. The platform was a 92 cm diameter round plastic board with 20 equally spaced 5 cm diameter holes at the border, with one hole that could be attached to the escape tunnel. The platform was held 80 cm above the ground. Three distal visual cues were placed surrounding the platform. The experiment room was well-lit at ~ 1000 lux. In the habituation phase (Day 1), the mouse was placed in the escape tunnel for 1 min and allowed to explore the platform for 5 min or until the mouse entered the tunnel. In the acquisition trial, mice were placed onto the platform with a transfer box and allowed to explore for 3 min or till the mice entered the tunnel. This procedure was conveyed twice daily with a 2 h interval, for a total of 4 days (Day 1–4). In the probe trial (Day 5), the escape tunnel was removed, and the mouse was allowed to explore the platform for 3 min.
Immunostaining
Mice were anesthetized by the i.p. injection of 20% urethane at 10 μL/g, followed by transcardial perfusion of PBS and 4% paraformaldehyde (PFA). The brains were then isolated and subjected to post-fixation with 4% PFA at 4 °C overnight, then immersed in 30% Sucrose for cryoprotection, before cryostat sectioning for 20 μm μm-thick slices. Brain slices were permeabilized with 0.5% Triton-X100 in PBS (0.5% PBST) for 1 h, blocked with 2% BSA in PBST for 1 h, and incubated with primary antibody overnight at 4 °C, followed by secondary antibody, counterstained with DAPI, and mounted for microscopic observation. Primary antibodies and dilution are listed below: Rabbit-anti-Cx43 (Merck, C6219, 1:200), Rabbit-anti-PANX1 (Thermo, 487900, 1:200), Rabbit-anti-Cx30 (Thermo, 712200, 1:200), Goat-anti-IBA1 (Abcam, ab5076, 1:400), Goat-anti-GFAP (Abcam, ab53554, 1:400), Rat-anti-GFAP (Abcam, ab279291, 1:400), Rabbit-anti-ARG1 (Thermo, PA529645, 1:200), Mouse-anti-Aβ 6E10 (Biolegend, 803001, 1:1000), Rat-anti-MBP (Millipore, MAB386, 1:200), Rabbit-anti-RTN3 (Millipore, ABN1723, 1:100), Lamp1 (Biolegend, 121602, 1:200), Rat-anti-Dectin-1(CLEC7A) (Invivogen, mabg-mdect-2, 1:200), Rabbit-anti-GRN (Abcam, ab187070, 1:200), Rabbit-anti-NeuN (Abcam, ab177487, 1:200), Rabbit-anti-cleaved caspase3 (CST, 9661, 1:200).
Aβ plaques were visualized either by anti-Aβ 6E10 antibody or Methoxy X04 staining or by autofluorescence at 405 nm89. For Methoxy X04 staining, sections were rinsed with ddH2O, and incubated with 34 μg/mL Methoxy-X04 (TOCRIS, 4920) diluted in 40% Ethanol for 15 min at room temperature, then rinsed twice with ddH2O and once with PBS, before subjecting to the blocking step.
Tunel labeling was performed using In Situ Cell Death Detection Kit, TMR red (Roche, 12156792910), according to the manufacturer’s protocol. The immunostaining procedures were performed after Tunel labeling.
For the APP/PS1:Cx43mgl-cKO and TAT-CX43@LNP experiment, mice were euthanized, and the brains were rapidly isolated, and separated into left and right halves. The left halves were used for acute brain slices experiments and the right halves were fixed by ice-cold PFA for immunostaining.
Images were captured using a VS200 slide scanner, FV3000 confocal microscope, or SpinSR spinning disk confocal microscope (Olympus). Super-resolution images were captured using a SpinSR spinning disk confocal microscope.
Microglia isolation and culture
Primary microglia were isolated and cultured as previously described90 with minor modifications. Briefly, postnatal day 7 mouse pups were subjected to hypothermia-induced anesthesia and euthanized by rapid decapitation with sharpened scissors. The brains were isolated, minced, and digested with 1 mg/mL papain for 1 h at 37 °C. Tissues were then triturated with 10 mL pipette, and the resulting cell suspension was passed through a 70 μm cell strainer and subjected to immunopanning with the rat-anti-mouse CD45 antibody (BD, 553076, 1:200) and the secondary goat-anti-rat antibody (Jackson ImmunoResearch, 112-005-003) to isolate microglia. The immunopanned microglia were trypsinized and subjected to culture in microglia growth medium supplemented with TGF-β2/IL-34/Cholesterol and 1% fetal calf serum.
Acute brain slice preparation and related experiments
The acute brain slices were prepared as previously described91. Briefly, mice were anesthetized by i.p. injection of 2.5% Avertin at 200 μL/10 g, and euthanized by rapid decapitation with sharpened scissors. Brains were rapidly isolated, stationed at the specimen holder, immersed in the ice-cold NMDG-based artificial cerebrospinal fluid (ACSF) with constant carbonation (95% O2, 5% CO2), and subjected to vibratome sectioning to obtain 280 μm slices. Slices were then recovered in NMDG-ACSF at 32 °C, followed by sequential Na+ spike-in addition. The slices were then transferred to the carbonated ACSF at room temperature and recovered for at least 1 h before experiments.
Dye uptake
The dye uptake experiment is a widely used assay to monitor the opening of hemichannels; this assay utilizes the poor selectivity of connexin hemichannels, which allow passage of small molecules with M.W. < 1.5 kDa64. Several dyes were used for dye uptake experiments, including EtBr, DAPI, carboxyfluorescein, etc., all of them cannot cross the lipid bilayer but permeate through connexin hemichannels. EtBr dye was selected over other dyes such as carboxyfluorescein and DAPI due to (i) carboxyfluorescein would diffuse across the cell body after entering through hemichannel, making it implausible to perform cell type specific dye uptake analysis; (ii) while both EtBr and DAPI would bind to DNA after entering the cell, which allow cell type specific analysis, the autofluorescence of Aβ plaque interferes with DAPI signal. Dye uptake experiment was performed on acute brain slices as previously described24. Briefly, acute brain slices were transferred onto 70 μm cell strainers put in the 6-well plate filled with the recording ACSF. Alternatively, CBX (200 µM), Gap26 (200 µM), and TAT-Cx43266-283 (50 μM) were added to the recording ACSF. After 15 min incubation, EtBr dye was added to the recording buffer at 5 µM, and incubated for 10 min. The slices were then rinsed 3 times with the recording ACSF (5 min each), fixed with 4% PFA for 1 h, and subjected to immunostaining.
For the dye uptake experiment in primary microglial culture, cells were rinsed twice with Hank’s balanced salt solution (HBSS) (Ca2+/Mg2+ free), and preincubated in HBSS with or without TAT-Cx43266-283 for 15 min. As a negative control, CaCl2 was added to the HBSS at 1.8 mM. EtBr was then added to 5 μM, and further incubated with cells for 10 min. Cells were then fixed with 4% PFA for 10 min and subjected to microscopic analysis.
Syncytial coupling - gap-FRAP
The gap-FRAP experiment was performed as previously described to analyze the connexin gap junction function92. Briefly, acute brain slices were incubated with Sulforhodamine 101 (SR101) in the recording ACSF at 32 °C for 30 min. After rinsing with recording ACSF, slices were transferred to a recording chamber on the FV3000 confocal microscope. The baseline fluorescence of SR101 in the target cell was monitored for 45 s (1 frame per 5 s), followed by a 15 s photobleaching with 80% laser power in a 20 × 20 μm area centered at the target cell. The SR101 fluorescence was subsequently monitored for another 9 min to quantify the fluorescence recovery due to the SR101 dye diffusion from adjacent astrocytes through gap junctions.
MitoSox staining
Acute brain slices were transferred onto 70 μm cell strainers put in the 6-well plate filled with the recording ACSF containing 5 μM MitoSox Red (Thermo, M36008), incubated for 20 min, then fixed with 4% PFA for 1 h, and subjected to immunostaining.
Supernatant collection and gliotransmitter detection
Acute brain slices were transferred to 1.5 mL microcentrifuge tubes containing 1 mL recording ACSF and incubated for 90 min. Supernatants were then collected for glutamate and D-serine measurement by commercial kits (Abcam, ab83389, ab241027), which were then normalized to the protein level of the slices measured by the BCA kit (Beyotime, P0012).
TNFα and IL1β measurement
Acute brain slices were incubated with ice-cold lysis buffer (in mM, 50 Tris-HCl, 100 NaCl, 2 EDTA, 1% Triton-X100, supplemented with protease inhibitor cocktail), sonicated for 10 s, and centrifugated at 12000 × g for 20 min. Supernatants were collected and stored at − 80 °C before measurement. TNFα and IL1β level was determined by ELISA kits (Abclonal, RK00027 and RK00006), which were then normalized to the protein level measured by the BCA kit (Beyotime, P0012).
RNA-sequencing
RNA-sequencing was performed on acute-isolated microglia from 9-month-old WT, APP/PS1, and APP/PS1:Cx43mgl-cKO mice. Three biological replicates were included for each group. Mice were anesthetized by the i.p. injection of 20% urethane at 10 μL/g and transcardially perfused with ice-cold HBSS, followed by rapid decapitation. Mice were sacrificed at 2 ~ 6 pm. The brains were isolated with the meninges removed, followed by dissection of the cortex and hippocampus, which were then minced and digested by 1% papain. Microglia were isolated from the resulting cell suspension via immunopanning with the rat-anti-mouse CD45 antibody (BD, 553076) and the secondary goat-anti-rat antibody (Jackson ImmunoResearch, 112-005-003), and directly lysed on the panning plate by Trizol (Thermo, 15596018), and stored at − 80 °C before RNA extraction. RNA was extracted with the Qiagen RNEasy Plus mini-isolation kit, and the RNA quality was examined by electrophoresis. The sequencing was performed using the DNBSEQ platform. The raw data were filtered with FastQC to remove reads of low quality, reads with adapter sequences and reads with high levels of N base. For these 9 samples, the clean reads ratio ranges from 98.65 to 99.96. The filtered reads were mapped to the reference Genome (Mus musculus, GCF_000001635.26_GRCm38.p6) using the HISAT-Bowtie2 pipeline. The average mapping ratio with reference genome is 98.74%, the average mapping ratio with gene is 81.80%; 18571 genes were identified. Differential expression analysis was performed on read counts using DESeq2. Pair-wise comparisons were performed between them. Differentially expressed genes were determined by a threshold of FC > 1.5, p < 0.05, and subjected to pathway enrichment analysis using DAVID.
TAT-Cx43@LNPs packaging and validation
TAT (YGRKKRRQRRR) and TAT-Cx43266-283 (YGRKKRRQRRR-AYFNGCSSPTAPLSPMSP) were synthesized by GL Biochem (Shanghai) Ltd. To package peptides into LNPs, 10 mg egg phosphatidylcholine (EPC) and 1 mg DOTAP were dissolved in 3 mL chloroform, followed by rotary evaporation at 40 °C, 150 rpm for 1 h to obtain a lipid thin layer. 30 mg Chitosan‑Methacryloyl (CMA) was dissolved in 6 mL 0.9% NaCl and 0.01 M HCl to obtain a homogenous mixture, followed by the addition of 10 mg/mL photo initiator I2959 and 5 mg/mL peptides. The solution was then used to hydrate the thin layer of lipid for 1 h. The resulting solution was filtered through 0.4 μm (3 times) and 0.2 μm (5 times) polycarbonate membranes (Whatman GE) to remove the multilamellar and unilamellar vesicles. The solution was then centrifuged at 300000 g for 1 h to pellet the uncrosslinked nano lipid gel, which was then resuspended in 8 mL PBS (pH 7.4). The solution was then subjected to UV illumination (365 nm, 1-2 mW/cm2) for 30 min to induce polymerization, resulting in the DOTAP-NLG. The size and zeta potential of NLGs was then analyzed using Malvern Zetasizer 2000. To evaluate the release of peptides from NLGs, 1 mL sample was added to a dialysis bag immersed in 14 mL PBS, and shaken at 100 rpm, 37 °C. The amount of peptide released into the PBS was then sampled and determined by spectrometry.
TAT-Cx43@LNPs in vivo treatment and analysis
TAT-Cx43266-283 peptide was labeled with Cy5.5 before being packaged into LNPs. TAT-CX43@LNP or TAT-CX43 alone are i.p. injected into mice at 8 μL/g. Biofluorescence analysis was performed at different time points to analyze Cy5.5 distribution. At different time points post-injection, mice were anesthetized, followed by cardiac puncture and serum preparation, and perfused with ice-cold PBS. The brain, heart, liver, and kidney were then collected and lysed with RIPA buffer and sonicated for 10 s. Lysates were then centrifuged at 12000 × g for 10 min, and the supernatants were collected. The Cy5.5 intensity was measured by a microplate reader, and the Cy5.5 intensity of the tissue lysate was normalized to the protein level measured by the BCA kit (Beyotime, P0012).
ECG recording
Mice were anesthetized by i.p. injection of Tribromoethanol at 400 mg/kg. The ECG recording was then performed using the small animal ECG monitor (Xinrun, RM6240E). The data digitalization and analysis were performed with the RM6240E 2.5 software.
Serum biochemistry analysis
Mice were anesthetized, and ~ 200 μL blood was collected via the retro-orbital sinus. The blood samples were then centrifuged at 12,000 × g for 10 min, and the serum samples collected from the supernatant were stored at − 80 °C until subjected to analysis using the Biostar V7 automated biochemistry analyzer (Seamaty, Ltd).
Hippocampal LTP recording
Mice were anesthetized by isoflurane inhalation and transcardially perfused with iced-cold sucrose ACSF. The brains were isolated and stationed onto the specimen holder, immersed in carbonated ice-cold sucrose ACSF, and subjected to vibratome sectioning to obtain 300 μm thick brain slices. Brain slices were then transferred to ACSF and recovered at 32 °C for 1 h, then at room temperature for 30 min before being subjected to LTP induction and recording. Slices were then transferred to the recording chamber with the ACSF flow rate of 1 mL/min. One monopolar electrode was positioned adjacent to the stratum radiatum of the CA1 region to stimulate the synaptic input and evoke the field excitatory post-synaptic potential from the Schaffer collateral-commissural-CA1 synapse. For recording, another electrode was placed in the CA1 apical dendritic layer. The signals were amplified by a differential amplifier (Axoclamp 700B, Axon, USA) and were digitized using a Digidata 1550B (Axon, USA). An input-output curve was plotted before LTP induction. To set the test stimulus intensity, a fEPSP of 50% of its maximal amplitude was determined. Biphasic constant-current pulses were used for stimulation. LTP was induced using a theta-burst stimulation (TBS) protocol, which consists of 50 bursts (consisting of 4 stimuli) at an inter-stimulus interval of 10 ms. The 50 bursts were applied for 30 s at 5 Hz (or at an inter-burst interval of 200 ms). The slopes of fEPSPs were monitored online. The baseline was recorded for 30 min. For baseline recording and testing at each time point, four 0.2 Hz biphasic constant-current pulses (0.1 ms/polarity) were used. fEPSPs were recorded every 5 min from 30 min before stimulation up to 240 min after stimulation across the CA1-CA3 Schaffer collaterals and normalized against t = 0. Data recording was performed using Clampex software, and data analysis was performed using Clampfit 10.8.
Quantification
Quantification of cell numbers
The number of IBA1+ and GFAP+ cells was manually counted and normalized to the area of the region of interest. Only cells with visible cell bodies were included. To quantify the distribution of cells around the plaques, the distance between cells and the plaque center was measured for each plaque, and only cells within 100 μm of the plaque center were included; the cell-plaque distances were then sorted into 5 μm bins, which were then normalized to the plaque number quantified and then to the area within the 5 μm bin. To quantify the periplaque microglia number, the number of IBA1 + cells within 50 μm of a target plaque was manually counted and normalized to the target plaque perimeter measured by Fiji.
Quantification of fluorescence intensity or positive area
Quantification of fluorescence intensity or positive area was performed using the Fiji software with or without macro scripts, where a threshold was set to distinguish the positive fluorescence signals, followed by the measurement of fluorescence intensity and positive area percentage within the region of interest. The same threshold was set for different experiment groups in the same batch. For images with high or uneven backgrounds, a subtract background function was performed with the rolling ball radius set at 50 pixels before quantification. To measure the fluorescence signal at different distances to the plaque, a serial of concentric circles with 5 μm or 1 μm escalating radius was drawn centering at the plaque center. The intensity or positive area was measured for each circle, and subtracted to that of the smaller circle. To measure the number of RTN3 and Homer1 puncta at different distances from the plaque, a Sholl analysis was performed to quantify the intersection between signals and the concentric circles at different distances.
Signal masking
Masking of Cx43, Cx30, and Panx1 signals by IBA1 and GFAP was performed using the 3D counter function of the Fiji software. Masking of Methoxy X04 signal by IBA1 was performed by Imaris 9.0. The ratio of masked Methoxy X04 volume and the total Methoxy X04 volume was quantified, or plotted as XY plot to analyze the correlation. Microglia morphology was evaluated using the Sholl analysis plugin in Fiji software.
Statistical analysis
Statistical significance between groups was determined with GraphPad Prism software. The unpaired t test was used to determine the difference between the two groups. One-way analysis of variance (ANOVA) was used to determine the difference among three and four groups. Two-way ANOVA was used to determine the difference between group variables in the curves. The Pearson correlation test was used to determine the correlation between variables. Data distribution was assumed to be normal, but this was not formally tested. A probability of p < 0.05 was considered statistically significant. All significant statistical results are indicated within the figures with the following conventions: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent ± standard deviation. No statistical methods were used to predetermine sample sizes. The sample size per group was determined from previous publications using a similar methodology. Control mice and their littermate mutant mice were collected from multiple litters and randomly selected for each experiment. Investigators were blinded to group allocation during data analysis. All experiments were performed at least three times, and the findings were replicated in individual mice and cell cultures in each experiment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data have been deposited in the NCBI GEO database under the accession number GSE272781. Source data are provided in this paper.
References
Scheltens, P. et al. Alzheimer’s disease. Lancet 397, 1577–1590 (2021).
Sheppard, O. & Coleman, M. in Alzheimer’s Disease: Drug Discovery (ed Huang X.) (2020).
Lannfelt, L., Relkin, N. R. & Siemers, E. R. Amyloid-β-directed immunotherapy for Alzheimer’s disease. J. Intern. Med. 275, 284–295 (2014).
Small, S. A. & Duff, K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron 60, 534–542 (2008).
Long, J. M. & Holtzman, D. M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179, 312–339 (2019).
DeTure, M. A. & Dickson, D. W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14, 32 (2019).
Bilbo, S. & Stevens, B. Microglia: The brain’s first responders.Cerebrum https://pmc.ncbi.nlm.nih.gov/articles/PMC6132046/ (2017).
Hanzel, C. E. et al. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 35, 2249–2262 (2014).
Dani, M. et al. Tau aggregation correlates with amyloid deposition in both mild cognitive impairment and Alzheimer’s disease subjects. J. Alzheimer’s Dis.70, 455–465 (2019).
Hamelin, L. et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264 (2016).
Femminella, G. D. et al. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 92, e1331–e1343 (2019).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290 (2017).
Hansen, D. V., Hanson, J. E. & Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472 (2018).
Deczkowska, A. et al. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 173, 1073–1081 (2018).
Zeng, H. et al. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat. Neurosci. 26, 430–446 (2023).
Leng, F. & Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here?. Nat. Rev. Neurol. 17, 157–172 (2021).
Bartels, T., De Schepper, S. & Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 370, 66–69 (2020).
Sebastian Monasor L., et al. Fibrillar Abeta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 9, https://doi.org/10.7554/elife.54083 (2020).
Giaume, C. & Naus, C. C. Connexins, gap junctions, and glia. 2, 133–142 (2013).
Koulakoff, A., Mei, X., Orellana, J. A., Saez, J. C. & Giaume, C. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Biophys. Acta 1818, 2048–2057 (2012).
Kajiwara, Y. et al. GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 6, 144 (2018).
Nagy, J. I., Li, W., Hertzberg, E. L. & Marotta, C. A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 717, 173–178 (1995).
Mei, X., Ezan, P., Giaume, C. & Koulakoff, A. Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 171, 92–105 (2010).
Yi, C. et al. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 23, 1691–1701 (2016).
Bai, B. et al. Proteomic landscape of Alzheimer’s Disease: novel insights into pathogenesis and biomarker discovery. Mol. Neurodegener. 16, 55 (2021).
Pechlivanidou, M. et al. Glial gap junction pathology in the spinal cord of the 5xFAD mouse model of early-onset Alzheimer’s disease. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms232415597 (2022).
Orellana, J. A. et al. Amyloid beta-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 31, 4962–4977 (2011).
Srinivasan, K. et al. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 31, 107843 (2020).
Johnson, E. C. B. et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780 (2020).
Jack, C. R. Jr. et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562 (2018).
Gajardo-Gomez, R., Labra, V. C. & Orellana, J. A. Connexins and pannexins: New insights into microglial functions and dysfunctions. Front. Mol. Neurosci. 9, 86 (2016).
Jackson, H. M., Soto, I., Graham, L. C., Carter, G. W. & Howell, G. R. Clustering of transcriptional profiles identifies changes to insulin signaling as an early event in a mouse model of Alzheimer’s disease. BMC Genom. 14, 831 (2013).
Minkeviciene, R. et al. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J. Neurochem. 105, 584–594 (2008).
Kinnunen, K. M. et al. Presymptomatic atrophy in autosomal dominant Alzheimer’s disease: A serial magnetic resonance imaging study. Alzheimers Dement 14, 43–53 (2018).
Lisman, J. et al. Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nat. Neurosci. 20, 1434–1447 (2017).
Wasseff, S. K. & Scherer, S. S. Activated microglia do not form functional gap junctions in vivo. J. Neuroimmunol. 269, 90–93 (2014).
Wang, N. et al. Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res. Cardiol. 107, 304 (2012).
Adlard, P. A. & Vickers, J. C. Morphologically distinct plaque types differentially affect dendritic structure and organisation in the early and late stages of Alzheimer’s disease. Acta Neuropathol. 103, 377–383 (2002).
Yuan, P. et al. TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739 (2016).
Spangenberg, E. et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10, 3758 (2019).
Huang, H. et al. Accumulation of neutral lipids in dystrophic neurites surrounding amyloid plaques in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1870, 167086 (2024).
Sharoar, M. G., Hu, X., Ma, X. M., Zhu, X. & Yan, R. Sequential formation of different layers of dystrophic neurites in Alzheimer’s brains. Mol. Psychiatry 24, 1369–1382 (2019).
APPswe/PSEN1dE9 (C57BL6), Retrieved Jan 1, 2024, from (https://www.alzforum.org/research-models/appswepsen1de9-c57bl6.). Alzforum Foundation (2019).
Pi, G. et al. Posterior basolateral amygdala to ventral hippocampal CA1 drives approach behaviour to exert an anxiolytic effect. Nat. Commun. 11, 183 (2020).
Kosel, F., Pelley, J. M. S. & Franklin, T. B. Behavioural and psychological symptoms of dementia in mouse models of Alzheimer’s disease-related pathology. Neurosci. Biobehav. Rev. 112, 634–647 (2020).
Wendt, S. et al. Simultaneous imaging of redox states in dystrophic neurites and microglia at Aβ plaques indicate lysosome accumulation not microglia correlate with increased oxidative stress. Redox Biol. 56, 102448 (2022).
Li, W. et al. Connexin 43 hemichannel as a novel mediator of sterile and infectious inflammatory diseases. Sci. Rep. 8, 166 (2018).
Roger, E. et al. Connexin-43 hemichannels orchestrate NOD-like receptor protein-3 (NLRP3) inflammasome activation and sterile inflammation in tubular injury. Cell Commun. Signal.21, 263 (2023).
Kobayashi, K. et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 4, e525 (2013).
Liu, S. et al. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J. Immunol. 188, 1098–1107 (2012).
El Khoury, J. et al. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382, 716–719 (1996).
Lau, S. F. et al. The VCAM1-ApoE pathway directs microglial chemotaxis and alleviates Alzheimer’s disease pathology. Nat. Aging 3, 1219–1236 (2023).
Minami, S. S. et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 20, 1157–1164 (2014).
Fairley, L. H. et al. Mitochondrial control of microglial phagocytosis by the translocator protein and hexokinase 2 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 120, e2209177120 (2023).
Zhang, H. et al. TSPO deficiency accelerates amyloid pathology and neuroinflammation by impairing microglial phagocytosis. Neurobiol. Aging 106, 292–303 (2021).
Zhao, X. et al. beta-amyloid binds to microglia Dectin-1 to induce inflammatory response in the pathogenesis of Alzheimer’s disease. Int. J. Biol. Sci. 19, 3249–3265 (2023).
Toral-Rios, D. et al. Cholesterol 25-hydroxylase mediates neuroinflammation and neurodegeneration in a mouse model of tauopathy. J. Exp. Med. 221, https://doi.org/10.1084/jem.20232000 (2024).
Haure-Mirande, J. V. et al. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol. 134, 769–788 (2017).
Abudara, V. et al. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia 63, 795–811 (2015).
Meunier, C. et al. Contribution of astroglial Cx43 hemichannels to the modulation of glutamatergic currents by D-serine in the mouse prefrontal cortex. J. Neurosci. 37, 9064–9075 (2017).
Gangoso, E., Thirant, C., Chneiweiss, H., Medina, J. M. & Tabernero, A. A cell-penetrating peptide based on the interaction between c-Src and connexin43 reverses glioma stem cell phenotype. Cell Death Dis. 5, e1023 (2014).
Gangoso, E. et al. A c-Src inhibitor peptide based on connexin43 exerts neuroprotective effects through the inhibition of glial hemichannel activity. Front. Mol. Neurosci. 10, 418 (2017).
Abudara, V. et al. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell. Neurosci. 8, 306 (2014).
Giaume, C., Naus, C. C., Saez, J. C. & Leybaert, L. Glial connexins and pannexins in the healthy and diseased brain. Physiol. Rev. 101, 93–145 (2021).
Mathur, D., Singh, S., Mehta, A., Agrawal, P. & Raghava, G. P. S. In silico approaches for predicting the half-life of natural and modified peptides in blood. PloS ONE 13, e0196829 (2018).
Cao, Y. et al. Designing siRNA/chitosan-methacrylate complex nanolipogel for prolonged gene silencing effects. Sci. Rep. 12, 3527 (2022).
Stalmans, S. et al. Cell-penetrating peptides selectively cross the blood-brain barrier in vivo. PloS one 10, e0139652 (2015).
Beauchamp, P. et al. Electrical propagation in synthetic ventricular myocyte strands from germline connexin43 knockout mice. Circ. Res. 95, 170–178 (2004).
Garg, S., Md Syed, M. & Kielian, T. Staphylococcus aureus-derived peptidoglycan induces Cx43 expression and functional gap junction intercellular communication in microglia. J. Neurochem. 95, 475–483 (2005).
Shaikh, S. B., Uy, B., Perera, A. & Nicholson, L. F. AGEs-RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem. Int. 60, 640–651 (2012).
Ma, Y. et al. Basal CD38/cyclic ADP-ribose-dependent signaling mediates ATP release and survival of microglia by modulating connexin 43 hemichannels. Glia 62, 943–955 (2014).
Eugenin, E. A. et al. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proc. Natl. Acad. Sci. USA 98, 4190–4195 (2001).
Barger, S. W. & Basile, A. S. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J. Neurochem. 76, 846–854 (2001).
Wu, S. Z. et al. Induction of serine racemase expression and D-serine release from microglia by amyloid beta-peptide. J. Neuroinflamm. 1, 2 (2004).
Cummings, J., Lee, G., Ritter, A., Sabbagh, M. & Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement 6, e12050 (2020).
De Strooper, B. & Karran, E. The cellular phase of Alzheimer’s disease. Cell 164, 603–615 (2016).
Howard, R. et al. Minocycline at 2 different dosages vs placebo for patients with mild Alzheimer disease: A randomized clinical trial. JAMA Neurol. 77, 164–174 (2020).
Yi, C. et al. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 65, 1607–1625 (2017).
National Library of Medicine. Effectiveness of oral boldo in women with overactive bladder.https://clinicaltrials.gov/study/NCT06162065 (2025).
Laird, D. W. & Lampe, P. D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 17, 905–921 (2018).
Su, Y., Verkhratsky, A. & Yi, C. Targeting connexins: Possible game changer in managing neuropathic pain?Trends Mol. Med.7, 642–659 (2024).
Evans, W. H. & Leybaert, L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun. Adhes. 14, 265–273 (2007).
Giaume, C., Leybaert, L., Naus, C. C. & Saez, J. C. Connexin and pannexin hemichannels in brain glial cells: properties, pharmacology, and roles. Front. Pharmacol. 4, 88 (2013).
Guo, A. et al. Inhibition of connexin hemichannels alleviates neuroinflammation and hyperexcitability in temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA 119, e2213162119 (2022).
Sela, M. et al. Brain-targeted liposomes loaded with monoclonal antibodies reduce alpha-synuclein aggregation and improve behavioral symptoms in Parkinson’s disease. Adv. Mater.35, e2304654 (2023).
Qian, Z., Li, Y. & Ye, K. Advancements and challenges in mouse models of Alzheimer’s disease. Trends Mol. Med. 30, 1152–1164 (2024).
Flurkey, K. M., Currer, J.& Harrison, D. E. in The Mouse in Biomedical Research (Second Edition) (Academic Press, 2007).
Yi, C. et al. Inactive variants of death receptor p75(NTR) reduce Alzheimer’s neuropathology by interfering with APP internalization. EMBO J. 40, e104450 (2021).
Fu, H. et al. Aβ-Aggregation-generated blue autofluorescence illuminates senile plaques as well as complex blood and vascular pathologies in Alzheimer’s disease. Neurosci. Bull. 40, 1115–1126 (2024).
Collins H. Y., Bohlen C. J. Isolation and culture of rodent microglia to promote a dynamic ramified morphology in serum-free medium. J. Vis. Exp. (2018).
Ting J. T., et al. Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method. J. Vis. Exp. 133, https://doi.org/10.3791/57122 (2018).
Yi, C., Teillon, J., Koulakoff, A., Berry, H. & Giaume, C. Monitoring gap junctional communication in astrocytes from acute adult mouse brain slices using the gap-FRAP technique. J. Neurosci. Methods 303, 103–113 (2018).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC 32170980 to C.Y.; 32271034 to J.N.; 32300791 to Y.S.), Chongqing Natural Science Fund for Distinguished Young Scholars (CSTB 2023NSCQ-JQX0030 to J.N.), National Key Research and Development Program of China (2021ZD0201703 to J.N.), Guangdong Basic and Applied Basic Research Foundation (2022B1515020012 to C.Y.; 2023A1515010817 to Y.S.), Science and Technology Planning Project of Guangdong Province (2023B1212060018 to C.Y.; 2021A1515110268 to Y.S.), Shenzhen Medical Research Fund (A2303014 to Y.S.), and Shenzhen Fundamental Research Program (RCJC20231211090018040 and ZDSYS20220606100801003 to C.Y.; JCYJ20210324121214039 and RCBS20210706092411028 to Y.S.; JCYJ20220530144816038 and RCBS20221008093118042 to Q.W.; JCYJ20240813150421029 to X.H.), Chinese Natural Science Foundation (Institute of brain and intelligence, CNSF 31921003 to J.N.). A.V. was supported by 2023 Key Support Project of the Liaoning Provincial Department of Science and Technology to Support the High-Quality Development of China Medical University ([2023]61-7), by Ciberned (CB06/05/00 76), by Spanish MICINN grant (PID2022-143020OB-I00), by Basque Government grant (IT1551-22), by Slovenian Research Agency grant J4-60077, by the Science and Technology Planning Project of Guangdong Province (No. 2021B1212040006), and by the Sanming Project of Medicine in Shenzhen (No. SZSM202411023; SZSM202411013. Human tissue was provided by the National Human Brain Bank for Development and Function, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. This study was supported by the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Neuroscience Center, and the China Human Brain Banking Consortium, by grants from STI2030-Major Project #2021ZD0201100 Task 1 #2021ZD0201101. We thank Professor Arantxa Tabernero for providing expert advice on the TAT-Cx43266-283 peptide. We thank Bioimaging Core of Shenzhen Bay Laboratory for providing imaging support. We also would like to acknowledge Bioimaging Core engineer Shixian Huang for assistance with the SpinSR spinning disk confocal microscopy.
Author information
Authors and Affiliations
Contributions
C.Y. and J.N. conceptualized the study with input from A.V., Y.S., Y.T., and H.C. C.Y., J.N., Z.Z., and A.V. supervised the study and ensured the coordination and resource support of the project. Y.S. performed most of the experimental work and analysis. H.L. assisted the acute brain slice experiments and performed the gap-FRAP experiments. Q.W. performed the histological analysis of human tissue samples and provided assistance with bioinformatics analysis, M.Z., X.H., and C.W. provide assistance with animal breeding, ECG measurement, and behavioral tests. W.Z., S.T., and X.Z. Synthesized lipid nanoparticles and performed related analysis with expertise and technical guidance from Z.Z. Y.T. and H.C. provided equipment and input for electrophysiology analysis. Y.S., C.Y., J.N., and A.V. wrote the manuscript. Y. T. and H.C. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Juan C Saez and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, Y., Li, H., Zhang, W. et al. Connexin43 hemichannel blockade turns microglia neuroprotective and mitigates cognitive deficits in a mouse model of amyloidosis. Nat Commun 16, 5621 (2025). https://doi.org/10.1038/s41467-025-60746-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60746-w
This article is cited by
-
Targeting Microglial Connexin43 Hemichannels: A Novel Therapeutic Avenue for Alzheimer’s Disease
Neuroscience Bulletin (2025)
