
Abstract
Temperature-dependent, selective molecular diffusion through porous materials is crucial for membrane separations and is typically modeled as an Arrhenius-type activated process. Although this dependence can be described phenomenologically by an activation energy, tracing its molecular origins is often difficult, hindering robust membrane design for practical applications. Here, we investigate gas transport across monolayer nanoporous graphene membranes and observe significant, reversible, temperature-robust, and gas species-selective activated transport, with increased selectivity at rising temperatures, unlike many conventional membranes. Combined experiment and modelling trace this behavior to graphene nanopore edge functional groups, whose thermal fluctuations modulate effective pore size. This activated transport remains stable with aging over 1 year and shows selectivity exceeding 70 for hydrogen/hydrocarbon mixture separation at 220 °C, representative of dehydrogenation reactor temperatures. Our results demonstrate the thermal and long-term robustness of nanoporous graphene membranes, suggesting potential for precise engineering of nanopore surface chemistries in membranes for challenging molecular separations.
Similar content being viewed by others


Introduction
Chemical separations are integral to industrial production and are intricately linked with reactor systems, as well as heat and energy utilities within process flow designs1. Operating these systems under matching conditions such as consistent pressures and temperatures enhances efficiency and reduces energy, infrastructure, and operational costs2. Unlike other separation processes such as adsorption and distillation, which require phase changes or pressure swings, membrane separations rely on selective transport across a barrier material3. This attribute, along with their compact footprint, modular configuration, and high energy efficiency, makes them increasingly attractive for designing production processes focused on process intensification and reductions in carbon and waste footprints4.
For example, thermally robust and chemically stable membranes that offer both high permeance and selectivity are sought after, providing distinct advantages across various industrial applications. These membranes can directly separate products downstream of, or within reactors, operating under harsh conditions involving high temperatures and aggressive chemicals. This capability eliminates the need for substantial changes in operating conditions and reduces the reliance on additional processes. Key applications include, but are not limited to, CO2/CH4 separation following biomass gasification, CO2 scrubbing from flue gas, H2/CH4 separation in hydrogen recovery, hydrogen production from hydrocarbon reforming, and the separation of active pharmaceutical ingredients or intermediates from high-temperature, harsh solvents5.
Across various molecular membrane separation mechanisms6—spanning solution-diffusion, sorption-diffusion, translational diffusion and surface diffusion in molecular sieving—diffusive processes play a crucial role (oftentimes the limiting process) in determining membrane separation performance7. These processes are significantly influenced by temperature, driven by diverse transport modes that vary depending on the class of membrane material, the length scales, and the polydispersity of membrane microstructures. Yet, temperature-dependent diffusive processes, pertaining to the energetic penalty of diffusive jumps, can be universally modeled as an activated process that have been well characterized phenomenologically by an activation energy. This empirical activation energy has been widely employed in describing and rationalizing the transport properties of membranes as a function of temperature, impacting the theoretical upper bound—the trade-off between membrane permeance and selectivity developed for gas separation—in dense polymeric membranes8, as well as molecular sieving across pores and surfaces in porous membranes6,9,10,11,12,13.
Although the activation energy of molecular permeation can be empirically determined quite easily by fitting permeance versus temperature data to an empirical Arrhenius-type model, identifying their molecular origins proves much more challenging. This complexity arises from the convoluted adsorption and solution behaviors of penetrants, and intricate structures and dynamics of membrane materials. Theoretical relationships based on chain fluctuations have been proposed for dense polymeric membranes, correlating the energy barrier of diffusion with penetrant molecular sizes—specifically, scaling with the square of the penetrant diameter14. Within this framework, larger molecules encounter higher energy barriers than smaller ones while diffusing across the membrane matrix. As temperature increases, the permeation rate for larger molecules escalates more significantly than for smaller ones, following an Arrhenius-type temperature dependence, inevitably leading to reduced membrane selectivity at higher operating temperatures8. This suggests a fundamental limitation in designing membranes that can maintain or improve selectivity at elevated temperatures, despite exhibiting high selectivity at ambient temperatures or ideal conditions typically employed in lab-scale tests for new membrane material development6. We collate temperature-dependent membrane selectivity among various existing and emerging membrane materials and indeed observe such a challenge in membrane design: membrane selectivity generally decreases with rising temperature, except for gas pairs involving a highly condensable gas, such as CO2 (Supplementary Fig. 1). Increased selectivity at higher temperatures is observed, though at much lower frequency, and is seen mostly in inorganic or composite membranes. However, understanding the molecular-level mechanisms behind temperature-robust selective mass transport and replicating these relationships in membranes remains elusive. The extensive reported values of activation energy among various membrane materials and structures, thus far, tend to be more indicative than predictive.
Atomically thin nanoporous membranes, featuring monolayer nanoporous graphene, present a fundamentally different approach to studying selective mass transport with molecular precision across a wide range of testing conditions and chemical environments5,10. Over the past decade, we and others have utilized nanopores suspended in monolayer crystals for various molecular separations10,15,16,17,18,19. This approach is driven by the atomic thinness of these nanopores that sets an upper bound on permeance achievable with molecular sieving, their high mechanical strength capable of withstanding high pressures for high recovery membrane processes, and broad chemical resistance that enables challenging molecular separations previously not feasible for membranes19,20. It has been demonstrated that high-density nanopores (~1 × 1013 cm‒2) can be effectively created on graphene lattices using a variety of techniques, including plasma etching, ozone/CO2 etching17, and ion irradiation21. These nanopores are capable of separating chemicals in gas phase10, ions in aqueous mixtures22, and molecules in organic solvents20. Expansion-shrinkage cycling processing techniques have been developed to fine-tune pore sizes and narrow size distributions with independently designable mean, standard deviation, and tail deviation at angstrom-level resolution22. Atom-resolved characterization and atomistic simulations have been employed to begin discerning the structure and catalog isomers for these nanopores17,23. These membranes are beginning to show functioning separation performance at coupon-scale, outperforming conventional membranes in ion, gas, and challenging molecular separations5,16,20,24,25.
Building on these advances, this work pushes the boundaries by exploring the possible molecular origins of temperature-dependent gas permeation through nanoporous membrane materials. We use nanoporous graphene membranes, with their relatively simple, 2D nanoporous structure, as an example system to carry out this study in the context of high-temperature, industrial hydrogen separations. We systematically conduct experiments and modeling of single and mixture gas permeation (including high boiling point and condensable hydrocarbons) across monolayer nanoporous graphene membranes with temperatures held above 200 °C. Significant, temperature-dependent, selective gas diffusion across graphene nanopores is revealed, which are edge-activated and translate into improved selectivity with rising temperatures. We demonstrate the thermal and long-term robustness (after over 1 year of aging) of nanoporous graphene membranes, achieving consistent hydrogen permeance (1.04 × 103 GPU, and 2.2 × 105 GPU when scaled for nanoporous graphene area at 220 °C), and selectivity of over 70 in high-temperature hydrogen separations relevant to dehydrogenation reactor conditions in petrochemical refining or liquid hydrogen carrier systems. We identify unexplored aspects of membrane design, specifically the role of designing the dynamics of the nanoporous materials and surface functional groups to enhance the performance of porous membranes, particularly in developing temperature-robust strategies using molecular sieving mechanisms.
Results
Membrane fabrication and gas permeation tests
Atomically thin nanoporous graphene membranes take the configuration of a thin film composite membrane (Fig. 1a, b). The membranes were fabricated by transferring monolayer graphene onto a porous support using a poly(methyl methacrylate)(PMMA)-assisted carrier process followed by nanopore creation (see details in Methods). Based on our prior work on engineering defect-tolerant membranes20,26, the porous supports chosen in this work are hydrophobic polyimide track-etched membranes (PITEM) with a nominal pore density of 6 × 109 cm‒2 and pore sizes of 10 nm or 20 nm, referred to as PITEM(10) and PITEM(20), respectively. Visually, graphene transferred to the PITEM is visible as a dark square (Fig. 1a, inset photo). Scanning electron microscopy (SEM) imaging of the membrane surface showed the top continuous and conductive graphene layer with underlying PITEM pores clearly visible (Fig. 1a).

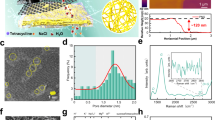
a A scanning electron microscopy image and a photograph of the surface of a polyimide track-etched membrane (PITEM) supported graphene, PITEM(10)_G membrane. b A schematic showing various transport pathways involved in gas permeation through nanoporous graphene membranes including (1) bulk impingement, (2) Surface diffusion, (3) Translational diffusion, and (4) Knudsen diffusion. c–e Temperature-dependent gas permeation through bare PITEM(10) support, graphene membranes PITEM(10)_G (without pore creation), and PITEM(10)_G_Pore (with pore creation) under thermal equilibrium. Six gas species were tested in this work, namely H2, He, CH4, N2, CO2, and SF6, respectively. Markers represent experimental data. The dashed lines in (c) represent the Knudsen diffusion model prediction of permeance as a function of temperature (\(T\)), i.e., scaling with 1/\(\sqrt{T}\), given the nominal pore size and density of the PITEM(10). In (d and e), the dashed lines represent the predicted permeance of PITEM(10)_G and PITEM(10)_G_Pore, obtained by multiplying the permeance of PITEM(10) for each gas at 300 K by the average coverage and scaling it by 1/\(\sqrt T\). Error bars on the x-axis represent the standard deviation of temperature values recorded during permeance tests. The error bars on the y-axis denote the standard deviation of permeance based on error propagation (Supplementary Note 2). f Knudsen selectivity (dash line) versus selectivity calculated by the ratio of single-gas permeance over that of H2 measured at 220 °C for three PITEM(10)_G_Pore samples, prepared under the same conditions, and with sample #1 measured again after aging for over 1 year without any interim measurements during this period. \(M\) denotes molecular mass, \(J\) denotes flux, and subscripts denote the gas. The numbers at the top left of (c−e) indicate the multiplication factor (e.g., 1e-6) applied to the y-axis values. Source data are provided as a Source Data file.
We measured gas permeation across monolayer graphene membranes, made by transferring graphene grown on copper onto PITEM support prior to pore creation, denoted as PITEM(20)_G and PITEM(10)_G, respectively (see Methods, Supplementary Figs. 2–5 and Supplementary Notes 1, 2). Given the pressure range in our single-gas measurements was <1 × 105 Pa (typically around 6 ~7 × 104 Pa), and temperature range tested was between 20 to 220 °C, the smallest mean free path of the gas molecules measured in our work is around 30.4 nm, larger than 20 nm nominal pore size of the PITEM(20) support. Therefore, gas transport across both bare PITEM(10) and PITEM(20) ought to fall under the Knudsen diffusion regime. We confirmed this with experiment (Fig. 1c and Supplementary Fig. 6a), where we show that the permeance of all gases scales with the inverse square root of temperature, 1/\({\surd {T}}\). This relationship arises from permeance scaling with the product of Knudsen diffusivity (scales with \(\surd T\)) and molar concertation at a given pressure (scales with 1/T as per the ideal gas law). We estimate the permeance using the Knudsen diffusion model using the nominal pore size and density of the PITEM supports; and the results are well aligned with the experimental measured values (dashed lines in Fig. 1c and Supplementary Fig. 6a).
The measured permeance of membranes PITEM(10)_G and PITEM(20)_G represent the leakage flow across PITEM pore areas that are not blocked by graphene, originating from: (i) defects and tears generated during membrane fabrication, and (ii) intrinsic defects present in the graphene produced by the chemical vapor deposition method. The graphene coverage, defined as the extent of blockage of transport by graphene, reached 98.4% and 99.3% (averaged over all gases), for PITEM(20)_G and PITEM(10)_G, respectively when measured at room temperature. We observe that the leakage permeance through PITEM(20)_G generally decreases as 1/\(\surd T\), falling within the Knudsen diffusion regime (Supplementary Fig. 6b). However, the leakage flow of all gas species through PITEM(10)_G increases as temperature rises (Fig. 1d), which is typical of activated gas transport. Activated gas transport has been experimentally observed in nanoporous graphene membranes by other research groups10. This occurs in the regime where gas permeates through pores that are approaching sizes below 2-3 times the diameter of the gas molecules27. We reason the observation of activated gas transport through PITEM(10)_G, but not through PITEM(20)_G, as follows26,28: Given the smaller support pore size, which imparts better isolation of large defects and tears, flow through these large defects, where the flows are in the Knudsen diffusion regime, is much reduced in PITEM(10)_G. The permeance from the activated transport, through small nanopores thought to be the intrinsic defects in graphene, dominates the flow through PITEM(10)_G. This is corroborated by the observation that the permeance of PITEM(10)_G is ~30 times lower than that of PITEM(20)_G measured at room temperatures, even though the open graphene area is only 4 times lower.
To investigate this seemingly activated transport through small nanopores on graphene, we purposefully increased the nanopore density by implementing nanopore creation in graphene using the method we developed and extensively characterized previously (see Methods). This method involves the use of ion irradiation to nucleate pore sites on the graphene lattice, followed by chemical etching to control pore growth20,21. As shown in Fig. 1e, significantly enhanced, activated gas transport was observed as a function of temperature through monolayer graphene membranes, PITEM(10)_G_Pore, after pore creation, confirming that the activated permeance is indeed from small graphene nanopores. The degree of permeance increase with temperature among different gas species varied, with small gas species like hydrogen and helium increasing more significantly, by 3-4 times, while large gases (e.g., SF6) showing relatively weak activated transport, with permeance increasing by <30% at 220 °C compared to room temperature. Activated transport was even observed for CO2, which has the largest enthalpy of adsorption on graphene surface (~ -17 to -21.7 kJ mol-1) among all gas species tested10,29. This sharply contrasts with previous molecular dynamics simulation results, where the heat of CO2 adsorption dominates, leading to a decrease in permeance as the temperature increases30. The degree of permeance enhancement (exceeding a factor of 4) at elevated temperatures for CO2 is even higher than H2 and He in some membranes (Supplementary Fig. 7). These results suggest that the permeance contribution from translational diffusion is dominant (Fig. 1b, pathway #3) in our systems, while surface pathways (Fig. 1b, pathways #2) to permeance are negligible as their contribution decreases with rising temperature, especially for gases with high adsorption enthalpy. Consequently, gas selectivity increases with rising temperatures (H2/SF6 selectivity reaches 39.4 at 220 °C versus 10.8 at room temperature). Such activated transport remained robust upon thermal cycling (Supplementary Fig. 8). In contrast, activated transport observed in previous reports on similar systems did not demonstrate thermal reversibility, or gas selectivity significantly declined after thermal treatment11,13; no robust evidence of reversible permeance across a temperature range up to 200 °C has been reported10,31. In addition, we fabricated multiple monolayer graphene membrane samples with the same fabrication and pore creation conditions, and consistently observed the same activated transport behavior and increased gas selectivity with rising temperatures (Fig. 1f, samples #1, #2, and #3). The membranes remained debris-free and visually intact both before and after gas permeation tests under thermal cycling at temperatures up to 220 °C (Supplementary Fig. 9). Furthermore, we remeasured gas permeance as a function of temperature for the membrane (sample #1) after over 1 year of aging and observed highly consistent, reversible, and significant activated gas permeation for small gas over large gases, exhibiting H2/SF6 selectivity of 42.8 at 220 °C (dark orange triangle in Fig. 1f).
Analysis of activated gas permeation through graphene nanopores
Given the membrane configuration and modes of gas transport (Fig. 1b), we modeled gas permeation across nanoporous graphene membranes by splitting it into two contributions to the permeance, \({P}_{m}\): (1) permeance through both created and intrinsic small nanopores on graphene, which are assumed to operate in the activated transport regime, and (2) permeance through large tears and defects constrained by the pore size of PITEM supports, which operate in the Knudsen diffusion regime and scale as 1/\(\surd T\) (equation (1)).
The model has three fitting parameters, namely, the activation energy, \(\left\langle {E}_{{act}}\right\rangle\) averaged over all nanopores in the activated transport regime, and two pre-factors \({N}_{p(A)}\) and \({N}_{p(n-A)}\) for the permeance contributed from the activated and Knudsen diffusion (non-activated) regimes, respectively. We fit the three parameters to the temperature-dependent permeance values, separately for each membrane and gas species, with the results represented by the dashed lines in Fig. 2a (without pore creation) and 2b (after pore creation). The model well captures the dependence of gas permeance on temperature for both membranes and all gas species. We plot \({E}_{{act}}\) and \({N}_{p(A)}\), which relate to the selective nanopore density in the membrane, as a function of gas kinetic diameter, \({d}_{k}\) (Fig. 2c,d). The success of creating selective nanopores on graphene is reflected by the significantly increased values of \({N}_{p(A)}\) for membranes after pore creation, specifically 3.26 and 3.73 times for He and H2 and almost unchanged for N2, CH4 and SF6 after pore creation (illustrated by the inset in Fig. 2d). We further plot the fitted values of \({N}_{p(n-A)}\) against 1/\(\sqrt{{M}_{i}}\). Except for CH4 (which may have stronger interactions with graphene and PITEM, as shown in Fig. 1c), the near linear dependence shows that transport falls in the Knudsen diffusion regime, validating that the non-activated permeance contribution is indeed from leakage of large defects constrained by the PITEM support pores. The larger values of \({N}_{p(n-A)}\) for the membrane without pore creation suggest that the defect levels are higher compared to the membrane after pore creation, due to variations in membrane fabrication across different batches.

Temperature-dependent gas permeance through nanoporous graphene before (a) and after (b) pore creation, on PITEM(10) supports. The gas permeance through nanoporous graphene is calculated by normalizing the membrane permeance by support porosity and removing the flow resistance from the support. The error bars on the y-axis denote the standard deviation of permeance based on error propagation (Supplementary Note 2). c Activation energy, error bars represent the standard error of the activation energy derived from permeance fit to Equation (1) and (d) pre-factor derived by fitting the transport model equation (1) to the gas permeation data, plotted as a function of gas kinetic diameter. Inset depicts membrane surface with intrinsic nanopores and created nanopores. e Gas permeation behaviors align with a Knudsen diffusion model through leakage in nanoporous graphene, i.e., gas transport through large defects that are not in the activated regimes, obtained by subtracting the total permeance by that from the activated transport according to equation (1). Molecular weight \({M}_{i}\) is in kg mol-1. Comparison of experimentally measured helium permeance across nanoporous graphene after pore creation against simulated helium permeance using the f, rigid pore model, g pore switching model, and h, edge flexible model (details in Supplementary Note 3). The minutes notation, i.e., 5, 13, 25, 60 and 120 min in the legend corresponds to various pore growth times. The numbers at the top left of (a−e) indicate the multiplication factor (e.g., 1e-6) applied to the y-axis values. Source data are provided as a Source Data file.
Of particular interest is that \(\left\langle {E}_{{act}}\right\rangle\) decreases monotonically with increasing kinetic diameter \({d}_{k}\) of the gas species (Fig. 2c) for both membranes before and after pore creation. This is counterintuitive to the energy barrier model developed for penetrants through a polymeric matrix, where larger penetrants ought to experience a higher \({E}_{{act}}\) to traverse14. The values of \(\left\langle {E}_{{act}}\right\rangle\) range from 1 to 10 kJ mol-1, which, although smaller, are largely within an order of magnitude as those typically observed in polymeric membranes (~30 kJ mol-1 and higher). This suggests that transport through the graphene nanopores appears like that through polymeric membranes, compared to pure inorganic sp2 hybridized carbon lattice found in graphene, in nature. Additionally, \(\left\langle {E}_{{act}}\right\rangle\) is quite similar for both membranes with and without pore creation, indicating that the intrinsic defects in the starting graphene materials28 and the purposely created nanopores exhibit rather similar behaviors in temperature-dependent, selective, activated gas transport.
We hypothesized that the discrepancies between model predictions using equation (1) and conventional understandings (Fig. 2c) likely arise from assuming a uniform energy barrier for all nanopores and not considering the finite pore size distribution. Our results suggest that pores responsible for the activation energy of smaller gases present a high barrier, preventing larger gases from passing through, while the activation energy of larger gases is governed by larger defects12. To resolve the discrepancy, better understand the molecular origin of the apparent activation energy \(\left\langle {E}_{{act}}\right\rangle\), and predict temperature-dependent permeance, we built a pore ensemble model that accounts for experimentally characterized pore size distribution20 and simulated gas permeance through such nanoporous graphene. This model attempts to mechanistically predict gas permeation across nanoporous graphene membranes as the sum of individual nanopores12,27 (Supplementary Note 3) following equation (2), rather than the empirical fitting used in equation (1). In such a pore ensemble model, named as the rigid pore model shown in Fig. 2f, the gas permeance across a nanopore ensemble is evaluated as
where \({f}_{{Dp}}\) is pore size distribution density function, \({D}_{p}\) is the pore diameter, \(\rho\) is the areal pore density. \(\varPi ({D}_{p},i,T)\) stands for the permeance per pore and was considered a function of pore diameter \({D}_{p}\), gas species i, and temperature T. \(\varPi ({D}_{p},{i},{T})\), in turn, is calculated using different pore-scale permeation models applied to the pore size distribution \({f}_{{Dp}}\). With a log-normal pore size distribution well characterized in our previous works20,21, and assuming the contribution of large defects is minimal (as Knudsen transport decreases with temperature), this model aims to capture the significant activated transport gas permeation behaviors observed in the experiment, focusing on explaining the mechanistic origin of activated transport, particularly for helium.
First, we tested a rigid pore model to predict pore-scale permeation. Here, the activation energy per pore \({E}_{a}\) is estimated by the Lennard-Jones (L-J) interactions of hard-sphere interaction between the gas molecule and all the pore edge atoms19 (Supplementary Eq. (12)), based on the assumption that pore-gas interactions are dominated by L-J interactions27. Although this is a simplified estimation of activation energy, the values obtained fall within the same range as those predicted by molecular dynamics simulations12. This approach has also been used in predicting the selectivity of various gas pairs across nanoporous graphene, aligning with experimental measurements19. The pore size distribution\({f}_{{Dp}}\) is simulated based on prior imaging characterizations and transport measurements21. To study the effect of \({f}_{{Dp}}\) on permeance, we simulated different \({f}_{{Dp}}\) corresponding to pore growth time from 5 min to 120 min (Supplementary Note 3).
We focus on comparing the predicted He permeance to experimental results (Fig. 2f) due to its inert nature and L-J potentials that are free from influences from specific interactions that other gas species might have with graphene surfaces. Although the total permeance (summing both activated and non-activated permeance) for a nanopore ensemble with an \({f}_{{Dp}}\) at ~20 min of pore growth matches the experimental values, the permeance from activated transport alone is predicted to be significantly lower—about an order of magnitude less than the experimentally measured values (results for other \({f}_{{Dp}}\) are provided in Supplementary Fig. 10). The overall permeance is dominated by non-activated transport, resulting in a decrease in permeance as the temperature rises. Unless pore size and size distribution are artificially adjusted to an unrealistic extreme (Supplementary Note 3), the rigid pore model fails to even qualitatively match the experimental results by a large margin.
Given the significant permeance from activated transport, it is impossible to rationalize, particularly for inert He, without invoking changes of some kind to the graphene nanopores as the temperature rises from room temperature to 220 °C27 (see Supplementary Note 5 for discussion on excluding contributions from other mechanisms). To explain the magnitude of such activated permeance, we hypothesized, based on previous experimental observations from our group and others, that graphene nanopores could transition from a partly or completely closed state to an open state due to (1) adsorption/desorption of heterogeneous materials, such as hydrocarbons or other surface contaminants, or (2) pore edge atom re-arrangements, which are temperature-activated10,12,32,33. However, the energy required for moving edge atoms of graphene is relatively high, ranging from 0.3–0.6 eV for taking out one hydrogen terminated carbon edge atom with 1 sigma bond and it is unlikely that such edge atom rearrangement would be temperature-reversible32,33. Nevertheless, we attempt to simulate He permeance as a function of temperature using two types of pore-scale permeation models: one assumes that graphene nanopores transition from a fully closed to a fully open state, denoted as the pore switching model; the other assumes that only the nanopore edges, defined as 1.2 times the van der Waals radius of a carbon atom, transition from a closed to an open state, denoted as the edge flexible model (see details in Supplementary Note 3).
Activated permeance is significantly enhanced after invoking the pore open/closed hypothesis, with the predicted He permeance increase as a function of temperature matching the experimental values. However, this requires a larger pore size and wider size distribution at a much longer pore creation condition (i.e., 120 min pore growth time, Fig. 2g and Supplementary Fig. 11) for the pore switching model. In contrast, the edge flexible model predicts He permeance as a function of temperature that matches well with experimental values, using pore size and distribution that align better with experimental conditions (i.e., 13 min pore growth time). Additionally, using the same set of model parameters, the pore switching model can only reasonably predict H2 permeance based on the pore size distribution at the 120-min pore growth mark (Supplementary Fig. 11). In contrast, the edge flexible model can predict temperature-dependent permeance for other gas species including H2 and CO2, closely matching our experimental results (Supplementary Fig. 12). While the model shows some deviations for CH4, N2, and SF6, the predicted permeance remains generally consistent (Supplementary Fig. 12). Next, we turn to the molecular mechanisms that may explain the edge flexible model.
Edge-activated gas permeation across graphene nanopores
The small activation energy (on the order of 1 \({k}_{B}T\)) in the model suggests that it is highly unlikely to arise from chemical bond breaking or rearranging (on the order of several hundreds to thousands of \({k}_{B}T\))32,33. Considering the close fit of the edge flexible model with experimental data, we hypothesized that the graphene nanopore edge dynamics could be the source of this activated transport. To further investigate the possible molecular origin of this edge-activated transport, we performed molecular dynamic (MD) simulations to study temperature-dependent helium permeation through graphene nanopores, accounting for the thermal vibration of the graphene lattice at different temperatures (see Methods). We first examined a series of graphene nanopores (carbon atom terminated) with different pore sizes. Given the possible isomers of graphene nanopores, we constructed them based on prior kinetic Monte Carlo studies into stable graphene nanopore isomers and selected the most stable nanopore isomers for our study23. Five nanopores with increasing pore sizes were chosen, built by removing 10, 12, 14, 16, and 20 carbon atoms from the graphene lattice (Fig. 3a). We started with Pore_10, based on prior simulations34 and our experimental observations that graphene nanopores in our systems are highly selective for He and H2 over N2, CH4, and SF6, particularly at elevated temperatures. We benchmarked the simulation results with our experimental data. The helium permeance per pore, for example, Pore_12 is 4.26 × 10−21 (mol s−1 Pa−1) at 300 K, consistent with prior simulation reports34. This value is higher than \(3.13\times {10}^{-22}\)(mol s−1 Pa−1) estimated from our experiments at 300 K and \(1.44\times {10}^{-21}\) (mol s−1 Pa−1) estimated at 500 K for nanoporous graphene created under ion irradiation and 20 min etch pore growth conditions (see details in Methods). Such differences could be attributed to the high pressure and force-field used in the simulation (Methods), as well as experimentally determined pore density variations, and possibly edge functional groups in real graphene nanopores. As the graphene lattice fluctuates, we define the instantaneous permeable pore size of graphene nanopores by drawing the largest possible circle enclosed by the van der Waals surface of the edge atoms of the graphene nanopores projected onto a plane. We plot the spread of permeable pore sizes at 300 K for different graphene nanopores in Fig. 3a. The size fluctuation follows a Gaussian distribution, with the mean permeable pore size increasing from 2.70 angstroms for Pore_10 to 4.74 angstroms for Pore_20. Our results show a general trend of helium permeance decreasing with increasing temperatures for large pores, which is understandable given the \(1/\sqrt{T}\) dependence of effusion. Helium permeance also decreases as the pore size decreases, showing a clear transition from the Knudsen effusion regime for Pore_20, Pore_16, and Pore_14 to the activated transport regime observed with Pore_10. However, the contribution of activated transport is small, consistent with the prediction of the ‘rigid pore model’ in Fig. 2f. This observation is corroborated by visualizing the spread of the permeable pore size as a function of temperature (Fig. 3c). Our results show that the spread of permeable pore size for carbon-terminated graphene nanopores increases slightly as temperature rises from 300 K to 500 K, but does not change significantly. These results suggest that the dynamics of simple carbon-terminated graphene nanopores alone are unlikely to explain the pronounced activated transport behaviors observed in the experiment.

a projected permeable pore size distribution of graphene nanopores due to thermal fluctuation at temperature of 300 K as a function of number of carbon atoms removed from graphene lattice. b helium permeance of graphene nanopores with varying sizes as a function temperature. The error bars represent the 95% confidence interval for the helium traverse rate across a graphene nanopore (see details in Methods). c permeable pore size distribution of graphene nanopores, namely Pore_10, Pore_14, and Pore_20 at 300 K, 400 K, and 500 K. The number at the top left of (b) indicates the multiplication factor (e.g., 1e-6) applied to the y-axis values. Source data are provided as a Source Data file.
As such, we further hypothesized that nanopore edge chemistry, particularly functional groups beyond carbon-termination34, could introduce additional dynamics influencing the permeable pore size as a function of temperature. We have previously analyzed the edge chemistry of nanoporous graphene created by ion irradiation and chemical etching methods21. Others have also pointed to the rich edge chemistries of graphene nanopores created by different methods17. As the pores grow, they are not merely carbon-terminated but stabilized by various functional groups. Specifically, our prior work shows the presence of C = O and C − O bonding after exposure to the etch solution (i.e., permanganate/sulfuric acid etchant used for pore growth), suggesting that pore edges could be terminated by ketone, quinone, hydroxyl, or carboxyl groups21. These edge functional groups have also been reported for carbon materials created under exposure to similar etching chemicals, particularly for graphene oxide systems35. Accordingly, we explored the addition of functional groups on graphene nanopore edges and investigated the thermal vibration of these functional groups along with the graphene lattice and their collective influence on temperature-dependent gas permeation (Fig. 4, temperature ranges from 300–500 K).

a projected permeable pore size distribution of graphene nanopores decorated with different edge functional groups as a function of temperature. Different nanopores are named as pore size (number of carbon atoms removed from graphene lattice), followed by the type of functional group and location of the functional group. b helium permeance through graphene nanopores with different edge functional groups. The error bars represent 95% confidence interval for the helium traverse rate across a graphene nanopore. Source data are provided as a Source Data file.
We make the following observations: (1) The spread of projected permeable pore size, once an edge functional group is added, can be significantly greater; for example, it can exceed 1.5 Å in cases like Pore_14_COOH (i.e., one –COOH group added to the edge of a Pore_14 nanopore). (2) The mean and spread of permeable pore size are functional group dependent. For example, with a hydroxyl group added to Pore_12, the permeable pore size spread as temperature rises is much less significant than with the addition of a carboxylic group. The mean permeable pore size decreases from 3.08 Å for Pore_12_OH_P2 to 1.75 Å for Pore_12_COOH at 300 K (P1, P2 denote pores with functional groups added at different locations). (3) The specific location where a functional group is added also influences the spread of the pore size; for example, comparing the size of Pore_12_OH_P1 over Pore_12_OH_P2 suggests that the separation mechanism of molecular sieving, particularly in membrane gas separation including small gases like H2, He, and CO2, is a highly discrete system and very sensitive to the nanopore local environment. (4) Importantly, we see that the permeable pore sizes correlate well with gas permeance. Particularly in cases like Pore_12_COOH or Pore_12_OH_P1, where the number of permeable pores above the kinetic diameter of He (2.6 Å) significant increases with temperature, we observe correlated, pronounced activated gas transport (Fig. 4b) as temperatures rises. We acknowledge that the thermal vibration between edge functional groups and the nanopore lattice, which leads to the size distribution of permeable nanopores, is dependent on the boundary conditions (see Methods for details of the model setup). Additionally, the as-defined permeable nanopore size may not comprehensively represent the total space available for gas permeation in a given nanopore, as the shape is non-spherical; and as pores become larger, such pore size is less accurately represented when considering the influence of functional groups. Nevertheless, our results provide clear evidence that the dynamics of edge functional groups, along with the graphene lattice to which they are attached, can significantly contribute to temperature-dependent activated permeance.
We also considered the possibility of surface contamination and its impact on the activated gas transport observed in monolayer nanoporous graphene membranes12. Surface contaminants, such as hydrocarbons or organic components absorbed on the surface covering the nanopores, have been observed by us and others12,21. These contaminants might desorb, become more mobile, or have moieties that fluctuate with increasing temperatures, unblocking the graphene nanopores and resulting in temperature-activated transport profiles. For the nanoporous graphene membranes fabricated in this work, the PMMA carrier layer-assisted transfer method will likely leave polymeric residue on the graphene surface, despite thorough washing (details provided in Methods). As such, we investigated whether PMMA residue could be the source of and contribute to the observed activated gas permeance for small gases, including He and H2. We prepared two types of membranes: one fabricated using the current method (i.e., PITEM(10)_G_Pore) and another with modified washing steps to mitigate PMMA residue (i.e., PITEM(10)_G_noPMMA_Pore). Gas permeation tests revealed that both membrane groups have comparable gas permeance and selectivity, with similar activated transport behavior at higher temperatures (Supplementary Figs. 13, 14). These findings suggest that PMMA residue on the graphene surface is not a major factor contributing to the activated transport of gas species through the membrane (details on PMMA washing steps and the permeation tests are provided in Supplementary Note 4 and Supplementary Table 1). We ruled out the possibility of gas flow through the non-porous regions of the PITEM beneath graphene nanopores, as well as lateral flow entering the graphene nanopores and passing through the PITEM-graphene gap as contributors to activated gas transport (see Supplementary Note 5). The activated permeance observed in our membranes remained highly reproducible after several thermal cycles and was stable after 1 year of aging (Fig. 1f). These observations further indicate that the level of reversible activated transport observed is unlikely to be due to random surface contamination, but rather due to the intrinsic properties of graphene nanopores.
Long-term, thermally robust hydrogen membrane separations
Next, we demonstrate the application of the activated permeation for high-temperature hydrogen separations that are used in petrochemical processes and considered key technologies for a clean energy future36. Specifically, catalytic dehydrogenation–hydrogenation of liquid organic hydrides (e.g., cyclohexane, methylcyclohexane, tetralin, decalin, and bicyclohexyl) has been proposed as a promising technology to store and supply hydrogen for fuel cell vehicles. The catalytic dehydrogenation of decalin uses platinum or palladium catalysts at 200 to 300 °C, and yields a mixture of hydrogen, naphthalene, tetralin, and unreacted decalin37. Since the reaction does not go to completion in the case of decalin, hydrogen must be removed from the reactor, which is achieved through condensation by cooling the gas stream followed by reheating. Employing membranes to separate hydrogen from the hydrocarbons at reactor temperatures will reduce energy consumption, infrastructure costs, and enable process intensification. Metal membranes, such as those made from palladium or its alloys38, can operate under reaction conditions but are sensitive to temperature variations, causing phase transformations, volume changes, and irreversible structural damage. Additionally, they are vulnerable to impurities commonly found in petrochemical gas streams, such as hydrogen sulfide, which lead to poisoning and degradation of separation performance. Meanwhile, supported nanoporous silica membranes have demonstrated a high hydrogen permeance of ~\(1\times {10}^{-6}\) mol m⁻² s⁻¹ Pa⁻¹, along with an exceptionally high selectivity of \({10}^{4}\) H₂/SF₆39. However, their separation mechanisms remain unclear, as H₂ permeance does not scale with temperature according to any known pore flow model, showing little variation up to 573 K. There is still a need for new membrane materials that can elucidate selective gas transport through nano-sized pores and effectively address these critical separation challenges.
To this end, we evaluated the thermal robustness, long-term stability, and chemical resistance of nanoporous graphene membranes, essential for high-temperature hydrogen separation downstream of dehydrogenation reactors. We first tested both single gas and gas mixture permeation through membranes aged for over 1 year at temperatures ranging from 300 K to 473 K. We observed highly consistent activated permeance as the temperature increased, indicated by the increasing H2/CH4 selectivity in both single and mixture gas testing scenarios (Fig. 5a, b). These temperature-dependent permeance results are consistent with measurements taken before the 1 year aging period. The highly reversible selectivity dependence on temperature further confirms that the activated permeance is an intrinsic property of the selective graphene nanopores.

a single gas permeance and corresponding hydrogen selectivity against methane at various temperatures. The temperature was increased from 300 K to 473 K and then returned to 300 K (as shown in the far-right group). b binary gas mixture permeance and hydrogen selectivity. The same membrane was used for both single and mixture tests. c permeances of hydrogen, tetralin, cis- and trans-decalin mixture, along with respective hydrogen selectivity at 500 K. d measurement of hydrogen permeance at 500 K over 40 days. The permeance shown is for nanoporous graphene, calculated by normalizing to the open graphene area, i.e., 0.47% of the total membrane area of PITEM(10)_G_Pore. The error bars on the y-axis denote the standard deviation of the measured permeance data. The numbers at the top left of (a, b) indicate the multiplication factor (e.g., 1e-8) applied to the y-axis values. Source data are provided as a Source Data file.
To further evaluate the chemical stability of nanoporous graphene membranes and their performance in hydrogen separation at high temperatures, we tested hydrogen separation from hydrocarbon mixture (decalin isomers and tetralin), simulating the gas mixture scenarios downstream of decalin dehydrogenation reactor at reactor temperatures (see Methods and Supplementary Note 6). The hydrogen permeance of nanoporous graphene, that is, permeance after normalized to the open graphene area (0.47 % of total membrane area), was approaching ~\(7\times {10}^{-5}\) mol m−2 s−1 Pa−1, or \(2\times {10}^{5}\) GPU, approximately 1000 times higher compared to typical polymeric membranes36. At a temperature of 500 K, the selectivity of hydrogen over hydrocarbons including tetralin and cis-decalin exceeded 70 (68.5 for trans-decalin), significantly higher than the corresponding Knudsen selectivity of ~8 (see Supplementary Fig. 16 and values in Supplementary Tables 3, 5). The hydrocarbon leakage that determines the selectivity is likely to be governed by defects in the graphene that are large enough to allow Knudsen diffusion of gas species such as SF6.
Interestingly, we observed varying permeances for hydrocarbons, specifically tetralin < cis-decalin < trans-decalin. This variation may result from the effects of surface interaction and steric hindrance. The hindered transport of tetralin through the graphene nanopores could be caused by favorable π-π interactions between tetralin and the graphene surface40 that increase the barrier to translocate across the pore by breaking the π-π interactions. Additionally, trans-decalin, with its symmetric and elongated structure, may experience lower steric hindrance compared to cis-decalin, which has an asymmetric and bent shape. This results in higher permeance for trans-decalin, consistent with shape-dependent hexane isomer separation observed in our previous work20. We anticipate that isomer separation of cis-decalin from trans-decalin at high temperatures may be possible by further optimizing the nanopore size and size distribution of nanoporous graphene membranes.
We further stress-tested the membrane at high temperatures for over 40 days. The permeation of H2, whether in a single or mixture feed, was periodically measured at a constant temperature of 500 K. The membrane exhibited stable H2 permeance without any degradation, as shown in Fig. 5d. Given that typical polymeric membranes cannot operate effectively at such high temperatures41, this result confirms the practically relevant hydrogen separation performance, thermal robustness and chemical resistance of the monolayer nanoporous graphene membrane. The chemical resistance is particularly significant as the membrane is free from plasticization, typically found in polymeric gas separation membranes, and from poisoning, hence associated performance deterioration in metal hydrogen separation membranes during high temperature operations.
In summary, gas permeation through graphene nanopores exhibits significant, reversible, gas-species selective activated transport, which theory and simulations show are properties of the nanopores originating from edge functional groups that are robust at high temperatures. The increase in selectivity between smaller and larger gas molecules with temperature is contrary to the behavior seen in most membranes (Supplementary Fig. 1) and prior simulation studies of graphene nanopores42,43. We attribute the differences in behavior to the smaller edge groups studied (–H and –OH) in prior works that did not protrude as much into the pore, whereas we observed increases in permeance when the functional groups were larger or protruded into the pores. Disaggregating the contributions of small nanopores with activated transport (but negligible transport of larger gases) and large defects with Knudsen diffusion together results in overall temperature-dependent gas permeation through monolayer graphene membranes. This pushes the boundaries of understanding in atomically thin membranes, particularly focusing on nanopore edge dynamics, which can be capitalized on enhancing high-temperature separation performance. Our findings indicate that nanoporous graphene membranes exhibit long-term stability, high-temperature resilience, and robust chemical resistance, making them suitable for demanding separation processes and addressing the key challenges faced by existing polymeric gas separation membranes.
A key insight from our research is the pivotal role that nanopore edges—and more broadly, the surface chemistry of pores in porous membranes—play in achieving effective separation, particularly in high-temperature and challenging applications. This understanding underscores the importance of the structure and configuration of the membrane’s selective layer pore structures. It also opens up possibilities for engineering flexible edge functional groups to fine-tune the transport behavior of atomically thin nanopores and membranes. Our work offers guidance for exploring dynamic interactions at nanopore edges and surfaces also in other membranes, creating opportunities for developing advanced, high-performance membranes by leveraging these dynamics.
Methods
Membrane fabrication
Graphene transfer
Nanoporous graphene membranes were fabricated by transferring graphene grown on copper to the surface of polyimide track etched membrane (PITEM) porous supports. Monolayer graphene on copper foils were purchased from Graphenea Inc., and hydrophobic PITEM supports with nominal pore size of 20 nm and 10 nm, 8 μm in thickness, \(6\times {10}^{9}\) cm-2 porosity were purchased from it4ip S.A. PITEM supports were washed with HF (5%) for 30 min prior to graphene transfer to remove silica particles.
We used a transfer method from our prior work that utilized a two-layer poly (methyl methacrylate) (PMMA) coating as a sacrificial carrier layer for clean transfer of graphene from the growth substrate to the porous support20. Typically, the two-layer PMMA coating was deposited on graphene via spin-coating: the first layer had a low molecular weight (Mw) of 15,000 g mol-1, and the second layer had a Mw of 495,000 g mol-1. The two casting solutions were 15 wt% (15k) and 4 wt% (495k) PMMA in anisole solvent. The 15 k PMMA powder was purchased from Sigma-Aldrich, and the 4 wt% 495 k PMMA solution was purchased from Kayaku Advanced Materials, Inc., and both were used as received. Spin-coating of both PMMA solutions was conducted at 2500 rpm for 1 min, with an additional 10 s ramp-up at the beginning. After coating with 15 k PMMA, the graphene sample was placed in an oven at 65 °C for 30 min before applying the 495 k PMMA layer. The final two-layer PMMA coating consisted of 3 coats of 15 k PMMA and one coat of 495 k PMMA. After the coating was fully dried in air, the copper substrate of graphene samples was removed via a two-step etch procedure by floating the sample on an ammonium persulfate etchant, APS-100 (Transene, Inc.). The first step was to remove graphene on the back side of the copper substrate (not coated with PMMA) by floating the sample on APS-100 for 7 min. After washing with deionized (DI) water thoroughly and completely drying in air, the graphene sample was etched overnight in 0.1 M APS aqueous solution to remove the copper completely. Then, the sample was again washed with DI water thoroughly, carefully deposited onto the PITEM support via direct fishing out of the DI water bath, followed by drying in air for at least 3 h. Subsequent removal of PMMA was conducted by immersing the membrane sample in acetone: The membrane sample, held by tweezers, was dipped in three sequential acetone baths: 35 mL for 5 min, 35 mL for 2 h, and 35 mL overnight. Finally, the PITEM-supported graphene membrane was thoroughly rinsed by dipping it several times in isopropanol, dried in air, and annealed at 130 °C for 1 h. It is important to note that the PITEM support used in this work, though useful for experiments, may not be suitable for practical application due to its high cost and low porosity, highlighting the need to identify or develop cost-effective porous supports with optimized pore size, density, adhesion to graphene, and robustness under varying conditions.
Pore creation
To create pores, the graphene membrane was irradiated with 8 keV gallium ions at a density of ~\(6\times {10}^{12}\) ions cm−2 and incident angle of 52° with an FEI Helios 600 NanoLab system. The membranes were subjected to chemical etching (i.e., pore growth) within 2 h of ion irradiation using an oxidizing etchant (1.875 mM KMnO4 in 6.25 wt% H2SO4) for cumulative durations of 20 min during which the etchant was refreshed after every 5 min.
Single-gas permeation tests
Gas permeation across membranes was measured using an in-house designed stainless-steel gas rig, which included a membrane holder module and upstream and downstream canisters with known and fixed volumes (Supplementary Fig. 2). Nanoporous graphene membranes were mounted on a copper gasket, sealed with carbon tape, and secured with a Kalrez gasket that had a hole punched for the membrane area (see schematic in Supplementary Fig. 3). The copper gasket-supported membrane stack was then sandwiched between two stainless steel flanges with knife edges to create a leak-tight metal-to-metal seal. Gas was slowly fed using a mass flow controller (MFC, Teledyne, HFC-D-303-A) to the upstream canister to reach a pressure of typically around \(6 \sim 7\times {10}^{4}\) Pa. At a given time, the valves on both the upstream and downstream sides of the membrane were opened, and membrane permeance was evaluated by monitoring the pressure rise in the permeate canister (or pressure drop in the feed canister) as gas molecules traversed the membrane (see Supplementary Note 2). We cross-checked and validated the accuracy of this method by measuring the flux under a set cross-membrane pressure difference (see the MFC method in Supplementary Note 2). Membrane permeance at different temperatures was measured by placing the membrane module in a furnace (FB1415M, Thermolyne Benchtop Muffle Furnaces). A Type-K thermocouple (MTI industrial) was attached to the membrane module surface to monitor the actual temperature during testing. Permeance measurements were conducted once the temperature reached the target value (20 to 220 °C) and stabilized. Six gas species were tested in this work: hydrogen (H2, UHP, Airgas), helium (He, UHP, Airgas), methane (CH4, 99.999%, Airgas), nitrogen (N2, 99.999%, Airgas), carbon dioxide (CO2, UHP, Airgas), and sulfur hexafluoride (SF6, 99.99%, Airgas). The gas rig was leak tested prior to membrane permeance measurements and found to be gas tight (<3.5 Pa mL s−1).
Mixed-gas permeation and hydrogen/hydrocarbon separation tests
Binary mixture test (H2/CH4)
The entire home-made gas rig was evacuated, and the membrane module in the furnace (FB1415M, Thermolyne Benchtop Muffle Furnaces) was heated to the desired temperature overnight. The two gases were fed into the feed side at the same molar ratio and flow rate (10 sccm) controlled using mass flow meters (Teledyne, HFC-D-303-A). The total feed pressure was kept constant (~70 kPa) using a back-pressure regulator (Swagelok). After reaching steady state, the permeate flow rate and the total pressures at feed and permeate sides were measured. Due to the low pressure on the permeate side, which was generally undetectable by gas chromatography (GC, Agilent 8890), the downstream was pressurized by allowing permeate gas to accumulate after passing through the membranes. This was achieved by closing the valve between the GC and the vacuum pump (XDS 5 scroll pump). Once the pressure reached ~20 kPa, the permeate gas mixture was sent to the GC to obtain the ratio of the two gases in the permeate using a thermal conductivity detector, which, combined with the total permeate flow rate, was used to calculate the permeance of each gas species assuming linear response of the mass flow meter to gas composition.
Hydrocarbon mixture test (H2/decalin isomers/tetralin)
The pathways for hydrocarbons in the gas rig were heated to ~500 K using heating tapes, fiberglass insulation, and thermocouples (Brisk heat controller). To eliminate contaminants and stabilize the temperature, the rig was kept under vacuum for over 12 h during the heating process. Then, H2 was fed into the feed side at a flow rate of 5 sccm. After reaching steady state, tetralin and decalin isomers (Sigma Aldrich, ReagentPlus 99%) were fed into the feed side using a syringe pump (Harvard Apparatus) at interjection rates of 4.5 µL min−1 and 0.5 µL min−1, respectively. The total feed pressures were kept constant at 160 kPa using a back-pressure regulator (Swagelok). The hydrogen permeate flow rate was measured using a mass flow meter placed downstream of a cold trap maintained at room temperature designed to condense the hydrocarbons. After measuring the permeate flow rate and the total pressures of both the feed and permeate, the cold trap and mass flow meter were bypassed and the permeate was directed to the GC for detection using a flame ionization detector. The permeate side was then allowed to reach 20 kPa by closing a valve downstream of the GC and isolating it from the vacuum. The permeate gas mixture was then sent to the GC to determine the flow rate and permeance of each hydrocarbon species (using a calibration curve obtained by flowing H2 and the hydrocarbons at known flow rates in the absence of a membrane in the module). Multiple measurements were performed with sufficient wait time to allow the system to reach steady state and average out any potential fluctuations in the hydrocarbon flow rates.
Molecular dynamic (MD) simulation of gas permeation across graphene nanopores
MD simulation was conducted to investigate helium permeation through graphene nanopore with various sizes and edge functional groups as a function of temperature. The simulations were performed using the LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator). For the temperatures of interest here, we expect quantum effects to be small. A periodic sheet of porous graphene was modeled by starting with a perfect unit cell of 4 × 4 Å. Graphene nanopores with various sizes (shown in Fig. 3a) were created by removing carbon atoms from the center of the graphene sheet according to the results of the most stable pore isomers of a kinetic Monte Carlo study conducted previously23. Two types of edge functional groups namely, hydroxyl and carboxylic groups, were attached to the graphene nanopores. The nanoporous graphene divided the simulation box (normal to the nanopore plane) of height 800 nm into two chambers of equal volumes, with the feed chamber containing 300 helium atoms while the permeate chamber contained 10 helium atoms. To avoid vertical displacement of the nanoporous graphene, the position of corner carbon atoms was fixed. Two identical, perfect graphene sheets were placed on each side of chamber with all atoms frozen acting as two pistons, pushing helium atoms flow through the nanopore. The pressure difference between the two pistons was ~100 bar. Interaction parameterization was modeled using the universal force field in the LAMMPS-interface package44. Each MD simulation was run in the NVT ensemble for \(6\times {10}^{7}\) timesteps with a time step of 0.05 fs. The Nosé-Hoover thermostat with a damping parameter of 100 was used to control the system’s temperature. The flux through nanoporous graphene was calculated by dividing the number of crossings by the product of the simulation duration and the membrane area. Given that helium crossings through the graphene nanopore occur at a constant mean rate and independently of time within fixed intervals, such permeation events follow a Poisson distribution. Therefore, the error in permeance represents a 95% confidence interval, calculated as \({P}_{m}\,\pm 1.96\frac{\sqrt{{P}_{m}}}{n}\), where \({P}_{m}\) is the permeance and n is the number of simulation runs.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data that support the findings of this study are available in the main text, figures and Supplementary Information. Source data are provided with this paper.
Code availability
The Python scripts and LAMMPS input scripts files used in simulating gas permeation through graphene nanopore are available at https://doi.org/10.5281/zenodo.15079630 (ref. 45).
References
Seader, J. D., Henley, E. J. & Roper, D. K. Separation Process Principles 3rd Edition, Vol. 848 (John Wiley Incorporated, 2010).
Biegler, L. T., Grossmann, I. E. & Westerberg, A. W. Systematic Methods of Chemical Process Design, Vol. 816 (Prentice Hall PTR, 1997).
Sholl, D. S. & Lively, R. P. Seven chemical separations to change the world. Nature 532, 435–437 (2016).
Torrente-Murciano, L. et al. The forefront of chemical engineering research. Nat. Chem. Eng. 1, 18–27 (2024).
Lu, Y. et al. Monolayer graphene membranes for molecular separation in high-temperature harsh organic solvents. Proc. Natl. Acad. Sci. USA 118, e2111360118 (2021).
Qian, Q. et al. MOF-based membranes for gas separations. Chem. Rev. 120, 8161–8266 (2020).
Kingsbury, R. S. et al. Kinetic barrier networks reveal rate limitations in ion-selective membranes. Matter 7, 2161−2183 (2024).
Rowe, B. W., Robeson, L. M., Freeman, B. D. & Paul, D. R. Influence of temperature on the upper bound: Theoretical considerations and comparison with experimental results. J. Membr. Sci. 360, 58–69 (2010).
de Lange, R. S. A., Keizer, K. & Burggraaf, A. J. Analysis and theory of gas transport in microporous sol-gel derived ceramic membranes. J. Membr. Sci. 104, 81–100 (1995).
Huang, S. et al. Single-layer graphene membranes by crack-free transfer for gas mixture separation. Nat. Commun. 9, 2632 (2018).
Yuan, Z., Benck, J. D., Eatmon, Y., Blankschtein, D. & Strano, M. S. Stable, temperature-dependent gas mixture permeation and separation through suspended nanoporous single-layer graphene membranes. Nano Lett. 18, 5057–5069 (2018).
Yuan, Z. et al. Predicting gas separation through graphene nanopore ensembles with realistic pore size distributions. ACS Nano 15, 1727–1740 (2021).
Yuan, Z. et al. Direct chemical vapor deposition synthesis of porous single-layer graphene membranes with high gas permeances and selectivities. Adv. Mater. 33, 2104308 (2021).
Freeman, B. D. Basis of permeability/selectivity tradeoff relations in polymeric gas separation membranes. Macromolecules 32, 375–380 (1999).
Koenig, S. P., Wang, L., Pellegrino, J. & Bunch, J. S. Selective molecular sieving through porous graphene. Nat. Nanotechnol. 7, 728–732 (2012).
Shen, L. et al. Highly porous nanofiber-supported monolayer graphene membranes for ultrafast organic solvent nanofiltration. Sci. Adv. 7, eabg6263 (2021).
Huang, S. et al. Millisecond lattice gasification for high-density CO 2 - and O 2 -sieving nanopores in single-layer graphene. Sci. Adv. 7, eabf0116 (2021).
Prozorovska, L. & Kidambi, P. R. State‐of‐the‐art and future prospects for atomically thin membranes from 2D materials. Adv. Mater. 30, 1801179 (2018).
Wang, L. et al. Fundamental transport mechanisms, fabrication and potential applications of nanoporous atomically thin membranes. Nat. Nanotechnol. 12, 509–522 (2017).
Cheng, C., Iyengar, S. A. & Karnik, R. Molecular size-dependent subcontinuum solvent permeation and ultrafast nanofiltration across nanoporous graphene membranes. Nat. Nanotechnol. 16, 989–995 (2021).
O’Hern, S. C. et al. Selective ionic transport through tunable subnanometer pores in single-layer graphene membranes. Nano Lett 14, 1234–1241 (2014).
Wang, J. et al. Cascaded compression of size distribution of nanopores in monolayer graphene. Nature 623, 956–963 (2023).
Govind Rajan, A. et al. Addressing the isomer cataloguing problem for nanopores in two-dimensional materials. Nat. Mater. 18, 129–135 (2019).
Hsu, K. -J. et al. Graphene membranes with pyridinic nitrogen at pore edges for high-performance CO2 capture. Nat. Energy 9, 964–974 (2024).
Kidambi, P. R. et al. Nanoporous atomically thin graphene membranes for desalting and dialysis applications. Adv. Mater. 29, 1700277 (2017).
Boutilier, M. S. H. et al. Molecular sieving across centimeter-scale single-layer nanoporous graphene membranes. ACS Nano 11, 5726–5736 (2017).
Yuan, Z. et al. Gas separations using nanoporous atomically thin membranes: recent theoretical, simulation, and experimental advances. Adv. Mater. 34, 2201472 (2022).
Boutilier, M. S. H. et al. Implications of permeation through intrinsic defects in graphene on the design of defect-tolerant membranes for gas separation. ACS Nano 8, 841–849 (2014).
Ehlert, C., Piras, A. & Gryn’ova, G. CO2 on graphene: benchmarking computational approaches to noncovalent interactions. ACS Omega 8, 35768–35778 (2023).
Yuan, Z. et al. Mechanism and prediction of gas permeation through sub-nanometer graphene pores: comparison of theory and simulation. ACS Nano 11, 7974–7987 (2017).
Sun, P. Z. et al. Exponentially selective molecular sieving through angstrom pores. Nat. Commun. 12, 7170 (2021).
Leuthner, G. T., Susi, T., Mangler, C., Meyer, J. C. & Kotakoski, J. Chemistry at graphene edges in the electron microscope. 2D Mater 8, 035023 (2021).
Girit, Ç. Ö. et al. Graphene at the edge: stability and dynamics. Science 323, 1705–1708 (2009).
Sun, C. et al. Mechanisms of molecular permeation through nanoporous graphene membranes. Langmuir 30, 675–682 (2014).
Dikin, D. A. et al. Preparation and characterization of graphene oxide paper. Nature 448, 457–460 (2007).
Adhikari, S. & Fernando, S. Hydrogen membrane separation techniques. Ind. Eng. Chem. Res. 45, 875–881 (2006).
Martynenko, E. A., Pimerzin, A. A., Savinov, A. A., Verevkin, S. P. & Pimerzin, A. A. Hydrogen release from decalin by catalytic dehydrogenation over supported platinum catalysts. Top. Catal. 63, 178–186 (2020).
Habib, M. A. et al. Palladium-alloy membrane reactors for fuel reforming and hydrogen production: a review. Energy Fuels. 35, 5558–5593 (2021).
Oda, K. et al. Dehydrogenation of methylcyclohexane to produce high-purity hydrogen using membrane reactors with amorphous silica membranes. Ind. Eng. Chem. Res. 49, 11287–11293 (2010).
Hunter, A. & Thornton, J. M. Pi-pi: Interactions: the geometry and energetics of phenylalanine-phenylalanine interactions in proteins. J. Mol. Biol. 218, 837−46 (1991).
Rezakazemi, M., Sadrzadeh, M. & Matsuura, T. Thermally stable polymers for advanced high-performance gas separation membranes. Prog. Energy Combust. Sci. 66, 1–41 (2018).
Vallejos-Burgos, F., Coudert, F.-X. & Kaneko, K. Air separation with graphene mediated by nanowindow-rim concerted motion. Nat. Commun. 9, 1812 (2018).
Guo, J., Galliero, G. & Vermorel, R. How membrane flexibility impacts permeation and separation of gas through nanoporous graphenes. Nano Lett. 24, 12292–12298 (2024).
Boyd, P. G., Moosavi, S. M., Witman, M. & Smit, B. Force-field prediction of materials properties in metal-organic frameworks. J. Phys. Chem. Lett. 8, 357–363 (2017).
Cheng, C., Kim, L., Persad, A. H., Chow, C. M., & Karnik, R. Code source files for the paper entitled “edge-activated graphene nanopores for thermally robust hydrogen membrane separations”. Zenodo https://doi.org/10.5281/zenodo.15079630 (2025).
Acknowledgements
R.K., C.C., L.K. and A.H.P. acknowledge financial support by Eni S.p.A. through the MIT Energy Initiative. C.C. is the recipient and acknowledges the financial support from the Australian Research Council Australian Future Fellowship Award (FT210100364, C.C.), funded by the Australian Government. Ion irradiation and SEM imaging were performed at the Materials Research Laboratory in MIT.nano. L.K. acknowledges the support from the Kwanjeong Educational Foundation (Award No. 19Am0145, L.K.); C.M.C. acknowledges the support from National Science Foundation Graduate Research Fellowship (Grant No. 1745302, C.M.C.) and the Croucher Foundation. We thank S. Carminati, L. Bonoldi, R. Field, Z. Yuan, and G. Jiang for valuable discussions.
Author information
Authors and Affiliations
Contributions
R.K. and C.C. conceived the project and designed the research. C.C. fabricated the membranes, performed imaging characterization, conducted gas transport measurements, theoretical modeling, and molecular dynamics simulation of gas transport, and analyzed the results. L.K. fabricated the membranes and performed the hydrogen hydrocarbon separation experiments, imaging characterization, and membrane contamination cleaning experiments. A.H.P. designed and built the gas transport measurement rigs. C.C., L.K., A.H.P., C.M.C. and R.K. participated in model development and the analysis and discussion of the results. C.C., L.K. and R.K. wrote the paper; all authors read and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: Some of the authors are co-inventors on patents and patent applications. (1) R.K. is co-inventor on patents US 11/135,546, CN 104,411,642, and AU 2017,210,547 that covers atomically thin membrane structures used in the present study, filed by the King Fahd University of Petroleum and Minerals and the Massachusetts Institute of Technology. (2) R.K. is co-inventor on patents US 11/717,791, AU 2013,231,930, and CN 104,411,642 that cover sealing of defects in atomically thin membranes, filed by the King Fahd University of Petroleum and Minerals and the Massachusetts Institute of Technology. (3) C.C. is co-inventor on a patent application US 63/502,064 filed by the Massachusetts Institute of Technology on formation of pores in graphene. (4) R.K., A.P., C.C., and L.K. are co-inventors on a patent application PCT/US2023/080744 filed by the Massachusetts Institute of Technology covering nanoparticle coatings to improve selectivity of nanoporous graphene membranes for hydrogen separation. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, C., Kim, L., Persad, A.H. et al. Edge-activated graphene nanopores for thermally robust hydrogen membrane separations. Nat Commun 16, 6052 (2025). https://doi.org/10.1038/s41467-025-61110-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61110-8
