
Abstract
Propane dehydrogenation (PDH) is a critical technology for propylene production, yet overcoming the trade-off between activity and stability remains a major challenge. Here, we engineer a robust Pt@Sn-MFI catalyst with a wormhole-type structure, featuring highly dispersed Pt clusters robustly anchored by open sites in Sn-MFI, i.e., [SiO]3 − Sn−O−Ptn, complemented by abundant zeolite defects (i.e., Si-OH) in the proximity. This architecture enables a near-thermodynamic equilibrium conversion and a propylene selectivity of ≥98.5%, with the high apparent forward rate coefficient of 1064.5 molC3H6 gPt−1h−1bar−1 and stability for at least 120 h without requiring H2 or CO2 co-feeding. Comprehensive characterization, isotope-labeling experiments and theoretical calculations reveal a plausible hydroxy-assisted PDH reaction pathway, wherein the synergy between Pt sites and neighboring hydroxyl groups (i.e., zeolite defects) significantly reduces the energy barrier for H2 formation via the combination of H in propane adsorbed on Pt sites with H in hydroxyl groups, thereby promoting the PDH process.
Similar content being viewed by others


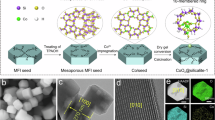
Introduction
Propane dehydrogenation (PDH) is a key process for converting propane directly into propylene, an essential chemical widely used in the industry1,2,3. Catalysts featuring platinum (Pt) as the active sites have been thoroughly researched for PDH due to their efficiency in activating paraffinic C-H bonds and their low tendency for C-C cleavage4. Given the intensely endothermic nature of PDH, it typically requires temperatures above 500 °C for operation. As a result, Pt sintering and coke formation tend to occur, which restricts the propylene selectivity and reduces the lifespan of the catalyst.
These challenges can be partially mitigated by incorporating a second metal, such as Sn5, Zn6, Ga7, Y8, Cu9, and Fe10, into the catalyst. The addition of the second metal (M) not only enhances Pt dispersion geometrically to prevent sintering but also tailors the electronic states of Pt to diminish side reactions that lead to coke formation. Despite these advantages, some PtM catalysts have been found to exhibit compromised PDH activity or undergo irreversible catalyst deactivation11,12.
Zeolites, as catalyst supports, can fine-tune the geometric and electronic structure of the active metal species to optimize their catalytic performance13. Zeolite-supported Pt-based bimetallic PDH catalysts, particularly those pairing Pt with Sn, have shown superior efficacy due to the advantages provided by the zeolite4,14. For all documented Pt-Sn-zeolite catalysts, Sn and Pt are introduced concurrently or sequentially onto pre-synthesized zeolites5,12,14,15,16,17,18,19,20, where Sn may form alloys with Pt14,21 or exist as discrete cations embedded within the zeolite framework18,19,22,23. In some catalysts, extra-framework SnOx species are also detected5,16,17. However, complex catalyst preparation procedures and the consequent diversity of Sn species pose challenges in identifying the catalytically active sites.
Hydroxyl species on the support surface have exhibited a synergetic effect with metal sites, thereby enhancing catalytic activity for hydrogenation24 and oxidative dehydrogenation reactions25. However, similar effects have not been discovered for PDH, although structural defects in zeolites, particularly silanol groups (Si-OH), have been utilized to modulate the forms of the supported metal species26,27. It is therefore expected that defect engineering of zeolite holds the potential to render efficient catalysts that can overcome the activity-stability trade-off in PDH.
In this work, we prepared a pure siliceous zeolite MFI-supported Pt-Sn catalyst, denoted as Pt@Sn-MFI, by a modified ligand-protected one-pot hydrothermal synthesis method followed by direct H2 reduction. The introduction of Sn in the zeolite synthetic system results in the creation of worm-like holes in the MFI crystals, wherein the Pt preferentially reside as ultrafine clusters. Concurrently, the integrated Sn species primarily exist at structural defects, serving as anchoring sites for the ultrafine Pt clusters by forming [SiO]3 − Sn−O−Ptn complexes, accompanied by abundant silanol groups (i.e., Si-OH) in the proximity.
The Pt@Sn-MFI catalyst can operate at close to thermodynamic equilibrium with high propylene selectivity of ≥98.5%, with the top-level apparent forward rate coefficient of 1064.5 molC3H6 gPt−1h−1bar−1, under a gas hourly space velocity (GHSV) of 9600 mL gcat−1 h−1 and 550 °C. Furthermore, long-term testing over 120 h indicated a high inverse deactivation coefficient of 844.2 h. Remarkably, it can achieve a high specific activity for propylene production, reaching 95.4 molC3H6 gcat−1 h−1, at a GHSV of 85700 mL gcat−1 h−1 and 600 °C. These performances indicate its superiority over various state-of-the-art catalysts, including PtSn/Al2O3, which is used in commercial PDH processes. Moreover, beyond PDH, Pt@Sn-MFI also exhibits high activity and exceptional stability for ethane dehydrogenation (EDH), showing no noticeable deactivation over 720 h of operation.
Comprehensive characterization, isotope-labeled experiments, and density functional theory (DFT) calculations collectively reveal that silanol groups (i.e., Si-OH) around the [SiO]3 − Sn−O−Ptn sites contribute to the catalytic activity by significantly facilitating C-H bond cleavage through cooperative interaction with Pt by a plausible hydroxy-assisted PDH reaction pathway. Utilizing an almost defect-free MFI as a support for PtSn resulted in a catalyst that showed significantly lower PDH activity compared to Pt@Sn-MFI, underscoring the essential role of silanol groups in enhancing this reaction. Additionally, the strong electronic interactions between ultrafine Pt clusters and atomically dispersed Sn, orchestrated by the Sn-O-Pt structure, are crucial for achieving high propylene selectivity and catalytic stability.
Results
Catalyst structure identification
Figure 1a shows the power X-ray diffraction (XRD) patterns of MFI, Pt@MFI, and Sn-containing MFI zeolites, all of which display well-defined crystalline features typical of MFI topology. The introduction of Sn and Pt led to a slight reduction in relative crystallinity. Notably, Sn-containing MFI exhibited a low 2θ shift relative to pure MFI in inserted images, suggesting unit cell expansion from the substitution of smaller Si atoms by Sn28. Chemical coordination of Sn species in the zeolite framework was investigated using Diffuse Reflectance Ultraviolet-Visible Spectroscopy (DR UV-Vis), as depicted in Fig. 1b. Both Sn-MFI and Pt@Sn-MFI displayed a distinct ligand-to-metal charge transfer band (O2−→Sn4+) centered at 210 nm, indicative of tetrahedrally coordinated framework Sn species29,30,31. Extra-framework SnO2 nanoclusters (band centered at >250 nm), although detected, appeared faintly but in negligible amounts. Quasi-in situ X-ray Photoelectron Spectroscopy (XPS) was utilized to analyze the chemical state of Sn species, as shown in Fig. 1d. The binding energy at 487.0 and 495.5 eV correspond to the 3d5/2 and 3d3/2 photoelectrons of tetrahedrally coordinated framework Sn species, confirming their integration into the zeolite framework in both Sn-MFI and Pt@Sn-MFI, consistent with previous studies19,28,32. In contrast, no signals corresponding to octahedrally coordinated extra-framework Sn species, typically observed at 486.0 and 494.4 eV, were detected19,33,34. This finding aligns with the aforementioned results from XRD and DR UV-Vis, further confirming the effective incorporation of Sn into the MFI framework.

a XRD patterns. b DR UV-Vis spectra. c Quasi-in situ Sn 3 d XPS spectra.
Furthermore, it is noteworthy that nonmetallic Sn species were found in Pt@Sn-MFI, indicating that the strong interaction between Sn and the zeolite framework stabilizes Sn in an oxidized state, preventing its reduction and subsequent alloy formation with Pt, even after interacting strongly with Pt species18,19. This differs from previous findings where a fraction of Sn could be reduced to metallic Sn0, forming an alloy or bimetallic cluster with Pt5,14,18,35.
The N2 adsorption-desorption isotherms of MFI zeolite materials are depicted in Fig. 2a. The isotherm of the Pt@MFI exhibited a typical type I, as per IUPAC classification, characteristic of microporous materials. Intriguingly, the isotherms of the Sn-MFI and Pt@Sn-MFI displayed a combination of type I and type IV, indicating the coexistence of microporous and disordered mesoporous domains. This observation, combined with the above discussion on the isomorphic substitution of Sn for tetrahedral Si, implies that the introduction of Sn into the synthetic gel significantly disrupts the regularity of the MFI structure by interacting with Si species during the crystallization process, resulting in the formation of disordered mesopores36,37,38. The resulting structural irregularity was reflected in the rougher morphology of Sn-containing zeolites, as observed in SEM images (Supplementary Fig. 1), while the presence of disordered mesopores was further confirmed by subsequent (S)TEM analysis. In addition, the slightly lower specific surface area of Pt@Sn-MFI compared to Sn-MFI, along with subsequent (S)TEM images, suggests that the highly dispersed Pt was confined within the zeolite channels, similar to Pt in Pt@MFI.

a N2 adsorption and desorption isotherms. b In-situ DRIFTS spectra with He flow. c119Sn MAS NMR spectra. The asterisk “ *” denotes spin-side bands in the spectrum of SnO2. d TEM image. e-f HAADF-STEM images. g Cluster size distribution of Pt@Sn-MFI.
Figure 2b illustrates the characteristic in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) spectra of MFI, Sn-MFI, and Pt@Sn-MFI zeolites in the range of 4000−3200 cm−1 under He flow. Three notable features were observed in this range. Firstly, the bands ranging from 3200−3600 cm−1 (centered at ~3460 cm−1) are typically attributed to silanol nests, involving multiple silanol groups interacting through extended hydrogen bonding, commonly occurring at crystal steps39,40. Secondly, the well-known band at 3740 cm−1 corresponds to isolated silanol groups, while the band at 3723 cm−1 corresponds to isolated stannol groups39,41. Notably, Sn-MFI exhibited a higher abundance of Si−OH/Sn−OH defects compared to MFI, indicating that the doping of Sn into the zeolite framework leads to the formation of a significant number of MFI defects. It is well consistent with the results from the N2 adsorption and desorption isotherm. These defects (i.e., Sn−OH/Si−OH) serve as anchoring sites for Pt species42,43, as further corroborated by the IR spectra of Pt@Sn-MFI with a lower absorption intensity of -OH groups in the 4000−3200 cm−1 range.
119Sn solid-state NMR experiments were employed to unambiguously characterize the Sn sites in Sn-containing zeolites. Enhanced NMR sensitivity was achieved by synthesizing ¹¹⁹Sn-enriched Pt@Sn-MFI using an EDTA-119Sn complex as the Sn precursor44. Generally, broad resonances appeared at ~400 ppm, which are assigned to framework Sn sites (i.e., open sites ((SiO)3 − Sn−OH) and closed sites ((SiO)4 − Sn))45,46. A resonance at ~608 ppm corresponds to octahedrally coordinated SnO2 aggregates, while the resonance at ~740 ppm is related to hexa-coordinate open Sn sites in the presence of water molecules. The octahedrally coordinated open Sn sites originate from the Sn−O−Si bonds of the closed Sn sites, which are broken by the attack of water molecules, forming open Sn sites with Sn−OH groups44. As revealed by 119Sn NMR spectra in Fig. 2c, the Sn coordination state of the Sn-MFI contained three types of tetrahedral-coordinated framework Sn site (i.e., (SiO)3 − Sn−OH and (SiO)4 − Sn) and an octahedral-coordinated framework open Sn site. However, for Pt@Sn-MFI, the characteristic resonance band of framework Sn sites in contrast to Sn-MFI weakened and even disappeared, while a new resonance band emerged at ~285 ppm, implying that Pt can be anchored by the open framework Sn sites (i.e., (Si−O)3Sn − O−Pt)19. The formation of the (Si−O)3Sn − O−Pt was further supported by the in-situ DRIFTS spectra (Supplementary Fig. 2), where a diminished signal of the framework Sn sites in Pt@Sn-MFI was observed compared with that of Sn-MFI using CD3CN as a probe molecule.
Figure 2d displays a TEM image of the Pt@Sn-MFI, showcasing a wormhole-type hierarchical pore structure without clearly visible metal nanoparticles. However, in HAADF-STEM images (Fig. 2e–g), no Pt nanoclusters were observed at the edges of the zeolite crystals. Instead, ultrafine Pt clusters with an average size of 1.1 nm were observed in areas away from the crystal edges, confined within the wormhole-type hierarchical pores of the MFI zeolite (indicated by the red area in Fig. 2e–f). This is smaller than the Pt clusters observed in Pt@MFI, which had an average size of 1.6 nm (Supplementary Fig. 3).
To obtain more reliable evidence on the interaction between Pt and framework Sn, fine structural details were elucidated using X-ray absorption spectroscopy (XAS) technique. Figure 3a displays the Pt L3-edge X-ray near-edge structure (XANES) spectra of Pt-MFI, Pt@Sn-MFI and references (i.e., Pt foil and PtO2). The XANES spectra indicated that the majority of Pt species in Pt@MFI and Pt@Sn-MFI catalysts were reduced to metallic Pt, as evidenced by the similar oscillation curves to the Pt foil. Correspondingly, the extended X-ray absorption fine structure (EXAFS) spectra (Fig. 3b) of these catalysts were dominated by a component at 2.75 Å, signifying Pt-Pt scattering. Notably, the higher intensity of white line in Pt@Sn-MFI, indicating a strong Pt-O-Sn interaction, aligns with the slightly higher Pt binding energy (71.2 eV) observed in the XPS spectrum of Pt 4 f compared to Pt foil (71.0 eV) in Supplementary Fig. 4. The interaction could be further supported by EXAFS spectroscopy, which identified a weak amplitude at 1.6 Å, indicative of Pt−O scattering due to a robust Pt-O-Sn interaction in the Pt@Sn-MFI. These findings further suggest that the integrated Sn species primarily exist at structural defects, serving as anchoring sites for the ultrafine Pt clusters by forming [SiO]3 − Sn−O−Ptn complexes. Additionally, the strong interaction, reflecting in electronic interactions, was also attested in the in-situ DRIFTS spectra using CO as a probe molecule (Fig. 3d). In Pt@Sn-MFI, the dominant peak corresponding to CO linear adsorption, located at 2082 cm−1, occurred in red-shifted compared to Pt@MFI (2088 cm−1), indicating the existence of electronic interactions between Pt and integrated Sn species. This interaction involves Sn donating electrons to Pt via O, resulting in an enrichment of the surface electron density of Pt and enhanced back-donation of electrons to the antibonding π* orbitals of CO. Furthermore, the absence of characteristic peaks of bridge-adsorbed CO in the range of 1800−1900 cm−1 for Pt@Sn-MFI indicated that Sn facilitates the dispersion of Pt and prevents the formation of bridge-adsorbed CO. These findings are consistent with the weaker amplitude observed at 2.75 Å in Pt@Sn-MFI, suggesting the presence of small-sized Pt clusters.

a XANES spectra. b EXAFS spectra. c WT-EXAFS spectra. d In-situ DRIFTS spectra using CO as a probe molecule. e, f Fitting EXAFS curves. e Pt@MFI; f, Pt@Sn-MFI. In each panel, the upper and lower lines are the FT magnitude and imaginary components, respectively. g Schematic atomic model of Pt@Sn-MFI. Yellow, red, white, gray, and navy-blue spheres represent Si, O, H, Sn and Pt atoms, respectively.
Wavelet Transform (WT) analysis of the EXAFS spectra was employed to differentiate overlapping atomic contributions in the catalysts47. Fig. 3c shows the WT-EXAFS contour plots of Pt foil, Pt-MFI and Pt@Sn-MFI. Similar to Pt foil, the most intense signal in the WT-EXAFS magnitudes of the Pt-MFI and Pt@Sn-MFI was centered at (10.5 Å−1; 2.75 Å), corresponding to the Pt−Pt scattering contribution. No shift of the most intense signal was observed for Pt@MFI versus Pt@Sn-MFI. Combined with the XPS results, it can be safely concluded that there is no PtSn alloy or PtSn bimetallic cluster in Pt@Sn-MFI.
Further quantitative analysis using least-squares EXAFS fitting revealed distinct differences between the catalysts. Unlike Pt@MFI, where only one Pt-Pt pathway was used for fitting (Fig. 3e), Pt@Sn-MFI included two paths (Pt−O and Pt−Pt) in the fitting due to the strong interaction of Pt with integrated Sn species in Pt@Sn-MFI (Fig. 3f). The corresponding fine structure parameters were summarized in Table 1. The attempt to fit Pt@Sn-MFI using only one Pt−Pt path in the EXAFS analysis resulted in a higher R-factor value of 0.014 (Supplementary Fig. 5 and Supplementary Table 1). The average Pt−Pt coordination numbers (CN) of Pt@MFI (RPt-Pt = 2.76 Å) and Pt@Sn-MFI (RPt-Pt = 2.76 Å) were 8.8 and 6.7, corresponding cluster sizes are ~1.6 nm and ~1.1 nm, respectively, assuming spherical nanocluster shapes48. The average size was consistent with STEM measurements (Fig. 2g and Supplementary Fig. 3d). In addition to the Pt−Pt shell, Pt in Pt@Sn-MFI was bonded to approximately one O atom from integrated Sn species in the first coordination shell (CNPt-O = 0.5) with a bond length of 2.04 Å. Based on the comprehensive spectral characterizations (e.g., XAFS, NMR, DRIFTS, etc.), we propose a probable atomic-level structure model for Pt@Sn-MFI, which combines the Pt structure ([SiO]3 − Sn−O−Ptn) with abundant zeolite defects (i.e., Si-OH) in the proximity, as shown in Fig. 3g.
Catalytic performance
Various catalysts for PDH were evaluated at 550 °C with a gas hourly space velocity (GHSV) of 9600 mL gcat−1 h−1 (10.0 vol.% C3H8 in N2), without H2 or CO2 co-feeding. Under these reaction conditions, the thermodynamic limit for propane conversion is 62.6% (Supplementary Fig. 6). In Fig. 4a, a high initial near-thermodynamic propane conversion (59.2 %) and high selectivity of propylene (98.5 %) were achieved over Pt@Sn-MFI. The catalytic performance of Pt@Sn-MFI was significantly superior to those of Pt@MFI (Con. 30.1 % & Sel. 95.1 %) and Sn-MFI (Con. 2.5 % & Sel. 91.3 %). The negligible catalytic performance of Sn-MFI and the high catalytic performance of Pt@MFI suggest that Pt primarily determines the dehydrogenation of propane rather than Sn. The unique and robust [SiO]3–Sn–O–Ptn structure in the Pt@Sn-MFI remarkably improves the propane conversion and propylene selectivity, attributed to the geometric and electronic effects of Sn. The small-sized Pt clusters not only provide more available active Pt sites but also effectively suppress side reactions, such as structure-sensitive C–C cracking and coke deposition1. Simultaneously, Sn contributes by donating electrons to Pt, which weakens the bonds of Pt-(C-C-C) and repels adsorbed propylene, thereby enhancing propylene selectivity. After 12 h on stream, Pt@Sn-MFI maintained a conversion of 59.2% and increased selectivity to 99.2%, with a consistent propylene yield of 58.7%. In contrast, the propane conversion and propylene yield over Pt@MFI rapidly decreased to 6.9 % and 6.7 % within 12 h.

a Various catalysts. b Different ratio of Pt with Sn. The Pt@Sn-MFI_3 marked here refers to Pt@Sn-MFI. c Different GHSV over Pt@Sn-MFI at 550 °C. d Long-term stability test for PDH conversion over Pt@Sn-MFI. e Comparison of the catalytic performance of the state-of-the-art catalysts. Reaction conditions of (a−d) catalyst mixed with 0.5 g of SiC, ambient pressure, GHSV = 9600 mL gcat−1h−1, 550 °C.
To elaborate on the impact of Sn content in Pt@Sn-MFI on catalytic performance, Pt@Sn-MFI_x with different Sn/Pt ratios (x = 1, 3, and 6) were synthesized while maintaining Pt content at 0.4 wt.%. TEM analysis (Supplementary Fig. 7) showed increasingly rougher MFI morphology with higher Sn content, with wormhole-type structures apparent at Sn/Pt ratios ≥3. As shown in Fig. 4b, catalytic tests identified a Sn/Pt ratio of 3 as optimal. At a Sn/Pt ratio of 6, the catalyst displayed a lower initial propane conversion of 57.8% and higher selectivity of 99.1% under the same reaction condition (Fig. 4b), which further increased to 99.7% selectivity after 12 h, but conversion dropped to 49.5%. This decline in conversion may be attributed to poor crystallinity of the MFI structure (Supplementary Fig. 8a) and excessive Sn (e.g., extra-framework Sn species, as indicated by DR UV-Vis, HAADF-STEM image and EDS-Mapping in Supplementary Figs. 8b–e) partially covering active Pt sites, while the rise in selectivity results from stronger Pt-Sn electronic interactions enhancing propylene desorption. Despite the high propylene selectivity of Pt@Sn-MFI_6, its propylene yield decreased from 54.2−49.3 %. In contrast, the catalyst with a low Sn/Pt ratio of 1, showed a similar initial propane conversion as the optimal catalyst (i.e., Pt@Sn-MFI) but exhibited lower propylene selectivity. This decrease in selectivity corresponds with weaker electronic interactions between Pt and Sn. During the reaction, the propylene yield of Pt@Sn-MFI_1 decreased from 57.5−55.2%. Notably, the absence of wormhole-type structures in Pt@Sn-MFI_1, which are conducive to effective mass transfer, might also contribute to its performance decline49,50 (Supplementary Fig. 7b). To further confirm the promoting effect of the wormhole-type structure on propane dehydrogenation, the 1/3Pt@Sn-MFI_3 was synthesized, maintaining the optimal Sn/Pt ratio of 3 but reducing the Sn content to 1/3 of the original. This modification led to the disappearance of the wormhole-type structure (Supplementary Fig. 7a) and was accompanied by a decrease in the propylene yield from 52.8−46.4%, reinforcing the significance of the structural features in enhancing catalytic performance.
Moreover, the catalytic performance of Pt@Sn-MFI for PDH was evaluated at 550 °C under different GHSVs, as shown in Fig. 4c. As the GHSVs increased from 9600−33000 mL gcat−1 h−1, the initial propane conversion decreased from 59.2 %−51.1%, while the initial propylene selectivity slightly increased from 98.5%−99.1%. Notably, the specific activity of propylene formation reached 19.6 molC3H6 gPt−1 h−1 at a GHSV of 33000 mL gcat−1 h−1, and further increased to 95.4 molC3H6 gPt−1h−1 at 600 °C with a GHSV of 85700 mL gcat−1 h−1. These values signify exceptionally high catalytic activities for propylene production (see Fig. 4e and Supplementary Table 2). Impressively, even after 120 h of continuous operation at a GHSV of 9600 mL gcat−1 h−1, the propane conversion remained at 55.7 % with 99.4 % selectivity for propylene, affording an extremely high inverse deactivation coefficient of 844.2 h (Fig. 4d). The overall performance of the catalyst surpasses the majority of state-of-the-art Pt-based catalysts (See Supplementary Table 2 for details). Considering the challenges of comparing catalytic activities across different studies due to variations in experimental conditions (e.g., temperature, propane partial pressures, and approach to equilibrium)23, all catalysts were compared based on the PDH apparent forward rate coefficient. The results in Supplementary Table 2 showed that Pt@Sn-MFI exhibited an impressive apparent forward rate coefficient of 1064.5 molC₃H₆ gPt⁻¹ h⁻¹ bar⁻¹ at 550 °C.
Additionally, the stability of the Pt@Sn-MFI catalyst was assessed under near-industrial reaction conditions (i.e., T = 550 °C, pure C3H8, GHSV = 9600 mL gcat−1h−1), as well. As shown in Supplementary Fig. 9, the Pt@Sn-MFI catalyst exhibited no significant signs of deactivation over 80 h of continuous operation. The HAADF-STEM image and corresponding Pt cluster size dispersion of the spent Pt@Sn-MFI catalyst following the pure propane dehydrogenation experiment showed no obvious aggregation of the Pt nanoclusters (Supplementary Fig. 10), demonstrating their robust structure and excellent anti-sintering properties.
Given the importance of catalyst reusability, we further evaluated its regeneration performance. As shown in Supplementary Fig. 11, after three regeneration cycles, the catalyst retained ~89.9% of its initial propane conversion while maintaining a high propylene selectivity of 99.7%. The HAADF-STEM image of spent Pt@Sn-MFI after three regeneration cycles showed that the majority of Pt species remained as clusters <2 nm in size (Supplementary Fig. 12), indicating minimal sintering and exceptional structural stability.
To eliminate the support effect, PtSn catalysts supported on SiO2 and MFI were synthesized using the traditional impregnation method. The PDH test results reveal superior catalytic performance for Pt@Sn-MFI compared to PtSn/SiO2 (Con. 53.4 % & Sel. 97.7 %). Similarly, the commercial mimic PtSn/Al2O3 catalysts showed moderate activity, with an initial propane conversion of 14.6 % and comparatively lower propylene selectivity of 97.1 %. Notably, although the C3H8 conversion and C3H6 selectivity of Pt@Sn-MFI (59.2% & 98.5%) and PtSn/MFI (58.3% & 98.2%) appear similar under initial reaction conditions, the key advantages of Pt@Sn-MFI lie in its enhanced stability and resistance to deactivation. Specifically, during 12 h of continuous operation, Pt@Sn-MFI maintained a stable C3H8 conversion of 59.2%, whereas PtSn/MFI showed a noticeable decline from 58.3−52.8%, indicating its higher susceptibility to deactivation. These results collectively underscore the outstanding catalytic efficacy of Pt@Sn-MFI for PDH, especially its notably high specific activity for propylene formation and robust long-term stability.
A parallel investigation was conducted for ethane dehydrogenation (EDH) at 550 °C, using a GHSV of 2400 mL gcat−1 h−1 (10.0 vol.% C2H6 in Ar), without H2 or CO2 co-feeding. Under these reaction conditions, the thermodynamic limit for ethane conversion is 26.3% (Supplementary Fig. 13). The Pt@Sn-MFI achieved a high initial conversion (23.9 %) close to the thermodynamic limit and a high selectivity of propylene (97.4 %) in Supplementary Fig. 14a. Further investigations, adjusting Sn contents (Supplementary Fig. 14b) and varying GHSVs (Supplementary Figs. 14c), confirmed an optimal Sn/Pt ratio of 3, and the catalyst with this ratio reached a remarkable specific activity of 8.4 molC2H4 gPt−1h−1 at a high GHSV of 60,000 mL gcat−1 h−1. Importantly, no deactivation trend was observed for the Pt@Sn-MFI catalyst during the 720 h operation at 550 °C, as indicated by an exceptionally high inverse deactivation coefficient of 4365.3 h (Supplementary Fig. 14d). Similarly, this accomplishment surpasses the catalytic activity reported for EDH, establishing a superhigh level and outperforming the majority of previously reported state-of-the-art EDH catalysts as detailed in Supplementary Fig. 14e and Supplementary Table 3. These results align with the previous results mentioned, confirming the robustness of the Pt@Sn-MFI catalyst.
Catalytic mechanism
In-situ DRIFTS measurements were conducted to investigate the reaction mechanism for PDH over the Pt@Sn-MFI. As shown in Supplementary Fig. 15a, as the PDH reaction proceeds, the -OH vibrational region showed the progressively increased intensity of the inverted peak at 3743 and 3718 cm−1, indicative of the consumption at defective MFI sites, i.e., Si-OH and Sn-OH, especially the former. And in response to the passage of the propane flow (Supplementary Fig. 15b), characteristic bands corresponding to propane (2996, 2912, and ~2861 cm−1) become gradually visible, followed by the appearance and enhancement of propylene bands (3012 and 2950 cm−1) as the reaction progressed51. Notably, upon switching the propane flow to helium, the characteristic bands in the -OH vibrational region disappeared completely within 12 min (Supplementary Fig. 15c), indicating rapid restoration of the MFI defects under helium flow conditions. Moreover, the observed sequence of propylene desorption preceding propane under helium flow purging suggests facile desorption of propylene on the Pt@Sn-MFI catalyst (Supplementary Fig. 15d), leading to an enhanced selectivity for propylene. This is corroborated by faster propylene desorption observed by in-situ DRIFTS spectra of C3H6-desorption experiments on Pt@Sn-MFI (Supplementary Fig. 16). The weak interaction between the electron-rich Pt sites and propylene is pivotal in preventing coking, thereby enhancing propylene selectivity and catalyst longevity. This is underscored by the absence of a clearly visualized characteristic band (1574 cm−1) associated with carbonaceous substances in Supplementary Fig. 17.
The obtained results prompt an exploration into the involvement of zeolite defects, specifically Si-OH, in the PDH process. In-situ DRIFTS spectra of Pt@Sn-MFI, pretreated with H2 and subjected to helium flow, showed the -OH vibrational region’s inverted peaks gradually diminished and disappeared (Supplementary Fig. 18a). H2 temperature-programmed desorption (H2-TPD) of Pt@Sn-MFI reveals the presence of numerous adsorbed H* species on the catalyst surface (Supplementary Fig. 18b). Based on these findings, it can be inferred that the H* chemisorbed by Pt sites spillover to the residual unsaturated O species caused by the combination of H in propane and H in Si-OH to form and release H2.
Isotope-labeled experiments were performed over the Pt@Sn-MFI to further substantiate the involvement of zeolite defects in the PDH process. Initially, an H-D exchange experiment was analyzed using in-situ DRIFTS while mass spectrometry monitored the exhaust gas in real time. As shown in Fig. 5a, the H atoms in the Si-OH of the Pt@Sn-MFI catalyst gradually underwent exchanged with D atoms, forming OD groups under a D2 flow at 550 °C, as evidenced by the inversion (or diminution) of OH bands (3400-3750 cm−1) and the emergence of new OD bands (2500-2800 cm−1)52. The presence of HD in the exhaust gas, monitored by MS (Fig. 5b), further confirms that the formation of OD groups on the catalyst occurs through the spillover of D formed by the dissociation of D2 on the Pt sites. In subsequent experiments, when the D2 bypassed the catalyst, HD was not detected by MS. However, once D2 re-contacted to the catalyst, HD was again detected by MS, confirming that the dissociation of D2 and the H-D exchange reactions occurred exclusively on the Pt@Sn-MFI catalyst. Next, the PDH reaction over the D-labeled Pt@Sn-MFI was recorded through in-situ DRIFTS and MS. In Fig. 5c, the OD bands on the catalyst surface gradually diminished while the OH bands gradually appeared during the PDH reaction. Concurrently, the gradual decrease of HD in the exhaust gas was monitored by MS (Fig. 5d), implying that the involvement of hydroxyl groups in the PDH reaction. Notably, in Fig. 5c, both isolated Si–OH and hydroxyl nests were detected, whereas Supplementary Fig. 15 showed only isolated Si–OH involvement in PDH. This distinction may arise from the facile H–D exchange between the generated H* and D-labeled Pt@Sn-MFI during PDH reaction. To further trace the dehydrogenation process, MS experiments were performed using isotopically labeled propane (C3D8) as the feed gas. As shown in Supplementary Fig. 19a, both D₂ and HD signals were detected upon introducing C3D8. The gradual decrease in HD signal over time, along with the rapid disappearance of both D2 and HD signals when C3D8 bypassed the catalyst, strongly supports the involvement of Si–OH groups in the dehydrogenation process.

a, b H-D exchange experiments on Pt@Sn-MFI. a, In-situ DRIFTS spectra of Pt@Sn-MFI pre-treated with H2 under the D2 flow. b, Mass spectra characterizing H-D exchange as DRIFTS data were recorded. c, d Propane dehydrogenation on D-labeled Pt@Sn-MFI. c, In-situ DRIFTS spectra of D-labeled Pt@Sn-MFI under the C3H8 flow. d, Mass spectra characterizing PDH as DRIFTS data were recorded. e Schematic diagram of the reaction mechanism.
To elucidate the advantage of Si-OH in the PDH reaction, an additional defect-free Pt@Sn-MFI, characterized by an almost negligible presence of hydroxyl groups, was synthesized (Supplementary Figs. 20a-c). Compared with the defect-free Pt@Sn-MFI catalyst, the defect-enriched Pt@Sn-MFI catalyst exhibited stronger signals for C3D6 and HD during C3D8 feeding (Supplementary Fig. 19), indicating that the Si-OH in the Pt@Sn-MFI catalyst facilitates the cleavage of C-D bonds of C3D8 and the formation of HD. Furthermore, the defect-free Pt@Sn-MFI catalyst displayed lower C3H8 conversion (Supplementary Fig. 20d), further confirming the crucial role of zeolite defects in enhancing PDH activity. Combined with the above results, a plausible reaction mechanism (i.e., hydroxy-assisted propane dehydrogenation) is proposed (Fig. 5e): propane is initially adsorbed onto the catalyst surface, followed by interaction with zeolite defects, i.e. Si-OH. This interaction leads to the combination of H from propane with H from Si-OH, releasing H2 and leaving unsaturated O species. Subsequently, propylene is desorbed from the catalyst, leaving behind the Pt-H* species. The spill-over of H* onto the previously unsaturated oxygen species then restores the Si-OH, continuing the catalytic cycle.
Density Functional Theory (DFT) was employed to gain deeper insights into the PDH pathway. Recognizing that experimental techniques such as XAFS and STEM provide only ensemble-averaged structural parameters rather than site-specific configurations, our model was not built to reproduce average values, but to represent a plausible local environment constrained by experimental trends. Guided by this rationale, a representative Sn-doped MFI model rich in hydroxyl groups was constructed by substituting Si atoms with Sn atoms, followed by bonding Pt to Sn via Sn-O-Pt bonds to achieve a stable structure of Sn-doped MFI with bonded Pt, guided by the principle of lowest energy (Supplementary Fig. 21). Notably, the average coordination numbers of Pt-Pt (4.8) and Pt-O (0.3) in the relaxed structure are reasonably consistent with EXAFS-derived averages, falling within their typical uncertainty ranges (20–30%), which supports the model’s structural plausibility. Using this representative structure, we systematically investigated the adsorption of methyl (or methylene) groups onto Pt sites, as well as the subsequent formation and release of H2 involving the dehydrogenation of propane, including the combination of H from the methyl and methylene groups, as well as the combination of H from the methyl or methylene groups with H from neighboring hydroxyl groups on the support material (Fig. 6). In the case of methyl adsorption on Pt sites, the energy relative to the initial state decreased to -1.799 eV. During the subsequent formation and release of H2, it was evident that the energy required for the combination of H from the methyl group in propane with H from neighboring hydroxyl groups (-2.470 eV) was significantly lower than the energy required for the combination of H from the methyl group and H from the methylene group in propane (-1.449 eV). Similar phenomenon was observed for the adsorption of methylene groups on Pt sites, where the energy required for the combination of H from the methylene group in propane with H from neighboring hydroxyl groups (-2.498 eV) was much lower than the energy required for the combination of H from the methyl group and H from the methylene group in propane (-0.615 eV). These results strongly emphasize the crucial role of neighboring hydroxyl groups in the propane dehydrogenation process. Moreover, after the desorption of propylene from the Pt sites (-3.147 eV for methyl_hydroxyl vs. -3.636 eV for methylene_hydroxyl), the H in the remaining Pt-*H species readily spillovers (-2.614 eV for methyl_hydroxyl vs. -2.619 eV for methylene_hydroxyl) to the previously unsaturated oxygen species, regenerating Si-OH. These computational findings are consistent with our experimental observations, providing further support for the proposed hydroxy-assisted reaction pathway.

The values above the solid line represent the energy relative to the initial state (defined as zero). Specifically, in the initial state, free propane was confined within MFI channel. In the propane adsorption stage, the adsorption of the methyl (or methylene) group in propane on Pt sites was considered. In the hydrogen formation and desorption stage, hydrogen from propane’s methyl and methylene groups, as well as from their combination (either methyl or methylene) with adjacent hydroxyl groups, was considered. In the propylene formation stage, the analysis focuses on two pathways (i.e., the methyl_hydroxyl and methylene_hydroxyl) that are considered more likely to occur in the reaction. In the rehydroxylation stage, H originating from propane on Pt sites spillovers to adjacent unsaturated oxygen species, resulting in the regeneration of hydroxyl groups. The green, white, gray, red, yellow, dimgrey and navy-blue spheres represent H in hydroxyl, H in propane, C, O, Si, Sn, and Pt, respectively.
Discussion
In summary, we have developed a robust Pt@Sn-MFI catalyst featuring a wormhole-like structure with enriched MFI defects (e.g., Si-OH), where highly dispersed Pt clusters are robustly anchored by Sn-OH in MFI crystal, forming [SiO]3 − Sn−O−Ptn complexes. This unique catalyst structure exhibits exceptional performance in propane dehydrogenation (PDH), achieving a high propane conversion (59.2 %) close to thermodynamic equilibrium, remarkable propylene selectivity ( ≥ 98.5 %) and superior catalytic stability (inverse deactivation coefficient: 844.2 h) under a GHSV of 9600 mL gcat−1 h−1 at 550 oC, without requiring H2 or CO2 co-feeding. Additionally, it demonstrates an impressive apparent forward rate coefficient of 1064.5 molC₃H₆ gPt⁻¹ h⁻¹ bar⁻¹. The high propylene selectivity and excellent catalytic stability are primarily related to the formation of the robust [SiO]3 − Sn−O−Ptn complexes, which facilitate electron-rich environments around Pt clusters via Sn donation. The impressive activity can be attributed to the synergistic interaction between active Pt sites and adjacent hydroxyl groups. Through a combination of comprehensive characterization, isotope-labeled experiments, and DFT calculations, we propose a plausible hydroxy-assisted PDH reaction pathway that effectively reduces the energy required for H2 formation by combining H in propane adsorbed on Pt sites with H of the adjacent hydroxyl group from zeolite defects, thereby significantly promoting PDH process. Our work not only highlights the synergistic interaction potential of metal sites with hydroxyl groups on support in heterogeneous catalysis but also paves the way for further advancements in the design of high-performance catalyst systems.
Methods
Catalyst synthesis
Synthesis of MFI zeolite
Nanosized MFI zeolite was prepared by a one-pot synthesis. Typically, the synthetic gel contains an aqueous tetrapropylammonium hydroxide (TPAOH) solution with components of 3.69 g of 20 wt% TPAOH (Sigma-Aldrich, containing ~0.6 wt% of K) and 3.0 g of 40 wt% TPAOH (Alfa-Aesar without K), 5.6 g of tetraethyl orthosilicate, 1.0 g of urea, and 0.05 g of isopropyl alcohol. The resulting solution was placed in a Teflon-lined autoclave for crystallization at 180 °C for 48 h under stirring at 50 rpm. The resulting crystals were collected by filtration, washed with deionized water and acetone, and dried at 60 °C. Subsequently, the solid sample was calcined under a gas mixture of 10 vol% H2 in Ar at 550 °C for 2 h with the heating rate of 2 °C min−1 to obtain the nanosized MFI zeolite. The content of K in MFI is negligible, approximately 0.04 wt%, which is different from previous work53, likely attributed to the different procedure of zeolite preparation.
Synthesis of Pt@MFI
Pt nanoclusters embedding in nanosized MFI zeolites were prepared by a one-pot synthesis. Typically, a solution consisting of 100 μL H2PtCl6 aqueous (0.44 mol L−1) containing 18.6 mg of EDTA was added to an aqueous mixture. The aqueous mixture contained tetrapropylammonium hydroxide (TPAOH) solution with components of 3.69 g of 20 wt% TPAOH (Sigma-Aldrich, containing ~0.6 wt% of K) and 3.0 g of 40 wt% TPAOH (Alfa-Aesar without K), 5.6 g of tetraethyl orthosilicate, 1.0 g of urea, and 0.05 g of isopropyl alcohol. The resulting yellow solution was stirred for 30 min and then transferred to a Teflon-lined autoclave for crystallization at 180 °C for 48 h under stirring at 50 rpm. The resulting crystals were collected by filtration, washed with deionized water and acetone, and dried at 60 °C. Subsequently, the solid sample was reduced under a gas mixture of 10 vol% H2 in Ar at 550 °C for 2 h with the heating rate of 2 °C min−1 to obtain the Pt@MFI catalyst. The amount of Pt in Pt@MFI is 0.4 wt%.
Synthesis of Sn-MFI
Nanosized Sn-containing MFI zeolite with a wormhole-like hierarchical pore structure was prepared by a one-pot synthesis. The synthesis of Sn-MFI followed the same procedure as the Pt@MFI catalyst, with the exception that 17.5 μL of SnCl4 aqueous solution containing EDTA was used instead of H2PtCl6 aqueous solution. The amount of Sn in Sn-MFI is 0.9 wt%.
Synthesis of Pt@Sn-MFI
Nanosized Pt@Sn-MFI with a wormhole-like hierarchical pore structure was prepared by a one-pot synthesis. Typically, 100 μL of H2PtCl6 aqueous (0.44 mol L−1) containing 18.6 mg of EDTA and 17.5 uL of SnCl4 aqueous containing 58.8 mg of EDTA were added to an aqueous mixture containing tetrapropylammonium hydroxide (TPAOH) solution with components of 3.69 g of 20 wt% TPAOH (Sigma-Aldrich, containing ~0.6 wt% of K) and 3.0 g of 40 wt% TPAOH (Alfa-Aesar without K), 5.6 g of tetraethyl orthosilicate, 1.0 g of urea, and 0.05 g of isopropyl alcohol. The resulting solution was stirred for 30 min and then transferred to a Teflon-lined autoclave for crystallization at 180 °C for 48 h under stirring at 50 rpm. The obtained crystals were collected by filtration, washed with deionized water and acetone, and dried at 60 °C. The solid sample was then reduced under a gas mixture of 10 vol% H2 in Ar at 550 °C for 2 h with the heating rate of 2 °C min−1 to obtain the Pt@Sn-MFI catalyst. The amount of Pt and Sn in Pt@Sn-MFI is 0.4 and 0.8 wt%, respectively. The Sn loading in Pt@Sn-MFI can be tuned by varying the amount of SnCl4 in the synthetic gel, while keeping the other conditions constant.
Synthesis of defect-free PtSn/MFI
The defect-free Pt@Sn-MFI was prepared by a modified hydrothermal synthesis method that minimizes the formation of framework defects by incorporating fluoride ions (F⁻) into the synthesis gel. Typically, 100 μL of H2PtCl6 aqueous (0.44 mol L−1) containing 18.6 mg of EDTA and 17.5 uL of SnCl4 aqueous containing 58.8 mg of EDTA were added to an aqueous mixture containing TPAOH solution with components of 3.69 g of 20 wt% TPAOH (Sigma-Aldrich, containing ~0.6 wt% of K) and 3.0 g of 40 wt% TPAOH (Alfa-Aesar without K), 5.6 g of tetraethyl orthosilicate, 0.35 g of HF, and 0.08 g of seeds. The resulting solution was stirred, transferred to a Teflon-lined autoclave, and crystallized at 170 °C for 7 days. The obtained crystals were filtered, washed, and dried, followed by reduction under 10 vol% H₂ in Ar at 550 °C for 2 h to obtain the defect-free Pt@Sn-MFI.
Synthesis of PtSn/MFI
The PtSn/MFI catalyst was prepared by incipient wetness impregnation. The PtSn mixture was obtained by thoroughly mixing 38.0 uL of H2PtCl6 aqueous (0.44 mol L−1) containing 6 mg EDTA and 6.3 uL of SnCl4 aqueous containing 16.5 mg EDTA. The mixture was then added dropwise to 0.8 g of MFI. After ultrasonic treatment for 2 h and subsequent static treatment for 24 h, the water was removed under vacuum at 60 °C. The PtSn/MFI catalyst was finally obtained after treatment at 550 °C for 2 h under a gas mixture of 10 vol% H2 in Ar.
Synthesis of PtSn/SiO2, PtSn/Al2O3
These reference catalysts were prepared by incipient wetness impregnation. As a typical run for the synthesis of PtSn-based catalysts, H2PtCl6·6H2O and SnCl2·2H2O were added to a 0.1 M HCl solution to achieve a final concentration of 51 mmol L−1for both Pt and Sn ions. A volume of 0.5 mL of the above solution was diluted with water to obtain the required volume for incipient wetness impregnation on 1 g of support (SiO2 and Al2O3). After ultrasonic treatment for 2 h and subsequent static treatment for 24 h, the water was removed under vacuum at 60 °C. The PtSn/SiO2 and PtSn/Al2O3 catalysts were finally obtained after treatment at 550 °C for 2 h under a gas mixture of 10 vol% H2 in Ar.
Characterization
Powder X-ray diffraction (Bruker D8 Advance) patterns were collected using Cu Kα source (λ = 1.5418 Å) at 40 kV and 40 mA to determine the crystalline phases present on the reduced catalyst. Diffuse reflectance UV visible (DR UV-Vis) spectra were recorded on a Varian Cary 5000 spectrophotometer equipped with a diffuse reflectance accessory (Praying Mantis, Harrick) at a scanning rate of 200 nm min−1. Fourier transform infrared (FT-IR) spectra were collected on a Nicolet iS10 FTIR spectrometer with the Thermo Scientific Smart iTX ATR sampling accessory. X-ray photoelectron spectroscopy (XPS) measurements were performed using a Kratos Axis Ultra DLD spectrometer with Al Kα radiation (hν = 1486.6 eV). Before Quasi-in situ XPS analysis, the samples were pre-reduced in a fixed-bed reactor under 10 vol% H2 in Ar for 2 h, then the gas inlet and gas outlet of the reactor were closed by shut-off valves. Afterward, the samples were taken out and loaded onto XPS sample holders in the glove box. All quasi-in situ XPS spectra were calibrated by shifting the detected adventitious carbon C 1 s peak to 284.4 eV. Nitrogen adsorption/desorption isotherms were measured using a Micromeritics ASAP 2420 instrument at 77 K after degassing the sample at 250 °C under vacuum for 6 h. 119Sn magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were recorded with a 400 MHz Bruker AVANCE III spectrometer. To prepare 119Sn-enriched samples, 119Sn precursor solutions were used in a similar route for the synthesis of Sn-MFI and Pt@Sn-MFI. The Pt and Sn contents of samples were determined using an inductively coupled plasma atomic emission spectrometry instrument (ICP-AES, Agilent 5110) after dissolving the samples in an HF solution. Transmission electron microscopy (TEM) images were obtained using a Tecnai Two electron microscope. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were captured using a Titan Themis Z microscope operating at 300 kV. Scanning electron microscopy (SEM) images were acquired using a FEI Teneo microscope.
X-ray absorption experiments at the Pt LIII edge (11,564 eV) were performed at the 1W1B station at the Beijing Synchrotron Radiation Facility (BSRF). Using a Si (111) double-crystal monochromator, the data were acquired in transmission mode using an ionization chamber for Pt foil and in fluorescence excitation mode using a Lytle detector for Pt-containing zeolites. The XANES data were analyzed using ATHENA software, while the Fourier-transformed EXAFS data were analyzed using ARTEMIS software and the wavelet-transformed EXAFS data were analyzed using MATLAB software. Propylene temperature-programed desorption (C3H6-TPD) experiments were conducted using a Micromeritics AutoChem 2950 apparatus. Prior to the TPD experiments, 100 mg of sample was pretreated at 550 °C for 2 h under a gas mixture of 10 vol% H2 in Ar (50 mL/min), following by switching the gas flow was switched to 20 mL min−1 of He flow to eliminate the adsorbed hydrogen completely and the sample was cooled down to 50 °C. C3H6 flow was introduced for 30 min, and subsequently, gaseous and weakly adsorbed C3H6 were purged with He for 60 min. Subsequently, the C3H6-TPD profile was recorded using a thermal conductivity detector at a rate of 10 °C min−1, with the sample heated up to 800 °C.
In-situ Diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) experiments were conducted on a Thermo Scientific Nicolet 6700 spectrometer equipped with a Harrick Scientific DRIFT cell and a mercury−cadmium−telluride (MCT) detector cooled by liquid nitrogen. For in-situ He-DRIFTS, the samples were pre-treated at 350 °C under He flow (50 mL min−1) for 3 h to desorb water and volatile compounds. The spectra of the dehydrated samples were then recorded at 120 °C, with 128 scans and a resolution of 1 cm−1. Background spectra were obtained using the KBr at identical conditions. All spectra were normalized by the peak area of the ν(Si–O–Si) overtone region (∼1700–2100 cm–1). For in-situ CD3CN-adsorbed DRIFTS, the sample was pre-treated at 350 °C under He flow for 3 h to desorb water and volatile compounds. After cooling the sample to the desired temperatures (i.e., 30 and 100 °C), the background spectra were recorded at these temperatures. The sample was then exposed to CD3CN by bubbling at room temperature for 15 min until saturation followed by heating to the target temperature and maintaining for 30 min before measurement. Spectra were recorded after 10 min at each temperature, with 32 scans and a resolution of 4 cm−1. In-situ CO-adsorbed DRIFTS was carried out to study the electronic interaction between Pt and Sn. Typically, the as-synthesized samples were pre-treated at 550 °C under H2 flow (20 mL mim−1) for 2 h. The gas flow was then switched to 20 mL min−1 of He flow to eliminate the adsorbed hydrogen completely and the sample was cooled down to ambient temperature. A background spectrum was collected, followed by the introduction of 5 vol% CO in He at 2 mL mim−1 was then introduced in the cell for 20 min. The cell was purged with 20 mL mim−1 of He for 30 min to desorb weakly adsorbed CO. The spectra were collected with 64 scans and a resolution of 4 cm−1. For in-situ C3H8-adsorbed/desorbed DRIFTS, the synthesized samples were pretreated at 550 °C for 2 h under an H2 flow (20 mL mim−1). The gas flow was then switched to 10 mL min−1 of He flow to completely eliminate adsorbed hydrogen and the samples were cooled to 350 °C. After collecting background spectra, the propane was introduced into the cell. Time-resolved spectra were monitored for 60 min by 64 scans and a resolution of 4 cm−1. Subsequently, the cell was purged with 10 mL mim−1 of He for 60 min, and time-resolved spectra were simultaneously collected by 64 scans and a resolution of 4 cm−1.
Catalytic evaluation
The catalytic PDH performance was evaluated using a four-channel Flowrence XD platform from Avantium, equipped with an online gas chromatograph, at 550 °C under ambient pressure, unless otherwise stated. Catalyst pellets weighing 20 mg and sized between 80-120 mesh, mixed with SiC, were loaded into the reactor and supported by quartz glass wool. Prior to the dehydrogenation reaction, the catalysts underwent a reduction step at 550 °C under a H2 flow of 20 mL min−1 for 2 h. Subsequently, the catalysts were fed with a mixture of 10.0 vol% C3H8 in N2 at a flow rate of 3.2 mL min−1. Unless specifically mentioned, a GHSV of 9600 mL gcat−1 h−1 was used for the reaction. The reaction products were analyzed using an on-line gas chromatograph, equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). The conversion of propane and selectivity to propylene were defined by the following equations based on a carbon atom balance method:
Specific activity of propylene formation was calculated as follows:
where (FC3H8)in is the flow rate of C3H8 inlet; X, Y, mcat and ωpt represent the conversion of C3H8, the selectivity of C3H6, the catalyst weight, and the content of supported Pt in the catalyst respectively.
Catalytic stability was evaluated by the deactivation rate constant (kd, h−1) based on a first-order deactivation model:
Where XC3H8,F and XC3H8,I are the final and initial C3H8 conversions of the PDH reaction, respectively, and t is the time on stream.
The apparent forward rate coefficient (kf) was calculated by the methodology from literature22,37:
Where the Rnet represents the net rate of reaction; PC3H80 is the inlet partial pressure of propane; X is the propane conversion; Ke is the equilibrium constant for PDH, obtained from Aspen Plus software.
Computational methods
The 275-atom MFI zeolite structure was used as the basis for our calculations. The geometric parameters of the model were determined as follows: a = 20.108 Å, b = 19.918 Å, c = 13.392 Å, α = β = γ = 90°. All the calculations were performed under the framework of density functional theory (DFT) implemented in the Vienna ab initio simulation package (VASP 5.4.4)54,55. The electronic exchange-correlation interactions were described using the Perdew-Burke-Ernzerhof (PBE) form of the generalized gradient approximation (GGA)56. In the calculations, the energy cut-off of the plane-wave basis was set to 520 eV. For ionic position relaxation, the Brillouin zone integration was carried out at Gamma point, and for the single point energy calculation, a 2 × 2 × 2 Monkhorst-Pack K-grid was employed. The models were optimized using the conjugate gradient algorithm, with the maximum force of 0.05 eV/Å. The total energy was well converged to <1\(\times {10}^{-5}\) eV.
A four-layer Pt nanocluster (Pt20) was constructed based on the previous literature57. This cluster configuration yields an average Pt–Pt coordination number (CN) of 4.8 and Pt–O CN of 0.3, which closely matches the experimentally EXAFS fitting data within acceptable margins ( ± 20–30%). Upon full relaxation, the Pt nanocluster retained a compact and thermodynamically stable geometry, exhibiting only minor deviations from the initial structure with fixed atomic positions. The final optimized nanocluster had a diameter of ~0.9 nm, closely matching the experimentally observed average cluster size of ~1.1 nm from STEM measurements, thereby validating the structural rationality of the model. Following the experimental guidance, Sn atoms were sequentially introduced into the optimized MFI framework as anchoring sites for the Pt nanoclusters. The entire system was then optimized using the energy minimization principle until a structurally stable model was achieved. Additionally, to realistically accommodate the Pt20 nanocluster within the catalyst’s wormhole-like structure, several Si atoms were deliberately removed from the MFI framework and the remaining dangling bonds were saturated with hydroxyl groups, thus creating sufficient internal space for stable incorporation of the Pt nanocluster. This final optimized model (Si61O156H32Sn6Pt20) exhibited high structural stability and accurately represented the catalytic system. Notably, the framework oxygen atoms coordinated to Sn were observed to play a critical role in stabilizing the Pt nanoclusters. This optimized structural model was then employed as the basis for subsequent mechanistic investigations, in which four distinct reaction pathways were systematically explored.
Data availability
Further data supporting the findings of this study are available in the Supplementary Information. Additional data are available from the corresponding author on request. Source data are provided with this paper.
References
Chen, S., Pei, C., Sun, G., Zhao, Z.-J. & Gong, J. Nanostructured catalysts toward efficient propane dehydrogenation. Acc. Mater. Res. 1, 30–40 (2020).
Otroshchenko, T., Jiang, G., Kondratenko, V. A., Rodemerck, U. & Kondratenko, E. V. Current status and perspectives in oxidative, non-oxidative and CO2-mediated dehydrogenation of propane and isobutane over metal oxide catalysts. Chem. Soc. Rev. 50, 473–527 (2021).
Liu, Z. et al. Highly stable propane dehydrogenation on a self-supporting single-component Zn2SiO4 catalyst. Angew. Chem. Int. Ed. 64, e20241329 (2025).
Sattler, J. J., Ruiz-Martinez, J., Santillan-Jimenez, E. & Weckhuysen, B. M. Catalytic dehydrogenation of light alkanes on metals and metal oxides. Chem. Rev. 114, 10613–10653 (2014).
Liu, L. et al. Structural modulation and direct measurement of subnanometric bimetallic PtSn clusters confined in zeolites. Nat. Catal. 3, 628–638 (2020).
Chen, S. et al. Propane dehydrogenation on single-site [PtZn4] intermetallic catalysts. Chem 7, 387–405 (2021).
Zhang, T. et al. Synergistic mechanism of latinum-GaOx catalysts for propane dehydrogenation. Angew. Chem. 134, e202201453(2022).
Ryoo, R. et al. Rare-earth-platinum alloy nanoparticles in mesoporous zeolite for catalysis. Nature 585, 221–224 (2020).
Han, Z. et al. Propane dehydrogenation over Pt-Cu bimetallic catalysts: the nature of coke deposition and the role of copper. Nanoscale 6, 10000–10008 (2014).
Cai, W. et al. Subsurface catalysis-mediated selectivity of dehydrogenation reaction. Sci. Adv. 4, eaar5418 (2018).
Ma, Y. et al. Germanium-enriched double-four-membered-ring units inducing zeolite-confined subnanometric Pt clusters for efficient propane dehydrogenation. Nat. Catal. 6, 506–518 (2023).
Sun, G. et al. Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation. Nat. Commun. 9, 4454 (2019).
Wang, Y., Wang, C., Wang, L., Wang, L. & Xiao, F.-S. Zeolite fixed metal nanoparticles: new perspective in catalysis. Acc. Chem. Res. 54, 2579–2590 (2021).
Zhu, J. et al. Ultrafast encapsulation of metal nanoclusters into MFI zeolite in the course of its crystallization: catalytic application for propane dehydrogenation. Angew. Chem. Int. Ed. 59, 19669–19674 (2020).
Zhang, Y., Zhou, Y., Tang, M., Liu, X. & Duan, Y. Effect of La calcination temperature on catalytic performance of PtSnNaLa/ZSM-5 catalyst for propane dehydrogenation. Chem. Eng. J. 181-182, 530–537 (2012).
Concepcion, P. et al. The promotional effect of Sn-beta zeolites on platinum for the selective hydrogenation of alpha,beta-unsaturated aldehydes. Phys. Chem. Chem. Phys. 15, 12048–12055 (2013).
Wang, Y. et al. Framework-confined Sn in Si-beta stabilizing ultra-small Pt nanoclusters as direct propane dehydrogenation catalysts with high selectivity and stability. Catal. Sci. Technol. 9, 6993–7002 (2019).
Xu, Z., Yue, Y., Bao, X., Xie, Z. & Zhu, H. Propane dehydrogenation over Pt clusters localized at the Sn single-site in zeolite framework. ACS Catal. 10, 818–828 (2019).
Ma, Y. et al. Skeleton-Sn anchoring isolated Pt site to confine subnanometric clusters within *BEA topology. J. Catal. 397, 44–57 (2021).
Xu, R. et al. Electronic metal-support interactions between zeolite and confined platinum-tin nanoalloy boosting propane dehydrogenation. Appl. Catal. B Environ. Energy 358, 124438 (2024).
Ma, S. & Liu, Z. P. Zeolite-confined subnanometric PtSn mimicking mortise-and-tenon joinery for catalytic propane dehydrogenation. Nat. Commun. 13, 2716 (2022).
Li, J. et al. Silanol-stabilized atomically dispersed Ptδ+-Ox-Sn active sites in protozeolite for propane dehydrogenation. J. Am. Chem. Soc. 146, 24358–24367 (2024).
Lefton, N. G. & Bell, A. T. Effects of structure on the activity, selectivity, and stability of Pt-Sn-DeAlBEA for propane dehydrogenation. ACS Catal. 14, 3986–4000 (2024).
Peng, Y. et al. Molecular-level insight into how hydroxyl groups boost catalytic activity in CO2 hydrogenation into methanol. Chem 4, 613–625 (2018).
Zhou, H. et al. Isolated boron in zeolite for oxidative dehydrogenation of propane. Science 372, 76–80 (2021).
Zhao, D. et al. In situ formation of ZnOx species for efficient propane dehydrogenation. Nature 599, 234–238 (2021).
Qi, L. et al. Propane dehydrogenation catalyzed by isolated Pt atoms in≡ SiOZn–OH nests in dealuminated zeolite Beta. J. Am. Chem. Soc. 143, 21364–21378 (2021).
Xia, C. et al. Confirmation of the isomorphous substitution by Sn atoms in the framework positions of MFI-typed zeolite. Catal. Today 316, 193–198 (2018).
Roy, S., Bakhmutsky, K., Mahmoud, E., Lobo, R. F. & Gorte, R. J. Probing Lewis acid sites in Sn-Beta zeolite. ACS Catal. 3, 573–580 (2013).
Corma, A., Domine, M. E. & Valencia, S. Water-resistant solid Lewis acid catalysts: Meerwein–Ponndorf–Verley and Oppenauer reactions catalyzed by tin-beta zeolite. J. Catal. 215, 294–304 (2003).
Van Der Graaff, W. N., Li, G., Mezari, B., Pidko, E. A. & Hensen, E. J. Synthesis of Sn-Beta with exclusive and high framework Sn content. ChemCatChem 7, 1152–1160 (2015).
Tang, B. et al. Improved postsynthesis strategy to Sn-Beta zeolites as Lewis acid catalysts for the ring-opening hydration of epoxides. ACS Catal. 4, 2801–2810 (2014).
Luo, H. Y., Bui, L., Gunther, W. R., Min, E. & Román-Leshkov, Y. Synthesis and catalytic activity of Sn-MFI nanosheets for the Baeyer–Villiger oxidation of cyclic ketones. ACS Catal. 2, 2695–2699 (2012).
Pachamuthu, M. P., Shanthi, K., Luque, R. & Ramanathan, A. SnTUD-1: A solid acid catalyst for three component coupling reactions at room temperature. Green. Chem. 15, 2158–2166 (2013).
Liu, L. et al. Regioselective generation and reactivity control of subnanometric platinum clusters in zeolites for high-temperature catalysis. Nat. Mater. 18, 866–873 (2019).
Yuan, E. et al. Facile synthesis of Sn-containing MFI zeolites as versatile solid acid catalysts. Microporous Mesoporous Mater. 270, 265–273 (2018).
Liu, X. & Zhu, Z. Synthesis and catalytic applications of advanced Sn-and Zr-Zeolites materials. Adv. Sci. 11, 2306533 (2024).
Kolyagin, Y. G., Yakimov, A. V., Tolborg, S., Vennestrøm, P. N. & Ivanova, I. I. Application of 119Sn CPMG MAS NMR for fast characterization of Sn sites in zeolites with natural 119Sn isotope abundance. J. Phys. Chem. Lett. 7, 1249–1253 (2016).
Barbera, K., Bonino, F., Bordiga, S., Janssens, T. V. & Beato, P. Structure–deactivation relationship for ZSM-5 catalysts governed by framework defects. J. Catal. 280, 196–205 (2011).
Bolis, V. et al. Calorimetric and IR spectroscopic study of the interaction of NH3 with variously prepared defective silicalites: Comparison with ab initio computational data. Appl. Surf. Sci. 196, 56–70 (2002).
Zecchina, A. et al. Silicalite characterization. 2. IR spectroscopy of the interaction of carbon monoxide with internal and external hydroxyl groups. J. Phys. Chem. 96, 4991–4997 (1992).
Medeiros-Costa, I. C. et al. Silanol defect engineering and healing in zeolites: opportunities to fine-tune their properties and performances. Chem. Soc. Rev. 50, 11156–11179 (2021).
Pan, Y. et al. Titanium silicalite-1 Nanosheet-supported platinum for non-oxidative ethane dehydrogenation. ACS Catal. 11, 9970–9985 (2021).
Qi, G. et al. Direct observation of tin sites and their reversible interconversion in zeolites by solid-state NMR spectroscopy. Commun. Chem. 1, 22 (2018).
Bermejo-Deval, R., Gounder, R. & Davis, M. E. Framework and extraframework tin sites in zeolite Beta react glucose differently. ACS Catal. 2, 2705–2713 (2012).
Bermejo-Deval, R. et al. Metalloenzyme-like catalyzed isomerizations of sugars by Lewis acid zeolites. Proc. Natl Acad. Sci. USA 109, 9727–9732 (2012).
Filez, M., Redekop, E. A., Poelman, H., Galvita, V. V. & Marin, G. B. Advanced elemental characterization during Pt–In catalyst formation by wavelet transformed X-ray absorption spectroscopy. Anal. Chem. 87, 3520–3526 (2015).
Frenkel, A. I., Hills, C. W. & Nuzzo, R. G. A view from the inside: complexity in the atomic scale ordering of supported metal nanoparticles. J. Phys. Chem. B 105, 12689−12703 (2001).
Song, S. et al. Recent progress in metal-molecular sieve catalysts for propane dehydrogenation. ACS Catal. 13, 6044–6067 (2023).
Lu, J. et al. Pt-Zn clusters encapsulated in hierarchical MFI zeolite for efficient propane dehydrogenation. Chem. Eng. J. 484, 149369 (2024).
Liu, G., Zhao, Z.-J., Wu, T., Zeng, L. & Gong, J. Nature of the active sites of VOx/Al2O3 catalysts for propane dehydrogenation. ACS Catal. 6, 5207–5214 (2016).
Guan, E. & Gates, B. C. Stable rhodium pair sites on MgO: influence of ligands and rhodium nuclearity on catalysis of ethylene hydrogenation and H–D exchange in the reaction of H2 with D2. ACS Catal. 8, 482–487 (2018).
Liu, L., Lopez-Haro, M., Calvino, J. J. & Corma, A. Tutorial: structural characterization of isolated metal atoms and subnanometric metal clusters in zeolites. Nat. Protoc. 16, 1871–1906 (2021).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Kishi, H. et al. Study of NO oxidation reaction over the Pt cluster supported on γ-Al2O3(111) surface. Curr. Appl. Phys. 12, S110–S114 (2012).
Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (Grant 22408275 to Q. C.), the Baseline Fund (BAS/1/1397-01-01 to J. G.) and KAUST CRG Fund (URF/1/4727-01-01 to Y. H.).
Author information
Authors and Affiliations
Contributions
Q. Cheng conceived the research idea. Q. Cheng performed experiments with assistance from X. Yao, Y. Pan and N. Morlanes. L. Ou and Z. Hu performed DFT calculations. L. Zheng performed the XAFS experiments. E. Abou-Hamad performed NMR experiments. X. Li and Y. Han contributed to discussions. Q. Cheng, Z. Hu and J. Gascon wrote and revised the manuscript with feedback from all authors. Q. Cheng and J. Gascon supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ning Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, Q., Yao, X., Ou, L. et al. Highly efficient catalytic propane dehydrogenation driven by MFI zeolite defect sites. Nat Commun 16, 5696 (2025). https://doi.org/10.1038/s41467-025-61182-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61182-6
This article is cited by
-
Review: Zeolite functional materials synthesized from coal gangue
Journal of Materials Science (2025)
