
Abstract
Sodium (Na)-air batteries show significant potential as alternatives to lithium-air batteries due to their high theoretical energy density and the abundant availability of sodium reserves. Nevertheless, the formation of complex products, specifically NaO2, Na2O2, Na2CO3·xH2O, during the multi-step reactions inevitably raises reconciled potential incompatibility that causes low efficiency and large overpotential. Here, we introduce a cascade electrocatalysis strategy that involves switchable metal and oxygen redox chemistry through electrochemical potential tuning. Leveraging the lithium ion spatial pinning effect, sodium ions trigger in the Na[Li1/3Ru2/3]O2 electrode system to toggle the geometric state at a low electrochemical potential and oscillate among different catalytic states to achieve sequential conversion of complicated multi-step intermediates. The Na[Li1/3Ru2/3]O2 catalyst effectively compartmentalizes the threshold potential that circumvents deactivating or competing pathways while coupling different catalytic cycles. As a result, the sodium-air battery employing this catalyst exhibits long-term reversibility over 1000 cycles with a decent catalysis efficiency exceeding 99%. Our results demonstrate that the cascade electrocatalysis strategy contributes to the design of integrated sodium-air batteries with long-term cycling stability.
Similar content being viewed by others
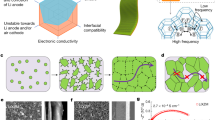


Introduction
Na-air batteries have garnered considerable attention as a promising and affordable energy storage solution owing to the abundance of sodium resources and high theoretical energy density1. However, the inadequate cycling stability of Na-air batteries, particularly on the electrode side, hinders their widespread implementation. Specifically, the reduction of O2 involves a multi-electron process, yielding multiple products such as NaO2, Na2O2, and Na2CO3·xH2O. While most electrode candidates exhibit selective catalysis for NaO2 conversion, they struggle to efficiently convert all products due to decomposing Na2O2 and Na2CO3·xH2O challenges, which consequently limit the cycling reversibility of a Na-air batteries2,3,4. To address this limitation, an ideal catalyst must moderate binding strength with multiple intermediates and promote their adsorption-desorption efficiency. Transition metal oxides (TMO), exemplified by RuO25, have recently emerged as promising catalysts in air batteries due to their abundant d orbitals and diverse valence distributions6,7. Nevertheless, TMO typically provides a single active center due to the fixed crystal structures, which is too limited for coupling electrons with adsorbates8,9,10. To overcome this challenge, designing two (or more) different reactive states of TMO is plausible to equilibrate the adsorption coverage of products and achieve high cycling stability in Na-air battery systems.
Cascade catalysis is an effective methodology for streamlining the multi-step conversion of complex products within a single reaction process through applying external stimuli11,12,13. Recently, with the light trigger, the alternatively changed TM-O bond has been proved to promote the oxygen evolution reactions (OER) efficiency by continuously regulating O-H bond cleavage and O-O bond formation11,12,13. Applying this cascade catalytic strategy to Na-air batteries requires two key design principles: dynamic active sites during cycling and alignment of electrochemical potential with redox components. Given the diversity of species generated during charging, active sites must adapt to the evolving redox process at varying voltages14. One approach is utilizing Na-containing layered oxides as catalysts, where Na ion extraction is voltage-dependent. From the perspective of d-band theory3, the d-electron-pair wave function properties of 4d/5d transition metal elements with a large spatial extent may offer opportunities for modulating the electronic structure to boost the catalytic activity. Then, Na⁺ electrochemical intercalation can induce various valence states in TM ions, allowing for tunable electronic and redox properties. Moreover, aligning active sites with the distinct electrochemical potentials of NaO₂, Na₂O₂, and their hydrates could maximize energy efficiency while minimizing parasitic reactions15,16,17. Although this strategy promises to enable the efficient multi-step reaction sequences, implementing this strategy and tuning electrochemical potentials in a Na-air battery remains poorly understood18.
In this study, we have synthesized a layered TMO catalyst, Na[Li1/3Ru2/3]O2, to facilitate the cascade conversion of mixed intermediates within Na-air batteries. The hybridization of the Ru-O bond in Na[Li1/3Ru2/3]O2, as illustrated in Fig. 1a, can be controlled by regulating alkali ions in the Ru layer. Accompanied by the variation of potential, the oriented orbital hybridizes in two axes between TM and O, resulting in varying degrees of the metal d band and oxygen p band broadening. Consequently, the catalyst with a different extent of d-p band broadening covers the intermediate and promotes the adsorption-desorption efficiency. This modification results in enhanced electron transfer efficiency at high electrochemical potentials and initiates the formation of dynamic TM active centers for sequential catalysis. The unit cell in the coupled integrated catalysis undergoes reversible geometric conversion of RuO6 with Li+ as spatial localization, promising the structural stability of the material. This modification results in enhanced electron transfer efficiency at high electrochemical potentials and initiates the formation of dynamic TM active centers for sequential catalysis. Consequently, the Na-air cell using Na[Li1/3Ru2/3]O2 as electrode exhibits an energy conversion efficiency exceeding 99%, coupled with a high discharge capacity of 15,000 mAh g−1. Furthermore, it demonstrates long-term reversibility over 1000 cycles with a minimal overpotential of 0.75 V. Compared with RuO2, Na[Li1/3Ru2/3]O2 showcases switchable active centers with good reversibility in electrochemical reactions. Notably, the catalysts present highly distinguishable threshold potentials for multiproduct decomposition in Na-air batteries, favoring the avoidance of deactivation or competing pathways. The sequential arrangement of potential windows, influenced by the dynamic transition metal valence state, could provide a practical framework for improving the performance of metal-air batteries.

a, b Ion-insertion catalysts may be more likely to induce cascade catalytic effects than conventional catalysts. c, d The cascade catalytic strategy to Na-air batteries requires two key design principles: dynamic active sites during cycling and alignment of electrochemical potential with redox components (Reaction 1–3). e Schematic illustration of electrochemical reactions of Na-air batteries at electrode–electrolyte interfaces, cascade catalytic strategy promises to enable the efficient multi-step reaction sequences. f The kinetic barrier of a reaction (ΔG) can be reduced owing to cascade catalysis. g, h Cascade catalysis changes the thermodynamic electrochemical driving force of the reaction (η = EF−Eeq, where EF is the Fermi level of the electrode and Eeq is the Fermi level of the reactant): pDOS, partial density of states.
Results
Sequential catalysis for improved rechargeability
The layered microparticles Na[Li1/3Ru2/3]O2 with a disordered TM/Li-intralayer arrangement were prepared by one-step solid-state reactions19 (Supplementary Fig. 1). The X-ray diffraction (XRD) pattern of Na[Li1/3Ru2/3]O2 and Rietveld refinement show that the adopts an O3-type layered structure (Supplementary Fig. 2 and Table S1). Na ions are situated within the AM (Alkali metal) layer, while a disordered Li/TM arrangement exists in the TM (transition metal) layer, delivering a Na−O−Li configuration20. Inductively coupled plasma optical emission spectroscopy (ICP-OES) affirms that the chemical composition of Na[Li1/3Ru2/3]O2 is consistent with the expected stoichiometry ratio (Supplementary Table 2).
A quasisolid-state polymer electrolyte (QPE) that consists of PVDF−HFP−10% SiO2−NaClO4−tetraethylene glycol dimethyl ether (TEGDME) as an electrolyte (Supplementary Fig. 3 and Fig. 4), both Na[Li1/3Ru2/3]O2 and commercial RuO2 were then utilized as catalysts for solid-state Na-air batteries (SSNAB) to evaluate the catalytic performances. In galvanostatic tests, Na[Li1/3Ru2/3]O2 indicates a low discharge-charge overpotential of 0.75 V while demonstrating a favorable discharge capacity of 15,000 mAh g−1 (Fig. 2a). As a reference, the commercial RuO2 catalyst has a high OER overpotential (0.97 V) with a poor discharge capacity (8500 mAh g−1). In contrast, the Na[Li1/3Ru2/3]O2 catalyst exhibits a considerably higher energy efficiency (over 99%) compared to the RuO2 catalyst (60%). Furthermore, both the two catalysts show similar discharge profiles with a single plateau at 2.3 V. Whereas, there are three different charging plateaus with Na[Li1/3Ru2/3]O2 catalyst at (i) C2.5 V, (ii) C3.5 V, and (iii) C4.5 V, respectively. It is known that plateau (i) originates from superoxide decomposition, and plateaus (the other two) can be ascribed to the decomposition of the peroxide phase and the by-product, respectively21. These results verify that the high Na[Li1/3Ru2/3]O2 activity could be attributed to the separation of reaction potentials. To further study the potential-dependent oxidation kinetics, the cyclic voltammetry (CV) of the two catalysts was collected (Fig. 2b). Compared with the multiple redox peaks of the RuO2 plots, Na[Li1/3Ru2/3]O2 catalyst also shows a multistage redox process. The larger redox current and more negligible polarization indicate that Na[Li1/3Ru2/3]O2 catalyst can tune redox reaction potential, which is essential for mediating the reaction kinetics22,23.

a Galvanostatic curves of SSNAB based on commercial RuO2 and Na[Li1/3Ru2/3]O2 sample at a current density of 100 mA g−1. b Contrast CV curves with the aforementioned two catalysts between 1.5 and 4.5 V, scan rate: 0.1 mV s−1. c The cycling performance with a 500 mAh g−1 capacity limitation. d The comparison with other notable published studies on the cycling performance of sodium-oxygen battery systems, the source of the literature data shown in this figure can be found in Supplementary Table 3. e, f In situ DEMS analysis of the gas consumption (e) and evolution (f) during SSNAB operation with RuO2 and Na[Li1/3Ru2/3]O2 electrodes. g, h SEM image of discharged electrode morphologies obtained after a full discharge during the galvanostatic test conducted at a current density of 100 mA g−1, as shown in Fig. 2a. i, j Synchrotron x-ray tomography reconstruction of discharged electrode utilizing volume rendering.
The difference in cycling stability between RuO2 and Na[Li1/3Ru2/3]O2 corroborates the cascade catalyst effect in the SSNAB. At a consistent capacity of 500 mAh g−1, the Na[Li1/3Ru2/3]O2 electrodes maintain stable discharge and charge voltages without significant degradation even after 1000 cycles, while the reaction voltage of RuO2 decreased after 350 cycles due to intense OER polarization (Fig. 2c). In addition, Na[Li₁/₃Ru₂/₃]O₂ and RuO₂ electrodes were further tested for stability at a high current density of 500 mA g⁻¹, with a capacity limit of 1000 mAh g−1. Notably, the Na[Li₁/₃Ru₂/₃]O₂ exhibited a stable specific capacity and maintained a discharge voltage above 1.5 V after 550 cycles. In contrast, RuO₂ showed significant degradation after only 120 cycles (Supplementary Fig. 5). The satisfied cycle stability and energy efficiency of Na[Li1/3Ru2/3]O2 electrodes show substantial advantages over previous reports of sodium-oxygen battery systems4,23,24,25,26,27,28,29,30,31,32,33,34,35,36 (Fig. 2d and Supplementary Table 3). These observations demonstrate that Na[Li1/3Ru2/3]O2 can be a suitable catalyst for coupling multiple consecutive catalytic reactions (Supplementary Figs. 6 and 7).
In ambient conditions, the pouch-type battery measures 10 × 10 cm² and achieves a capacity of 650 mAh at 4.5 A. Its impressive cycling stability over 40 cycles highlights the potential of Na[Li₁/₃Ru₂/₃]O₂ for large-scale Na−air battery applications (Supplementary Fig. 8). The flexible pouch-type solid-state Na-air battery can constantly power a micro-robot, indicating the great prospect of the Na[Li1/3Ru2/3]O2 in promoting the practical application of Na-air batteries (Supplementary Fig. 9). These results suggest the importance of the Na[Li1/3Ru2/3]O2 in constructing a highly safe and stable. To investigate whether the redox potential of the catalyst aligns with the voltage window of Na-air batteries, charge and discharge tests of Na[Li1/3Ru2/3]O2 and RuO₂ as electrodes were conducted in an Ar atmosphere. The charge-discharge curves of Na[Li₁/₃Ru₂/₃]O₂ revealed significant oxygen redox processes. In contrast, RuO₂ lacked effective redox activity and alignment with the sodium-oxygen battery voltage window, limiting its effectiveness as a cascade catalyst for energy efficiency optimization. (Supplementary Fig. 10).
To further understand the discharge-charge mechanism, the gas evolution was probed with in situ DEMS analysis (Fig. 2e, f). During discharge-charge processes, the e−/O2 > 2 ratios of Na[Li1/3Ru2/3]O2 can be obtained, which suggests that more than one electron is transferred or multiple steps are involved in the complex electrode reaction37. Scanning electron microscopy (SEM) was employed to understand the specific morphology of the discharge products. (Fig. 2g, h). The two distinct forms of the deposited product (nanorod-like products for RuO2 and thin-layer ones for Na[Li1/3Ru2/3]O2) indicate a strong correlation between electrode surfaces and the thermodynamics/kinetics of product deposition. Using the high spatial resolution and high contrast of synchrotron X-ray computed tomography, the active components in the electrode can be separated from the reaction products, enabling the examination of the product formation and decomposition processes38. The reconstructed 3D volume rendering images of coexisting products with Na[Li1/3Ru2/3]O2 (Displayed in purple) precipitate massively within the electrode with a homogeneous distribution. Additionally, the green 3D visualization image roughly retrieved the scattered product distribution in the RuO2 electrode (Fig. 2i, j). The 3D analysis measures the volume variation of the deposited products between the electrodes above (Supplementary Fig. 11), indicating that electrochemical ion insertion may lead to uniform product deposition by expanding the electrochemically active surface area beyond the electrode-electrolyte interface in the host material39.
Spatiotemporal control of complicated products
SEM and Time-of-flight secondary ion mass spectrometry (TOF-SIMS) further analyzed the morphological evolution of the discharge/charge products to explore the underlying catalysis process (Fig. 3a). At stage I, typical micrometer-scale cubic products of NaO2, approximately 1 μm in size, were observed randomly scattered on the surface of Na[Li1/3Ru2/3]O2 (Fig. 3b). Following stage II, many nanorod-like products of Na2-xO2 and a few aggregated particles (Na2CO3) appear on the Na[Li1/3Ru2/3]O2 surface. TOF-SIMS images show the in-plane and 3D distribution of the Na− signal at the various potentials (Fig. 3a and Supplementary Fig. 12)40. The color bar from black to white represents the relative intensity variation of the Na− signal. The distribution and size of these white particles are similar to those of product particles in the SEM images (Fig. 3c). Furthermore, SEM images of stages III–V confirmed that most cubic and nanorod-shaped products gradually decompose. As the charging progresses, the Na− signal intensity gradually decreases in stages III–V, indicating that there were almost no residual oxygen-containing products in the electrode post-charging at a fully charged electrode. This is consistent with the TOF-SIMS depth profile results (Supplementary Fig. 13). In contrast, for commercial RuO2 electrodes, the sporadic products during the discharge process and the incomplete decomposition of products after charging indicate their inferior catalytic capability (Supplementary Fig. 14).

a Charge–discharge profile at a current density of 200 mA g−1, with a capacity limit of 1000 mAh g−1 corresponding to b Ex-situ SEM analyses, and c corresponding the TOF-SIMS of element mappings of Na− secondary ion distribution on the surface of cycled Na[Li1/3Ru2/3]O2 electrodes. d Ex situ Raman spectra of Na[Li1/3Ru2/3]O2 at the different voltage states in the SSNAB. e O K-edge XAS under the TEY pattern for cycled Na[Li1/3Ru2/3]O2 electrode.
The enhancement of catalytic activity could be associated with the hierarchical architecture of the catalyst. Na[Li1/3Ru2/3]O2 catalyst may undergo structural transformation and play a dynamic catalytic effect. Therefore, ex-situ XRD is employed to inquiry the electronic and configurational evolution of Na[Li1/3Ru2/3]O2 at one catalysis cycle (Supplementary Fig. 15). In the discharged state (stage II), several new diffraction peaks are attributed to the formation of NaO2 (JCPDS 77-0207), Na2O2 (JCPDS 74-0895), and parasitic product Na2CO3 (JCPDS 77-2082), respectively41. During the subsequent charge process (stage III–V), the (003) diffraction peak of Na[Li1/3Ru2/3]O2 continuously shifts owing to the O3 − O1′ − O1 phase change42. The intensity of all diffraction peaks from NaO2, Na2O2, to Na2CO3 gradually weakens and completely disappears at the end of the charging process. The high-resolution XPS spectra obtained for the O 1 s region of the fully discharged electrodes, revealed Na2O2 is the main discharge product accounts for approximately 47.6% (Supplementary Fig. 16). The phase transition reveals that the reductive coupling mechanism may occur at high voltage regions for TM, favoring the activation of new O redox-active centers for sequential catalysis.
The progression of the electrode reaction during the cycled system was further disclosed in detail with Raman analysis. Fig. 3d shows two typical Raman vibration shifts of 485 and 595 cm−1 at the pristine electrode (I), which can stem from the O-Ru-O bending (Eg) and another Ru-O stretching (A1g) in Na[Li1/3Ru2/3]O2, respectively43. After discharge (stage II), the additional peaks are observed at 1107 cm−1 and 1146 cm−1 (O2−*,NaO2), 800 cm−1 (Na2O2), and additional 1080 cm−1 (Na2CO3)44. During charging (stage III-V), the vibration of Eg and A1g of Na[Li1/3Ru2/3]O2 gradually weakened, and the vibration peaks from NaO2, Na2O2 to Na2CO3 gradually disappeared. At high potential (stage V), the new Raman vibration of peroxo-species (O2)n– at 850 cm−1 and superoxide-species O-O bond at around 1100 cm−1 was observed in charged Na[Li1/3Ru2/3]O245. The results unequivocally validate that the reversible evolution of composite products is closely related to the structural changes of Na[Li1/3Ru2/3]O2. Potential-driven formation of (O2)n– species in catalysts may contribute to dramatic promotion in the catalytic activity.
To further elucidate the chemical compositions of the Na[Li1/3Ru2/3]O2 electrodes, O K-edge XAS spectra were obtained (Fig. 3e). Three distinct absorption peaks at 532.0, 532.9, and 534.4 eV can be identified, corresponding to NaO2, Na2O2, and Na2CO3, respectively. Additionally, a substantial amount of Na2CO3 can be fully decomposed even after charging 4.5 V. These findings align well with the previously discussed XRD and Raman results. In contrast, for the structurally stable RuO2 catalyst, the XRD analyses indicate the presence of undecomposed byproducts after deep charging (Supplementary Fig. 17). The Distribution of Relaxation Times model deriving from the EIS spectra also proves the above conclusion. (Supplementary Figs. 18 and 19 and Table 4).
Structural reversibility
A cascade pathway driven by electrochemical potential has been substantiated for Na[Li1/3Ru2/3]O2 serving as switchable electrocatalysts for the SSNAB. Hence, it is crucial to comprehend the underlying mechanism of its electrochemical reversibility. The inherent 3D-disordered cationic framework of Na[Li1/3Ru2/3]O2 could stabilize structures at highly charged states (Fig. 4a). High-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) further validated the O3-type structure of pristine Na[Li1/3Ru2/3]O2 with lattice spacings of 0.527 nm (Fig. 4b and Supplementary Fig. 20). Furthermore, the O1 phase along the [010] plane is captured at the 4.5 V-charged state (Supplementary Fig. 21a)46. No noticeable contrast change was observed in the Na layers, which aligns closely with the limited migration of Ru ions to the Na sites at a charge of 4.5 V. Therefore, Na ions of Na[Li1/3Ru2/3]O2 always occupy the octahedral sites in the Na layer instead of prismatic sites. TM/Na sites are aligned on top of each other. The O3-type layered structure of pristine Na[Li1/3Ru2/3]O2, which was also preserved in the samples discharged to 1.5 V (Supplementary Figs. 21b and 22). Furthermore, the phase transition of O3 − O1′ − O1 could minimize the electrostatic repulsions between Na and TMs. Therefore, the appearance of O1′ and O1 intermediate phases is crucial to trigger the anionic redox and enable the reversible oxygen evolution.

a 3D-disordered cation framework of Na[Li1/3Ru2/3]O2 with randomly distributed unhybridized O 2p orbitals. b HAADF-STEM images and crystal structure diagram of Na[Li1/3Ru2/3]O2 along the [010] zone axis, showing layer spacings for pristine Na[Li1/3Ru2/3]O2. c STEM-HAADF image and corresponding EELS spectra from the selected regions in C-4.5 V of Na[Li1/3Ru2/3]O2. d Reciprocal susceptibility χ−1, with dashed lines indicating linear fits per the Curie−Weiss law; inset shows the temperature dependence of magnetic susceptibility in the Na[Li1/3Ru2/3]O2-cycled system.
The voltage-dependent structural and chemical transformations for Na[Li1/3Ru2/3]O2 are investigated by spectroscopy characterizations. The Ru N-edge electron energy loss spectra (EELS) of charged Na[Li1/3Ru2/3]O2 at 4.5 V display a redshift (Fig. 4c) as one moves from the bulk to the surface, indicating that Ru is oxidized at the surface. Additionally, the pre-edge peak transfers 0.5 eV to a higher energy level from the bulk to the surface for the O K-edge, consistent with previous reports regarding the presence of O2n− species47.
To further examine the electron-spin configurations of the catalysts, ex-situ magnetic susceptibility experiments were performed on Na[Li1/3Ru2/3]O2 samples at various charge and discharge states from 0 to 200 K (Fig. 4d and Supplementary Table 5). The pristine sample exhibited an effective magnetic moment (μeff) of about 2.78 μeff, indicating high spin (t2g3eg1) Ru4+. In contrast, the charged sample showed a sharp decrease in μeff to 1.45 μeff, suggesting low-spin Ru5+ and internal charge transfer between Ru and O during oxidation48. Considering the empty eg orbitals in higher energy levels, the electronic hopping between half-filled t2g levels and unoccupied eg levels leads to ferromagnetic behavior. This implies the involvement of anions in the redox activity of Na[Li1/3Ru2/3]O2, as the coupling of lone-pair electrons on Ru and O gradually reduces the cell’s overall magnetization. Additionally, changes in electrochemical potential can influence orbital occupation, regulating the dynamic catalytic process that relies on electron orbital occupancy49. The EPR measurements and projected density of states (DOS) also support this conclusion (Supplementary Figs. 23 and 24).
To uncover the origin of the structural reversibility, the chemical environment of Li was investigated to reveal its effect on electron and lattice alteration. The 7Li solid-state NMR spectra illustrated two signals at around 150 and 800 ppm, which are ascribed to Li+ within the alkali metal layers (LiAM) layer and the pristine sample’s transition metal layer (LiTM), respectively50. Following charging to 4.5 V, almost all the Li ions reside in the AM layer, suggesting the migration of LiTM into the AM layer. The migrated Li into the AM layer could resist lattice gliding by the “pinning effect”, ensuring excellent structure endurance. Furthermore, when discharging to 1.5 V, a reversible migration of Li contributes to stabilizing the crystalline structure (Supplementary Fig. 25a). According to the 23Na NMR spectra (Supplementary Fig. 25b), Na migration within the AM layers of Na[Li1/3Ru2/3]O2 delivers a reversible evolution, enhancing the reversibility of the Na−O−Li (Fig. 4f)51. In addition, Na[Na₁/₃Ru₂/₃]O₂ as a reference sample further confirms the key role of Li+ in the sodium-air battery electrode (Supplementary Figs. 26–28). According to the reference model put forth by ref. 52, We can justifiably ascribe the variations in migration mechanisms to the existence of lone-pair electrons in the t2g and eg occupation orbitals of TM. Given this evidence, we conclude that the reversible redox process, as an external stimulus, could tune the electronic state of catalysis for the sequential electrocatalysis reaction process.
Switchable metal and oxygen redox center
The interplay between structural evolution and cascade catalysis is explored in one catalysis cycle of battery operation with layered Na[Li1/3Ru2/3]O2. We select five state-of-charge (SOC) states, namely the pristine state(I), 2.5 V(II), 3.5 V(III), 4.5 V(IV), and discharged state 1.5 V (V). It is challenging to detect the oxidation state of oxygen and distinguish it from the strong signals from Ru–O hybridization through traditional X-ray absorption spectrum. Thus, we studied the oxygen states before and after the charging via mapping of resonant inelastic X-ray scattering (mRIXS), a unique method for detecting oxygen oxidation to a non-divalent state. By measuring signals with emission energy near 523.7 eV and another excitation energy arround 531.0 eV (indicated by white arrows in Fig. 5a for the IV stage), we confirmed the oxidation of lattice O. The signal appeared at 3.5 V, intensified while charging to 4.5 V, and reversed upon discharging to 1.5 V (Fig. 5a). The evolution of the mRIXS characteristic on a catalytic cycle offers conclusive experimental certification for an invertible oxygen-redox reaction53. The oxygen oxidation signal reveals the lattice oxygen reactivity of catalysts, where (O2)n– species are identified under a high voltage as marked in the white dashed circle. In the charged state, the signal of (O2)n– species completely vanishes, which serves as the infrastructure for reversible electrocatalysis.

a O K-edge mRIXS results at various electrochemical states and homologous sPFY spectra integrated over the 523.7 eV emission-energy range. b Schematic representation of STXM-based soft X-ray Ptychography. c STXM morphology image of the charged Na[Li1/3Ru2/3]O2 electrode, PCA appraisal (top), and RGB maps of oxygen (bottom). d STXM − XANES analysis of the O K-edge in different regions from (c).
The spatial and chemical element distributions and nanoscale morphological information provide crucial insights into the dynamic catalytic mechanism. Scanning transmission X-ray microscopy (STXM) delivers significant evidence regarding the local bonding environment of components from the surface to the core within the charged electrode54, as shown in Fig. 5b. The O K-edge STXM of charged Na[Li1/3Ru2/3]O2 verifies the existence of oxygen-containing species. Principal Component Analysis (PCA) and RGB mapping of the absorption image stack from STXM further visualized the differences in the chemical distribution of oxygen between the above-mentioned electrode surface and bulk (Fig. 5c). The comparison of O K-edge STXM XAS at the different areas of the charged electrodes Na[Li1/3Ru2/3]O2 is shown in Fig. 5d. The peaks observed around 529 eV and 532 eV are attributed to the t2g and eg unoccupied orbitals of hybridized O 2p–Ru, respectively55. The eg/t2g ratio in the surface region (region 1) was lower than in the bulk (region 3), indicating fewer unoccupied orbitals. The gradual increase in O K-edge intensity from the surface to the bulk can be linked to the anionic redox, which differentiates it from RuO2 (Supplementary Fig. 29). Partially occupied orbitals are crucial for improving electrocatalyst activity by promoting receiving and offering of electron, hence allowing the stimulation of molecular bonds for reaction intermediate.
The emergence of O-redox during electrochemical cycling could influence the cationic redox reactions. The alterations in the Ru valence state of Na[Li1/3Ru2/3]O2 were assessed by analyzing the ex-situ X-ray absorption near edge structure XANES spectra (Supplementary Fig. 30). Based on the white-line energy of the standard reference samples (Supplementary Fig. 31), we found that Ru in Na[Li1/3Ru2/3]O2 progressively oxidized from its initial 4+ state to 4.45+ at 3.5 V56. However, a shift of the absorption edge back to lower energy after charging to 4.5 V is attributed to the reductive coupling mechanism (RCM) of oxygen redox (Supplementary Fig. 32). Following discharging to 1.5 V, the edge returned to its original position, indicating that Ru was reduced to its initial valence state. With potential variations, when the metal d band intersects the oxygen p band at a higher potential, the redox center of the catalyst is no longer solely defined by the metal. Consequently, the catalytic activity could be continually enhanced by transitioning the active center of catalysts from primarily cationic to anionic redox activation.
Extended X-ray absorption fine structure (EXAFS) further indicates that Ru–O interatomic distances decreased. In contrast, Ru-Ru distances remained nearly constant when Na[Li1/3Ru2/3]O2 was charged to high voltages (Supplementary Figs. 33 and 34). The EXAFS fitting results revealed a reduction in the Ru-O coordination number from 6 to 5 (Supplementary Fig. 35 and Table S6). The overall coordination number of the first Ru–O shell was restored during cycling, suggesting that the stretching vibration O–Ru–O arrangement is reversible57. This is consistent with the XPS analysis (Supplementary Fig. 36). With changes in potential, the oriented orbitals hybridize in two axes between the transition metal (TM) and oxygen (Supplementary Table S6). The unit cell in the coupled integrated catalysis undergoes reversible geometric transformation between octahedral and tetragonal forms of RuO6, with Li+ providing spatial localization (Fig. 5a, inset). Ru acts as a positive center throughout the cycles, forming a strong bond with the oxygen framework, which helps prevent oxygen loss at high cutoff voltages and maintains structural integrity during long-term cycling (Supplementary Table S7). The active metal sites in the cascade catalysts would experience a redox shift in their chemical states in response to reversing current during the charge and discharge processes.
The mechanism and reversibility of RuO2 in sodium-air batteries using commercial RuO2 as the electrode were investigated by ex-situ XRD, TEM, and XAS analyses, which revealed that crystalline RuO2 transforms into an amorphous Ru/Na2O mixture during discharge, and recharges back to amorphous RuO2, highlighting its straightforward conversion behavior. This observation highlights the conversion-type behavior of RuO2. It is important to note that the reversibility of this material is inferior to that of intercalated materials Na[Li1/3Ru2/3]O2 (Supplementary Figs. 37–39).
Mechanism analysis
The experimental results unambiguously verify that the unusual reactivity is ascribed to the switchable active center for the redox of cations (Mn+ ⇄ M(n+1)+) and anions (2O2− ⇄ (O2)n−) in Na[Li1/3Ru2/3]O2. However, the relationship between its catalytic performance and the unique crystal/electronic structure remains unraveled. Transforming the M–O covalency by an in situ electrochemistry process can help adjust the d-band center (Ɛd) energy level, a classical indicator for binding strength58. We thus established a correlation between overpotential and adsorption energy to act as a descriptor of the catalytic performance of the Na[Li1/3Ru2/3]O2 catalysts. Density functional theory (DFT) calculations were implemented to evaluate the origin of the activity and scrutinize the adsorption energies between Na[Li1/3Ru2/3]O2 and various products. The integrated-crystal orbital Hamilton population (ICOHP) represents the strength of chemical bonds59. The value of Ru-O adsorbed in Na2CO3 is −2.15 eV for C-4.5 V (Fig. 6a), which obvious is less than that of Ru-O in underpotential C-3.5 V with Na2O2 (−1.87) and C-2.5 V with NaO2 (−1.55), indicating that Na2CO3 forms more stable and strong chemical bonds when adsorbed on the Na[Li1/3Ru2/3]O2 surface at the charge 4.5 V.

a COHP of active Ru atom and coordination atom of different adsorbed species for C-2.5 V, C-3.5 V, and C-4.5 V of Na[Li1/3Ru2/3]O2 electrodes. b The PDOS of adsorbed NaO2, Na2O2, Na2CO3, and the 4d orbital of the Ru atom that is directly engaged in charging for C-2.5 V, C-3.5 V, and C-4.5 V, with corresponding Ru 4d-band center denoted by dashed lines. c Schematic representation of orbital interactions between NaO2/ Na2CO3 and 4d orbital for C-2.5 V and C-4.5 V of Na[Li1/3Ru2/3]O2 catalysts, respectively. d Gibbs free energy and activation energy analysis of varying electrochemical states Na[Li1/3Ru2/3]O2 (left) and RuO2 (right) electrodes. The insets are the optimized molecular structures at various steps, see Supplementary Data 1 and 2. e Schematic of a gradient catalytic system for regulating integrated SSNAB batteries. The divided squares in the schematic indicate the dynamic evolution process of different atomic valences.
The decomposition of adsorbed Na2CO3 can be understood as breaking the Na-O bond, resulting in the formation of individual Na atoms and NaCO3 clusters. In this context, the COHP profiles have proved that the interaction between oxygen atoms in Na2CO3 and Ru atoms in Na[Li1/3Ru2/3]O2 is quite strong. This strong interaction will stretch the Na-O bond in Na2CO3 and make it easier to be broken (Supplementary Fig. 40 and Table S8). Additional calculations were conducted on the density of states (DOS) of adsorbed several products and the 4d orbital and d band center for the Ru atom60. The d-band center of Na[Li1/3Ru2/3]O2-4.5 V adsorbing Na2CO3 is at −1.60 eV, which is nearer to the Fermi energy in comparison to that of Na[Li1/3Ru2/3]O2-3.5 V adsorbing Na2O2 (−1.73 eV) and Na[Li1/3Ru2/3]O2-2.5 V adsorbing NaO2 (−1.97 eV). In brief, the upwards shift of the Ru d-band center of Na[Li1/3Ru2/3]O2 can reduce the electronic orbital occupation of antibonding states, resulting in a gradually increased adsorption energy for NaO2, Na2O2, and Na2CO3 (Fig. 6b). The catalytic performance of RuO2 through DFT calculations was also investigated. The d-band centers of RuO2 at various charging states: −1.57 eV (C-2.5 V), −1.55 eV (C-3.5 V), and −1.51 eV (C-4.5 V). This stability suggests that the electronic properties of Ru remain consistent, limiting the formation of dynamic active centers. Density of states analysis indicates that the covalency of Ru-O bonds is also stable across charging states, with ICOHP values of −1.54, −1.68, and −1.71, confirming similar Ru-O interactions(Supplementary Fig. 41).
As adsorbent containing oxygen generally interacts with the d band of Ru atoms, the binding energies of NaO2 and Na2CO3 depend proportionally on the d-band centers of catalyst active sites (Fig. 6c). The elevated position of dz2 electrons is ascribed to eg orbitals, which orient vertically toward the O ligands of the adsorbate, promoting a significant spatial overlap between the O 2p orbital and the partially filled dz2 occupation orbital61 (Supplementary Figs. 42 and 43). Hence, the dz2 electrons of the octahedral active site are crucial for the catalytic activity of SSNAB. Fig. 6d illustrates each reaction step’s free energy competition for electron involvement (Supplementary Data 1 and 2). For the cascade catalyst, two catalytic models were designed to match the stepwise decomposition of Na2O2 and Na2CO3. Throughout the charging process, Na[Li1/3Ru2/3]O2 exhibits a lower overpotential (0.207 V) compared to RuO2 (~0.855 V), indicating that the dynamic variation of the d-band center in the cascade catalyst active sites can facilitate the decomposition of complex products62. Furthermore, charge density difference implying that the reaction occurs on the Na[Li1/3Ru2/3]O2 sites with an abundance of electron transfers compared with the RuO2 surface (Supplementary Figs. 44 and 45), which is coincident with dissociation energy of different adsorbent on Na[Li1/3Ru2/3]O2 and RuO2 (Supplementary Fig. 46). Moreover, a similar cascade catalytic reaction mechanism may also be achieved in other sodium-containing layered oxides (such as NLMO), confirming the universality of the Na-O-Li structural configuration of the electrode in realizing this mechanism in sodium-air batteries (Supplementary Figs. 47 and 48).
The investigation of redox switchable catalysis enables the formation of O-O bonds and provides ligand vacancies that extend beyond conventional metal sites, which is anticipated to enhance the catalytic efficiency for the complex products of SSNAB. Specifically, the high-spin active center, characterized by weak adsorption strength on NaO2, facilitates subsequent desorption transformations. A moderately positioned d-band center with appropriate adsorption strength for Na2O2 helps to maintain a balance between absorption and desorption. In contrast, the low-spin state active center can stabilize the hard-to-decompose Na2CO3 through strong covalent interactions, allowing continuous transformation63 (Fig. 6e). This approach introduces a versatile strategy for optimizing geometrical configurations to lower each oxygen redox step’s energy barrier.
Discussion
In conclusion, our study has illustrated the merits of Na[Li1/3Ru2/3]O2 as an efficient cascade catalyst for SSNAB. The noteworthy enhanced reactivity and reversibility observed in this system are achieved through modulation of Ru–O covalent interactions. Introducing Li dopants has proven effective in fine-tuning the RuO6 distortion, forming varied degrees of eg* band broadening through controlled Na+ occupation. The dynamic active sites could align their potential to the redox process during cycling, leading to the favourable catalytic reactivity for all the products. Within the potential region, the eg* band broadening could facilitate the affinity for NaO2 desorption, thereby bolstering adsorption-desorption efficiency. Meanwhile, at elevated potentials where the rate-determining step involves Na2O2/Na2CO3 decomposition, the high-spin Ru4+ state transitions to a low-spin Ru5+ state in the eg orbitals due to the loss of one electron. This transition enhances the adsorption capacity for oxygen-containing intermediates, reducing the thermodynamic barrier for the oxygen evolution reaction while improving the kinetic properties. Cascade catalysts have the potential to position different functionalities, which may help in controlling reaction intermediates. This could lead to unique characteristics in the catalysis process in SSNAB and may also open up opportunities for improvements in other energy storage areas.
Methods
Material synthesis
The layered Na[Li1/3Ru2/3]O2 was prepared using conventional solid-state techniques19. Stoichiometric quantities of Na2CO3(Aladdin, 99%), Li2CO3 (Aladdin, 99%), and RuO2 (Aladdin, 99.9%) were thoroughly blended and ground for thirty minutes using a mortar and pestle. The resultant powder was then shaped into pellets and calcined at 750 °C for 10 h in air, whereafter a second calcination at 900 °C in air for another 10 h, after which it was allowed to cool to room temperature naturally before being transferred into the glove box for further electrode preparation. Na[Li1/3Mn2/3]O2 material was prepared from Na2CO3(Aladdin, 99%), Li2CO3 (Aladdin, 99%), and MnO2 (Aladdin, 99.9%) precursor; the specific operation steps can be referred to in the above process.
Na[Na1/3Ru2/3]O2 was synthesized through solid-state reactions using stoichiometric amounts of Na2CO3 (Aladdin, 99%) and RuO2 (Aladdin, 99.9%), with an additional 5 mol% Na2CO3. These components were thoroughly combined and ground. The resulting powder was pelletized and calcined at 900 °C for 10 h under an argon atmosphere51.
Electrolyte preparation
Poly(vinylidene fluoride-co-hexafluoropropylene)(PVDF-HFP) powders and sodium salts were dried at 100 °C for 12 h before use to remove trapped water. PVDF-HFPpolymer (1.5 g; Sigma-Aldrich) was mixed in 15 mL of acetone(Aladdin, ≥98%) containing 10 wt% fumed silica and heated at 50 °C for 2 h with stirring, then rested for 1 h to remove bubbles. The mixture solution was distributed as a film on a watch glass and dried at 60 °C overnight using a vacuum chamber. In another beaker, liquid electrolytes were prepared by dissolving NaClO4 salts (Aladdin, ≥99.99%) in tetraethylene glycol dimethyl ether (TEGDME, ≥99.5%) by magnetic stirring at 700 rpm for 12 h in a glovebox under argon atmosphere. Molarities were adjusted to 1 M electrolyte. Subsequently, TEGDME with dissolved sodium salts and PVDF-HFP solution was mixed, where the mass ratio of PVDF-HFP solution to liquid electrolyte was adjusted as 7:2, and a quasi-solid electrolyte (QPE) membrane was obtained.
Electrochemical measurements
Nickel foam was punched into circular electrode sheets with a diameter of 12 mm, cleaned in 1 M HCl solution for 30 min, and then thoroughly washed with deionized water/ethanol successively to remove the residual chloride ions. A pristine electrode was produced by coating nickel foam with a uniform ink containing 90 wt% Na[Li1/3Ru2/3]O2 powder and 10 wt% PVDF-HFP binder mixture in 5 ml N-Methyl pyrrolidone (Aladdin, 98%) to ensure uniform dispersion, enhancing battery performance. This solution was dispersed evenly with a nickel foam collector (Youveim, purity:>99.9%, thickness mm, porosity:98%, areal density: 280 gm-2) by ultrasonic instrument (KQ-400KDE), dried under vacuum for 12 h, achieving a mass loading of approximately 0.5 mg cm−2. Post-casting, the electrodes were dried at 80 °C overnight inside a vacuum environment. One side of the air electrode was in contact with the electrolyte, and the other was exposed to ambient air as an oxygen source. Batteries were assembled in an argon-filled glovebox (O2 and H2O contents <0.1 ppm) using 2032-type coin cells, layering the Na metal sheet (China Energy Lithium Co., Ltd, diameter:14 mm, thickness: 0.45 mm), a 16 mm composite electrolyte film containing NaClO4/TEGDME, and the electrode sequentially. The electrochemical tests were conducted using a NEWARE Battery Test System (CT-4008T-5V50mA-164, Shenzhen, China) under various parameters in ambient air (25 °C, relative humidity:20%)64. The CV tests were conducted over a potential scan range of 1.5 to 4.5 V (vs. Na+/Na) at a scan rate of 0.1 mVs-1, with a sodium foil serving as the counter electrode and reference electrode. Electrochemical impedance spectroscopy (EIS) was carried out with an impedance analyser at open circuit voltage with an AC oscillation of 0.01 V amplitude over frequencies from 850 kHz to 0.1 Hz.
Assembly of solid-state pouch batterie
The solid-state soft pack battery assembled in this study is pouch-type. Foam nickel coated (8 × 8 cm²) with a catalyst serves as the air electrode (mass loading: 10 mg cm−2), while sodium strips (8 × 8 cm²) are used as the anode. Aluminum tabs are used for the positive electrode, and nickel tabs for the negative electrode. Quasi-solid polymer electrolyte acts as the solid electrolyte (10 × 10 cm²). The entire battery assembly process is conducted in an argon-filled glove box, where the electrode sheet, solid electrolyte, and anode sheet are stacked in order and vacuum sealed with a perforated aluminum-plastic film. The performance testing of the completed solid-state soft pack battery is carried out under ambient air conditions.
Material characterization
The substance’s composition was analyzed using a range of characterization techniques, including XRD (PANalytical Empyrean), XPS (KRATOS AXIS Ultra DLD), Raman (inVia-Reflex), SEM (ZEISS Merlin Compact; Phenom Pro), in situ differential electrochemical mass spectrometry (DEMS, Hiden Analytical), and ICP-MS (PerkinElmer, Optima 5300DV).
O K-edge resonant inelastic X-ray scattering (RIXS)
mRIXS was performed at Beamline BL20U1 and BL20U2 of the Shanghai Synchrotron Radiation Facility (SSRF). The mapping data were obtained using a highly efficient modular spectrometer with an excitation energy increment of 0.2 eV. The final two-dimensional maps were generated through a multistep data processing approach, as detailed in a prior study65.
Scanning transmission X-ray microscopy (STXM)
The STXM measurements were conducted at the beamline. BL08U1A of SSRF. The grids were placed in a sample holder, sealed in an aluminum pouch, and transported to the beamline. STXM images were obtained with an 8 × 8 μm² scanned (200 × 200 points) image size with a step size of 0.04 μm, and setting a dwell time of 1 ms per pixel, followed by PCA-CA analysis to identify natural spectrum groups and calculate average spectra66.
XAFS measurements
XAS spectra were recorded at beamline BL14W1 of SSRF for the Ru K-edge. Samples were prepared by cycling the electrode to the desired voltage and transporting them to the beamline in an argon-filled bag to protect against moisture and oxygen. Data analysis was conducted with the Athena software, with EXAFS fittings done via the Artemis program51.
HAADF-STEM and EELS
TEM, HR-TEM, atomic resolution-HAADF-STEM images, and EELS were obtained employing the aberration-corrected JEM-ARM200F(C)67.
TOF-SIMS
The Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) (IONTOF GmbH, Münster, Germany) was employed. The 3D data was obtained in an area of 15 × 15 μm. The measurements were conducted in negative modes40.
SQUID magnetometry
Susceptibility experiments were implemented in zero-field cooling mode by a SQUID XL magnetometer with 1 T implemented fields from 0 to 200 K. Powder samples were sealed in vacuum quartz tubes with cotton to prevent movement. EPR spectra were recorded on a Bruker Elexsys E580 spectrometer (ω = 9.36 GHz)48.
Solid-state 23Na and 7Li MAS NMR spectroscopy
Solid-state NMR experiments for 23Na were conducted on a WB 14.1 T Bruker ADVANCE III spectrometer. Spectra were collected at spinning frequencies of 15 kHz using the Hahnecho pulse sequence. For 7Li, measurements were conducted on a WB 11.75 T spectrometer with a Larmor frequency of 194.3 MHz52.
Computational details
DFT calculations were implemented employing the Vienna ab initio simulation package with the Perdew-Burke-Ernzerhof functional for exchange-correlation interactions. A plane-wave basis set featuring a kinetic energy cutoff of 400 eV, energy convergence criterion of 10−5 eV, force convergence criterion of 0.02 eV Å−1, and (2 × 2 × 1) Monkhorst–Pack k-point sampling were adopting for structure relaxation. The Na[Li1/3Ru2/3]O2 system was modeled by substituting Ru ions with Li ions in a cell containing 16 formula units60.
Data availability
All data supporting this work are available in the article and its Supplementary Information or are available from the corresponding authors upon request. Source data are provided with this paper.
References
Bruce, P. G., Freunberger, S. A., Hardwick, L. J. & Tarascon, J.-M. Erratum: Li–O2 and Li–S batteries with high energy storage. Nat. Mater. 11, 172–172 (2011).
Hartmann, P. et al. A rechargeable room-temperature sodium superoxide (NaO2) battery. Nat. Mater. 12, 228–232 (2012).
Xia, C., Black, R., Fernandes, R., Adams, B. & Nazar, L. F. The critical role of phase-transfer catalysis in aprotic sodium oxygen batteries. Nat. Chem. 7, 496–501 (2015).
Ma, J. -l. et al. Prevention of dendrite growth and volume expansion to give high-performance aprotic bimetallic Li-Na alloy–O2 batteries. Nat. Chem. 11, 64–70 (2018).
Hu, Y. Y. et al. Origin of additional capacities in metal oxide lithium-ion battery electrodes. Nat. Mater. 12, 1130–1136 (2013).
Qiao, Y., Jiang, K., Deng, H. & Zhou, H. A high-energy-density and long-life lithium-ion battery via reversible oxide–peroxide conversion. Nat. Catal. 2, 1035–1044 (2019).
Wang, X. et al. Achieving a high-performance sodium-ion pouch cell by regulating intergrowth structures in a layered oxide electrode with anionic redox. Nat. Energy. 9, 184–196 (2024).
Mortemard de Boisse, B. et al. Intermediate honeycomb ordering to trigger oxygen redox chemistry in layered battery electrode. Nat. Commun. 7, 11397 (2016).
Huang, Z.-F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992 (2021).
Ye, L. et al. A rechargeable calcium–oxygen battery that operates at room temperature. Nature 626, 313–318 (2024).
Ben Yahia, M., Vergnet, J., Saubanère, M. & Doublet, M.-L. Unified picture of anionic redox in Li/Na-ion batteries. Nat. Mater. 18, 496–502 (2019).
Dong, T. et al. How do polymer binders assist transition metal oxide electrodes to address the challenge of high-voltage lithium battery applications?. Electrochem. Energy Rev. 4, 545–565 (2021).
Wang, X.-X. et al. Metal–organic framework-based mixed conductors achieve highly stable photo-assisted solid-state lithium–oxygen batteries. J. Am. Chem. Soc. 145, 5718–5729 (2023).
Hao, H. et al. Molybdenum carbide electrocatalyst in situ embedded in porous nitrogen-rich carbon nanotubes promotes rapid kinetics in sodium-metal–sulfur batteries. Adv. Mater. 34, e2106572 (2022).
Liu, S. et al. Atomically dispersed iron sites with a nitrogen–carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat. Energy 7, 652–663 (2022).
Park, H. et al. Activating reversible carbonate reactions in Nasicon solid electrolyte-based Na-air battery via in-situ formed catholyte. Nat. Commun. 15, 2952 (2024).
Xiong, Q., Huang, G. & Zhang, X. B. High-capacity and stable Li-O2 batteries enabled by a trifunctional soluble redox mediator. Angew. Chem. Int. Ed. 59, 19311–19319 (2020).
Xie, H. et al. Ta–TiOx nanoparticles as radical scavengers to improve the durability of Fe–N–C oxygen reduction catalysts. Nat. Energy 7, 281–289 (2022).
Wang, Q. et al. Unlocking anionic redox activity in O3-type sodium 3 d layered oxides via Li substitution. Nat. Mater. 20, 353–361 (2021).
Wu, Z. et al. Realizing high capacity and zero strain in layered oxide electrodes via lithium dual-site substitution for sodium-ion batteries. J. Am. Chem. Soc. 145, 9596–9606 (2023).
Zhao, S. et al. Bifunctional effects of cation additive on Na-O2 batteries. Angew. Chem. Int. Ed. 60, 3205–3211 (2020).
Liu, Y. et al. Metal–organic framework-derived hierarchical Co3O4@MnCo2O4.5 nanocubes with enhanced electrocatalytic activity for Na–O2 batteries. Nanoscale 11, 5285–5294 (2019).
Li, N. et al. Enabling pyrochlore-type oxides as highly efficient electrocatalysts for high-capacity and stable Na–O2 batteries: the synergy of electronic structure and morphology. ACS Catal. 7, 7688–7694 (2017).
Kwak, W.-J. et al. Nanoconfinement of low-conductivity products in rechargeable sodium–air batteries. Nano Energy 12, 123–130 (2015).
Hu, Y. et al. Porous perovskite calcium–manganese oxide microspheres as an efficient catalyst for rechargeable sodium–oxygen batteries. J. Mater. Chem. A 3, 3320–3324 (2015).
Yin, W.-W. et al. A long-life Na–air battery based on a soluble NaI catalyst. Chem. Commun. 51, 2324–2327 (2015).
Chang, S. et al. High-performance quasi-solid-state Na-air battery via gel electrode by confining moisture. Adv. Funct. Mater. 31, 2011151 (2021).
Zhang, S. et al. Graphene nanosheets loaded with Pt nanoparticles with enhanced electrochemical performance for sodium–oxygen batteries. J. Mater. Chem. A 3, 2568–2571 (2015).
Ma, J. -l. et al. Synthesis of porous and metallic CoB nanosheets towards a highly efficient electrocatalyst for rechargeable Na–O2 batteries. Energy Environ. Sci. 11, 2833–2838 (2018).
Khan, Z. et al. Three-dimensional SnS2 nanopetals for hybrid sodium-air batteries. Electrochim Acta. 257, 328–334 (2017)
Kim, M., Ju, H. & Kim, J. Single crystalline Bi2Ru2O7 pyrochlore oxide nanoparticles as efficient bifunctional oxygen electrocatalyst for hybrid Na-air batteries. Chem. Eng. J. 358, 11–19 (2019).
Kang, Y. et al. Novel high-energy-density rechargeable hybrid sodium–air cell with acidic electrolyte. ACS Appl. Mater. Interfaces 10, 23748–23756 (2018).
Lin, X. et al. Reviving anode protection layer in Na-O2 batteries: failure mechanism and resolving strategy. Adv. Energy Mater. 11, 2003789–2003797 (2021).
Sun, B. et al. Hierarchical porous carbon spheres for high-performance Na–O2 batteries. Adv. Mater. 29,1606816–1606823 (2017).
Liu, X., Lei, X., Wang, Y.-G. & Ding, Y. Prevention of Na corrosion and dendrite growth for long-life flexible Na–air batteries. ACS Cent. Sci. 7, 335–344 (2021).
Yin, W.-W. & Fu, Z.-W. A highly efficient bifunctional heterogeneous catalyst for morphological control of discharged products in Na–air batteries. Chem. Commun. 53, 1522–1525 (2017).
Liu, Y. et al. Uncovering the geometry activity of spinel oxides in Li-CO2 battery reactions. ACS Energy Lett. 9, 2173–2181 (2024).
Su, Z. et al. X-ray nanocomputed tomography in Zernike phase contrast for studying 3D morphology of Li-O2 battery electrode. Acs Appl. Energy Mater. 3, 4093–4102 (2020).
Pang, Y., Pan, J., Yang, J., Zheng, S. & Wang, C. Electrolyte/electrode interfaces in all-solid-state lithium batteries: a review. Electrochem. Energy Rev. 4, 169–193 (2021).
Yu, W. et al. A volatile redox mediator boosts the long-cycle performance of lithium-oxygen batteries. Energy Storage Mater. 38, 571–580 (2021).
Bi, X., Wang, R., Amine, K. & Lu, J. A critical review on superoxide-based sodium–oxygen batteries. Small Methods. 3, 1800247–1800254 (2018).
Dubouis, N. et al. Extending insertion electrochemistry to soluble layered halides with superconcentrated electrolytes. Nat. Mater. 20, 1545–1550 (2021).
Ning, F. et al. Inhibition of oxygen dimerization by local symmetry tuning in Li-rich layered oxides for improved stability. Nat. Commun. 11, 4973–4984 (2020).
Yadegari, H. et al. On rechargeability and reaction kinetics of sodium–air batteries. Energy Environ. Sci. 7, 3747–3757 (2014).
Gao, R. et al. Probing the self-boosting catalysis of LiCoO2 in Li–O2 battery with multiple in situ/operando techniques. Adv. Funct. Mater. 30, 2002223–2002232 (2020).
Wang, C. et al. Resolving complex intralayer transition motifs in high-Ni-content layered electrode materials for lithium-ion batteries. Nat. Mater. 22, 235–241 (2023).
Li, L. et al. Fluorination-enhanced surface stability of disordered rocksalt electrodes. Adv. Mater. 34, e2106256 (2022).
Zuo, Y. et al. Regulating the potential of anion redox to reduce the voltage hysteresis of Li-rich electrode materials. J. Am. Chem. Soc. 145, 5174–5182 (2023).
Zhong, H. et al. Key role of eg* band broadening in nickel-based oxyhydroxides on coupled oxygen evolution mechanism. Nat. Commun. 14, 7488 (2023).
Yao, Y. et al. The effect of electrochemically inactive Ti substituted for Ru in Li2RuTiO3 on structure and electrochemical performance. J. Energy Chem. 60, 222–228 (2021).
Cao, X. et al. Triggering and stabilizing oxygen redox chemistry in layered Li[Na1/3Ru2/3]O2 enabled by stable Li–O–Na configuration. ACS Energy Lett. 7, 2349–2356 (2022).
Carlier, D., Ménétrier, M., Grey, C. P., Delmas, C. & Ceder, G. Understanding the NMR shifts in paramagnetic transition metal oxides using density functional theory calculations. Phys. Rev. B 67, 174103 (2003).
Zhuo, Z. et al. Cycling mechanism of Li2MnO3: Li–CO2 batteries and commonality on oxygen redox in electrode materials. Joule 5, 975–997 (2021).
Mefford, J. T. et al. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 593, 67–73 (2021).
Eum, D. et al. Coupling structural evolution and oxygen-redox electrochemistry in layered transition metal oxides. Nat. Mater. 21, 664–672 (2022).
Wang, H., An, H. W., Shan, H. M., Zhao, L. & Wang, J. J. Research progress on interfaces of all-solid-state batteries. Acta Phys.-Chim. Sin. 37, 2007070 (2021).
Sun, X. et al. Tailoring electronic-ionic local environment for solid-state Li-O2 battery by engineering crystal structure. Sci. Adv. 8, eabq6261 (2022).
Hwang, J. et al. Perovskites in catalysis and electrocatalysis. Science 358, 751–756 (2017).
Zhu, Y. et al. Lattice engineering on Li2CO3-based sacrificial electrode prelithiation agent for improving the energy density of Li-ion battery full-cell. Adv. Mater. 36, e2312159 (2023).
Tian, J. et al. Sabatier relations in electrocatalysts based on high-entropy alloys with wide-distributed d-band centers for Li-O2 batteries. Angew. Chem. Int. Ed. 62, e202310894 (2023).
Wang, X. et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 611, 702–708 (2022).
Benti, N. E. et al. The effect of CO2 contamination in rechargeable non-aqueous sodium–air batteries. J. Chem. Phys. 152, 074711 (2020).
Fu, Y. et al. dz2 band links frontier orbitals and charge carrier dynamics of single-atom cocatalyst-aided photocatalytic H2 production. J. Am. Chem. Soc. 145, 28166–28175 (2023).
Zhu, Y., Yang, F., Guo, M., Chen, L. & Gu, M. Real-time imaging of the electrochemical process in Na–O2 nanobatteries using Pt@CNT and Pt0.8Ir0.2@CNT air electrodes. ACS Nano 13, 14399–14407 (2019).
Ji, H. et al. Ultrahigh power and energy density in partially ordered lithium-ion electrode materials. Nat. Energy 5, 213–221 (2020).
Li, M. et al. Ion–dipole-interaction-induced encapsulation of free residual solvent for long-cycle solid-state lithium metal batteries. J. Am. Chem. Soc. 145, 25632–25642 (2023).
Zhang, F. et al. Surface regulation enables high stability of single-crystal lithium-ion electrodes at high voltage. Nat. Commun. 11, 3050 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 92372110 and No. 22075063), the Chinesisch-Deutsches Mobilitätspropgamm (M-0281), the Fundamental Research Funds for the Central (Grant No. HIT.OCEF.2023039), the Heilongjiang Touyan Team (HITTY-20190033), the ‘Young Scientist Studio’ of Harbin Institute of Technology (HIT), the funds from Chongqing Research Institute of HIT, and funds from Suzhou Research Institute of HIT. We thank the National Key Research and Development Program of China(2022YFE0138900), the Natural Science Fund for Distinguished Young Scholars of Chongqing (cstc2021jcyj-jqX0003), “Young Scientist Studio” of Harbin Institute of Technology (HIT), and funds from Chongqing Research Institute of HIT. We thank the beamline BL13W1 at SSRF for micro-CT measurements, beamline BL20U1 and BL20U2 at SSRF for RIXS measurements, beamline BL08U1A at SSRF for STXM, and 4W1A beamline of Beijing Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
J.W. conceived and designed the experiments. X.S. undertook the materials synthesis, characterization, and performance testing. X.L. and P.J. performed the HAADF-STEM. Y.S. contributed to the DFT calculations. J.L. and H.L. assisted with the RIXS and STXM. B.D. and Q.L. assisted with the micro-CT test. M.L. assisted with the NMR. X.Z. assisted with the XAFS analysis. J.W. and D.S. assisted with data analysis and paper revision. All the authors discussed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Feng Liang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, X., Li, H., Song, Y. et al. Cascade reactors for long-life solid-state sodium–air batteries. Nat Commun 16, 5814 (2025). https://doi.org/10.1038/s41467-025-60840-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60840-z
