
Abstract
Development of contra-steric alkene hydrometallation method is one of the keys in branched product synthesis, especially when a bulky ligand steric environment is essential at other stages of the catalytic cycle. This study presents our efforts in this regard with broad scope using N-heterocyclic carbene (NHC), and establishes a chiral (NHC)Ni(II)-catalyzed intermolecular cross-hydroalkenylation system of enamine (alkenyl-carbamate/phthalimide) and α-olefin for the synthesis of highly substituted branched allylamines. A sterically adaptable chiral NHC-Ni(II) catalyst design with an open quadrant enhances interaction with the heteroatom on enamine, offering a precise chemo-, regio-, and enantioselective insertion control throughout the process in a mixture of olefins and allowing convergent synthesis. Besides, α-olefin serves as both terminal and internal 2-alkenyl reagents, yielding a 1,2,3-substituted allylamine by regio- and E-selective isomerization of the 1,2-disubstituted allylamine. Overall, the success of this alkene-based strategy addresses the intrinsic problems associated with alkyne- and alkenyl halide reagent-based synthesis, demonstrates good versatility and scope, and addresses the shortcomings of conceivable approaches based on rigid and sterically bulky NHCs.
Similar content being viewed by others
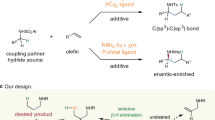
Introduction
The catalytic enantioselective synthesis of allylamines from basic substrates is an essential tool in organic synthesis1. Despite recent advances from alkyne-/alkenyl reagent-based C-C cross-couplings (Fig. 1a, i-ii)2,3,4,5,6,7,8,9,10,11,12,13 and related C-N forming aminations (Fig. 1b, i)1,14,15,16,17,18,19,20,21,22, the catalytic asymmetric synthesis of allylamines with two adjacent branched carbon centers (double branched product) is a resilient problem. Remarkable examples were demonstrated by Ni hydride catalyst on a terminal enamine with o-methoxybenzamide and indenyl halide11 in 77% e.e. or with carbazole and cyclohexenyl triflate13 in 96% e.e., while a Pd catalyzed intramolecular Heck reaction was applied on the cyclic enamine with a tether23 in 86% e.e. Regrettably, both of the above approaches rely on the stoichiometric synthesis of terminal and internal 2-alkenyl halide/ metallic reagents as alkenyl group donors and specialized N-protective groups, often reducing the overall atom- and step-economy. Replacing the preactivated alkenyl reagents or alkynes with simple alkenes is an ideal solution in allylamine synthesis. However, the scope is limited to aromatic olefins in both acyclic imine cross-coupling24,25,26 and asymmetric aza-Prins reaction with benzofused N-sulfonyl iminoester27 (single branched product except in an indene case). Hence, developing streamlined alkene-based intermolecular methods that can offer highly branched chiral allylamines generally from simple alkenes and cyclic/acyclic enamines is crucial for the complementary and sustainable growth of the field.

a Alkyne based strategies. b Alkene based strategies. c Contra-steric, chemo- & regio-selective hydrometallation (HM, iv vs i-iii).
In essence, achieving a highly selective and redox-neutral cross-hydroalkenylation (cross-HA)28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43 of enamine 1 (alkenyl-phthalimide/carbamate as olefin acceptor) and α-olefin 2 (as olefin donor) presents an ideal route for synthesizing 1,2- and 1,2,3-substituted allylamines (Fig. 1b, ii). However, this remains as a tough mission despite recent advances in NHC ligand designs28,31,32,34,35,36,39,40,41,42,44,45,46,47,48,49,50,51,52,53,54 and the methods in controlling olefin isomerization55,56,57,58. The primary challenges lie in guiding the catalytic enantio-, chemo-, and regio-selective insertion reactivity in a complex alkene mixture while simultaneously upholding high product selectivity and stability amidst several competitive and reversible insertions29,30,31,33,43,44,59,60. In particular, a contra-steric chemoselective b-HM of a sterically bulkier alkene (enamine 1) over a number of sterically readily accessible competitors is critical (Fig. 1c). Maintaining a high branch over linear carbometallation regioselectivity is crucial in order to give a sterically more demanding allylamine 3 with two consecutive branched centers. Complicating matters further, a slightly less selective HM can consume the substrate by isomerization55,56,57,58 and oligomerization61 undesirably. These complications have dragged the cross-HA progress as they render typical optimizations by managing substrate ratios from time to time ineffective.
In this work, we present our efforts in developing a contra-steric and chemo-selective HM strategy for NHC-Ni catalysts. It simplifies the synthesis of allylamines with adjacent branched carbon centers, offering applications for the synthesis of allylamines 3 by chiral NHCs and optionally E-3' by E-selective isomerization.
Results
System development
We began by evaluating achiral NHCs for the desired cross-HA reactivity of the sterically bulky vinylphthalimide 1a and 1-octene 2a (Table 1 and Fig. 2). This study unveiled a cross-HA condition of 1a and 2a, yielding 1,2-substituted allylamine 3aa featured with two consecutive branched carbon centers and a gem-olefin selectively, without noticeable 1a homo-dimerization (cross-:homo-HA of 1a > 95:5 in all entries). Our findings underscored the influence of both the o- and p-substituents of the N-aryl NHC on this reactivity. First, the size of the N-aryl o-substituents is one of the keys in controlling the b-/l-HM, as reflected by the ratio of allylamine 3aa to homoallylamine 4aa in cases of L1-4 (See Supplementary Information pages 19-20 for a detailed discussion)32,34,35,36,37,38,39,45,52. These structural demands essentially eliminated several privileged chiral NHCs46 with bulky rigid N-aryls6 during the enantioselective version development, showing that unlike styrene, diene, or vinylether40,58, the polarized enamine alone is inadequate to promote a contra-steric chemoselective b-HM with bulky NHCs. Second, we discovered that even a small p-Me group on N-aryl could alter the preferred substitution pattern of allylamine products, and the use of saturated NHC core could limit the isomerization to some extent (L4 vs L5). Overall, these two sets of discoveries laid the foundation for the divergent olefin synthesis of 1,2- and 1,2,3-substituted allylamines from the same set of substrates on demand (up to 94% yield of 3aa, and 87% yield of E−3'aa by isomerization cascade), representing a highly selective intermolecular cross-HA example using an α-olefin as both terminal and internal 2-alkenyl reagents (Fig. 1b, ii), and offering hints for the chiral version development.

a NHCs. b Enamine derivatives.
Next, we noted the broad applicability of our findings across challenging scenarios (Table 2). First, our investigation focused on exploring the impact of α-olefin donors on the cross-HA scope, as well as the contra-steric HM using 1a as an acceptor (Table 2a). The system exhibited robust performance across a diverse array of donors, often achieved at low substrate ratios (1:2 = 1:1.2 to 1:2). Representative α-olefins with distinct characteristics, like linear (2a), α-branched (2b, c), β-branched (2d–g), γ-branched (2h, i), and heteroatom-substituted cases (2j–r, Bpin, ether, dialkylamines, and tosylamines), consistently showed high reactivity, yield, and selectivity (3:4 = 90:10 to 95:5; 3:E-3' > 95:5 mostly. Cyclic alkene like cyclopentene is a possible donor for 1h in this reaction. However, it provided a homoallylamine in 23% yield (70% yield based on conversion) at 5 mol% [(L3)Ni(allyl)Cl]/NaBArF condition at 50 degree, instead of the allylamine target in this work). Since a lower concentration of 2 could facilitate the chemoselective HM of 1 as the plan suggested (Fig. 1c), a batch addition of 2 was a more effective way than a higher 2:1 ratio at the start to give 3 when the cross-HA was challenged by nonselective isomerization and homo-HA of 2 (e.g., 56 vs 70% yield of 3ak, from 38% under standard condition, 100% conversion of 2 was observed in all cases). Yet, the success relied on a slow homo-HA of 1 in this system. Second, we evaluated the scope of the enamines and the effect of N-protective groups (N-PG) (Table 2b). Unlike systems with nucleophilic alkenyl-M reagents based on bidentate and rigid ligands, the monodentate NHC and the non-aggressive alkenyl group donor employed here accommodated structurally diverse cyclic and internal enamines (Enamines with high Z-configuration were used directly in most cases. It was formed as major isomer according to the literature synthetic procedure).62 with sensitive (alkenyl)NR3R4 readily (Fig. 2b, e.g., those with phthalimides, NH, and NHBoc) by offering ample coordination space and mild C-C bond forming condition. Interestingly, the study also showed that the size of N-PG was not as crucial as expected for favoring the desired product selectivity. By using L3, 3 was always preferred over 4, even when using 1 with dissimilar steric demands and coordination ability (set 6-7), suggesting the chemoselective contra-steric HM step was directed mostly by matching the electronic property of the Ni(II)H and the polarized 1. Thus, the steric repulsion with N-PG created by bulkier NHC like L1 was tolerated in a number of cases. As such, L3 was replaced by L1 to suppress the undesired consumption of 2a when HM of cyclic and internal enamine was more difficult (e.g., 1c and 1h), and to increase the cross-t-t:t-h selectivity (e.g., 3ea and 3e'a). Yet, an optimal N-PG coordination to the Ni(II) catalyst was also essential (set 6 vs 7 vs 8). Despite the olefins on vinylamides and vinylcarbamates 1e-1e''' showed very similar 1H NMR chemical shift, no 3 was obtained from vinylamide cases. As there was no conversion of 2a, the result suggested a deactivation of catalyst by vinylamide. Third, we examined the scope of the one-pot cross-HA and gem-olefin isomerization (Fig. 3). The phenomenon as discovered in the divergent preparation of 3aa and E-3'aa by L4 in Table 1 at 80 °C proved to be quite general. The amounts of 3 (3:E-3' up to <5:95) and 4 ( < 8%) were maintained at low levels55. Overall, our one-step synthesis stands out for its scope and directness, especially when compared to stepwise approaches that using internal/ terminal 2-alkenyl halides and also having the regio- and stereo-selectivity challenge in subsequent isomerization of isolated allylamines (e.g., E- vs Z- and alkenyl- vs allyl-amines).

a Condition in Table 1 was applied, except using 10 mol% [(L4)Ni(allyl)Cl]/NaBArF with 1.3 equiv. of 2 at 80 °C. No homo-HA of 1 was noted. b 5 mol% cat., at 50 °C, 2 equiv. of 2a. c 15 mol% cat. d 88% enamine conversion.
Enantioselective system
Based on our experiences from Tables 1–2, we postulated that a chiral C1 NHC design with an undecorated quadrant for enhancing hemilabile coordination and adapting steric demand might hold the key to overcoming the aforementioned challenges34,37,38,39. Indeed, our study revealed that both the cyclic and acyclic chiral allylamine syntheses became quite straightforward when we replaced the OTf anion used in styrene cross-HA42 before with BArF to enhance the desired reactivity (Table 3). First, we examined cyclic enamine 1h as the acceptor (Table 3a), envisioning streamlined syntheses of (-)-isoretronecanol, (-)-trachelanthamidine, and (-)−5-epitashiromine63 by offering 3hk as a key common intermediate in just 1 versus 6 linear steps. After a brief screening of chiral NHCs (L6-8) and catalyst generation methods (From allyl to cinnamyl-Cl. The reason is not entirely clear for why cinnamyl-Cl is better. The change provided a better material balance of substrate in the cross-HA. It probably facilitated the desired catalyst formation via a sterically more crowded allyl environment)42,51, we achieved the enantio- and chemo-selective contra-steric HM, and maintained high t-t to t-h cross-HA selectivity (i.e., product selectivity with two over one branched carbon centers) by 1h and 2a as the substrate pair. The use of a less coordinating anion was critical for achieving the desired reactivity, and the use of cinnamyl-Cl in catalyst preparation was essential for good reproducibility. Notably, the in situ generated C2 symmetric L6 and C1 L8 NHC catalysts yielded 3 in equally high e.e. at the same (R)-absolute configuration, despite L8 catalyst being used as a mixture of atropisomers ( ~ 80% yield for both Cl complexes by NMR). This suggested that those two catalysts shared a similar C2/pseudo-C2 chiral environment (not a full story, see discussion later), and the extra Me group on the L8 vs L6 was the key to give a higher yield based on the conversion of 1h. Next, the system was further challenged by various α-olefins 2, and we were delighted to see its high versatility with linear, α-/β-branched, and hetero-structures (3ha-3hs, 85–98% e.e.). Moreover, despite challenges posed by larger-sized rings (1h' and 1h'') and extra substituents (1i and 1j, a spiro-ring and an alkene substituent,), high e.e. values were consistently obtained using our strategy under slightly modified conditions (3h'a, 3h''a, 3h''k, 3ik, and 3jk, up to 99% e.e.).
For acyclic enamine cases, L8 continued to perform well (Table 3b). A representative acyclic internal enamine Z-1g featured a medium-sized and easily removable Boc on enamine, gave the best performance (3ga in 97% e.e. vs 3aa and 3ea in 4 and 75% e.e. from terminal enamines, and 3fa in 61% e.e. from internal enamine with NR3R4). Also, the e.e. was not so sensitive to the enamine E/Z-stereopurity (3ga, 96–97% e.e., using either Z−1g or a 9:1 mixture of Z/E−1g, no E−1g was left). These features removed two major restrictions associated with specialized N-protective groups and Z/E-olefin separation required in certain systems relying on rigid ligand environment. Remarkably, comparing the result from terminal enamine 1e and internal enamine E/Z-1g, the Me on E/Z-1g was one of the keys for achieving high e.e. and was not unnecessary (e.g., 3ga vs 3ea or 3gp vs 3ep, see later for discussion). The high e.e. could be recovered by using allylether as donor (94% e.e. in 3ek vs 75–78% e.e. in 3ea and 3ep), presumably due to a better control of chiral NHC N-aryl conformation via a Ni(II)-ether coordination. Next, Z-1g was selected as the representative acceptor for exploring the α-olefin scope. Gratifyingly, except for 2b with an α-branch, it demonstrated good to excellent reactivity, functional group compatibility (amide and ether), and chemo-, regio-, enantio-selectivity under our condition. Moreover, a high 3:E-3' ratio was generally obtained except for 1a with a phthalimide, and no drop was noticed when NHBoc was replaced by a sterically bulkier NMeBoc (Z-1f ~ Z-1g). This set of results suggested that the E-3' formation might be favored by a bigger N-PG but more likely relied on the optimal N-PG coordination to catalyze. Indeed, the in situ isomerization of the chiral allylamine 3 to E-3' by L8 catalyst was ineffective even it was run at a higher temperature when Boc was employed, and the chiral 3 to E-3' isomerization with high enantio-, regio- and E-selectivity required to be conducted by L4 under isolated condition and extended reaction time (Fig. 4). This result showed that the contra-steric HM efficiency of 1 over 3 could be distinguished by NHC and N-PG (Boc) combinations, ensuring a high 1,2-substituted allylamine purity in Table 3b.

a 3ga to E−3'ga. b 3hm to E−3'hm.
Discussion
The highly selective synthesis of the allylamine 3' involved two contra-steric chemoselective b-HM steps, with a strong steric repulsion requirement during the olefin carbometallation step in between. This was exceptional not only due to the precise and sequential insertion steric preference swaps, but also the remarkable chemo-, stereo-, and regioselective isomerization contrary to conventional expectations within a mixture of olefins56. At this stage, the results were explained by the N-PG hemilabile coordination to the (NHC)Ni(II) center (Fig. 5). First, the Ni(II)H was generated by a typical olefin insertion to the complex and β-H elimination. Then, enabled by slimmer and adaptive N-aryl NHC designs (We didn’t observe a significant difference between L3 and L4 in preliminary modeling of the isomerization or α-olefin oligomerization. The N-aryl p-Me group seems too far away to be responsible for making a decisive differences), this hemilabile coordination assisted a contra-steric chemoselective b-HM of the polarized enamine and saved the α-olefin from undesired consumption in the absence of benzylic36/allylic-stabilization seen in other methods (Fig. 5a). It also prevented the NHC from the undesired NHC-alkyl reductive elimination53, and compensated the loss of NHC steric volume required in chemo- and regio-selective α-olefin carbometallation. Conceivably, it further guided a cyclic hydrometallated species formation (Fig. 5b), creating higher conformational demands in β-Hendo vs β-Hexo elimination, causing steric repulsion among the exo-cyclic chain and the NHC, and finally favoring highly regio- and stereo-selective isomerization of 3 to E-3' (Additional isomerization experiments using isolated 3 as substrate were carried out. The sterically smaller NMeBoc and substrate lacking a Me group also strongly favor the E−3' over 3'' selectivity under forcing conditions, supporting the working hypothesis. See Supplementary Information pages 21-22 for more discussion). Consequently, the cross-HA of 1 and 2 as well as the isomerization of 3 to E-3' could occur in one-pot even though the isomerization of the sterically smaller α-olefin 2 was generally faster in other systems (Table 1 by L4 at r.t., 3' was detected even though the conversion of 1 and 2 was incomplete), and even most of the bulky L-Ni(I) hydride catalyzed gem-olefin hydrofunctionalizations strongly disfavor the contra-steric b-HM28. Overall, this catalytic cycle highlighted the great ability of our approach to differentiate among at least five types of olefins (1, 2, 3, 3', and 2 dimers/isomers) throughout the process (Fig. 5c), effortlessly merging two highly challenging synthetic sequences into one.

a Hemilabile coordination effect on selective hydrometallation. b Hemilabile coordination effect on regio- & E-selective isomerization. c Proposed NHC-controlled cross-HA and 3 to 3' isomerization catalytic cycle illustrated by enamine 1 and α-olefin 2.
The enantioselective version by L8 exemplified a prominent enamine structure-product e.e. relationship, encompassing a broad enamine scope for this reaction, including terminal, substituted cyclic, and E/Z-internal acyclic enamines. First, based on the similar e.e. obtained from C2 NHC L6 and C1 L8 in (R)-3ha synthesis (Table 3a), the prevalent preference from (L8)Ni(II)H was explained by the model in Fig. 6a41,42. Minimizing undesired steric interactions among the pseudo-C2 symmetric N-aryl o-Cy substituents on atropisomer A1 and the bulky N-PG on the substrate was crucial (Fig. 6a, i). Such an atropisomeric catalyst A1 with a single open quadrant design also facilitated a desirable coordination of the N-Boc to the catalyst, fulfilling the steric demands in various scenarios41,42 by tilting the N-aryl planes (Fig. 6a, i-iii). The E/Z-configuration of the enamine 2-position was not the key to control the e.e. according to the models (Fig. 6a, ii-iii), thereby simplifying efforts in preparing substrate at high E/Z-purity and furnishing a convergent synthesis.

a By sterically adaptive L8. b Effect of pseudo s-trans to s-cis configuration ratio of acyclic enamine.
Intriguingly, the above is a simplified model since L8 catalyst was not formed as a single atropisomer that shared the same anti-configuration as L6, and 4 possible isomers were involved (i.e., 2 × pseudo-C2 symmetric cases (A1 and A2) and 2 x cases with two o-Cy placed on the same side (A3 and A4); (A1 + A4)/(A2 + A3) = 58/42 was deduced based on initial L8 anti:syn-ratio and an assumption in the literature)47,48,54. This result suggested that A2-A4 were ineffective for catalyzing minor enantiomer (S)-3 formation. In particular, A1 and A3 favored (R)-3 by sharing a similar open quadrant for the N-PG coordination (Fig. 6a, i-iii c.f. iv, for cyclic and E/Z-1), while the o-Cy on A2 and A4 blocked (S)-3 formation by creating severe substituent repulsions (internal vs terminal-1 cases in Fig. 6a, v). Besides, A2 and A4 were deduced as minor isomers in the [(L8)Ni(cinnamyl)Cl] synthesis by crude NMR (Due to significant overlap of the 1H NMR signals and the difficulties in A1-4 complexes isolation, the atropisomeric ratio was deduced by the characteristic peaks at the upfield region (0 to -0.5 ppm, o-Cy CH). See Supplementary Information page 27 for details, (A1 + A3)/(A2 + A4) = 84/16). Consequently, the direct application of this in situ generated catalyst mixture did not harm the e.e. of internal enamine cases and it practically saved us from challenges associated with the preparation and separation of the atropisomeric L8 catalysts as well as the further complications related to the cinnamyl isomers (L6 complex ~ 44:40:16 by NMR, L8 complex was not resolved). Nevertheless, A2 and A4 could lower the e.e. by offering more space for adapting the N-PG excessive steric demand (Fig. 6a, v), especially when no substituent R on 1 was placed for o-Cy repulsions (e.g. terminal enamine with Nphth vs NHBoc in Table 3b, 3aa vs 3ea, 4% vs 75% e.e.) and when larger steric effect was applied on the same type of N-PG (Z-1g with N(Me)Boc vsZ-1f with NHBoc, 61% vs 97% e.e.). Based on the above, the e.e. change was not just a result of fine-tuning the N-PG coordination ability and size, but was also affected by the chiral NHC atropisomeric ratio and their relative reactivity towards different alkene substitutions. Second, the e.e. trend of acyclic 3 did not directly correlate to the steric bulk of NR3R4 on 1 (Fig. 6b, e.e. of 3ga > 3fa, but size 1f > 1g), since it could also be favored by a more defined enamine structure when a less rigid ligand design was used. A larger 1,3-allylic strain difference was conceivable to favor a higher pseudo s-trans to s-cis configuration ratio created by the enamine carbamate partial double bond. Hence, acyclic Z-1g and cyclic enamine 1h gave high e.e. by sharing a similar s-trans configuration, whereas Z-1f gave much lower e.e., despite being sterically closer to 1h than Z-1g. Similarly, as an extended pseudo s-trans configuration was favored in E-1g, there was only a slight e.e. decrease when a mixture of Z/E-1g vs pure Z-1g was used ( ~ 1% e.e. drop in 3ga). This s-trans demand contrasted with the s-cis demand in a number of enantioselective transition-metal catalyzed 1,3-diene hydrovinylation14,29,30,31,41,59,60,64, complementing approaches based on a dormant steric repulsion between the rigid ligand and N-protective group. And Figs. 6a and 6b together could better account for the dramatic e.e. drop when 1h was replaced by Z-1f. Overall, the above two models illustrate the aspects of expanding the highly enantioselective HM scope of enamines.
Lastly, the versatilities and scope of this simple alkene-based strategy are expected to streamline the bioactive amine synthesis and offer gem-olefins for Ni/NHC-catalyzed enantioselective hydroalkylation, or hydroarylation49,50 as well as other olefin post-modifications (Fig. 7, such as epoxidation, ozonolysis, and bromohydrin, see Supplementary Information pages 23–26 for details).

a Ketone. b Epoxide. c Bromohydrin.
In conclusion, herein we report a contra-steric, chemo- and enantio-selective HM strategy by enhancing interaction between the heteroatom on enamine and a sterically adaptable NHC-Ni(II) catalyst design with an open quadrant. By maintaining a sterically demanding regioselective carbometallation step, it achieves a catalytic intermolecular cross-HA of acyclic and cyclic enamine 1 and α-olefin 2 with broad scope under redox-neutral condition and yields two types of allylamines with two consecutive branched carbon centers 3 and 3'. Overall, this strategy emphasizes the needs of flexible chiral NHC designs in adapting to diversity challenges such as added steric repulsions and altered carbon frameworks in alkene insertion.
Methods
General procedure of the cross-hydroalkenylation (HA) for allylamine 3 synthesis
In a glove box, 1 (0.25 mmol) and 2 (0.5 mmol or other indicated amount) were added to the in situ generated [(L3 or L8)Ni(cinnamyl)]BArF catalyst solution sequentially. The mixture was stirred at 35 °C for 24 h. After work up, the yield, structure and selectivity of allylamine 3 were determined by NMR, HPLC, HRMS and isolation.
General procedure of the one-pot cross-HA and isomerization for allylamine E-3’ synthesis
The above procedure was followed except [(L4)Ni(allyl)]BArF was used as catalyst and the mixture was stirred at 80 °C for 24 h.
Data availability
The data generated in this study are provided in the Supplementary Information. The procedure and characterization data of new compounds that support the findings of this study are included in this article and its supplementary files. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Trowbridge, A., Walton, S. M. & Gaunt, M. J. New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem. Rev. 120, 2613–2692 (2020).
Standley, E. A., Tasker, S. Z., Jensen, K. L. & Jamison, T. F. Nickel catalysis: synergy between method development and total synthesis. Acc. Chem. Res. 48, 1503–1514 (2015).
Ohashi, M., Hoshimoto, Y. & Ogoshi, S. Aza-nickelacycle key intermediate in nickel(0)-catalyzed transformation reactions. Dalton Trans. 44, 12060–12073 (2015).
Ortiz, E., Shezaf, J., Chang, Y. H. & Krische, M. J. Enantioselective metal-catalyzed reductive coupling of alkynes with carbonyl compounds and imines: convergent construction of allylic alcohols and amines. ACS Catal. 12, 8164–8174 (2022).
Wang, Y., He, Y. L. & Zhu, S. L. NiH-catalyzed functionalization of remote and proximal olefins: new reactions and innovative strategies. Acc. Chem. Res. 55, 3519–3536 (2022).
He, Y. L., Chen, J., Jiang, X. L. & Zhu, S. L. Enantioselective NiH-Catalyzed reductive hydrofunctionalization of alkenes. Chin. J. Chem. 40, 651–661 (2022).
Patel, S. J. & Jamison, T. F. Catalytic three-component coupling of alkynes, imines, and organoboron reagents. Angew. Chem. Int. Ed. 42, 1364–1367 (2003).
Patel, S. J. & Jamison, T. F. Asymmetric catalytic coupling of organoboranes, alkynes, and imines with a removable (trialkylsilyloxy)ethyl group-direct access to enantiomerically pure primary allylic amines. Angew. Chem. Int. Ed. 43, 3941–3944 (2004).
Zhou, C. Y., Zhu, S. F., Wang, L. X. & Zhou, Q. L. Enantioselective nickel-catalyzed reductive coupling of alkynes and imines. J. Am. Chem. Soc. 132, 10955–10957 (2010).
Li, L., Liu, Y. C. & Shi, H. Nickel-catalyzed enantioselective α-Alkenylation of N-Sulfonyl amines: modular access to chiral α-branched amines. J. Am. Chem. Soc. 143, 4154–4161 (2021).
Cuesta-Galisteo, S., Schorgenhumer, J., Wei, X., Merino, E. & Nevado, C. Nickel-catalyzed asymmetric synthesis of α-arylbenzamides. Angew. Chem. Int. Ed. 60, 1605–1609 (2021).
Wang, J. W. et al. Nickel-catalyzed switchable site-selective alkene hydroalkylation by temperature regulation. Angew. Chem. Int. Ed. 61, e202205537 (2022).
Liu, C. F. et al. Synthesis of tri- and tetrasubstituted stereocentres by nickel-catalysed enantioselective olefin cross-couplings. Nat. Catal. 5, 934–942 (2022).
Flaget, A., Zhang, C. & Mazet, C. Ni-catalyzed enantioselective hydrofunctionalizations of 1,3-dienes. ACS Catal. 12, 15638–15647 (2022).
Li, G., Huo, X., Jiang, X. & Zhang, W. Asymmetric synthesis of allylic compounds via hydrofunctionalisation and difunctionalisation of dienes, allenes, and alkynes. Chem. Soc. Rev. 49, 2060–2118 (2020).
Chen, J. H., Guo, J. & Lu, Z. Recent advances in hydrometallation of alkenes and alkynes via the first row transition metal catalysis. Chin. J. Chem. 36, 1075–1109 (2018).
Chen, J. H. & Lu, Z. Asymmetric hydrofunctionalization of minimally functionalized alkenes via earth abundant transition metal catalysis. Org. Chem. Front. 5, 260–272 (2018).
Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287–4296 (2020).
Pawlas, J., Nakao, Y., Kawatsura, M. & Hartwig, J. F. A general nickel-catalyzed hydroamination of 1,3-dienes by alkylamines: catalyst selection, scope, and mechanism. J. Am. Chem. Soc. 124, 3669–3679 (2002).
Kaper, T. et al. Intermolecular hydroaminoalkylation of propadiene. Chem. Eur. J. 26, 14300–14304 (2020).
Bahena, E. N., Griffin, S. E. & Schafer, L. L. Zirconium-catalyzed hydroaminoalkylation of alkynes for the synthesis of allylic amines. J. Am. Chem. Soc. 142, 20566–20571 (2020).
Kang, T. et al. Alkene difunctionalization directed by free amines: diaminesynthesis via nickel-catalyzed 1,2-carboamination. ACS Catal. 12, 3890–3896 (2022).
Kiewel, K., Tallant, M. & Sulikowski, G. A. Asymmetric heck cyclization route to indolizidine and azaazulene alkaloids: synthesis of (+)-5-epiindolizidine 167B and indolizidine 223AB. Tetrahedron Lett. 42, 6621–6623 (2001).
Liu, X. H. & Feng, X. M. Dual nickel and brønsted acid catalysis for hydroalkenylation. Angew. Chem. Int. Ed. 57, 16604–16605 (2018).
Xiao, L. J. et al. Nickel(0)-catalyzed hydroalkenylation of imines with styrene and its derivatives. Angew. Chem. Int. Ed. 57, 3396–3400 (2018).
Li, Y., Zhang, X. S., Zhu, Q. L. & Shi, Z. J. Olefinic C–H bond addition to aryl aldehyde and its N-sulfonylimine via rh catalysis. Org. Lett. 14, 4498–4501 (2012).
Liu, R. R. et al. Nickel-catalyzed enantioselective addition of styrenes to cyclic N-sulfonyl α-ketiminoesters. ACS Catal. 5, 6524–6528 (2015).
Lee, B. C. et al. N-Heterocyclic carbenes as privileged ligands for nickel-catalysed alkene functionalisation. Chem. Soc. Rev. 52, 2946–2991 (2023).
Hilt, G. Hydrovinylation reactions – atom-economic transformations with steadily increasing synthetic potential. Eur. J. Org. Chem. 2012, 4441–4451 (2012).
RajanBabu, T. V. Asymmetric hydrovinylation reaction. Chem. Rev. 103, 2845–2860 (2003).
Hirano, M. Recent advances in the catalytic linear cross-dimerizations. ACS Catal. 9, 1408–1430 (2019).
Ho, C.-Y., He, L. S. & Chan, C. W. NHC-NiH reactions with simple olefins: advances in hydroalkenylation, polymerization, and ionic-liquid synthesis. Synlett 12, 1649–1653 (2011).
Ho C.-Y., Raja, D. Alkene hydrovinylation, hydroalkenylation, and cycloisomerization. In: Science of Synthesis. (Georg Thieme Verlag KG, 2023).
Ho, C.-Y. & He, L. S. Medium-sized heterocycle synthesis by the use of synergistic effects of NI-nhc and γ-coordination in cycloisomerization. J. Org. Chem. 79, 11873–11884 (2014).
Ho, C.-Y. & He, L. S. Shuffle off the classic β-Si elimination by Ni-NHC cooperation: implication for C–C forming reactions involving Ni-alkyl-β-silanes. Chem. Commun. 48, 1481–1483 (2012).
Ho, C.-Y. & He, L. S. Catalytic intermolecular tail-to-tail hydroalkenylation of styrenes with α olefins: regioselective migratory insertion controlled by a nickel/N-heterocyclic carbene. Angew. Chem. Int. Ed. 49, 9182–9186 (2010).
Biswas, S., Zhang, A., Raya, B. & RajanBabu, T. V. Triarylphosphine ligands with hemilabile alkoxy groups: ligands for Nickel(II)-catalyzed olefin dimerization reactions. Hydrovinylation of vinylarenes, 1,3-Dienes, and cycloisomerization of 1,6-Dienes. Adv. Synth. Catal. 356, 2281–2292 (2014).
Zhang, A. B. & Rajanbabu, T. V. Fine-tuning monophosphine ligands for enhanced enantioselectivity. Influence of chiral hemilabile pendant groups. Org. Lett. 6, 1515–1517 (2004).
Gao, Y., Houk, K. N., Ho, C.-Y. & Hong, X. The mechanism and regioselectivities of (NHC) nickel(II) hydride-catalyzed cycloisomerization of dienes: a computational study. Org. Biomol. Chem. 15, 7131–7139 (2017).
Chen, W. H., Li, Y., Chen, Y. & Ho, C.-Y. NHC)NiH-catalyzedregiodivergent cross-hydroalkenylation of vinyl ethers with α-olefins:syntheses of 1,2- and 1,3-disubstituted allyl ethers.Angew. Chem. Int. Ed. 57, 2677–2681 (2018).
Chen, Y., Dang, L. & Ho, C.-Y. NHC-Ni catalyzed enantioselective synthesis of 1,4-dienes by cross-hydroalkenylation of cyclic 1,3-dienes and heterosubstituted terminal olefins. Nat. Commun. 11, 2269 (2020).
Ho, C.-Y., Chan, C. W. & He, L. S. Catalytic asymmetric hydroalkenylation of vinylarenes: electronic effects of substrates and chiral N-heterocyclic carbene ligands. Angew. Chem. Int. Ed. 54, 4512–4516 (2015).
Li, Z. Q., Apolinar, O., Deng, R. H. & Engle, K. M. Directed Markovnikov hydroarylation and hydroalkenylation of alkenes under nickel catalysis. Chem. Sci. 12, 11038–11044 (2021).
Chen, Y., Chen, W. H., Gu, X. & Ho, C.-Y. Our voyage from catalytic cross-hydroalkenylation to transfer-dehydroaromatization of cyclic π-systems: reactivity and selectivity changes enabled by NHC-Ni and NHC-Pd hydride equivalents. Synlett 34, 1639–1654 (2023).
Cavell, K. J. & McGuinness, D. S. Redox processes involving hydrocarbylmetal (N-heterocyclic carbene) complexes and associated imidazolium salts: ramifications for catalysis. Coord. Chem. Rev. 248, 671–681 (2004).
Janssen-Muller, D., Schlepphorst, C. & Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 46, 4845–4854 (2017).
Luan, X. et al. Matching the chirality of monodentate n-heterocyclic carbene ligands: a case study on well-defined palladium complexes for the asymmetric α-arylation of amides. Org. Lett. 10, 5569–5572 (2008).
Xi, J., Ng, E. W. H. & Ho, C.-Y. Unsymmetric N-aryl substituent effects on chiral NHC-Cu: enantioselectivity and reactivity enhancement by ortho-H and Syn-configuration. ACS Catal. 13, 407–421 (2023).
Ma, J.-B., Zhao, X., Zhang, D. & Shi, S.-L. Enantio- and regioselective Ni-catalyzed para-C–H alkylation of pyridines with styrenes via intermolecular hydroarylation. J. Am. Chem. Soc. 144, 13643–13651 (2022).
Wang, Z.-C. et al. Enantioselective C–C cross-coupling of unactivated alkenes. Nat. Catal. 6, 1087–1097 (2023).
Fernández-Salas, J. A., Marelli, E., Cordes, D. B., Slawin, A. M. Z. & Nolan, S. P. General and Mild Ni0-catalyzed α-arylation of ketones using aryl chlorides. Chem. Eur. J. 21, 3906–3909 (2015).
Altenhoff, G., Goddard, R., Lehmann, C. W. & Glorius, F. An N-heterocyclic carbene ligand with flexible steric bulk allows suzuki cross-coupling of sterically hindered aryl chlorides at room temperature. Angew. Chem. Int. Ed. 42, 3690–3693 (2003).
Clement, N. D. & Cavell, K. J. Transition-metal-catalyzed reactions involving imidazolium salt/N-heterocyclic carbene couples as substrates. Angew. Chem. Int. Ed. 43, 3845–3847 (2004).
Chaulagain, M. R., Sormunen, G. J. & Montgomery, J. New N-heterocyclic carbene ligand and its application in asymmetric Nickel-catalyzed aldehyde/alkyne reductive couplings. J. Am. Chem. Soc. 129, 9568–9569 (2007).
Vasseur, A., Bruffaerts, J. & Marek, I. Remote functionalization through alkene isomerization. Nat. Chem. 8, 209–219 (2016).
Kapat, A., Sperger, T., Guven, S. & Schoenebeck, F. E-Olefins through intramolecular radical relocation. Science 363, 391–396 (2019).
Hilt, G. Double bond isomerisation and migration-new playgrounds for transition metal-catalysis. ChemCatChem 6, 2484–2485 (2014).
Wang, J. W. et al. Nickel-catalyzed remote asymmetric hydroalkylation of alkenyl ethers to access ethers of chiral dialkyl carbinols. J. Am. Chem. Soc. 145, 10411–10421 (2023).
Zhang, Z., Chen, Y., Gu, X. & Ho, C.-Y. NHC)Ni(II)-directedinsertions and higher substituted olefin synthesis from simple olefins.Acc. Chem. Res. 56, 1070–1086 (2023).
Biswas, S. et al. A new paradigm in enantioselective cobalt catalysis: cationic cobalt(I) catalysts for heterodimerization, cycloaddition, and hydrofunctionalization reactions of olefins. Acc. Chem. Res. 54, 4545–4564 (2021).
Nakamura, A., Ito, S. & Nozaki, K. Coordination-insertion copolymerization of fundamental polar monomers. Chem. Rev. 109, 5215–5244 (2009).
Hashimoto, T., Nakatsu, H., Takiguchi, Y. & Maruoka, K. Axially chiral dicarboxylic acid catalyzed activation of quinone imine ketals: enantioselective arylation of enecarbamates. J. Am. Chem. Soc. 135, 16010–16013 (2013).
Reddy, K. K. S., Rao, B. V. & Raju, S. S. A common approach to pyrrolizidine and indolizidine alkaloids; formal synthesis of (−)-isoretronecanol, (−)-trachelanthamidine and an approach to the synthesis of (−)-5-epitashiromine and (−)-tashiromine. Tetrahedron:Asymmetry 22, 662–668 (2011).
Duvvuri, K. et al. Cationic Co(I)-intermediates for hydrofunctionalization reactions: regio- and enantioselective cobalt-catalyzed 1,2-hydroboration of 1,3-dienes. J. Am. Chem. Soc. 141, 7365–7375 (2019).
Acknowledgements
We thank the National Natural Science Foundation of China (22471116, CYH and ZZ) and the Guangdong Provincial Key Laboratory of Catalysis (2020B121201002, CYH) for financial support. All the authors acknowledge the assistance of SUSTech Core Research Facilities.
Author information
Authors and Affiliations
Contributions
W. C. and Z. Z. performed the experimental studies and analysis. C.-Y. H. supervised the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, W., Zhang, Z. & Ho, CY. A contra-steric chemoselective hydrometallation strategy with bulky NHC for catalytic asymmetric branched allylamine synthesis. Nat Commun 16, 5670 (2025). https://doi.org/10.1038/s41467-025-60932-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60932-w
