
Abstract
Biomolecular condensates are droplet-like membrane-less compartments in cells that can sequester proteins. Modulating these condensates offers a promising way to durably inhibit disease-driving proteins that lack enzymatic activity and thus elude traditional drug targeting. However, many such proteins remain beyond the reach of current condensate-modulating strategies. Here we show an alternative approach: by destabilizing target proteins, we directly induce their liquid–liquid phase separation (LLPS), causing them to form condensates. Using this strategy, we develop a small molecule RQ that forces β-catenin (an oncogenic protein in liver cancer) into cytoplasmic condensates. This sequestration prevents β-catenin from entering the nucleus and activating cancer-promoting genes. In nanoparticle form (albumin-bound Abroquinone), RQ is selectively taken up by β-catenin-driven liver cancer cells and kills them while sparing normal cells. This approach suppresses β-catenin-driven tumor growth and overcomes immune evasion, demonstrating a promising paradigm for targeting previously untargetable proteins by inducing their phase separation.
Similar content being viewed by others

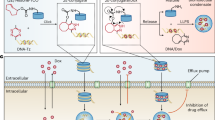
Introduction
Biomolecular condensates, membrane-less assemblies that compartmentalize specific proteins in subcellular environments, have emerged as crucial players in various cellular functions1,2. Moreover, mounting evidence supports the notion that these condensates form through liquid-liquid phase separation (LLPS), wherein a single-phase system undergoes demixing to yield a two-phase system comprising a condensed phase and a dilute phase of specific biomolecules2,3. The evolving comprehension of condensate function in both normal and aberrant cellular states, coupled with the identification of aberrant behaviors linked to disease states, underscores the paramount importance of biomolecular condensates for drug discovery4,5. In this context, numerous therapeutics that modify condensates have been identified or developed6, encompassing but not limited to: the anticancer drugs cisplatin and tamoxifen, which can partition into transcriptional condensates7; bis-ANS, a modulator of protein condensates with great therapeutic potential in neurodegenerative diseases involving TDP43 and FUS8; and Anti-Coronavirus small molecules termed CP21R7 that modulate nucleocapsid protein condensation9. Although these condensate-modifying therapeutics (c-mods) can demonstrate remarkable efficacy in spatiotemporally modulating the composition of condensates, sustaining modulation of conformational and interaction landscapes, and even degrading specific components within the condensate itself6,10, these strategies do not apply to regulating all pathogenic proteins, particularly those unrelated to inherent biomolecular condensates. One such example is β-catenin, for which no LLPS regulatory molecule has been discovered thus far to modulate its biofunction or suppress its carcinogenesis.
The β-catenin protein, when combined with members of the T cell factor/lymphoid enhancer factor (TCF/LEF) family of DNA-binding proteins, acts as a potent transcriptional activator11. It plays a crucial role in regulating the Wnt signaling pathway, an evolutionarily conserved cascade that is essential for development and adult stem cell maintenance11. However, this pathway also contributes to the progression of various cancers, including breast cancer, gastric cancer, bone cancer, colorectal cancer, and hepatocellular carcinoma12,13. It does so by promoting cell proliferation13, mediating epithelial-mesenchymal transition14, facilitating Oncofetal reprogramming15 and enhancing cell resistance to chemo-/radio-/immune-therapy16. Consequently, there is a significant interest in inhibitors targeting this pathway. Several peptide and small-molecule inhibitors that target the Wnt/β-catenin signaling cascade have demonstrated efficacy in vivo, but only at high concentrations and always accompanied by adverse events17,18,19. As a result of these limitations, none of them have progressed into the clinical phase of drug discovery yet17,18.
To effectively and expeditiously impede the carcinogenicity of β-catenin via LLPS, it is imperative to develop a condensate-inducing therapeutic (c-inds) rather than a condensate-modifying therapeutic (c-mods), which can compartmentalize cytoplasmic β-catenin within the condensed phase, thereby constricting its nuclear translocation and transcriptional activity. Consequently, this potent approach will robustly suppress the oncogenic Wnt/β-catenin signaling cascade. In this work, a pharmaceutical strategy employing c-inds is provided to harness chemical molecules with high-affinity binding capabilities to the target protein β-catenin and partially disrupt its conformation, thereby inducing a localized transformation from the well-defined topological structure to a random coil and/or amorphous state that closely resembles intrinsically disordered regions (IDRs). The partial structural damage of β-catenin in this case bestows upon it the theoretical propensity to spontaneously form non-stoichiometric macromolecular assemblies through spontaneous LLPS6,20,21. The screening of β-catenin c-inds involves the establishment of a sophisticated Circular Dichroic (CD)-assisted quantization scheme, which precisely quantifies the stability of protein structure by fitting the enthalpy-dependent curve of temperature and conformation parameter during the thermal denaturation process of β-catenin/c-inds condensates. In this way, Rosmanol quinone (RQ) was discovered as β-catenin c-inds, which can induce the LLPS of β-catenin in cells at low micromolar scale concentration and potently inhibit the Wnt/β-catenin signaling pathway and the consequent cell proliferation. Additionally, in order to enhance its translational potential, RQ was conjugated with albumin to generate a spherical Abroquinone with a hydrodynamic diameter of approximately 163 nm. This innovative formulation exhibits specific uptake by β-catenin-hyperactivated hepatoma carcinoma cells (HCC) through β-catenin-accelerated macropinocytosis. As anticipated, Abroquinone significantly enhanced the formation of β-catenin condensates and, more notably, exhibited tumor cell-specific cytotoxicity. Consequently, Abroquinone not only suppresses in vivo carcinogenesis of β-catenin but also rectifies the Wnt/β-catenin-mediated immune evasion in HCC while maintaining favorable biosafety profiles.
Results
Construction of a screening system for condensate-inducing therapeutics (c-inds) of β-catenin
The canonical β-catenin/Wnt signaling pathway predominantly regulates target gene expression by modulating the cellular stability of cytoplasmic β-catenin22,23. In its inactive state, cytoplasmic β-catenin undergoes phosphorylation and ubiquitination mediated by the destruction complex, leading to continuous degradation of β-catenin by the proteasome24,25. Conversely, in its active state, the destruction complex can be restrained, resulting in stabilization and accumulation of cytoplasmic β-catenin22,25. During carcinogenesis, hyperactivation leads to facilitated nuclear translocation of cytoplasmic β-catenin through binding with overexpressed BCL9, subsequently activating its transcriptional activity and promoting carcinogenesis26,27. As per our hypothesis, the Achilles’ Heel of the carcinogenic Wnt signaling pathway lies in the cytoplasmic accumulation of β-catenin, which can potentially be induced by c-inds to undergo LLPS, thereby leading to physical interphase blocking and subsequent hardening effect of β-catenin nuclear translocation.
To test it, the BCL9 binding domain of β-catenin (residue 137–245, referred to as BBD) was chemically synthesized for screening the c-inducers of β-catenin. Utilizing native chemical ligation, a powerful technique enabling complete synthesis of protein molecules, BBD was synthesized using a four-segment ligation strategy (Fig. 1a). Subsequently, it was purified through reversed-phase HPLC and its eligibility in terms of purity and molecular weight was confirmed by LC–MS analysis (Fig. 1b). Following Guanidine hydrochloride (GuHCL) dialysis-induced refolding renaturation, CD spectroscopy revealed that BBD exhibited the required α-helical secondary structure characteristic, while Bcl9-p had no obvious α-helix secondary structure. (Fig. 1c). Furthermore, this helicity could be further enhanced upon binding with the BCL9 peptide (Fig. 1c). Additionally, consistent with previous reports28, BBD demonstrated almost unanimous binding affinity towards BCL9 peptide (Supplementary Fig. 1A). Collectively, these findings validate the successful synthesis and folding of BBD, rendering it suitable for subsequent research.

a Chemical synthesis strategy of the BCL9 binding domain of β-catenin (residue 137–245, referred to as BBD). b LC–MS analysis of the synthetic BBD. c CD spectroscopy of 1 μM BBD or 1 μM BBD with 2 μM 351−376BCL9 peptide (Bcl9-p) in standard PB buffer. d Schematic diagram of BBD condensates. e Thermal denaturation of BBD and BBD with 0.5 M GuHCL, each BBD at 10 μM in PBS, pH 7.4, as monitored by CD spectroscopy at 222 nm. The normalized experimental data were fitted into a two-state protein thermal denaturation curve.
The next step involved the addition of GuHCL to the BBD solution, reaching a concentration of 0.5 M, in order to destabilize the structure of BBD and promote its oligomerization (Fig. 1d and Supplementary Fig. 1B). To accurately assess the stability of the BBD structure, a thermal-induced denaturation experiment was conducted by gradually increasing the temperature while monitoring changes in ellipticity at the characteristic α-helix peak at 222 nm using CD spectroscopy.
The denaturation process can be described using the thermodynamic formulas (1–4) (Supplementary Fig. 1C). Additionally, the ellipticity (Sθ) in this process can be expressed as an additive combination of the ellipticity at the native state multiplied by a percentage of native protein (\({\alpha }_{n}{f}_{n}\)), and the ellipticity at the denatured state multiplied by a percentage of denatured protein (\({\alpha }_{d}{f}_{d}\)) (formula (5)). According to formulas (2–4), both the percentage of native protein (\({f}_{n}\)) and the percentage of denatured protein (\({f}_{d}\)) can be expressed as functions related to temperature (T). Consequently, we can express the ellipticity (Sθ) as shown in equation on right side in formula (5). It is important to note that proteins in their native state exist as monomers, while in the denatured state they tend to aggregate. Consequently, their respective ellipticities (\({\alpha }_{n}\) and \({\alpha }_{d}\)) during thermal denaturation may vary independently with temperature (T). Therefore, we can describe (\({\alpha }_{n}\)) as \(\left({\alpha }_{n1}T+{\alpha }_{n2}\right)\) and (\({\alpha }_{d}\)) as \(({\alpha }_{d1}T+{\alpha }_{d2})\), which accounts for potential oligomerization of proteins. This leads us to derive Eq. (6), representing a partition function that describes the equilibrium between the native (monomeric) state and the denatured (aggregated) state at a given temperature. Fitting formula (6) to the CD monitoring data allows for determination of the melting temperature (Tm), which is positively correlated with protein structure stability. This enables quantitative comparison of protein structure stability through Tm comparison. The thermal-induced denaturation process of BBD and BBD with 0.5 M GuHCL was recorded, as shown in Fig. 1e. Fitting formula (6) resulted in the quantized stability of protein structure, indicating that 0.5 M GuHCL decreased Tm from 49.9 to 42.7 °C. Further derivation yielded an expression relating temperature (T) to protein denaturation ratio as formula (7) (Supplementary Fig. 1C). By this expression, the temperature-dependent BBD denaturation ratio can be found in Fig. 1e, and it is evident that the Ability of BBD to maintain natural structure against thermal-induced denaturation was significantly reduced by 0.5 M GuHCL, suggesting a lowered stability of protein structure. The above results demonstrate that this CD-assisted quantization scheme can effectively assess the stability of protein structure by fitting the enthalpy-dependent curve of temperature and conformation parameter during the thermal denaturation process of target proteins.
The screening of β-catenin c-inds
Targeting the bc19 binding domain of β-catenin (BBD), we constructed a receptor-based pharmacophore model using Discovery Studio 2019 software (Fig. 2a). Using this pharmacophore model, we employed virtual screening to identify 319 small-molecule drugs from the open DrugBank 5.1.8 database29 that exhibit potential binding affinity with BBD, as presented in Supplementary Table 1. Subsequently, we predicted the binding conformations of the top ten pharmacophore-scoring molecules at the bc19 binding domain of β-catenin via AutoDock Vina 1.2.0 software (Fig. 2b and Supplementary Fig. 2A). To validate the computational predictions and further screen potential candidates, we quantitatively assessed the binding affinity of BBD with the ten candidate molecules using isothermal titration calorimetry (ITC) (Supplementary Fig. 2B). The results indicated that rosmarinic quinone (RQ) demonstrated the highest binding energy with BBD, exhibiting a dissociation constant of 1.09 μM (Supplementary Fig. 2B). The predicted binding sites of β-catenin for rosmarinic quinone were identified as Arg190, Pro154, and Ile153 (Supplementary Fig. 2C). Mutations were introduced at these three sites and the mutant R101A/P154A/I153A BBD protein was expressed. As expected, this mutant showed very low affinity to RQ (Supplementary Fig. 2D), which supports the predicted results of binding sites. Notably, both β-catenin and its mutant remain in a monomeric state prior to the addition of RQ (Supplementary Fig. 2D). This suggests that the differing affinity is attributable to the loss of the binding site rather than insufficient exposure of binding sites due to aggregation.

a 3D structure diagram of BBD. b Schematic representation of the three-dimensional molecular structure with a high predicted binding affinity for β-catenin. c Tm screening of BBD with the ten candidate molecules. 10 μM BBD with 20 μM candidate molecules was investigated in PBS buffer (pH 7.4). n = 3 biological replicates; The error bars in C are presented as mean values ± SD. d The thermal denaturation of BBD, 10 μM BBD with 20 μM RQ was investigated in PBS buffer (pH 7.4) using CD spectroscopy at the wavelength of 222 nm. The obtained experimental data were normalized and fitted to generate a two-state protein thermal denaturation curve. e The experimental SAXS patterns (green) of 10 μM BBD (Ctrl) and 10 μM BBD with 20 μM RQ (RQ) showed together with their calculated scattering curves (orange) based on simulated models.
Afterward, the ten candidate molecules were screened using the aforementioned CD-assisted quantization scheme to evaluate the structural stability of BBD. Considering that most of these molecules exhibited binding affinities at the micromolar level, a concentration of 20 μM was employed to interact with 10 μM BBD to ensure optimal conformation of the molecule-BBD complex. As illustrated in Fig. 2c, we successfully fitted the experimental data and determined the melting temperature (Tm) of BBD in response to each candidate molecule. Consistent with expectations, amodiaquine, N-acetylglucosamine, bortezomib, and cisatracurium had negligible impact on the Tm of BBD (Fig. 2c). This outcome can be attributed to their low binding affinity with BBD (Supplementary Fig. 2B), which maintained BBD as a monomer within the small-molecule mixture. In contrast, Rosmanol quinone (RQ) and Ritonavir displayed a significant decrease in Tm exceeding 3 °C. It is worth mentioning that under these conditions, RQ statistically significantly reduced the Tm of BBD from 49.2 to 41.3 °C (Fig. 2d). We subsequently utilized small-angle X-ray scattering (SAXS) to examine the tendency of BBD to form condensates following RQ treatment (Supplementary Table 2). As depicted in Fig. 2E, under the tested conditions in the absence of RQ, the experimental data points (green) were closely aligned with the theoretical curve for the monomer (orange), indicating that the measured radius of gyration (Rg) of 14.9 Å corresponds to the monomeric state. Following RQ treatment, the Rg value increased from 14.9 to 59.8 Å, suggesting that RQ markedly enhances the tendency of BBD monomers to form condensates.
Identifying Rosmanol quinone (RQ) as a condensate-inducing therapeutic (c-inds) of β-catenin
To investigate the condensation behavior, we expressed and purified β-catenin protein to conduct in vitro phase separation experiments. By employing RQ to induce protein aggregation, we observed the formation of characteristic droplets (Supplementary Fig. 3A). As depicted, in the absence of RQ induction, the Cy3-labeled protein solution exhibited a diffuse fluorescence pattern; however, following RQ treatment, spherical droplets approximately 1 μm in diameter were formed. To determine whether these spherical droplets represent typical liquid-liquid phase separation (LLPS), we characterized their properties. Given that LLPS is often influenced by ionic strength, we analyzed the turbidity of the solution at varying concentrations of NaCl and RQ. Our findings indicated that as the concentration of RQ increased, so did the turbidity of the solution; conversely, an increase in NaCl concentration resulted in decreased turbidity (Supplementary Fig. 3B). This behavior aligns with established characteristics of LLPS, where both inducing agents and ion concentrations significantly impact the phase separation process. Subsequently, we performed a fluorescence recovery after photobleaching (FRAP) assay in vitro. In comparison to single protein solutions’ fluorescence recovery rates, spherical droplets demonstrated notable recovery within bleached regions (Supplementary Fig. 3C, D). However, due to some degree of fluorescence quenching within these droplets, overall fluorescence did not fully return to its initial state. Furthermore, in addition to observing the fusion of β-catenin droplets induced by 10 μM RQ, we also noted instances of droplet fission (Supplementary Fig. 3E), further highlighting their fluidic nature. While droplet fusion is a well-characterized phenomenon in LLPS systems, droplet fission is less commonly reported and remains less well understood. However, recent studies suggest that fission events can still arise in passive systems due to local concentration fluctuations, differences in component diffusion rates, or spontaneous nucleation effects5. Additionally, internal rearrangements within protein droplets may create transient instabilities, which could lead to fission as a means of minimizing surface energy21. Moreover, despite our system being passive, minor external influences such as solvent convection and thermal fluctuations may also play a role in modulating droplet stability and facilitating splitting6. Taken together, these factors offer a possible explanation for the droplet dynamics we observed and reinforce our conclusion that these condensates exhibit liquid-like properties.
Furthermore, we stably expressed a C-terminal EGFP-fused green fluorescent β-catenin in U293T cells (Fig. 3a). Under control conditions without RQ treatment, β-catenin exhibited diffuse intracellular localization, while both 5 and 10 μM RQ treatments induced the formation of spherical condensates (Fig. 3a). By randomly quantifying the number of spherical condensates in ten cells (Fig. 3b), it was observed that the number of condensates significantly increased with 10 μM RQ treatment compared to 5 μM RQ treatment. Furthermore, we investigated the dynamics of β-catenin condensates by monitoring fluorescence recovery after photobleaching (FRAP). As depicted in Fig. 3a, c, untreated β-catenin showed faint bleaching and nearly complete fluorescence recovery after photobleaching, whereas RQ-treated β-catenin exhibited slower and less than half recovery of fluorescence after photobleaching, consistent with behaviors associated with LLPS. Furthermore, the fluorescence polarization of U293T cells containing EGFP-fused green fluorescent β-catenin further supported this result. In particular, RQ-free cells exhibited a low polarization value of green fluorescence, while increasing concentrations of RQ led to a dose-dependent increase in polarization values (Supplementary Fig. 3f). The observed increase in polarization values in Supplementary Fig. 3F suggests that RQ promotes molecular aggregation of EGFP-fused green fluorescent β-catenin due to the principle of fluorescence polarization, where larger particles in solution exhibit higher polarization values as a result of slower molecular rotation.

a–c Fluorescence recovery after photobleaching (FRAP) of EGFP-fused β-catenin condensates in U293T cells was analyzed. Data were collected from three independent experiments. a Representative time-lapse microscopy images show the effects of 0, 5, and 10 μM RQ on β-catenin condensates after 6-h incubation. Scale bars: 2 μm. Bleached areas are marked with blue circles. b The number of β-catenin condensates was quantified in ten randomly selected cells per sample. c Mean fluorescence intensity plots represent FRAP results for three condensates per condition (error bars: mean ± SD). d Fluorescence intensity analysis of green, fluorescent β-catenin in cytoplasm and nucleus in response to the 6-h incubation with 0, 5, and 10 μM RQ. The number of β-catenin levels was quantified in a random selection of 30 cells from each sample. e Schematic diagram of RQ-induced LLPS of β-catenin. RQ can induce liquid-liquid phase separation (LLPS) of cytoplasmic β-catenin, resulting in physical interphase blocking and subsequent inhibition of nuclear translocation. f The Western blot analysis was performed to examine the expression levels of β-catenin (β-cat), active β-catenin (Aβ-cat), c-myc (MYC), and Cyclin D1 (CTND1) in three Wnt-hyperactivated HCC cell lines, Hep3B, HepG2, and Hepa1-6, after treatment with 10 μM RQ for 24 h. n = 3 biological replicates. Data were presented as mean values ± SD. g The cell viability of three Wnt-hyperactivated HCC cell lines, Hep3B, HepG2, and Hepa1-6, was assessed after 48 h of treatment with gradient-diluted RQ. The median inhibitory concentrations (IC50) were determined by averaging the results from four independent experiments (n = 4). Data were presented as mean values ± SD. All box-whisker plots B, D center on the median; the bounds of the boxes mark the upper and lower quartile; the whiskers extend to the upper and lower extremes (1.5x interquartile range from the upper and lower quartiles). The p < 0.001 in b, d represents p = 10E-15. p value in b, d calculated by two-tailed t-test.
We subsequently quantified the distribution of β-catenin in nuclei and cytoplasm using these fluorescence photographs and observed a reduction in intranuclear β-catenin percentage from 53.7 to 20.5% following treatment with 5 μM RQ (Fig. 3d and Supplementary Fig. 4A). Importantly, treatment with 10 μM RQ very significantly suppressed the nuclear translocation of β-catenin (Fig. 3d). Similarly, the content of β-catenin protein in the nucleus exhibited a significant decrease after RQ incubation, aligning with the observed trend in fluorescence photographs (Supplementary Fig. 4B). Simultaneously, considering the low expression levels of bcl9 protein in Hep3B cells (Supplementary Fig. 4C), we successfully established Hep3B cell lines with bcl9 overexpression (Supplementary Fig. 4D) and conducted RQ drug intervention. Notably, RQ was still able to induce β-catenin protein condensation, indicating that RQ can effectively achieve stable inhibition of β-catenin protein (Supplementary Fig. 4E–G). These findings support our hypothesis that c-inds can induce liquid-liquid phase separation (LLPS) of cytoplasmic β-catenin, resulting in physical interphase blocking and subsequent inhibition of nuclear translocation (Fig. 3e). Consequently, the absence of β-catenin nuclear translocation results in the suppression of the Wnt/β-catenin signaling pathway, as evidenced by decreased protein levels of β-catenin (β-cat), active β-catenin (Aβ-cat), and two other key proteins in this pathway, c-myc (MYC) and Cyclin D1 (CTND1), observed in three Wnt-hyperactivated HCC cell lines Hep3B, HepG2, and Hepa1-6 following RQ treatment (Fig. 3f and Supplementary Fig. 5A). As a result, dose-dependent growth inhibitions induced by RQ are observed in these three cell lines (Fig. 3g). Simultaneously, we further elucidated that RQ can inhibit β-catenin through luciferase reporter gene assay. As illustrated in Supplementary Fig. 5B, the relative TOP/FOP luciferase activity of the RQ group exhibited a significant decrease, which is consistent with the inhibition conclusion demonstrated in Supplementary Fig. 5C. At the same time, RQ also exhibited significant dose-dependent growth inhibition in cell lines with mutations in APC or β-catenin. Additionally, RQ can downregulate target genes downstream of the β-catenin protein, such as AXIN2, ASCL2, and MYC. Collectively, the identification of RQ as a condensate-inducing therapeutic of β-catenin and its potential as an inhibitor of the Wnt/β-catenin signaling pathway for anti-tumor treatment has been revealed by all these findings.
The construction of albumin-binding rosmanol quinone (abroquinone) enables targeted internalization into tumor cells
Macropinocytosis plays a pivotal role in the acquisition of proteins by cancer cells, and an increasing body of research has revealed its association with the activation of the Wnt/β-catenin signaling pathway30,31. Furthermore, this association was once again demonstrated in HCC (Fig. 4a), where single-sample geneset enrichment analysis (ssGSEA) scores for macropinocytosis were significantly higher in patients with hyperactive Wnt/β-catenin signaling compared to those with hypoactive Wnt/β-catenin signaling (Fig. 4b). Additionally, ssGSEA scores for macropinocytosis were found to be significantly higher in tumor tissue than para-carcinoma tissue (Supplementary Fig. 6A). Next, we substantiated this association by conducting a comparative cellular uptake assay using β-catenin-overexpressing (OE) and normal-expressing Hepa1-6 cells with 70 Kd dextran, which is known to be internalized into cells via macropinocytosis32. As depicted in Fig. 4c, the intracellular uptake of dextran in β-catenin-overexpressing cells was found to be 2.05 times higher than that in normal cells. Furthermore, this enhanced cellular internalization could be attenuated by the macropinocytosis inhibitor EIPA, indicating that β-catenin activates the process of macropinocytosis in cancer cells. Consequently, these findings, along with previous reports33,34, suggest that macropinocytosis holds exceptional potential for achieving the tumor cell-specific uptake of Wnt/β-catenin therapeutics.

a The comparative diagram illustrates the protein expression profiles associated with macropinocytosis in hepatocellular carcinoma (HCC) samples exhibiting hyperactive (Wnt/β-cat high) or hypoactive (Wnt/β-cat low) Wnt/β-catenin signaling. The sample data were obtained from the Cancer Genome Atlas (TCGA, LIHC). b Gene set enrichment analysis scores for macropinocytosis were determined using single-sample geneset enrichment analysis (ssGSEA). The p < 0.001 represents p = 10E-15. c Cellular uptake of the macropinocytosis indicator rhodamine -labeled dextran (70 kd) was assessed in β-catenin-overexpressing Hepa1-6β-catenin OE) and normal-expressing Hepa1-6 cells measured by CLSM. EIPA, a specific inhibitor of macropinocytosis. Semi-quantitative results shown in the right panel represent the mean values from three independent experiments (the error bars are presented as mean ± SD). d Cellular uptake of the four high-concentration proteins (albumin, HSA; fibrin, FBL; immune globulin, IGG; hemoglobin, HGB) at tumor sites into Hepa1-6β-catenin OE. The four proteins were labeled with FITC, and their cellular internalization was quantified using flow cytometry. e The hydrodynamic diameter of tunable-sized Abroquinone (AQ) was determined by dynamic light scattering (DLS), with adjustments made to the molar ratio between HSA and RQ. f The cellular internalization of FITC-labeled AQs into β-catenin-overexpressing (OE) HCC cell line Hepa1-6β-cateninOE, β-catenin-normal-expressing HCC cell line Hepa1-6, T lymphocyte cell line Jurkat, and macrophage cell line RAW264.7 was investigated using flow cytometry. Internalization was quantified following a 6-h incubation of cells with 1 μM AQs. g The FITC-labeled AQ with a size of 163.0 nm was internalized into Hepa1-6β-catenin OE cells through cellular endocytosis, which was inhibited by a series of inhibitors. The quantification of internalization was performed after incubating the cells with 1 μM AQs and inhibitors for 12 h. All box-whisker plots B center on the median; the bounds of the boxes mark the upper and lower quartile; the whiskers extend to the upper and lower extremes (1.5x interquartile range from the upper and lower quartiles). The experiments were replicated independently three times with consistent results. p value in b, c calculated by two-tailed t-test.
To investigate the macropinocytosis propensity of HCC cells with hyperactive Wnt/β-catenin signaling, we assessed the uptake of four high-concentration proteins (albumin, HSA; fibrin, FBL; immune globulin, IGG; hemoglobin, HGB) at tumor sites in Hepa1-6β-catenin OE. As depicted in Fig. 4d by flow cytometry results, HSA exhibited the highest degree of cellular internalization, while its uptake could be significantly inhibited by the macropinocytosis inhibitor EIPA (Supplementary Fig. 6B). The HSA macropinocytosis tendency in Wnt/β-catenin-hyperactivated tumor cells have inspired us to consider it as a potential delivery vector for RQ. To investigate this, we utilized molecular protein-ligand docking employing CB-Dock2 to predict the binding interaction between HSA and RQ. This analysis unveiled 18 distinct potential binding sites, each comprising three to five amino acid residues as depicted in Supplementary Fig. 6C. To validate these findings, we initially expressed and purified three mutant human serum albumins (HSAs), wherein specific mutations were introduced: HSA MS1–5 with all potentially binding amino acid residues in Site1-5 mutated to alanine; HSA MS6–10 with all potentially binding amino acid residues in site 6–10 mutated to alanine; and HSA MS10–18 with all potentially binding amino acid residues in site 11–8 mutated to alanine. As illustrated in Supplementary Fig. 6D, only HSA MS1–5 exhibited a significantly reduced affinity towards RQ, while both HSA MS6–10 and HSA MS11–18 displayed nearly identical affinity toward RQ as wild-type HAS done, suggesting that the actual affinity sites are located within sites 1 to 5. Furthermore, we expressed and purified HSA MS1–3 and HSA MS4-5 separately. It was observed that while HSA MS1–3 lost its affinity for RQ, HSA MS4–5 maintained this affinity (Supplementary Fig. 6D), indicating that site 4 and site 5 do not contribute to the affinity for RQ. Subsequently, we expressed and purified individual mutants - namely, HSA MS1, HSA MS2, and HSA MS3 -to assess their respective interactions with RQ. The results revealed that all three sites contributed towards the affinity for RQ (Supplementary Fig. 6D). These findings demonstrate that HSAs can engage with RQ through a trivalent mechanism.
As shown in Supplementary Fig. 7, another simulation results suggest that HSA has the capability to self-assemble with RQ into a higher-order structure, which were obtained by theoretical feasibility studies using the coarse-grained model of HSA and RQ implemented in the GROMACS software package (version 2023.1) along with the MARTINI force field (Supplementary Table 3). In the simulation, HSA and RQ were randomly dispersed within a cubic box measuring 25 × 25 × 25 nm3 and filled with coarse-grained water. Cl− ions were added to neutralize the system’s charge. After undergoing 1000 ns of unrestricted movement, HSA and RQ successfully formed an aggregate structure (Supplementary Fig. 7A). After enlarging the box size and increasing the number of molecules, molecular simulations with three different initial positions all consistently demonstrated aggregation behavior (Supplementary Fig. 7B). Consistent with this finding, the binding of HSA with RQ resulted in the formation of tunable-sized abroquinone (AQ) by adjusting the molar ratio between HSA and RQ. Using this approach, three AQ structures were constructed with sizes measuring 91.6, 163.0, and 247.3 nm, respectively (Fig. 4e and Supplementary Fig. 7C).
Next, the cellular internalization of these three AQs into β-catenin-overexpressing (OE) HCC cell line Hepa1-6β-cateninOE, β-catenin-normal-expressing HCC cell line Hepa1-6, T lymphocyte cell line Jurkat, and macrophage cell line RAW264.7 was investigated. After a 6-h incubation period, AQ-3 with a size of 247.3 nm exhibited the highest level of cellular internalization in all four cell lines (Fig. 4f). In contrast, AQ-2 with a size of 163.0 nm showed satisfactory cellular internalization specifically in Hepa1-6β-cateninOE cells but had limited uptake by RAW264.7 and jurkat cells (Fig. 4f). Considering the need to avoid potential reduction in blood concentration due to uptake by T lymphocytes and macrophages, AQ-2 was used as the standard abroquinone for subsequent studies. Afterward, in order to further investigate the endocytosis species of AQ, we employed a clathrin-mediated endocytosis inhibitor chlorpromazine (CPZ), a dynamin-mediated endocytosis inhibitor Dynasore (DYN), two caveolae-mediated endocytosis inhibitors Filipin and Genistein (GEN), as well as two macropinocytosis inhibitors EIPA and cytochalasin D (Cyto D) to suppress the internalization of AQ (Fig. 4g). The uptake of AQ was significantly suppressed by both macropinocytosis inhibitors EIPA and Cyto D, while other inhibitors had minimal effects (Fig. 4g), indicating a reliance on macropinocytosis for AQ uptake. In combination, the binding of RQ with human serum albumin (HSA) enhances its exploitation of tumor-specific macropinocytosis, thereby enabling precise suppression of the Wnt/β-catenin cascade in tumor cells. Additionally, the distribution of RQ in the lysosome and cytoplasm was quantified using LC–MS. It was observed that there was a peak accumulation of RQ in the lysosome at 6 h after AQ incubation, followed by a rapid decline thereafter (Supplementary Fig. 7D). Conversely, the concentration of RQ in the cytoplasm exhibited a continuous increase during the 12-h period of AQ incubation (Supplementary Fig. 7D). These findings suggest that HSA within AQ can undergo degradation within the lysosome, leading to the release of RQ which subsequently diffuses into the cytoplasm and induces β-catenin condensate formation.
Abroquinone (AQ) demonstrates tumor-targeting properties and exceptional biosafety
The compelling in vitro tumor-specific macropinocytosis of AQ prompted us to investigate its in vivo biodistribution. To this end, AQ was labeled with trivalent gold ions, and the concentration of 197 Au in tissue homogenates from various organs was measured using inductively coupled plasma mass spectrometry (ICP-MS). Following intravenous injection of Au-labeled AQ into mice bearing orthotopic HCC, the accumulation of AQ in tumor-bearing liver increased over time within the first eight hours, while the concentration of AQ in other organs exhibited a declining trend (Fig. 5a). Further analysis of AQ accumulation through organ-to-liver ratios revealed progressively increasing values for all organs within the initial eight-hour period, indicating a pronounced preference for tumor accumulation of AQ (Fig. 5b).

a The biodistribution of AQ was assessed in the heart, liver, spleen, lung, kidney, and intestine at various times after intravenous injection at 5 mg/kg. b Tumor-bearing liver to indicated organ ratios were calculated based on data from (a). c–f Acute toxicity was evaluated after a 10-day treatment. c Mouse body weights, n = 6/group. d Blood analysis included white blood cell count (WBC), lymphocyte count (Lymph#), monocyte count (Mon#), granulocyte count (Gran#), red blood cell count (RBC), hemoglobin (HGB), platelet count (PLT), and mean platelet volume (MPV). e Hepatotoxicity was assessed by H&E staining and serum AST/ALT levels. (f) Nephrotoxicity was assessed by H&E staining and serum CRE/BUN levels. g–i Fluorescence recovery after photobleaching (FRAP) of EGFP-fused green fluorescent β-catenin condensate in Hep3B cells was performed. g Representative images obtained from time-lapse microscopy during FRAP experiments are presented, illustrating the effects of 6-h incubation with 0, 2.5, and 5 μM AQ on β-catenin. Scale bars: 2 μm. The bleached area is indicated by blue circles. h The number of β-catenin condensates was quantified in a random selection of ten cells from each sample. The p < 0.001 represents p = 10E-15. i The plot of mean fluorescence intensity demonstrates the FRAP results for three condensates per condition. j, k The cell viability of various cell types was evaluated following a 48-h treatment with gradient-diluted AQ (j) and HSA (k) (n = 3). HCC cell lines (Hep3B, HepG2, Sk-Hep1 HuH7, Hepa1-6, SNU878, SNU739, and SNU368), normal liver cell lines (AML12, LO2, HUVEC, and LSEC), primary stem cells (BMSCs, UC, and PMSC) and immune cell lines (RAW264.7, jurkat, and DC2.4). All box-whisker plots H center on the median; the bounds of the boxes mark the upper and lower quartile; the whiskers extend to the upper and lower extremes (1.5x interquartile range from the upper and lower quartiles). The error bars in a, c, e, f, i are presented as mean values ± SD. p value in Fig. 5e, f calculated by two-tailed t-test. The experiments in g, i were independently replicated three times, yielding consistent results.
Subsequently, the biosafety of AQ was evaluated in healthy C57/B6 mice. Due to its half-life period of approximately 40 h in tumor-bearing liver (Supplementary Fig. 8A), the drug injection interval was set at 48 h. Following a 10-day treatment regimen with either 10 mg/kg RQ, 10 mg/kg AQ, or 50 mg/kg AQ administered once every two days, both groups treated with AQ maintained their body weight similar to that of the mock-treated group; however, RQ treatment resulted in a weight loss of about 20% (Fig. 5c). Routine blood analysis revealed no impact on hemocytes for both doses of AQ treatment, which is in stark contrast to myelosuppression and anemic symptoms observed following RQ treatment (Fig. 5d). The analysis of hepatotoxicity further revealed that only treatment with RQ resulted in liver side effects, while mice treated with AQ maintained healthy livers (Fig. 5e). Additionally, neither AQ nor RQ treatment induced nephrotoxicity (Fig. 5f) or pathological injuries in the lung, heart, and spleen (Supplementary Fig. 8B). Given that beta-catenin plays a crucial role in bone homeostasis and that inhibiting this pathway in vivo can increase the risk of fractures, we evaluated the osteotoxicity of HSA-RQ. Twenty female C57 mice were randomly assigned to four groups, as depicted in Supplementary Fig. 8C–F. After treatment, we performed micro-CT on the femurs of the mice. As anticipated, there were no significant differences in the number of trabecular bones among the groups in the CT images (Supplementary Fig. 8C). Quantitative analysis also revealed no significant differences in trabecular and cortical bone parameters (such as BV/TV, BMD, TB.th, and Ct.Th) across the groups (Supplementary Fig. 8D). This indicates that RQ is safe and non-toxic. Consistent with this conclusion, STEM and HE staining also produced similar findings (Supplementary Fig. 8E, F). Thus, RQ has no obvious bone toxicity and has good biosafety. These findings collectively indicate that HSA delivery mitigates the toxicity and adverse effects associated with RQ, thereby enhancing its potential for clinical translation.
Abroquinone (AQ) also induced the LLPS of β-catenin and exhibited selective inhibition against tumor cells
To investigate the condensation behavior induced by AQ in tumor cells, we stably expressed a C-terminal EGFP-fused green fluorescent β-catenin in Hep3B HCC cells. Under control conditions without AQ treatment, β-catenin exhibited diffuse intracellular localization (Fig. 5g). As anticipated, the addition of 2.5 μM AQ resulted in the formation of agglutinating droplets of β-catenin (Fig. 5g, h), which displayed challenging recovery of their green fluorescence after photobleaching (Fig. 5g, i). Furthermore, treatment with 5 μM AQ further enhanced these distinctive phenomena associated with liquid-liquid phase separation (Fig. 5g–i). These findings demonstrate that AQ inherits the biofunction of RQ as c-inds for β-catenin.
Next, we assessed the cytotoxicity of AQ against a variety of cell types, including HCC cells, normal liver cells, primary stem cells, and immune cells. As depicted in Fig. 5j and Supplementary Fig. 9A, AQ exhibited dose-dependent suppression of viability in ten HCC cell lines (Hep3B, HepG2, Sk-Hep1 HuH7, Hepa1-6, SNU878, SNU739, HUH6, SNU398, and SNU368), while showing minimal cytotoxicity towards normal liver cell lines (AML12, LO2, HUVEC, and LSEC), primary stem cells (BMSCs UC and PMSC) as well as immune cell lines (RAW264.7, jurkat and DC2.4). As shown in Supplementary Fig. 9A, a similar concentration-dependent growth inhibition was observed in β-catenin active cell lines (HCT116 and CT26 cells). Importantly, HSA alone had no impact on the viability of these cells, suggesting that all the cytotoxic effects observed with AQ were attributed to RQ (Fig. 5k). Additionally, we established representative cell lines with either overexpression or knockout of β-catenin (CTNNB1) to assess the specific targeting of AQ toward β-catenin. As depicted in Supplementary Fig. 9B–F, the upregulation of β-catenin augmented the susceptibility of tumor cells to AQ, as evidenced by a reduction in the IC50 value for cell viability derived from AQ treatment. Conversely, the knockout of β-catenin resulted in a significant increase in the AQ-derived IC50 value, indicating resistance to AQ (Supplementary Fig. 9B–F). These findings suggest that AQ exerts its suppressive effects on tumor cells through a mechanism dependent on β-catenin. Collectively, AQ demonstrated selective inhibition against tumor cells, likely due to its tumor-specific macropinocytosis.
Abroquinone (AQ) inhibited the hyperactivated Wnt/β-catenin pathway in an orthotopic HCC mouse model and stimulated T-cell-mediated immunity
To challenge the biofunction of AQ, a reconstructive Hepa1-6 cell line was established, wherein stable expression of the S37A/S33A mutant β-catenin was achieved. It is worth noting that this specific mutation effectively abolishes phosphorylation by GSK3β, which in turn prevents degradation of β-catenin35. Consequently, there is cytoplasmic accumulation of β-catenin and subsequent hyperactivation of the Wnt/β-catenin pathway. The orthotopic HCC mouse model was established by subcapsular injection of 1 × 105 hyperactivated Wnt/β-catenin Hepa1-6 cells into the liver of normal C57/B6 mice through anesthesia laparotomy. A 10-day treatment regimen with a dosage of 5 mg/kg AQ administered once every two days was initiated at 1 week post-HCC cell transplantation. Following the 10-day treatment, significantly suppressed liver cancer was observed in AQ-treated mice (Fig. 6a), and AQ exhibited a statistically significant downregulation effect on β-catenin levels at tumor sites (Fig. 6b). Importantly, minimal β-catenin staining was observed in the nuclear region of the liver in AQ-treated mice (Supplementary Fig. 10A), indicating effective inhibition of β-catenin’s translocation into the nucleus. Subsequently, scRNA-seq analysis was employed to elucidate the cellular and molecular characteristics of cells in this hyperactivated Wnt/β-catenin Hepa1-6 cell line upon AQ treatment. In Fig. 6c of the single-cell expression atlas (n = 3/group), a remarkable diversity of immune cells, consisting of three distinct types, along with four populations of tumor-related cells were identified within the two contrasting Hepa1-6 tumors. After analyzing the cluster of tumor cells, it was observed that AQ significantly downregulated the expression of β-catenin and its downstream protein Cyclin D1, as well as Wnt5a (Fig. 6d). Furthermore, single-sample geneset enrichment analysis (scGSEA) revealed that the two most prominently suppressed pathways were the Wnt/β-catenin signaling pathway (Fig. 6e) and the cell cycle pathway (Fig. 6f), while the top upregulated pathway was found to be the apoptosis signaling pathway (Fig. 6g), indicating that AQ induced cell cycle arrest and apoptosis in tumor cells. Additionally, this finding was further supported by a decrease in IHC staining for the proliferation marker Ki-67 (Fig. 6h) and an increased number of apoptotic cells in AQ-treated tumors (Fig. 6i, j).

a The representative photograph of the mice liver subjected to either AQ or mock treatment. b The immunohistochemical (IHC) staining and its semi-quantitative scoring of the β-catenin. c t-Distributed stochastic neighbor embedding (t-SNE) plots of broad cell types in mice (n = 3/group). d The expression of β-catenin, Cyclin D1, and Wnt 5a in the cluster of tumor cells. e–g The scGSEA was performed to analyze the enrichment of single-sample genesets in the Wnt/β-catenin signaling pathway (e) cell cycle pathway (f) and apoptosis signaling pathway (g) between AQ-treated mice and Control-treated mice within the tumor cell cluster. h The immunohistochemical (IHC) staining and its semi-quantitative scoring of the ki-67. i The TUNEL staining and its semi-quantitative scoring. j The representative H&E staining. The experiments in 6h–j were replicated independently three times with consistent results. k Two distinct subclusters: CD8 T cells and Treg, subdivided from T cell cluster. l Significant differential genes between AQ-treated mice and mock-treated mice within the CD8 T cells subcluster. m t-SNE plots showing the expression of selected marker genes in cytotoxic T cells. n Multichannel flow cytometry analysis of cytotoxic T lymphocytes (CTLs) in tumors with different treatments. CTLs were identified by CD45+/CD3+/CD8+ (n = 3 biological replicates, the error bars are presented as means ± SD). The gating strategy for CTL involves two sequential steps: (1) identification of CD45+ expressing cells and (2) characterization of CTL as cells positive for both CD8+ and CD3+ within the CD45+ population. Subsequently, GzmB+ or IFN γ+ CTL were identified in this population. o Survivorship curve of C57/B6 mice bearing orthotopic HCC tumors in response to either the AQ treatment or mock treatment (n = 8/group). All box-whisker plots h, i center on the median; the bounds of the boxes mark the upper and lower quartile; the whiskers extend to the upper and lower extremes (1.5x interquartile range from the upper and lower quartiles). The p < 0.001 in b, e–i represents p = 10E-15. p value in b, e–i, o calculated by two-tailed t-test.
Moreover, an increasing number of reports have revealed that the inhibition of carcinogenic Wnt/β-catenin signaling pathway can restore T lymphocyte-mediated anti-tumor adaptive immunity36,37. Thus, further clustering was performed within the T cell population, leading to the identification of two distinct subclusters: Regulatory T cells (Treg) expressing CD4, CD25, and Foxp3 as positive markers; and CD8 + T cells expressing CD3 and CD8 as positive markers. As anticipated, there was a significant upregulation in the proportion of CD8 T cells in AQ-treated samples (Fig. 6k). Additionally, AQ treatment promoted the expression of various cytotoxic factors, including granzyme B (Gzmb), perforin 1 (Prf1), and interferonγ (Ifng) in CD8 T cells (Fig. 6l, m). Furthermore, these findings were corroborated by multichannel flow cytometry analysis, which revealed a more than threefold increase in cytotoxic T lymphocytes following AQ treatment (Fig. 6n). The proportion of Treg was downregulated by AQ treatment, as evidenced by scRNA-seq analysis (Fig. 6k) and multichannel flow cytometry (Supplementary Fig. 10B), demonstrating the collapse of Treg-mediated immunosuppression. The administration of AQ resulted in a statistically significant extension of overall survival in mice with hyperactivated Wnt/β-catenin orthotopic HCC tumors. In the mock-treated group, the median survival was merely 43 days, and all mice had perished by day 60 (Fig. 6o). However, in the AQ-treated group, an impressive 87.5% of mice were still thriving at day 90 (Fig. 6o). The above results demonstrate that AQ effectively inhibits the hyperactivated Wnt/β-catenin pathway in an orthotopic HCC mouse model and enhances T-cell-mediated immunity, thereby achieving potent tumor suppression.
AQ effectively suppressed the hyperactivated Wnt/β-catenin pathway in a humanized mice model of HCC, while also enhancing T-cell-mediated immunity
The potency of AQ was further challenged by establishing a humanized mouse model of hepatocellular carcinoma (HCC) in NSG. Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice through subcutaneous inoculation of engineered HCC tumor cells Hep3BAPC-KD. These cells were generated by stable transfection of APC shRNA into the Hep3B cell line, resulting in hyperactivation of the Wnt/β-catenin pathway. Humanized immunity was established by sub-lethal irradiation (0.5 Gy), followed by transplantation of 5 × 106 human peripheral blood mononuclear cells (PBMCs) obtained from a healthy donor who passed HLA typing. Once achieving >1% engraftment of human CD45-positive cells in peripheral blood, the mice can be used for tumor cell inoculation. When the tumor reaches a volume of 100–150 mm3, twelve mice were randomly divided into two groups and received intravenous injections of N.S. or AQ every other day at a dosage of 5 mg/Kg per dose (Fig. 7a). As expected, AQ significantly suppressed β-catenin expression at tumor sites by 82.1% (Fig. 7b), particularly in the intranuclear localization of β-catenin. Additionally, LC–MS-mediated proteomics analysis revealed a decrease in proteins associated with the Wnt/β-catenin pathway (Fig. 7c, d). Additionally, multichannel flow cytometry (Fig. 7e) and ssGSEA of the proteomics data (Fig. 7f) revealed an increase in both the quantity and activity of cytotoxic T cells, along with a decrease in the number of Tregs. Consequently, AQ exhibited remarkable tumor suppression capabilities (Fig. 7g), resulting in a substantial reduction of tumor weights by 94.3% (Fig. 7h) and a significant decrease in tumor volume by 91.7% (Fig. 7i). Concurrently, we assessed the tumor inhibitory effects of AQ in the patient-derived orthotopic xenograft (PDOX) model of colon cancer. As illustrated in Supplementary Fig. 11, AQ demonstrates comparable and notable tumor inhibition along with significant immune-activating properties.

a Schematic of the humanized CDX model development and AQ therapy experimental protocols by figdraw.com. b IHC staining and semi-quantitative scoring of β-catenin; experiments were replicated three times with consistent results. c LC–MS-based targeted protein analysis of the Wnt/β-catenin pathway in AQ-treated and mock-treated tumors. d GSEA analysis of Wnt/β-catenin pathway data from AQ-treated and mock-treated tumors; p value calculated by permutation test. e Multichannel flow cytometry was used to analyze Treg and CTL populations in tumors from two treatment groups (n = 3 biological replicates per group). CTLs were identified as CD45 + CD8 + CD3+ cells, with GzmB+ or IFNγ + CTLs further characterized. Tregs were identified as CD45+ CD4+ CD25+ cells, with FOXP3+ Tregs further characterized. f ssGSEA of the proteomics data about Treg cells and CTLs. g, h Tumor photos (g) and tumor weights (h) from tumors. i The tumor growth curve upon treatment with AQ or a control substance (n = 5/group). j Schematic illustrating the development of a humanized PDX model and the experimental protocols for AQ and Anti-PD1 combo-therapy in this model by figdraw.com. k RNA-seq analysis of Wnt/β-catenin pathway in the four groups. l ssGSEA of tumor samples from four groups about Wnt/β-catenin pathway, cell cycle arrest and apoptosis (n = 3/group). m ssGSEA of tumor samples from four groups about activated CD8 T cell, CD8 TCR, and Tregs. (n = 3/group). n The tumor photos depict tumors treated with mock-treated, AQ-treated, anti-PD1-treated, and AQ/Anti-PD1 combination therapy. o The tumor growth curve is plotted for the treatment groups (n = 5/group). All box-whisker plots B&H center on the median; the bounds of the boxes mark the upper and lower quartile; the whiskers extend to the upper and lower extremes (1.5x interquartile range from the upper and lower quartiles). The error bars in e, i, l, m are presented as means ± SD. The p < 0.001 in b, h, i represents p = 10E-15. p value in b, e, h, i calculated by two-tailed t-test.
AQ effectively inhibited the hyperactivated Wnt/β-catenin pathway in a humanized patient-derived xenograft (PDX) model of HCC, while enhancing the sensitivity of anti-PD1 therapy
Next, to further validate the clinical translational potential of AQ, a Hu-PDX model of hepatocellular carcinoma (HCC) was established in NSG mice by engrafting a postsurgical tumor mass obtained from an HCC patient carrying an APC loss-of-function mutation. PDX tumors were implanted in the iliac fossa of NSG mice with successful human immune remodeling. When the tumor reached a volume of 100 ± 30 mm3, 20 mice were randomly divided into four groups for the indicated treatments as illustrated in Fig. 7j. As anticipated, there was a significant downregulation of β-catenin levels observed in both the AQ monotherapy group and the AQ/Anti-PD1 combo-therapy group (Supplementary Fig. 12). Furthermore, RNA-seq analysis revealed the suppression of the hyperactivated Wnt/β-catenin pathway in both groups (Fig. 7k). Additionally, ssGSEA of tumor samples demonstrated that combo treatment and AQ monotherapy exhibited almost identical abilities to suppress the Wnt/β-catenin pathway; however, combo treatment showed a stronger effect in inducing cell cycle arrest and apoptosis (Fig. 7l). Further ssGSEA analysis revealed a more robust CD8 T cell response and suppression of Tregs in the combination treatment group compared to AQ monotherapy (Fig. 7m), which is consistent with the observed tumor inhibition efficiencies (Fig. 7n, o). AQ monotherapy exhibited a remarkable 67.0% tumor inhibition, surpassing the 30.3% achieved by Anti-PD1 monotherapy, while AQ/Anti-PD1 combination therapy demonstrated an impressive 80.0% tumor inhibition (Fig. 7n, o), indicating a highly favorable synergistic effect. Overall, these findings demonstrate that AQ effectively inhibits the hyperactivated Wnt/β-catenin pathway in a Hu-PDX model of HCC while enhancing the sensitivity of anti-PD1 therapy.
Discussion
The aberrant activation of the Wnt/β-catenin signaling pathway plays a pivotal role in the pathogenesis of various malignancies11. A comprehensive landscape analysis investigating the mechanisms and patterns of somatic alterations in key oncogenic signaling pathways across all cancer types within the Cancer Genome Atlas (TCGA) dataset reveals that persistent activation mutation in the Wnt/β-catenin pathway, including APC, CTNNB1 (β-catenin), and GS3Kβ, represents the most variable gene alterations across all cancer types38. During tumor progression, these mutations always result in the dysfunction of the β-catenin destruction complex and subsequent cytoplasmic accumulation of β-catenin, as well as constitutive activation of nuclear β-catenin translocation11,39. This aberrant activation of the Wnt/β-catenin signaling pathway not only promotes tumor cell proliferation and metastasis but also importantly facilitates immune exclusion through T-cell immunity inhibition11,40,41. Although the broad outline and important tumorigenesis of this pathway are known, specific targeting of components for therapeutic intervention has proven difficult due to the toxic and adverse effects from Wnt inhibitors17. Additionally, since the Wnt/β-catenin signaling pathway is evolutionarily conserved with a crucial role in embryonic development and adult stem cell homeostasis regulation, systemic therapy using Wnt inhibitors cannot be tolerated by the human body12,42.
To address this issue, a hierarchical targeting strategy was devised in this study, whereby AQ initially executed primary targeting by inducing the LLPS of cytoplasmic β-catenin accumulation and subsequently performed secondary targeting through selective cell internalization via β-catenin-activated macropinocytosis of tumor cells. It is noteworthy that cytoplasmic accumulation of β-catenin represents a prominent feature specific to tumor cells due to aberrantly continuous activation of the carcinogenic Wnt/β-catenin signaling pathway, which does not occur in normal or stem cells upon controlled activation of Wnt/β-catenin signaling. Additionally, β-catenin-activated macropinocytosis is an exclusive characteristic of tumor cells, enabling them to exploit nutrients to meet the increased material demands for cell replication and migration derived from β-catenin31. With this tumor-targeting design, RQ with primary tumor targeting has already demonstrated manageable side effects in vivo, while AQ with hierarchical tumor targeting exhibits favorable biosafety profiles even at concentrations as high as 10 times the therapeutic concentration up to 50 mg/Kg (Fig. 5C–F). The AQ developed herein, so to speak, possesses the potential to triumph over the long-standing dose-dependent side effects of Wnt inhibitors, thus exhibiting clinical translational prowess in β-catenin c-inds.
Furthermore, the AQ-based approach established in this study broadens the potential applications of biomolecular condensate modulation by introducing the concept of condensate-inducing therapeutics (c-inds). Traditionally, condensate-modifying therapeutics (c-mods) have been explored through methods that depend on identifying specific pre-existing condensates to target. While these efforts have opened new avenues for treating human diseases, they are inherently tied to known condensates. In contrast, our c-inds strategy represents a significant departure from this dependency. By employing small molecules that reduce the stability of previously stable protein regions, we effectively mimic intrinsically disordered regions (IDRs), inducing LLPS de novo. This mechanism bypasses the need for pre-identified condensates, instead focusing on identifying small molecules capable of directly driving phase separation in target proteins. Consequently, this approach leads to rapid compartmentalization of the target proteins within condensed phases, effectively neutralizing their biofunction more swiftly and directly than traditional small-molecule antagonists, which rely solely on competitive inhibition. In doing so, this study not only introduces a novel perspective on drug discovery but also provides a practical framework for identifying and validating therapeutic agents capable of initiating or exploiting disease-specific condensates.
Methods
Ethical statement
For human study participants, ethical approval was obtained from the Medical Ethics Committee of Xi’an Jiaotong University (approval number: 2021-056) and written informed consent was secured from all participants prior to tissue collection and use in this research. Human tumor tissues used in this study were obtained from a 50-year-old patient. Human peripheral blood mononuclear cells (PBMCs) used for humanized mouse models were collected from one healthy volunteer donor following the same ethical guidelines (approval number: 2021-056), and the donor provided written informed consent specifically for research purposes; confidentiality and anonymity of the donor were strictly maintained. Animal studies complied with institutional guidelines approved by the Medical Ethics Committee of Xi’an Jiaotong University (approval number: 2021-056).
Human tissue/PBMCs collection
Human tumor tissues used in this study were obtained from a 50-year-old patient with hepatocellular carcinoma (HCC) who underwent surgery at the First Affiliated Hospital of Xi’an Jiaotong University. PBMCs were isolated using density-gradient centrifugation with Ficoll-Hypaque.
Animals studies
Laboratory animals, including C57BL/6 mice and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, were obtained from the Laboratory Animal Center of Xi’an Jiaotong University. All mice were housed under specific pathogen-free (SPF) conditions, maintained at controlled temperature (20–24 °C) and humidity (40–60%), with a standard 12-h light/dark cycle. Field-collected animal samples were not employed in this study. The maximum tumor size/tumor burden in this study was permitted by the Medical Ethics Committee of Xi'an Jiaotong University, and it was confirmed that the tumor size/tumor burden in the study did not exceed the standard.
Native chemical ligation, desulfurization, and deprotection of BBD
Native chemical ligation reactions were carried out in 0.1 M phosphate buffer (pH 7.4) containing 6 M guanidine hydrochloride (GuHCl), 200 mM MPAA, and 20 mM TCEP. The ligation products were purified to homogeneity by preparative C18 reversed-phase HPLC. Following the first and second ligation steps, the thiazolidine (Thz) moiety in the intermediate products was converted to a cysteine (Cys) residue by overnight treatment with 0.2 M methoxylamine hydrochloride (pH 4.0).
For desulfurization, the fully assembled peptide was dissolved at 5–20 mg/mL in 1 mL of 0.2 M NaH2PO4 buffer (pH 6.5) containing 6 M GuHCl and 0.25 M TCEP. Subsequently, 250 μL of 40 mM reduced glutathione and 200 μL of 0.2 M VA-044 were added. The reaction was quenched by adding 7.5 mL of 0.5% TFA in water, and the products were purified by reversed-phase HPLC on a C4 column. Acm (acetamidomethyl) deprotection was performed by adding silver acetate (AgOAc) to the peptide-containing HPLC fractions. The concentration may vary independently with temperature to a final concentration of 20 mM, followed by overnight incubation at 25 °C with gentle agitation. DTT was then added to a final concentration of 24 mM to quench the reaction, resulting in a metal–DTT precipitate. After centrifugation, the supernatant was diluted with 0.1% TFA in water (to reduce the acetonitrile concentration) and further purified by reversed-phase HPLC.
Folding of BBD
The BBD was first dissolved at 1 mg/mL in 6 M guanidine hydrochloride (GuHCl). This denatured protein solution was then diluted sixfold into phosphate-buffered saline (PBS, pH 7.4) containing 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), followed by overnight dialysis against the same buffer to allow refolding. The protein concentration after refolding was determined by UV absorbance at 280 nm using a molar extinction coefficient of 1490.
CD spectroscopy and thermal denaturation
CD spectra of BBD (1 μM in 10 mM phosphate buffer, pH 7.4) were recorded at room temperature using a J-810 spectropolarimeter (JASCO Inc., Easton, MD) with a 1 mm path-length quartz cuvette. Thermal denaturation of BBD was monitored on the same instrument equipped with a Peltier temperature-control device.
For thermal unfolding experiments, a 2.5 mL sample of BBD (5 μM in PBS, pH 7.4) was loaded in a 3 mL cuvette and heated from 25 to 90 °C at a rate of 1 °C/min, with CD readings taken at 1 °C intervals under continuous stirring. After reaching each temperature increment, the sample was equilibrated for 20 s before the ellipticity at 222 nm was recorded over 16 s.
Data processing of thermal denaturation
Thermal denaturation data were analyzed by fitting to a two-state unfolding model in order to determine the midpoint temperature of denaturation (Tm). Specifically, the data were fit using a six-parameter equation (Eq. (7) below) derived from a two-state equilibrium between the native (N) and denatured (D) protein states. The derivation of Eq. (7) is as follows. For the equilibrium N ↔ D, the change in Gibbs free energy is given by:
where ∆G0 is the standard Gibbs free energy difference, R is the gas constant, T is the absolute temperature, and J is the reaction quotient (a dimensionless parameter). At equilibrium (achieved at each temperature step when ΔGND = 0), J equals the equilibrium constant K, leading to:
The observed CD signal at temperature T, Sθ, is the weighted average of the native and denatured state signals:
where fn and fd are the fractions of the protein in the native and denatured states, respectively, and αn and αd are the CD ellipticity values for the fully native and fully denatured states. By definition of the equilibrium constant, these fractions are given by:
Substituting Eqs. (2, 4) into Eq. (3) yields:
During thermal unfolding, ΔGND varies with temperature and can be expressed as:
where Tm is the midpoint (melting) temperature and ΔHm is the enthalpy change at Tm. To account for the slight temperature dependence of the baseline ellipticity, αn and αd are each represented as linear functions of T in the fitting model. Substituting these baseline terms into Eq. (5) gives the final fitting function:
where αn1, αn2, αd1, and αd2 are fitted baseline coefficients for the native and denatured states. Equation (7) contains six fitting parameters in total (αn1, αn2, αd1, αd2, Tm, and ΔHm). Nonlinear regression of the CD signal (Sθ) as a function of temperature to this equation provided the best-fit values of Tm and ΔHm.
Once Tm and ΔHm were determined, the fraction of protein in the native state at any temperature was calculated from the CD signal using:
From Eq. (6) (setting ΔGND = 0 at equilibrium), the equilibrium constant as a function of temperature is:
Substituting this expression for K into Eq. (4) yields:
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence data generated in this study have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, that are publicly accessible at GSA accession number CRA019212 [https://ngdc.cncb.ac.cn/gsub/submit/bioproject/subPRO045151]. All the other data generated in this study are provided in the Supplementary Information/Source Data file. Unless otherwise stated, all data supporting the results of this study can be found in the article, supplementary, and source data files. Source data are available for Figs. 1b, e, 2c–e, 3b–d, f, g, 4c, e, f, 5a–f, h–k, 6a, h, i, n, o, 7b, c, e, f, h, i, k–m, o, as well as all numerical data presented in the Supplementary Figs. within the associated source data file. Source data are provided with this paper.
References
Boeynaems, S. et al. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435 (2018).
Qian, Z.-G., Huang, S.-C. & Xia, X.-X. Synthetic protein condensates for cellular and metabolic engineering. Nat. Chem. Biol. 18, 1330–1340 (2022).
Brangwynne, C. P. et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 (2009).
Cai, D., Liu, Z. & Lippincott-Schwartz, J. Biomolecular condensates and their links to cancer progression. Trends Biochem. Sci. 46, 535–549 (2021).
Alberti, S. & Hyman, A. A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 22, 196–213 (2021).
Mitrea, D. M., Mittasch, M., Gomes, B. F., Klein, I. A. & Murcko, M. A. Modulating biomolecular condensates: a novel approach to drug discovery. Nat. Rev. Drug Discov. 21, 841–862 (2022).
Klein, I. A. et al. Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392 (2020).
Babinchak, W. M. et al. Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun. 11, 5574 (2020).
Quek, R. T. et al. Screen for modulation of nucleocapsid protein condensation identifies small molecules with anti-coronavirus activity. ACS Chem. Biol. 18, 583–594 (2023).
Garcia-Cabau, C. & Salvatella, X. Regulation of biomolecular condensate dynamics by signaling. Curr. Opin. Cell Biol. 69, 111–119 (2021).
Jackstadt, R., Hodder, M. C. & Sansom, O. J. WNT and β-catenin in cancer: genes and therapy. Annu. Rev. Cancer Biol. 4, 177–196 (2020).
Zhang Y. & Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 13, 165 (2020).
Moon, R. T., Kohn, A. D., Ferrari, G. V. D. & Kaykas, A. WNT and β-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701 (2004).
Zhang, J., Tian, X.-J. & Xing, J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J. Clin. Med. 5, 41 (2016).
Sharma, A., Blériot, C., Currenti, J. & Ginhoux, F. Oncofetal reprogramming in tumour development and progression. Nat. Rev. Cancer 22, 593–602 (2022).
Zhong, Z. & Virshup, D. M. Wnt signaling and drug resistance in cancer. Mol. Pharmacol. 97, 72–89 (2020).
Chatterjee, A. et al. Advances in targeting the WNT/β-catenin signaling pathway in cancer. Drug Discov. Today 27, 82–101 (2022).
Morris, A., Pagare, P. P., Li, J. & Zhang, Y. Drug discovery efforts toward inhibitors of canonical Wnt/β-catenin signaling pathway in the treatment of cancer: a composition-of-matter review (2010–2020). Drug Discov. Today 27, 1115–1127 (2022).
Verkaar, F. & Zaman, G. J. New avenues to target Wnt/β-catenin signaling. Drug Discov. Today 16, 35–41 (2011).
Dignon, G. L., Zheng, W., Kim, Y. C. & Mittal, J. Temperature-controlled liquid–liquid phase separation of disordered proteins. ACS Cent. Sci. 5, 821–830 (2019).
Dignon, G. L., Zheng, W. & Mittal, J. Simulation methods for liquid–liquid phase separation of disordered proteins. Curr. Opin. Chem. Eng. 23, 92–98 (2019).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012).
MacDonald, B. T., Tamai, K. & He, X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 (2009).
Aberle, H., Bauer, A., Stappert, J., Kispert, A. & Kemler, R. β-catenin is a target for the ubiquitin–proteasome pathway. Trends Cell Biol. 16, 3797–3804 (1997).
Li, V. S. et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 149, 1245–1256 (2012).
Krieghoff, E., Behrens, J. & Mayr, B. Nucleo-cytoplasmic distribution of β-catenin is regulated by retention. J. Cell Sci. 119, 1453–1463 (2006).
Townsley, F. M., Cliffe, A. & Bienz, M. Pygopus and legless target armadillo/β-catenin to the nucleus to enable its transcriptional co-activator function. Nat. Cell Biol. 6, 626 (2004).
Sampietro, J. et al. Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 24, 293–300 (2006).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Redelman-Sidi, G. et al. The canonical Wnt pathway drives macropinocytosis in cancer. Cancer Res. 78, 4658–4670 (2018).
Tejeda-Muñoz, N., Albrecht, L. V., Bui, M. H. & De Robertis, E. M. Wnt canonical pathway activates macropinocytosis and lysosomal degradation of extracellular proteins. Proc. Natl Acad. Sci. USA 116, 10402–10411 (2019).
Jones, A. T. Macropinocytosis: searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med. 11, 670–684 (2007).
Yan, J. et al. Rewiring chaperone-mediated autophagy in cancer by a prion-like chemical inducer of proximity to counteract adaptive immune resistance. Drug Resist. Update 73, 101037 (2024).
Liu, H. & Qian, F. Exploiting macropinocytosis for drug delivery into KRAS mutant cancer. Theranostics 12, 1321 (2022).
Hernández, A. R., Klein, A. M. & Kirschner, M. W. Kinetic responses of β-catenin specify the sites of Wnt control. Science 338, 1337–1340 (2012).
Kerdidani, D. et al. Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat. Commun. 10, 1405 (2019).
Spranger, S. & Gajewski, T. F. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J. Immunother. Cancer 3, 43 (2015).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337. e310 (2018).
Harb, J., Lin, P.-J. & Hao, J. Recent development of Wnt signaling pathway inhibitors for cancer therapeutics. Curr. Oncol. Rep. 21, 1–9 (2019).
Yan, J. et al. Carnosic acid-induced co-self-assembly of metal-peptide complexes into a nanocluster-based framework with tumor-specific accumulation for augmented immunotherapy. Chem. Eng. J. 416, 129141 (2021).
Liu, T. et al. A tumor-targeting metal-organic nanoparticle constructed by dynamic combinatorial chemistry toward accurately redressing carcinogenic Wnt cascade. Small 18, 2104849 (2022).
Parsons, M. J., Tammela, T. & Dow, L. E. WNT as a driver and dependency in cancer. Cancer Discov. 11, 2413–2429 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 32171256 and 82272782) and the National Key Research and Development Program of China (No. 2022YFE0133500), Thousand Talents Plan of Shaanxi Province (W. He), The Young Talent Support Plan of Xi’an Jiaotong University (W. He). We thank Instrument Analysis Center of Xi’an Jiaotong University for their assistance with TEM, DLS, FT-IR, and XPS analysis. We also appreciate the help of proteomic analysis and Single-cell RNA sequencing analysis from Tgene Biotech (Shanghai) Co., Ltd.
Author information
Authors and Affiliations
Contributions
W.H. and J.Y. designed the study and analyzed the data. H.L., J.W., S.Y., N.L., T.L., Z.L., and W.Y. performed the experiments. W.H. wrote the manuscript. J.Y. revised the manuscript. W.H. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dennis Della Corte, Ron Firestein, Fajun Zhang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, J., Liu, H., Yang, W. et al. Small-molecule-induced liquid-liquid phase separation suppresses the carcinogenesis of β-catenin. Nat Commun 16, 5997 (2025). https://doi.org/10.1038/s41467-025-61112-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61112-6
