
Abstract
Single-atom catalysts (SACs) enable atomic-level control over active sites, but orbital-level manipulation to steer catalytic behavior remains challenging. Here, we address this issue through d-orbital engineering of Cu SACs, achieving simultaneous control over coordination geometry (Cu-N3) and high metal loading (33.2 wt%) for direct benzene-to-phenol oxidation with H2O2. The tri-coordinated Cu SAC (Cu-N3-33.2) exhibits the highest performance with 85.8% benzene conversion and a turnover frequency of 680.3 h−1 at 60 oC, ranking it among the best metal-based catalysts. In-situ ATR-IR spectroscopy and DFT calculations reveal that dynamically formed Cu-O intermediates, driven by p-d orbital hybridization between Cu (d orbitals) and O (p orbitals), lower the H2O2 activation barrier by 0.98 eV compared to Cu-N4 sites. High-density atomic Cu sites prevent over-oxidation by consuming singlet oxygen (1O2). This work establishes a dual-parameter optimization paradigm, including orbital configuration and site density, redefining design principles for selective oxidation SACs.
Similar content being viewed by others

Introduction
The benzene oxidation reaction (BOR) holds a pivotal position in organic synthetic chemistry, given that phenol serves as a crucial raw material and chemical intermediate for numerous industrial applications1,2,3. Currently, the traditional three-step cumene process dominates global phenol production due to its established scalability. However, this process suffers from significant drawbacks, including complex production workflows, low phenol selectivity, high energy demand, and considerable environmental impact4,5. Consequently, direct oxidation of benzene to phenol using hydrogen peroxide (H2O2) has emerged as a promising yet challenging topic in the pursuit of green and sustainable chemistry, with profound industrial and societal implications6,7. The development of innovative and efficient catalysts for BOR is therefore of paramount importance.
Single-atom catalysts (SACs) have garnered substantial attention as an emerging class of materials due to their unique electronic structure and maximum atom utilization, which enable exceptional catalytic performance across a diverse range of reactions8,9,10,11,12,13. Furthermore, the atomic dispersion of active sites in SACs facilitates mechanistic investigations, enabling atomic-level insights into structure-activity relationships14. Nevertheless, the inherent variability in the electronic structures of SACs presents challenges in elucidating the reaction mechanism, thereby hindering the systematic study of structure-activity relationships15,16,17,18,19. Among the many factors governing the catalytic performance, the d-orbital of the metal center plays a decisive role. Adjustments to the coordination structure can alter the electronic state and spatial configuration of the exposed d-orbitals, which in turn modulate the adsorption energies, activation barriers, and reaction pathways. Such precise tuning allows for the stabilization of key intermediates and the suppression of undesired side reactions, offering a pathway to enhanced activity and selectivity20,21,22,23,24.
To further optimize SACs, the interplay between metal loading and coordination structure must be systematically explored. Modulating metal loading significantly impacts the intrinsic activity of active sites by altering the local geometry and d-orbital of the metal centers through mechanisms such as spin coupling, electron transfer, and charge redistribution25,26. SACs with distinct coordination structures can be synthesized through controlled pyrolysis, yet this approach introduces complexities such as the formation of structurally diverse active sites and modifications to the carbon substrate, including defect formation. These challenges complicate the identification of original active sites and the mechanistic understanding of catalytic processes. Consequently, the development of high-loading SACs and the systematic investigation of orbital catalysis are essential for guiding the rational design of more effective catalysts and deepening our understanding of atomic-level catalytic mechanisms27,28.
Herein, we resolve these challenges through a self-assembly strategy that decouples coordination engineering from metal loading optimization, enabling the systematic exploration of the structure–activity relationship at the orbital level. The optimized Cu-N3-33.2 catalyst exhibits highly efficient catalytic performance in the selective oxidation of benzene, achieving a high benzene conversion (85.8%) and turnover frequency (680.3 h−1) at 60 oC within 1 h. In addition, the utilization of water as a bi-phase solvent effectively inhibits excessive oxidation of phenol, thereby enhancing selectivity. Mechanistic investigations using quenching experiments and electron paramagnetic resonance (EPR) spectroscopy reveal, that singlet oxygen (1O2) plays a key role in phenol over-oxidation to p-benzoquinone, providing critical insights into the reaction pathway. Furthermore, density functional theory (DFT) calculations and in-situ attenuated total reflection-infrared (ATR-IR) spectroscopy elucidates the mechanism of H2O2 activation and benzene oxidation, highlighting the role of the unique d-orbital environment of Cu-N3 in promoting H2O2 adsorption and activation. This study offers a simple and efficient strategy for the synthesis of high-loading SACs with well-defined coordination structures, advancing both the understanding of reaction mechanisms and the rational design of selective BOR catalysts.
Results
Structural and morphology characterizations of catalysts
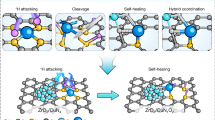
Due to the presence of multiple hydrogen bonds, guanine molecules can act as both hydrogen bond donors and acceptors. This property facilitates the formation of precursors with layered structures through supramolecular self-assembly and π-π interactions (Supplementary Fig. 1)29. Moreover, the abundance of N sites in guanine enables the binding of a large number of metal ions, making it possible to prepare SACs with high metal loading. Inspired by this, a series of Cu SACs (Cu-Nx-y, where x represents the coordination number and y denotes the Cu content) were prepared via a two-step process involving the self-assembly of guanine molecules and a subsequent high-temperature carbonization process (Fig. 1a), where the contents of Cu SACs and their coordination numbers can be easily tuned by adjusting the amounts of added Cu2+ ions. Meanwhile, the neat N-doped carbon (NC) was also prepared using a similar process without the addition of Cu2+ ions. As shown in Supplementary Fig. 2a, b, the powder X-ray diffraction (PXRD) patterns and Fourier Transform Infrared (FT-IR) spectra of guanine self-assembly precursors (Cu-Nx-y-p) with different Cu2+ contents closely resemble those of pristine guanine, indicating that the presence of Cu2+ does not affect the structural integrity of guanine. Subsequently, Cu-Nx-y was prepared through a high-temperature pyrolysis process under N2 environment. Based on inductively coupled plasma optical emission spectrometry (ICP-OES) analysis

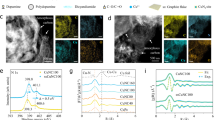
a Schematic illustration for the synthesis of Cu-Nx-y catalysts. b TEM image of Cu-N3-33.2. HAADF-STEM images of (c) Cu-N4-3.3 and (d) Cu-N3-33.2. e EDX elemental mapping images of Cu-N3-33.2. All images in Fig. 1a are original drawings by the authors, created using Autodesk 3ds Max 2025 software. No Autodesk or third-party images were used; permissions not required.
(Supplementary Table 1), the Cu content in the as-prepared Cu-Nx-y ranges from 3.3 wt% (Cu-N4-3.3), 9.7 wt% (Cu-N4-9.7), 15.8 wt% (Cu-N3-15.8) to 33.2 wt% (Cu-N3-33.2), indicating the successful preparation of Cu SACs with controlled metal loading. Notably, only a broad peak centered at 2θ ≈ 26° is observed in the PXRD patterns of both NC and all Cu-Nx-y samples, which corresponds to the (002) plane of graphitized carbon (Supplementary Fig. 2c), indicating that Cu might be atomically dispersed over the support. Similarly, during pyrolysis, the active groups of the precursors decompose while retaining certain stable functional groups like C = O and C-N (Supplementary Fig. 2d), which confirms the viability of the in-situ N-doping strategy. Moreover, scanning electron microscopy (SEM, Supplementary Fig. 3) and transmission electron microscopy (TEM, Fig. 1b and Supplementary Fig. 4) images demonstrate that the as-prepared NC and all Cu-Nx-y have a layered nanosheet morphology with
wrinkles and lateral size in microscale. Notably, as shown in Fig. 1c, d and Supplementary Fig. 5, plenty of atomically-scale bright spots, rather than Cu nanoparticles or clusters, were observed in the aberration-corrected high-angle annular dark-field scanning TEM (AC HAADF-STEM) images, which intuitively reflects the atomic dispersion of Cu atoms in Cu-N4-3.3, Cu-N4-9.7, Cu-N3-15.8, and even Cu-N3-33.2. Meanwhile, energy-dispersive X-ray (EDX) elemental mappings show that C, N, and Cu elements are uniformly distributed throughout the Cu-Nx-y samples (Fig. 1e and Supplementary Fig. 6). Furthermore, Raman spectra of NC and all Cu-Nx-y show the similar D- and G-band intensity ratios (ID/IG), suggesting that the introduction of Cu has no effect on the degree of disorder of the samples (Supplementary Fig. 7). Additionally, N2 adsorption–desorption isotherms (Supplementary Fig. 8) reveal that all Cu-Nx-y samples possess similar BET-specific surface areas, further indicating that the introduction of Cu has no obvious influence on the structure of NC support, which is beneficial for exploring the underlying structure-activity relationship.
The surface chemical compositions and electronic states of NC and Cu-Nx-y were investigated by X-ray photoelectron spectroscopy (XPS). Survey spectra show that C, O and N are present in all samples, while Cu is only present in Cu-Nx-y (Supplementary Fig. 9). In the high-resolution N 1 s spectra of all samples (Supplementary Fig. 10), the peaks at around 398.5, 399.9, 401.0, and 402.5 eV are ascribed to pyridinic-N, pyrrolic-N, graphitic-N, and N-oxides, respectively30,31,32. Regarding Cu 2p spectra (Supplementary Fig. 11), the peaks at around 932.6 and 934.5 eV are attributed to the Cu 2p3/2 electronic configurations with oxidation states of +2 and +1/0, respectively. This result indicates the oxidation state of Cu atoms in Cu-Nx-y, which is further corroborated by the analysis of the corresponding Cu LMM Auger spectra (Supplementary Fig. 12)33,34. Moreover, it is noteworthy that the proportion of Cu+ species in Cu-Nx-y increases with higher Cu loading, suggesting a gradual reduction in oxidation state. In other words, varying Cu contents in Cu-Nx-y can significantly affect the electronic structure of the atomic Cu centers, which is expected to influence catalytic performance.
To further reveal the local electronic and geometric structures of the Cu centers in Cu-Nx-y, X-ray absorption spectroscopy (XAS) analysis was performed. As shown in the Cu K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 2a), the absorption energy edges of all Cu-Nx-y samples fall between those of Cu foil and CuO, implying that the oxidation state of the atomic Cu centers should be between 0 and +235,36. Meanwhile, it can also be observed that the pre-edge absorption energy of Cu-Nx-y shifts towards lower energies with increasing Cu content, suggesting a decrease in the average valence state of Cu centers, which is consistent with the XPS results. Moreover, the k2-weighted Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra show that all Cu-Nx-y samples display one primary peak at ~1.47 Å, which is attributed to the Cu-N scattering in the first coordination shell (Fig. 2b and Supplementary Fig. 13a). As compared to CuO and Cu foil, the absence of Cu-O-Cu (2.5 Å) and Cu-Cu (2.2 Å) coordination confirms the atomic dispersion of Cu atoms in all Cu-Nx-y samples.

a Normalized Cu K-edge XANES and (b) FT-EXAFS spectra of different Cu-Nx-y catalysts. c FT-EXAFS spectra and corresponding fitting EXAFS spectra of Cu foil and different Cu-Nx-y catalysts. d Wavelet transform EXAFS plots of different Cu-Nx-y catalysts. All the FT-EXAFS data are presented without phase correction. The grayish-white, light blue and dark blue refer to C, N, and Cu atoms, respectively.
Therefore, it can be concluded that Cu centers in all Cu-Nx-y samples are coordinated with N atoms. Quantitative fitting of the EXAFS spectra (Fig. 2c and Supplementary Table 2) shows that the Cu centers in Cu-N4-3.3 and Cu-N4-9.7 are tetra-coordinated with Cu-N4 configuration, while those in Cu-N3-15.8 and Cu-N3-33.2 are tri-coordinated with Cu-N3 configuration, revealing that changing the metal loading in Cu SACs leads to different coordination structures of the Cu centers, thereby affecting the valence state and electronic structure of the Cu atoms. Furthermore, the wavelet transform (WT) EXAFS contour plot of Cu foil shows an intensity maximum at about 7.8 Å−1 (Supplementary Fig. 13b), which is attributed to the Cu-Cu coordination. In contrast, all Cu-Nx-y samples only present an intensity maximum at around 4.8 Å−1 (Fig. 2d and Supplementary Fig. 13c, d), which can be attributed to the Cu-N path, further endorsing the atomic dispersion of Cu atoms.
Benzene oxidation performances of catalysts
The benzene oxidation reaction (BOR) using H2O2 as the oxidant was conducted at 60 oC with Cu-Nx-y catalysts. Before evaluating the catalytic performance of these catalysts in BOR, several control experiments were performed, and the results were displayed in Supplementary Table 3. The conversion of benzene hardly occurred in the absence of H2O2, indicating that H2O2 is essential for the selective oxidation of benzene to phenol. Moreover, the conversion of benzene was almost 0 in the absence of Cu SACs or when using NC as the catalyst, demonstrating that the Cu sites are the authentic active sites for BOR. Therefore, it can be concluded that BOR is a Cu-catalyzed process, with H2O2 as an essential reactant. Considering the high Cu loadings (up to 33.2 wt%) of the as-prepared Cu-Nx-y, these catalysts are expected to provide a substantial number of active sites for the efficient oxidation of benzene to phenol. Figure 3a displays the catalytic activity of various Cu SACs at 60 °C for 1 h using acetonitrile (MeCN) as the solvent. As expected, the benzene conversion gradually increased from 19.6% to 78.7% with the increase of Cu loading from 3.3 to 33.2 wt%. To explore the relationship between BOR performance and H2O2 concentration, the BOR performance of Cu-N3-33.2 was evaluated using different amount of H2O2. The results show that increasing the H2O2 concentration will increase the benzene conversion but will also slightly sacrifice the selectivity (Supplementary Fig. 14a). Moreover, the utilization of H2O2 of different catalysts was calculated under the same reaction conditions (i.e., adding 5 mL H2O2). The results show that Cu-N3-33.2 with best catalytic activity also exhibits the highest H2O2 utilization, reaching 32.6% (Supplementary Fig. 14b), indicating that catalysts with higher BOR activity have higher H2O2 activation efficiency. To further reveal whether the improvement in catalytic performance is solely due to the increase in Cu loading, we kept the Cu content in the catalysts consistent and further evaluated their BOR performance. Surprisingly, as shown in Supplementary Table 4, the Cu SACs with Cu-N3 centers exhibited much higher activity compared to those with Cu-N4 centers. Correspondingly, the calculated turnover frequency (TOF) and mass activity of the Cu-Nx-y catalysts also increased with higher Cu loading. Particularly, Cu-N3-33.2, with a Cu loading of 33.2 wt%, exhibited impressive TOF and mass activity values of 680.3 h−1 and 1927.6 mmol·g−1·h−1, respectively, distinguishing it among previously reported Cu SACs for the selective oxidation of benzene with H2O2 (Supplementary Fig. 15 and Supplementary Table 5)1,16,37,38,39,40,41,42,43,44,45. Meanwhile, under the same coordination structure, Cu-Nx-y catalysts with higher Cu loading also showed higher activity, indicating that the exceptional catalytic activity of Cu-N3-33.2 is attributed to its unique low-coordination structure and higher number of active sites. Besides its highly efficient catalytic activity, Cu-N3-33.2 also exhibited remarkable stability under the test conditions. As shown in Fig. 3b, both benzene conversion and phenol selectivity have no significant changes after six cycles. More importantly, TEM image, PXRD patterns, as well as normalized Cu K-edge XANES and FT-EXAFS spectra reveal that after the reaction, the Cu atoms in Cu-N3-33.2 remain atomically dispersed without aggregation, and both the morphology and coordination structure are preserved compared to their original states (Supplementary Fig. 16), exclude the formation of CuOx cluster after reaction. In addition, ICP-OES analysis result shows that the Cu content (29.5 wt%) in Cu-N3-33.2 after the stability test is comparable to that before the reaction, further demonstrating its satisfactory stability.

a, c Catalytic performance of the Cu-Nx-y catalysts for the selective oxidation of benzene to phenol. b Recycling performance of Cu-N3-33.2. d Temperature-dependent BOR performance of Cu-N3-33.2 using different solvents. Catalytic performance of Cu-N3-33.2 for (e) benzene oxidation and (f) phenol oxidation in the presence of different quenchers. Reaction conditions: 5 mg catalyst, 0.3 mL benzene, 6 mL solvent (MeCN in (a), H2O in (c)), 5 mL H2O2, 60 oC, 1 h. *Extend the reaction time to 5 h. MeCN: Acetonitrile. TBA: Tert-butanol.
Although the Cu-Nx-y catalysts exhibit excellent catalytic activity and stability in BOR, their selectivity for phenol is poor and decrease significantly with the increase of Cu loading (Fig. 3a). Specifically, the phenol selectivity of Cu-N3-33.2 is only 52.9%. Since MeCN and H2O2 are miscible, the catalyst and oxidant in the reaction system are in full contact with benzene, thereby efficiently converting it to phenol. However, the instability of phenol and its high solubility in MeCN make it easy to be further oxidized to p-benzoquinone. Therefore, timely separation of the generated phenol is expected to significantly improve its selectivity. Considering the poor solubility of benzene in water, BOR will proceed at the benzene-water interface when water is used as the solvent46. Although this may affect catalytic activity, this unique interfacial reaction is advantageous for product separation, thereby improving phenol selectivity. As predicted, the benzene conversion of all Cu-Nx-y catalysts decreased significantly when using water as the solvent. In contrast, the phenol selectivity of all Cu-Nx-y catalysts dramatically increased (Fig. 3c). Especially for Cu-N3-33.2, the phenol selectivity increased from 52.9% to 91.6%. More notably, when the reaction time was extended to 5 h, the conversion of benzene reached 85.9%, which is comparable to the activity observed when MeCN was used as the solvent. Meanwhile, the phenol selectivity remains as high as 96%, which is significantly better than the latter, revealing that the interfacial effect is conducive to the separation of products, thereby improving the selectivity of products. Since the generated phenol is slightly soluble in water, a small amount of p-benzoquinone byproduct can also be detected when water is used as the solvent. To gain a deeper understanding of the relationship between the interfacial effect and phenol selectivity, temperature-dependent catalytic activity tests were conducted. As shown in Fig. 3d, the activity of Cu-N3-33.2 in both MeCN and water solvents gradually decreased as the reaction temperature decreased. It is noteworthy that the phenol selectivity hardly changes with temperature when MeCN is used as the solvent. In contrast, when water is used as the solvent, the phenol selectivity gradually increase with decreasing reaction temperature, and impressively, the phenol selectivity can reach 100% at 40 oC, which is attributed to the decrease in the solubility of phenol in water as the temperature decreases. Notably, as shown in Supplementary Fig. 17a, both benzene conversion and phenol selectivity of Cu-N3-33.2 have no significant changes after five cycles using H2O as solvent. Moreover, the PXRD pattern and HRTEM image after stability testing showed that no Cu-based metal clusters or nanoparticles were formed in Cu-N3-33.2 (Supplementary Fig. 17b, c), demonstrating its excellent structural and activity stability in water. This study highlights the selective oxidation of benzene to phenol in a thermocatalytic system using water as solvent, offering cost-effective and environmentally sustainable pathways for advancing industrial phenol synthesis.
BOR mechanism study
Previous studies have shown that Cu SACs are able to activate H2O2 and generate reactive oxygen species (ROSs) including ·OH and O2·- radicals, thus initiating the efficient oxidation of benzene to phenol12,41,47. Unfortunately, the resulting phenol is also susceptible to further oxidation by these ROSs. To further reveal the underlying reaction mechanism, quenching experiments and electron paramagnetic resonance (EPR) spectra were employed to detect the ROSs during the reaction. As a kind of quencher for ·OH radicals, the addition of tert-butanol (TBA) influences the benzene conversion but has no significant effect on phenol selectivity (Fig. 3e), suggesting that ·OH radicals are absent in the reaction system or is hardly responsible for the over-oxidation of phenol. Furthermore, it is well known that EPR spectroscopy are usually employed to detect the ·OH and O2·- radicals using 5, 5-dimethyl-1-pyrroline-noxide (DMPO) as the scavenger48. However, as shown in Supplementary Fig. 18a, no obvious signals belonging to DMPO-OH· or DMPO-O2·- species are observed, which means that almost no ·OH and O2·- radicals are generated in the reaction system. Moreover, L-histidine is usually used to trap holes singlet oxygen (1O2) species. It is noteworthy that although the addition of L-histidine reduces the conversion of benzene, it significantly improves the phenol selectivity. Impressively, after adding 2 mmol of L-histidine into the reaction system, the phenol selectivity can even reach 100%, indicating that 1O2 species are primarily responsible for the over-oxidation of phenol to p-benzoquinone. Furthermore, EPR spectroscopy solidly confirms the presence of 1O2 species in the reaction system (Supplementary Fig. 18b). To further reveal the roles of 1O2 species in the BOR process, the catalytic performance of Cu-N3-33.2 toward phenol oxidation reaction was evaluated under similar reaction conditions. As displayed in Fig. 3f, the phenol conversion is 43.5% in the absence of L-histidine. However, with the addition of L-histidine, the phenol conversion dropped sharply, further confirming that the 1O2 species is the authentic ROS caused further oxidation of phenol.
To elucidate the reaction mechanism of the selective oxidation of benzene to phenol over Cu single-atom catalysts (SACs), a detailed pathway is proposed based on experimental observations and previous reports41,47. As illustrated in Fig. 4a, the reaction starts with the adsorption and activation of H2O2 on the Cu-Nx active sites to form O-Cu-Nx intermediates (step I). Subsequently, the C6H6 molecules adsorbed on the Cu-O sites (step II) and forms the Cu-Nx-C6H5OH intermediates via a hydrogen transfer process (step III). Finally, the catalytic cycle is completed after the desorption of C6H5OH molecules and the regeneration of the Cu-Nx sites (step IV). Besides, to gain a deeper understanding of the effect of coordination structures of Cu SACs on the catalytic performance, the catalytic activities of all Cu-Nx-y samples were evaluated by adjusting the catalyst amount to maintain a consistent Cu content. As clearly shown in Supplementary Fig. 19, the samples with Cu-N3 coordination configuration exhibit much higher activity compared to those with Cu-N4 coordination configuration under identical reaction conditions. According to the XPS and XANES results, both Cu2+ and Cu+ species exist in all Cu-Nx-y samples, and the proportion of Cu+ species increases with the increase of Cu loading. Further in-depth XPS analysis showed that the Cu+/Cu2+ ratio was positively correlated with the BOR activity (Supplementary Fig. 20). Moreover, we therefore investigated the genuine active species indirectly by studying the BOR activity of Cu SACs with pure Cu (II)-N4 structure. Results show that 2.4-Cu1/NOC (a previous reported Cu SACs with pure Cu (II)-N4 structure)12 and commercial copper phthalocyanine (CuPc, with pure Cu (II)-N4 structure) cannot effectively activate benzene under the same reaction conditions of the Cu-Nx-y catalysts (Supplementary Table 6), confirming that low-coordinated Cu (I)-N3 structures should be the genuine active species in our reaction system despite the minor presence of Cu (II)-N4 species. To further reveal the effect of the coordination configuration of atomic Cu centers on the catalytic performance, in-situ IR spectroscopy analysis was first performed. As shown in Fig. 4b and Supplementary Fig. 21, characteristic peaks corresponding to Cu-O species (1045, 1380 and 1480 cm−1) were detected during the reaction, indicating the formation of Cu-O species in presence of H2O249,50,51. According to previous reports, the in-situ formed Cu-O species should be the authentic active species for selective oxidation of selective oxidation of benzene to phenol41,47. It is noteworthy that the in-situ IR signals of Cu-O species from Cu-N3-33.2 are stronger than those of Cu-N4-3.3, indicating that the Cu-N3 coordination configuration is prone to produce more Cu-O active species, which is beneficial to promote the reaction. In addition, control experiments showed that CuO or metallic Cu loaded on NC support (CuO/NC and Cu NPs/NC) exhibit much inferior BOR performance compared to Cu-N3-33.2 (Supplementary Fig. 22 and Table 6), confirming that the in-situ generated O-Cu-Nx species should be the real active site for the efficient conversion of benzene to phenol.

a The proposed BOR mechanisms over the Cu-Nx-y catalyst. b In-situ IR spectra of Cu-N3-33.2. c, d Projected density of states (pDOS) of the Cu centers on the CuN3 and CuN4 interfaces before and after H2O2 adsorption. e Free energy diagrams of benzene oxidation to phenol on Cu-N3 and Cu-N4 sites. e1-e6 The charge density difference of Cu-N3, Cu-N4 in BOR. Yellow and cyan isosurfaces indicate the electron accumulation and depletion, respectively.
To further corroborate these experimental findings and elucidate the underlying mechanisms, density functional theory (DFT) calculations were performed. Analysis of the electronic structure of Cu atoms in the Cu-N4 and Cu-N3 centers (Fig. 4c, d and Supplementary Fig. 23), suggests that the electronic interactions between the O atom in H2O2 and Cu atom in these two centers are different. Specifically, For the CuN4 configuration, the relaxed structure reveals a Cu atom embedded within four N atoms, exhibiting D4h symmetry and strong crystal field effects. Notably, the four coordinated N atoms interact with the dxy orbitals of the Cu atom in a “head-to-head” manner, resulting in d-orbital energy splitting, where the dxy orbitals is located at a higher energy level than the remaining four orbitals. Moreover, the calculation results further show that the dxy orbitals located at the Fermi level determine the physicochemical properties of the Cu site. However, despite the energy level alignment, the steric mismatch between the dxy orbitals of the Cu atom and the p orbitals O atoms prevents effective bonding, while other d orbitals suitable for bonding are located at significantly deeper energy levels. Therefore, as shown in Fig. 4c, the energy levels and density of states (DOS) of the dxy orbitals of Cu atom show almost no changes even after H2O2 adsorption, indicating weak electron exchange due to orbital incompatibility. In contrast, for the CuN3 coordination configuration, the theoretically optimized structure shows that the Cu atom is embedded in a trigonal planar configuration (C3v symmetry) formed by three N atoms, with an average Cu–N bond length of 1.90 Å, which is highly consistent with the experimental EXAFS results (1.89–1.91 Å, Supplementary Table 2) (deviation < 0.01 Å), verifying the reliability of the calculation model. Compared with the planar D4h symmetry of CuN4, the reduction in the N coordination number in CuN3 causes the Cu atom to protrude slightly from the carbon substrate plane, forming a more open coordination environment. This geometric distortion not only shortens the distance between Cu and adsorbates (such as H2O2), but also promotes the more efficient hybridization of the d orbitals of Cu atom and the p orbitals of O atom. As shown in Fig. 4d, part of the d orbitals of Cu atom in the Cu-N3 coordination configuration are pushed above the Fermi level after H2O2 adsorption, which indicates that these d orbitals match the p orbitals of O atom in H2O2, resulting in a strong electronic interaction. Through the pDOS analysis, it was found that the p orbitals of the O atom in the H2O2 molecule and the d orbitals of the Cu atom (especially the dz2 orbitals) show obvious overlap in energy distribution, indicating that there is a strong electronic coupling between O atom and the Cu atom. Furthermore, through the two-dimensional (2D) charge density difference analysis (Supplementary Fig. 24), it can be intuitively observed that H2O2 molecule gains electrons from the Cu atom during the adsorption process, which directly confirms the orbital matching mechanism and the redistribution of interface charges. Meanwhile, it seems like the Cu dz2 orbitals signatures shifted below the Fermi energy upon the adsorption of the H2O2 molecule. We believe that this phenomenon may originate from the strong hybridization between the p orbital of the O atom in the H2O2 molecule and the dz2 orbital of Cu, which causes the energy of some electronic states to shift downward. This interaction not only changes the d orbitals distribution of Cu atoms but also reflects the dynamic adjustment of the electronic structure during the adsorption process. Ultimately, the strong electronic interaction between the d orbitals of Cu atom in the Cu-N3 coordination configuration and the p orbitals of O atom in H2O2 endow it with stronger adsorption of H2O2 compared with that in the Cu-N4 coordination configuration, which is confirmed with the much lower adsorption energy of H2O2 on the CuN3-slab (Supplementary Fig. 25). The mechanism of the difference in adsorption strength was further revealed by calculating the d-band center of the Cu atom. The results show that the d-band centers of the Cu-N3 and Cu-N4 coordination structures are located at −1.5673 and −2.7682 eV, respectively. Since the d-band center of Cu-N3 is closer to the Fermi level, it exhibits a significantly enhanced H2O2 adsorption ability, which is highly consistent with experimental observations and theoretical predictions. This finding clearly reveals the crucial role of the coordination structure on the electronic structure and catalytic activity of Cu atoms in Cu-N-C catalysts. The adsorption energy of H2O2 on CuN3 (−117.87 kJ/mol) is significantly lower than on CuN4 (-24.27 kJ/mol) (Supplementary Fig. 25), directly linked to the electronic structure differences. The higher d-band center of CuN3 (−1.5673 eV vs. −2.77 eV for CuN4) enhances H2O2 adsorption by strengthening the p-d hybridization between O (p orbitals) and Cu (d orbitals), as evidenced by the overlapping pDOS (Fig. 4d and Supplementary Fig. 23). This electronic synergy lowers the H2O2 activation efficiency, rationalizing the superior activity of CuN3. Furthermore, the Bader charge analysis and calculated adsorption energies of these systems were further performed. For the interaction with C6H6O molecules, the charge transfer amounts is 0.09 e− in Cu-N3, while that in Cu-N4 is 0.04 e−. For the case of H2O2, Cu-N3 exhibits a charge transfer of 1.86 e−, which is significantly higher than the 0.17 e− observed in Cu-N4. These quantitative results confirm that the Cu-N3 configuration has superior charge transfer activity compared to the Cu-N4 configuration, which is consistent with its higher adsorption energy for H2O2, indicating an optimized electronic interaction with the reactants. This difference may originate from the lower coordination number of the Cu-N3 configuration, where the unsaturated coordination structure enhances the tunability of the reactants to the electronic structure of Cu atoms, thereby improving the catalytic activity.
Moreover, As shown in Supplementary Table 7, Bader charge analysis reveals that the oxidation states of Cu in CuN3, CuN4 and CuO are +0.52 (11 − 10.4775 = 0.5225), +0.82 (11 − 10.1828 = 0.8172) and +1.00 (11 − 9.996 = 1.004), respectively. Bader charge analysis, while not equivalent to formal oxidation states, reveals the relative electron density distribution around Cu centers, corroborating the XANES-derived oxidation state trends. Moreover, the projected density of states (pDOS) analysis shows that the d orbitals of Cu atom in CuO display broader and flatter features, which is attributed to the significantly enhanced ligand field strength at high oxidation states, thereby strengthening the hybridization between d orbitals of Cu atom and ligands (e.g., O atoms). In contrast, the sharper pDOS peak of Cu in CuN3 suggest stronger electron localization, which may be due to its lower oxidation state and weaker ligand field effects, thereby retaining d electrons closer to the Cu atoms(Supplementary Fig. 26). Combined with the catalytic performance, the localized electronic structure in CuN3 can promote the generation of O-Cu-N3 active intermediates, thus improving the BOR performance. Kinetic isotope experiment revealed that the conversion of deuterated benzene was comparable to that of untreated benzene, indicating that the activation of C-H bonds in BOR is not the rate-determining step (Supplementary Fig. 27). Notably, the free-energy diagram of the pathways for the generation of phenol shows that the adsorption of H2O2 is crucial for the oxidation of benzene to phenol, and Cu SACs with Cu-N3 coordination configuration are more conducive to promoting the reaction (Fig. 4e). Furthermore, the electron density difference maps show that the Cu-N3 coordination configuration is more responsive to electron transfer than the Cu-N4 coordination configuration (Fig. 4e1-e6 and Supplementary Fig. 28), indicating that the Cu SACs with Cu-N3 coordination configuration has a stronger and more favorable interaction with the reactants. Consequently, the open coordination environment and unique electron density distribution of the Cu-N3 coordination configuration enable it to promote electron transfer and enhance the interaction with reactants, resulting in better catalytic performance. We further investigated the H2O2 activation pathways, including the generation of 1O2 species. As shown in Supplementary Fig. 29, the energy barrier for the formation of 1O2 on the Cu-N3 site is very high, implying that the in-situ generated O-Cu-N3 should preferentially react with benzene to form phenol, while excess H2O2 may promote the generation of 1O2, leading to formation of over-oxidation products (e.g., benzoquinone), which is consistent with the experimental results shown in Supplementary Fig. 14a. Although Cu-N4 exhibits a high tendency to form 1O2, its actual generation is restricted by the inherently high energy barrier for the formation of O-Cu-N4 formation from Cu-N4, thereby limiting the generation of 1O2 in Cu-Nx-y catalysts with Cu-N4 structure. The combination of in-situ IR spectroscopy and DFT calculations provides compelling evidence that the Cu-N3 coordination configuration in Cu SACs is more effective for the selective oxidation of benzene to phenol, due to its ability to generate more active Cu-O species and facilitate stronger electronic interactions with reactants.
Discussion
In summary, we have successfully designed and synthesized tri- and tetra-coordinated Cu SACs with ultra-high metal loadings via a simple two-step process involving supramolecular self-assembly and high-temperature carbonization. The results show that the electronic states at the orbital level can be manipulated to optimize catalytic performance by controlling metal loading within carbon-based supports. The optimized tri-coordinated Cu SAC with ultra-high metal loading of up to 33.2 wt% exhibited a superior catalytic performance for the selective oxidation of benzene to phenol. We have revealed that the in-situ generated Cu-O species are revealed to be the active centers for the oxidation of benzene to phenol, while the in-situ generated 1O2 species are a decisive factor in the undesirable over-oxidation of phenol to p-benzoquinone. This work provides deep insights into the precise tuning of catalytic reactivity of SACs by electron orbital engineering.
Methods
Synthesis of Cu-Nx-y
Guanine (5 g) and Cu(NO3)2·3H2O (0.21/0.67/0.95/1.63 g) were uniformly dispersed in 50 mL of deionized water and heated at 100 oC for 12 h in an oil bath. The resulting suspension was rapidly frozen using liquid nitrogen and subsequently freeze-dried to yield the precursor. The precursor was then subjected to pyrolysis at 700 oC for 2 h under protection of N2, with a heating rate of 1 oC min–1, producing Cu single-atom catalysts, designated as Cu-N4-3.3, Cu-N4-9.7, Cu-N3-15.8, and Cu-N3-33.2. Neat nitrogen-doped carbon material (NC) was synthesized using the same procedure without the addition of Cu(NO3)2·3H2O.
Catalytic test
For a typical liquid phase benzene oxidation reaction, 5 mg catalyst, 0.3 mL benzene, 5 mL H2O2 and 6 mL solvent were mixed in a 48 mL thick-walled pressure flask and reacted at 60 oC at a stirring speed of 500 rpm for 1 h. Reactant and product concentrations were analyzed by gas chromatography-mass spectrometer (GC-MS) using tetradecane as the internal standard.
Where Δnbenzene is converted benzene (mol); t is the reaction time (h); nCu is the amount of Cu-based active sites (mol).
X-ray absorption spectra analysis
The obtained XAS spectra was processed in Athena (version 0.9.26) for background, pre-edge line and post-edge line calibrations. Then Fourier transformed fitting was carried out in Artemis (version 0.9.26). The k2 weighting, k-range of 3–14 Å–1 and R range of 1 ~ 3 Å were used for the fitting of Cu foil; k-range of 2–10 Å–1 and R range of 1 ~ 2 Å were used for the fitting of samples. The four parameters, coordination number, bond length, Debye-Waller factor and E0 shift (CN, R, ΔE0) were fitted without anyone was fixed, the σ2 was set. For Wavelet Transform analysis, the χ(k) exported from Athena was imported into the Hama Fortran code. The parameters were listed as follow: R range, 1–4 Å, k range, 0–15 Å–1 for samples; k weight, 2; and Morlet function with κ = 10, σ = 1 was used as the mother wavelet to provide the overall distribution.
Density functional theory calculation details
First-principle Density functional theory (DFT) calculations with the spin-polarized consideration were carried out within the density functional theory framework. The projector-augmented wave (PAW)52 method and the generalized gradient approximation (GGA) for the exchange correlation energy functional, as implemented in the Vienna ab initio simulation package (VASP 5.4.4)53,54 were used. The GGA calculation was performed with the Perdew–Burke–Ernzerhof (PBE) exchange-correlation potential55. A plane-wave cutoff energy of 400 eV was used. Grimmes’s DFT-D3 dispersion correction was used to describe the van der waals interaction56. A 6 × 6 graphene supercell with 15 Å vacuum layer was first constructed. A 3 × 3 K-points was used in all these calculations. Dipole correction was taken into consideration in the slab calculation57.
The adsorption free energy (ΔGads) is obtained by:
Where ΔE is the energy difference of the reactants and products, obtained from DFT calculations; ΔEZPE and ΔS are the contributions to the free energy from the zero-point vibration energy and entropy, respectively. T is the temperature (298 K). The free energy for gas phase water is calculated at 0.035 bars. The free energy for other molecules is calculated at 1 bar.
Data availability
The source data are provided in the Source data file. The data supporting this study are available for the corresponding author upon request. Source data are provided with this paper.
References
Pan, Y. et al. Regulating the coordination structure of single-atom Fe-NxCy catalytic sites for benzene oxidation. Nat. Commun. 10, 4290 (2019).
Chen, X., Zhang, J., Fu, X., Antonietti, M. & Wang, X. Fe-g-C3N4-Catalyzed oxidation of benzene to phenol using hydrogen peroxide and visible light. J. Am. Chem. Soc. 131, 11658–11659 (2009).
Niwa, S.-i et al. A one-step conversion of benzene to phenol with a palladium membrane. Science 295, 105–107 (2002).
Morimoto, Y., Bunno, S., Fujieda, N., Sugimoto, H. & Itoh, S. Direct hydroxylation of benzene to phenol using hydrogen peroxide catalyzed by nickel complexes supported by pyridylalkylamine ligands. J. Am. Chem. Soc. 137, 5867–5870 (2015).
Tanev, P. T., Chibwe, M. & Pinnavaia, T. J. Titanium-containing mesoporous molecular sieves for catalytic oxidation of aromatic compounds. Nature 368, 321–323 (1994).
Yang, J.-H. et al. Direct catalytic oxidation of benzene to phenol over metal-free graphene-based catalyst. Energy Environ. Sci. 6, 793–798 (2013).
Zhang, H. et al. Enhancing chemical reactions in a confined hydrophobic environment: an NMR study of benzene hydroxylation in carbon nanotubes. Chem. Sci. 4, 1075–1078 (2013).
Qiao, B. et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011).
Kaiser, S. K., Chen, Z., Faust Akl, D., Mitchell, S. & Pérez-Ramírez, J. Single-atom catalysts across the periodic table. Chem. Rev. 120, 11703–11809 (2020).
Liu, H. et al. Reconstructing the coordination environment of Fe/Co dual-atom sites towards efficient oxygen electrocatalysis for Zn–air batteries. Angew. Chem. Int. Ed. 64, e202419595 (2024).
Zhang, H. et al. Tailoring oxygen reduction reaction kinetics of Fe−N−C catalyst via spin manipulation for efficient zinc–air batteries. Adv. Mater. 36, 2400523 (2024).
Jin, H. et al. Understanding the density-dependent activity of Cu single-atom catalyst in the benzene hydroxylation reaction. ACS Catal. 13, 1316–1325 (2023).
Hai, X. et al. Geminal-atom catalysis for cross-coupling. Nature 622, 754–760 (2023).
Yin, P. et al. Single cobalt atoms with precise N-coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. 55, 10800–10805 (2016).
Wang, X. et al. Regulation of coordination number over single Co sites: triggering the efficient electroreduction of CO2. Angew. Chem. Int. Ed. 57, 1944–1948 (2018).
Zhu, Y. et al. A cocoon silk chemistry strategy to ultrathin N-doped carbon nanosheet with metal single-site catalysts. Nat. Commun. 9, 3861 (2018).
Wan, J. et al. IIn situ phosphatizing of triphenylphosphine encapsulated within metal–organic frameworks to design atomic Co1–P1N3 interfacial structure for promoting catalytic performance. J. Am. Chem. Soc. 142, 8431–8439 (2020).
Zhang, J. et al. Tuning the coordination environment in single-atom catalysts to achieve highly efficient oxygen reduction reactions. J. Am. Chem. Soc. 141, 20118–20126 (2019).
Rong, X., Wang, H.-J., Lu, X.-L., Si, R. & Lu, T.-B. Controlled synthesis of a vacancy-defect single-atom catalyst for boosting CO2 electroreduction. Angew. Chem. Int. Ed. 59, 1961–1965 (2020).
Guo, H. & Guo, S. Strain engineering of multimetallic nanomaterials for advanced electrocatalysis. CCS Chem. 7, 326–344 (2025).
Qi, C. et al. Modulating electronic structures of iron clusters through orbital rehybridization by adjacent single copper sites for efficient oxygen reduction. Angew. Chem. Int. Ed. 62, e202308344 (2023).
Li, X., Chen, T., Yang, B. & Xiang, Z. Fundamental understanding of electronic structure in FeN4 site on electrocatalytic activity via dz-orbital-driven charge tuning for acidic oxygen reduction. Angew. Chem. Int. Ed. 62, e202215441 (2023).
Ramaswamy, N., Tylus, U., Jia, Q. & Mukerjee, S. Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to coordination chemistry. J. Am. Chem. Soc. 135, 15443–15449 (2013).
Yuan, K. et al. Boosting oxygen reduction of single iron active sites via geometric and electronic engineering: nitrogen and phosphorus dual coordination. J. Am. Chem. Soc. 142, 2404–2412 (2020).
Shan, J. et al. Metal-metal interactions in correlated single-atom catalysts. Sci. Adv. 8, eabo0762 (2022).
Jin, Z. et al. Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction. Nat. Catal. 4, 615–622 (2021).
Chang, J. et al. Synthesis of ultrahigh-metal-density single-atom catalysts via metal sulfide-mediated atomic trapping. Nat. Synth. 3, 1427–1438 (2024).
Wang, Y. et al. General negative pressure annealing approach for creating ultra-high-loading single atom catalyst libraries. Nat. Commun. 15, 5675 (2024).
Otero, R. et al. Guanine quartet networks stabilized by cooperative hydrogen bonds. Angew. Chem. Int. Ed. 44, 2270–2275 (2005).
Ran, J. et al. Endowing g-C3N4 membranes with superior permeability and stability by using acid spacers. Angew. Chem. Int. Ed. 58, 16463–16468 (2019).
Wang, D. et al. Coordination-engineered Cu–Nx single-site catalyst for enhancing oxygen reduction reaction. ACS Appl. Energy Mater. 2, 6497–6504 (2019).
Zhang, H. et al. Single atomic iron catalysts for oxygen reduction in acidic media: particle size control and thermal activation. J. Am. Chem. Soc. 139, 14143–14149 (2017).
Cao, J. et al. Strongly coupled 2D transition metal chalcogenide-MXene-carbonaceous nanoribbon heterostructures with ultrafast ion transport for boosting sodium/potassium ions storage. Nano-Micro Lett. 13, 113 (2021).
Wagh, N. K. et al. Densely colonized isolated Cu-N single sites for efficient bifunctional electrocatalysts and rechargeable advanced Zn-air batteries. Appl Catal. B. Environ. 268, 118746 (2020).
Bao, H. et al. Isolated copper single sites for high-performance electroreduction of carbon monoxide to multicarbon products. Nat. Commun. 12, 238 (2021).
Wang, X. et al. Uncoordinated amine groups of metal–organic frameworks to anchor single Ru sites as chemoselective catalysts toward the hydrogenation of quinoline. J. Am. Chem. Soc. 139, 9419–9422 (2017).
Shen, Q. et al. Ionic-liquid-assisted synthesis of metal single-atom catalysts for benzene oxidation to phenol. Sci. China Mater. 65, 163–169 (2022).
Deng, D. et al. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 1, e1500462 (2015).
Chen, W. et al. Dynamic evolution of nitrogen and oxygen dual-coordinated single atomic copper catalyst during partial oxidation of benzene to phenol. Nano Res. 15, 3017–3025 (2022).
Liu, J. et al. Direct observation of metal oxide nanoparticles being transformed into metal single atoms with oxygen-coordinated structure and high-loadings. Angew. Chem. Int Ed. 60, 15248–15253 (2021).
Che, W. et al. Out-of-plane single-copper-site catalysts for room-temperature benzene oxidation. Angew. Chem. Int Ed. 63, e202403017 (2024).
Liu, Y. et al. Atomically dispersed Cu anchored on nitrogen and boron codoped carbon nanosheets for enhancing catalytic performance. ACS Appl Mater. Interfaces 13, 61047–61054 (2021).
Zhang, T. et al. Preassembly strategy to fabricate porous hollow carbonitride spheres inlaid with single Cu–N3 sites for selective oxidation of benzene to phenol. J. Am. Chem. Soc. 140, 16936–16940 (2018).
Zhou, H. et al. Cation-exchange induced precise regulation of single copper site triggers room-temperature oxidation of benzene. J. Am. Chem. Soc. 142, 12643–12650 (2020).
Zhang, M. et al. Metal (Hydr)oxides@polymer core–shell strategy to metal single-atom materials. J. Am. Chem. Soc. 139, 10976–10979 (2017).
Xie, J. et al. Highly selective oxidation of benzene to phenol with air at room temperature promoted by water. Nat. Commun. 14, 4431 (2023).
Zhang, T. et al. Regulating electron configuration of single Cu sites via unsaturated N,O-coordination for selective oxidation of benzene. Nat. Commun. 13, 6996 (2022).
Li, S., Ke, Y., Zhang, X., Wu, S. & Xie, Z. Iron carbide nanoparticles encapsulated in guanine-derived carbon for peroxymonosulfate activation. Sep Purif. Technol. 338, 126570 (2024).
Dong, S. & Spiro, T. G. Ground- and excited-state mapping of plastocyanin from resonance raman spectra of isotope-labeled proteins. J. Am. Chem. Soc. 120, 10434–10440 (1998).
Liu, C. et al. In situ reconstruction of Cu–N coordinated MOFs to generate dispersive Cu/Cu2O nanoclusters for selective electroreduction of CO2 to C2H4. ACS Catal. 12, 15230–15240 (2022).
Tao, W. et al. Reductive NO coupling at dicopper center via a [Cu2(NO)2]2+ diamond-core intermediate. J. Am. Chem. Soc. 144, 22633–22640 (2022).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B 48, 13115–13118 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Neugebauer, J. & Scheffler, M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al (111). Phys. Rev. B 46, 16067–16080 (1992).
Acknowledgements
This work was financed by the National Natural Science Foundation of China (22372039) and the Natural Science Foundation of Fujian Province (2021J06010).
Author information
Authors and Affiliations
Contributions
S.L. conceived and performed the experiments, collected and analyzed data and wrote the paper. J.C. and Y.L. conceived and performed the DFT calculations and wrote the DFT section. W.W., L.C. and X.Z. joined the discussion of data and gave useful suggestions. C.C., X.H., Y.T. and Z.X. conceived the idea, supervised the project work and led the data analysis and discussion and prepared and finalized the paper. All the authors commented on the manuscript and have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Aiqin Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Cao, C., Chen, J. et al. Manipulating d-orbital of Cu single atom site by coordination engineering for selective oxidation of benzene. Nat Commun 16, 5790 (2025). https://doi.org/10.1038/s41467-025-61198-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61198-y
