
Abstract
1,3-Butadiene (C4H6), the main raw material for producing important chemicals (nylon, synthetic resin, rubber), relies on petroleum cracking with intensive carbon emissions. The electrocatalytic dimeric hydrogenation of natural gas/coal-derived C2H2 to C4H6 provides a nonpetroleum pathway. However, C4H6, as a byproduct of C2H2 hydrogenation, is usually neglected because of its very low Faradaic efficiency. Here, we theoretically and experimentally report a mechanism comprising acetylene dimerization and subsequent hydrogenation. The first dimerization process can be accelerated under appropriate coverage of acetylene and water. A ligand-modifying strategy is subsequently proposed to regulate the wettability of Cu nanoarrays to enable suitable coverages. The optimized 1-dodecanethiol-modified Cu nanoarrays deliver a 65.3% C4H6 Faradaic efficiency at 100 mA cm−2. The C4H6 formation kinetics become sluggish at the two ends of the surface C2H2/H2O ratios, and moderate C2H2/H2O coverage accelerates the C‒C coupling process to promote C4H6 production. Moreover, life cycle assessment demonstrates its sustainability.
Similar content being viewed by others


Introduction
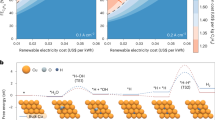
The reliance on limited petroleum reserves, the tilt of ethylene (C2H4) in the petrochemical industry, and the frequent fluctuations in crude oil prices threaten the supply of 1,3-butadiene (C4H6)1,2,3,4. In addition, the high energy requirement of the traditional oil-dependent pathway is environmentally unfriendly (Fig. 1a),5,6,7,8, making it highly desirable to seek an alternative green and petroleum-independent synthesis strategy9,10. Acetylene (C2H2), obtained from natural gas/coal and acting as one of the primary feedstocks for commodity chemicals in the first half of the last century1, has gradually become recognized as a nonpetroleum raw material with the development of arc-plasma technology11,12 since it is highly active and can be hydrogenated and/or dimerized to C2H4 and/or C4H6 under specific conditions (Fig. 1b)13. A Ru-based homogeneous catalyst can drive controlled dimerization of C2H2 under high pressure (10−50 bar) to obtain C4H614, but the reliance on noble metals and their subsequent separation makes this process costly. For the reported heterogeneous catalysts, high pressures and temperatures are also needed, accompanied by safety risks and technical difficulties15,16,17. Therefore, further development of a petroleum-independent, sustainable, and mild strategy for the hydrodimerization of C2H2 to C4H6 is urgently needed.

a Traditional methods for C4H6 synthesis include thermal cracking of petroleum and extractive distillation of C4 fractions, which recycle large quantities of organic solvents to separate products with close boiling points. b This work describes the development of an electrochemical method for synthesizing C4H6 that overcomes previous challenges by relying on petroleum and organic solvents, which favor the green and sustainable production of chemical products.
With advancements in electrocatalytic C2H2 hydrogenation (EAH), the electrorefining of rough ethylene (C2H4)18,19,20 and the electrosynthesis of C2H421,22 are currently achieved over copper-based materials. Although trace amounts of the C4H6 product have been observed in EAH, it is always neglected as a byproduct because of its extremely low yield and selectivity (C4H6 Sele. <10%)13,20,23,24. Very recently, a well-designed iodide-induced Cuδ+-Cu0 site was reported to promote C4H6 electrosynthesis25. However, the effects of the interface properties of acetylene and water on the C−C coupling synthetic mechanism are still not clear. Inspired by studies on CO2 electroreduction coupling to C2H426,27, it is reasonable to infer that multiple C−C coupling pathways exist in the EAH process and that appropriate regulation of the coupling behavior will benefit C4H6 electrosynthesis. In addition, the surface coverage of acetylene is speculated to influence the C−C coupling process. Thus, understanding the influence of acetylene/water coverage on regulating the coupling process in EAH to increase the activity and selectivity of C4H6 is highly desirable for synthesizing C4H6 under mild conditions.
In this work, our preliminary experiments and theoretical calculations of the EAH process over commercial Cu nanoparticles (Cu NPs) model catalysts demonstrate that C4H6 generation via the precoupling and rehydrogenation pathways benefits from moderate coverage of C2H2 and H2O. Next, we construct ligand-modified Cu nanoarrays with continuously tunable wettability, which are suitable models for researching the effects of surface coverage. We find that 1-dodecanethiol-modified Cu nanoarrays (denoted as Cu-12SH-NAs) deliver a C4H6 Faradaic efficiency (FE) of 65.3% at a current density of 100 mA cm−2 with a C4H6 production rate of 0.49 mmol mg−1 h−1, substantially outperforming other counterparts. Cu-12SH-NAs with moderate wettability are revealed to enrich surface C2H2 and accelerate the coupling kinetics, thus enabling high C4H6 FE over a wide potential range. As a proof-of-concept application, a 50 cm2 reactor with a Cu-12SH-NAs catalyst is implemented and maintains stable operation for 13 h at a current of 1.0 A, with a C4H6 production of 325 mmol. Furthermore, a cradle-to-gate life cycle assessment confirms the sustainability of the proposed C4H6 electrosynthesis strategy.
Results
Calculation-assisted reaction analysis
Since C2H4 and C4H6 are the main C2H2-based products produced during the EAH process over commercial Cu NPs model catalysts (Fig. 2a and Supplementary Figs. 1, 2), reaction analysis is necessary to selectively increase the yield of C4H6 for the rational design of catalysts. First, the stoichiometric ratios of C2H2 and *H aimed at the production of one molecule of C2H4 and C4H6 are 0.5 and 1, respectively (Eqs. 1, 2), which account for the relatively high C4H6 selectivity in the low current density region with slow hydrogenation kinetics (Fig. 2a). That is, maintaining the required high *C2H2/*H ratio under negatively shifted potentials is a promising way to simultaneously increase the activity and selectivity of the target C4H6. In addition, exploration of the mechanism of C−C coupling is also important for the production of C4H6. Then, the reaction pathway involving the coupling process was further analysed. Specifically, for the electrocatalytic dimeric hydrogenation of C2H2 to C4H6, there are two kinds of reaction modes: (i) the precoupling and rehydrogenation (C−H) pathways (Eq. 3) and (ii) prehydrogenation and recoupling, the latter of which can be further divided into two pathways (H−C−1 & H−C−2) (Eqs. 4, 5). In this regard, primary density functional theory (DFT) calculations are conducted to evaluate the three proposed reaction pathways over periodic Cu slabs. The energy barriers of the coupling steps are higher than those of hydrogenation for all three paths (Fig. 2b, c; Supplementary Figs. 3−6; and Supplementary Note 1 and Supplementary Table 1), indicating that the C−C coupling process can be regarded as the rate-determining step (RDS) to some extent. In this case, the C‒H pathway is proposed as the theoretical optimum path because it results in the lowest coupling and hydrogenation barrier (Fig. 2c). To further distinguish these pathways, the Butler‒Volmer function is applied to deduce the three theoretical rate expressions28,29, taking the coupling process as the RDS (details in Supplementary Note 2). As shown in Fig. 2d, the reaction order of H+/H2O in the three pathways is 0, 1, and 2, respectively. Thus, the reaction pathway can be ascertained through experimental analysis of the reaction order towards H+/H2O.

a Linear-sweep voltammetry of commercial Cu NPs at 5 mV s−1 in a 1 M KOH electrolyte (no iR compensation) and the corresponding Operando DEMS signals. Free energy diagram of the C2H2 dimeric hydrogenation process over Cu NPs (b) and the corresponding activation energy Ea of each elementary reaction (c). Green: Cu, red: C, blue: H. Visuals were produced with VESTA44 and blender. d Expression of the reaction control steps and reaction order for the C‒H, H‒C‒1, and H‒C‒2 pathways. Potential-dependent partial current density of C4H6 over commercial Cu NPs under different pH values (e) and isotope-substituted conditions (f). Free energy diagram of the first step of the C2H2 dimeric hydrogenation process under different coverage conditions (g) and the corresponding activation energy difference values under different coverage conditions (h). The error bars correspond to the standard deviation of at least three independent measurements. Source data are provided as a Source Data file.
Accordingly, pH-dependent and kinetic isotope effect (KIE) experiments efficiently reflect the relevance of H+/H2O in electrocatalytic hydrogenation reactions and the corresponding experimental reaction order28,30,31. Thus, to determine the influence of the H+ concentration and H2O dissociation on the activity of C4H6 electrosynthesis, potential-dependent C4H6 partial current densities (jC4H6) with different pH values and KIEs were explored over a Cu NPs model catalyst. As shown in Fig. 2e, the apparent activity of C4H6 electrosynthesis is almost the same for electrolytes with different pH values (Supplementary Figs. 7, 8). Similarly, \({j}_{{C}_{4}{H}_{6}}\) remains unchanged within the error range when D2O is used as a replacement for H2O; that is, the KIE value is approximately close to 1 (Fig. 2f and Supplementary Figs. 9, 10). These results demonstrate that the reaction order of C4H6 electrosynthesis through the EAH process to H+/H2O is 028,32, which is consistent with the theoretical value of the C‒H pathway, revealing that C4H6 electrosynthesis follows the C‒H pathway. Considering the aforementioned *C2H2/*H stoichiometric ratio discussion, all three C–C coupling pathways were further optimized through DFT calculations. The results suggest that the C–H pathway exhibits advantages over H–C–1 and H–C–2 pathways in terms of its lower coupling barriers under varying coverage of *C2H2 and *H. In addition, moderate coverage of C2H2 and *H is promising for reducing the barrier difference between coupling and hydrogenation and even achieving a lower coupling barrier than hydrogenation (Fig. 2g, h and Supplementary Figs. 11‒25). Owing to the *H sources from H2O during the EAH process, modulating the microenvironment of the Cu-based catalyst, increasing the mass transfer of the C2H2 feedstock, and adjusting its wettability to enable an optimal C2H2/H2O ratio can efficiently boost the activity and selectivity of the target C4H6.
Wettability tuning electrocatalyst synthesis and optimization
To increase the mass transfer of gaseous C2H2 feedstocks, Cu-based hydroxide nanoarrays (denoted as Cu NAs) were designed and synthesized through reported liquid deposition on a Cu mesh. The successful formation and nanoarray-like morphology were characterized by X−ray diffraction (XRD) patterns and scanning electron microscopy (SEM) images (Supplementary Fig. 26). Since alkanethiols with different alkyl chain lengths are efficient at continuously tuning the surface wettability, Cu-xSH-NAs, where x represents the number of carbon atoms in the alkyl chain, are designed and synthesized for microenvironment modulation (Supplementary Figs. 27, 28). The increasing trend of the contact angle, along with the growth of the carbon chain, suggests reduced wettability (Fig. 3a)33. In addition, confocal laser scanning microscopy (CLSM) was performed to explore the gas‒liquid distribution principle over the interface between the electrolyte and electrode34. Specifically, the height of the electrolyte over the pores of the Cu mesh tends to decrease with increasing length of the carbon chain (Fig. 3b and Supplementary Fig. 29), indicating that the permeability decreases. In addition, since the light absorption and blocking effects of the gas and solid phases are extremely different (solid > gas)34, the tail effect of the fluorescent agent in the electrolyte over the electrolyte‒electrode interface is used to describe the C2H2/H2O distribution behavior. The decay distances of the fluorescence intensity through the z-axis direction monotonically increase with increasing growth of the carbon chain (Fig. 3c and Supplementary Figs. 30, 31), indicating enhanced hydrophobicity. This means that the liquid‒solid interface is gradually transformed into a gas‒solid interface after wettability modulation, which is promising for controlling the C2H2/H2O ratio. For gas-involved electrocatalytic reactions, the gaseous reactants are always impeded by aqueous H2O with a dense cluster structure in the electrical double layer (EDL); thus, breaking the hydrogen bond (HB) in the H2O cluster creates channels for gases; that is, the statistical ratios of H2O with saturated (4−HB H2O) and unsaturated (1, 2, 3−HB H2O) coordination in the EDL could be used as a descriptor of gas accessibility35,36. Therefore, molecular dynamics simulations were conducted to obtain corresponding H2O-type information after ligand modification. As shown in Fig. 3d, the relative presence of 4−HB H2O decreases from 27% to 23% and 12% as the number of carbon chains increases from 2 to 6 and 12, whereas that of 1, 2, 3−HB H2O increases from 72% to 77% and 87%, respectively, indicating that the continuity of HB in H2O clusters has been disturbed. Moreover, both the oxygen and hydrogen density distributions along the z direction, which are regarded as measurements of gas diffusion tunnels, tend to decrease with increasing carbon chain length (Fig. 3e–g), suggesting broadened channels for gas diffusion37,38. In addition, the decreased statistical number of HBs over the whole model with increasing carbon number of the ligands further verifies that the continuity of the HBs has been reduced (Supplementary Fig. 32, 33). Taken together, these results demonstrate that the accessibility of C2H2 could be increased via alkanethiol modification, thus enabling a moderate C2H2/H2O ratio. Therefore, Cu-xSH-NAs catalysts are suitable for C4H6 synthesis through the EAH process.

a The average electrolyte (1 M KOH) droplet contact angle of the ligand-modified Cu NAs. The insets show photographs of the water droplets. b 3D reconstructed microstructure derived from fluorescence spectroscopy of Cu-based catalysts with different modified ligands. The fluorescence intensity represents the height of the penetrated electrolytes. Scale bars, 100 μm. c Corresponding z-axis fluorescence intensity line scans of labeled regions. All “a.u.” represents arbitrary units. d Percentages of different types of interfacial water structures over different catalysts determined from the MD models. e–g Schematic of the interfacial model of the Cu-xSH-NA (x = 2, 6, 12) surface and the corresponding O and H densities along the z-axis direction. Green: Cu, red: C, blue: H, yellow: S, light orange: O. Visuals were produced with VESTA44, blender, and origin. h Contour plots of C4H6 FE over a Cu-based catalyst with different modified ligands. Partial current density (i) and Tafel analysis (j) for C2H4 and C4H6 production over Cu-based catalysts with different modified ligands. The error bars represent the standard deviation from at least three independent measurements. (R: 1.3 ± 0.3 Ω). Source data are provided as a Source Data file.
The optimization of the aforementioned catalysts is executed in a flow cell with a gas diffusion electrode under potentiostatic conditions using pure C2H2 as the feed gas. First, the performance of the Cu-xSH-NAs was evaluated to explore the appropriate length of carbon chains (Supplementary Figs. 34−36). As shown in Fig. 3h, i, the Cu-12SH-NAs deliver the highest FE and \({j}_{{C}_{4}{H}_{6}}\). In addition, although both \({j}_{{C}_{2}{H}_{4}}\) and \({j}_{{C}_{4}{H}_{6}}\) exhibit a volcano-like profile along with the length of the carbon chains, the peak of \({j}_{{C}_{2}{H}_{4}}\) is located at a carbon number of 10 (Fig. 3i), which is more hydrophilic than that of \({j}_{{C}_{4}{H}_{6}}\), responding to the aforementioned difference in the required *C2H2/*H ratio towards C2H4 and C4H6 in the reaction analysis. Next, to further link moderate wettability to optimum C4H6 production, an electrokinetic evaluation was conducted through Tafel analysis (Supplementary Figs. 37, 38)33. As shown in Fig. 3j, the Tafel slopes for both C2H4 and C4H6 first decrease but then increase with increasing hydrophobicity. The decreasing region indicates faster C2H2 hydrogenation kinetics because of a more sufficient C2H2 supply. Similarly, in the increasing section, the greater the Tafel slope is, the slower the kinetics of the EAH are, which is caused by an insufficient *H supply33. Note that the fastest kinetics for C2H4 and C4H6 are located at the same position as the peak point of \({j}_{{C}_{2}{H}_{4}}\) and \({j}_{{C}_{4}{H}_{6}}\) in Fig. 3i, further confirming that a gap exists in the required *C2H2/*H ratio toward the electrosynthesis of C2H4 and C4H6. Thus, the moderate wettability generated by the Cu-12SH-NAs could lead to a predominance of C4H6 production.
Electrocatalytic acetylene dimeric hydrogenation towards C4H6
Since a suitable C2H2/H2O ratio is highly important for C4H6 production, acetylene at different concentrations (60%−100%) was introduced to optimize the performance of C4H6 electrosynthesis over the selected Cu-12SH-NAs (Supplementary Figs. 39−43). First, with 80% C2H2 as the optimized feed gas, the highest C4H6 FE of 65.3% was achieved at −0.4 V vs. RHE, and the corresponding C4H6 yield rate reached 0.49 mmol mg−1 h−1 with a current density of 100 mA cm−2 (Fig. 4a), which is competitive with recent reported electrocatalytic processes regarding C4H6 as a byproduct and comparable to the most recently reported case for C4H6 production25 (Supplementary Figs. 44, 45 and Supplementary Tables 2, 3). Notably, the quantification of the EAH process revealed that C4H6 was the main C4 product and that the total selectivity of the generated butene and butane was ~1% (Supplementary Table 4). In addition, the FE of C4H6 reached above 40% in the potential range from −0.3 to −0.7 V vs. RHE over the Cu-12SH-NAs but was generally below 40% across the full test range of the Cu NAs (Supplementary Fig. 46), again supporting the conclusion that tailoring the C2H2/H2O ratio is quite important in the proposed C4H6 electrosynthesis strategy (Fig. 4b, c). Online differential electrochemical mass spectrometry (DEMS) was conducted in linear sweep voltammetry (LSV) mode to further analyse the EAH process. First, the LSV curves of the Cu-12SH-NAs and Cu NAs exhibited lower overpotentials than those of the commercial Cu NPs (Figs. 2a, 4d), suggesting that the nanoarray morphology significantly improves the mass transfer of the gaseous C2H2. The signal monitored by mass spectrometry (MS) provides more information about the origin of the electric current23,39. As shown by the MS signals at m/z = 39 and m/z = 28, the signals of C4H6 begin to increase (−0.23 V vs. RHE) after the C2H4 onset potential (−0.22 V vs. RHE) over the Cu NAs. However, the Cu-12SH-NAs deliver a more positive onset potential for C4H6 (−0.18 V vs. RHE) than for C2H4 (−0.19 V vs. RHE), indicating that the coupling kinetics increase after ligand modification. In addition, the slope of the ion current for C4H6 (1.51 × 10−12 V−1) over the Cu-12SH-NAs is greater than that over the Cu NAs (1.29 × 10−12 V−1), and the corresponding slope of C2H4 for the Cu-12SH-NAs (2.12 × 10−12 V−1) is lower than that for the Cu NAs (2.42 × 10−12 V−1), demonstrating the better ability for C4H6 generation over the Cu-12SH-NAs (Fig. 4e, f and Supplementary Fig. 47). Moreover, the MS signal of C4H6 over the Cu NAs displays a volcanic shape, which is similar to that of commercial Cu NPs and begins to decrease at −0.55 V vs. RHE (Figs. 2a, 4e), indicating that the coupling becomes kinetically unoptimized in the rapid hydrogenation region. Conversely, the C4H6 signal of the Cu-12SH-NAs presented a nearly monotonically increasing trend with negatively shifted potentials (Fig. 4f), demonstrating that tailoring the wettability of the catalyst surface could efficiently expand the kinetically favorable region of C4H6 generation. In addition, the results of the DEMS test under square wave potentials show that the C4H6 signal over the Cu-12SH-NAs remained almost unchanged with negatively shifted potentials, which is quite different from the decreased C4H6 signal observed for the Cu NAs, indicating better C4H6 selectivity and stability of the Cu-12SH-NAs, which is suitable for practical application under fluctuating renewable electricity (Fig. 4g and Supplementary Fig. 48). Furthermore, both the FE of C4H6 and the current density over the Cu-12SH-NAs remained unchanged within the error range during the 13 h continuous test at a potential of −0.4 V vs. RHE, suggesting robust durability (Fig. 4h and Supplementary Fig. 49).

a FE ratios of C4H6 to C2H4 at different concentrations. b, c Potential-dependent current densities and FEs of the obtained products over the Cu-NAs and Cu-12SH-NAs under 80% C2H2. d LSV curves of different catalysts. DEMS signals for C2H4 and C4H6 of the Cu NAs (e) and Cu-12SH-NAs (f) in LSV mode. The insets are partial enlarged detail of the onset potentials (g) DEMS signals for C4H6 of the Cu NAs and Cu-12SH-NAs under square wave potentials. All “a.u.” represents arbitrary units. h Continuous tests of the Cu-12SH-NAs catalyst for the EAH process for 13 h. The error bars correspond to the standard deviation of at least three independent measurements. Source data are provided as a Source Data file.
Mechanism of the high selectivity for C4H6
To determine the reasons for the increased C4H6 FE and selectivity after ligand modification, a series of characterizations was performed. Specifically, in situ Raman and in situ attenuated total reflectance−Fourier transform infrared (ATR−FTIR) spectroscopy with high surface species sensitivity was conducted to evaluate the status and coverage of C2H2 and the corresponding intermediates on the catalytic surface. As shown in Fig. 5a, b, the signals of both ν(C≡C) (~1700 cm−1)10,40 and ν(π bond of C≡C) (~1950 cm−1)23,41 over the Cu-12SH-NAs remained more notable than their Cu NAs counterparts at each potential, indicating improved C2H2 mass transfer and enrichment effects over the Cu-12SH- NAs due to the designed hydrophobic−aerophilic interface. In addition, a characteristic peak located at ~2100 cm−1, attributed to a CH2=CH‒C≡CH species (Supplementary Fig. 50)42, an isomer of the directly coupled CH=CH−CH=CH intermediate, could be observed only over the Cu-12SH-NAs, experimentally confirming the C‒H pathway and suggesting that the coupling kinetics are enhanced after ligand modification (Fig. 5a, b). In addition, the signals of reactants (C2H2, H2O) and product-relevant intermediates (*C4H6, and *C2H3) were also detected via in situ ATR−FTIR spectroscopy (Fig. 5c, d and Supplementary Figs. 51, 52). Accordingly, the integrals of the IR bands are directly proportional to the coverage of the respective surface adsorbate, and the area ratio between ν(C≡C) and δ(H−O−H) has been used as a descriptor of the relative coverage ratio of C2H2 and H2O over the catalyst surface23,43. The plot of ν(C≡C)/δ(H−O−H) over the Cu-12SH-NAs was larger than that over the Cu NAs, demonstrating that the coverage ratio between C2H2 and H2O after ligand modification is greater and more suitable for C4H6 generation over the whole applied potential range (Fig. 5e)23. Similarly, the higher value of ν(*C4H6)/ν(C C), accompanied by the lower value of ν(*C2H3)/ν(C≡C) over the Cu-12SH-NAs than over their Cu NAs counterparts, indicates that more C2H2 feedstocks are converted to the target C4H6 after modification, further accounting for the enhanced C4H6 FE and selectivity (Supplementary Fig. 53). Finally, the in-depth origins of the enhanced C4H6 selectivity obtained over the Cu-12SH-NAs are summarized in Fig. 5f. The C2H2 feedstocks can be enriched over the hydrophobic−aerophilic interface caused by ligand modification, consequently resulting in moderate C2H2/H2O coverage. The lower C‒H pathway energy barrier due to the suitable C2H2/H2O ratio accelerated the C‒C coupling process, thus leading to satisfactory C4H6 selectivity and FE over Cu-12SH-NAs.

In situ Raman spectra of the Cu-12SH-NAs (a) and Cu NAs (b) in a C2H2-saturated electrolyte (633 nm laser, from −0.3 V to −0.8 V versus RHE in 1 M KOH). In situ ATR−FTIR spectra over the Cu-12SH-NAs (c) and Cu NAs (d) when C2H2 was used as the feed gas (from −0.3 V to −0.8 V versus RHE in 1 M KOH). All “a.u.” represents arbitrary units. e Area ratios of absorbed C2H2 to H2O, C4H6 to C2H2, and C2H4 to C2H2. f Schematic illustration of the mechanism of enhanced C4H6 production over Cu-12SH-NAs. Green: Cu, red: C, blue: H, light orange: O. Visuals were produced with VESTA44 and blender. Source data are provided as a Source Data file.
A sustainable proof-of-concept application in a 50 cm2 two-electrode system
Encouraged by this, we implemented the Cu-12SH-NAs catalyst in a reactor with a 50 cm2 geometric electrode (Fig. 6a). Notably, moist C2H2, which benefits C2H2/H2O coverage regulation, is directly injected into the cathode chamber. As a result, the C4H6 FE approached and remained above 50% in the voltage range from 1.9 to 2.2 V (Fig. 6b). This performance (~2.0 V at 1.0 A) is retained for at least 13 h of continuous operation and 325 mmol C4H6 was obtained (Fig. 6c and Supplementary Figs. 54, 55). Note that the observed C4H6 FE fluctuations arise as a result of the permeation of the anode electrolyte following a simple drying regeneration process (Supplementary Figs. 56, 57). Although the 13 h stability is far from the criterion for industrial application, the periodic cleaning and drying operation could refresh the selectivity without obvious attenuation, verifying the reliability of the proposed selectivity modulation principles. Given the promising performance of the Cu-12SH-NAs catalyst, a life cycle assessment (LCA) of CO2 emissions from cradle to gate was then performed to evaluate the environmental feasibility of this new oil-independent C4H6 synthesis technology (Supplementary Note 3 and Supplementary Table 5). The amount of CO2 emissions per kilogram of C4H6 as a function of electricity carbon intensity is shown in Fig. 6d. Fortunately, our strategy is greener than traditional petroleum-cracking strategies when the carbon intensity of the electricity is lower than 110 g of CO2 equivalent per kWh, which can be reached via the use of renewable electricity (such as solar and wind electricity)21. In addition, the results of the preliminary techno-economic analysis (TEA) show that the C4H6 electrosynthesis process from coal-derived acetylene, which uses the parameters achieved in the present work, is profitable (Supplementary Fig. 58, Supplementary Note 4 and Supplementary Table 6). The abovementioned results indicate that the proposed C4H6 electrosynthesis strategy is environmentally friendly and has economic potential, and deserves to be developed.

a Illustration of the enlarged two-electrode system. b Performance of the Cu-12SH-NAs (without iR correction) over the two-electrode reactor. Commercial Ti felt coated with iridium oxide was used as the cathodic material. c Chronopotentiometric stability test of the Cu-12SH-NAs at a current of 1.0 A in the enlarged two-electrode reactor at room temperature for 13 h. d CO2 emission of C4H6 production through the EAH process for coal-derived acetylene (red line) with electricity from different sources and different carbon intensities, and the reference CO2 emission of C4H6 production via the traditional oil route. CO2e, CO2 equivalent. Source data are provided as a Source Data file.
Discussion
In summary, a primary reaction analysis based on a commercial Cu model catalyst is first proposed both theoretically and experimentally for C4H6 electrosynthesis through the EAH process. The combined results of the DFT calculations and controlled experiments show that C4H6 is generated by acetylene dimerization and the subsequent dimer hydrogenation process. In addition, further theoretical prediction demonstrates that the kinetics of the rate-limiting dimerization process is likely accelerated under the appropriate coverage of C2H2 and *H, leading to increased C4H6 production. A ligand-modifying strategy is then designed to enable an optimum *C2H2/*H ratio by adjusting the coverage of C2H2 and H2O. As a result, the selected Cu-12SH-NAs catalyst delivers up to 65.3% C4H6 Faraday efficiency at a current density of 100 mA cm−2, with a C4H6 production rate of 0.49 mmol mg−1 h−1. In addition, the mechanism exploration indicates that the C2H2 feedstocks are enriched, consequently resulting in moderate C2H2/H2O coverage suitable for C4H6 generation. As a proof-of-concept application, 325 mmol C4H6 is obtained through the proposed electrosynthesis strategy under 1.0 A for 13 h over Cu-12SH-NAs in a 50 cm−2 reactor. Furthermore, the results of cradle-to-gate LCA indicate that the proposed strategy is a potential alternative or complementary method to the traditional route from the view of carbon emission reduction. Our work not only provides a case study of a mechanism analysis-inspired method for selectively modulating chemical reactions but also demonstrates a facile petroleum-independent C4H6 electrosynthesis pathway.
Methods
Materials
Cu nanoparticles (10−30 nm, 99.9%, Meryer (Shanghai) Biochemical Technology Co., Ltd.), deuterium oxide (99.9%, Aladdin Co., Ltd), KOH (95%, Aladdin Co., Ltd), NaOH (99%, Aladdin Co., Ltd.), (NH4)2S2O8 (99%, Aladdin Co., Ltd), Cu mesh (99.95%), ethanol (99.99%, Aladdin Co., Ltd), ethanethiol (liquid, 98%, Aladdin Co., Ltd), 1-hexanethiol (liquid, 96%, Aladdin Co., Ltd), 1-decanethiol (liquid, 96%, Aladdin Co., Ltd), 1-dodecanethiol (liquid, 98%, Aladdin Co., Ltd.), 1-tetradecanethiol (liquid, 97%, Aladdin Co., Ltd), and 1-hexadecanethiol (solid, 97%, Aladdin Co., Ltd). Rhodamine B (99.7%, Aladdin Co., Ltd), gas diffusion layer carbon paper (GDL-CP, 28BC), proton exchange membranes (Nafion 117), and anion exchange membranes (FAA-3-PK-130) were purchased from Suzhou Sinero Technology Co., Ltd Ar and pure acetylene gas were purchased from Tianjin Taiya Gas Sales Co., Ltd All the chemicals used were of analytical grade and were used without further purification. Aqueous solutions were prepared by using ultrapure water (18.2 MΩ cm).
Materials characterization
The Bruker D8 Focus Diffraction System (Germany), using a Cu Kα radiation source (λ = 0.154178 nm), was conducted on the powder X-ray diffraction (XRD) acquisition. Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) were performed on an FEI Apreo S LoVac microscope (10 kV). The Raman spectra were obtained with a HORIBA LabRAM Odyssey Raman microscope under excitation with a 532 nm laser and a 633 nm laser at a power of 20 mW. Fourier transform infrared spectroscopy (FTIR) was performed on a Nicolet IS50 instrument with an MCT/A detector. The fluorescence spectra were obtained with a Nikon A1R+ microscope. The contact angles were measured via a Powereach JC2000C1 contact angle system (Shanghai Zhongchen Digital Technology Apparatus Co., Ltd.) at ambient temperature.
Synthesis of Cu NAs
The Cu meshes were ultrasonicated successively with acetone, diluted with hydrochloric acid for 15 min, and then with ethanol for 5 min 3 times. The pretreated Cu meshes were immersed in a mixture of aqueous solutions of 2.5 M NaOH and 0.13 M (NH4)2S2O8 for 10 min. Cu NAs with a typical diameter of ca. Two hundred nanometer grew on the Cu meshes. The obtained Cu NAs were then removed and washed several times with ultrapure water. Finally, the Cu NAs were dried in a 60 °C vacuum drying oven overnight.
Synthesis of Cu-xSH-NAs (where x represents the number of carbon atoms in the alkyl chain)
The Cu NAs were submerged in a 10 mM thiol ethanol solution for 1 min to form a thiol layer. The obtained Cu-xSH-NAs were then removed and washed several times with ethanol. Finally, the Cu-xSH-NAs were dried in a 60 °C vacuum drying oven overnight.
Fabrication of the electrodes
The electrodes used in this work were fabricated via a cold pressing method. Commercial GDL-CP and Cu mesh with catalysts were cut into a square shape with a size of 2.5 × 2.5 cm2 and 2.2 × 2.2 cm2, respectively. Then, one piece of Cu mesh and GDL-CP were compressed together under 1 MPa pressure at room temperature for 20 s to form the electrode. The average catalyst loading was determined to be ~2−4 mg via the subtraction method. Besides, the commercial Cu NPs electrodes were fabricated via the traditional spin-casting method. Specifically, 2 mg of Cu NPs were dissolved in 1 mL of ethanol with sonication for 1 h. Then, 4 μL of Nafion solution (5 wt%) was added to the solution with sonication for another 30 min. The as-prepared solution was then spin-coated on the GDL-CP substrates with 1 mL on each substrate under a constant spin speed of 500 rpm.
Electrochemical measurements in the flow cell
Before the electrochemical measurements in the flow cell, the commercial GDL-CP and the Cu-xSH-NAs (or Cu NAs) were cut into square shapes with a size of 2.5 × 2.5 cm2 and then pressed together via a heating press. Electrochemical measurements were carried out as the steps reported in our previous work21,23. Specifically, the electrocatalytic performance was evaluated via a CS150H electrochemical workstation in a typical three-electrodes flow cell consisting of a working electrode (Cu NPs, Cu-xSH-NAs, or Cu NAs), a counter electrode(Pt foil), and a reference electrode (Hg/HgO, which was periodically calibrated with respect to a reversible hydrogen electrode in a high-purity hydrogen-saturated electrolyte). In addition, The Nafion 117 proton exchange membrane was used to separate the cathode and anode cells. The 1 M KOH solution was routinely prepared immediately before experimentation to prevent chemical degradation, to make sure that the pH value is around 13.6 ± 0.3, and a peristaltic pump was used to circulate the liquid phase. The mass flowmeter was conducted to control the gas flow rate. The working electrode was firstly fixed at the interface between the gas flow block and the cathodic electrolyte by conductive copper tape before the performance tests. Then, to achieve relatively stable and reliable performance evaluation, the electrochemical semihydrogenation of acetylene was conducted at different applied potentials (−0.3 to −0.8 V vs. RHE) for 10−20 min before injecting the outlet gases into the GC for quantitative analysis. Before LSV, the resistance (R) was measured first using a CS150H electrochemical workstation, and the R values were 1.3 ± 0.3 Ω. The current density of the Tafel slopes acquisition was iR corrected with a compensation level of 80%. The electrode potentials were rescaled to the RHE via the following Eq. (6):
Quantitative analysis of the C2H2 conversion, evolution rate, and FEs of the obtained products
The products were subjected to an Agilent 7890 A gas chromatograph equipped with columns, including plot-Q and 5 A molecular sieves; two detectors, a flame ionization detector (FID) and a thermal conductivity detector (TCD); and He as the carrier gas. The evolution rates of the different products were calculated via Eq. (7−9), and the FEs of the different products were calculated via Eq. 10. All the experiments with error bars were repeated three times.
X : The feedstock and products, including H2, C2H2, C2H4, C2H6, C4H6, and C4H8 (including 1-C4H8, (Z)-2-C4H8, and (E)-2-C4H8). p : Peak area of feedstock and products. t : The continuous time of electrocatalytic C2H2 hydrogenation (EAH). c : Concentrations of feedstock and products. m: The mass of the catalyst over the electrode. n: moles of feedstock and products. k: The slope of the calibration curves for feedstock and products. S: The gas flow rate. a : Electron transfer number. F : Faraday constant. Q: The total Coulomb number of the EAH.
Electrochemical operando online DEMS analysis
Operando online DEMS analysis was conducted according to our previous work21,23 and performed on the QAS 100 instrument provided by Linglu Instruments (Shanghai) Co., Ltd To clarify the selectivity issues more directly and clearly, operando DEMS experiments were conducted to monitor the distribution of the products during the on-stream reaction. Specifically, a negatively pressured quartz capillary was put into the gas channel of the flow cell used in the performance evaluation to insure that the gas at the flow cell outlet was directly injected into the DEMS instrument. Note that all the ion currents plotted in this work are provided without any correction or subtraction. The LSV test and rectangular wave potentials were applied from −0.1 to −0.9 V vs. RHE with a constant interval of 95 s using a CS150H electrochemical workstation. During the experiment, the flow rates of C2H2 gas and the electrolyte were set the same as those used for the electrochemical measurements parts.
Electrochemical in situ ATR−FTIR measurements
According to our previous work21,23, the in situ ATR−FTIR measurements were performed on a Nicolet 6700 FTIR spectrometer equipped with an MCTA detector using silicon deposited with gold film as the prismatic window and an ECIR-II cell purchased from Linglu Instruments. The Cu NAs or Cu-12SH-NAs served as the working electrode. Pt foil and Hg/HgO with an internal reference electrolyte were used as the counter and reference electrodes, respectively. A 1 M KOH solution was used as the electrolyte. The electrolyte was presaturated with pure C2H2 gas, and the gas was continuously bubbled through during the entire measurement. The spectrum was recorded every 30 s under an applied potential ranging from −0.3 to −0.8 V vs. RHE.
Electrochemical in situ Raman measurements
In situ electrochemical Raman spectra were recorded via an electrochemical workstation on a HORIBA LabRAM Odyssey Raman microscope under 633 nm laser excitation under controlled potentials following the procedure in our previous reports21,23. The homemade Teflon electrolytic cell equipped with a piece of round quartz glass for the incidence of lasers and protection of the tested samples was used in the experiments. The electrolyte was pretreated with pure C2H2 gas to obtain C2H2-saturated KOH before the experiments. In addition, the working electrode was parallel to the quartz glass to maintain the plane of the sample perpendicular to the incident laser. The Pt wire was rolled to a circle around the working electrode to serve as the counter electrode. The reference electrode was Hg/HgO with an internal reference electrolyte of 1 M KOH. The spectrum was recorded under applied potentials ranging from −0.3 to −0.8 V vs. RHE.
Confocal laser scanning microscopy (CLSM)
The confocal laser scanning microscopy (Nikon A1R+, Japan) equipped with a confocal microscope was conducted to estimate the variation in available water at the interface. A 561 nm laser was used as the excitation source, the microscope was equipped with a ×20 objective lens, and the step size was 0.5 μm. One hundred microliters of fluorescein-labeled 1 M KOH was added to the confocal dishes, and then 5 × 5 mm2 Cu NAs, Cu-12SH-NAs, and Cu-16SH-NAs were placed on the liquid droplet with the catalyst side in contact with the electrolyte. The interfacial contact state of catalysts with different wettabilities can be directly reconstructed by analyzing a series of CLSM images at different depths within the catalyst layer.
Data availability
All data are available in the main text or the supplementary materials. Source data and supplementary data for optimized computational models are provided with this paper. Source data are provided with this paper.
References
Trotuş, I.-T., Zimmermann, T. & Schüth, F. Catalytic reactions of acetylene: a feedstock for the chemical industry revisited. Chem. Rev. 114, 1761–1782 (2014).
Qi, L. et al. Ethanol conversion to butadiene over isolated zinc and yttrium Sites grafted onto dealuminated beta zeolite. J. Am. Chem. Soc. 142, 14674–14687 (2020).
Chung, S.-H. et al. Origin of active sites on silica–magnesia catalysts and control of reactive environment in the one-step ethanol-to-butadiene process. Nat. Catal. 6, 363–376 (2023).
Makshina, E. V. et al. Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene. Chem. Soc. Rev. 43, 7917–7953 (2014).
Lee, M. G. et al. Bimetallic metal sites in metal–organic frameworks facilitate the production of 1-butene from electrosynthesized ethylene. J. Am. Chem. Soc. 146, 14267–14277 (2024).
Goldemberg, J. Ethanol for a sustainable energy future. Science 315, 808–810 (2007).
Chai, Y. et al. Control of zeolite pore interior for chemoselective alkyne/olefin separations. Science 368, 1002–1006 (2020).
Gomez, E., Yan, B., Kattel, S. & Chen, J. G. Carbon dioxide reduction in tandem with light-alkane dehydrogenation. Nat. Rev. Chem. 3, 638–649 (2019).
Wi, T.-U. et al. Upgrading carbon monoxide to bioplastics via integrated electrochemical reduction and biosynthesis. Nat. Synth., https://doi.org/10.1038/s44160-44024-00621-44166 (2024).
Xue, W. et al. Electrosynthesis of polymer-grade ethylene via acetylene semihydrogenation over undercoordinated Cu nanodots. Nat. Commun. 14, 2137 (2023).
Bond, R. L., Galbraith, I. F., Ladner, W. R. & McConnell, G. I. T. Production of acetylene from coal, using a plasma jet. Nature 200, 1313–1314 (1963).
Wang, S. et al. Highly efficient ethylene production via electrocatalytic hydrogenation of acetylene under mild conditions. Nat. Commun. 12, 7072 (2021).
Zhang, L. et al. Deprotonated 2-thiolimidazole serves as a metal-free electrocatalyst for selective acetylene hydrogenation. Nat. Chem. 16, 893–900 (2024).
Trotuş, I.-T., Zimmermann, T., Duyckaerts, N., Geboers, J. & Schüth, F. Butadiene from acetylene–ethylene cross-metathesis. Chem. Commun. 51, 7124–7127 (2015).
Shi, X. et al. Copper catalysts in semihydrogenation of acetylene: from single atoms to nanoparticles. ACS Catal. 10, 3495–3504 (2020).
Schobert, H. Production of acetylene and acetylene-based chemicals from coal. Chem. Rev. 114, 1743–1760 (2014).
Agbaba, Ö, Trotuş, I.-T., Schmidt, W. & Schüth, F. Light olefins from acetylene under pressurized conditions. Ind. Eng. Chem. Res. 62, 1819–1825 (2023).
Bu, J. et al. Selective electrocatalytic semihydrogenation of acetylene impurities for the production of polymer-grade ethylene. Nat. Catal. 4, 557–564 (2021).
Shi, R. et al. Room-temperature electrochemical acetylene reduction to ethylene with high conversion and selectivity. Nat. Catal. 4, 565–574 (2021).
Bai, L. et al. Efficient industrial-current-density acetylene to polymer-grade ethylene via hydrogen-localization transfer over fluorine-modified copper. Nat. Commun. 14, 8384 (2023).
Zhao, B.-H. et al. Economically viable electrocatalytic ethylene production with high yield and selectivity. Nat. Sustain. 6, 827–837 (2023).
Huang, L., Bao, D., Jiang, Y., Zheng, Y. & Qiao, S.-Z. Electrocatalytic acetylene hydrogenation in concentrated seawater at industrial current densities. Angew. Chem. Int. Ed. 63, e202405943 (2024).
Chen, F. et al. Ethylene electrosynthesis from low-concentrated acetylene via concave-surface enriched reactant and improved mass transfer. Nat. Commun. 15, 5914 (2024).
Bai, R. et al. Weak acetylene adsorption terminated carbon–carbon coupling kinetics on silver electrocatalysts. CCS Chem. 5, 200–208 (2022).
Teh, W. J. et al. Selective electroreduction of acetylene to 1,3-butadiene on iodide-induced Cuδ+–Cu0 sites. Nat. Catal. https://doi.org/10.1038/s41929-41024-01250-41920 (2024).
Fan, L. et al. Proton sponge promotion of electrochemical CO2 reduction to multi-carbon products. Joule 6, 205–220 (2022).
Crandall, B. S. et al. Kilowatt-scale tandem CO2 electrolysis for enhanced acetate and ethylene production. Nat. Chem. Eng. 1, 421–429 (2024).
Deng, W., Zhang, P., Seger, B. & Gong, J. Unraveling the rate-limiting step of two-electron transfer electrochemical reduction of carbon dioxide. Nat. Commun. 13, 803 (2022).
Su, L. et al. Electric-double-layer origin of the kinetic pH effect of hydrogen electrocatalysis revealed by a universal hydroxide adsorption-dependent inflection-point behavior. J. Am. Chem. Soc. 145, 12051–12058 (2023).
Goyal, A. & Koper, M. T. M. The interrelated effect of cations and electrolyte pH on the hydrogen evolution reaction on gold electrodes in alkaline media. Angew. Chem. Int. Ed. 60, 13452–13462 (2021).
Strmcnik, D. et al. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 5, 300–306 (2013).
Varela, A. S. et al. PH Effects on the selectivity of the electrocatalytic CO2 reduction on graphene-embedded Fe–N–C motifs: bridging concepts between molecular homogeneous and solid-state heterogeneous catalysis. ACS Energy Lett. 3, 812–817 (2018).
Zhuansun, M. et al. Promoting CO2 electroreduction to multi-carbon products by hydrophobicity-induced electro-kinetic retardation. Angew. Chem. Int. Ed. 62, e202309875 (2023).
Shi, R. et al. Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces. Nat. Commun. 11, 3028 (2020).
Ram, R. et al. Water-hydroxide trapping in cobalt tungstate for proton exchange membrane water electrolysis. Science 384, 1373–1380 (2024).
Li, P. et al. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat. Catal. 5, 900–911 (2022).
Wu, Y. et al. Tuning NO coverage promotes ampere-level electrosynthesis of a nylon-6 precursor. Preprint at researchsquare https://doi.org/10.21203/rs.3.rs-4791713/v1 (2025).
Wang, Y.-H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Cheng, C., Chen, F., Zhang, B., Zhao, B.-H. & Du, X. Promoting water dissociation and weakening active hydrogen adsorption to boost the hydrogen transfer reaction over a Cu-Ag superlattice electrocatalyst. Angew. Chem. Int. Ed. e202413897 (2024).
Matsuda, R. et al. Highly controlled acetylene accommodation in a metal–organic microporous material. Nature 436, 238–241 (2005).
Heaviside, J., Hendra, P. J., Tsai, P. & Cooney, R. P. Adsorption and polymerization of acetylene on oxide surfaces. A Raman study. J. Chem. Soc. Faraday Trans. 1. 74, 2542–2549 (1978).
Sheppard, N. The assignment of the vibrational spectra of the C4 hydrocarbons butyne−1, butene−1, and vinyl acetylene, to the normal modes of vibration of these molecules. J. Chem. Phys. 17, 74–78 (1949).
Moon, J. et al. Discriminating the role of surface hydride and hydroxyl for acetylene semihydrogenation over ceria through in situ neutron and infrared spectroscopy. ACS Catal. 10, 5278–5287 (2020).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
The authors are grateful to the National Key Research and Development Program of China (2024YFA1510100 to B.Z. and B.H.Z.) and the National Natural Science Foundation of China (22209120 to B.H.Z.). This work is also supported by the Fundamental Research Funds for the Central Universities of China.
Author information
Authors and Affiliations
Contributions
B.Z. conceived the idea and directed the research. B.H.Z. and B.Z. designed the experiments. C.C., F.C., and J.W. synthesized the materials and carried out most of the experiments. J.W., Y.Han., and Y.He. assisted with the experiments. C.C. contributed to the theoretical calculations. B.H.Z., C.C., and B.Z. analysed the data. B.H.Z. and F.C. wrote the paper. B.Z. Revised the paper with comments from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, C., Wang, J., Chen, F. et al. Selective electrosynthesis of 1,3-butadiene by tailoring the coverage of acetylene and water. Nat Commun 16, 5685 (2025). https://doi.org/10.1038/s41467-025-60881-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60881-4
