
Abstract
With the rise of antibiotic-resistant bacteria, non-antibiotic therapies like gallium gain increasing attention. Intravenous gallium nitrate is under Phase II clinical trials to treat chronic Pseudomonas aeruginosa infections in cystic fibrosis patients. However, its clinical efficacy is constrained by the achievable peak concentration in human tissue. To address this limitation, we apply a genome-wide CRISPR interference approach (CRISPRi-seq) to identify potential synergistic targets with gallium. We classify the essential genes by response time and growth reduction, pinpointing the most vulnerable therapeutic targets in this species. In addition, we identify a highly conserved gene, fprB, encoding a ferredoxin-NADP⁺ reductase, whose deletion sensitizes P. aeruginosa to gallium, lowering its MIC by 32-fold and shifting mode of action from bacteriostatic to bactericidal. Further investigation reveals that FprB plays a critical role in modulating oxidative stress induced by gallium, via control of iron homeostasis and reactive oxygen species accumulation. Deleting fprB enhances gallium’s efficacy against biofilm formation and improves outcomes in a murine lung infection model of P. aeruginosa, suggesting FprB is a promising drug target in combination with gallium. Overall, our data show CRISPRi-seq as a powerful tool for systematic genetic analysis of P. aeruginosa, advancing the identification of novel therapeutic targets.
Similar content being viewed by others

Introduction
Pseudomonas aeruginosa is an opportunistic Gram-negative pathogen that frequently causes infections, particularly in immunocompromised individuals, including those with cystic fibrosis (CF) and chronic obstructive pulmonary disease1,2,3. It is responsible for a range of infections, including pneumonia, urinary tract infections, bloodstream infections, and wound infections, which result in considerable morbidity and mortality4. Due to its extensive resistance, P. aeruginosa has been classified by the World Health Organization as a high priority pathogen in the “ESKAPE” panel5. Therefore, the considerable threat posed by multidrug-resistant P. aeruginosa highlights the urgent need for new antimicrobial strategies.
Identifying the essential genes is a crucial step in developing novel anti-infective agents against multidrug-resistant pathogens like P. aeruginosa6. A variety of techniques such as transposon sequencing (Tn-seq), transposon-directed insertion site sequencing (traDIS), high-throughput insertion tracking by deep sequencing (HITS) and insertion sequencing have been employed to globally identify essential genes through high-throughput transposon mutagenesis and sequencing6,7,8,9. However, these approaches have inherent technical constraints, such as requirement of large libraries to fully cover the genome because not all transposon insertions resulting in gene inactivation, and their inability to simultaneously investigate the relative importance of essential genes under the conditions tested. In contrast, CRISPR interference (CRISPRi), which employs a catalytically inactive form of Cas9 nuclease (dCas9) in conjunction with single guide RNAs (sgRNAs) to inhibit gene expression, offers several advantages over these traditional genetic methods for analyzing essential and conditionally essential gene phenotypes10. Furthermore, quantitative CRISPRi facilitates the pinpointing of vulnerabilities in essential genes, furnishing crucial insights to fine-tune targets for drug discovery11. Recently, we and other groups developed CRISPRi-seq, CRISPRi in conjunction with next-generation sequencing, in multiple bacterial species, enabling genome-wide screenings to identify essential genes and factors relevant to antibiotic or phage sensitivity11,12,13,14,15,16,17. However, a genome-wide CRISPRi-seq toolbox to study P. aeruginosa gene fitness is still lacking. While systems like Mobile-CRISPRi18,19 streamlined the use of this technique and addressed some sgRNA cloning challenges in P. aeruginosa, efficient and scalable sgRNA cloning remains a limitation for the broader application of genome-wide CRISPRi in this organism. In this study, we addressed these challenges by establishing a rigorously controlled tetracycline (tet)-inducible CRISPRi system in P. aeruginosa, demonstrating its efficiency and tunability across a wide range of strains. Furthermore, we employed a ccdB-based counter selection system for efficient sgRNA cloning, and developed a comprehensive genome-wide CRISPRi library that targets 98% of the genetic elements in P. aeruginosa PA14.
Gallium has been approved by the U.S. Food and Drug Administration (FDA) as a therapy for cancer-related hypercalcemia20, and has also been recognized as a promising antibacterial agent21,22. Recently, a proof-of-principle Phase I clinical trial of gallium in CF patients with chronic P. aeruginosa lung infections proved its safety and efficacy23. This study used intravenous gallium nitrate, which was already approved by the U.S. FDA for hypercalcemia of malignancy. A follow-up Phase II clinical trial in 2016 (NCT02354859) showed a marked reduction in P. aeruginosa in the sputum of CF patients receiving gallium nitrate versus the placebo cohort, despite not meeting the primary endpoint24. The efficacy of gallium therapy for P. aeruginosa CF patients is limited by the peak plasma and sputum concentrations, which are 8–12 µM23. Gallium, which has an ionic radius almost identical to that of iron, can be taken up by bacteria in place of iron25. Once inside the cell, gallium is incorporated into iron-containing proteins but cannot substitute for iron in its essential biological functions, such as redox cycling. In P. aeruginosa, loss-of-function mutations that confer increased tolerance to gallium therapy have been identified. Notably, HitA and HitB, which form the major transporters of Fe3+, have emerged as key factors in gallium tolerance due to their role in Ga3+ uptake23,26. However, the genes potentially associated with increased sensitivity upon loss of function remain largely unexplored, impeding the advancement of synergistic strategies to enhance the efficacy of gallium treatment. In this study, we used a genome-wide CRISPRi library screening to identify the genetic determinants of gallium tolerance in P. aeruginosa.
Using the CRISPRi library, we developed and implemented quantitative CRISPRi-seq to generate a comprehensive list of essential genes and assess their vulnerability and responsiveness in P. aeruginosa PA14. In this context, vulnerability refers to the extent to which gene silencing impairs bacterial fitness, while responsiveness captures the timing and dynamics of fitness loss following gene knockdown. Next, we applied CRISPRi-seq to systematically identify genes influencing gallium susceptibility in P. aeruginosa. The screen confirmed known gallium tolerance determinants such as HitAB and uncovered novel insights, most notably the critical role of the conserved ferredoxin-NADP+ reductase FprB in gallium resistance. This work demonstrates the power of CRISPRi-seq for mechanistic discoveries and pinpoints FprB as a promising target for enhancing gallium efficacy against resilient P. aeruginosa infections.
Results
A titratable tet-inducible CRISPR interference system for P. aeruginosa
CRISPR interference (CRISPRi) utilizes a nuclease-deficient Cas9 protein (dCas9) in combination with a single guide RNA (sgRNA) to bind to a specific DNA sequence. This binding forms a complex that blocks the transcriptional machinery, leading to reduced or silenced expression of the target gene. To achieve tunable CRISPRi in P. aeruginosa, we engineered an optimized tetracycline-inducible (tet-inducible) genetic circuit encompassing Ptet and a repressor gene tetR, codon-adapted for P. aeruginosa (Supplementary Fig. 1a). This configuration enables regulation of downstream gene expression by addition of a tetracycline derivative - doxycycline (Dox). This inducer system was refined over IPTG- or arabinose-inducible systems due to its greater potential for application for in vivo infection studies. Doxycycline offers stable, tunable induction with consistent performance, as demonstrated in our previous murine CRISPRi models27.
To evaluate the induction efficiency of the developed system in P. aeruginosa, we used the LuxABCDE luminescence reporter system (Fig. 1a). Real-time monitoring of Ptet-driven luxABCDE expression in PA14 was performed in LB broth with varying concentrations of inducer. Increasing Dox concentrations (0 to 100 ng/mL) resulted in a dose-dependent increase of luminescence (Fig. 1a), without influence on growth (Supplementary Fig. 1b). The maximum induction led to approximately a 108-fold increase in expression at 20 h post-induction, without any discernible growth delay, confirming the system’s efficiency and titratability. In parallel, the well-characterized arabinose-inducible system in P. aeruginosa28 showed a dynamic range of approximately 29-fold induction under the same conditions (Fig. 1a).

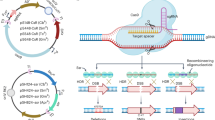
a Evaluation of the tetracycline- and arabinose-inducible reporter system in P. aeruginosa. The efficacy was assessed by measuring luminescence from luxABCDE expression induced by varying concentrations of doxycycline (Dox) or L-arabinose. b Schematic representation of the tet-inducible CRISPRi system. The system employs a SpydCas9-sgRNA complex, that is directed to DNA target region by a 20-base pair sequence in the sgRNA inhibiting transcription via steric hindrance. dcas9 expression is driven by a Ptet promoter, and the sgRNA is expressed from a constitutive promoter. Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. c Pyocyanin measurements of the strain with CRISPRi targeting phzM. The up panel shows photographs of culture tubes and lower panel for pyocyanin quantification. d, e Repression of swarming motility by CRISPRi. d presents the swarming colonies of indicated strains on medium with indicated concentration of Dox, and e presents the measurements the surface diameter of the colony. f The reversibility of tet-inducible CRISPRi system in P. aeruginosa. Repression of luxABCDE was induced by 25 or 100 ng/mL Dox at OD600 = 0.001. Following Dox removal and dilution at 7.5 h, luminescence was measured in recovering cultures. LB medium served as negative control. g Growth of CRISPRi knockdown strains targeting ftsZ in various P. aeruginosa backgrounds cultured in LB medium with increasing concentrations of Dox. h CcdB-based sgRNA cloning strategy. The ccdB gene is flanked by BsaI sites for Golden Gate assembly; replacement with sgRNA allows colony formation on LB agar in CcdB sensitive WM3064 strain. i Evaluation of the sgRNA cloning efficiency with the CcdB counter selection system. Three sgRNAs were cloned, and ten colonies from each cloning were randomly picked and sequenced. Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. Data from all growth and measurement assays are presented as mean ± SD from three biological replicates. Statistical analysis for panel c and e was performed using Two-way ANOVA and Tukey’s multiple-comparison test. Source data are provided as a Source Data file.
Next, we employed the tet-inducible system to control the expression of S. pyogenes dcas9, to construct a titratable CRISPRi system for P. aeruginosa using the Tn7 integrative vector pJMP284629 (Fig. 1b). Previous studies indicated that overexpression of S. pyogenes dCas9 is toxic in certain bacteria, including P. aeruginosa30,31. To determine if the expression of dCas9 and integration of the CRISPRi system impacts P. aeruginosa growth, we introduced the CRISPRi system into the chromosome of PAO1 and PA14 strains, utilizing an sgRNA targeting the lucff gene, which is absent in both of the P. aeruginosa strains. No significant growth inhibition was observed across the tested Dox concentrations (Supplementary Fig. 1c). We subsequently evaluated the efficiency of the tet-inducible CRISPRi system by targeting phzM and flgK. Gene phzM encodes a SAM-dependent methyltransferase, an enzyme in the biosynthetic pathway of pyocyanin, the compound responsible for the distinctive blue-green pigmentation of P. aeruginosa32. As shown in Fig. 1c, CRISPRi-mediated silencing of phzM transcription resulted in markedly reduced pyocyanin production, with an observable phenotype at Dox concentrations as low as 12.5 ng/mL. The second gene flgK, encoding a flagellar hook-associated protein, was reported to be required for the swarming motility of P. aeruginosa33. CRISPRi-mediated repression of flgK resulted in dose-dependent inhibition of the swarming ability (Fig. 1d, e). Thus, CRISPRi-mediated silencing recapitulates the phenotypes of deletion strains of non-essential genes in P. aeruginosa.
To assess the reversibility of the tet-inducible CRISPRi system, we performed a time-course analysis of CRISPRi-mediated regulation of luxABCDE. The results demonstrated that expression of the previously repressed luxABCDE operon resumed and became detectable within approximately 1.5 h following Dox removal (Fig. 1f). To assess the versatility of our CRISPRi system across various P. aeruginosa strains, we tested the CRISPRi effectiveness in reference strain PAO134, the mucoid strain PDO30035, and one of our clinical isolates named P5. A CRISPRi vector was engineered to target the conserved essential gene ftsZ, which is crucial for Z ring formation during cell division36. Repression of ftsZ is expected to lead to a growth defect, which was used to evaluate the efficiency of the CRISPRi for an essential gene37. As shown in Fig. 1g, efficient CRISPRi knockdown of ftsZ was achieved in all these strains in a titratable way. However, the effective Dox concentration for CRISPRi knockdown varied among the strains, likely due to differences in cell membrane permeability for the inducer (Fig. 1g). Taken together, these results suggest that the developed tet-inducible CRISPRi system is fully functional and can serve as a foundation for genome-wide CRISPRi libraries in P. aeruginosa.
To enable efficient sgRNA cloning for the construction of a genome-wide CRISPRi library, we designed a ccdB counter selection system in combination with Golden Gate assembly. This approach facilitates the seamless insertion of the 20 bp base-pairing region of sgRNAs (see “Methods”) (Fig. 1h). The ccdB gene, encoding a toxin, was inserted into the cloning site for the sgRNA’s base-pairing region, and the resulting vector was named pCRISPRi-ccdB. pCRISPRi-ccdB was maintained in the Escherichia coli WM3064 with a GyrA462Arg-Cys mutation (SZU148), conferring resistance to CcdB toxicity. The ccdB gene was flanked by BsaI to enable its replacement by spacer sequence during Golden Gate assembly, providing positive selection for vectors in which the sgRNA had successfully replaced the ccdB gene upon transformation into E. coli WM3064. In three sgRNA cloning tests, a 100% success rate in replacing ccdB with sgRNAs was achieved (Fig. 1i and Supplementary Fig. 1d, e). This indicates that the strategy is effective for generating low background sgRNA pools, facilitating the construction of compact CRISPRi libraries.
Genome-wide CRISPRi-seq allows identification of essential genes in P. aeruginosa PA14
Next, we constructed a genome-wide CRISPRi library using our previously developed sgRNA design pipeline, prioritizing for each genetic element in the P. aeruginosa PA14 genome the selection of an sgRNA with minimal expected maximum off-target activity, and a binding site close to the target start codon9 (Fig. 2a). To improve screening robustness, we created two such sgRNA sets using the top two selected sgRNAs per genetic element, comprising 5981 and 5971 sgRNAs, respectively, exhibiting comparable expected specificities (Supplementary Fig. 1f). The spacers were synthesized as one oligo chip. The two spacer pools were cloned into pCRISPRi-ccdB vector and maintained in E. coli WM3064. CRISPRi libraries were constructed by triparental conjugation, introducing the sgRNA libraries into PA14 with the aid of a helper strain harboring the Tn7 transposase plasmid. Protocols for sgRNA cloning in E. coli WM3064 and the subsequent conjugal transfer to P. aeruginosa were optimize to ensure high colony yield, resulting in more than 50-fold coverage of sgRNA diversity. Subsequent Illumina sequencing followed by 2FAST2Q analysis38 indicated establishment of libraries with 5832 sgRNAs in set 1 and 5863 sgRNAs in set 2, covering 97.5% and 98.2% of the genetic features of PA14 strain, respectively (Supplementary Data 4).

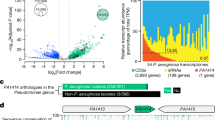
a Workflow of the CRISPRi library construction and fitness evaluation. Oligos (set 1 and 2) were synthesized to create sgRNA libraries targeting 5981 and 5971 genes, respectively. These oligos were then amplified by PCR to form double-strand DNA, which was subsequently cloned into pCRISPRi-ccdB. The resulting plasmids were transformed into E. coli WM3064 to generate sgRNA libraries, which were integrated in to the PA14 chromosome via triparental mating. Effect of CRISPRi on strain fitness in pool was evaluated after approx. 7 (G7, ~5 h incubation in exponential phase), 14 (G14, ~10 h), and 21 (G21, ~15 h) using Illumina sequencing and DEseq2 comparison of spacer abundance in libraries with and without CRISPRi activation (Dox +/−). Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. b Genome localization of essential genes identified by CRISPRi-seq and Tn-seq. The outer blue, red and cyan tracks represent genes annotated as essential in previous Tn-seq screens reported by refs. 40,41,37, respectively, whereas purple track represents genes identified in this study using CRISPRi-seq. c Comparative analysis of candidate essential gene sets from CRISPRi-seq and Tn-seq screens. d, e Line plots showing the log2FC trends in CRISPRi data for the non-essential (d) and essential (e) genes. Black dots represent sgRNA log2FC values. The solid black line represents the locally estimated scatterplot smoothing fit of the individual mean linear regression at different generations (0, 7, 14, and 21). Bar chart displays Functional classification (top 5) of genes with fitness changes for each cluster, based on available COG terms and manual classifications defined in Supplementary Fig. 2c. Source data are provided as a Source Data file.
To demonstrate the utility of our approach, we carried out CRISPRi-seq screens to assess the essential gene landscape in P. aeruginosa PA14 (Fig. 2a). The pooled libraries were cultured in LB broth and maintained in exponential phase by 1:100 back-dilution for 7, 14, and 21 generations with and without Dox induction. Through a single-step PCR amplification followed by Illumina sequencing, we profiled the abundance of each sgRNA in both induced and non-induced libraries. The fitness quantified by the log2FC in sgRNA abundance following induction versus non-induction conditions was then analyzed with the DESeq2 package in R39. A target gene was defined as essential if either of its two sgRNAs showed a fitness value of log₂FC <−1 with an adjusted P-value < 0.05 (see “Methods”). The performance of sgRNAs from set 1 and set 2 is shown in Supplementary Fig. 2a. According to this definition, our CRISPRi library screening across 7, 14, and 21 generations revealed 618 candidate essential genes in PA14 strain (Supplementary Data 5 and 6). The essential genes delineated by our CRISPRi-seq study exhibits significant overlap with those previously identified using Tn-seq screening methodologies37,40,41 (Fig. 2b, c and Supplementary Fig. 2b). However, differences exist among the Tn-seq studies, potentially due to variations in culture conditions or cut-offs for essential gene definition. In addition, CRISPRi-specific effects—such as transcriptional silencing at the operon level or polar effects may contribute to the identification of a distinct subset of essential genes not captured by transposon mutagenesis (Supplementary Data 6). Notably, some essential genes, such as tRNAs and the essential housekeeping gene tuf encoding translation elongation factor Tu42, through previous individual mutant studies, were identified exclusively by CRISPRi-seq (Supplementary Data 6). This underscores the utility of using CRISPRi-seq to generate a comprehensive list of essential genes in this pathogen.
As expected, the log2FC of the majority of sgRNAs targeting essential genes showed a gradual decline with increasing induction generations, indicating that the growth defects caused by the repression of these genes became more pronounced with prolonged CRISPRi repression, clear difference in depletion kinetics across targets were observed (Supplementary Data 5). Upon silencing, some genes exhibited significant growth defects after just 7 generations of induction, while others only displayed such defects at 14 or 21 generations. This variation suggests that P. aeruginosa essential genes respond differently to repression. Based on the kinetics, we classified them into five categories based on their vulnerability and responsiveness (Fig. 2d, e, Supplementary Fig. 2c, and Supplementary Data 6): cluster (1) vulnerable and quick-responsive genes, cluster (2) vulnerable and slow-responsive genes, cluster (3) invulnerable and quick-responsive genes, cluster (4) invulnerable and slow-responsive genes, and cluster (5) non-essential genes. Here, vulnerability is defined as a strong depletion phenotype (specifically genes with log₂FC < −4 at any time point), indicating a more severe fitness defect than the essentiality threshold (log₂FC < −1). Responsiveness reflects the timing of the fitness defect appearance, with “quick-responsive” genes showing substantial depletion (log₂FC < −2 for vulnerable; log₂FC < −1 for invulnerable) by generation 7, while “slow-responsive” genes reach similar levels only at later time points (generation 14 or 21). To evaluate the performance of sgRNAs, we compared the distribution of essential genes identified by each set across the vulnerability and responsiveness clusters. Set 1 consistently identified a greater number of essential genes—particularly those in clusters 1 and 2, which represent the most vulnerable and stringently defined categories—indicating higher knockdown efficacy in line with our sgRNA design strategy (Supplementary Fig. 2d). The representative genes from each category were selected and tested in CRISPRi knockdown growth assays, confirming that the vulnerable and quick-responsive category exhibited the most pronounced and rapid growth inhibition (Supplementary Fig. 3). Functional enrichment, based on COG (Clusters of Orthologous Groups), for the genes classified into different categories showed that the “translation, ribosomal structure, and biogenesis” and “cell wall/membrane/envelope biogenesis” COG categories were predominantly represented in the vulnerable classes (Fig. 1e). This highlights essential genes in these two categories as the most effective therapeutic targets, consistent with the fact that most used antibiotics target genes in these COGs43. Taken together, this proof-of-concept study with CRISPRi-seq in PA14 demonstrated the utility of this methodology for functional genomic analysis in P. aeruginosa.
Chemical genetic profiling of gallium in P. aeruginosa by CRISPRi-seq
Gallium therapy has been considered a potential treatment for P. aeruginosa infections, but its action mechanisms remain unclear23. To elucidate the genetic factors that modulate gallium’s effectiveness and identify potential targets for enhancing therapeutic efficacy in P. aeruginosa, we employed CRISPRi-seq screening to conduct a genome-wide investigation (Fig. 3a). The PA14 CRISPRi library was cultured in LB broth with Dox for 14 generations (preinduction), before shifting to medium with Ga(NO3)3 and culturing for another 7 generations (Fig. 3a). The concentration of Ga(NO3)3 used for the CRISPRi-seq was 200 µM, which leads to 70%–80% of growth reduction (Fig. 3b). By using a post-hoc filter of |log2FC| > 2, we identified 98 sgRNAs whose targeted repression correlated with increased fitness, while 9 sgRNAs were associated with decreased fitness, after the treatment with Ga(NO3)3 (Fig. 3c and Supplementary Data 7). A parallel screen with only 7 generations of pre-induction yielded fewer candidate hits with low confidence (Supplementary Fig. 4a). Therefore, we focused our analysis on 14 generation pre-induction dataset, which provided higher-resolution phenotypic profiles. Gene ontology (GO) enrichment analysis was utilized to discern the distinctive biological characteristics of gallium sensitivity-associated genes, which were assorted into various functional categories. There were 98 genes whose repression caused increased bacterial fitness under gallium treatment, enriched with various translation-related GO terms, such as the large ribosomal subunit, structural constituents of ribosomes, and translation processes (Supplementary Fig. 4b and Supplementary Data 8). This suggests that the deceleration of protein biosynthesis could potentially mitigate the deleterious effects induced by gallium treatment. Conversely, the genes whose repression led to reduced fitness were found to be involved in various metabolic processes, including energy metabolism and redox reactions. The enriched ontologies were GTP cyclohydrolase II activity, dihydrolipoyllysine-residue succinyltransferase activity, the oxoglutarate dehydrogenase complex, P450-containing electron transport chain, ferredoxin-NADP+ reductase activity, and riboflavin biosynthetic process (Supplementary Fig. 4b and Supplementary Data 8).

a Workflow for screening genes involved in sensitivity to Ga(NO3)3 treatment. The CRISPRi library was induced with Dox (25 ng/ml) for 14 generations, followed by culture with or without 200 μM Ga(NO3)3. Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. b Growth curves of PA14 in LB broth with various concentrations of Ga(NO3)3. c Volcano plots showing the identified genes involved in Ga(NO3)3 tolerance. d Growth of control strains PA14, and PA14 harboring CRISPRi system with non-targeting sgRNA (lucff). e Validation of targets associated with increased fitness: hitA, hitB, PA14_67550, PA14_56670, and thiE using CRISPRi with individual sgRNAs. f Schematic depicting HitAB involved in Ga3+ and Fe3+ uptake in P. aeruginosa. g Validation of screening hits with decreased fitness. Knockdown strains targeting PA14_22320, fdx2, fprB were constructed and their growth was tested in medium containing the indicated concentrations of gallium and Dox. Representative phenotypes under selected concentrations of Ga(NO₃)₃, chosen to best illustrate differential sensitivity, are shown. h Schematic illustrating the role of FprB in NADP(H)-dependent electron transfer. Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. i Plate spot assays showing sensitivity of PAO1 mutants to gallium. All growth data are presented as mean ± SD from three biological replicates. Source data are provided as a Source Data file.
To further explore the molecular mechanisms of P. aeruginosa’s response towards gallium treatment, we validated the top CRISPRi-seq hits by assessing the growth of the corresponding single guide RNA containing knockdown strains in the presence or absence of Ga(NO3)3. PA14 wild-type and PA14 harboring CRISPRi targeting lucff served as controls (Fig. 3d). Among selected 11 top-ranked targets, 8 were successfully validated using individual sgRNAs, showing the reliability of the screening. The top hit, hitA, conferred increased fitness under gallium treatment upon repression (Fig. 3e), and it encodes a ferric iron-binding periplasmic protein. Notably, our screening also identified hitB (Fig. 3e), a gene located in the same operon as hitA. The sgRNA targeting hitA likely also represses hitB due to the polar effects of the CRISPRi system, which may account for the more pronounced phenotype observed upon hitA repression. Consistent with our findings, a previous study identified a loss-of-function mutations in hitAB as a major contributor to increased gallium tolerance23. HitAB are involved in Fe3+ uptake, and were shown to provide the ion binding sites for internalization of gallium in P. aeruginosa44 (Fig. 3f). Other Fe3+ transportation systems were not directly identified in the screening. However, genes whose knockdown increased fitness under gallium stress included thiE, in the same operon as hemL, required for biosynthesis of heme, as well as PA14_56670, encoding an uncharacterized transcriptional regulator and being part of operon encompassing feoAB genes, encoding iron transport system. The deletion of hitA led to drastically increased survival of P. aeruginosa upon treatment with gallium (Fig. 3i and Supplementary Fig. 4c), indicating that HitAB could be the major transportation system utilized by Ga3+ for internalization.
The genes whose knockdown led to decreased fitness in gallium treatment included fprB, encoding a ferredoxin-NADP+ reductase; fdx2, encoding the ferredoxin 2Fe-2S; and PA14_22320, encoding a small hypothetical protein (Fig. 3g). Notably, Fdx2 and FprB are involved in the same biochemical reaction in P. aeruginosa (Fig. 3h). To validate these hits and additionally test the conservation of the observed phenotype in other P. aeruginosa strains, we engineered deletion mutants of PA3237 (ortholog of PA14_22320) and fprB (PA4615) in the PAO1 strain. We found fdx2 to be essential in our CRISPRi-seq screening, and it could not be deleted. Although individual CRISPRi strain targeting PA14_22320 confirmed the results of CRISPRi-seq screening, deletion of this gene did not decrease the tolerance to gallium treatment (Supplementary Fig. 4c, PA3237). Strikingly, the effect of fprB on enhancing P. aeruginosa sensitivity to gallium was abolished by deletion of hitAB (Fig. 3i), demonstrating that HitAB is the primary gallium transporter under these conditions and the gallium sensitivity induced by fprB deletion depends on active gallium uptake.
The ferredoxin-NADP+ reductase FprB is crucial for P. aeruginosa survival under gallium stress
The deletion of fprB did not cause any growth defect in LB agar or LB broth without Ga(NO3)3 (Fig. 4a). However, upon treatment with 5 or 10 μM Ga(NO3)3 severely impaired the growth of the ΔfprB mutant on LB agar and in LB broth, while the wild-type or complemented ΔfprB strain showed no growth inhibition (Fig. 4a, b). Minimum inhibitory concentration (MIC) measurements in Mueller–Hinton broth medium showed that deletion of fprB decreased the MIC of gallium from 320 µM to 10 µM (Supplementary Fig. 5a). Remarkably, time-killing assays showed that Ga(NO3)3 was bacteriostatic at all tested concentrations, as high as 1600 µM, towards the wild-type P. aeruginosa and complemented ΔfprB strain. However, for the ΔfprB mutant, as low as 12.5 µM Ga(NO3)3 led to a clear bactericidal effect (Fig. 4c). These results suggest that fprB plays an important role in maintaining the survival of P. aeruginosa under gallium treatment. Deletion of fprB improved the efficacy of gallium treatment against P. aeruginosa not only in terms of inhibition concentration, but also by shifting the killing mode from bacteriostatic to bactericidal. This study identifies an association between fprB and gallium susceptibility in P. aeruginosa, not previously reported.

a Growth curve of PAO1 wild type, ΔfprB and complemented strain (ΔfprB-Com) in the absence and presence of 12.5 μM Ga(NO3)3. b Plate spot assays showing sensitivity of PAO1 wt, ΔfprB, and ΔfprB-Com strains to gallium. c Time-kill assay of P. aeruginosa treated with Ga(NO3)3 in LB broth. Bacterial counts were determined at indicated timepoints by serial dilutions and plating on LB agar plates. d Reaction kinetic traces for determining FprB activity by 2,6-dichlorophenolindophenol (DCPIP)-diaphorase assay. e Molecular docking of FprB with FAD. Structure of FprB was predicted by Alphafold3 following molecular dynamics refinement. FAD is shown in Corey-Pauling-Koltun (CPK) colored sticks with carbons in light blue. The FprB amino acids predicted near FAD are indicated. f, g Impact of putative FAD binding disrupting amino acid changes (A57G, Y58A, F72A, and I74A) in FprB on P. aeruginosa’s resistance to Ga(NO3)3 treatment. Growth characteristics were determined by serial dilutions followed by dropping onto LB agar plates (f) or by growth using a microplate reader (g). Data from all growth and measurement assays are presented as mean ± SD from three biological replicates. Source data are provided as a Source Data file.
fprB encodes a ferredoxin-NADP+ reductase (Fpr) that facilitates the reversible electron transfer between NADPH and electron carrier proteins, including [Fe-S] clusters ferredoxin and flavodoxin45,46,47. To confirm its biochemical activity, we purified the P. aeruginosa PAO1 FprB protein and performed the NADPH-dependent 2,6-dichlorophenolindophenol (DCPIP) reduction assay as previously described48. The results showed that FprB efficiently catalyzed the reaction (Fig. 4d). Bacterial Fprs contain a prosthetic flavin adenine dinucleotide (FAD), which acts as an electron carrier essential for their catalytic activity49,50. Consistently, AlphaFold3 predicted FprB to bind FAD51 (Supplementary Fig. 5b). To test whether the observed phenotype of FprB is attributable to its biochemical function, we introduced point mutations into conserved amino acids within the FAD-binding domain of FprB. The mutations A57G, Y58A, F72A, and I74A were selected for analysis based on their proximity to the FAD binding site (Fig. 4e). As shown in Fig. 4f, g, the strains expressing mutated FprB variants exhibited increased sensitivity to gallium compared to both the wild-type and the strain complemented with wild-type FprB. These findings confirm that the FprB-mediated resistance to gallium therapy depends on its biochemical activity.
Mechanistic exploration of the increased sensitivity of the ΔfprB mutant to gallium
We next sought out to uncover the mechanism of increased sensitivity of P. aeruginosa towards gallium caused by inactivation of fprB. As a first step, we demonstrated that gallium didn’t inhibit the biochemical activity of FprB with an in vitro assay (Supplementary Fig. 5c). Next, we tested whether deletion of fprB leads to altered accumulation of intracellular gallium. Surprisingly, the fprB knockout strains displayed notably reduced intracellular gallium levels compared to both wild-type and complemented ΔfprB strains (Supplementary Fig. 5d). Given the antibacterial properties of gallium ions, which interfere with iron-dependent biological processes by attaching to iron-utilizing proteins and competing with iron for bacterial siderophore-mediated uptake52, we hypothesized that the deletion of fprB may influence the concentration of intracellular iron. Our results demonstrated that in the absence of fprB expression, P. aeruginosa accumulates higher intracellular Fe and ferrous iron (Fe2+) levels compared to wild-type and complemented strains (Fig. 5a). The data showed that increased gallium sensitivity of ΔfprB was not due to higher intracellular gallium, but rather to severe interference with an iron-dependent biological processes.

a Intracellular total iron and Fe2+ accumulation. Bacteria were cultured for 6 h in LB broth with or without Ga(NO3)3, starting at an OD600 of 0.05. Total iron or Fe2+ were measured as described in Methods, and normalized to OD600. b, c ROS and H2O2 accumulation in bacteria grown for 4 h in the absence or presence of Ga(NO3)3, detected by flow cytometry using carboxy-H2DCFDA and Peroxy Orange-1, respectively. d, e Plate spot assays for sensitivity of PAO1 wt, ∆fprB and ∆fprB-Com strains to H2O2 and Plumbagin. All strains were initially adjusted to an OD600 of 0.003 and then serially diluted tenfold before spotting onto LB agar plates (10−2 to 10−6). f Volcano plot of differential expression between ∆fprB and wt transcriptomes. Statistical analysis was performed using a two-sided Wald test implemented in DESeq2, with Benjamini-Hochberg correction for multiple comparisons. Significant difference was defined as (|log2FC|> 2 with Padj < 0.05). Red (upregulated); cyan (downregulated), grey (no difference). g The role of downregulated NO reductase (NorBC), N2O reductases (NosZ) and NosR, and upregulated PA2691 and CyoABCDE in bacterial aerobic respiration. h NO levels in the supernatant of bacterial cultures. i Proposed mechanism of gallium-induced cell death in wt and ∆fprB mutant. Statistical analysis for (a, c, and h) was performed using Two-way ANOVA and Tukey’s multiple-comparison test. All data are presented as mean ± SD from three biological replicates. Source data are provided as a Source Data file.
Increased intracellular ferrous ions can react with hydrogen peroxide via the Fenton reaction, leading to the accumulation of reactive oxygen species (ROS), which may cause severe oxidative damage and cell death53,54. To test whether the increased gallium sensitivity of the ΔfprB mutant is linked to increased ROS, we measured intracellular hydrogen peroxide using Peroxy Orange-1 and general ROS using carboxy-H2DCFDA, the two specific fluorescent indicator dyes (Fig. 5b, c and Supplementary Fig. 5e). The analysis showed that gallium treatment induced hydrogen peroxide accumulation in both wild-type and ΔfprB mutant strains, with higher levels observed in ΔfprB mutant. Specifically, the wild-type strain treated with 50 µM Ga(NO3)3 exhibited similar levels of hydrogen peroxide as the ΔfprB mutant treated with 12.5 µM Ga(NO3)3. No detectable hydrogen peroxide was observed in the fprB-complemented strains, possibly due to FprB overexpression. These findings indicate that FprB is involved in modulating hydrogen peroxide production under gallium treatment. Furthermore, ROS accumulation was observed exclusively in the ΔfprB mutant, but not in the wild-type or complemented strains, under the tested gallium concentrations. This finding underscores the role of FprB in regulating ROS levels and its contribution to gallium sensitivity. Consistently, intracellular concentrations of NADP+ and NADPH were significantly increased in the ΔfprB mutant under gallium treatment compared to the wild-type or complemented strain (Supplementary Fig. 6a), indicating elevated oxidative stress in the knockout strain55. The elevated NADP+/NADPH ratio in the ΔfprB mutant under gallium stress further demonstrated the loss of a reduced redox state (Supplementary Fig. 6a). Notably, supplementing NADPH to the medium increased the survival of the ΔfprB mutant upon gallium exposure (Supplementary Fig. 6b). Further supporting the role of FprB in controlling ROS, ΔfprB mutant exhibited increased sensitivity to hydrogen peroxide and plumbagin, a known ROS inducer56 (Fig. 5d, e). Conversely, exogenous supplementation of the hydroxyl radical scavenger dimethyl sulfoxide (DMSO) partially restored gallium-impaired growth in the ΔfprB background (Supplementary Fig. 6c). To test if FprB can serve as a synergistic target for antibiotics that induce ROS, we treated the ΔfprB mutant with colistin (membrane disruptor), ciprofloxacin (DNA gyrase inhibitor), and apramycin (protein synthesis inhibitor). However, no increased synergistic or even additive effect was observed (Supplementary Fig. 6d), indicating ROS potentiation by FprB deficiency may be pathway-specific, which likely requires direct oxidative stressors rather than antibiotic-induced ROS under these conditions. In addition, checkerboard assays demonstrated the absence of synergistic interactions between gallium and colistin, ciprofloxacin, or apramycin (Supplementary Fig. 6e).
To further elucidate the mechanism by which FprB controls ROS production, we performed RNA-seq analysis on wild-type and ΔfprB mutant strains grown in LB medium to an OD600 of 0.5–0.7 (Fig. 5f and Supplementary Data 9). The analysis revealed that several genes related to nitric oxide reduction, such as norB, norC, nosZ, and nosR, were significantly downregulated in the ΔfprB mutant. Notably, while these genes exhibited substantial fold-changes (|log2FC|> 2), their adjusted P-values approached the significance threshold (Padj < 0.05), indicating a trend-level association that merits further validation. NorBC catalyzes the two-electron reduction of NO to N2O, while NosZ is the only enzyme known to reduce N2O to N2 with NosR as the electron donor for N2O reduction (Fig. 5g, upper panel). Inhibition of the denitrification respiratory pathway can elevate NO levels, a reactive nitrogen species free radical, leading to nitrosative stress and subsequent cell injury and death57,58. Consistent with the transcriptional downregulation of norBC, nosZ, and nosR, we observed elevated NO levels in the ΔfprB mutant compared to wild-type (Fig. 5h). This phenotype was further exacerbated under gallium treatment, with the ΔfprB mutant exhibiting significantly higher NO accumulation than wild-type (Fig. 5h), suggesting a synergistic interaction between FprB deficiency and gallium-induced nitrosative stress. In addition, mgtA (PA4825) and the neighboring genes, which might be in the same operon (PA4822-24) were also found to be significantly downregulated in the ΔfprB mutant. The genes upregulated in the ΔfprB mutant includes PA2691, cyoA, cyoB, cyoC, cyoD, and cyoE. PA2691 is an NADH dehydrogenase that catalyzes electron transfer from NADH to quinone, serving as a key entry point for electrons in the respiratory chain59. The cytochrome o complex (CyoABCDE) is an ubiquinol oxidase involved in bacterial aerobic respiration. It catalyzes the two-electron oxidation of ubiquinol-8 and the four-electron reduction of O2 to H2O, coupling electron flux to proton motive force generation across the membrane60 (Fig. 5g, lower panel). Based on the above information, we propose a model to elucidate the increased gallium sensitivity and bactericidal effect observed in the ΔfprB mutant (Fig. 5i): Upon uptake by P. aeruginosa, gallium disrupts iron-dependent enzymes and metal homeostasis, resulting in hydrogen peroxide production. In the wild-type strain, detoxification enzymes degrade hydrogen peroxide, preventing further ROS generation and subsequent cell death. However, in the ΔfprB mutant, gallium-induced hydrogen peroxide reacts with increased intracellular iron, fueling the Fenton reaction and leading to ROS accumulation. Concurrently, the mutant exhibits a higher basal level of nitric oxide due to downregulation of nor genes and an enhanced respiratory chain from upregulated electron transfer genes, potentially exacerbating ROS leakage. The accumulation of ROS likely hastens cell death, thereby explaining the pronounced bactericidal action of gallium against the ΔfprB mutant.
Notably, there is another ferredoxin-NADP+ reductase encoding gene fprA, which is an essential gene (Supplementary Fig. 7a), in the chromosome of P. aeruginosa, encoding a protein which shares 42% amino acid sequence identity with FprB in both PAO1 and PA14 strains (Supplementary Fig. 7b, c). However, neither FprA nor its substrate Fdx1 was identified in our CRISPRi-seq screen under gallium stress (Supplementary Fig. 7d). Introducing the fprA coding sequence under the control of the original fprB promoter into the ΔfprB mutant led to a slight but statistically significant increase in gallium tolerance at later timepoints, though the biological effect was limited (Supplementary Fig. 7e, f). These findings suggest that although FprA and FprB are both ferredoxin-NADP+ reductases, their function are only partially overlapping.
Targeting FprB may enhance the efficacy of gallium-based therapies against P. aeruginosa
Given the hypersensitivity of the P. aeruginosa ΔfprB mutant to gallium-based treatment in vitro, we investigated FprB as a potential synergistic target for gallium therapy. A comprehensive genome analysis of all the 981 P. aeruginosa strains with full genomes available in NCBI revealed that all but one contained the fprB ortholog, with each strain harboring a single copy (Fig. 6a). The genomic context of fprB exhibited conserved synteny across different genomes, with the order and transcriptional orientation of flanking genes remaining highly consistent (Fig. 6b). The protein sequences of FprB showed minimal variance (Supplementary Fig. 7g, h). Despite minor variations, the genes exhibited significant collinearity (Supplementary Fig. 7g), indicating a shared evolutionary origin. The FprB protein sequences were highly conserved, with only 20 polymorphic sites detected among the 258 amino acid positions in representative strains from various phylogenetic sub-clusters, indicating that even the largest variation still maintained 95.7% identity with the consensus sequence (Supplementary Data 10). These findings demonstrate that FprB is highly conserved in P. aeruginosa and represents a universal applicable target.

a Genome analysis of 981 P. aeruginosa strains from NCBI shows the presence of the fprB gene in most strains except GCA_00473745.3 with a single copy. b Genomic context analysis of the fprB gene in 981 P. aeruginosa strains reveals consistent gene order across different genomes. c Biofilm formation analysis in P. aeruginosa static cultures incubated 24-h incubation at 37 °C with or without Ga(NO₃)₃. Data are presented as mean ± SD from triplicates. d, e Survival of mice infected with different strains and treated with gallium. BALB/c mice were intranasally administrated with P. aeruginosa (2.5 × 107 c.f.u.). At 3 h.p.i., mice were treated with 50 µL of PBS or 250 mM Ga(NO₃)₃ via intraperitoneal injections. (n = 6 per group). Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. f, g Bacterial load in lungs of infected mice. BALB/c mice (female, 6–8 weeks) were intranasally administrated with P. aeruginosa (1.0 × 107 c.f.u.). At 3 h.p.i., mice were treated with 50 µL of PBS or 250 mM Ga(NO3)3 via intraperitoneal injections. At 27 h.p.i., mice were sacrificed to assess the bacteria load in the lung. Created in BioRender. Liu, X. (2025) https://BioRender.com/kcvjnbt. g Bars represent mean ± SD from six biologically independent samples per group (n = 6). Individual data points are shown. Statistical analysis was performed by default two-sided Gehan-Breslow-Wilcoxon test (e) or Two-way ANOVA and Tukey’s multiple-comparison test (c, g). Source data are provided as a Source Data file.
P. aeruginosa is known to form biofilms in CF airways61. We tested the efficiency of gallium in inhibiting biofilm formation in both wild-type and ΔfprB strains. Gallium effectively inhibited biofilm formation in the ΔfprB mutant at concentrations as low as 5–10 µM, levels comparable to peak plasma and sputum concentrations reported in humans23 (Fig. 6c). Biofilm formation in the wild-type and complemented strain was not affected (Fig. 6c). To further evaluate FprB as a synergistic target for gallium therapy in vivo, we utilized a murine lung infection model (Fig. 6d, f). Deletion of fprB in P. aeruginosa significantly increased animal survival and reduced pulmonary bacterial burden upon gallium therapy (Fig. 6e, g). These results underscore the need for further investigation into FprB as a therapeutic target.
Discussion
Our study establishes and validates a tetracycline-inducible pooled CRISPRi-seq screening strategy to uncover the genetic basis of growth-related traits in the human pathogen P. aeruginosa. By applying CRISPRi-seq to profile the chemical genetic landscape of gallium treatment, we identified genes contributing to gallium susceptibility at a genome-wide scale. Notably, FprB emerged as a synergistic target for such therapy, demonstrating the effectiveness of CRISPRi-seq based genome-wide forward genetic screenings. Future research should prioritize the development of adjuvants targeting FprB to improve the selectivity and efficacy of gallium-based therapeutics.
The CRISPRi technique has become an increasingly popular tool for exploring gene functions in various bacteria under given conditions16,62,63,64,65. However, its application in P. aeruginosa is rare, and no genome-wide scale study employing CRISPRi-seq has been described. In contrast, several genome-wide analyses of gene function in P. aeruginosa employing Tn-seq techniques have been reported6,7,41,66. Transposon-based techniques have limitations, such as inability to insert at defined sites, restriction to analysis of only non-essential genes, and a propensity for polar effects8,13. The engineered tet-inducible CRISPRi system presented in this study could provide robust reinforcement for the transposon-based techniques. This study presents a genome-wide identification of essential genes in P. aeruginosa PA14 using CRISPRi-seq. By designing sgRNAs for all annotated genetic features and constructing compact CRISPRi libraries, we achieved high coverage and robustness in our study. Through CRISPRi-seq screenings, we identified 618 candidate essential genes, many of which overlap with essential genes previously identified by Tn-seq screenings in the same strain37,40,41 (Fig. 2b, c). Notably, some essential genes were uniquely identified through CRISPRi-seq screening, highlighting the distinct advantages of this approach. However, the potential for polar effects of the CRISPRi system on genes within the same transcriptional unit30,62, which may lead to false positives, must be considered. To address this, we incorporated operon information for each gene in P. aeruginosa PA14 (Supplementary Data 6), enabling a more accurate interpretation of the CRISPRi-seq data. Compared to previous Tn-seq studies, our CRISPRi-seq screen identified a broader set of essential and fitness-contributing genes. This divergence likely reflects differences in experimental conditions, including medium composition and stress exposure, as well as the tunable and conditional nature of CRISPRi-based repression. In addition, we used relatively inclusive thresholds to capture both core essential and context-dependent fitness genes, thereby generating a more comprehensive resource. Reanalysis with stricter cutoffs showed greater convergence with prior studies, supporting the robustness of the core gene set. These results demonstrate that CRISPRi-seq complements transposon-based approaches by uncovering additional layers of genetic vulnerability.
Subdividing essential genes into “vulnerable” and “invulnerable” categories—and further into “quick” versus “slow” responders—captures the temporal dynamics of bacterial cell fitness response to gene silencing. A rapid, pronounced drop implies acute dependence on the targeted gene, whereas a delayed depletion may reflect metabolic buffering or slower turnover of functional gene products. Conversely, genes showing moderate yet consistent depletion were classified as invulnerable, highlighting subtler contributions to cellular fitness. Such categorization of essential genes provides valuable insights into their potential as therapeutic targets11,12. Nevertheless, it is important to recognize that sgRNA efficiency may confound these classifications, and its contribution requires further systematic investigation in P. aeruginosa. Notably, genes of the “translation, ribosomal structure, and biogenesis” and “cell wall/membrane/envelope biogenesis” COGs were generally identified as vulnerable. These findings highlight these genes as the most promising targets for antimicrobial development. In fact, most clinically used antibiotics act on targets within these two categories43. Additionally, 28 genes categorized as “vulnerable” fall under the “Function unknown” COG category (Supplementary Data 6). Further research is needed to elucidate the functions of these genes or the associated operons, as they could represent important therapeutic targets in P. aeruginosa. In summary, this proof-of-concept study highlights the efficacy of CRISPRi-seq in dissecting the genetic landscape of P. aeruginosa, paving the way for novel therapeutic strategies. Future studies should focus on refining the gene vulnerability profiles under diverse growth conditions, particularly those relevant in vivo.
In this study, we focused on chemical genetic profiling via CRISPRi-seq to elucidate genetic factors that influence the efficacy of gallium therapy against P. aeruginosa. We identified genes whose repression either enhanced or diminished bacterial fitness under gallium treatment. We acknowledge that gallium-induced growth reduction in the CRISRPi-seq screen, resulting in lower cell density, may have decreased competition among strains and potentially reduce the sensitivity of the assay. This limited competition might mask fitness defects in mutants with moderate sensitivities. However, subsequent validations with individual knockdowns of key candidates validated the existence of the gallium-induced phenotypes, reinforcing the robustness of our screening outcomes despite these limitations. Key findings include the identification of hitA and hitB, both involved in Fe3+ uptake, whose repression enhances bacterial fitness, in agreement with previous findings in P. aeruginosa44,67. The identification of ferric ion-associated mechanisms is not surprising, as gallium and ferric ion share comparable radii and chemical properties, allowing gallium ion to be incorporated via the route of the P. aeruginosa iron uptake system involving HitAB and to act as an iron mimic in the biological processes26,67. The exclusive identification of HitAB, but not other iron uptake systems, indicates HitAB is the primary transport system utilized by gallium.
CRISPRi screening also allowed identification of genes whose loss of function results in increased sensitivity to gallium. Notably, the deletion of fprB led to a substantial reduction in the minimum inhibitory concentration (MIC) of gallium from 320 µM to 10 µM in Mueller–Hinton broth, a nutrient-rich medium widely used as the standard for antibiotic susceptibility testing. It is worth noting that lower MIC values for gallium have been reported in low-iron media22,26, suggesting that iron availability plays a critical role in modulating gallium toxicity. This reduced MIC falls within the range of peak plasma and sputum concentrations observed in humans (8–12 µM)23, underscoring the potential of fprB as a promising therapeutic target. It has been demonstrated that gallium can prompt an increased intracellular ROS level68,69. FprB is a ferredoxin-NADP+ reductase that binds FAD and NADP(H) via its N- and C-terminal domains and plays a crucial role in detoxifying ROS and redox homeostasis47,49. Mechanistic investigation showed the ΔfprB mutant displayed elevated intracellular iron and ferrous iron (Fe2+) levels upon gallium treatment (Fig. 5a), which may react with the gallium-induced ROS, especially hydrogen peroxide, leading to production of highly reactive radical species via the Fenton reaction53,54. This is consistent with the observed increase in hydrogen peroxide and ROS levels in the ΔfprB mutant under gallium treatment, compared to wild-type and complemented strains (Fig. 5b, c). RNA-seq analysis of the transcriptomes of the ΔfprB and wild-type strains revealed upregulation of genes associated with aerobic respiration and downregulation of those involved in nitric oxide reduction in the ΔfprB mutant. These transcriptional changes likely exacerbate ROS production, thereby promoting damage to cellular constituents. Increased oxidative stress responses, such as upregulation of NADPH, glutathione, or superoxide dismutase, have been reported to enhance P. aeruginosa tolerance to gallium70. In our study, supplementation with 1 mM NADPH slightly improved the survival of the ΔfprB mutant but did not restore its MIC (Supplementary Fig. 6b), suggesting that FprB deficiency disrupts both iron homeostasis and redox balance. This complex sensitivity phenotype cannot be fully rescued by enhancing the antioxidant response alone. Notably, FprA and FprB, the two ferredoxin-NADP⁺ reductases in P. aeruginosa, exhibit functional divergence in their response to gallium stress. While FprB emerged as a critical modulator of Ga(NO₃)₃ sensitivity in this study, FprA displayed no discernible role under these conditions. The inability of FprA overexpression to fully rescue ΔfprB gallium hypersensitivity underscores their only partially redundant roles. FprA is essential for normal growth of P. aeruginosa, indicating that this protein may underpin fundamental redox homeostasis processes that are indispensable for viability, potentially masking its involvement in gallium-specific resistance mechanisms.
Previous studies showed that gallium exhibits a bacteriostatic mode of action against P. aeruginosa by affecting the key enzymes in multiple metabolic pathways71. Remarkably, our findings revealed a shift in gallium’s mode of action from bacteriostatic to bactericidal upon the loss of the fprB gene. This highlights how the genetic background of bacteria can significantly influence the effectiveness of an antimicrobial, transforming its impact from merely inhibiting growth to actively killing the pathogen. These insights offer a promising strategy to enhance gallium therapy by exploiting specific genetic vulnerabilities in P. aeruginosa. Lastly, considering that gallium therapy for CF patients suffering from chronic P. aeruginosa infections is currently in Phase II clinical trials and has shown promising outcomes, we explored the potential of FprB as a synergistic target to enhance the efficacy of this treatment with CF infection related models. Given that P. aeruginosa forms resilient biofilms in the CF airway, which are a major obstacle to effective treatment61, we performed biofilm inhibition assays and utilized a murine lung infection model to evaluate the impact of targeting FprB. Our results demonstrated that P. aeruginosa strains lacking fprB showed significantly reduced biofilm formation, even at low gallium concentrations (5–10 μM). In the murine lung infection model, the absence of fprB significantly enhanced the efficacy of gallium. Furthermore, suboptimal concentrations of gallium (25 μM) have been reported to enhance the virulence of P. aeruginosa in vitro, raising concerns about the potential side effects of gallium therapy72. We propose that simultaneously targeting FprB with gallium therapy could mitigate this issue. The reduced gallium MIC (10 μM) observed in ΔfprB mutants enables effective therapeutic outcomes at concentrations lower than those required to induce virulence factor expression. Together, this study provides deeper insight into the genetic interactions with gallium, paving the way for more effective therapeutic strategies against P. aeruginosa infections.
Methods
Ethical compliance
All animal experiments were conducted in accordance with relevant ethical regulations. The study protocol was approved by the Institutional Animal Care and Use Committee of Shenzhen University Medical School (Approval Number: IACUC-202400111).
Bacterial strains and growth conditions
All strains and plasmids utilized in this study are listed in Supplementary Data 1, while primers are in Supplementary Data 2. Pseudomonas strains used in this study includes PAO1, PA14, P5, and PDO300. Routine cultures for Pseudomonas strains were performed at 37 °C in Luria-Bertani (LB) broth or on LB agar plates with antibiotic where necessary: 20 μg/mL gentamicin and 30 μg/mL apramycin. For gallium treatment CRISPRi-seq screen, PA14 were cultured in M9 minimal medium containing Na2HPO4·7H2O (47.8 mM), KH2PO4 (22 mM), NaCl (8.6 mM), NH4Cl (18.6 mM), MgSO4 (1.0 mM), CaCl2 (0.1 mM), and sodium citrate (20 mM) as the carbon source at 37 °C under vigorous aeration (250 rpm). E. coli WM3064 represents a diaminopimelic acid (DAP)-auxotrophic strain73, offering significant utility for conjugation experiments and replication of plasmids necessitating Pir protein. E. coli strains were cultivated in LB broth at 37 °C, supplemented with antibiotics where necessary: 20 μg/mL gentamicin and 100 μg/mL ampicillin. Cultures were subjected to CRISPRi repression, primarily induced with 25 ng/mL doxycycline (Dox), unless stated differently.
Development of a tetracycline (tet)-inducible platform in P. aeruginosa
The tet-inducible system for P. aeruginosa was developed based on our previously published tet-inducible system in Streptococcus pneumoniae, which proved to be very tightly controlled13,74. A gblock (gblock 6-TetPt5) containing Pseudomonas codon-optimized tetR (termed tetR-opt), a constitutive Pcon promoter and tet-inducible Ptet promoter were synthesized by Sangon (Sangon, China). The sequence of gblock 6-TetPt5 is shown in Supplementary Data 3. The tet-inducible system uses a modified pUC18T-mini-Tn7T-P3-luxABCDE-based integration vector (Supplementary Data 1), incorporating a constitutively expressed tetR-opt driven by the Pcon promoter, amplified with primers OXL384 and OXL567 (Supplementary Data 2). The P3 promoter was substituted with a Ptet (amplified with OXL387 and OXL568) to drive luxABCDE reporter gene, enabling precise quantification of tet-regulated gene expression. Herein, we designated the plasmid containing the tet-inducible system as Ptet-lux-Tn7T-Gm. All PCR fragments were amplified using Phanta DNA Polymerase (Vazyme, P515-03, China) in preparation for In-Fusion cloning with the ClonExpress Ultra One Step Cloning Kit V2 (Vazyme, C116-02, China). The Ptet-lux-Tn7T-Gm plasmid can be introduced into P. aeruginosa via triparental conjugation18, resulting in Tn7 transposition of the Ptet-luxABCDE fragment and its integration into the chromosome downstream of glmS.
Construction of the ccdB counterselection system for sgRNA cloning
The ccdB counterselection system75 was employed to enhance the efficiency of sgRNA cloning through negative selection against non-modified plasmids. Firstly, E. coli WM3064 with mutated gyrA (462Arg→Cys) was engineered via CRISPR/Cas9 editing as previously reported76, to confer resistance to the CcdB toxin77. Sequences of the sgRNA and single strand DNA repair template designed to introduce the gyrA462Arg→Cys mutation by CRISPR/Cas9 editing were listed in the oligo list (Supplementary Data 2). The obtained E. coli WM3064 gyrA462Arg→Cys was confirmed by Sanger sequencing, used as the host for construction of sgRNA cloning vector with ccdB counter selection system. The ccdB gene flanked by two BsaI sites, ordered as the gBlock “gblock4-ccdB-CSY” (Supplementary Data 3) was amplified using primers OXL289 and OXL290, and then cloned into the Mobile-CRISPRi plasmid pJMP284629 (Addgene#160676) via BsaI-mediated Golden Gate Assembly. The product of Golden Gate Assembly was transformed into E. coli WM3064 gyrA462Arg→Cys followed by selection with 30 μg/mL apramycin on LB agar plates supplied with 300 μM DAP. The resulting plasmid is named pJMP2846-ccdB, which serves as the vector for constructing the CRISPRi system and subsequent sgRNA cloning.
Construction of the tet-inducible CRISPRi system
Our CRISPRi system was primarily constructed based on pJMP2846-ccdB. The tet-inducible element mentioned above was amplified and introduced to replace the native IPTG-inducible part (PLlacO1-LacI) that drives dCas9 expression in pJMP2846-ccdB, using Primers OXL657, OXL658, OXL659, and OXL660 (Supplementary Data 2). The cassette containing Illumina read 1, P3 promoter, BsaI site-flanked mCherry, sgRNA scaffold, sgRNA terminator, and Illumina read 2 was generated from plasmid pPEPZ-sgRNAclone (Addgene#141090)9 using primers OXL226 and OXL227. The kanamycin resistance gene segment was replaced with one conferring apramycin resistance gene using primers OXL483 and OXL484. The construct was then transformed into E. coli WM3064 and selected with 30 μg/mL apramycin on LB agar plate supplied with 300 μM DAP. The resulting plasmid was named pCRISPRi-ccdB. To test the efficiency of pCRISPRi-ccdB for sgRNA cloning and counter selection, sgRNA targeting different genes were cloned by BsaI mediated Golden Gate Assembly. Two 24-nt oligonucleotides with 5′-ends of TATA and AAAC, respectively, were synthesized for each guide sequence and annealed in TEN buffer (10 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 8.0) in a thermocycler. The annealing process involved incubation at 95 °C for 5 min and then slowly cooling them to room temperature to allow for proper pairing. The annealed oligonucleotides and pCRISPRi with ccdB vector were assembled based on the One-step Golden Gate protocol76, using DNA restriction enzyme BsaI (Lablead, F5518S, China) and ligase T4 (Vazyme, C301-01, China). The assembly mixtures were transformed into E. coli WM3064 and plated on LB agar plates containing 30 μg/mL apramycin and 300 μM DAP for selection of successful assembly clones. As a control to test the efficiency of CcdB counter selection, the intact pCRISPRi-ccdB plasmid without sgRNA cloning was transformed into E. coli WM3064 in parallel.
Construction of pooled CRISPRi libraries in P. aeruginosa PA14
sgRNA design
The sgRNAs were designed by our previously published R script for automatic sgRNA selection (https://github.com/veeninglab/CRISPRi-seq) using the option “HawkinsEcoMedian” for the reprAct_penalties input parameter, and otherwise default parameters9. All sgRNAs were designed to target the coding strand of the genes. Briefly, all possible candidate sgRNAs for each gene are scored on the basis of (1) distance of sgRNA target binding site to target start codon (on-target score), and (2) number and within-spacer position of mismatches with all other potential binding sites in the genome, i.e., all other Protospacer Adjacent Motifs sites (off-target score). These scores are weighted, prioritizing candidates with a minimal maximum expected off-target effect. Given this metric, the two best-ranking sgRNAs were selected for this study and assigned to set 1 (5981 sgRNAs) and 2 (5971 sgRNAs), respectively. The 20-nt spacer sequences for all designed sgRNAs and their targets, as well as the 16 non-targeting (NT) control sgRNAs, are provided in Supplementary Data 4.
sgRNA cloning
The set 1 and set 2 spacer containing oligo pools were synthesized on chip by GenScript (Nanjing, China). The oligo sequences were listed in Supplementary Data 4. Each oligo contains a 20-nt sgRNA spacer sequence flanked with 2 BsaI sites, the set specific sequence for amplification of either set 1 or set 2 sgRNAs, and the universal sequence for amplification of all the sgRNAs in one chip. Set 1 and set 2 sgRNA pools were amplified by PCR with primer pairs OXL409/OXL410 and OXL411/OXL412 (Supplementary Data 2), respectively. The amplicons were purified with a Monarch DNA Cleanup kit (NEB, T1030S, USA). The purified amplicons were cloned into the plasmid pCRISPRi-ccdB by Golden Gate Assembly with use of BsaI. Specifically, for each Golden Gate Assembly reaction, 300 ng of the amplicons were mixed with 500 ng of the pCRISPRi-ccdB plasmid in a 200-μL PCR tube, followed by addition of 1 μL of BsaI (Lablead, F5518S, China), 1 μL of T4 ligase (Vazyme, C301-01, China) and 1 µL of 10× T4 ligation buffer. MiliQ water was added into the PCR tube to bring the final volume to 20 µL. The reaction mixture was incubated in a thermocycler with the following program: 150 rounds of 37 °C for 1.5 min, 16 °C for 3 min, and 1 round of 37 °C for 5 min, 80 °C for 10 min. The product of the Golden Gate Assembly reaction was then transformed into chemical competent E. coli WM3064 and the successful transformants were selected on LB agar plates with 30 μg/mL apramycin and 300 μM DAP. In total, such 20-μL Golden Gate Assembly mixtures were performed and transformed 60 times. Over 250,000 transformant colonies were harvested for each sgRNA pool, summing to more than 50 times average sgRNA library coverage.
Construction of the CRISPRi library in P. aeruginosa PA14 via triparental conjugation
The E. coli WM3064 with sgRNA library were served as donor strain and subsequently conjugated into P. aeruginosa PA14 strain with a helper E. coli WM3064 strain harboring a plasmid (pJMP1039, Addgene#119239)18 that allows expression of TnsABCD transposase complex. The triparental conjugation was performed as describe previously29. The successful transconjugants with integration of Tn7 cassette into PA14 chromosome were selected on LB agar plates supplemented with 30 μg/mL apramycin at 37 °C. In total, the CRISPRi library construction involved collecting around 300,000 colonies for the set 1 sgRNA pool and 570,000 colonies for the set 2 sgRNA pool.
CRISPRi-seq Screen
For the gene essentiality screen of P. aeruginosa PA14, the pooled libraries were cultured in LB broth at 37 °C until they reached an OD600 of 0.6, then diluted 1:100 into fresh LB containing or lacking 25 ng/mL Dox to induce the dCas9 expression. Samples were collected after approximately 7, 14, and 21 generations of exponential growth. For the 7-generation sample, cultures were harvested at OD600 = 0.6 following the first dilution. To obtain samples at 14 and 21 generations, cultures were subjected to two additional 1:100 dilutions, with harvesting at OD600 = 0.6 after each round. At each time point, 5 mL of culture was collected. Bacterial pellets were used for genomic DNA isolation using the FastPure Bacteria DNA Isolation Mini Kit (Vazyme, DC103-01, China), following the manufacturer’s instructions. All experiments were performed in four independent biological replicates.
For the CRISPRi-seq screen under Ga(NO3)3 treatment, pooled libraries were pre-induced in LB broth with Dox for 14 generations, followed by treatment with Ga(NO3)3 (100 or 200 μM) plus Dox in M9 medium and the culture continued growing until 21 generations. The spacer-encompassing region was amplified from genomic DNAs using a one-step PCR process, which incorporated Illumina barcodes N701-N712 and N501-N512 as index 1 and index 2, respectively, yielding 303 bp products9. The amplicons were sequenced on an Illumina NovaSeq system by Haplox (Shenzhen, China).
CRISPRi-seq data analysis
The relative abundance of each sgRNA per condition from the raw pair-end sequencing data of the CRISPRi-seq screen was determined with 2FAST2Q (v2.5.0)38. Using the default configuration of 2FAST2Q, a nucleotide-based quality filtering step discards all trimmed reads with a Phred score below 30. The depletion or enrichment of sgRNAs was analyzed using DESeq2 (Version 1.40.2) in R (version 4.3.1)39. In the gene essentiality screen, fitness values were calculated by comparing the CRISPRi induction with Dox to the non-induction condition (+Dox Vs -Dox). The analysis was performed with DESeq2 default parameters with alpha = 0.05. Gene essentiality was further determined by the calculated log2FC values from both set 1 and set 2 screening results over 7, 14, and 21 generations. Essential genes were defined as those for which at least one sgRNA (from either set 1 or set 2) showed a log₂FC ≤ −1 at any tested generation (7, 14, or 21). The results from individual analyses of set 1 and set 2 across 7, 14 and 21 generations are displayed in Supplementary Data 5. For vulnerable/invulnerable categorization, we adopted a two-tiered classification system: (1) initial grouping based on log2FC thresholds (Supplementary Fig. 2c), which was optimized to maximize biological relevance while minimizing false positives; and (2) temporal stratification into quick/slow responders using 7-generation log2FC cutoffs (−2 for vulnerable, −1 for invulnerable), reflecting distinct kinetic profiles of gene repression impact on fitness. The essential gene list was compared with three Tn-seq studies conducted in P. aeruginosa PA1437,40,41. Core essential genes identified by Poulsen et al.37 were highlighted in the list (Supplementary Data 6). Operon information was extracted from the study by ref. 78. The generated results, including the classification of genes into five categories, comparison with Tn-seq studies, and operon information, are presented in Supplementary Data 6.
For the gallium treatment CRISPRi-seq screen, fitness values were determined by comparing the Ga(NO3)3-treated condition with Dox to the untreated condition [+Ga(NO3)3 Vs -Ga(NO3)3]. Differential genes were identified by DEseq2 analysis with default parameters with alpha = 0.05. A posterior filtering for log2FC ≥ 2 was used to define sgRNA targets as increased fitness genes, and a log2FC ≤ −2 as decreased fitness genes. The results are provided in Supplementary Data 7. GO enrichment analysis for the differential essential targets was conducted and is presented in Supplementary Data 8.
Construction of knockdown and knockout strains
The gene knockdown strains were engineered using a method akin to library construction but utilized a singular targeted sequence. The genes of interest in P. aeruginosa PAO1 were deleted via CRISPR-Cas9 genome editing with pCasPA/pACRISPR system as previously described76. In brief, the pCasPA plasmid expressing Cas9 was first introduced and colonies were selected with 100 μg/mL tetracycline. The pACRISPR plasmid containing sgRNA was then electroporated with homologous repair templates flanking the target gene into PAO1 cells harboring pCasPA. The homologous regions were PCR amplified and then purified through gel extraction. Putative knockout mutants were isolated on LB agar with 100 μg/mL tetracycline and 200 μg/mL carbenicillin selection and validated by sanger sequencing. To achieve gene complementation, the Ptet-lux-Tn7T-Gm plasmid, which facilitates gene expression under a tet-inducible promoter, was utilized. The gene’s open reading frame was PCR-amplified from PAO1 genomic DNA using primers with 15 bp extensions and subsequently cloned into the ptet-lux-Tn7T-Gm vector through In-Fusion cloning technology (Vazyme, China). This recombinant plasmid was introduced into the WM3064 strain via transformation and selected using 20 mg/mL gentamicin and 300 μM DAP. Following triparental conjugation, the fragment with gene complementation was incorporated into the genome of the PAO1 knockout strain. Complemented mutants were selected on LB agar containing 20 μg/mL gentamicin and further confirmed by PCR and sequencing.
Growth and luminescence measurements
For each strain examined, working stocks were inoculated and diluted 1:100 in fresh LB broth with or without varying concentrations of Ga(NO3)3 or Dox, to initiate the cultures. The starting OD600 was standardized to 0.003 for all cultures. A total of 100 µL of the bacterial culture was dispensed into 96-well flat-bottom cell culture plates (NEST Biotechnology, 701011, China) in triplicates. Bacterial growth was monitored at 37 °C using a Tecan Spark microplate reader (Tecan, Switzerland) with optical density readings taken every 10 min. For the detection of bioluminescent signals, the cultures to black 96-well flat clear bottom microplates (αPLUS, WP96-48CMED, China) and signal was collected at 10-min intervals using the Tecan Spark reader.
Swarming motility assays
Swarming motility assays were conducted as described before79 with specific modifications. The swarming plates were composed of M8 medium, 0.5% agar, 100 µM CaCl2, 2 mM MgSO4, and 0.4% glucose. Each plate was centrally inoculated with 2 µL of bacterial suspension in LB broth, with the culture’s OD600 adjusted to 0.2. Plates were then incubated at 37 °C for overnight. Swarming proficiency was quantified by measuring the diameter of the minimal bounding circle enclosing the swarming area on plates. The average diameter was determined from three independent experiments conducted under standardized conditions.
Pyocyanin concentration quantification
Pyocyanin quantification was conducted as previously described80. Bacteria were cultured overnight in LB broth and adjusted to an OD600 of 0.2. Subsequently, 50 µL of the standardized culture was inoculated into 5 mL LB broth and incubated at 37 °C with shaking at 250 rpm for 24 hours. Following incubation, cultures were centrifuged to obtain cell-free supernatants, which were extracted with chloroform. The chloroform layer was acidified with 0.2 N HCl, resulting in the formation of a pink aqueous phase. The absorbance of this pink layer was measured at 520 nm using a Tecan Spark microplate reader (Tecan, Switzerland).
Determination of minimum inhibitory concentration of Ga(NO3)3
Minimum inhibitory concentrations (MICs) of Ga(NO3)3 were determined using the broth microdilution assay in 96-well polystyrene plates (NEST Biotechnology, 701011, China) as previously described81. Bacterial suspensions, adjusted to an OD600 of 0.0015, were inoculated into Mueller–Hinton broth (Oxoid, CM0405, USA) containing Ga(NO3)3 at concentrations ranging from 0 to 2560 μM. The plates were incubated at 37 °C for 24 h, followed by OD600 measurement using Tecan Spark.
Gallium time-killing assay
For gallium killing assays, bacterial cultures (OD600 = 0.5–0.7) were diluted to an OD600 of 0.003 in LB broth and incubated at 37 °C, with or without Ga(NO3)3 supplementation. An aliquot was plated to determine the colony-forming units (CFU) at Time 0, prior to the addition of Ga(NO3)3. At specified time points, aliquots were taken and washed with sterile PBS. The cells were then serially diluted and plated on LB agar to quantify the surviving bacteria.
Intracellular gallium and iron profiling
The methodology for assessing gallium levels in PAO1 strains, including PAO1ΔfprB and complemented strain, was based on the previously established protocol70. Bacterial cultures (OD600 = 0.003) were grown in 6 mL of LB broth, either with or without 10 µM Ga(NO3)3 for 16 h. Cells were collected and washed twice using 50 mM HEPES buffer (pH 7.2). The samples were resuspended in 50 mM HEPES buffer and adjusted to an OD600 of 0.6, followed by centrifugation at 8000 × g for 5 min to pellet the cells. Lysis was performed using 70% nitric acid at 100 °C for 1 h. Sample volumes were then normalized to 2 mL and determined by inductively coupled plasma mass spectrometry with a NexION 300× instrument (PerkinElmer, USA). To detect intracellular iron, bacterial cultures (20 mL, initial OD600 = 0.05) were cultivated with or without Ga(NO3)3 at concentrations of 12.5 and 50 µM, and harvested after 16 h of incubation. These cultures were then sonicated at 200 W for 1 min using the SCIENTZ-IID ultrasonic homogenizer (Scientz, China). Subsequently, cell lysates were prepared for intracellular iron content analysis, employing a previously established method with slight modifications82. The Total Iron Colorimetric Assay Kit (E-BC-K772-M, Elabscience, China) was used to measure the total iron content in bacteria, and the Ferrous Iron Colorimetric Assay Kit (E-BC-K773-M, Elabscience, China) was utilized to determine the levels of intracellular ferrous iron (Fe2+) through spectrophotometric measurements at 593 nm using a microplate reader (Tecan Spark, Switzerland).
Quantification of intracellular ROS and H2O2 by flow cytometry
Carboxy-H2DCFDA and PeroxyOrange-1 (Thermo Fisher Scientific, USA) fluorescent probes were employed for detection of total intracellular reactive oxygen species (ROS) and hydrogen peroxide (H2O2), respectively. Upon oxidation, these probes generate fluorescent signals that can be quantified by flow cytometry, thereby indicating the extent of oxidative stress within cells83. Bacterial cultures (OD600 = 0.5–0.7) were diluted to an OD600 of 0.01 in LB broth, either in the absence or presence of Ga(NO3)3, and incubated for 4 h at 37 °C. Following this incubation, Carboxy-H2DCFDA (final concentration of 10 μM) or PeroxyOrange-1 (final concentration of 5 μM) was added to the growth medium, and the cultures were further incubated in the dark for 2 h at 37 °C with shaking at 250 rpm. An unstained control sample was included to assess the background fluorescence. For each sample, a total of 100,000 ungated events were recorded for each sample. The NovoCyte Advanteon Flow Cytometer (Agilent, USA) was used to measure carboxy-H2DCFDA fluorescence, while the FACSAria II Flow Cytometer (BD Biosciences, USA) was employed for Peroxy Orange-1 fluorescence detection. Data analysis was conducted utilizing FlowJo software version 10 (BD Biosciences).
Quantification of nitric oxide (NO) in bacterial supernatants
NO levels in bacterial supernatants were measured using 2,3-diaminonaphthalene (DAN, Aladdin, D110922, China), a fluorometric assay targeting nitrite (NO₂−) as the stable end-product of NO metabolism84. Bacterial cultures grown to mid-log phase (OD600 = 0.5–0.7) under experimental conditions were centrifuged at 10000 × g for 5 min at 4 °C, and resulting supernatants were filtered through 0.22 μm membranes to remove cells. To quantify nitrite, 150 μL of supernatant was mixed with 75 μL of DAN solution (158 μM in 0.62 N HCl) in a 96-well black microplate, incubated in the dark for 30 min at 37 °C, and reacted further with 10 μL of 3.0 N NaOH to stabilize fluorescence. Fluorescence intensity was measured at excitation/emission wavelengths of 365/450 nm using a Tecan Spark microplate reader (Tecan, Switzerland). A sodium nitrite standard curve (0–10 μM) was used for quantification, and non-treated medium served as the blank control. Data were expressed by dividing measured fluorescence intensity by the corresponding culture OD600, yielding normalized NO quantitation results.
NADP+ and NADPH detection
Total oxidized and reduced nicotinamide adenine dinucleotide phosphates (NADP+ and NADPH) were quantified using the NADP/NADPH-Glo™ Assay (Promega, G9081, UK) according to the manufacturer’s protocol. Bacterial cultures were diluted to an OD600 of 0.01 and treated with or without Ga(NO3)3 for 4 h at 37 °C with shaking at 250 rpm. The bacteria were harvested by centrifugation at 7000 × g for 2 min and washed with distilled water at 4 °C. To ensure complete cell lysis, an equal volume of 0.2 N NaOH containing 1% DTAB (Sangon, A600431, China) was added. Following the manufacturer’s guidelines, NADP/NADPH-Glo™ Detection Reagent was introduced, and luminescence was measured using a Tecan Spark microplate reader.
Protein purification of FprB
The fprB coding sequence was cloned into the pGood_6p vector and transformed into E. coli Rosetta (DE3) cells, allowing expression of a GST-tagged protein. Single colony was picked and cultured overnight in LB with 100 µg/mL ampicillin at 37 °C and 220 rpm, then diluted 1:100 into 200 mL of LB broth and grown to an OD600 of 0.8. Protein expression was induced with 1 mM IPTG, and cultures were incubated overnight at 16 °C with shaking 220 rpm. Cells were harvested by centrifugation, resuspended in buffer (20 mM Tris-HCl, 150 mM NaCl, pH 8.0), and lysed by sonication. The lysate was centrifuged, and the supernatant was incubated with Glutathione Sepharose 4B (Cytiva, 17075605, USA) beads for 4 h at 4 °C. After washing with buffer (20 mM Tris-HCl, 1 M NaCl, pH 8.0), protein was eluted with buffer (20 mM Tris-HCl, 150 mM NaCl, 20 mM GSSH). The GST tag was removed using homemade P3c protease. FprB protein was further purified by size-exclusion chromatography and stored in buffer (20 mM Tris-HCl, 150 mM NaCl, pH 8.0, 20% glycerol).
FprB enzyme activity assay in vitro
FprB enzyme activity was assessed using an NADPH-dependent 2,6-dichlorophenolindophenol (DCPIP, FD32424, Biosynth, Switzerland) reduction assay as previously described48. Briefly, a reaction mixture was prepared in a clear 96-well microplate, containing 800 nM FprB, 400 μM NADPH, 800 µM DCPIP, 2% DMSO, and 50 mM Tris/HCl buffer (pH 8.0). Absorbance at 620 nm was measured using a Tecan Infinite M100PRO reader at 25 °C. Negative control wells contained the reaction mixture without FprB. Each condition was tested in triplicate.
RNA-Sequencing
P. aeruginosa strains (PAO1 and ΔfprB) grown in LB medium were harvested at an OD600 of 0.5–0.7. Total RNA was isolated using a RNAprep Pure Cell/Bacteria Kit (TIANGEN, DP430, China). Transcriptome sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina, USA) by Novogene Technology Co., Ltd. (Beijing, China). Raw data of fastq format were firstly processed through in-house perl scripts. The filtering criteria included: (1) removal of reads containing adapter sequences; (2) removal of reads containing undetermined bases (N bases); (3) removal of low-quality reads, defined as those in which bases with Phred quality scores ≤ 5 accounted for more than 50% of the read length. For read alignment, the reference genome and gene annotation files of P. aeruginosa PAO1 (accession number AE004091.2) were downloaded from NCBI. The clean reads were aligned to the reference genome using Bowtie2 (version 2.2.3)85 with default parameters. Differentially expressed genes between the two groups were analyzed using the DESeq2 package with default parameters (alpha = 0.05).
Distribution, evolution analysis and sequence comparison of FprB
Distribution analysis
Full chromosome sequences of 981 P. aeruginosa strains were downloaded from the NCBI Genome database (https://www.ncbi.nlm.nih.gov/genome, accessed [03/04/2024]). To detect the FprB in the P. aeruginosa genomes, the sequence of NP_253305.1 was used to align against with the peptide sequences derived from the genome sequences of the P. aeruginosa strains. BLASTP was used for the sequence alignment. The criterion for homology was set as ‘coverage * identity’ ≥ 0.6.
Gene collinearity analysis
The nucleotide sequence containing the fprB gene orthologs and flanking sequences (5-kb for the upstream and downstream from the center of gene), were retrieved from the analyzed P. aeruginosa genomes. PipMaker was used to make the collinearity analysis between each pair of the sequences (http://pipmaker.bx.psu.edu/pipmaker/)86.
Multiple sequence alignment and phylogenetic analysis
CLUSTALW was used for FprB protein sequence alignment (https://www.ebi.ac.uk/jdispatcher/msa/clustalo)87. The FprB multiple sequence alignment results were used for phylogenetic analysis using MEGA 7.0 with the Neigbor-Joining algorithm (https://www.megasoftware.net/)88.
Biofilm measurements
Biofilm quantification was evaluated in 96-well microtiter plates following established protocols89. P. aeruginosa cultures were grown to an OD600 of 0.6. The cultures were then diluted 1:200 in LB broth. Subsequently, 100 μL of the diluted cultures were added to each well, with triplicates for each condition. Following a 24-h incubation, the media supernatant was discarded, and the wells were washed with distilled water. Biofilms were then fixed with 95% methanol for 15 min and stained with 0.5% crystal violet for 10 min. The wells were rinsed with distilled water, and 75% ethanol was added to dissolve the dye. Absorbance was measured at 590 nm.
Mouse infections
The study protocol of animal experiments was approved by the Institutional Animal Care and Use Committee of Shenzhen University Medical School (Approval Number: IACUC-202400111). Female BALB/c mice (18–22 g, 6–8 weeks old) were purchased from Guangdong Medical Laboratory Animal Center and maintained under specific pathogen-free (SPF) conditions. The mice were housed under SPF conditions with a 12-h light/dark cycle, ambient temperature of 22 ± 2 °C, and 40–60% relative humidity. Prior to infection, mice were randomly assigned to either treatment or control groups. Mice were anesthetized and intranasally inoculated with 107 CFU in 40 µL of cell suspension. After 3 h, mice were treated intraperitoneally with either 50 µL of PBS or a 250 mM Ga(NO3)3 solution in water (approximately 160 mg/kg), in accordance with the dosages used in a previous study23. At 27 h post-infection (h.p.i.), mice were sacrificed, necropsied, and lungs were collected for bacteriological analysis. Lung tissue samples were homogenized, serially diluted in sterile PBS, and plated onto LB agar to quantify bacterial load.
Statistical analysis
GraphPad Prism (version 8.4.3) and R (version 4.3.1) were used for all statistical analyses. The experiments were conducted in at least triplicate to ensure biological reproducibility. Reported values represent the mean and standard deviation across the three replicates. Statistical significance between groups was determined by Two-way analysis of variance and the Turkey’s multiple comparisons test at a p-value threshold of 0.05. The illustrative diagrams were created using Adobe Illustrator 2021 and BioRender (https://www.biorender.com/).
Materials availability
The vector ptet-lux-Tn7T-Gm was deposited at Addgene (https://www.addgene.org/226434/). The pooled CRISPRi sgRNA libraries (set 1 and set 2) for P. aeruginosa PA14 were deposited at Addgene (https://www.addgene.org/pooled-library/liu-crispri-p-aeruginosa/). All other materials supporting the findings of this study are available from the corresponding author, Xue Liu (liuxuescience@gmail.com), upon request.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequencing data supporting this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession PRJNA1063516. The dataset spans SRA accessions SRR27547817-SRR27547879 and SRR32432725-SRR32432777, with corresponding BioSample accession (SAMN39398487-SAMN39398491, SAMN46956771-SAMN46956772) available through the NCBI BioSample database. Other data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
Code availability
Custom scripts used in this study for sgRNA abundance analysis, data quality assessment, and differential gene expression analysis are publicly available on Zenodo at https://doi.org/10.5281/zenodo.15179309.
References
Shi, Y. et al. The opportunistic pathogen Pseudomonas aeruginosa exploits bacterial biotin synthesis pathway to benefit its infectivity. PLoS Pathog. 19, e1011110 (2023).
Rossi, E. et al. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 19, 331–342 (2021).
Martínez-Solano, L., Macia, M. D., Fajardo, A., Oliver, A. & Martinez, J. L. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin. Infect. Dis. 47, 1526–1533 (2008).
Qin, S. et al. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target Ther. 7, 199 (2022).
Miller, W. R. & Arias, C. A. ESKAPE pathogens: antimicrobial resistance, epidemiology, clinical impact and therapeutics. Nat. Rev. Microbiol. 22, 598–616 (2024).
Lee, S. A. et al. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 112, 5189–5194 (2015).
Gallagher, L. A., Shendure, J. & Manoil, C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio 2, e00315–00310 (2011).
Cain, A. K. et al. A decade of advances in transposon-insertion sequencing. Nat. Rev. Genet. 21, 526–540 (2020).
de Bakker, V., Liu, X., Bravo, A. M. & Veening, J. W. CRISPRi-seq for genome-wide fitness quantification in bacteria. Nat. Protoc. 17, 252–281 (2022).
Peters, J. M. et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165, 1493–1506 (2016).
Bosch, B. et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 184, 4579–4592.e4524 (2021).
Hawkins, J. S. et al. Mismatch-CRISPRi reveals the Co-varying expression-fitness relationships of essential genes in Escherichia coli and Bacillus subtilis. Cell Syst. 11, 523–535.e529 (2020).
Liu, X. et al. Exploration of bacterial bottlenecks and Streptococcus pneumoniae pathogenesis by CRISPRi-Seq. Cell Host Microbe 29, 107–120 e106 (2021).
Liu, X. et al. Genome-wide CRISPRi screens for high-throughput fitness quantification and identification of determinants for dalbavancin susceptibility in Staphylococcus aureus. mSystems 9, e0128923 (2024).
Jiang, W., Oikonomou, P. & Tavazoie, S. Comprehensive genome-wide perturbations via CRISPR adaptation reveal complex genetics of antibiotic sensitivity. Cell 180, 1002–1017.e1031 (2020).
Wang, T. et al. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nat. Commun. 9, 2475 (2018).
Zhu, Q. et al. Construction and application of the conditionally essential gene knockdown library in Klebsiella pneumoniae to screen potential antimicrobial targets and virulence genes via Mobile-CRISPRi-seq. Appl. Environ. Microbiol. 89, e0095623 (2023).
Peters, J. M. et al. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat. Microbiol 4, 244–250 (2019).
Yu, M. A. et al. Investigating Pseudomonas aeruginosa gene function during pathogenesis using mobile-CRISPRi. Methods Mol. Biol. 2721, 13–32 (2024).
Leyland-Jones, B. Treating cancer-related hypercalcemia with gallium nitrate. J. Support. Oncol. 2, 509–516 (2004).
Chatterjee, M. et al. Antibiotic resistance in Pseudomonas aeruginosa and alternative therapeutic options. Int. J. Med. Microbiol. 306, 48–58 (2016).
Kaneko, Y., Thoendel, M., Olakanmi, O., Britigan, B. E. & Singh, P. K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Invest. 117, 877–888 (2007).
Goss, C. H. et al. Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci. Transl. Med. 10, eaat7520 (2018).
Reig, S., Le Gouellec, A. & Bleves, S. What is new in the anti-pseudomonas aeruginosa clinical development pipeline since the 2017 WHO alert? Front Cell Infect. Microbiol 12, 909731 (2022).
Chitambar, C. R. & Narasimhan, J. Targeting iron-dependent DNA synthesis with gallium and transferrin-gallium. Pathobiology 59, 3–10 (1991).
García-Contreras, R. et al. Isolation and characterization of gallium resistant Pseudomonas aeruginosa mutants. Int. J. Med. Microbiol. 303, 574–582 (2013).
Liu, X. et al. A conserved antigen induces respiratory Th17-mediated broad serotype protection against pneumococcal superinfection. Cell Host Microbe 32, 304–314.e308 (2024).
McMackin, E. A. W. et al. Cautionary notes on the use of arabinose- and rhamnose-inducible expression vectors in Pseudomonas aeruginosa. J. Bacteriol. 203, e0022421 (2021).
Banta, A. B., Ward, R. D., Tran, J. S., Bacon, E. E. & Peters, J. M. Programmable gene knockdown in diverse bacteria using mobile-CRISPRi. Curr. Protoc. Microbiol. 59, e130 (2020).
Cui, L. et al. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat. Commun. 9, 1912 (2018).
Qu, J. et al. Modulating Pathogenesis with Mobile-CRISPRi. J. Bacteriol. 201, e00304–e00319 (2019).
Wang, K. et al. Overexpression of phzM contributes to much more production of pyocyanin converted from phenazine-1-carboxylic acid in the absence of RpoS in Pseudomonas aeruginosa. Arch. Microbiol. 202, 1507–1515 (2020).
Rattanachak, N. et al. High-throughp ut transcriptomic profiling reveals the inhibitory effect of hydroquinine on virulence factors in Pseudomonas aeruginosa. Antibiotics 11, 1436 (2022).
Stover, C. K. et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 (2000).
Pan, J., Song, F. & Ren, D. Controlling persister cells of Pseudomonas aeruginosa PDO300 by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. Bioorg. Med. Chem. Lett. 23, 4648–4651 (2013).
Margolin, W. FtsZ and the division of prokaryotic cells and organelles. Nat. Rev. Mol. Cell Biol. 6, 862–871 (2005).
Poulsen, B. E. et al. Defining the core essential genome of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 116, 10072–10080 (2019).
Bravo, A. M., Typas, A. & Veening, J. W. 2FAST2Q: a general-purpose sequence search and counting program for FASTQ files. PeerJ 10, e14041 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Liberati, N. T. et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA 103, 2833–2838 (2006).
Skurnik, D. et al. A comprehensive analysis of in vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog. 9, e1003582 (2013).
Whitney, J. C. et al. An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163, 607–619 (2015).
Kohanski, M. A., Dwyer, D. J. & Collins, J. J. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8, 423–435 (2010).
Guo, Y., Li, W., Li, H. & Xia, W. Identification and characterization of a metalloprotein involved in gallium internalization in Pseudomonas aeruginosa. ACS Infect. Dis. 5, 1693–1697 (2019).
Yeom, J., Jeon, C. O., Madsen, E. L. & Park, W. Ferredoxin-NADP+ reductase from Pseudomonas putida functions as a ferric reductase. J. Bacteriol. 191, 1472–1479 (2009).
Romsang, A., Duang-Nkern, J., Wirathorn, W., Vattanaviboon, P. & Mongkolsuk, S. Pseudomonas aeruginosa IscR-regulated ferredoxin NADP(+) reductase gene (fprB) functions in iron-sulfur cluster biogenesis and multiple stress response. PLoS ONE 10, e0134374 (2015).
Carrillo, N. & Ceccarelli, E. A. Open questions in ferredoxin-NADP+ reductase catalytic mechanism. Eur. J. Biochem. 270, 1900–1915 (2003).
Martinez-Julvez, M. et al. Identification of inhibitors targeting ferredoxin-NADP(+) reductase from the Xanthomonas citri subsp. Citri Phytopathogenic Bacteria. Molecules 23, 29 (2017).
Monchietti, P., López Rivero, A. S., Ceccarelli, E. A. & Catalano-Dupuy, D. L. A new catalytic mechanism of bacterial ferredoxin-NADP(+) reductases due to a particular NADP(+) binding mode. Protein Sci. 30, 2106–2120 (2021).
Tondo, M. L. et al. Crystal structure of the FAD-containing ferredoxin-NADP+ reductase from the plant pathogen Xanthomonas axonopodis pv. citri. Biomed. Res. Int. 2013, 906572 (2013).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Choi, S. R., Britigan, B. E. & Narayanasamy, P. Dual inhibition of Klebsiella pneumoniae and Pseudomonas aeruginosa iron metabolism using gallium porphyrin and gallium nitrate. ACS Infect. Dis. 5, 1559–1569 (2019).
Belenky, P. et al. Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep. 13, 968–980 (2015).
Vilchèze, C., Hartman, T., Weinrick, B. & Jacobs, W. R. Jr. Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat. Commun. 4, 1881 (2013).
Singh, R., Mailloux, R. J., Puiseux-Dao, S. & Appanna, V. D. Oxidative stress evokes a metabolic adaptation that favors increased NADPH synthesis and decreased NADH production in Pseudomonas fluorescens. J. Bacteriol. 189, 6665–6675 (2007).
Hong, Y., Zeng, J., Wang, X., Drlica, K. & Zhao, X. Post-stress bacterial cell death mediated by reactive oxygen species. Proc. Natl. Acad. Sci. USA 116, 10064–10071 (2019).
Corpas, F. J., Río, L. A. D. & Palma, J. M. Impact of nitric oxide (NO) on the ROS metabolism of peroxisomes. Plants (Basel) 8, 37 (2019).
Zhang, L., Wüst, A., Prasser, B., Müller, C. & Einsle, O. Functional assembly of nitrous oxide reductase provides insights into copper site maturation. Proc. Natl. Acad. Sci. USA 116, 12822–12827 (2019).
Kordes, A. et al. Genetically diverse Pseudomonas aeruginosa populations display similar transcriptomic profiles in a cystic fibrosis explanted lung. Nat. Commun. 10, 3397 (2019).
Salunkhe, P., Töpfer, T., Buer, J. & Tümmler, B. Genome-wide transcriptional profiling of the steady-state response of Pseudomonas aeruginosa to hydrogen peroxide. J. Bacteriol. 187, 2565–2572 (2005).
Bjarnsholt, T. et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 44, 547–558 (2009).
Qi, LeiS. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Bikard, D. et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437 (2013).
Liu, X. et al. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 13, 931 (2017).
Lee, H. H. et al. Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol 4, 1105–1113 (2019).
Poulsen, B. E., Clatworthy, A. E. & Hung, D. T. The use of Tn-Seq and the FiTnEss analysis to define the core essential genome of Pseudomonas aeruginosa. Methods Mol. Biol. 2377, 179–197 (2022).
Zemke, A. C., Madison, C. J., Kasturiarachi, N., Pearce, L. L. & Peterson, J. Antimicrobial synergism toward Pseudomonas aeruginosa by Gallium(III) and inorganic nitrite. Front. Microbiol. 11, 2113 (2020).
Li, L. et al. Superior antibacterial activity of gallium based liquid metals due to Ga(3+) induced intracellular ROS generation. J. Mater. Chem. B 9, 85–93 (2021).
Guo, M. et al. Gallium nitrate enhances antimicrobial activity of colistin against Klebsiella pneumoniae by inducing reactive oxygen species accumulation. Microbiol. Spectr. 11, e0033423 (2023).
Tovar-García, A. et al. Characterization of gallium resistance induced in a Pseudomonas aeruginosa cystic fibrosis isolate. Arch. Microbiol. 202, 617–622 (2020).
Wang, Y. et al. Comparative proteomics unveils the bacteriostatic mechanisms of Ga(III) on the regulation of metabolic pathways in Pseudomonas aeruginosa. J. Proteom. 289, 105011 (2023).
García-Contreras, R. et al. Gallium induces the production of virulence factors in Pseudomonas aeruginosa. Pathog. Dis. 70, 95–98 (2014).
Wang, P. et al. Development of an efficient conjugation-based genetic manipulation system for Pseudoalteromonas. Micro. Cell Fact. 14, 11 (2015).
Sorg, R. A., Gallay, C., Van Maele, L., Sirard, J. C. & Veening, J. W. Synthetic gene-regulatory networks in the opportunistic human pathogen Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 117, 27608–27619 (2020).
Bernard, P. Positive selection of recombinant DNA by CcdB. Biotechniques 21, 320–323 (1996).
Chen, W. et al. CRISPR/Cas9-based genome editing in Pseudomonas aeruginosa and cytidine deaminase-mediated base editing in Pseudomonas Species. iScience 6, 222–231 (2018).
Bernard, P. & Couturier, M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 226, 735–745 (1992).
Wurtzel, O. et al. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 8, e1002945 (2012).
Simanek, K. A. et al. Quorum-sensing synthase mutations re-calibrate autoinducer concentrations in clinical isolates of Pseudomonas aeruginosa to enhance pathogenesis. Nat. Commun. 14, 7986 (2023).
Al-Maddboly, L. A. et al. Anti-biofilm and anti-quorum sensing activities of galloylquinic acid against clinical isolates of multidrug-resistant Pseudomonas aeruginosa in open wound infection: in vitro and in vivo efficacy studies. BMC Microbiol. 25, 206 (2025).
Dousa, K. M. et al. Synergistic effects of sulopenem in combination with cefuroxime or durlobactam against Mycobacterium abscessus. mBio 15, e0060924 (2024).
Zhong, Z. X. et al. Natural flavonoids disrupt bacterial iron homeostasis to potentiate colistin efficacy. Sci. Adv. 9, eadg4205 (2023).
Hong, Y., Li, L., Luan, G., Drlica, K. & Zhao, X. Contribution of reactive oxygen species to thymineless death in Escherichia coli. Nat. Microbiol. 2, 1667–1675 (2017).
Nussler, A. K., Glanemann, M., Schirmeier, A., Liu, L. & Nüssler, N. C. Fluorometric measurement of nitrite/nitrate by 2,3-diaminonaphthalene. Nat. Protoc. 1, 2223–2226 (2006).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Schwartz, S. et al. PipMaker–a web server for aligning two genomic DNA sequences. Genome Res. 10, 577–586 (2000).
Madeira, F. et al. The EMBL-EBi job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525 (2024).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Haney, E. F., Trimble, M. J. & Hancock, R. E. W. Microtiter plate assays to assess antibiofilm activity against bacteria. Nat. Protoc. 16, 2615–2632 (2021).
Acknowledgements
We thank Prof. Sisi Li, Dr. Xuan Du, and Dr. Fan Zou from Shenzhen University for helpful discussions and technical support, Dr. Xueling Lu from Shenzhen Children’s Hospital for help in data analysis. Furthermore, we thank Prof. Xilin Zhao from Xiamen University and Pengbo Cao from Westlake University for insightful discussions. This work was supported by the National Key Research and Development Program of China (2023YFD 10800100) to X.L. and Y.Z., National Natural Science Foundation of China (82200047) to Y.Z. and (82270012) to X.L., the Science and Technology Project of Shenzhen (JCYJ20220818095602006) to X.L., Shenzhen Science and Technology Program (KQTD20200909113758004) to L.Yang, Pearl River Talent Project of Guangdong Province (2021QN02Y283) to X.L., Shenzhen University 2035 Program for Excellent Research (86901-00000216) to X.L., the Program for Youzuzhikeyan of Shenzhen University to X.L. and L.Ye. J.W.V. and X.L. were supported by NCCR AntiResist grant 51NF40_180541 from the Swiss National Science Foundation. A.K. was supported by the Polish National Science Centre funds no. 2021/43/D/NZ2/02151.
Author information
Authors and Affiliations
Contributions
Y.Z. and X.L. designed the research. Y.Z., T.T.Z., X.X., J.Z.O., A.M.R., W.H.S., Y.J.L., and Y.L.L. performed the experiments and analyzed the data. A.K., V.D.B., L.Y. (Liang Yang), L.Y. (Liang Ye), N.J., J.W.V., Y.J.W., and X.L. contributed to data analysis. Y.Z. and T.T.Z. analyzed the sequencing data. Y.J.W. performed the evolutionary conservation analysis. Y.Z. and X.L. wrote the manuscript. X.L. supervised the project. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
Yu Zhang and Xue Liu have filed a patent application (ZL202311534857.7) on aspects of the reported findings. Jan-Willem Veening is a scientific advisory board member at i-Seq Biotechnology. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mark Broenstrup, Silvia Cardon, Rodolfo García-Contreras, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Zhang, T., Xiao, X. et al. CRISPRi screen identifies FprB as a synergistic target for gallium therapy in Pseudomonas aeruginosa. Nat Commun 16, 5870 (2025). https://doi.org/10.1038/s41467-025-61208-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61208-z
This article is cited by
-
CRISPRi-based phenotypic screening in bacteria: technologies, applications, and prospects
Biotechnology and Bioprocess Engineering (2025)
