
Abstract
Peptide-based materials offer unique advantages for constructing supramolecular chiral systems due to their bioactivity and intrinsic chirality. However, precise control over the expression, transfer, and amplification of chirality from the molecular to supramolecular level remains a significant challenge in developing high-performance chiral materials. In this study, we demonstrate that achiral anions regulate hydrogen bonding interactions in dipeptide assembly, leading to the formation of chiral microrolls composed of two-dimensional nanosheets. These microrolls exhibit pH-dependent chiroptical switching and intense circularly polarized luminescence with a dissymmetry factor (|glum|) of 0.062. Furthermore, the chiral ultraviolet emission from these microrolls enables enantioselective polymerization of diacetylene, offering potential applications in chiral catalysis. These findings enhance our understanding of chirality modulation in biomolecular assemblies and provide a pathway toward the development of high-performance chiral biomaterials.
Similar content being viewed by others
Introduction
Supramolecular assembly offers a powerful strategy for constructing well-ordered functional architectures via a bottom-up approach1,2,3. Peptides, as fundamental constituents of biological organisms, serve as essential building blocks for fabricating chiral supramolecular architectures due to their intrinsic molecular chirality4,5,6. The handedness of peptide assemblies is predominantly determined by the chirality of their constituent amino acids7. Even a single alteration in the chirality of an amino acid within a peptide sequence can significantly affect the resulting supramolecular chirality8. Furthermore, the amino acid sequence plays a crucial role in regulating molecular packing modes, which in turn influences the manifestation of supramolecular chirality9,10,11.
Beyond intrinsic molecular factors, the assembly environment critically influences peptide assembly chirality4. Our research group previously demonstrated that bipyridine derivatives with varying structures and molar ratios can induce dipeptides to form either right- or left-handed helices12,13. Among external additives, metal ions are particularly effective in directing chiral peptide assembly14. For example, Feng et al. showed that the synergistic effects of hydrogen bonding and metal-ion coordination enable precise control over the supramolecular chirality of L-phenylalanine-based hydrogels15. Additionally, variations in pH value can alter the charge states of peptide residues, dynamically modulating the handedness of the assemblies16. Qi et al. reported that tripeptides self-assemble into highly uniform left-handed helical ribbons in alkaline solutions, whereas right-handed helical structures are favored in acidic environments17.
Circularly polarized luminescence (CPL) refers to the phenomenon in which chiral luminescent materials emit left- or right-handed circularly polarized light with different intensities upon excitation18. Materials exhibiting CPL properties have important applications in areas such as 3D displays, optical anti-counterfeiting, and photocatalytic asymmetric synthesis19,20,21. Biomolecule-based CPL materials, synergistically integrating the inherent bioactivity, exceptional biocompatibility, and biodegradability of biomolecules with the unique optical advantages of CPL, have garnered increasing research interest for their potential applications in biomedical imaging, biosensing, and environmentally sustainable optoelectronic devices22. The performance of CPL is quantified by the dissymmetric factor glum, defined as:
where IL and IR represent the left- and right-handed CPL intensities respectively23. Compared with high-performance systems such as liquid crystals, helicenes, lanthanide complexes, and inorganic nanocrystals, biomaterials universally exhibit significantly lower glum value (10-5–10-3) that fall short of practical application requirements24,25,26. At present, developing CPL systems with high glum value in biomolecular assemblies for real-world applications remains a significant challenge27.
Anions play versatile and crucial roles in biological systems by mediating structural integrity, function activity, and signaling processes28,29,30. However, their involvement in regulating molecular chiral assembly is rarely known31. In this study, we demonstrate that achiral anions co-assembly can effectively drive the dipeptide formation of chiral hierarchical nanoarchitectures, leading to a significant enhancement in CPL emissions. Specifically, SO42− anion regulates hydrogen bonding interactions in dipeptide assemblies, directing them from achiral microbelts to chiral microrolls rolled by two-dimensional (2D) nanosheets. The resulting microrolls exhibit strong CPL emissions, with an amplified |glum| value of up to 0.062, and demonstrate the ability to induce enantioselective photopolymerization of diacetylene. Furthermore, the microrolls exhibit pH-dependent morphological transitions, which are accompanied by reversible switching of their chiroptical signals. This work provides new insights into the role of anions in the formation of supramolecular chiral structures and offers a robust strategy to engineer chirality in dipeptide assemblies, paving the way for the development of high-performance CPL biomaterials.
Results
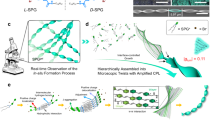
The dipeptide N-fluorenylmethoxycarbonyl-tyrosinyl-lysine (Fmoc-YK) was strategically selected for its rationally designed amphipathic architecture, which combines a hydrophobic Fmoc-tyrosine motif with a hydrophilic and cationic lysine residue. This molecular duality drives both spontaneous aqueous self-assembly and anion-responsive phase behavior. It spontaneously formed hydrogels at a concentration of 20 mM under acidic conditions (pH 3.1) (Fig. S1). Throughout the gel formation, the pH remained essentially constant. Cryo-transmission electron microscopy (cryo-TEM) observed that Fmoc-YK molecules initially underwent liquid-liquid phase separation (LLPS), resulting in high-concentration droplets that acted as nucleation sites. The droplets underwent the nucleation–elongation process to form beaded chain-like arrangements, and subsequently evolved into thermodynamically favorable nanofibers ~10 nm in diameter and several micrometers in length (Fig. 1a, b and S2). The three-dimensional (3D) entanglement of these nanofibers gave rise to the macroscopic hydrogel structure. However, the Fmoc-YK hydrogel was metastable and transitioned spontaneously into a more thermodynamically stable crystalline state after ~6 h (Fig. S1). TEM and scanning electron microscopy (SEM) images showed that during the gel-to-crystal phase transition, the nanofibers transformed into microbelts, with widths of ~1 μm and lengths extending to millimeter scale (Fig. 1c, Figs. S3a and S4). Due to the cationic lysine residue in Fmoc-YK, this transition was believed to be regulated by anions, with different anions exerting distinct effects on the phase transition.

(a) Schematic representation of the microbelts and microrolls formation via LLPS and gel-to-crystal translation process; b formation process of gels via LLPS (scale bar: 500 nm); (c) TEM images of the aged Fmoc-YK microbelts (scale bar: 2 μm); (d, e) formation process of microrolls via LLPS (scale bar: 500 nm, 1 μm), yellow arrows indicating 2D nanosheets.; (f) PXRD, (g) SAED (scale bar: 2 nm−1), and (h) FTIR spectra of Fmoc-YK nanofibers, microbelts and Fmoc-YK/SO42− microrolls.
Our findings demonstrated that introduction of dihydrogen phosphate (H2PO4−), acetate (CH3COO−), or citrate ions had minimal impact on the phase transition, with the resulting crystals retaining their original belt-like morphology (Figs. S1 and S3b). In contrast, peroxydisulfate (S2O82−) ions stabilized the metastable gel, enabling it to remain stable for over 6 months while maintaining its fibrous nanostructure (Fig. S5). The presence of SO42− ions, however, significantly accelerated the phase transition, reducing the transition time to < 1 h (Fig. S6). Cyro-TEM monitoring revealed that Fmoc-YK/SO42− also went through LLPS process to form droplets rich in SO42− and Fmoc-YK, which subsequently acted as nucleation sites to generate nanofibers (Fig. 1a, d). Then these nanofibers gradually transitioned into 2D nanosheets within 8 min. As they grew, the nanosheets curled, eventually forming microrolls with outer diameters of ~1 μm (Fig. 1d, e and S7).
Powder X-ray diffraction (PXRD) analysis revealed that the nanofibers in Fmoc-YK gels exhibited poor crystallinity, as indicated by the flat diffraction patterns. In contrast, the sharp diffraction peaks of the spontaneously formed microbelts and co-assembled microrolls indicated their strong crystallinity (Fig. 1f). Additionally, the distinct diffraction peak positions of the microbelts and microrolls suggested different molecular packing arrangements. Selected-area electron diffraction (SAED) results corroborated the PXRD observations, showing a diffuse spot pattern for nanofibers, while microbelts and microrolls displayed distinct and regular diffraction spots, indicative of single-crystal structures following the phase transition (Fig. 1g).
Fourier transform infrared (FTIR) spectroscopy further revealed that the C = O stretching vibration peaks of the amide bonds in Fmoc-YK were observed in the range of at 1620–1640 cm⁻¹ and 1670–1690 cm⁻¹, demonstrating that all three samples exhibited β-sheet secondary structures (Fig. 1h and S8)32,33,34,35. This result was further proven by the characteristic negative Cotton effect at around 210 nm in circular dichroism (CD) spectra (Fig. S9a). Besides, a strong absorption peak at 1140 cm⁻¹ appeared in the spectrum of Fmoc-YK/SO42−, attributed to the S–O stretching vibrations of SO42− 36. This suggested that SO42− ions were incorporated into the microroll structure, rather than merely modulating the Fmoc-YK self-assembly process (Fig. 1h).
Ultraviolet-visible (UV-vis) spectroscopic analysis revealed that Fmoc-YK/SO42− microrolls displayed broad absorption spanning from 260 nm to 300 nm, coincided with those of Fmoc-YK microbelts and monomeric Fmoc-YK both with and without SO42− (Fig. S10a). This indicated that SO42− had minimal impact on absorption of the Fmoc-YK. However, fluorescence spectroscopy (FL) revealed that upon 260 nm excitation, the microrolls exhibited a red-shifted emission maximum at 330 nm, contrasting with the 317 nm emission observed for Fmoc-YK microbelts, monomeric Fmoc-YK and Fmoc-YK/SO42− (Fig. S10b). This bathochromic shift demonstrated the SO42− could stabilize the excited Fmoc-YK in the aggregated state.
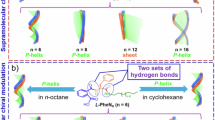
To determine the molecular packing of Fmoc-YK microbelts and Fmoc-YK/SO42− microrolls, single crystal X-ray diffraction (SXRD) and Rietveld powder diffraction refinement were employed to resolve their crystal structures (Fig. S11). Both crystal types exhibited a layered arrangement, where hydrogen bonds between peptide molecules, or between peptides and SO42− ions, formed 2D molecular layers. These layers were interconnected through hydrophobic interactions between the Fmoc groups (Fig. 2a, b). In the Fmoc-YK crystals, two primary types of hydrogen bonding interactions were observed within the molecular layers. The first involved NH…O = C hydrogen bonds between the amino and carboxyl groups, with bond lengths of 1.8–1.9 Å (Fig. 2c). Along the b axis, the peptide molecules adopted a β-sheet configuration, with NH…O = C hydrogen bonds between two amide bonds at distances of 2.0 Å and 2.3 Å, respectively (Fig. 2d). Additionally, the Fmoc groups aligned in parallel along the b direction. However, the centroid-to-centroid distance between them was ~5.0 Å, exceeding the 4.0 Å threshold required for π-π interactions, thus preventing such stacking within the layer33.

a, b 2D molecular layer structure of microbelts and microrolls; (c) hydrogen bonding between amino and carboxyl groups in microbelts; (d) β-sheet structure of Fmoc-YK microbelts; (e) hydrogen bonding between SO42− and various groups; (f) Fmoc groups arrangement in adjacent layers.
In the Fmoc-YK/SO42− microrolls, in addition to the hydrogen bonds observed in Fmoc-YK (i.e., between amino and carboxyl groups, and between amide bonds), additional hydrogen bonding occurred between the SO42− ions and various functional groups. These included OH…OS hydrogen bonds between the sulfate and carboxyl groups, NH…OS hydrogen bonds with the amino groups, and OH…OS interactions with phenolic hydroxyl groups. These interactions worked synergistically to stabilize the 2D molecular layers (Fig. 2e).
In both Fmoc-YK microbelts and Fmoc-YK/SO42− microrolls, hydrophobic Fmoc groups were located at the top and bottom edges of the molecular layers, with hydrophobic interactions between these groups promoting interlayer cohesion. Notably, although the Fmoc groups in adjacent layers were nearly perpendicularly in alignment, the direction of interaction was offset from the centroids-to-centroid vector. This suggested that the interlayer interactions were predominantly hydrophobic rather than T-shaped π-π stacking (Fig. 2f).
To investigate the formation mechanism of Fmoc-YK/SO42− microrolls, SEM was employed to observe their growth process. As illustrated in Fig. 3a, 2D nanosheets began to precipitate from the Fmoc-YK/SO42− gel within the first minute. Unlike the predominantly flat nanosheets reported in previous studies, these nanosheets displayed a distinct curvature. As the maturation time increased to 3 min, the curvature intensified, leading to the spontaneous formation of semi-tubular and even tubular structures. These tubular structures subsequently served as templates, around which additional nanosheets continued to wrap and grow along the tube walls while extending radially. This process culminated in the formation of multi-walled microrolls within 16 min. The curling mechanism was believed to be driven by the asymmetric stacking of Fmoc-YK and SO42− molecules, which adopted a superhelical arrangement (Fig. 3b)37. It is noteworthy that the chiral assembly of Fmoc-YK was dominated by the SO42− and was independent of the cation. The chiral assembly architecture remained unchanged when sodium ions (Na+) were replaced with potassium (K+), magnesium (Mg2+), or iron ions (Fe2+) (Fig. S12).

a Morphological evolution of microrolls (scale bar: 2 μm); (b) supercoiled arrangement of Fmoc-YK/SO42− microrolls; (c) circularly polarized luminescence (CPL) value of Fmoc-YK/SO42− microrolls and other Fmoc-YK assemblies with various anions; (d) the dissymmetric factor (glum) value of micobelts and microrolls (error bars, mean ± SD); (e) CPL, direct current (DC), (f) circular dichroism (CD) spectra and (g) SEM images of Fmoc-YLKL/SO42− and Fmoc-YDKD/SO42− microrolls (scale bar: 1 μm).
The chiral structure of the Fmoc-YK/SO42− microrolls exhibited remarkable chiral optical properties. CD spectroscopy which measured the differential absorption of left- and right-circularly polarized light in chiral systems, revealed a strong CD signal between 280 and 320 nm in microrolls samples (Fig. S9a, b). This strong absorption was ascribed to the π-π* transitions of the fluorenyl moieties in helically arranged state38,39. In marked contrast, control groups including individual Fmoc-YK (both in molecular and aggregated states) and Fmoc-YK/SO42− in molecular state exhibited negligible absorption within this region (Fig. S10c). This comparative analysis illustrated that the chiroptical activity of the microrolls stems from their supramolecular chiral architecture rather than intrinsic molecular chirality. CPL spectroscopy, which measured the emission of left- or right-circularly polarized light from chiral materials, showed that the Fmoc-YK/SO42− microrolls emitted a robust CPL signal near 340 nm under 260 nm excitation (Fig. 3c, d). By contrast, Fmoc-YK alone and combined with other anions (H2PO4−, CH3COO−, Citrate and Cl−) exhibited very weak CPL signals, demonstrating that microrolls substantially amplified the chiral luminescence signal of Fmoc-YK (Fig. 3c, d, Figs. S9c, S13 and S14). Additionally, the CPL and CD signals from the two enantiomers of Fmoc-YK (Fmoc-YLKL and Fmoc-YDKD) displayed opposite signs (Fig. 3e, f). This symmetry originated from the opposite helical handedness of microrolls assembled from enantiomers. SEM analysis revealed that Fmoc-YLKL/SO42− and Fomc-YDKD/SO42− microrolls adopted M-type (left-handed helix) and P-type (right-handed helix) arrangements, respectively, with diameters of ~1 μm (Fig. 3g, and Figs. S15–S17). These results suggested that the supramolecular chiral and polarization direction of light emitted by the microrolls could be modulated by molecular chirality. Notably, the Fmoc-YLKL/SO42− and Fomc-YDKD/SO42− microrolls exhibited an asymmetric factor glum as high as −0.062 ± 0.017 and 0.028 ± 0.003, respectively (Fig. S18). The Fmoc-YLKL/SO42− microrolls exhibit remarkably high glum values compared to other peptide-based assemblies (Fig. S19)40,41. The high value underscores that anion co-assembly is an effective strategy for achieving chiral amplification at the supramolecular level.
The dynamic and reversible nature of non-covalent interactions enables the modulation of supramolecular assembly structures in response to environmental stimuli42,43,44. In this study, the Fmoc-YK molecule contains pH-responsive amino and carboxyl groups. We hypothesized that altering the solution pH could modulate the chiral supramolecular structure of the Fmoc-YK/SO42− system. To avoid introducing extraneous ions that might interfere with the self-assembly process, H2SO4 and NaOH were used to adjust the pH of the system. Experimental results confirmed our hypothesis. At pH <4, the Fmoc-YK/SO42− system stabilized as microrolls. When the pH increased to the range of 4−10.3, the microrolls dissolved and reassembled into nanobelts (Fig. 4a and S20a, c). The nanobelts, which were intertwined, formed macroscopic hydrogels with an average diameter of ~200 nm, bridging the gap between the microbelts and nanofibers formed by individual Fmoc-YK molecules. Upon the pH dropping below 4.0, the system underwent a reassembly into microrolls, accompanied by the rapid collapse of the nanobelt gel (Fig. S20b, d). This morphological transition from microrolls to nanobelts was reversible, with over three cycles achieved through alternating additions of NaOH and H2SO4 (Figs. S21, S22), demonstrating a clear “pH switch” property. However, when the pH exceeded 10.3, the nanobelts disassembled rapidly, resulting in a transparent solution.

a pH-responsive morphology changes and underlying mechanism (scale bar: 2 μm); (b) CD spectra of microrolls and nanobelts; (c–e) cyclic CPL spectrum and glum (error bars, mean ± SD).
This reversibility in supramolecular morphology was accompanied by concurrent transformations of chiroptical signals45. A comparative CD spectral analysis revealed that the nanobelts exhibited an inverted and attenuated Cotton effect at 260 nm relative to the microrolls (Fig. 4b). Further, under 260 nm excitation, the microrolls emitted intense left-handed CPL at 340 nm with a glum value of −0.077. In contrast, the nanobelts emitted right-handed CPL, with the glum value decreasing by an order of magnitude to ~0.0078 (Fig. 4c, d). Repeated pH-induced switching between microrolls and nanobelts enabled in situ inversion of the CPL signal for at least three cycles (Fig. 4e). This inversion highlighted a strong correlation between the supramolecular arrangement and CPL properties, rather than a direct dependence on the inherent molecular chirality. Unlike most previous reports, where opposite CPL signals arise from different chiral assembly building blocks, our study demonstrated CPL inversion and amplification via the pH switch mechanism, which controlled the insertion and extraction of SO42−. This approach not only provides insights into self-assembly pathways and chirality switching phenomena in natural systems but also offers a framework for designing intelligent chiral optical materials based on switchable CPL luminescence.
The pH-responsive molecular mechanism was attributed to changes in the protonation state of the Fmoc-YK functional groups. At low pH (< 4.0), the amino group was protonated, causing the molecule to adopt a cationic form. To balance the charge, Fmoc-YK co-assembled with SO42−, forming microrolls. As the pH increased (4 < pH < 10.3), the amino group remained protonated, while the carboxyl group deprotonated, resulting in a zwitterionic form of Fmoc-YK that achieved charge balance. In this state, the peptide self-assembled into nanobelts (Fig. S23). CD spectra showed that all samples at different pH (3.1, 6.0, 7.2, 9.5) exhibited negative bands at 210 nm, a typical signature of β-sheet structures (Fig. S24). However, in the presence of sulfate, strong CPL emission was observed only when pH < 4.0 (typical at 3.1), where Fmoc-YK co-assembled with SO42− to form microrolls. Under other pH conditions, merely weak CPL signals were detected due to the failure of effective chiral assembly (Fig. S25). In strongly alkaline conditions, Fmoc-YK existed as a deprotonated carboxyl anion, forming soluble salts with cations present in the solution.
Chirality and energy transfer play a crucial role in biological systems and offer valuable strategies for constructing high-performance chiral optical materials46. Building on this concept, we investigated whether Fmoc-YK/SO42− microrolls could serve as templates to transfer both chiral and energy signals to Thioflavin T (ThT), an achiral fluorescent dye. ThT selectively bound to β-sheet regions in peptide assemblies, leading to fluorescence emission (Fig. 5a). As shown in Fig. 5b, the absorption spectrum of ThT (330–475 nm) overlapped with the fluorescence emission spectrum of Fmoc-YK/SO42− microrolls (320–425 nm), suggesting that ThT could act as an energy acceptor when the microrolls were excited. To further explore this, we measured the fluorescence spectra of the microrolls and ThT. At a constant Fmoc-YK/SO42− concentration of 20 mM, we gradually increased the ThT concentration from 0.001 mg/mL to 0.15 mg/mL. Under 260 nm excitation, the emission peaks of the microrolls (320–425 nm) progressively decreased, while the emission peaks of ThT (470–600 nm) increased (Fig. 5c and S26a). According to the definition of energy-transfer efficiency:
where IDA and ID are the fluorescence intensities of the donor in the presence and absence of the acceptor, respectively47,48, the microrolls achieved an outstanding energy transfer efficiency of 97% (ThT 0.15 mg/mL), rivaling high-performance systems such as liquid crystals, inorganic nanomaterials, and polymeric materials49,50,51. Notably, in the absence of microrolls, ThT exhibited minimal emission in the 470–600 nm range under identical excitation at concentrations ranging from 0.001 mg/mL to 0.15 mg/mL (Fig. S26b). These results suggested the occurrence of fluorescence resonance energy transfer (FRET) between the Fmoc-YK/SO42− microrolls and ThT.

a Schematic diagram of the energy and chiral transfer in Fmoc-YK/SO42⁻ microrolls upon doping with Thioflavin T (ThT); (b) normalized fluorescence (FL) spectrum of Fmoc-YLKL/SO42⁻ microrolls and UV absorption (abs) spectrum of 0.1 mg/mL ThT solution (the shaded area indicating the overlap between the microrolls’ emission peak and ThT’s absorption peak); (c) FRET in Fmoc-YLKL/SO42⁻ microrolls after doping with varying concentrations of ThT solution; (d–f) the CD spectra, CPL spectra, and glum of Fmoc-YLKL/SO42⁻ and Fmoc-YDKD/SO42⁻ microrolls after doping with 0.1 mg/mL ThT; (g) schematic setup for enantioselective photopolymerization of 2,4-heneicosadiynoic acid (HA); (h) CD spectra of poly(diacetylene) (PDA) films after exposing to CPL generated from the Fmoc-YK/SO42− microrolls. The inset shows the photographs of HA and PDA films.
It is important to note that the incorporation of ThT is constrained by a threshold concentration. Below 0.1 mg/mL, the microrolls retained their structural integrity, indicating that ThT was likely localized on the surface or within the voids of the coiled structures without disrupting the assembly (Figs. S27 and S28). However, at a ThT concentration of 0.15 mg/mL, there was a significant reduction in the microroll population, and fibrous assemblies emerged. This transition was likely due to the positive charge of ThT disrupting the electrostatic balance between Fmoc-YK and SO42−, which led to the disassembly of the microrolls and the formation of a fibrous network. Based on these observations, we conclude that 0.1 mg/mL is the optimal ThT doping concentration.
To investigate whether the achiral ThT could adopt the chirality of the microrolls, we analyzed the CD spectra of the microrolls in the presence of 0.1 mg/mL ThT. A new CD signal at 470 nm (Fig. 5d), corresponding to the absorption peak of ThT, was observed, indicating that the microrolls induced a transfer of supramolecular chirality to ThT. Furthermore, after doping with ThT, mirror-symmetric CPL signals in the 400–600 nm range were observed for both Fmoc-YLKL/SO42− and Fmoc-YDKD/SO42− microrolls (Fig. 5e). This finding demonstrated that the exceptional chirality and excited-state energy of the microrolls were transferred to the achiral ThT, resulting in the appearance of a strong new CPL signal. Remarkably, ThT exhibited a glum value on the order of 10−2 (–0.012) (Fig. 5f), which was at a high level for chirality and energy transfer systems (typically limited to 10⁻³)52,53,54. This result underscores the high efficiency of chirality and energy transfer mediated by the microrolls. Our additional experiments demonstrate that microrolls can be further templated to transfer chirality to conventional fluorescent dyes. When achiral rhodamine Rhodamine B was mixed with the microscrolls, it exhibited positive Cotton effects at around 570 nm and CPL at 620 nm (Fig. S29).
CPL has emerged as one of the most promising sources for initiating enantioselective polymerization reactions, enabling the production of optically active polymers from non-chiral monomers without the need for chiral dopants or catalysts55,56. The CPL emission from Fmoc-YK/SO42− microrolls falls within the high-energy ultraviolet (UV) range, allowing it to trigger various polymerization reactions. Additionally, the large glum value of these microrolls could enhance the stereoselectivity of the resulting products, positioning them as potential candidates for a new generation of environmentally friendly, biodegradable CPL light sources.
In this study, we investigated the use of CPL from microrolls as a chiral light source to initiate enantioselective polymerization of 2,4-heneicosadiynoic acid (HA). We developed an irradiation platform (Fig. 5g) containing Fmoc-YLKL/SO42− and Fmoc-YDKD/SO42− microrolls encapsulated within a quartz cell. Upon irradiation with 275 nm UV light, the Fmoc-YK/SO42− microrolls emitted chiral CPL signals with opposite handedness in the 300–400 nm wavelength range. A short-pass filter was used to isolate the CPL, which was then directed onto a quartz slide coated with HA film. Within 3 min of CPL irradiation, the initially white HA film transformed into blue, indicating the successful polymerization of the non-chiral HA monomer into poly(diacetylene) (PDA) (Fig. 5h inset).
Remarkably, PDA films exposed to CPL with opposite handedness displayed inverse Cotton effects. As illustrated in Fig. 5h, exposure to left-handed CPL from Fmoc-YLKL/SO42− microrolls resulted in a negative Cotton effect at 680 nm, while exposure to right-handed CPL from Fmoc-YDKD/SO42− microrolls induced a positive Cotton effect. This observation demonstrated that the chirality of the resulting PDA polymer could be modulated by the optical activity of the CPL. Notably, when the HA film was irradiated with conventional UV light (without CPL), no significant signals were detected in the CD spectra of the resulting PDA, confirming that the chirality of the PDA originated solely from the CPL emitted by the Fmoc-YK/SO42− microrolls (Fig. 5h).
To evaluate the emission performance of the microrolls, the circularly polarized luminescence brightness (CPB) which integrates circular polarization (glum) and brightness (fluorescence quantum yield) of microrolls is calculated57. The CPB of the aggregated samples is defined as:
where the \({\xi }_{{abs}}\) is the absorption efficiency, which is the ratio between the number of photons absorbed over the number of incident photons, \(\phi\) is the fluorescence quantum yield, and \(\left|{g}_{{lum}}\right|\) is the dissymmetry factor. We have measured the absorption efficiency and fluorescence quantum yield of the microrolls at 260 nm by using the integrating sphere. The \({\xi }_{{abs}}\) was determined to be 67.2%, while the \(\phi\) was 0.087. Combining these values with the previously measured \(\left|{g}_{{lum}}\right|\) (0.062), we calculated the CPB to be 0.0017. The CPB value is primarily constrained by the photophysical properties of the Fmoc group. In the future, substitution of Fmoc with high-performance fluorescent chromophores might revolutionize the CPB performance in this system.
Discussion
In conclusion, we introduce an effective anion co-assembly strategy for engineering supramolecular chiral structures in dipeptide assemblies. By leveraging sulfate ions, we successfully transform Fmoc-YK assemblies into chiral microrolls, significantly enhancing their CPL performance with a glum value as high as −0.062. The dynamic responsiveness of these assemblies to pH variations highlights their potential for developing switchable chiral materials that can adapt to changing environmental conditions. Additionally, these chiral microrolls show promise as efficient chiral light sources for enantioselective polymerization, opening new pathways for the application of peptide-based materials in chiral catalysis. This work not only provides valuable insights into the role of anions in regulating supramolecular chirality but also offers a versatile strategy for engineering chirality in dipeptide assemblies, paving the way for the development of high-performance CPL biomaterials.
Methods
Materials
Fmoc-YLKL and Fmoc-YDKD with enantiomeric chirality were synthesized by GL Biochem Ltd. (Shanghai, China), with a purity exceeding 98%. Analytical-grade salts (Na2SO4, MgSO4, FeSO4, NaH2PO4, Na2S2O8, CH3COONa and Sodium Citrate) were procured from Beijing Chemical Company (Beijing, China). NaOH and H2SO4 for pH adjustments were obtained from HuShi Company. 2,4-heneicosadiynoic acid (HA) was supplied by TCI (Tokyo, Japan). Custom 300–400 nm short-pass filters were acquired from Shenzhen Zhonglai Technology Co., Ltd. (Shenzhen, China), and 275 nm UV lamps from Zhongshan Yanxi Zao Lighting Electrical Factory (Zhongshan, China). Water was purified by an ELGA PURELAB purification system (High Wycombe, UK) with a resistivity of 18.2 M·cm.
Sample preparation
Fmoc-YK gel
A 20 mM solution of Fmoc-YK (5.32 mg, 500 μL) was prepared by dissolving the compound in deionized water with the aid of ultrasonication. After the ultrasound stopped, the clear solution gradually solidified into a translucent gel (pH 3.1). pH changes in the system were continuously monitored using a pH meter (Mettler Toledo, FE28, Switzerland). Results demonstrated that the pH remained essentially constant throughout the gel formation process.
Fmoc-YK microbelts
The aforementioned Fmoc-YK gel (20 mM) gradually develops white crystals in the room temperature. After 6 h of aging, the gel fully transforms into crystalline microbelts (pH 3.1).
Fmoc-YK microrolls
Fmoc-YK solution (5.32 mg, 400 μL, pH 3.1) was mixed with sodium sulfate solution (50 mM, 100 μL, pH 3.1) to form the hydrogel. The pH of the solution was precisely adjusted through the addition of H2SO4 prior to the mixing process. After 1 h of aging, the hydrogel fully transforms into crystalline microrolls (20 mM, pH 3.1).
The influence of other anions (H2PO4 −, CH3COO−, citrate ions, and Cl−) on the assembly process
Adjust the pH of the sodium salt solution to 3.1 by using the conjugate acids of these anions, and take 100 μL of the adjusted sodium salt solution and mix it thoroughly with 400 μL of Fmoc-YK (5.32 mg, pH 3.1), ensuring the final assembly system reaches a peptide concentration of 20 mM at pH 3.1.
Fmoc-YK nanobelts
pH of the Fmoc-YK microrolls (20 mM) was adjusted to the range of 4.0−10.3 by adding NaOH (1 M). The Fmoc-YK microrolls were completely transformed into Fmoc-YK nanobelts after aging for about 2 h. When the pH was reduced below 4.0 by adding H2SO4 solution (1 M), the nanobelts reassembled into microrolls.
In the subsequent characterization procedures, unless otherwise specified, gel-type samples (e.g., nanofiber and nanobelt) were directly collected from the as-prepared hydrogel, while precipitated samples (e.g., microbelt and microroll) were obtained directly from the above prepared suspension following vigorous agitation to ensure homogeneity.
HA Membrane
HA powder was dissolved in toluene, subjected to ultrasonication, and filtered using a 5 μm organic filter to remove undissolved particles. The resulting solution was applied to a quartz slide and left to air-dry, producing a white HA membrane. The membrane was subsequently stored under dark conditions for future use.
Chiral PDA Triggered by CPL
Chiral micro-rolls were placed in a quartz cuvette with a 0.2 mm pathlength, positioned behind a 300–400 nm short-pass filter. A white HA membrane was put after the filter. After setting up the irradiation platform, the cuvette was exposed to 275 nm UV light until the HA-coated quartz slide exhibited a blue color. The irradiation was carried out in a light-proof environment to minimize external light interference.
Characterization
Scanning Electron Microscopy (SEM)
For liquid-state samples, 5 μL of the solution was deposited onto a clean silicon wafer, and residual moisture was removed under vacuum. For powder samples, the samples were dried under low-temperature vacuum conditions. Both samples were sputter-coated with a thin platinum layer to enhance conductivity. The micromorphology was imaged using a Hitachi SU8020 SEM at 10 kV and 10 μA.
Transmission Electron Microscopy (TEM)
A 5 μL volume of sample was dropped to a carbon-coated copper grid (300 mesh) and left to settle briefly. Excess liquid was blotted with filter paper. After vacuum drying, the samples were imaged using a JEOL JEM-2100(UHR) TEM at 100–120 kV.
Cryo-Transmission Electron Microscopy (cryo-TEM)
Samples for cryo-TEM were prepared using an immersion freezing technique. A 3 μL droplet of Fmoc-YK solution (dissolved in 80 °C ddH2O) was deposited onto a carbon-coated multi-hole TEM grid. Excess liquid was removed with filter paper, leaving a thin film that was rapidly cooled by liquid nitrogen. The samples were then transferred to a low-temperature holder and visualized using a Thermo Scientific Themis300 cryo-TEM at 120 kV.
Selected Area Electron Diffraction (SAED)
SAED patterns were captured using the FEI Tecnai G2 20 S-TWIN TEM, with sample preparation performed as described for TEM.
Fourier Transform Infrared Spectroscopy (FTIR)
Powder samples were vacuum-dried at low temperatures, then finely ground with dry potassium bromide (KBr) in an agate mortar. The mixture was pressed into thin, transparent pellets. FTIR spectra were recorded on a Thermo NICOLET iS 50 spectrometer.
X-ray Diffraction (XRD)
Powder samples were placed on a clean silicon wafer and pressed flat to ensure even sample distribution. XRD patterns were recorded using a PANalytical B.V. X’Pert PRO MPD diffractometer.
Single Crystal X-ray diffraction (SXRD)
Crystals were first transferred to polyisobutylene for protection, after which a suitably sized crystal was selected and mounted on the diffraction device. Data were collected at 170 K using a Rigaku XtaLAB Synergy-R diffractomete with CuKα radiation (λ = 1.54184 Å). Structure solution and refinement were performed using the Olex2 software package.
Circular Dichroism (CD)
The sample was prepared by uniformly suspending the precipitated material (generated from a 20 mM dipeptide solution after shaking) to ensure consistency during measurements. Subsequently, a 100 μL aliquot of the suspension was drop-cast onto the quartz plate with the depth of 0.2 mm groove, followed by clamping the two quartz plates together and mounting them on a holder for CD spectroscopic characterization. CD spectra were recorded using a JASCO J-1700 spectrophotometer.
Circularly Polarized Luminescence (CPL)
A 100 μL aliquot of homogenously vortexed dipeptide solution (20 mM) was drop-cast onto 0.2 mm path length quartz cuvettes. The CPL measurements were conducted using a JASCO CPL-300 spectrophotometer.
Absorption efficiency
The Fmoc-YK/SO42− microrolls were rapidly frozen by immersion in liquid nitrogen for 5 min, followed by freeze-drying under vacuum (10 Pa) for 24 h to obtain a powder. The resulting powder was uniformly packed into a powder sample holder. UV-Vis diffuse reflectance and transmittance spectra were recorded using a spectrophotometer (Lambda750) equipped with an integrating sphere (60 mm), with a 1.25-in White Calibration Material (PerkinElmer) as a 100% reflectance reference. Spectra were collected from 200 to 800 nm. The absorption percentage (A%) was calculated as A% = 100% – R% – T%, where R% and T% represent the reflectance and transmittance percentages, respectively. All measurements were performed in triplicate to ensure reproducibility.
Fluorescence quantum yield
The fluorescence quantum yield of Fmoc-YK/SO42− microrolls under 260 nm excitation was measured using an integrating sphere with the C11347-11 detector (Hamamatsu, Japan). Measurements were repeated three times independently.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information files. Crystallographic data of the Fmoc-YK has been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition number 2446035. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. The data that support the findings of this study are also available from the authors on request. Source data are provided with this paper.
References
Aida, T., Meijer, E. W. & Stupp, S. I. Functional supramolecular polymers. Science 335, 813–817 (2012).
Jia, Y. & Li, J. Reconstitution of FoF1-ATPase-based biomimetic systems. Nat. Rev. Chem. 3, 361–374 (2019).
Ariga, K. Nanoarchitectonics: the method for everything in materials science. Bull. Chem. Soc. Jpn. 97, uoad001 (2024).
Sang, Y. & Liu, M. Hierarchical self-assembly into chiral nanostructures. Chem. Sci. 13, 633–656 (2022).
Liu, M., Zhang, L. & Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 115, 7304–7397 (2015).
Chen, Z., Chi, Z., Sun, Y. & Lv, Z. Chirality in peptide-based materials: from chirality effects to potential applications. Chirality 33, 618–642 (2021).
Qi, K. et al. Chiral inversion induced by aromatic interactions in short peptide assembly. Nat. Commun. 15, 6186 (2024).
Xu, H. et al. Controlling 1D nanostructures and handedness by polar residue chirality of amphiphilic peptides. Small 20, e2304424 (2024).
Wang, M. et al. Unexpected role of achiral Glycine in determining the suprastructural handedness of peptide nanofibrils. ACS Nano 15, 10328–10341 (2021).
Sangji, M. H. et al. Supramolecular interactions and morphology of self-assembling peptide amphiphile nanostructures. Nano Lett. 21, 6146–6155 (2021).
Li, Q. et al. Self-Assembly of peptide hierarchical helical arrays with sequence-encoded circularly polarized luminescence. Nano Lett. 21, 6406–6415 (2021).
Wu, A. et al. Co-assembled supramolecular gel of dipeptide and pyridine derivatives with controlled chirality. Angew. Chem. Int. Ed. 60, 2099–2103 (2021).
Wu, A. et al. Tunable chirality of self-assembled dipeptides mediated by bipyridine derivative. Angew. Chem. Int. Ed. 62, e202314368 (2023).
Ji, W. et al. Metal-ion modulated structural transformation of amyloid-like dipeptide supramolecular self-assembly. ACS Nano 13, 7300–7309 (2019).
Wang, F. & Feng, C. L. Metal-ion-mediated supramolecular chirality of l-phenylalanine based hydrogels. Angew. Chem. Int. Ed. 57, 5655–5659 (2018).
Zhang, G. et al. Role of molecular chirality and solvents in directing the self-assembly of peptide into an ultra-pH-sensitive hydrogel. J. Colloid Interface Sci. 577, 388–396 (2020).
Xie, Y. et al. Reconfigurable chiral self-assembly of peptides through control of terminal charges. Small 13, 1700999 (2017).
Sang, Y., Han, J., Zhao, T., Duan, P. & Liu, M. Circularly polarized luminescence in nanoassemblies: generation, amplification, and application. Adv. Mater. 32, e1900110 (2020).
Zhao, T. & Duan, P. Photon upconversion cooperates with downshifting in chiral systems: modulation, amplification, and applications of circularly polarized luminescence. Angew. Chem. Int. Ed. 63, e202406524 (2024).
Jiang, S. & Kotov, N. A. Circular polarized light emission in chiral inorganic nanomaterials. Adv. Mater. 35, e2108431 (2023).
Wade, J. et al. 500-fold amplification of small molecule circularly polarised luminescence through circularly polarised FRET. Angew. Chem. Int. Ed. 60, 222–227 (2021).
Jiang, J. et al. Aqueous circularly polarized luminescence induced by homopolypeptide self-assembly. J. Am. Chem. Soc. 145, 27282–27294 (2023).
Lu, J., Jung, H. J., Kim, J.-Y. & Kotov, N. A. Bright, circularly polarized black-body radiation from twisted nanocarbon filaments. Science 386, 1400–1404 (2024).
Liu, J., Molard, Y., Prévôt, M. E. & Hegmann, T. Highly tunable circularly polarized emission of an aggregation-induced emission dye using helical nano- and microfilaments as supramolecular chiral templates. ACS Appl. Mater. Interfaces 14, 29398–29411 (2022).
Gowda, A. et al. Organic chiral nano- and microfilaments: types, formation, and template applications. Mater. Horiz. 11, 316–340 (2024).
Zhang, Y. et al. Circularly polarized luminescence in chiral materials. Matter 5, 837–875 (2022).
Lu, J. et al. Enhanced optical asymmetry in supramolecular chiroplasmonic assemblies with long-range order. Science 371, 1368–1374 (2021).
Li, S., Du, L. & Wang, W. Impact of anions on the surface organisation of lipid monolayers at the air–water interface. Environ. Chem. 14, 407–416 (2017).
Geng, Y. et al. Controllable preparation and formation mechanism of highly ordered hydroxyapatite nanofibers: Effect of PO43−/CO32−. J. Mater. Res. Technol. 30, 7125–7133 (2024).
Castillo, G. M., Lukito, W., Wight, T. N. & Snow, A. D. The sulfate moieties of glycosaminoglycans are critical for the enhancement of β-amyloid protein fibril formation. J. Neurochem. 72, 1681–1687 (1999).
Zhang, W., Zhao, J. & Yang, D. Anion-coordination-driven assembly: from discrete supramolecular self-assemblies to functional soft materials. Chempluschem 87, e202200294 (2022).
Yang, S. et al. Progress in infrared spectroscopy as an efficient tool for predicting protein secondary structure. Int. J. Biol. Macromol. 206, 175–187 (2022).
Li, X., Li, Q., Wu, A. & Li, J. CO2 induces symmetry breaking in layered dipeptide crystals. Angew. Chem. Int. Ed. 62, e202214184 (2022).
Ghosh, G. et al. Control over multiple nano- and secondary structures in peptide self-assembly. Angew. Chem. Int. Ed. 61, e202113403 (2022).
Ghosh, G., Barman, R., Sarkar, J. & Ghosh, S. pH-responsive biocompatible supramolecular peptide hydrogel. J. Phys. Chem. B 123, 5909–5915 (2019).
Manoilova, O. V., Olindo, R., Areán, C. O. & Lercher, J. A. Variable temperature FTIR study on the surface acidity of variously treated sulfated zirconias. Catal. Commun. 8, 865–870 (2007).
Bera, S. et al. Rigid helical-like assemblies from a self-aggregating tripeptide. Nat. Mater. 18, 503–509 (2019).
Xing, Q. et al. Aromatic motifs dictate nanohelix handedness of tripeptides. ACS Nano 12, 12305–12314 (2018).
Ryan, K. et al. Nanoscale piezoelectric properties of self-assembled Fmoc–FF peptide fibrous networks. ACS Appl. Mater. Interfaces 7, 12702–12707 (2015).
Lee, H. E. et al. Cysteine-encoded chirality evolution in plasmonic rhombic dodecahedral gold nanoparticles. Nat. Commun. 11, 263 (2020).
Shen, Y. et al. Strong CPL-active liquid crystal materials induced by intermolecular hydrogen-bonding interaction and a chirality induction mechanism. Soft Matter 18, 477–481 (2022).
Li, X. et al. A photoinduced reversible phase transition in a dipeptide supramolecular assembly. Angew. Chem. Int. Ed. 57, 1903–1907 (2018).
Li, X. et al. Self-assembled dipeptide aerogels with tunable wettability. Angew. Chem. Int. Ed. 59, 11932–11936 (2020).
Li, X. et al. Three-dimensional (3D) bioprinting of medium toughened dipeptide hydrogel scaffolds with Hofmeister effect. J. Colloid Interface Sci. 639, 1–6 (2023).
Ramesh, A., Das, T. N., Maji, T. K. & Ghosh, G. Unravelling denaturation, temperature and cosolvent-driven chiroptical switching in peptide self-assembly with switchable piezoelectric responses. Chem. Sci. 15, 16355–16366 (2024).
Shen, B. W., Kim, Y. & Lee, M. Supramolecular chiral 2D materials and emerging functions. Adv. Mater. 32, 1905669 (2020).
Peng, H.-Q. et al. Biological applications of supramolecular assemblies designed for excitation energy transfer. Chem. Rev. 115, 7502–7542 (2015).
Ji, L. et al. Dimension-tunable circularly polarized luminescent nanoassemblies with emerging selective chirality and energy transfer. ACS Nano 14, 2373–2384 (2020).
Yao, M. et al. Orthogonal integration of holographic and fluorescent dual images based on energy transfer from liquid crystals to a photocleavable AIEgen. J. Mater. Chem. C. 11, 3504–3512 (2023).
Cortes-Villena, A., Caminos, D. A., Galian, R. E. & Perez-Prieto, J. Singlet sensitization of a BODIPY rotor triggered by marriage with perovskite nanocrystals. Adv. Opt. Mater. 11, 2300138 (2023).
Wang, Y. et al. Quantitative examination and mechanistic insights of polymer chain conformation confined in nanopores by time-resolved fluorescence resonance energy transfer. ACS Macro Lett. 13, 1584–1590 (2024).
Xu, L. et al. Solvent-modulated chiral self-assembly: selective formation of helical nanotubes, nanotwists, and energy transfer. ACS Appl. Mater. Interfaces 14, 1765–1773 (2022).
Ji, L. et al. Cooperative chirality and sequential energy transfer in a supramolecular light-harvesting nanotube. Angew. Chem. Int. Ed. 58, 844–848 (2019).
Deng, M., Zhang, L., Jiang, Y. & Liu, M. Role of achiral nucleobases in multicomponent chiral self-assembly: purine-triggered helix and chirality transfer. Angew. Chem. Int. Ed. 55, 15062–15066 (2016).
Furlan, F. et al. Chiral materials and mechanisms for circularly polarized light-emitting diodes. Nat. Photonics 18, 658–688 (2024).
Wang, Z., Hao, A. & Xing, P. Charge-transfer complex doped photothermal hydrogels for discriminating circularly polarized near-infrared light. Angew. Chem. Int. Ed. 62, e202214504 (2023).
Arrico, L., Di Bari, L. & Zinna, F. Quantifying the overall efficiency of circularly polarized emitters. Chem. Eur. J. 27, 2920–2934 (2021).
Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB 0520300 to S.B.), the Chinese Major Program for the National Key Research and Development Project (2020YFA0112603 to S.B., 2023YFC3904601 to Q.L.), the National Natural Science Foundation (22277121 to S.B., 22307117 to S.L., 22407126 to Y.T., 22193031 to Junbai Li, 22193032 to Y.Y., 22402040 to Xianbao Li), the China Postdoctoral Science Foundation (No. 2023M730816 to Xianbao Li), the Shandong Provincial Laboratory Project (No. SYS202202 to C.J.), the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (No. JNL-2023005Q to Xin Li), the Shandong Provincial Natural Science Foundation (No. ZR2024QC163 to Xin Li). The authors are deeply grateful to Dr. Bo Guan for providing guidance on the LLPS experiments.
Author information
Authors and Affiliations
Contributions
Xin Li, Xianbao Li and Q.L. conceived the project. Xin Li, Q.H., Xianbao L, and R.L. prepared samples, performed characterizations and analyzed the results. S.L., Y.S., Jieling Li and Y.T. helped with the sample fabrication and characterizations. A.W., Q.L. and Y.Y. analyzed data. S.B. and Junbai Li provided technical supervision and critical revisions. All authors discussed the results and contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Goutam Ghosh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Han, Q., Li, X. et al. Chiroptical switching and strong circularly polarized luminescence in peptide supramolecular assemblies. Nat Commun 16, 6044 (2025). https://doi.org/10.1038/s41467-025-61007-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61007-6
