
Abstract
Covalent organic frameworks (COFs) are promising materials for photocatalytic hydrogen peroxide (H2O2) production. However, optimizing their electronic structures to enhance charge separation, oxygen adsorption, and reaction efficiency remains a challenge. Here we show that incorporating thiophene and furan isomeric units into the side chains of COFs enables precise tuning of their electronic structures and photocatalytic activity. Thiophene-containing frameworks exhibit superior charge separation and photocatalytic performance compared to those with furan, owing to stronger donor–acceptor interactions. A 2-substituted thiophene-based COF (DT2TA-TAPB), synthesized from 1,3,5-tris(4-aminophenyl)benzene and 2,5-di(thiophen-2-yl)terephthalaldehyde, exhibits reduced exciton binding energy, extended electron lifetime, and improved spatial charge separation. Mechanistic analysis reveals that the sulfur and adjacent carbon atoms within the thiophene of DT2TA-TAPB stabilize the endoperoxide intermediate, promoting a one-step, two-electron pathway for H2O2 generation. Consequently, DT2TA-TAPB achieves H2O2 yields of 10972 and 8587 μmol g-1 h-1 in 10% ethanol and pure water, respectively, outperforming most reported COF-based photocatalysts.
Similar content being viewed by others
Introduction
Hydrogen peroxide (H2O2) is an essential chemical compound with extensive applications in the chemical industry and environmental treatment1,2. At present, the most mature industrial method for H2O2 production—the anthraquinone process—has high energy consumption and inevitably generates hazardous organic waste, which poses an environment threat3. By contrast, H2O2 generation via artificial photosynthesis from water and oxygen requires mild reaction conditions and offers energy-saving advantages, making it a highly promising alternative method4,5,6. Recently, researchers have developed a series of photocatalysts to facilitate H2O2 production, including carbon nitrides (g-C3N4)7,8,9, polymer resins10, and covalent triazine frameworks11. Nevertheless, the majority of these photocatalysts have unclear structures and lack flexibility for structural modification, severely restricting the in-depth understanding of the underlying mechanisms associated with their use in photocatalytic H2O2 generation.
Covalent organic frameworks (COFs), a category of porous crystalline materials linked by covalent bonds, have garnered widespread attention among researchers for their tunable structures, high specific surface areas, and semiconducting characteristics12,13,14. COFs were applied in photocatalytic H2O2 synthesis in 202015, and the filed has witnessed rapid development. Precise control of the COF skeleton offers an attractive platform for exploring the structure-performance relationship during photocatalytic H2O2 production. The molecular structure of COFs affects the electronic environment of the active site16,17, thereby impacting catalytic activity and the reaction pathway during photocatalytic H2O2 production. To this aim, researchers have recently proposed various strategies to develop advanced COF-based photocatalysts, including introducing functional groups18,19, designing donor-acceptor separated centers20,21,22,23, and doping24,25. Among them, designing COFs with different building blocks or linkers, such as sulfone units26, cyanide groups18, acetylene27, alkyl chain28, and s-heptazine20, to enhance catalytic activity has attracted great interest among researchers. Unfortunately, precisely modulating the electronic structure of the active sites to optimize the catalytic activity of COFs remains challenging.
Aromatic heterocycles with conjugated π-systems and excellent electron affinity provide superior optical and electrical properties compared to other structures29,30,31. The heteroatoms with lone pairs of electrons in the aromatic heterocycle interferes with sp2 hybridization of neighboring carbon atoms, altering the charge distribution of the heterocycle and facilitating the oxygen reduction reaction (ORR)32,33. The incorporation of aromatic heterocycles into COFs, such as bipyridines34, triazines11, and [1,2,4]-triazolo-[1,3,5]-triazines35, enhances photocatalytic activity during the photosynthesis of H2O2. Furthermore, the varying heteroatom positions within aromatic heterocycles provide opportunities to explore the influence of isomers on the photocatalytic activity of COFs. For instance, Xi et al. studied the impact of the relative position of nitrogen in N-heterocycles (pyridazine) on photocatalytic activity during ORR36, and Zhao et al. subsequently reported modulation of the photocatalytic ORR pathway by changing the relative position of nitrogen in bipyridine37. Owing to the diversity of N-heterocycles, current research has primarily focused on the effects of N-heterocyclic isomers on catalytic activity38. Nevertheless, aromatic heterocycles extend beyond N-heterocycles, and other heteroatoms (e.g., sulfur and oxygen) also influence the electronic structure and catalytic activity of COFs through different electronic effects and geometrical structures. In particular, the unique electronic properties of thiophene and furan, as representative sulfur and oxygen heterocycles, have contributed to their wide application in polymer semiconductors39,40. The incorporation of thiophene units effectively broadened the light absorption range and facilitated charge carrier separation41,42. In addition, thiophene isomers modulated electron transfer direction in electrocatalysis33, while benzofuran improved COFs structural stability43. However, despite the promising potential of thiophene and furan in photocatalytic H2O2 generation, the simultaneous regulation of heterocyclic conformation and heteroatom type to precisely tune the electronic structure has yet to be explored.
Herein, we propose the introduction of two pairs of thiophene and furan isomers (2- and 3-substituted) into the side chains of COFs to precisely modulate their electronic structure, thereby achieving high photocatalytic efficiency for H2O2 production. Four types of COFs were successfully synthesized by combining 1,3,5-tris(4-aminophenyl)benzene (TAPB) with various monomers, including 2,5-di(thiophen-2-yl)terephthalaldehyde (DT2TA), 2,5-di(thiophen-3-yl)terephthalaldehyde (DT3TA), 2,5-di(furan-2-yl)terephthalaldehyde (DF2TA), and 2,5-di(furan-3-yl)terephthalaldehyde (DF3TA), named DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB, respectively (Fig. 1). The introduction of heterocyclic isomers enabled effective regulation of the electronic structure of the COFs, their oxygen affinity, and the ORR pathway. The hole-electron theory calculations indicated that order of the Sr index was DT2TA-TAPB < DT3TA-TAPB < DF2TA-TAPB < DF3TA-TAPB, with DT2TA-TAPB exhibiting excellent carrier separation efficiency, which was ascribed to the forming of a better-matched donor-acceptor structure compared to the other developed COFs. The calculations indicated that the sulfur and neighboring carbon atoms in DT2TA-TAPB acted as active sites to stabilize the endoperoxide intermediate, directly generating H2O2 and improving ORR selectivity. As a result, DT2TA-TAPB exhibited a record-high H2O2 yield of 10972 μmol g-1 h-1 H2O2, surpassing the performance of most previously reported COF-based photocatalysts. This study offers fresh insight and inspiration for the rational design of functional COFs for photocatalytic H2O2 synthesis.

Schematic of the synthesis of DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB and the corresponding S1 excited-state electron distributions (blue: electrons, green: holes).
Results
Characterization of COFs
The crystal structures of the COFs were elucidated via powder X-ray diffraction (PXRD) spectroscopy and simulations conducted in Materials Studio44. The PXRD patterns revealed that most of the COFs demonstrated high crystallinity, as evidenced by the presence of sharp diffraction peaks. As depicted in Fig. 2a, the characteristic peak of DT2TA-TAPB was located at 3.20°, which was assigned to the (100) reflection. The crystallinity of DT2TA-TAPB was decreased and its characteristic peak was shifted compared to the other developed COFs. Additionally, several weak and unassigned diffraction peaks were observed, likely attributable to the significant steric hindrance of DT2TA molecules38,45. This steric effect interfered with molecular arrangement, disrupted the crystallization process and ultimately led to reduced crystallinity46. The PXRD pattern of DT3TA-TAPB exhibited diffraction peaks at 2.78°, 5.68°, and 7.55° (Fig. 2b). Similarly, peaks at 2.72°, 5.48°, and 7.26° are attributed to the PXRD pattern of DF2TA-TAPB (Fig. 2c), while those at 2.78°, 5.62°, and 7.46 are assigned to the PXRD pattern of DF3TA-TAPB (Fig. 2d). These peaks corresponded to the (100), (110), and (210) facets, respectively. Additionally, the diffraction peaks aligned well with the simulated structures obtained in Materials Studio using the AA stacking mode. Pawley refinements of the PXRD patterns of DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB exhibited the same unit cell parameters of a = b = 35.87 Å, c = 3.60 Å, α = β = 90°, and γ = 120°, while those of DT2TA-TAPB were a = b = 31.0 Å, c = 4.0 Å, α = β = 90°, and γ = 120°. The Brunauer–Emmett–Teller (BET) surface area and porosity of the COFs were determined by performing N2 adsorption-desorption measurements at 77 K. The adsorption-desorption isotherms of the COFs were classified as type-II curves, indicative of mesoporous characteristics (Fig. 2e). The calculated BET surface areas and pore size distribution fitted by nonlocal density functional theory (DFT) models were 296.9, 445.1, 479.8, and 1182.0 m2 g-1 and 1.48, 2.34, 2.34, and 2.34 nm for DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB, respectively (Supplementary Fig. 1)47.

Experimental and simulated PXRD patterns of a DT2TA-TAPB, b DT3TA-TAPB. c DF2TA-TAPB, and d DF3TA-TAPB. e N2 sorption isotherms of the COFs. f FT-IR spectra of the COFs.
The chemical structures of the COFs were identified by Fourier transform infrared (FT-IR) and solid-state 13C nuclear magnetic resonance (13C NMR) spectroscopies. The FT-IR spectra of the COFs exhibited the disappearance of C=O stretching bands at 1685 cm–1 and appearance of C=N stretching vibrations at approximately 1620 cm–1, confirming successful formation of C = N bonds (Fig. 2f and Supplementary Fig. 2). However, a weak C=O stretching vibrations peak observed in DT2TA-TAPB was likely a consequence of the significant steric from the 2-position thiophene units, which hindered the complete extension of framework, leading to the retention of a small fraction of aldehyde groups at the terminal positions of the COFs. Furthermore, the solid-state 13C NMR spectra exhibited signals at approximately 160 ppm, which corresponded to the C atoms in the C=N bonds of the COFs. Meanwhile, the solid-state 13C NMR spectra also exhibited the characteristic peaks of the heterocycle isomers, indicating their successful integration into the COFs (Supplementary Fig. 3). The scanning electron microscopy images revealed nanofiber and nanosheet morphologies in the COFs (Supplementary Fig. 4). Furthermore, the clear lattice stripes of the COFs were observed via high-resolution transmission electron microscopy, wherein the lattice spacings of 0.27, 0.35, 2.1, and 2.3 nm were assigned to the (100) plane of DT2TA-TAPB (Supplementary Fig. 5a), (201) plane of DT3TA-TAPB (Supplementary Fig. 5b), (110) plane of DF2TA-TAPB (Supplementary Fig. 5c), and (110) plane of DF3TA-TAPB (Supplementary Fig. 5d), respectively.
Photoelectrochemical properties of COFs
The separation of photogenerated charge carriers in the COFs was investigated via photoluminescence (PL) spectroscopy and photoelectrochemical measurements. The steady-state PL spectra revealed that DT2TA-TAPB had weaker emission intensity than DF2TA-TAPB, DF3TA-TAPB and DT3TA-TAPB (Supplementary Fig. 6). The charge carrier lifetimes were elucidated via time-resolved photoluminescence (TR-PL) spectroscopy. As depicted in Supplementary Fig. 7, the PL lifetimes of DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB were 0.60, 0.45, 0.35, and 0.29 ns, respectively. Notably, the longest PL lifetime observed for DT2TA-TAPB was attributed to its effective suppression of non-radiative recombination, which enhanced charge separation and facilitated the migration of photogenerated carriers to catalytic sites, thereby promoting photocatalytic activity48,49. Moreover, DT2TA-TAPB exhibited a markedly higher photocurrent density than the other COFs, highlighting its superior capability in facilitating the transport of photogenerated charge carriers (Supplementary Fig. 8). Electrochemical impedance spectroscopy further revealed that DT2TA-TAPB had lower charge transference resistance than the other COFs, which was consistent with the PL, and TR-PL measurement results (Supplementary Fig. 9). The above phenomena indicated that carrier separation and transportation was more favorable in the thiophene isomer-based COFs than in the furan isomer-functionalized COFs, whereas DT2TA-TAPB possessed more prominent carrier utilization than DT3TA-TAPB. Together, these results suggested that the separation efficiency of photogenerated carriers could be optimized by regulating the heteroatom type and heterocyclic conformation in COFs.
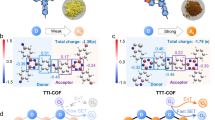
To interpret the discrepancy in electron transfer between the different COFs, we calculated the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of all relevant monomers. As presented in Supplementary Fig. 10, the HOMO and LUMO of the heterocycle-modified aldehyde monomers were lower than the corresponding orbitals of TAPB, enabling the construction of staggered structures for the donor-acceptor pairs50. Moreover, different heteroatom types and heterocyclic conformations of the aldehyde monomers affected the delocalized distribution of the molecule orbitals, which modulated the charge transfer efficiency and energy levels of the COFs. The thiophene isomer-based COFs exhibited more attractive catalytic activities than the furan isomer-based COFs, which was attributed to the thiophene-modified aldehyde monomers forming better-matched donor-acceptor structures with TAPB than the furan-modified aldehyde monomers, thus improving the carrier migration efficiency. Furthermore, to better understand the electron transfer of the COFs, their S1 excited-state electron distributions were calculated based on hole-electron theory. The distributions of electrons and holes of the developed COFs are displayed in Fig. 3a, revealing that the heteroatom position and type effectively regulate the electron and hole distributions. The calculated Sr index values of DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB were 0.068, 0.241, 0.367, and 0.697, respectively, with a lower Sr index reflecting higher carrier separation efficiency51. Therefore, the experimental and computational results indicated that the thiophene isomer-modified COFs exhibited superior electron separation ability compared to their furan isomers-functionalized counterparts.

a S1 excited-state electronic structures of DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB (blue: electrons, green: holes). Arrhenius fitting curves for b DT2TA-TAPB and c DT3TA-TAPB. Two-dimensional pseudo-color of fs-TA spectra of d DT2TA-TAPB and e DT3TA-TAPB. TA decay kinetic curves of f DT2TA-TAPB (Instrument response functions: 0.249 ps) and g DT3TA-TAPB (Instrument response functions: 0.2615 ps).
Subsequently, temperature-dependent PL spectroscopy was performed to analyze the exciton dissociation energies of the COFs52. As the temperature increased, thermal excitation weakened the Coulombic attraction between electrons and holes, thereby facilitating exciton dissociation and reducing PL intensity53,54,55,56. This phenomenon was analyzed using the Arrhenius equation, which was employed to estimate the exciton binding energy (Eb) from temperature-dependent PL variations57,58,59. Temperature-dependent PL spectroscopy of the COFs revealed that the integrated PL intensity decreased gradually as the temperature increased from 100 to 300 K. Fitting the experimental data using the Arrhenius equation yielded Eb values of 126.6, 137.8, 136.7, and 139.1 meV for DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB, respectively (Fig. 3b, c and Supplementary Figs. 11 and 12). These results suggested that the excitons in DT2TA-TAPB dissociate more readily than those in other COFs, thereby increasing the proportion of free carriers60. In addition, femtosecond time-resolved transient absorption (fs-TA) spectroscopy was performed to thoroughly elucidate the real-time photogenerated free charge separation and migration behaviors in the COFs. The fs-TA spectra of COFs exhibited a broad ground-state bleach signal at 450-680 nm, attributed to the combined contributions of ground-state bleaching and stimulated emission (Fig. 3d–g and Supplementary Figs. 13–17). Furthermore, a positive absorption signal was observed at the wavelengths >700 nm, which stemmed from excited-state absorption resulting from photogenerated electrons. Fitting the decay curves at a probe wavelength of 580 nm with a bi-exponential model revealed two distinct relaxation pathways for the photogenerated electrons: a short-lived component (τ1) corresponding to electron trapping and a long-lived component (τ2) associated with electron–hole recombination61,62. The τ1 values for the four COFs were similar, probably because of their structural similarity. In contrast, DT2TA-TAPB exhibited a longer τ2 (2677 ps) than the other COFs, indicating an extended carrier lifetime. The average carrier lifetimes (τave) were determined to be 2677.65, 1618.55, 903.67, and 823.14 ps for DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB, respectively. These findings revealed that DT2TA-TAPB effectively suppressed charge recombination, thereby substantially enhancing photocatalytic H2O2 generation. The influence of external environmental factors—including pump irradiance63, atmospheric conditions64, and solvent effect65,66,67—on charge transfer dynamics was systematically studied. At a pump irradiance of 20 μJ, carrier lifetimes were prolonged, whereas at 2 μJ, the transient signal was considerably weaker68. This behavior was likely attributed to the weakening of Coulomb interactions between charge carriers at higher pump intensities, which facilitated exciton dissociation and enhanced the generation of free charge carriers. Additionally, increased pump fluence improved carrier concentration, fostering stimulated emission and partially suppressing non-radiative recombination, which enhanced electron-hole separation. Moreover, the excited-state lifetime of photogenerated charge carriers was notably longer in air than in a nitrogen atmosphere, likely because oxygen acted as an electron acceptor, captured photogenerated electrons and prolonged their lifetime69. Similarly, the excited-state lifetime in ethanol exceeded that in pure water, due to ethanol served as a sacrificial hole scavenger, thus suppressing charge recombination. These findings highlighted the critical role of environmental factors in exploring the dynamics of photogenerated charge carriers.
Photocatalytic H2O2 production by COFs
The UV–vis diffuse reflectance (DRS) absorption spectra of the developed COFs exhibited a strong and broad absorption peak in the visible light region, revealing superior light-harvesting capability (Fig. 4a). The intrinsic bandgap energies of DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB were estimated to be 2.48, 2.48, 2.47, and 2.35 eV, respectively, in the corresponding Tauc Plots (Supplementary Fig. 18). Furthermore, Mott–Schottky (MS) measurements were performed to evaluate the conduction band (CB) positions. As depicted in Supplementary Fig. 19, the MS plots of the COFs exhibited positive slopes, signifying n-type semiconductor properties. The flat band potentials of DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB were determined to be −1.27, −1.14, −1.36, and −1.24 eV, respectively. The energy band structure alignments of the COFs were determined by combining the measured optical bandgaps and CB positions, as shown in Fig. 4b. The CB positions of the COFs were more negative than the standard potential of oxygen reduction, indicating their feasibility for visible-light-catalyzed H2O2 production.

a UV-vis DRS spectra of the COFs. b Band gaps of the COFs. c Photocatalytic H2O2 yield of the COFs. Reaction condition: 300 W Xenon lamp λ > 420 nm, 10% EtOH solution. d H2O2 yields of DT2TA-TAPB and DT3TA-TAPB under different atmospheres. e Photocatalytic H2O2 production rates of DT2TA-TAPB and DT3TA-TAPB in pure water, 10% EtOH, and seawater. f Wavelength-dependent H2O2 production rates of DT2TA-TAPB. g Performance comparison of DT2TA-TAPB with other recently reported COF-based photocatalysts in 10% EtOH or pure water. h Long-term photocatalytic H2O2 production by DT2TA-TAPB (Reaction conditions: 10 mg catalyst in 40 mL pure water, under air atmosphere, irradiated by a 300 W Xenon lamp (light intensity: 100 mW cm−2)). Error bars indicate the standard deviations of four replicate measurements.
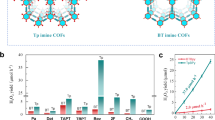
The photocatalytic H2O2 production performance of the developed COFs was evaluated in the presence of 10% ethanol (EtOH) as sacrificial agent and under O2 atmosphere. The generated H2O2 was detected by the titanium sulfate colorimetric method (Supplementary Fig. 20). As presented in Fig. 4c and Supplementary Fig. 21, the developed COFs exhibited excellent photocatalytic H2O2 production, with yields of 10972, 8171, 5886, and 5514 μmol h–1 g–1 for DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB, respectively. The excellent catalytic activity of DT2TA-TAPB was attributed to its low exciton binding energy and the generation of free carriers with sufficiently long lifetimes. Notably, the thiophene isomer-based COFs displayed significant photocatalytic activity relative to the furan isomer-functionalized COFs, which aligned with the electron-hole theory calculations results. Thereafter, the thiophene isomer-based COFs were considered as the main objects of the in-depth investigation of the impact of heterocyclic isomers on photocatalytic H2O2 production. The photocatalytic H2O2 production capacity of the COFs was investigated under O2, air, and Ar atmosphere to confirm that H2O2 was derived from the photocatalytic reduction of O2. As depicted in Fig. 4d and Supplementary Fig. 22, the H2O2 yield of DT2TA-TAPB and DT3TA-TAPB under an O2 atmosphere were obviously higher than that obtained under air (DT2TA-TAPB: 5223 μmol h–1 g–1, DT3TA-TAPB: 3617 μmol h–1 g–1) and Ar (DT2TA-TAPB: 885 μmol h–1 g–1, DT3TA-TAPB: 544 μmol h–1 g–1) atmosphere, and almost no H2O2 was detected under the Ar atmosphere, indicating that O2 was an indispensable reactant in H2O2 generation.
In addition, the photocatalytic H2O2 production performance of DT2TA-TAPB and DT3TA-TAPB was evaluated in pure water and seawater. As illustrated in Fig. 4e and Supplementary Fig. 23, DT2TA-TAPB and DT3TA-TAPB generated 8587 and 5143 μmol g−1 h−1 H2O2 in pure water and 7245 and 3381 μmol g−1 h−1 H2O2 in seawater. The results indicated that the H2O2 yield in seawater was inferior to that in pure water, which was attributed to seawater containing large quantities of ionic components and impurities that may affect H2O2 formation or lead to its decomposition70. Simultaneously, the photocatalytic H2O2 production performance of DT2TA-TAPB was evaluated under nature condition. DT2TA-TAPB generated 1476.2 μmol g−1 h−1 H2O2 under visible light irradiation (λ > 420 nm, 100 mW cm−2), without the use of sacrificial agents and under air atmosphere (Supplementary Fig. 24). In addition, the catalyst amount played a crucial role in determining photocatalytic efficiency. As shown in Supplementary Fig. 25, the H2O2 yields using catalyst masses of 1, 5, 10, 20, and 25 mg are 13880.1, 11377.9, 7195.3, 2472.3, and 942.5 μmol g-1 h-1, respectively. The highest H2O2 yield of 13880.1 μmol g-1 h-1 was attained with 1 mg of catalyst. However, further increasing catalyst mass resulted in a gradual decline in yield70, likely stemmed from excessive catalyst which limited light absorption and utilization26, while simultaneously reducing active site exposure, ultimately impaired catalytic performance. These results indicated that reducing the catalyst concentration was more favorable for the improvement of H2O2 yield.
The wavelength-dependent apparent quantum yield (AQY) of DT2TA-TAPB was assessed under various bandpass filters. Notably, DT2TA-TAPB exhibited a high AQY value of 3.3% at 380 nm, and its wavelength-dependent H2O2 production matched well with its absorption spectrum, indicating that H2O2 production was a photoinduced chemical reaction process (Fig. 4f)71. Furthermore, we synthesized a benchmark sample, g-C3N4 (Supplementary Fig. 26)72, and compared its H2O2 production performance with that of DT2TA-TAPB. The results indicated that the H2O2 production rate of DT2TA-TAPB was approximately 23- and 41-times higher than that of g-C3N4 in 10% EtOH and pure water, respectively (Supplementary Fig. 27). Meanwhile, the H2O2 yield of DT2TA-TAPB was far superior to that of the majority of reported COF-based photocatalysts without adding a sacrificial agent or in pure water (Fig. 4g and Supplementary Table 1), highlighting its remarkable performance of photocatalytic H2O2 production. To assess the practical application of DT2TA-TAPB, long-term photocatalytic H2O2 production experiments were conducted under simulated natural conditions. The results revealed that DT2TA-TAPB continuously and stably produced H2O2 over the initial 24 h, followed by a plateau that remained stable up to 28 h, indicating the establishment of a dynamic equilibrium between H2O2 formation and decomposition (Fig. 4h). The performance of DT2TA-TAPB remained stable over five successive cycles (Supplementary Fig. 28), highlighting its sustained photocatalytic activity. Additionally, the recovered DT2TA-TAPB exhibited minor variations in RXRD and FT-IR spectra, with PXRD exhibited a slight decline in peak intensity, presumably owing to catalyst delamination, while minor changes in the FT-IR spectra suggested partial oxidation (Supplementary Fig. 29). In contrast, SEM morphology remained largely unchanged (Supplementary Fig. 30). These findings imply that the catalyst preserved its structural stability despite minor changes induced during cycling. To elucidate the underlying mechanism, the kinetics of H2O2 formation and decomposition were systematically studied. H2O2 formation followed zero-order kinetics, whereas its decomposition adhered to first-order kinetics14,73. Accordingly, the overall H2O2 generation process could be described as follows:
where t denotes the reaction time, [H2O2] represents the concentration of generated H2O2, and kf and kd correspond to the rate constants for its formation and decomposition, respectively. The kinetic constants were derived by fitting the H2O2 formation curve, yielding a formation rate constant (kf) of 186.3 μM h-1 and a decomposition rate constant (kd) of 0.131 h-1 (Supplementary Fig. 31). These findings indicated that, in the initial stage, H2O2 formation rate constant exceeded its decomposition. As the reaction progressed, the two rate constants gradually approached equilibrium, resulting in a stabilized concentration of H2O2. In addition, H2O2 decomposition measurements were performed under visible-light irradiation in the presence of DT2TATAPB. As shown in Supplementary Fig. 32, more than 98% H2O2 was maintained after irradiation for 1 h, revealing its excellent stability in the presence of DT2TA-TAPB and supporting sustained photocatalytic production.
Photocatalytic mechanisms of COFs
Given the remarkable photocatalytic H2O2 production performance of DT2TA-TAPB and DT3TA-TAPB, we investigated their photocatalytic reaction pathways. Quenching experiments were conducted using a series of scavengers, including p-benzoquinone (p-BQ, •O2– scavenger), EtOH (h+ scavenger), and KBrO3 (e- scavenger), which were respectively added to the photocatalytic system to determine the possible reaction intermediates involved. As presented in Fig. 5a, the addition of 10% EtOH accelerated H2O2 production, whereas almost no H2O2 was detected after the addition of KBrO3. Moreover, almost no H2O2 was generated in the DT3TA-TAPB system with the addition of p-BQ, whereas a small amount of H2O2 was detected in the DT2TA-TAPB system. These results confirmed the involvement of photogenerated electrons in ORR and generation of superoxide radicals as intermediates. The reaction intermediates of H2O2 generation were determined via in situ electron paramagnetic resonance (in situ EPR) spectroscopy, employing 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the spin-trap agent to recognize active oxygen species. The in situ EPR spectra of DT2TA-TAPB and DT3TA-TAPB exhibited a typical sixfold characteristic peak for DMPO-•O2–, the intensity of which increased and then decreased over time (Fig. 5b, c)74. A slight shift in the DMPO-•O2– peaks was observed in the DT3TA-TAPB, suggesting a potential conversion of DMPO-•O2– to DMPO-•OOH75. Notably, the weaker DMPO-•O2– signal in the DT2TA-TAPB suspension was likely owing to the predominant formation of an endoperoxide in DT2TA-TAPB via a one-step, two-electron pathway, directly promoting H2O2 generation26. Subsequently, the reaction intermediates were monitored during the reaction process via in situ diffuse reflectance infrared flourier transform (in situ DRIFT) spectroscopy. The in situ DRIFT spectra of DT2TA-TAPB and DT3TA-TAPB exhibited three peaks at 910 ~ 990, 1141, and 1230 cm−1, corresponding to O-O vibrations, •O2–, and •OOH intermediate, respectively (Supplementary Figs. 33 and 34 and Fig. 5d, e). However, the above results did not provide sufficient evidence to discern the ORR pathway.

a Photocatalytic H2O2 generation rates of DT2TA-TAPB and DT3TA-TAPB with different scavengers. Time-dependent in situ EPR spectra of b DT2TA-TAPB and c DT3TA-TAPB. In situ DRIFT spectra of d DT2TA-TAPB and e DT3TA-TAPB at 1152 ~ 1126 cm−1. f Koutecky-Levich plots of DT2TA-TAPB and DT3TA-TAPB obtained by RDE measurements at −0.75 V (vs. Ag/AgCl). g The •O2− yields of DT2TA-TAPB and DT3TA-TAPB detected by the NBT method. h Adsorption energy of O2 on optimum site of DT2TA-TAPB and DT3TA-TAPB. i Calculated Gibbs free energy diagrams for the reduction of O2 to H2O2 on DT2TA-TAPB and DT3TA-TAPB.
Therefore, we conducted oxygen reduction linear sweep voltammetry at various rotational speeds using a rotating disk electrode (RDE) to thoroughly investigate the number of electrons transferred during the ORR process (Supplementary Fig. 35). The numbers of electron transfers in DT2TA-TAPB and DT3TA-TAPB were determined to be 1.92 and 1.34, respectively, using the Koutecky-Levich method, with the former being closer to 2 (Fig. 5f). Rotating ring-disk electrode (RRDE) measurements further revealed that DT2TA-TAPB exhibited higher selectivity for H2O2 production than DT3TA-TAPB (Supplementary Figs. 36 and 37). These findings provided compelling evidence that DT2TA-TAPB facilitated efficient H2O2 generation under visible light through a direct one-step, two-electron ORR pathway, whereas the step-wise one-electron pathway played a more dominant role in the DT3TA-TAPB system. Moreover, the amount of •O2– formed in the catalytic systems was quantified by the nitro blue tetrazolium (NBT) method (Supplementary Fig. 38)71. As depicted in Fig. 5g, the •O2– yield of DT3TA-TAPB was 72.2 mM, which was superior to that of DT2TA-TAPB (65.9 mM), suggesting that the capability of DT2TA-TAPB to generate superoxide radicals was opposite to that of generating H2O2. Taken together, the number of electron transfers and NBT results indicated that H2O2 production by DT3TA-TAPB was a two-step two-electron ORR process, while DT2TA-TAPB dramatically increased ORR selectivity and H2O2 yield mainly through a direct two-electron pathway. The results demonstrated that the introduction of thiophene isomers could modulate the ORR pathway, thereby improving selectivity and efficiency towards H2O2. To further elucidate the mechanism of photocatalytic H2O2 generation by DT2TA-TAPB, isotopic labeling experiments using 18O2 and H218O were conducted. Upon introducing 18O2 and utilizing MnO2 to decompose the generated H218O2, both 18O2 and 16O2 were detected by gas chromatography-mass spectrometry (GC-MS), confirming that H2O2 formation involved both the ORR and the water oxidation reaction (WOR) (Supplementary Fig. 39). Furthermore, photocatalytic reactions in pure H218O, 18O2 was detected in the gas-phase products of the reaction system by GC-MS, indicating a four-electron WOR pathway in DT2TA-TAPB. Notably, the addition of MnO2 to the reaction solution led to the detection of 18O2, revealing that 18O2 originated from water oxidation and subsequently participated in ORR, thus facilitating H218O2 formation (Supplementary Fig. 40). These results demonstrated that H2O2 was generated via a synergistic mechanism involving both water oxidation and oxygen reduction during the photocatalysis over DT2TA-TAPB, with water simultaneously acting as the electron and oxygen source in the absence of a sacrificial reagent. This dual-pathway mechanism not only improved the utilization efficiency of photogenerated charge carriers but also effectively mitigated catalyst degradation associated with hole accumulation.
To gain deeper insight into the effect of isomers on the ORR mechanism at the atomic scale, DFT calculations were performed. Previous studies have reported that the carbon atom adjacent to the sulfur atom in thiophene is a possible active site29,76. As illustrated in Fig. 5h, the calculated oxygen adsorption energies for DT2TA-TAPB and DT3TA-TAPB were −5.33 eV and −4.58 eV, respectively. The preferred affinity of DT2TA-TAPB for oxygen was attributed to the sulfur atom in thiophene and its adjacent carbon atom serving as active sites in DT2TA-TAPB, which were conducive to Yeager-type adsorption. Notably, oxygen adsorbed onto DT2TA-TAPB via Yeager-type adsorption formed the endoperoxides (*OO*) intermediate with a lower Gibbs free energy (−2.62 eV), which was crucial for improving ORR selectivity36. Furthermore, the Gibbs free energy for the formation of *OOH via Pauling-type adsorption on DT3TA-TAPB was −2.89 eV, indicating that O2 adsorbed on DT3TA-TAPB was prone to be activated into *OOH (Fig. 5i). Consequently, compared to DT3TA-TAPB, DT2TA-TAPB favored the formation of the *OO* intermediate, resulting in the production of H2O2 through direct one-step two-electron pathway, while DT3TA-TAPB favored the formation of the *OOH intermediate through two-step one-electron pathway. Together with the previous experimental findings, the results indicated that DT2TA-TAPB facilitated H2O2 production through dual channels during ORR. Thereby, introduction of the thiophene isomer modulated the ORR pathway and improved H2O2 yield.
Discussion
In summary, we designed and fabricated four COFs functionalized with thiophene and furan isomers to tune the electronic structure and achieve enhanced photocatalytic H2O2 production. The results confirmed that the thiophene isomer-functionalized COFs exhibited superior performance compared to furan isomer–based COFs, which was primarily attributed to the formation of a better-matched donor-acceptor system. In addition, alterations in thiophene conformation modulated the electronic structure of the COFs, resulting in DT2TA-TAPB demonstrating a reduced exciton binding energy and prolonged photogenerated electrons lifetime compared to DT3TA-TAPB. The potential active sites were explored through calculations, which revealed that the sulfur atom of thiophene and its neighboring carbon atom in DT2TA-TAPB acted as active sites for stabilization of the *OO* intermediate, thereby promoting the direct reduction of O2 to H2O2. Under visible light irradiation, DT2TA-TAPB exhibited high yield of 10972 μmol g-1 h-1 H2O2, ranking it as one of the best COF-based photocatalysts. The study findings offer a fresh perspective for in-depth investigations of the structure-performance relationship of photocatalysts for H2O2 synthesis.
Methods
Synthesis of 2,5-di(thiophen-2-yl)terephthalaldehyde
A mixture of 2,5-dibromoterephthalaldehyde (292 mg, 1 mmol), thiophene-2-boronic acid pinacol ester (525 mg, 2.5 mmol), and Pd(PPh3)4 (20 mg) was added to 16 mL of 1 mol L-1 aqueous sodium carbonate solution and 20 mL of 1,2-dimethoxyethane. The reaction was conducted under a N2 atmosphere at 100 °C for 24 h. After the reaction, an aqueous workup is performed, followed by extraction with dichloromethane, and the solvent is then evaporated using a rotary evaporator. The crude product was purified by silica gel column chromatography with a dichloromethane and petroleum ether (2:1) mixture as the eluent, yielding pure 2,5-di(thiophen-2-yl)terephthalaldehyde (DT2TA) (yield, 58.0%). 1H NMR (400 MHz, CDCl3): δ 10.51 (s, 2H), 8.25 (s, 1H), 7.65 (d, 2H), 6.80 (d, 2H), 6.61-6.10 (q, 2H).
Synthesis of 2,5-di(thiophen-3-yl)terephthalaldehyde
A mixture of 2,5-dibromoterephthalaldehyde (292 mg, 1 mmol), 4,4,5,5-tetramethyl-2-(3-thienyl)-1,3,2-dioxaborolane (525 mg, 2.5 mmol), and Pd(PPh3)4 (20 mg) was added to16 mL of 1 mol L-1 aqueous sodium carbonate solution and 20 mL of 1,2-dimethoxyethane. The reaction was conducted under a N2 atmosphere at 100 °C for 24 h. After the reaction, an aqueous workup is performed, followed by extraction with dichloromethane, and the solvent is then evaporated using a rotary evaporator. The crude product was purified by silica gel column chromatography with a dichloromethane and petroleum ether (2:1) mixture as the eluent, yielding pure 2,5-di(thiophen-3-yl)terephthalaldehyde (DT3TA) (yield, 56.0%). 1H NMR (400 MHz, CDCl3): δ 10.21-10.20 (t, 2H), 8.11-8.10 (t, 2H), 7.52-7.50 (qd, 2H), 7.40-7.38 (m, 2H), 7.25-24 (t, 2H).
Synthesis of 2,5-di(furan-2-yl)terephthalaldehyde
A mixture of 2,5-dibromoterephthalaldehyde (292 mg, 1 mmol), 2-furanboronic acid (280 mg, 2.5 mmol), and Pd(PPh3)4 (20 mg) was added to16 mL of 1 mol L-1 aqueous sodium carbonate solution and 20 mL of 1,2-dimethoxyethane. The reaction was conducted under a N2 atmosphere at 100 °C for 24 h. After the reaction, an aqueous workup is performed, followed by extraction with dichloromethane, and the solvent is then evaporated using a rotary evaporator. The crude product was purified by silica gel column chromatography with a dichloromethane and petroleum ether (2:1) mixture as the eluent, yielding pure 2,5-di(furan-2-yl)terephthalaldehyde (DF2TA) (yield, 72.0%). 1H NMR (400 MHz, CDCl3): δ 10.51 (s, 2H), 8.25 (s, 2H), 7.65 (d, 2H), 6.80 (d, 2H), 6.61-6.60 (q, 2H).
Synthesis of 2,5-di(furan-3-yl)terephthalaldehyde
A mixture of 2,5-dibromoterephthalaldehyde (292 mg, 1 mmol), 3-furanboronic acid (280 mg, 2.5 mmol), and Pd(PPh3)4 (20 mg) was added to 16 mL of 1 mol L-1 aqueous sodium carbonate solution and 20 mL of 1,2-dimethoxyethane. The reaction was conducted under a N2 atmosphere at 100 °C for 24 h. After the reaction, an aqueous workup is performed, followed by extraction with dichloromethane, and the solvent is then evaporated using a rotary evaporator. The resulting crude product was purified by silica gel column chromatography using a dichloromethane and petroleum ether (2:1) mixture as the eluent, yielding pure 2,5-di(furan-3-yl)terephthalaldehyde (DF3TA) (yield, 78.0%). 1H NMR (400 MHz, CDCl3): δ 10.30 (s, 2H), 8.05 (d, 2H), 7.62-7.61 (q, 2H), 7.60-7.59 (q,2H), 6.65-6.64 (q, 2H).
Synthesis of DT2TA-TAPB
A 10 mL glass ampoule was charged with DT2TA (0.089 mmol, 26.52 mg) and TAPB (0.059 mmol, 20.73 mg). Subsequently, 0.9 mL of o-DCB and 0.1 mL of n-BuOH were added to the ampoule, and the mixture was sonicated to ensure uniform dispersion. Following this, 0.1 mL of 6 mol L-1 AcOH aqueous solution was rapidly introduced. The ampoule tube was then sealed after three freeze-pump-thaw cycles. The reaction mixture was heated at 120 °C for three days. After cooling, the precipitate was collected by centrifugation, washed sequentially with DMF, THF, and hexane, and dried under vacuum (yield, 82.0%).
Synthesis of DT3TA-TAPB
A 10 mL glass ampoule was charged with DT3TA (0.089 mmol, 26.52 mg), TAPB, (0.059 mmol, 20.73 mg), and 1 mL of o-DCB. The mixture was sonicated to ensure uniform dispersion. Subsequently, 0.1 mL of 6 mol L-1 AcOH aqueous solution was rapidly added. The ampoule was then sealed after three freeze-pump-thaw cycles. The reaction mixture was heated at 120 °C for three days. After cooling, the precipitate was collected by centrifugation, washed sequentially with DMF, THF, and hexane, and dried under vacuum (yield, 83.6%).
Synthesis of DF2TA-TAPB
A 10 mL glass ampoule was charged with DF2TA (0.089 mmol, 23.67 mg), TAPB (0.059 mmol, 20.73 mg), 0.1 mL of o-DCB, and 0.9 mL of n-BuOH. The mixture was sonicated to achieve uniform dispersion. Subsequently, 0.1 mL of a 6 mol L-1 AcOH aqueous solution was rapidly added. The ampoule was sealed after three freeze-pump-thaw cycles. The reaction mixture was then heated at 120 °C for three days. After cooling, the precipitate was collected by centrifugation and washed sequentially with DMF, THF, and hexane, and then dried under vacuum (yield, 91.2%).
Synthesis of DF3TA-TAPB
A 10 mL glass ampoule was charged with DF3TA (0.089 mmol, 23.67 mg), TAPB (0.059 mmol, 20.73 mg), 0.3 mL of o-DCB, and 0.7 mL of n-BuOH. The mixture was sonicated to achieve uniform dispersion. Subsequently, 0.1 mL of a 6 mol L-1 AcOH aqueous solution was rapidly added. The ampoule was then sealed after three freeze-pump-thaw cycles. After heating at 120 °C for three days, the precipitate was collected by centrifugation, washed sequentially with DMF, THF, and hexane, and then dried under vacuum (yield, 92.3%).
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2323749 (DT2TA), 2323748 (DT3TA), 2339826 (DF2TA), and 2323750 (DF3TA). These data can be accessed free of charge from the CCDC at www.ccdc.cam.ac.uk. All data supporting the findings of this study are available within the article and its Supplementary Information. Source data are provided with this paper.
References
Mase, K. et al. Seawater usable for production and consumption of hydrogen peroxide as a solar fuel. Nat. Commun. 7, 11470 (2016).
Das, P. et al. Integrating bifunctionality and chemical stability in covalent organic frameworks via one-pot multicomponent reactions for solar-driven H2O2 production. J. Am. Chem. Soc. 145, 2975–2984 (2023).
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006).
Freese, T. et al. An organic perspective on photocatalytic production of hydrogen peroxide. Nat. Catal. 6, 553–558 (2023).
Wang, S. et al. Efficient photocatalytic production of hydrogen peroxide using dispersible and photoactive porous polymers. Nat. Commun. 14, 6891 (2023).
Tang, Y. et al. The evolution of photocatalytic H2O2 generation: from pure water to natural systems and beyond. Energy Environ. Sci. 17, 6482–6498 (2024).
Zeng, X. et al. Simultaneously tuning charge separation and oxygen reduction pathway on graphitic carbon nitride by polyethylenimine for boosted photocatalytic hydrogen peroxide production. ACS Catal. 10, 3697–3706 (2020).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374–384 (2021).
Li, Q. et al. Shear stress triggers ultrathin-nanosheet carbon nitride assembly for photocatalytic H2O2 production coupled with selective alcohol oxidation. J. Am. Chem. Soc. 145, 20837–20848 (2023).
Shiraishi, Y. et al. Resorcinol–formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985–993 (2019).
Wu, C. et al. Polarization engineering of covalent triazine frameworks for highly efficient photosynthesis of hydrogen peroxide from molecular oxygen and water. Adv. Mater. 34, 2110266 (2022).
Geng, K. et al. Covalent organic frameworks: design, synthesis, and functions. Chem. Rev. 120, 8814–8933 (2020).
Qin, L. et al. Structural motifs in covalent organic frameworks for photocatalysis. Adv. Funct. Mater. 34, 2401562 (2024).
Zhang, X. et al. Keto-anthraquinone covalent organic framework for H2O2 photosynthesis with oxygen and alkaline water. Nat. Commun. 15, 2649 (2024).
Krishnaraj, C. et al. Strongly reducing (diarylamino)benzene-based covalent organic framework for metal-free visible light photocatalytic H2O2 generation. J. Am. Chem. Soc. 142, 20107–20116 (2020).
Yue, J.-Y. et al. Regulating the H2O2 photosynthetic activity of covalent organic frameworks through linkage orientation. ACS Catal. 14, 4728–4737 (2024).
Yong, Z. & Ma, T. Solar-to-H2O2 catalyzed by covalent organic frameworks. Angew. Chem. Int. Ed. 62, e202308980 (2023).
Zhou, E. et al. Cyanide-based covalent organic frameworks for enhanced overall photocatalytic hydrogen peroxide production. Angew. Chem. Int. Ed. 63, e202400999 (2024).
Li, L. et al. Custom-design of strong electron/proton extractor on COFs for efficient photocatalytic H2O2 production. Angew. Chem. Int. Ed. 63, e202320218 (2024).
Cheng, H. et al. Rational design of covalent heptazine frameworks with spatially separated redox centers for high-efficiency photocatalytic hydrogen peroxide production. Adv. Mater. 34, 2107480 (2022).
Liu, R. et al. Linkage-engineered donor–acceptor covalent organic frameworks for optimal photosynthesis of hydrogen peroxide from water and air. Nat. Catal. 7, 195–206 (2024).
Qin, C. et al. Dual donor-acceptor covalent organic frameworks for hydrogen peroxide photosynthesis. Nat. Commun. 14, 5238 (2023).
Chang, J. N. et al. Oxidation-reduction molecular junction covalent organic frameworks for full reaction photosynthesis of H2O2. Angew. Chem. Int. Ed. 62, e202218868 (2023).
Shu, C. et al. Mixed-linker strategy for the construction of sulfone-containing D–A–A covalent organic frameworks for efficient photocatalytic hydrogen peroxide production. Angew. Chem. Int. Ed. 63, e202403926 (2024).
Wang, H. et al. A crystalline partially fluorinated triazine covalent organic framework for efficient photosynthesis of hydrogen peroxide. Angew. Chem. Int. Ed. 61, e202202328 (2022).
Luo, Y. et al. Sulfone-modified covalent organic frameworks enabling efficient photocatalytic hydrogen peroxide generation via one-step two-electron O2 reduction. Angew. Chem. Int. Ed. 62, e202305355 (2023).
Chen, L. et al. Acetylene and diacetylene functionalized covalent triazine frameworks as metal-free photocatalysts for hydrogen peroxide production: a new two-electron water oxidation pathway. Adv. Mater. 32, 1904433 (2020).
Xie, Z. et al. Variation of chemical microenvironment of pores in hydrazone-linked covalent organic frameworks for photosynthesis of H2O2. Angew. Chem. Int. Ed. 63, e202410179 (2024).
Li, D. et al. Metal-free thiophene-sulfur covalent organic frameworks: precise and controllable synthesis of catalytic active sites for oxygen reduction. J. Am. Chem. Soc. 142, 8104–8108 (2020).
Hou, Y. et al. Rigid covalent organic frameworks with thiazole linkage to boost oxygen activation for photocatalytic water purification. Nat. Commun. 15, 7350 (2024).
Yang, T. et al. Covalent furan-benzimidazole-linked polymer hollow fiber membrane for clean and efficient photosynthesis of hydrogen peroxide. Adv. Funct. Mater. 33, 2300714 (2023).
Long, X. et al. Heterocyclization strategy for construction of linear conjugated polymers: efficient metal-free electrocatalysts for oxygen reduction. Angew. Chem. Int. Ed. 58, 11369–11373 (2019).
Wang, Q. et al. Positional thiophene isomerization: a geometric strategy for precisely regulating the electronic state of covalent organic frameworks to boost oxygen reduction. Angew. Chem. Int. Ed. 63, e202320037 (2024).
Kou, M. et al. Molecularly engineered covalent organic frameworks for hydrogen peroxide photosynthesis. Angew. Chem. Int. Ed. 61, e202200413 (2022).
Zhang, Z. et al. Tris(triazolo)triazine-based covalent organic frameworks for efficiently photocatalytic hydrogen peroxide production. Angew. Chem. Int. Ed. 63, e202411546 (2024).
Liao, Q. et al. Regulating relative nitrogen locations of diazine functionalized covalent organic frameworks for overall H2O2 photosynthesis. Angew. Chem. Int. Ed. 62, e202310556 (2023).
Wu, W. et al. Pyridine-based covalent organic frameworks with pyridyl-imine structures for boosting photocatalytic H2O2 production via one-step 2e− oxygen reduction. Angew. Chem. Int. Ed. 63, e202404563 (2024).
Tong, L. et al. Atomically precise regulation of the N-heterocyclic microenvironment in triazine covalent organic frameworks for coenzyme photocatalytic regeneration. J. Am. Chem. Soc. 146, 21025–21033 (2024).
Fan, W. et al. Efficient hydrogen peroxide synthesis by metal-free polyterthiophene via photoelectrocatalytic dioxygen reduction. Energy Environ. Sci. 13, 238–245 (2020).
Chen, L. et al. An isomeric solid additive enables high-efficiency polymer solar cells developed using a benzo-difuran-based donor polymer. Adv. Mater. 35, 2301231 (2023).
Fang, Y. et al. Design and synthesis of broadband absorption covalent organic framework for efficient artificial photocatalytic amine coupling. Nat. Commun. 15, 4856 (2024).
Bao, R. et al. Designing thiophene-enriched fully conjugated 3D covalent organic framework as metal-free oxygen reduction catalyst for hydrogen fuel cells. Angew. Chem. Int. Ed. 62, e202216751 (2023).
Su, Y. et al. Crystalline and stable benzofuran-linked covalent organic frameworks from irreversible cascade reactions. J. Am. Chem. Soc. 142, 13316–13321 (2020).
Zhao, W. et al. Accelerated synthesis and discovery of covalent organic framework photocatalysts for hydrogen peroxide production. J. Am. Chem. Soc. 144, 9902–9909 (2022).
Hao, F. et al. Photo-driven quasi-topological transformation exposing highly active nitrogen cation sites for enhanced photocatalytic H2O2 production. Angew. Chem. Int. Ed. 62, e202315456 (2023).
Pelkowski, C. E. et al. Tuning crystallinity and stacking of two-dimensional covalent organic frameworks through side-chain interactions. J. Am. Chem. Soc. 145, 21798–21806 (2023).
Wang, X. et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 10, 1180–1189 (2018).
Hojo, R. et al. Imidazophenothiazine-based thermally activated delayed fluorescence materials with ultra-long-lived excited states for energy transfer photocatalysis. J. Am. Chem. Soc. 145, 18366–18381 (2023).
Zhang, L. et al. Structurally locked high-crystalline covalent triazine frameworks enable remarkable overall photosynthesis of hydrogen peroxide. J. Am. Chem. Soc. 146, 29943–29954 (2024).
Qian, Y. et al. Computation-based regulation of excitonic effects in donor-acceptor covalent organic frameworks for enhanced photocatalysis. Nat. Commun. 14, 3083 (2023).
Chen, Z. et al. Tuning excited state electronic structure and charge transport in covalent organic frameworks for enhanced photocatalytic performance. Nat. Commun. 14, 1106 (2023).
Yan, H. et al. Enhancing photosynthesis efficiency of hydrogen peroxide by modulating side chains to facilitate water oxidation at low-energy barrier sites. Adv. Mater. 36, 2311535 (2024).
Alvertis, A. M. et al. Phonon screening and dissociation of excitons at finite temperatures from first principles. Proc. Natl. Acad. Sci. USA 121, e2403434121 (2024).
Jiang, Y., Wang, X. & Pan, A. Properties of excitons and photogenerated charge carriers in metal halide perovskites. Adv. Mater. 31, 1806671 (2019).
Grancini, G. et al. Hot exciton dissociation in polymer solar cells. Nat. Mater. 12, 29–33 (2013).
Dimitrov, S. D. et al. On the energetic dependence of charge separation in low-band-gap polymer/fullerene blends. J. Am. Chem. Soc. 134, 18189–18192 (2012).
Ru, C. et al. Regulation of exciton effects in functionalized conjugated polymers by B-N Lewis Pairs for visible-light photocatalysis. Angew. Chem. Int. Ed. 64, e202417712 (2025).
Fu, G. et al. Construction of thiadiazole-bridged sp2-carbon-conjugated covalent organic frameworks with diminished excitation binding energy toward superior photocatalysis. J. Am. Chem. Soc. 146, 1318–1325 (2024).
Li, C. et al. Covalent organic frameworks with high quantum efficiency in sacrificial photocatalytic hydrogen evolution. Nat. Commun. 13, 2357 (2022).
Huang, Y. et al. Achieving a solar-to-chemical efficiency of 3.6% in ambient conditions by inhibiting interlayer charges transport. Nat. Commun. 15, 5406 (2024).
Qiu, J. et al. COF/In2S3 S-scheme photocatalyst with enhanced light absorption and H2O2-production activity and fs-TA investigation. Adv. Mater. 36, 2400288 (2024).
Cheng, C. et al. Verifying the charge-transfer mechanism in S-scheme heterojunctions using femtosecond transient absorption spectroscopy. Angew. Chem. Int. Ed. 62, e202218688 (2023).
Godin, R. et al. Time-resolved spectroscopic investigation of charge trapping in carbon nitrides photocatalysts for hydrogen generation. J. Am. Chem. Soc. 139, 5216–5224 (2017).
Lin, W. et al. Decoupled artificial photosynthesis via a catalysis-redox coupled COF||BiVO4 photoelectrochemical device. J. Am. Chem. Soc. 145, 18141–18147 (2023).
Venkatraman, R. K. & Orr-Ewing, A. J. Solvent effects on ultrafast photochemical pathways. Acc. Chem. Res. 54, 4383–4394 (2021).
Corp, K. L. & Schlenker, C. W. Ultrafast spectroscopy reveals electron-transfer cascade that improves hydrogen evolution with carbon nitride photocatalysts. J. Am. Chem. Soc. 139, 7904–7912 (2017).
Deng, Z. et al. Revealing the excited-state mechanisms of the polymorphs of a hot exciton material. Nat. Commun. 16, 258 (2025).
Liu, Y. et al. Tuning intermediate-band Cu3VS4 nanocrystals from plasmonic-like to excitonic via shell-coating. Chem. Mater. 32, 224–233 (2020).
Li, W. et al. Unsymmetric protonation driven highly efficient H2O2 photosynthesis in supramolecular photocatalysts via one-step two-electron oxygen reduction. Angew. Chem. Int. Ed. 64, e202421356 (2025).
Liu, F. et al. Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight. Nat. Commun. 14, 4344 (2023).
Zhi, Q. et al. Piperazine-linked metalphthalocyanine frameworks for highly efficient visible-light-driven H2O2 photosynthesis. J. Am. Chem. Soc. 144, 21328–21336 (2022).
Tan, F. et al. Aqueous synthesis of covalent organic frameworks as photocatalysts for hydrogen peroxide production. CCS Chem. 4, 3751–3761 (2022).
Chakraborty, A. et al. Enhancing photocatalytic hydrogen peroxide generation by tuning hydrazone linkage density in covalent organic frameworks. Nat. Commun. 16, 503 (2025).
Liu, Y. et al. Fluorination of covalent organic framework reinforcing the confinement of Pd nanoclusters enhances hydrogen peroxide photosynthesis. J. Am. Chem. Soc. 145, 19877–19884 (2023).
He, J. et al. Piezo-catalysis mechanism Elucidation by tracking oxygen reduction to hydrogen peroxide with in situ EPR spectroscopy. Angew. Chem. Int. Ed. 63, e202410381 (2024).
Yue, J. Y. et al. Thiophene-containing covalent organic frameworks for overall photocatalytic H2O2 synthesis in water and seawater. Angew. Chem. Int. Ed. 62, e202309624 (2023).
Acknowledgements
Y.T. gratefully acknowledges support from the National Natural Science Foundation of China (22221001, 22131007), the Major Science and Technology Projects in Gansu Province (23ZDGA012), and the Fundamental Research Funds for the Central Universities (lzujbky-2023-stlt01, lzujbky-2024-jdzx13). F.C. acknowledges support from the Major Science and Technology Projects in Gansu Province (22ZD6GD060) and the Key Research and Development Program in the Tibet Autonomous Region (XZ202401ZY0075). We are also grateful to the Electron Microscopy Centre and the Analysis and Testing Centre of Lanzhou University for their assistance.
Author information
Authors and Affiliations
Contributions
Y.J.: conceptualization, methodology, investigation, writing—original draft. H.L.: conceptualization, methodology, writing—review & editing. G.T.: software, data curation. P.S.: writing—review & editing. Z.W. and C.H.: data curation. R.H. and T.H.: resources. F.C. and Y.T.: funding acquisition, supervision, writing—review & editing. All authors read and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wonyong Choi, Mei-Yan Gao and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ju, Y., Lin, H., Tan, G. et al. Regulating the electronic structure of covalent organic frameworks via heterocyclic isomers for highly efficient photocatalytic H2O2 generation. Nat Commun 16, 5658 (2025). https://doi.org/10.1038/s41467-025-60960-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60960-6
